 Paola Lisotto1
Paola Lisotto1 Natacha Couto1,2
Natacha Couto1,2 Sigrid Rosema1Mariëtte Lokate1
Sigrid Rosema1Mariëtte Lokate1 Xuewei Zhou1
Xuewei Zhou1 Erik Bathoorn1
Erik Bathoorn1 Hermie J. M. Harmsen1
Hermie J. M. Harmsen1 Alexander W. Friedrich1
Alexander W. Friedrich1 John W. A. Rossen1,3,4Monika A. Chlebowicz-Fliss1*
John W. A. Rossen1,3,4Monika A. Chlebowicz-Fliss1*- 1Department of Medical Microbiology and Infection Prevention, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 2The Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, United Kingdom
- 3Department of Pathology, University of Utah School of Medicine, Salt Lake City, UT, United States
- 4IDbyDNA Inc., Salt Lake City, UT, United States
Background: Vancomycin-resistant Enterococcus faecium (VREfm) is a successful nosocomial pathogen. The current molecular method recommended in the Netherlands for VREfm typing is based on core genome Multilocus sequence typing (cgMLST), however, the rapid emergence of specific VREfm lineages challenges distinguishing outbreak isolates solely based on their core genome. Here, we explored if a detailed molecular characterisation of mobile genetic elements (MGEs) and accessory genes could support and expand the current molecular typing of VREfm isolates sharing the same genetic background, enhancing the discriminatory power of the analysis.
Materials/Methods: The genomes of 39 VREfm and three vancomycin-susceptible E. faecium (VSEfm) isolates belonging to ST117/CT24, as assessed by cgMLST, were retrospectively analysed. The isolates were collected from patients and environmental samples from 2011 to 2017, and their genomes were analysed using short-read sequencing. Pangenome analysis was performed on de novo assemblies, which were also screened for known predicted virulence factors, antimicrobial resistance genes, bacteriocins, and prophages. Two representative isolates were also sequenced using long-read sequencing, which allowed a detailed analysis of their plasmid content.
Results: The cgMLST analysis showed that the isolates were closely related, with a minimal allelic difference of 10 between each cluster’s closest related isolates. The vanB-carrying transposon Tn1549 was present in all VREfm isolates. However, in our data, we observed independent acquisitions of this transposon. The pangenome analysis revealed differences in the accessory genes related to prophages and bacteriocins content, whilst a similar profile was observed for known predicted virulence and resistance genes.
Conclusion: In the case of closely related isolates sharing a similar genetic background, a detailed analysis of MGEs and the integration point of the vanB-carrying transposon allow to increase the discriminatory power compared to the use of cgMLST alone. Thus, enabling the identification of epidemiological links amongst hospitalised patients.
Introduction
Enterococci are Gram-positive cocci commonly present in the human and animal gastrointestinal tract. Amongst them, Enterococcus faecium has been increasingly recognised as one of the leading causes of healthcare-associated infections (HAIs) due to its intrinsic and acquired resistance to several antibiotics, such as ampicillin, gentamicin, and vancomycin (Arias and Murray, 2012; García-Solache and Rice, 2019). Multidrug resistant E. faecium strains represent a threat for patients with a compromised immune system making them particularly susceptible to infections. Hence, in a joint effort to fight nosocomial infections, health agencies included E. faecium in the list of ESKAPE pathogens for which is urgently required the development of novel therapeutics drugs (Rice, 2008). Vancomycin-resistant E. faecium (VREfm) can arise from HA-vancomycin-susceptible E. faecium (VSEfm) by acquiring the vanA and/or vanB operons present in enteric anaerobic bacteria (Stinear et al., 2001; Donskey, 2004; Domingo et al., 2005; Howden et al., 2013). Our hospital mainly encounters the vanB positive VREfm (Zhou et al., 2018), where the vanB gene is located on the conjugative transposon Tn1549, usually integrated into the bacterial chromosome (Bender et al., 2016; Freitas et al., 2016). On the contrary, in vanA positive E. faecium, the transposon is generally associated with Tn1546 located on plasmids (Novais et al., 2008).
The high genome plasticity that characterises hospital-associated E. faecium, also referred to as clade A1, is an important factor contributing to their acquisition of mobile genetic elements (MGEs; Lebreton et al., 2013). This population not only is resistant to multiple antibiotics, but it also holds a repertoire of virulence factors that, together with the increasing tolerance towards disinfectants and higher tenacity to survive on environmental surfaces, makes VREfm a successful nosocomial pathogen (Werner et al., 2013; Gao et al., 2018; Pidot et al., 2018; García-Solache and Rice, 2019; Gorrie et al., 2019; de Maat et al., 2020).
The application of next generation sequencing (NGS) in clinical microbiology and infection prevention has proved successful for outbreak investigation (Deurenberg et al., 2017). In this scenario, whole-genome sequencing (WGS) provides high resolution regarding the relatedness of the isolates. It allows for a detailed analysis of the MGEs content that can point to a possible epidemiological link amongst the patients (Deurenberg et al., 2017; Raven et al., 2017; Pinholt et al., 2019). However powerful, its discriminatory power is not fully deployed by core genome Multilocus sequence typing (cgMLST), currently the recommended molecular typing method of VREfm in the Netherlands. This method is based on a gene-by-gene approach where a well-established set of core genes, 1,423 for E. faecium, are compared amongst the isolates and differences are then translated into allelic distances (de Been et al., 2015). By cgMLST, very closely related genomes are then grouped to form a Complex Type (CT). Because this method is based on comparing a well-established set of core genes, it only allows for clonal spread investigation. In contrast, horizontal gene transfer of MGEs, such as those carrying the Van operon, could be herewith missed (Raven et al., 2017). Therefore, considering the high genomic plasticity characterising E. faecium, additional investigation of accessory genes and plasmids could provide the required discrimination (Schürch et al., 2018).
Here, we applied WGS to characterise and compare vanB-carrying VREfm isolates belonging to ST117/CT24 circulating in the University Medical Center Groningen (UMCG) between 2011 and 2017, causing two major outbreaks. We aim to analyse the genetic relatedness amongst isolates involved in the same outbreaks and, by including VSEfm and non-outbreak VREfm isolates of the same genetic background, investigate the relative contribution of the MGEs in distinguishing outbreaks from each other.
Materials and Methods
Study Settings and Bacterial Isolates
This study retrospectively analysed 39 VREfm and three VSEfm strains isolated at UMCG between 2011 and 2017 solely belonging to the ST117/CT24-vanB clone as assessed by cgMLST typing. Upon admission in our hospital, rectal swab screening for VRE is performed as described before (Zhou et al., 2018) on: (i) patients who were admitted to a hospital abroad in the last year; (ii) patients transferred from another hospital in the Netherlands; (iii) patients admitted to the intensive care and haematology wards; (iv) children adopted from abroad. VRE positive patients are treated in contact isolation and, if nosocomial acquisition is suspected, screening of contact patients is performed. These measures are lifted if at least five negative rectal swabs are obtained. In general, suspected VRE colonies were confirmed by MALDI-TOF Mass Spectrometry (Bruker), and antibiotic susceptibility testing was conducted as part of the routine screening procedures by VITEK®2 using the AST card P-586 (bioMérieux). All first VRE isolates of individual cases were sequenced as described below. In the time span analysed in this study, two outbreaks caused by this specific lineage took place, one in 2014 and one in 2017.
From the 2014 outbreak, 12 isolates were included, of which 10 have been described previously by Zhou et al. (2018). Only three isolates caused infections, whilst the other nine were isolated during preventive screening procedures from rectal swabs. Twenty-one isolates were collected during the outbreak in 2017 from rectal swabs (16), faeces (1), and environmental samples (4). Six isolates that belonged to several sporadic VREfm carriages and/or infection cases between 2011 and 2015 have been retrospectively assessed by WGS, which has been implemented in our laboratory for outbreak investigation since 2014. Finally, three VSEfm isolates were collected in 2016 from pus, pleural fluid, and rectal swabs. Finally, the genome of a fully sequenced E. faecium strain from Spain (Tedim et al., 2017) belonging to ST117/CT24 was used as an external reference and was referred to here as E1 strain. In total, 43 isolates were included in this study.
WGS and Isolates Characterisation
Bacterial isolates were grown overnight on blood agar plates at 37°C. Genomic DNA was extracted using the Ultraclean Microbial DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, United States) according to the manufacturer’s instructions. DNA concentration and purity were measured by the Qubit dsDNA HS assay kit (Life Technologies, Carlsbad, CA, United States). Preparation of the DNA libraries was performed with the Nextera XT v2 kit (Illumina, San Diego, CA, United States) and sequenced in a MiSeq platform (Illumina). Trimming and de novo assemblies were performed in CLC Genomics Workbench v12 (QIAGEN, Hilden, Germany). Two isolates of interest, samples A12 and B11, and single representatives of the outbreaks in 2014 and 2017, were also sequenced using the MinION (Oxford Nanopore Technologies, Oxford, United Kingdom). For these two strains, base calling was performed using Albacore (v1.2.2). Data quality was analysed through Poretools (v0.6.0; Loman and Quinlan, 2014). Hybrid assemblies were performed using Unicycler (v0.4.1; Wick et al., 2017). Assembly statistics are provided in Supplementary Table 1.
Multilocus sequence typing Sequence Types (STs) and cgMLST Complex Types (CTs) were extracted from the assembled genomes using Ridom SeqSphere+ (v5.1.0; Ridom GmbH, Münster, Germany) and the E. faecium curated scheme published previously (de Been et al., 2015). The vanB-carrying transposons were identified by BLASTn comparisons of de novo and hybrid assemblies with the reference sequence of Tn1549 (GenBank AF192329.1). Detailed analysis of the integration points was performed by BLASTn comparison with the E1 strain using Artemis Comparison Tool (ACT; Carver et al., 2005) and Artemis (Carver et al., 2012). IS finder (Kichenaradja et al., 2010) was used to investigate the presence of insertion sequences in the vanB-carrying transposons.
The genomes were characterised in silico using the online tools VirulenceFinder (v2.0; Joensen et al., 2014) and ResFinder (v3.2; Zankari et al., 2012). Additionally, the genomes were investigated for the presence of bacteriocins using BAGEL4 (van Heel et al., 2018) and prophages using PHASTER (Arndt et al., 2016). The predicted prophage regions were manually extracted using Artemis (Carver et al., 2012), and multiple BLASTn analyses were performed to compare the isolates. Finally, BRIG (Alikhan et al., 2011) was used for data visualisation. Plasmid analysis was performed on the hybrid assemblies, first analysed with mlplasmids (Arredondo-Alonso et al., 2018) to predict plasmid- and chromosome-derived sequences. Afterwards, multiple BLASTn analyses were performed to compare the predicted plasmid sequences, and the results were visualised with the R package Circlize (Gu et al., 2014). Abricate (https://github.com/tseemann/abricate, version 1.0.1) was used to query (> 80% identity and > 60% coverage) the predicted plasmid sequences vs. a curated database of known relaxases proteins from Enterococcus (Clewell et al., 2014), PlasmidFinder database (Carattoli et al., 2014) was used to identify replication initiator proteins (RIPs). The pangenome analysis was performed using Roary (Page et al., 2015).
Results
In this study, WGS was applied to characterise and compare VREfm sharing the same genetic background (ST117/CT24) and isolated between 2011 and 2017 from hospitalised patients and environmental samples. We analysed the genetic relatedness amongst isolates, including VSEfm isolates, and characterised MGEs and accessory genes that could potentially distinguish them. Detailed information on the investigated isolates is presented in Table 1 and Supplementary Figure 1.
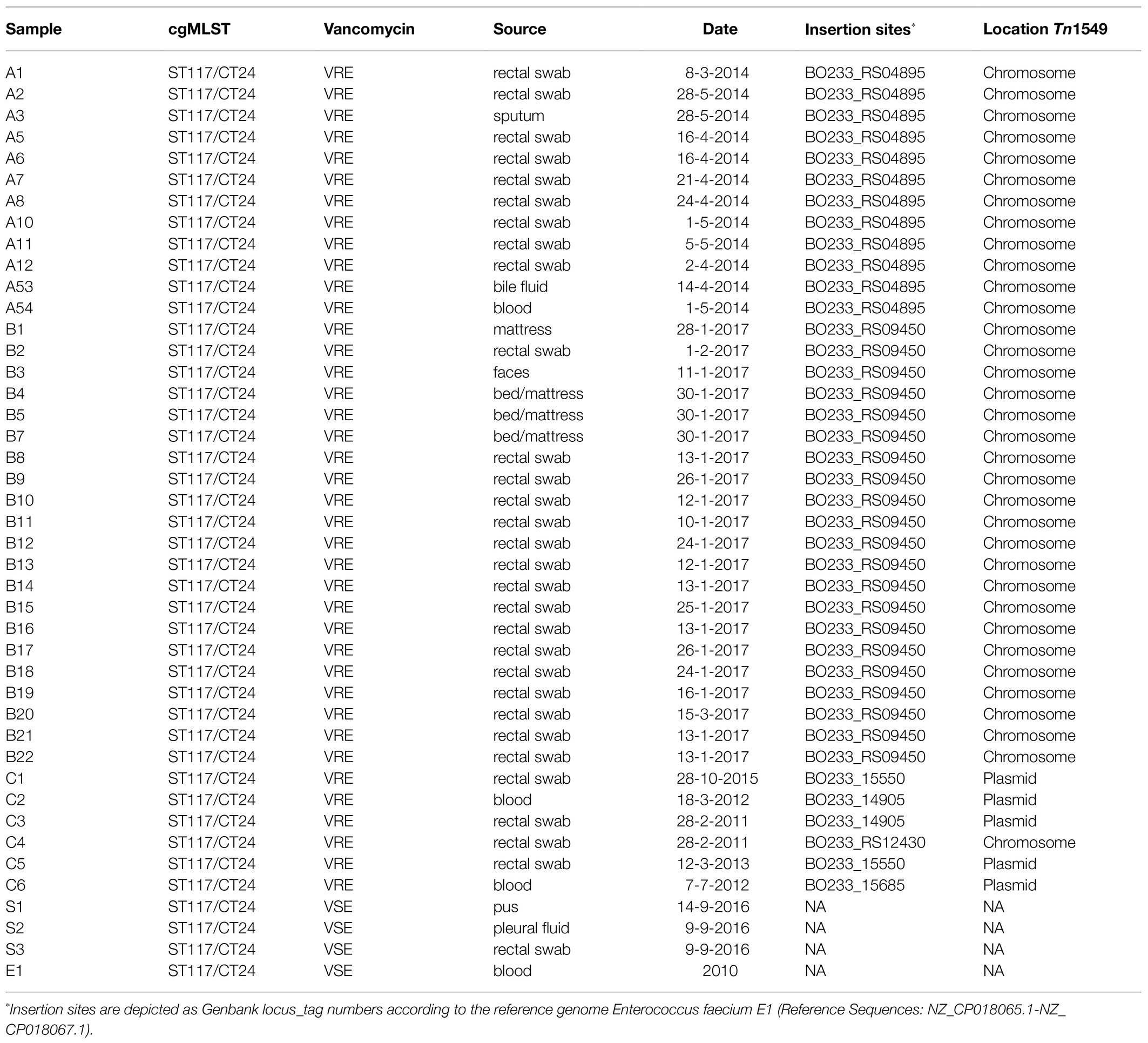
Table 1. Epidemiological and molecular data of the isolates included in this study.
Molecular Epidemiology of the Isolates
Core genome analysis showed that the isolates are closely related, with a minimal allelic difference lower than 10 between each cluster’ nearest isolates (Figure 1). The two major clusters correspond to outbreak events in 2014 and 2017. The former includes 10 samples previously described by Zhou et al. (2018) belonging to an outbreak (A) in April 2014. Two isolates (A53 and A54) that clustered separately were not included in the previous investigation. These were obtained from bile (A53) and blood samples (A54) of the same patient within 2weeks. Few allele differences separate this cluster from the sporadic cases collected from rectal swabs (n=4) or bloodstream infections (n=2) in different patients between 2011 and 2015. The outbreak in 2017 was presumably initiated by a patient transferred from an external hospital who was accidentally not screened for the presence of VREfm at admission nor during the stay in the previous hospital. Only after 10days, a rectal swab was collected from the patient from which a VREfm could be cultured. A subsequent screening identified 15 patients carrying an ST117/CT24 VREfm. Noteworthy, a few months before the outbreak in 2017, three VSE isolates were sequenced, two (S1 and S2) were isolated from infection sites and one (S3) from a rectal swab. These three VSE isolates turned out to be closely related to the outbreak cluster in 2017.
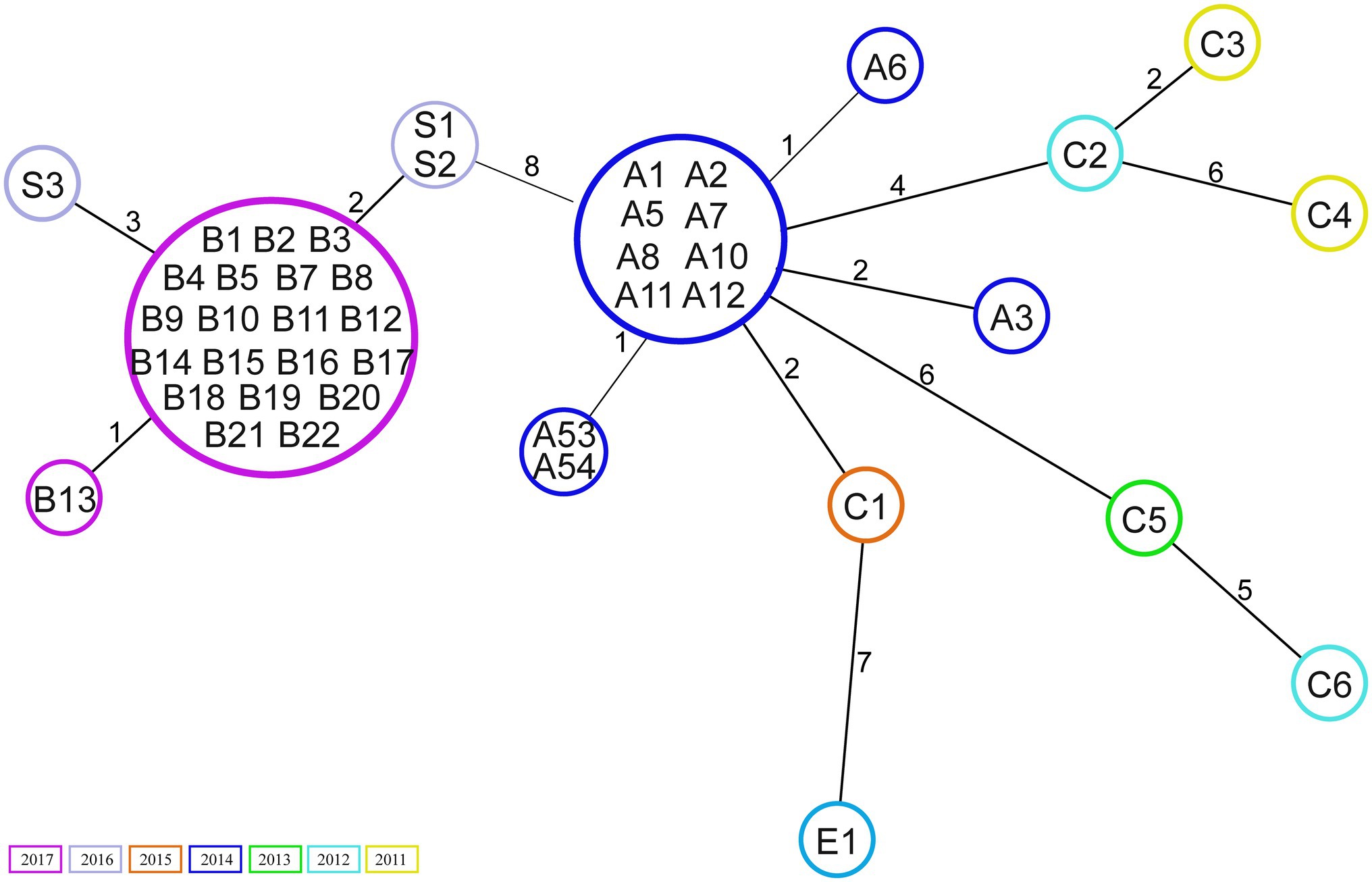
Figure 1. Minimum spanning tree based on core genome Multilocus sequence typing (cgMLST; 1,423 target genes). Numbers within the circle indicate the isolates. The numbers next to the lines correspond to allele differences between the isolates. Isolates are coloured by year of collection.
Characterisation of vanB-Carrying Transposon Tn1549
The vanB-carrying transposon was analysed in detail using Tn1549/Tn5382 (Genbank AF192329.1) as a reference that we referred to throughout the paragraph.
First, the vanB operon alone was investigated by single-nucleotide polymorphism (SNP) analysis, which showed 9–10 SNP-difference between the operons of the majority of the isolates and the reference. A unique pattern was found in the operons of isolates C5 and C6. Despite being isolated in different years, they shared the same 41 SNPs. Both the nucleotide and the amino acid alignments are shown in Supplementary Figure 2.
Next, the entire sequence of the vanB-carrying transposon Tn1549 was analysed. All isolates contained the transposon. However, its genomic position was different, as indicated in detail in Table 1. The transposons of the isolates involved in the two outbreaks were characterised by the integration into the chromosome. In contrast, those of the sporadic isolates were mainly linked to plasmids except for one case (C4) in which the transposon was integrated into the chromosome. The transposon of the 12 isolates from the 2014 outbreak, 10 of which previously described by Zhou et al. (2018) was located on the bacterial chromosome, integrated into the metal-dependent phosphoesterase gene (GenBank locus tag: BO233_RS04895). The overall sequence of this transposon had a similar structure as the previously described reference transposon with 99 SNP differences.
In the 21 isolates from the 2017 outbreak, the transposon was located on the bacterial chromosome, but it was integrated upstream of the putative permease gene (GenBank locus tag: BO233_RS09450) with 98 SNP differences compared to the transposon reference sequence.
In one of the sporadic isolates collected in 2011 (C4), the transposon was integrated into the chromosome in the proximity of the sufB gene (GenBank locus tag: BO233_RS12430) and differed 108 SNPs from the reference sequence.
The sporadic cases of VREfm that occurred between 2011 and 2015 belonged to three separated clusters and showed significant differences: (i) in two isolates, from 2015 (C1) and 2013 (C5), the transposon integration point was located in the plasmid DNA invertase Pin gene (GenBank locus taq: BO233_15550), and their transposons differed by 101 and 260 SNPs from the reference, respectively; (ii) in one single isolate from 2012 (C6), the transposon integrated into a peptidoglycan-binding protein gene (GenBank locus taq: BO233_15685) located on the plasmid with 260 SNPs; and (iii) the vanB transposon found in isolates C2 (2012) and C3 (2011) was located on another plasmid and integrated upstream of the gene coding for DNA helix-turn-helix (Genbank locus taq: BO233_14905). Comparing these transposons with the reference resulted in 103 and 76 SNP differences, respectively. No IS elements were identified in the vanB-transposon carried by the isolates.
Pangenome and Accessory Genome Analysis
The pangenome analysis (Supplementary Figure 1; Supplementary Table 2) revealed that 2,460 genes were present in all 43 isolates (core genes) whilst 1,433 were assigned to the accessory genome and were variably present amongst the isolates. The samples belonging to the 2017 outbreak had a different profile than the 2014 outbreak, with apparent dissimilarities in the presence/absence of accessory genes. These differences were further investigated, resulting in 362 and 216 unique genes present in 2017 and 2014 outbreak isolates, respectively. Despite half of the unique genes identified in 2017 outbreak strains encoded for hypothetical proteins, these isolates carried genes involved in mannose-specific metabolism. On the contrary, 2014 isolates had a lower proportion of genes with unknown function (n=73), and phage-related genes were identified in eight isolates (A5, A6, A7, A8, A10, A11, A53, and A54). A more scattered and unique pattern was observed in the single sporadic cases.
A high degree of similarity in virulence genes was observed amongst the isolates (Supplementary Figure 1). We detected in all isolates several genes encoding cell surface virulent factors involved in cell adhesion and attachment, such as acm, ecbA, efaAfm, scm, sgrA, and espfm; and pili gene cluster (PGC) proteins pilA and pilB. Amongst the secreted virulence factors that VREfm can produce, all the isolates were found to be positive for sagA, a major secreted antigen correlated with the binding of extracellular matrix proteins and cell growth. The hyl gene, a glycosyl hydrolase that belongs to the same class of secreted factors, was identified only in one of the sporadic cases isolated in 2013. Finally, in all the isolates, the gene encoding a putative transmembrane protein PTSclin, which phosphorylates and translocate specific carbohydrates across the cell membrane, was identified.
All VREfm carried genes associated with resistance to glycopeptides, trimethoprim (dfrG), macrolide [erm(B), mrs(C)], and aminoglycosides [aac(6')-Ii, aac(6')-aph(2''), ant(6)-Ia, aph(2'')-Ia, and aph(3')-III] (Supplementary Figure 1). These genes, except for VanHBX, were also detected in the vancomycin-susceptible isolates S1, S2, and S3. A gene conferring resistance to chloramphenicol (>99% of the amino acid sequence identical to cat, chloramphenicol O-acetyltransferase type A [EC:2.3.1.28]), was exclusively detected in the 2014 isolates and in the isolates C1, C5, and C6. Genes or chromosomal mutations associated with linezolid resistance were not detected in any of the isolates. Isolate A6 showed resistance to tigecyclin, but no gene associated with tetracycline resistance was found. Except for this isolate, the phenotypic antibiotic susceptibility patterns (Supplementary Table 3) align with the predicted resistome. Isolates C2, C3, and C6 were vanB-carrying VREfm with low-level resistance. All other VREfm were measured as vancomycin-resistant by the susceptibility tests.
The in silico analysis of the bacteriocin genes (Supplementary Figure 1) showed that enterocin A (GenBank: AAD29132.1) was detected in all isolates whilst three bacteriocins were differentially present in the investigated isolates. Lactococcin 972 (GenBank: TNX50477.1) was detected in the 2014 outbreak, C1 and C6 isolates, whilst two additional bacteriocins were present in the genome of 2017 outbreak strains and VSE isolates, namely enterocin P (GenBank: AF005726.1) and bacteriocin T8 (GenBank: DQ402539.1).
The prophage content of the isolates was investigated; a total of six prophages sequence were identified and used to generate a concatenated reference sequence (Figure 2). Phages 1, 2, and 5 were identified in all but two isolates (C5 and C6). A different profile was observed for phages 3 and 6: the isolates carrying phage 3 lacked phage 6 and vice versa. The former was missing in the C1, C5 and in seven isolates of the 2014 outbreak, which were instead carrying the latter; the epidemiological data analysis confirmed a possible connection between patients. Based on the bed occupancies, we observed that the patient infected with isolate A53 carried phages 3 and 6. The former was also found in 2014 outbreak isolates A1 and A12 obtained from different patients who most probably had contact. The latter phage was found in isolate A54 as well as in eight isolates of the 2014 outbreak cluster. Noteworthy isolate A54 was isolated 2weeks after A53 from the same patient, suggesting that phage 3 was lost over time. Finally, phage 4 was uniquely present in reference strain E1. Based on PHASTER analysis, the putative phages were shown to be related to phages of Enterococcus, Bacillus, and Listeria. However, when compared to the predicted references, the overall sequence homology was low. The highest score was observed for phage 3 that shared 17% DNA similarity with Enterococcus phage vB_EfaS_IME197 (Genbank NC_028671.2). The DNA similarity in the other phages was ranging between 3 and 11%. Detailed information on the presence of these phages and the PHASTER output is presented in Supplementary Table 4.
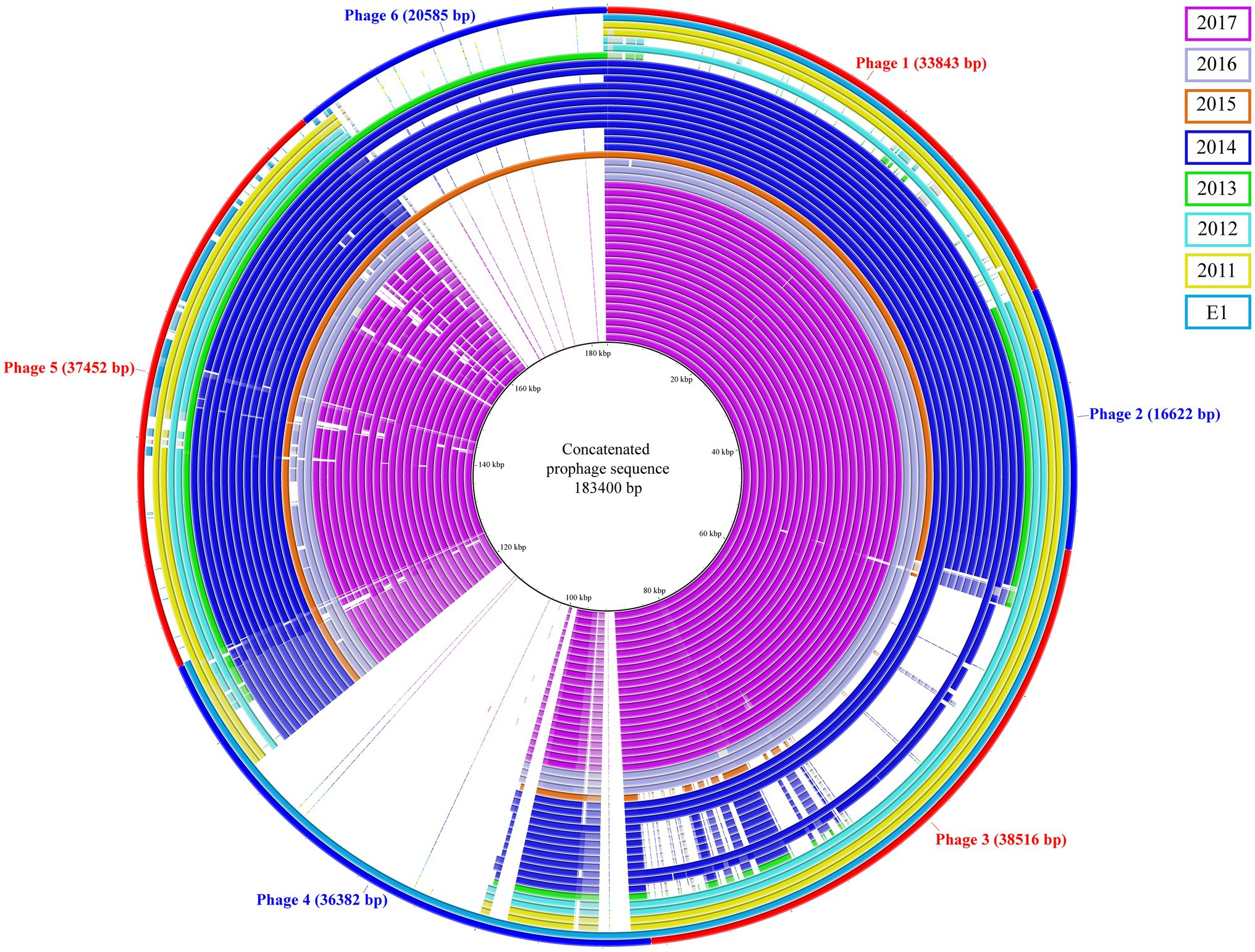
Figure 2. Concatenated prophage sequences identified in ST117/CT24 isolates. The concatenated reference sequence is indicated as the red-blue outer circle, and the inner circles represent the ST117/CT24 isolates coloured by the year in which they were isolated.
The hybrid assemblies A12 and B11 were analysed in more detail for plasmids as they were the representative isolates of the two outbreaks in 2014 and 2017, respectively. The isolates were predicted to have each six plasmid contigs (Supplementary Table 5), which were characterised based on their RIPs and relaxase proteins. The content of these sequences was concatenated and compared. Our analysis revealed that they shared 75% of genomic similarity. Moreover, to understand if DNA rearrangements occurred in plasmids, we performed a BLAST analysis of the contigs separately (Figure 3). In both isolates, plasmid 1 was the largest (mean=223.6kpb) and contained the RepA_N initiator sequence, which showed similarity with pDO3 megaplasmid (GenBank CP003586.1). The BLAST analysis revealed a high similarity between plasmid 1 of both isolates, which shared 72% of DNA identity. One of the differences observed was the presence of enterocin P and the high-level gentamicin resistant gene [aac(6')-aph(2'')] in the plasmid of the B11 isolate. The second plasmid detected in isolates A12 and B11 was medium size (mean=28.9kbp) and carried a RIP protein belonging to the Inc18 family similar to pRE25 (GenBank X92945.2). This plasmid was very conserved in both isolates, which shared more than 90% similarity. In both isolates, the predicted plasmid 3 presented a variable degree of rearrangements and did not carry any RIPs. In A12, however, the gene conferring resistance to chloramphenicol was identified in this contig. The small size plasmid 4 (mean=7.8kbp) of both isolates contained a RepA protein similar to pB82 (GenBank AB178871), which belong to the Rep_3 family. The main difference between the isolates was the distinct presence of two bacteriocins: Lactococcin 972 and bacteriocin T8 were found in A12 and B11, respectively. Plasmid 6 in A12 and plasmid 5 in B11 were also classified as small plasmids (mean=5.7kbp) and shared 70% of DNA identity. They carried the RepA protein similar to pEF418 (GenBank AF408195), which belong to the Rep_3 family. Finally, plasmid 5 in A12 and plasmid 6 in B11 presented a variable degree of rearrangements and could not be assigned to any plasmid family due to the lack of replicon initiator proteins. The only relaxase family identified in the plasmids belonged to the MOB_P group. A relaxase similar to pEF1 (GenBank DQ198088.1) was found on plasmids 1 and 3 in A12 and B11, respectively. Finally, a MobA protein was found in plasmid 4 of both isolates where the closest match was with pHY (GenBank AB570326.1) and pB82 (GenBank AB178871) for A12 and B11, respectively.
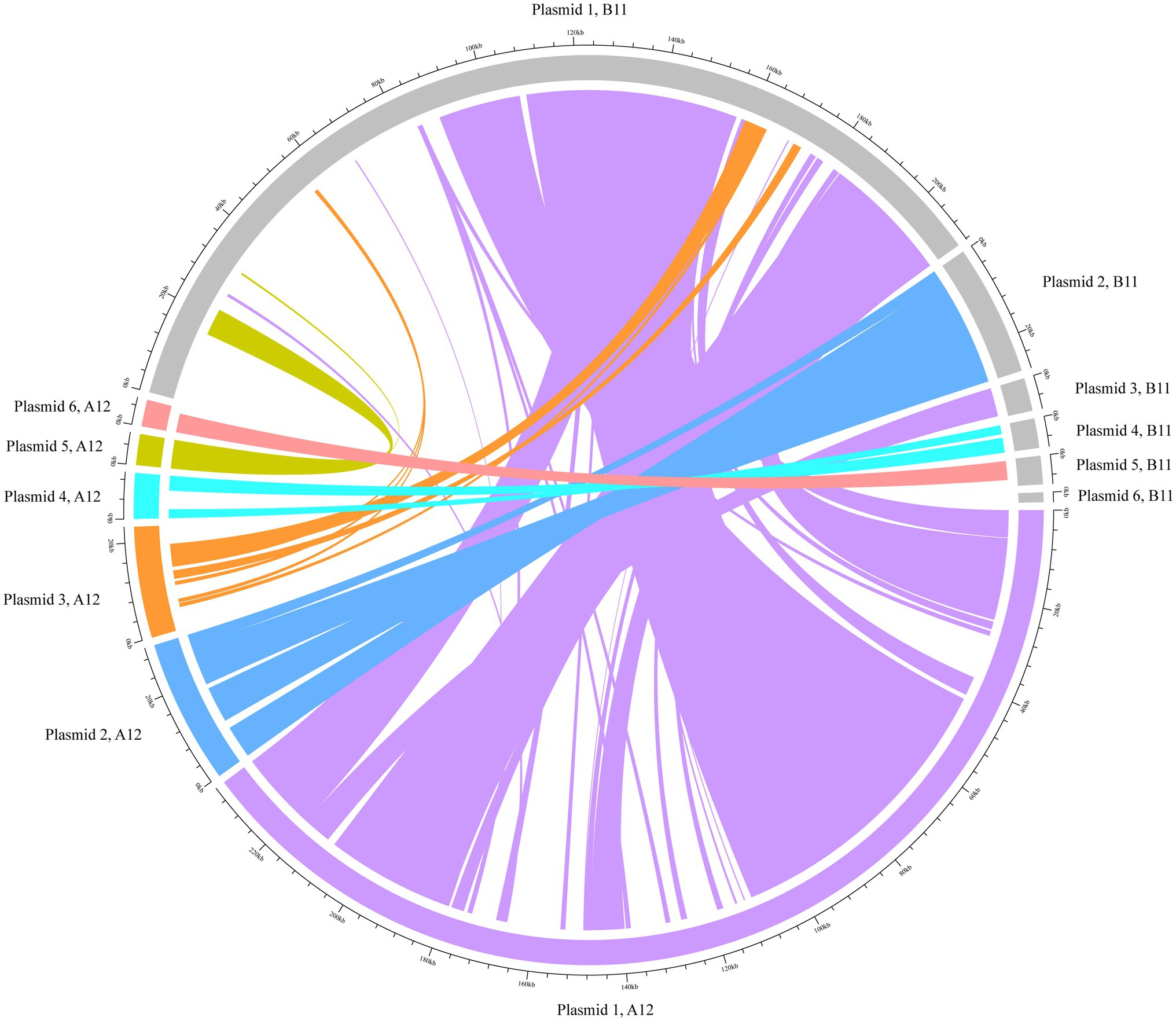
Figure 3. BLAST analysis of the plasmids using Circlize. The plasmid contigs from the hybrid assembles A12 and B11 were used as internal reference for the BLAST analysis. Each of A12 predicted plasmid contigs are presented in colours whilst B11 is depicted in grey.
Discussion
Here, we describe the genomic makeup of 39 VREfm strains isolated from 2011 until 2017, including two consecutive outbreak isolates sharing the same genetic background and compare them with VSEfm isolates. These isolates belonged to ST117, a lineage frequently associated with nosocomial outbreaks (Papagiannitsis et al., 2017; Zhou et al., 2018; Abdelbary et al., 2019; Falgenhauer et al., 2019; Liese et al., 2019; Pinholt et al., 2019; Eisenberger et al., 2020; Weber et al., 2020) and classified as part of the hospital-associated clade A1 (Lebreton et al., 2013). A previous study showed that strains belonging to this clade have a larger genome size suggesting that the acquisition of new traits occurred whilst persisting in the hospital environment. The ability to acquire mobile elements and the flexibility to adapt under harsh conditions led to the rise of this clade (Lebreton et al., 2013). Therefore, an in-depth analysis was performed on our collection of isolates to characterise these elements and investigate if they could be used to type closely related strains belonging to ST117/CT24-vanB clone.
The genome analysis revealed that the isolates were closely related with only a few allele differences. According to the typing scheme proposed by de Been et al. (2015), these isolates are likely to belong to the same outbreak. In such a scenario where isolates share the same genetic background, Zhou et al. (2018) proposed to perform a detailed transposon investigation. This approach proved to increase the discriminatory power by providing additional information compared to cgMLST analysis alone (Zhou et al., 2018). Our results support this recommendation. We observed diversity in the integration point of the Tn1549, which suggests independent acquisitions of vanB-carrying transposons in investigated VREfm isolates (Howden et al., 2013; Sadowy, 2021). Moreover, by including in our analysis vancomycin-susceptible isolates as recommended in other studies (Howden et al., 2013; Bender et al., 2016; van Hal et al., 2016; Pinholt et al., 2019), we confirmed the exchange of genomic material between VSE and VRE belonging to the outbreak in 2017. Alongside, Schürch et al. (2018) also suggested the inclusion of accessory genes and plasmid analysis in addition to a SNP or gene-by-gene typing approach. Our pangenome analysis revealed that one-third of the genes could be classified as part of the accessory genome, where most of the differences amongst the isolates were noticed. The main contributors that could differentiate our isolates were plasmids, with bacteriocins and resistance genes being the main driver of these differences, and prophages.
A recent analysis of the complete plasmid sequences in a vast collection of E. faecium isolates (Arredondo-Alonso et al., 2020) confirmed that bacteriocins, ribosomally synthesised peptides with antibacterial activity directed against bacteria (Franz et al., 2007), were observed in hospital-associated plasmidome populations and were potentially involved in niche adaptation. Similarly, a recent study, which revealed some unpublished data on German VREfm isolates from the 1990s, classified ST117/CT24 strains as “strong producers of bacteriocins” (Werner et al., 2020). Werner et al. (2020) suggested that, through these molecules, commensal enterococci could be wiped out, leading to the expansion of this specific lineage in the gastrointestinal tract. Our data add to these findings by providing insight into the role of VSE. We observed in vancomycin-susceptible isolates the same set of bacteriocins identified in VRE isolates from the 2017 outbreak, indicating that this trait has been acquired and persisted successfully in this lineage.
The hospital-associated plasmidome populations of E. faecium described by Arredondo-Alonso et al. (2020) were found to carry some antibiotic resistance genes in their core structure. We observed the co-occurrence of the erythromycin resistance gene (erm) with aminoglycoside resistance genes in both outbreaks. Additionally, in 2014, a chloramphenicol resistance gene (cat) was identified in the same predicted plasmid contig with aminoglycoside resistance genes. Considering that chloramphenicol use in humans has been limited to a small number of diseases (Schwarz et al., 2004), these findings highlight the promiscuous nature that characterises hospital-associated E. faecium in acquiring genetic elements via horizontal-gene transfer (HGT; Mikalsen et al., 2015). Contrary to Arredondo-Alonso et al. (2020), our study did not identify multi-replicon plasmids; instead, single RIP families belonging to theta-replicating plasmids were detected. These findings could be explained by the limited plasmidome population investigated, which focused only on the hybrid assembly isolates.
Similar to other studies (Abdelbary et al., 2019; Falgenhauer et al., 2019; Eisenberger et al., 2020; Weber et al., 2020), we identified several virulence factors involved in surface adhesion and biofilm formation in almost all ST117 isolates investigated. Two of these genes, pilA and hyl, were carried on plasmids as described in the literature (Freitas et al., 2018; Gao et al., 2018). Our data are in line with the observation that the pattern of virulence and resistance genes herewith detected is linked to “high risk” Enterococcus strains (Weber et al., 2020) and is consistent with the classification within the major putative virulence markers typical of clade A1 as proposed by Freitas et al. (2018).
Although the phage analysis in this study did not allow us to clearly distinguish between closely related isolates, it revealed the constant gain and loss of genomic material that characterises E. faecium. For the 14 isolates collected between 2013 and 2015, we observed differential presence/absence of two prophages. However, the small set of isolates herewith included does not allow us to speculate on the stability of these elements in the enterococci genome. Despite this, previous observations suggested that bacteriophages represent the main cause of genomic diversity in E. faecium (van Schaik et al., 2010). One possible reason is the absence of a functional CRISPR-Cas system, which would make enterococci susceptible to phage attacks (Palmer and Gilmore, 2010; van Schaik et al., 2010). The presence of phage-like elements frequently identified in the genome of hospital-associated E. faecium is well documented (van Schaik et al., 2010; Mikalsen et al., 2015; Pinholt et al., 2019; Arredondo-Alonso et al., 2020). However, it is virtually impossible to classify these putative elements giving the high mosaicism that characterise their genomes (Yasmin et al., 2010; Shamia and Horsburg, 2016). Therefore, it is not surprising that the closest match identified by the web-tool PHASTER is just partially matching some elements in the predicted prophage sequences identified in the isolates of this study.
Whilst the amount of literature concerning E. faecium increases, less attention has been paid to understanding the mechanisms behind its ability to persist in the environment. A set of genes responsible for the survival of this opportunistic pathogen under nutrient depletion has been described in a recent study. Most of these genes were located in the core genome, where usp, a putative universal stress protein, was identified as a candidate that would allow the survival of E. faecium in PBS (de Maat et al., 2020). In our pangenome analysis, we confirmed the presence of such elements in the core genome, including in the environmental samples taken during the 2017 outbreak. Since eradicating this pathogen in hospital settings is crucial, future studies should pick up the work done by de Maat and colleagues to elucidate and further characterise the role of these genes (de Maat et al., 2020).
We acknowledge the relatively small sample size of this single-centre retrospective study, which solely focus on an individual lineage where mainly vancomycin-resistant strains have been described. Currently, in our hospital, VSEfm are not included in the sequencing surveillance, but, at the clinician’s discretion, some isolates were sequenced and have been included in this study. The lack of this data for isolates collected before 2016 does not allow us to conclude whether the outbreaks prior to this moment arose de novo from hospital-associated VSEfm, either by transposition of the Tn1549 from anaerobic gut commensals or by homologous recombination of Tn1549 between vanB VREfm and VSEfm. Despite these limitations, our study shed lights on the factors responsible for the recent expansion of specific sub-populations of VREfm within clade A1 that were not explained so far. Recently, other studies reported the spread of specific lineages throughout Europe (Bender et al., 2016; Papagiannitsis et al., 2017; Abdelbary et al., 2019; Falgenhauer et al., 2019; Liese et al., 2019; Pinholt et al., 2019; Eisenberger et al., 2020; Weber et al., 2020), amongst which ST117/CT24 has been sporadically identified carrying the vanA operon (Kjær Hansen et al., 2021). However, to the best of our knowledge, this is the first study reporting the molecular characterisation of the ST117/CT24-vanB clone and the relative contribution of its MGEs and accessory genes, therefore broadening the currently available epidemiological data.
In conclusion, our results show the importance of an extended molecular investigation that targets the core and the set of accessory genes acquired by hospital adapted strains of E. faecium. The analysis of the transposon and its integration point and the examination of the pangenome allowed us to better characterise VREfm clones in our hospital and conclude that identified VREfm outbreaks were caused by closely related isolates where significant differences were associated with MGEs. Thus, by employing these analyses, it is possible to gain insights into horizontal gene transfer in a selective environment such as the gastrointestinal tract of hospitalised patients.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena (PRJEB42347, PRJEB41626, and PRJEB25590) and https://www.ncbi.nlm.nih.gov/ (PRJNA725797).
Author Contributions
MC-F, JR, XZ, AF, and EB conceived the study. PL performed the formal analysis and visualisation of the data and wrote the manuscript. NC analysed the data and revised the manuscript. SR analysed the data. ML was responsible for the clinical data. XZ and EB revised the manuscript and commented on the interpretation of the results. HH, JR, and AF contributed to the interpretation of data and revised the manuscript. MC-F contributed to the analysis and supervised the study throughout. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Marie Skłodowska-Curie Actions (Grant Agreement number: 713660 – PRONKJEWAIL – H2020-MSCA-COFUND-2015).
Conflict of Interest
JR is currently employed by IDbyDNA Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.728356/full#supplementary-material
Supplementary Figure 1 | Overview of the pangenome and accessory genes analysis. The neighbour-joining tree is based on cgMLST (1,423 target genes). The presence and absence of the predicted resistances genes, bacteriocin, and virulence factors are indicated with coloured and white cells, respectively. Similarly, the presence and absence of a gene in the pangenome matrix is depicted with blue and white lines, respectively. On the pangenome matrix, the in silico predicted bacteriocins and resistance genes which could distinguish the clusters have been highlighted with orange and red lines, respectively.
Supplementary Figure 2 | Nucleotide and amino acid alignment of vanB gene in the representative isolates. Clonal isolates (100% identity) from 2014 and 2017 outbreaks are not depicted in the alignments and were represented by isolates A12 and B11, respectively.
Supplementary Table 1 | Assembly statistics.
Supplementary Table 2 | Pangenome analysis results. List of all the genes identified in the isolates and those uniquely reported in the 2014 and 2017 outbreak.
Supplementary Table 3 | Summary of the phenotypic antibiotic susceptibility test in the isolates included in the study.
Supplementary Table 4 | Summary of the putative phages identified in this study. The presence and absence of the phages have been indicated with 1 and 0, respectively. The output generated by PHASTER is ordered according to the phage ID reported in the study.
Supplementary Table 5 | mlplasmid results of the hybrid assemblies A12 and B11. The predicted plasmid contigs have been renamed in ascending order. Detailed information concerning the RIP families and relaxase protein is herewith summarised.
References
Abdelbary, M. H. H., Senn, L., Greub, G., Chaillou, G., Moulin, E., and Blanc, D. S. (2019). Whole-genome sequencing revealed independent emergence of vancomycin-resistant Enterococcus faecium causing sequential outbreaks over 3 years in a tertiary care hospital. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1163–1170. doi: 10.1007/s10096-019-03524-z
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Arias, C. A., and Murray, B. E. (2012). The rise of the enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278. doi: 10.1038/nrmicro2761
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arredondo-Alonso, S., Rogers, M. R. C., Braat, J. C., Verschuuren, T. D., Top, J., Corander, J., et al. (2018). Mlplasmids: a user-friendly tool to predict plasmid- and chromosome-derived sequences for single species. Microb. Genom. 4:e000224. doi: 10.1099/mgen.0.000224
Arredondo-Alonso, S., Top, J., McNally, A., Puranen, S., Pesonen, M., Pensar, J., et al. (2020). Plasmids shaped the recent emergence of the major nosocomial pathogen Enterococcus faecium. mBio 11, e03284–e03219. doi: 10.1128/mBio.03284-19
Bender, J. K., Kalmbach, A., Fleige, C., Klare, I., Fuchs, S., and Werner, G. (2016). Population structure and acquisition of the vanB resistance determinant in German clinical isolates of Enterococcus faecium ST192. Sci. Rep. 6:21847. doi: 10.1038/srep21847
Carattoli, A., Zankari, E., Garciá-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: The Artemis comparison tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Clewell, D. B., Weaver, K. E., Dunny, G. M., Coque, T. M., Francia, M. V., and Hayes, F. (2014). “Elements in enterococci: transmission, maintenance, and epidemiology,” in Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. eds. M. S. Gilmore, D. B. Clewell, Y. Ike, and N. Shankar (Boston: Massachusetts Eye and Ear Infirmary).
de Been, M., Pinholt, M., Top, J., Bletz, S., Mellmann, A., van Schaik, W., et al. (2015). Core genome multilocus sequence typing scheme for high- resolution typing of Enterococcus faecium. J. Clin. Microbiol. 53, 3788–3797. doi: 10.1128/JCM.01946-15
de Maat, V., Arredondo-Alonso, S., Willems, R. J. L., and van Schaik, W. (2020). Conditionally essential genes for survival during starvation in Enterococcus faecium E745. BMC Genomics 21:568. doi: 10.1186/s12864-020-06984-2
Deurenberg, R. H., Bathoorn, E., Chlebowicz, M. A., Couto, N., Ferdous, M., García-Cobos, S., et al. (2017). Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 243, 16–24. doi: 10.1016/j.jbiotec.2016.12.022
Domingo, M. C., Huletsky, A., Bernal, A., Giroux, R., Boudreau, D. K., and Picard, F. J. (2005). Characterisation of a Tn5382-like transposon containing the vanB2 gene cluster in a clostridium strain isolated from human faeces. J. Antimicrob. Chemother. 55, 466–474. doi: 10.1093/jac/dki029
Donskey, C. J. (2004). The role of the intestinal tract as a reservoir and source for transmission of nosocomial pathogens. Clin. Infect. Dis. 39, 219–226. doi: 10.1086/422002
Eisenberger, D., Tuschak, C., Werner, M., Bogdan, C., Bollinger, T., Hossain, H., et al. (2020). Whole-genome analysis of vancomycin-resistant Enterococcus faecium causing nosocomial outbreaks suggests the occurrence of few endemic clonal lineages in Bavaria, Germany. J. Antimicrob. Chemother. 75, 1398–1404. doi: 10.1093/jac/dkaa041
Falgenhauer, L., Fritzenwanker, M., Imirzalioglu, C., Steul, K., and Scherer, M. (2019). Rhine-Main VREfm study group, Heudorf U, Chakraborty T. near-ubiquitous presence of a vancomycin-resistant Enterococcus faecium ST117/CT71/vanB-clone in the Rhine-Main metropolitan area of Germany. Antimicrob. Resist. Infect. Control. 8:128. doi: 10.1186/s13756-019-0573-8
Franz, C. M., van Belkum, M. J., Holzapfel, W. H., Abriouel, H., and Gálvez, A. (2007). Diversity of enterococcal bacteriocins and their grouping in a new classification scheme. FEMS Microbiol. Rev. 31, 293–310. doi: 10.1111/j.1574-6976.2007.00064.x
Freitas, A. R., Tedim, A. P., Francia, M. V., Jensen, L. B., Novais, C., Peixe, L., et al. (2016). Multilevel population genetic analysis of vanA and vanB Enterococcus faecium causing nosocomial outbreaks in 27 countries (1986-2012). J. Antimicrob. Chemother. 71, 3351–3366. doi: 10.1093/jac/dkw312
Freitas, A. R., Tedim, A. P., Novais, C., Coque, T. M., and Peixe, L. (2018). Distribution of putative virulence markers in Enterococcus faecium: towards a safety profile review. J. Antimicrob. Chemother. 73, 306–319. doi: 10.1093/jac/dkx387
Gao, W., Howden, B. P., and Stinear, T. P. (2018). Evolution of virulence in Enterococcus faecium, a hospital-adapted opportunistic pathogen. Curr. Opin. Microbiol. 41, 76–82. doi: 10.1016/j.mib.2017.11.030
García-Solache, M., and Rice, L. B. (2019). The enterococcus: a model of adaptability to its environment. Clin. Microbiol. Rev. 32, e00058–e00018. doi: 10.1128/CMR.00058-18
Gorrie, C., Higgs, C., Carter, G., Stinear, T. P., and Howden, B. (2019). Genomics of vancomycin-resistant Enterococcus faecium. Microb. Genom. 5:e000283. doi: 10.1099/mgen.0.000283
Gu, Z., Gu, L., Eils, R., Schlesner, M., and Brors, B. (2014). Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. doi: 10.1093/bioinformatics/btu393
Howden, B. P., Holt, K. E., Lam, M. M., Seemann, T., Ballard, S., Coombs, G. W., et al. (2013). Genomic insights to control the emergence of vancomycin-resistant enterococci. mBio 4, e00412–e00413. doi: 10.1128/mBio.00412-13
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Kichenaradja, P., Siguier, P., Pérochon, J., and Chandler, M. (2010). ISbrowser: an extension of ISfinder for visualizing insertion sequences in prokaryotic genomes. Nucleic Acids Res. 38, D62–D68. doi: 10.1093/nar/gkp947
Kjær Hansen, S., Andersen, L., Detlefsen, M., Holm, A., Roer, L., Antoniadis, P., et al. (2021). Using core genome multilocus sequence typing (cgMLST) for vancomycin-resistant Enterococcus faecium isolates to guide infection control interventions and end an outbreak. J. Glob. Antimicrob. Resist. 24, 418–423. doi: 10.1016/j.jgar.2021.02.007
Lebreton, F., van Schaik, W., McGuire, A. M., Godfrey, P., Griggs, A., Mazumdar, V., et al. (2013). Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4, e00534–e00513. doi: 10.1128/mBio.00534-13
Liese, J., Schüle, L., Oberhettinger, P., Tschörner, L., Nguyen, T., Dörfel, D., et al. (2019). Expansion of vancomycin-resistant Enterococcus faecium in an academic tertiary Hospital in Southwest Germany: a large-scale whole-genome-based outbreak investigation. Antimicrob. Agents Chemother. 63, e01978–e01918. doi: 10.1128/AAC.01978-18
Loman, N. J., and Quinlan, A. R. (2014). Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401. doi: 10.1093/bioinformatics/btu555
Mikalsen, T., Pedersen, T., Willems, R., Coque, T. M., Werner, G., Sadowy, E., et al. (2015). Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics 16:282. doi: 10.1186/s12864-015-1407-6
Novais, C., Freitas, A. R., Sousa, J. C., Baquero, F., Coque, T. M., and Peixe, L. V. (2008). Diversity of Tn1546 and its role in the dissemination of vancomycin-resistant enterococci in Portugal. Antimicrob. Agents Chemother. 52, 1001–1008. doi: 10.1128/AAC.00999-07
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palmer, K. L., and Gilmore, M. S. (2010). Multidrug-resistant enterococci lack CRISPR-cas. mBio 1, e00227–e00210. doi: 10.1128/mBio.00227-10
Papagiannitsis, C. C., Malli, E., Florou, Z., Medvecky, M., Sarrou, S., Hrabak, J., et al. (2017). First description in Europe of the emergence of Enterococcus faecium ST117 carrying both vanA and vanB genes, isolated in Greece. J. Glob. Antimicrob. Resist. 11, 68–70. doi: 10.1016/j.jgar.2017.07.010
Pidot, S. J., Gao, W., Buultjens, A. H., Monk, I. R., Guerillot, R., Carter, G. P., et al. (2018). Increasing tolerance of hospital Enterococcus faecium to handwash alcohols. Sci. Transl. Med. 10:eaar6115. doi: 10.1126/scitranslmed.aar6115
Pinholt, M., Bayliss, S. C., Gumpert, H., Worning, P., Jensen, V. V. S., Pedersen, M., et al. (2019). WGS of 1058 Enterococcus faecium from Copenhagen, Denmark, reveals rapid clonal expansion of vancomycin-resistant clone ST80 combined with widespread dissemination of a vanA-containing plasmid and acquisition of a heterogeneous accessory genome. J. Antimicrob. Chemother. 74, 1776–1785. doi: 10.1093/jac/dkz118
Raven, K. E., Gouliouris, T., Brodrick, H., Coll, F., Brown, N. M., Reynolds, R., et al. (2017). Complex routes of nosocomial vancomycin-resistant Enterococcus faecium transmission revealed by genome sequencing. Clin. Infect. Dis. 64, 886–893. doi: 10.1093/cid/ciw872
Rice, L. B. (2008). Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197, 1079–1081. doi: 10.1086/533452
Sadowy, E. (2021). Mobile genetic elements beyond the VanB-resistance dissemination among hospital-associated enterococci and other gram-positive bacteria. Plasmid 114:102558. doi: 10.1016/j.plasmid.2021.102558
Schürch, A. C., Arredondo-Alonso, S., Willems, R. J. L., and Goering, R. V. (2018). Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene-based approaches. Clin. Microbiol. Infect. 24, 350–354. doi: 10.1016/j.cmi.2017.12.016
Schwarz, S., Kehrenberg, C., Doublet, B., and Cloeckaert, A. (2004). Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 28, 519–542. doi: 10.1016/j.femsre.2004.04.001
Shamia, A., and Horsburg, M. (2016). Comparative genomics of Enterococcus faecium bacteriophages. ASRJETS 26, 69–90.
Stinear, T. P., Olden, D. C., Johnson, P. D. R., Davies, J. K., and Grayson, M. L. (2001). Enterococcal vanB resistance locus in anaerobic bacteria in human faeces. Lancet 357, 855–856. doi: 10.1016/S0140-6736(00)04206-9
Tedim, A. P., Lanza, V. F., Manrique, M., Pareja, E., Ruiz-Garbajosa, P., Cantón, R., et al. (2017). Complete genome sequences of isolates of Enterococcus faecium sequence type 117, a globally disseminated multidrug-resistant clone. Genome Announc. 5, e01553–e01516. doi: 10.1128/genomeA.01553-16
van Hal, S. J., Ip, C. L. C., Ansari, M. A., Wilson, D. J., Espedido, B. A., Jensen, S. O., et al. (2016). Evolutionary dynamics of Enterococcus faecium reveals complex genomic relationships between isolates with independent emergence of vancomycin resistance. Microb. Genom. 2:e000048. doi: 10.1099/mgen.0.000048
van Heel, A. J., de Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 46, W278–W281. doi: 10.1093/nar/gky383
van Schaik, W., Top, J., Riley, D. R., Boekhorst, J., Vrijenhoek, J. E., Schapendonk, C. M., et al. (2010). Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics 11:239. doi: 10.1186/1471-2164-11-239
Weber, A., Maechler, F., Schwab, F., Gastmeier, P., and Kola, A. (2020). Increase of vancomycin-resistant Enterococcus faecium strain type ST117 CT71 at Charité—Universitätsmedizin Berlin, 2008 to 2018. Antimicrob. Resist. Infect. Control 9:109. doi: 10.1186/s13756-020-00754-1
Werner, G., Coque, T. M., Franz, C. M., Grohmann, E., Hegstad, K., Jensen, L., et al. (2013). Antibiotic resistant enterococci-tales of a drug resistance gene trafficker. Int. J. Med. Microbiol. 303, 360–379. doi: 10.1016/j.ijmm.2013.03.001
Werner, G., Neumann, B., Weber, R. E., Kresken, M., Wendt, C., Bender, J. K., et al. (2020). Thirty years of VRE in Germany—"expect the unexpected": The view from the National Reference Centre for staphylococci and enterococci. Drug Resist. Updat. 53:100732. doi: 10.1016/j.drup.2020.100732
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Yasmin, A., Kenny, J. G., Shankar, J., Darby, A. C., Hall, N., Edwards, C., et al. (2010). Comparative genomics and transduction potential of Enterococcus faecalis temperate bacteriophages. J. Bacteriol. 192, 1122–1130. doi: 10.1128/JB.01293-09
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: vancomycin-resistant, Enterococcus faecium, outbreak, mobile genetic elements, typing, cgMLST
Citation: Lisotto P, Couto N, Rosema S, Lokate M, Zhou X, Bathoorn E, Harmsen HJM, Friedrich AW, Rossen JWA and Chlebowicz-Fliss MA (2021) Molecular Characterisation of Vancomycin-Resistant Enterococcus faecium Isolates Belonging to the Lineage ST117/CT24 Causing Hospital Outbreaks. Front. Microbiol. 12:728356. doi: 10.3389/fmicb.2021.728356
Edited by:
Mattias Collin, Lund University, SwedenReviewed by:
Babak Haghshenas, Kermanshah University of Medical Sciences, IranMichael Kemp, University of Southern Denmark, Denmark
Copyright © 2021 Lisotto, Couto, Rosema, Lokate, Zhou, Bathoorn, Harmsen, Friedrich, Rossen and Chlebowicz-Fliss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monika A. Chlebowicz-Fliss, bS5hLmNobGVib3dpY3pAdW1jZy5ubA==