 Arley Calle-Tobón1,2
Arley Calle-Tobón1,2 Andres F. Holguin-Rocha1
Andres F. Holguin-Rocha1 Celois Moore1
Celois Moore1 Meagan Rippee-Brooks3
Meagan Rippee-Brooks3 Paula Rozo-Lopez1Jania Harrod1Soheila Fatehi1Guillermo L. Rua-Uribe2
Paula Rozo-Lopez1Jania Harrod1Soheila Fatehi1Guillermo L. Rua-Uribe2 Yoonseong Park1
Yoonseong Park1 Berlin Londoño-Rentería1*
Berlin Londoño-Rentería1*- 1Department of Entomology, Kansas State University, Manhattan, KS, United States
- 2Grupo Entomología Médica, Universidad de Antioquia, Medellín, Colombia
- 3Department of Biology, Missouri State University, Springfield, MO, United States
The Asian “tiger mosquito” Aedes albopictus is currently the most widely distributed disease-transmitting mosquito in the world. Its geographical expansion has also allowed the expansion of multiple arboviruses like dengue, Zika, and chikungunya, to higher latitudes. Due to the enormous risk to global public health caused by mosquitoes species vectors of human disease, and the challenges in slowing their expansion, it is necessary to develop new and environmentally friendly vector control strategies. Among these, host-associated microbiome-based strategies have emerged as promising options. In this study, we performed an RNA-seq analysis on dissected abdomens of Ae. albopictus females from Manhattan, KS, United States fed with sugar and human blood containing either normal or heat-inactivated serum, to evaluate the effect of heat inactivation on gene expression, the bacteriome transcripts and the RNA virome of this mosquito species. Our results showed at least 600 genes with modified expression profile when mosquitoes were fed with normal vs. heat-inactivated-containing blood. These genes were mainly involved in immunity, oxidative stress, lipid metabolism, and oogenesis. Also, we observed bacteriome changes with an increase in transcripts of Actinobacteria, Rhodospirillaceae, and Anaplasmataceae at 6 h post-feeding. We also found that feeding with normal blood seems to particularly influence Wolbachia metabolism, demonstrated by a significant increase in transcripts of this bacteria in mosquitoes fed with blood containing normal serum. However, no differences were observed in the virome core of this mosquito population. These results suggest that heat and further inactivation of complement proteins in human serum may have profound effect on mosquito and microbiome metabolism, which could influence interpretation of the pathogen-host interaction findings when using this type of reagents specially when measuring the effect of Wolbachia in vector competence.
Introduction
Currently, Aedes (Stegomyia) albopictus (Skuse, 1894) is considered the medically important most invasive mosquito species in the world (Benedict et al., 2007). In the last 30 years, it has rapidly spread from its native Asian continent across the world and it can be found in countries on every continent except Antarctica (Enserink, 2008; Ding et al., 2018). The expansion from its traditional habitat of tropical and sub-tropical regions to much cooler temperate regions has been attributed to its ecological plasticity, its ability to enter diapause to escape unfavorable seasonal conditions (Kraemer et al., 2019). These factors have further increased the risk of outbreaks in areas where mosquito-borne viral diseases are uncommon, such as Northern America and temperate Europe (Moreno-Madriñán and Turell, 2017, 2018; Barzon, 2018; Adams et al., 2019; Robert et al., 2020). In the United States, Ae. albopictus was first registered in 1987 and its presence has been reported in at least 40 States (Moore and Mitchell, 1997; Hahn et al., 2017). Needless to say that Ae. albopictus has also been responsible for several outbreaks of dengue (DENV) and chikungunya virus (CHIKV) in tropical areas around the globe (Rezza, 2012; Luo et al., 2017; Lindh et al., 2019).
This mosquito has an opportunistic feeding pattern (Delatte et al., 2010; Kamgang et al., 2012), which increases the risk of transmission of zoonotic diseases to humans. Due to the lack of therapeutic options or widely available vaccines against human arbovirus infections, control of mosquito population is the most used way to prevent diseases transmission (Mahmud et al., 2019). To halter expansion of this mosquito and control pathogen transmitted by it, we need a better understanding of its biology that will lead to innovative and effective control strategies. Pathogens transmission to and from the vertebrate occurs mainly through the bite of female mosquitoes while obtaining their blood meal required to produce eggs. This blood meal is then digested in the midgut to provide the nutrients, but may also increase oxidative stress and cell damage by human active components included in it (Pakpour et al., 2014). To prevent significant damage, mosquitoes adopt different strategies (Khattab et al., 2015) and such active blood derived factors are often, neutralized by enzymes and other molecules in the midgut (Khattab et al., 2015). More importantly, the midgut represents the first barrier against infection with pathogens, so that midgut epithelial integrity as well as proper cellular regenerative process play important roles in pathogen permissiveness and vector competence. Specifically, a delay in the proliferative repair system to preserve the midgut barrier may increase susceptibility to infection (Taracena et al., 2018; Hixson et al., 2021).
Also, because blood meal factors, can remain active for several hours interacting with mosquito cells, human pathogens or even its microbiome mosquito physiology may be affected (Margos et al., 2001; Pakpour et al., 2014). For instance, active complement in midgut decreases pathogen populations present in the blood meal, representing one of the main factors of reduction in Plasmodium population in Anopheles mosquitoes (Smith et al., 2014), and previous studies also suggest that the presence of active complement factors in a blood meal modifies gene expression of multiple genes related to the immune system in Aedes aegypti mosquitoes (Giraldo-Calderón et al., 2020). However, the latest study did not explore the effect of active complement on the microbial communities present in mosquitoes. Blood meal acquisition induces drastic modifications in the mosquito bacterial communities that can be observed from 24 h after feeding (Hegde et al., 2018). Although the microbiome of mosquitoes can be highly variable, both within and between species, it is often dominated by relatively few genera. For mosquitoes, microbiota-host interactions can influence a diverse number of physiological traits including immunity, reproduction, survival, and vector competence (Scolari et al., 2019; Seabournid et al., 2020). Due to this effect, the interest in mosquitoes microbiome manipulation to control the transmission of vector borne diseases has increased in recent years. For example, some endosymbionts Alphaproteobacteria Wolbachia sp. strains, when introduced into non-natural hosts, show the ability to decrease susceptibility of Aedes sp. and Anopheles sp. mosquitoes to arboviruses and Plasmodium sp., respectively (Ippolito et al., 2018; Kalappa et al., 2018; O’Neill, 2018). The exact mechanism that allows Wolbachia to decrease vector competence is still unclear. However, factors such as a rise in host immune response, elevation in host methyltransferase to reduce viral production and suppression of key lipids required for viral replication have been identified as potentially important mechanisms (Caragata et al., 2016; Bhattacharya et al., 2017; Geoghegan et al., 2017).
To date, most mosquito microbiota studies have been focused on the bacterial composition and their role in mosquitoes biology. However, numerous insect-specific viruses (ISV) have been reported in culicids with increasing evidence suggesting that ISVs influence mosquito physiology as well as its ability to transmit important arboviruses (Öhlund et al., 2019). In this regard, virome studies in Ae. albopictus virome revealed that this species harbors viruses from multiple families such as Flaviviridae, Totiviridae, Iflaviridae, Virgaviridae, Rhabdoviridae, and Nodaviridae, that remain stable in mosquito populations, apparently transmitted vertically (Zakrzewski et al., 2018; Kubacki et al., 2020; Shi et al., 2020). Hence, investigating whether active blood factors in the mosquito midgut affect mosquito microbiota, virome and their transcriptome may provide new strategies for mosquito and arbovirus control (Gao et al., 2020). The objective of this study was to evaluate whether the transcriptome of Ae. albopictus is affected by feeding with normal or heat-inactivated serum-containing blood, and to determine if different type of meals are associated with modifications in the microbial communities within the mosquito. We consider our approach as a part of meta-transcriptomics (Cox et al., 2017), a fast and robust protocol for meta-taxonomic analysis using RNA-seq data, that provide the guideline for further study (Cox et al., 2017). The results generated from this study provide new information on the effect of human blood on this species and should form the basis for future investigations on the biology, microbiome, and vector competence of Ae. albopictus.
Results
Differential Gene Expression After Blood Meal
A total of 12 libraries were generated and sequenced from Ae. albopictus abdomens. Mosquitoes were divided according to the meal they received into sugar-fed (SF), inactivated blood-fed (IB), or normal blood-fed (NB). Each feeding treatment was represented by four libraries, and a total of 314,422,593 single-reads were retained after read cleaning with a length average of 137 base pairs (bp). Differential expression analyses showed significant differences in the expression level among treatments (Differentially Expressed Genes or DEG). Specifically, we observed that when comparing IB and SF, a total of 1,527 genes were found differentially expressed (Log2FC > | 1| and p ≤ 0.05) (Figure 1A) with 752 upregulated and 775 downregulated. The comparison between NB and SF groups showed a total of 1,038 DEG of which 612 were upregulated and 426 downregulated (Figure 1B). Within the genes observed with differential expression, 447 upregulated and 414 downregulated were shared between IB vs. SF and NB vs. SF comparisons. RT-qPCR was performed on eight selected genes (AALF001757, AAEL004052, AAEL004318, AALF008822, AALF010682, AALF011004, AALF011200, and AALF012673) to validate the results observed in the RNA-seq experiments (Figure 1C).
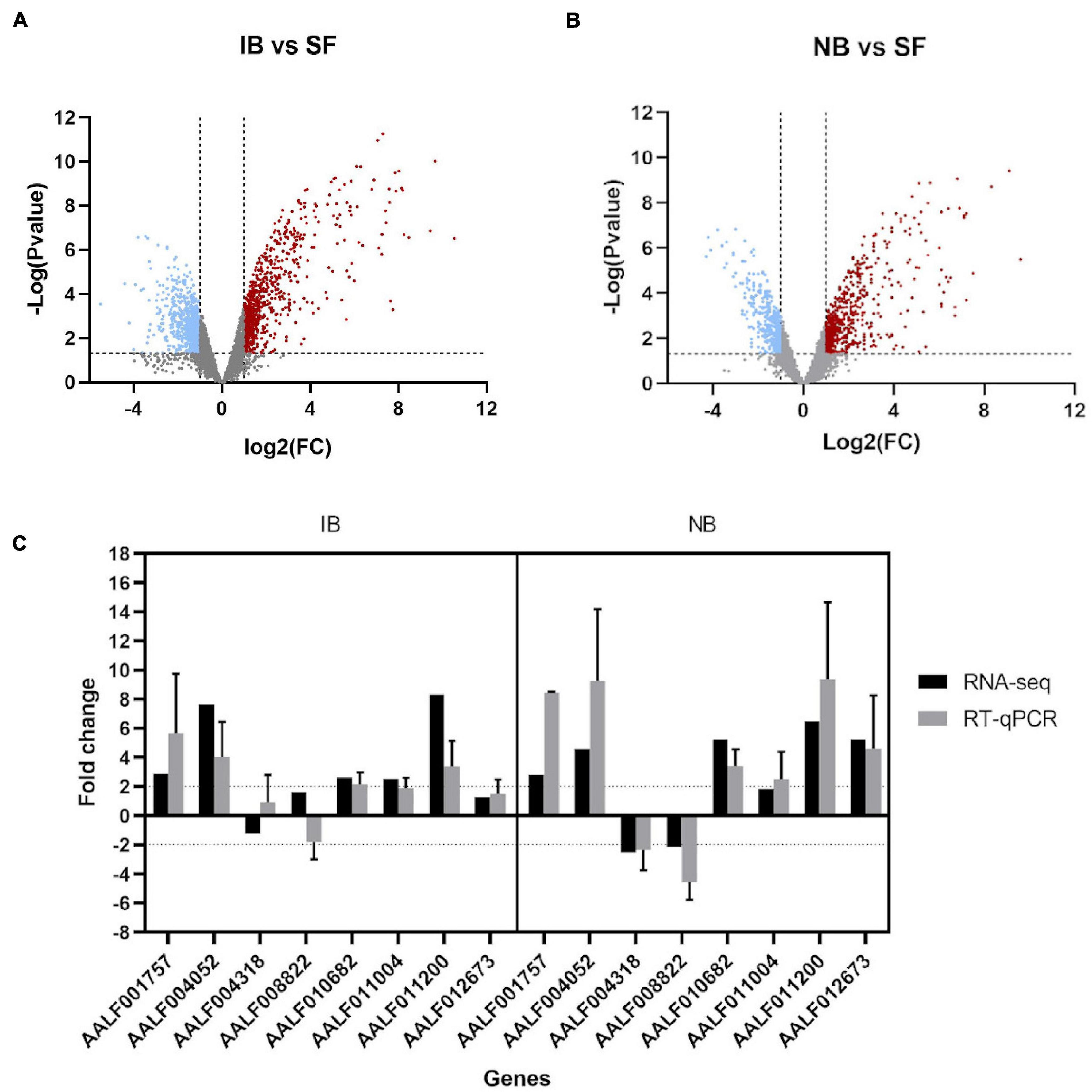
Figure 1. Transcriptome analysis of Ae. albopictus fed with inactivated blood and normal blood. (A) Volcano plots for the differentially expressed genes (DEGs), Inactivated Blood vs. Sugar Fed. Upregulated (red plots) and downregulated (blue plots) DEGs. (B) Volcano plots for the DEGs, Normal Blood vs. Sugar Fed. (C) Fold change of eight immune-related genes expression for RNA-seq and qPCR analysis. qPCR data are presented as FC means (±SE) of three technical and three biological replicates.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis in the Search Tool for the Retrieval of Interacting Genes (STRING) platform was performed after the differentially expressed Ae. albopictus genes were assigned for the Ae. aegypti orthologs. Although the current annotation is relying upon the computational predictions, it was found that comparing IB vs. SF groups, forty-one upregulated and five downregulated metabolic pathways were enriched [False Discovery Rate (FDR) < 0.05]. Yet, in the NB vs. SF group, forty-one metabolic pathways were upregulated and eight downregulated (Supplementary Table 1) and thirty-one upregulated pathways were shared between both comparisons, involving into amino acid or lipid metabolism and oxidative stress; while two downregulated enriched pathways involving pentose and glucuronate interconversions, and nitrogen metabolism were common in both treatments (Figure 2). Furthermore, the IB group showed a significant decrease in expression of genes related to autophagy and the FoxO pathways. Meanwhile, the NB group showed significant changes in the metabolism of sphingolipids and pathways related to cytochrome P450 (Supplementary Table 1).
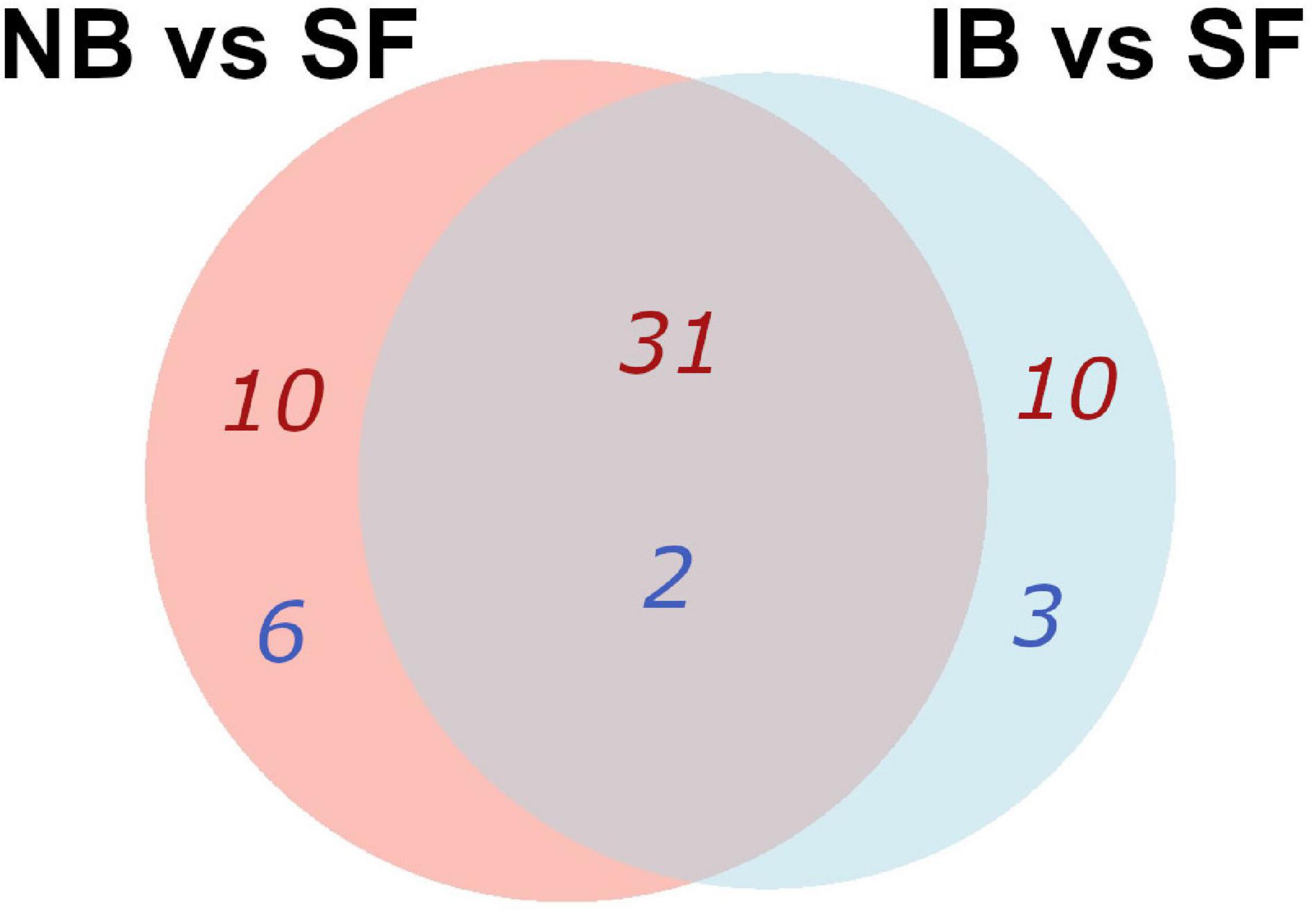
Figure 2. Venn diagram of KEGG pathways enrichment that were common and specific for the pairwise comparisons, using the differentially expressed genes (DEGs) orthologs of Ae. aegypti in the two comparison groups. The number of pathways up- in red, down- in blue.
Also, while identifying which genes showed significant differences between NB vs. IB to determine if heat inactivation of serum influences the expression of those genes, we observed 600 genes differentially expressed between these two groups, of which 481 (80.2%) were upregulated in the NB group (Figure 3A). Furthermore, the KEGG pathways analysis revealed four overexpressed and one downregulated metabolic pathways in the NB group (Figure 3B) one of these downregulated genes in the NB group is apoptosis while autophagy was found downregulated in the IB group. The DEG analysis showed forty-seven immune related genes, with thirty-one of those over-expressed in the NB group (Figure 4); twelve genes were related to lipid metabolism (eleven upregulated and one downregulated), ten genes were related to the cytochrome P450 family (five upregulated and five downregulated), while ten genes were associated with oogenesis (eighth upregulated and two downregulated).
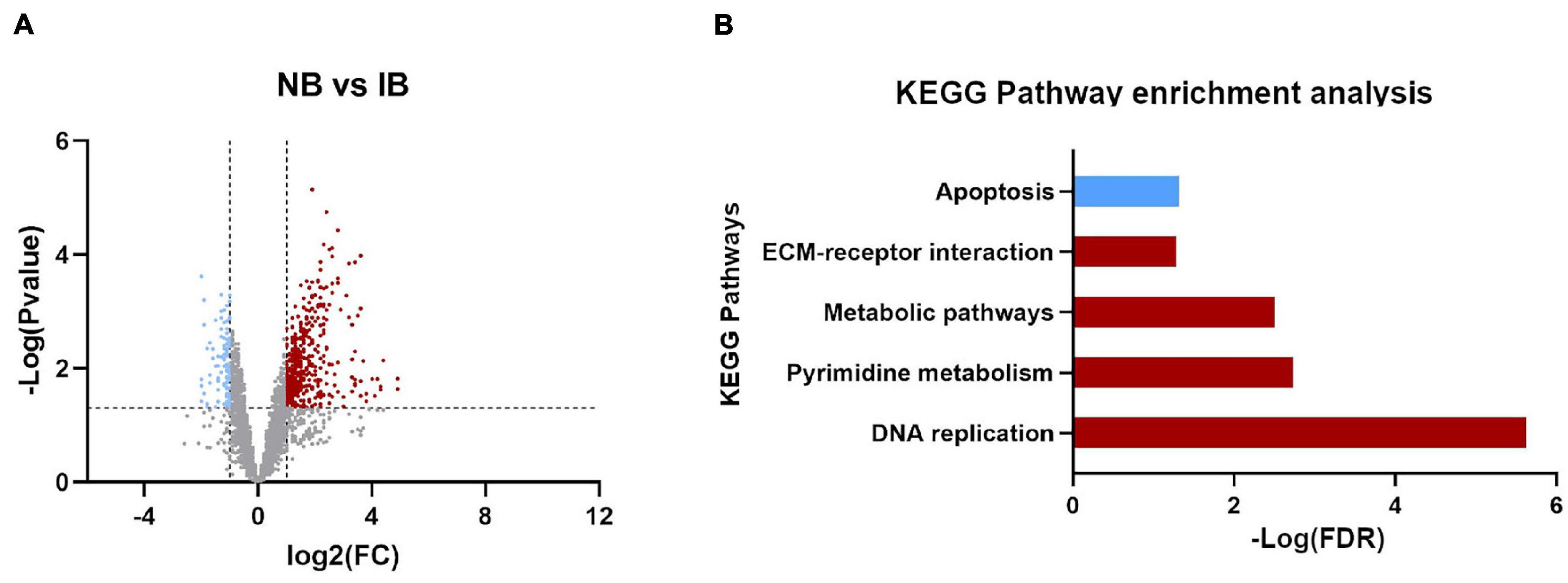
Figure 3. Differentially expressed genes influenced by the presence of active complement. (A) Volcano plot for the differentially expressed genes (DEGs), Normal Blood vs. Inactivated Blood. Upregulated (red plots) and downregulated (blue plots) DEGs. (B) KEGG pathways enrichment identified in DEGs between NB vs. IB. In red the upregulated pathways and in blue downregulated.
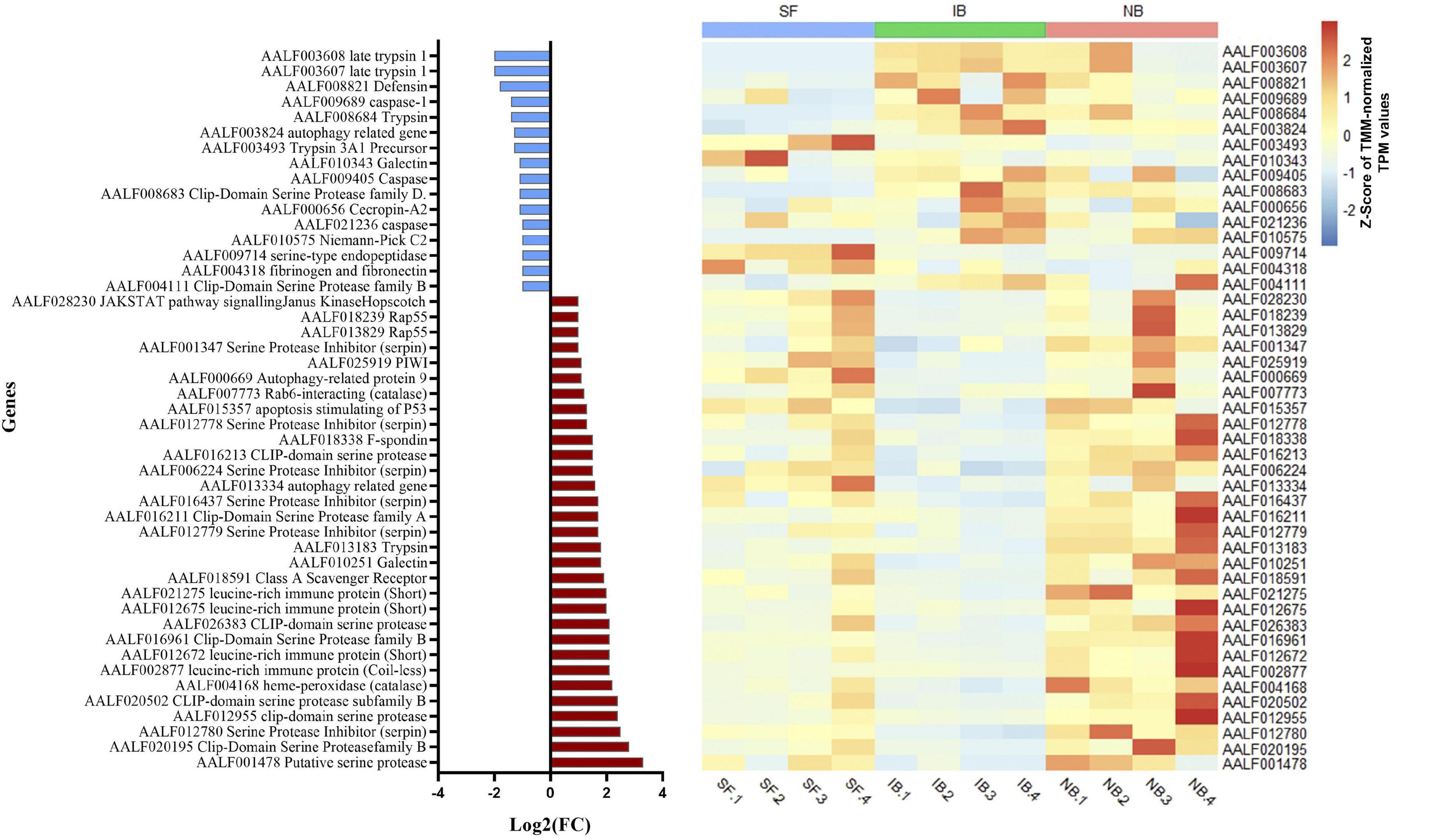
Figure 4. Fold change expression of immune related genes differentially expressed between NB and IB. Upregulated genes (red) and downregulated genes (blue).
Gut Damage Detection
To determine if active complement protein was causing damage in mosquito guts, we evaluated the expression of two genes involved into molecular signaling pathways implicated in regeneration. The gene Socs36E is a target and negative regulator of the JAK/STAT pathway, and the Keren gene, which is one of the known epidermal growth factor receptor (EGFR) ligands, these ligands are highly expressed in the Drosophila midgut following stress-induce damaged. The qRT-PCR assays on dissected guts fed with the treatments showed that the Socs36E gene had no differences in its expression between the treatments, however, the Keren gene had a significant increase in the NB treatment compared to IB (Figure 5).
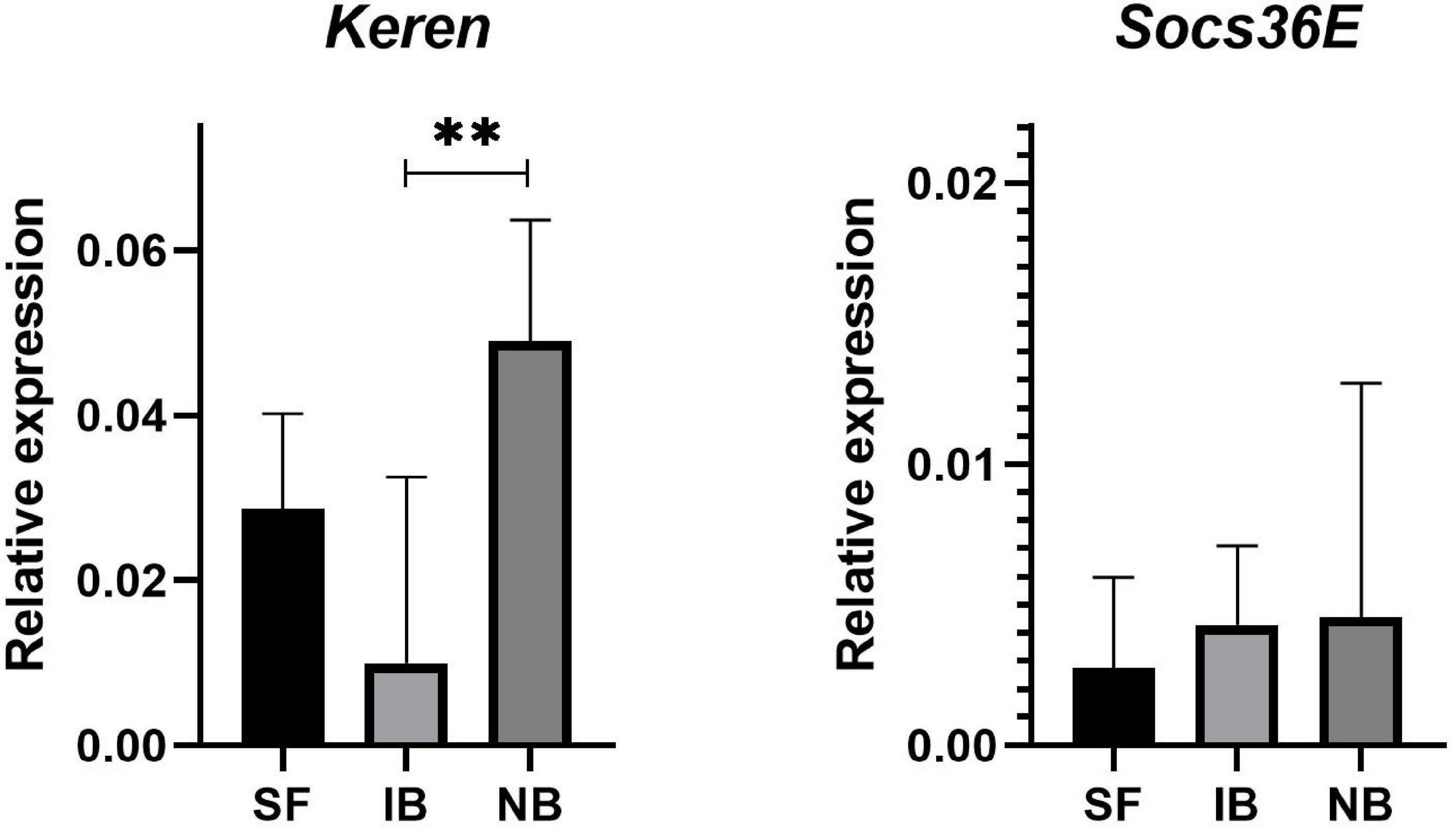
Figure 5. Relative expression of Keren and Socs36E compared to RP49 in midguts of Ae. albopictus mosquitoes sugar fed (SF), blood with heat-inactivated serum (IB), or normal serum (NB). ∗∗P < 0.01; ANOVA.
Changes in the Bacteriome After Blood Meal
After mapping the RNA-seq libraries to the Ae. albopictus genome, cleaning the presence of rRNA and assembling the remaining sequences to perform the microbiome identification, we found that 314,417 out of 314,422,593 (0.0001%) reads corresponded to bacteria. Then, we identified the presence of thirty-one operational taxonomic units (OTUs) (Supplementary Table 2). The number of sequences for each OTU was normalized as reads per million mapped reads (RPM). The values obtained were then compared between treatments to identify variations in the abundance of the different taxa sequences (Figure 6). The comparison test between all treatments showed significant differences in the number of sequences for Anaplasmatacea, Actinobacteria, and Rhodospirillaceae. Also, we observed that after 6 h post-blood feeding, Actinobacteria and Rhodospirillaceae showed an increase regardless the type of blood meal. However, further analysis of the Anaplasmatacea suggests a complex behavior, since in the IB group RPM decreased and the NB group presented a significant increase. Interestingly, the family Anaplasmataceae includes the genus Wolbachia, Ehrlichia, and Neorickettsia, so, we performed individual search to determine which of these families were the ones showing variation. The search revealed alterations in abundancy of Wolbachia reads abundance (Figure 7A). To validate the observed results, a PCR-based assay was performed to measure transcripts levels of the Wolbachia groEL gene, a single copy gene constitutively expressed (Chen et al., 2019). The relative levels of Wolbachia transcripts were normalized with respect to the Ae. albopictus Ribosomal protein 49 (RP49) gene expression (Figure 7B; Janeh et al., 2017). The detection of Wolbachia was in concordance with the transcripts results obtained from the abundance determination after taxonomic classification, confirming that the feeding the blood meal type influences the abundance of Wolbachia. Also, contigs mapped on the Wolbachia surface protein gene (Wsp) were used for a phylogenetic analysis using reference sequences of the Wsp gene from the different groups of this genus. The non-rooted tree obtained allowed us to determine the natural presence of Wolbachia from clade A and B in this population of Ae. albopictus studied (Figure 8).
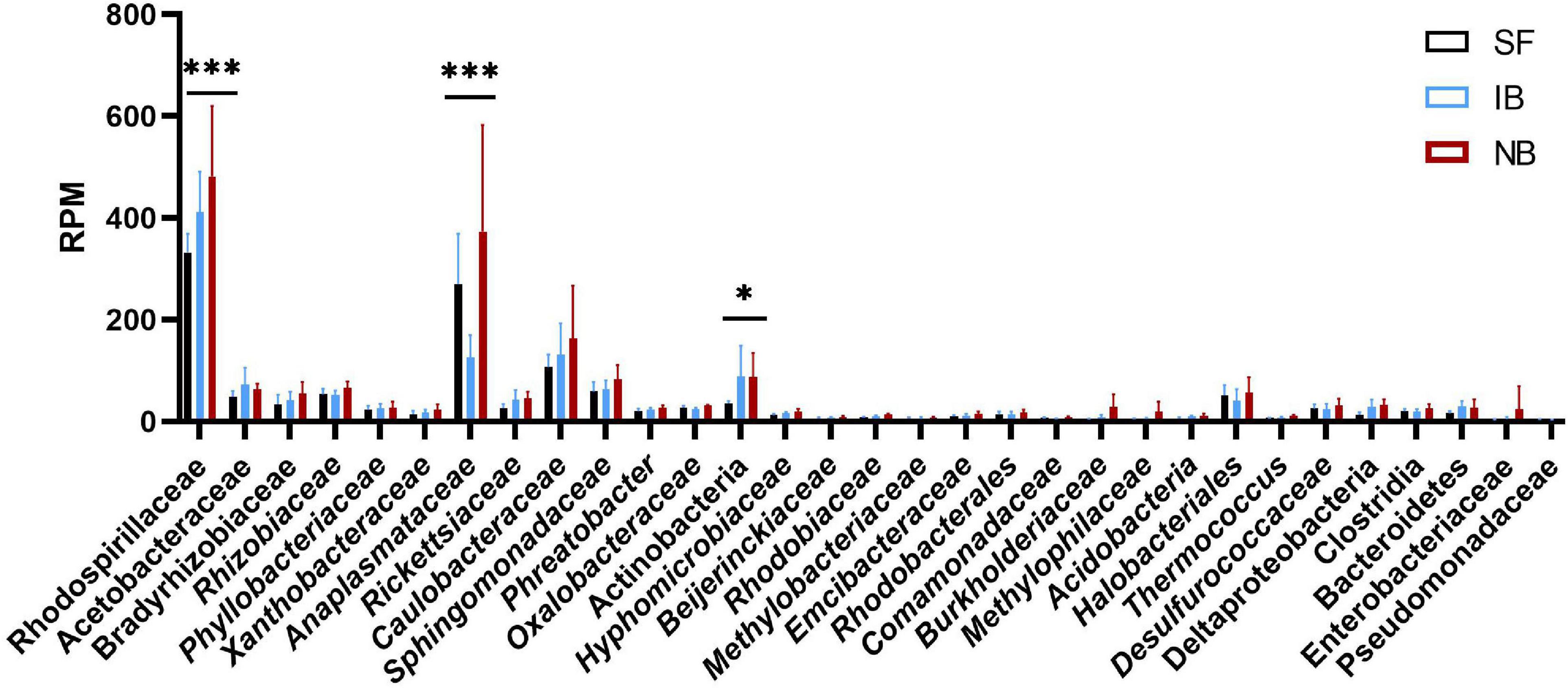
Figure 6. Composition of the Ae. albopictus bacteriome after feeding with sugar, inactivated blood, and normal blood. ∗P < 0.05, ∗∗∗P < 0.001; ANOVA.
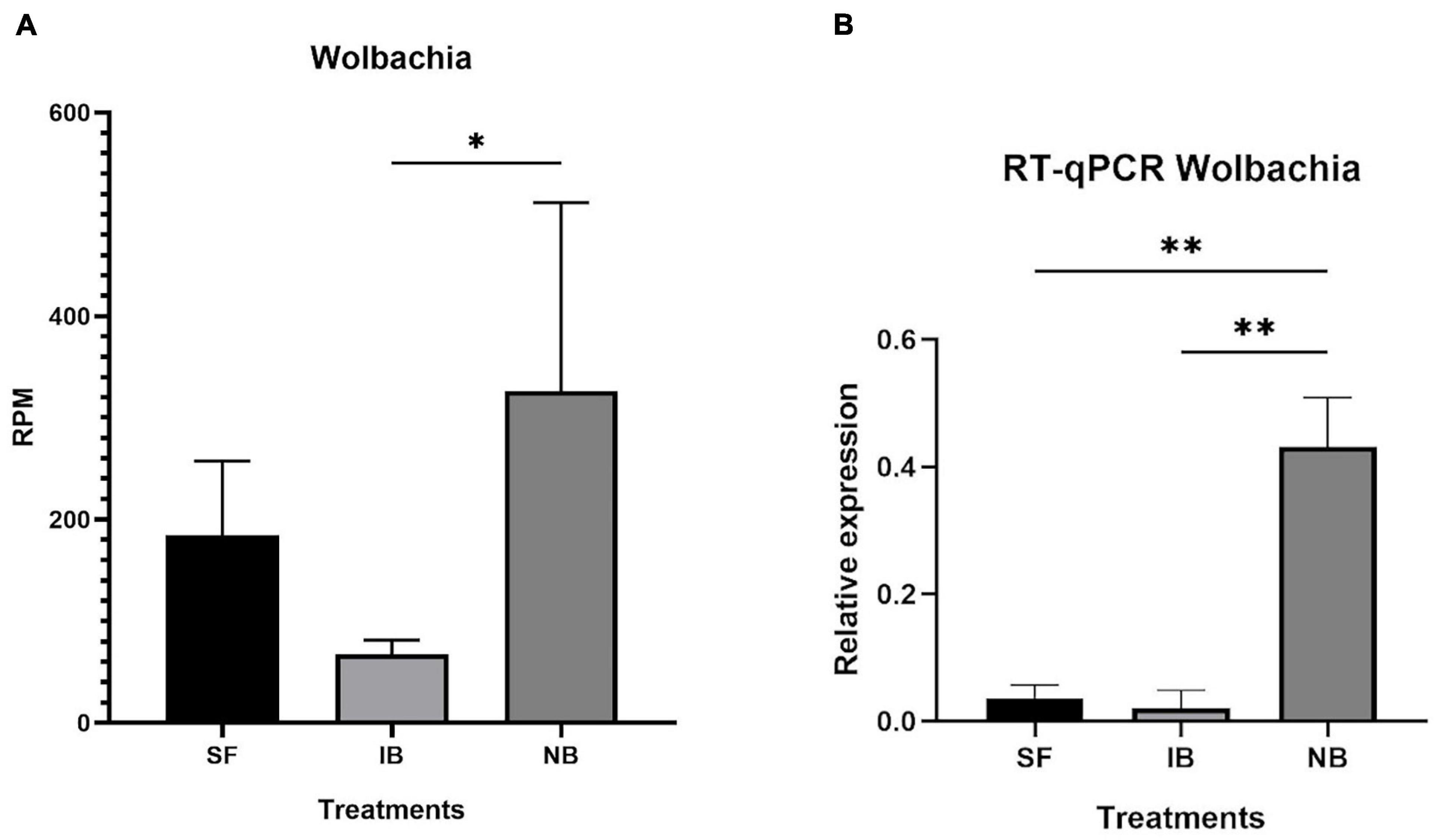
Figure 7. Relative abundance of Wolbachia in Ae. albopictus after each feeding treatment. (A) Relative abundance of Wolbachia reads normalize as reads per million reads mapped in the Ae. albopictus genome. (B) RT-qPCR of relative abundance of Wolbachia groEL transcript compared to Ae. albopictus RP49. ∗P < 0.05, ∗∗P < 0.01; ANOVA.
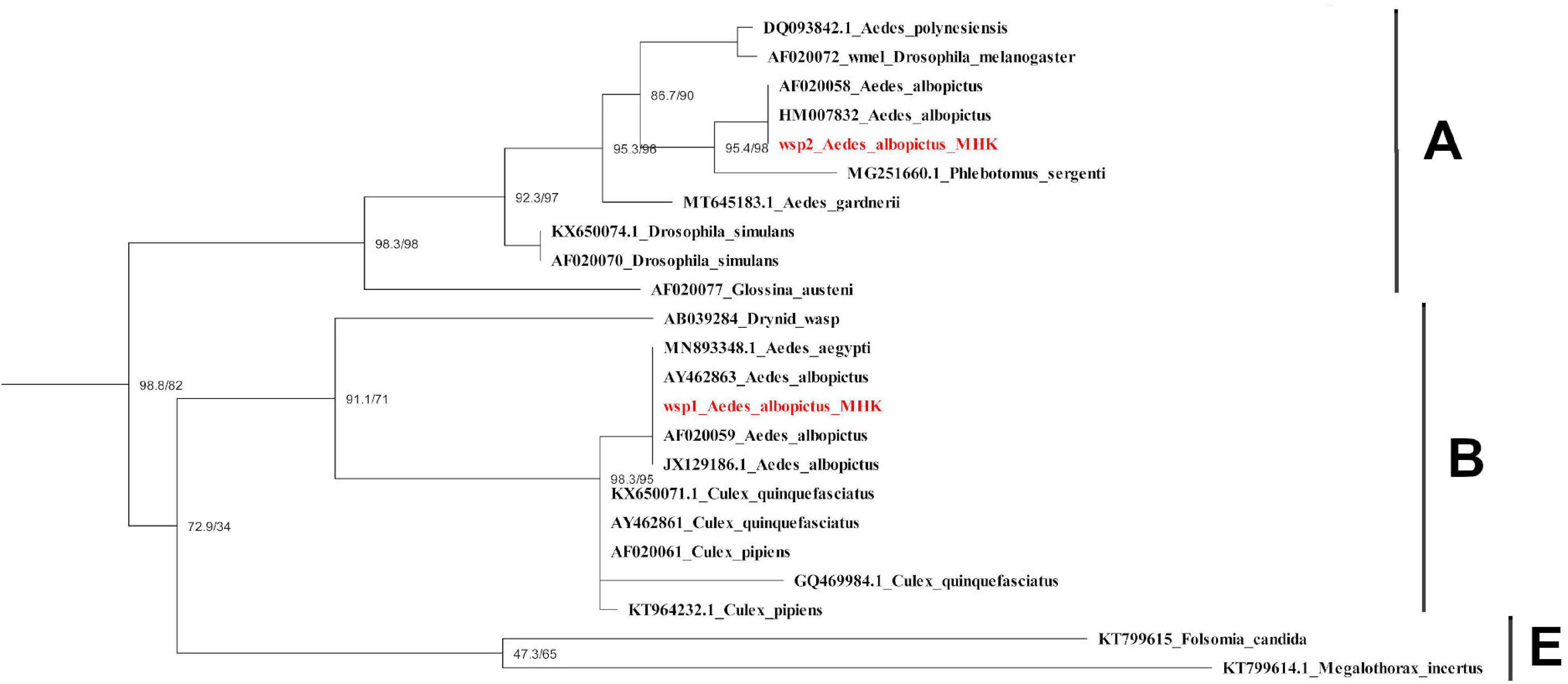
Figure 8. Phylogenetic tree of Wolbachia Wsp gene obtained from Ae. albopictus mosquitoes in Manhattan, KS, United States. Sequences identified in this study are labeled in red. Scale bar indicates the number of substitutions per site. Wsp: Wolbachia surface protein. KS: kansas.
Virome Diversity and Stability After Blood Meal
Insect specific viruses (ISV) have been studied as potential modulators of immune responses in mosquitoes influencing vector competence (Öhlund et al., 2019). Previous studies have demonstrated that the presence of active serum factors influence pathogen populations in the midgut. Thus, we explored the possibility that the presence of active complement in mosquito midgut had any effect on ISV presence. To test this assumption, we used an RNA-seq approach to describe the RNA virome of Ae. albopictus. After removal of mosquito and ribosomal sequences, 103,384 contigs greater than 450 bp were obtained and were taxonomically assigned using DIAMOND and only eukaryotic viruses were taken into consideration. Eighty contigs were identified as viral OTUs, which were classified into six viral species belonging to four families (Flaviviridae, Totiviridae, Xinmoviridae, and Phenuiviridae) (Supplementary Table 2). The individual abundance of each viral species, measured as the number of reads mapped to each contig divided by the total amount of ribosomal RNA-depleted reads in the library and multiplied for 1,000,000 (i.e., reads per million, RPM), showed that the Aedes albopictus anphevirus (Xinmoviridae) was the most abundant virus in average (Figure 9). This virus was recently suggested and has been identified in RNA-seq samples from Ae. albopictus in multiple regions such as North America, Europe, and Asia (Manni and Zdobnov, 2020). The Australian Anopheles totivirus (Totiviridae) was the second most abundant in the samples evaluated. The most diverse family was Flaviviridae, in which we found the presence of Aedes flavivirus, Kamiti River virus, and Cell fusing agent virus. The Phasi Charoen-like phasivirus (Phenuiviridae) was the least abundant among the viral sequences found. A contrasting result with what is observed in Ae. aegypti, in which this virus superinfects it (Zhang et al., 2018). An overview of the metavirome visualized using Krona is shown in Supplementary Figure 1. The comparison among feeding treatments did not show significant changes in the abundance or the diversity of the virome (ANOVA p-value > 0.05), suggesting that blood feeding did not modify the viral community identified in the post-feeding time studied here (6 h).
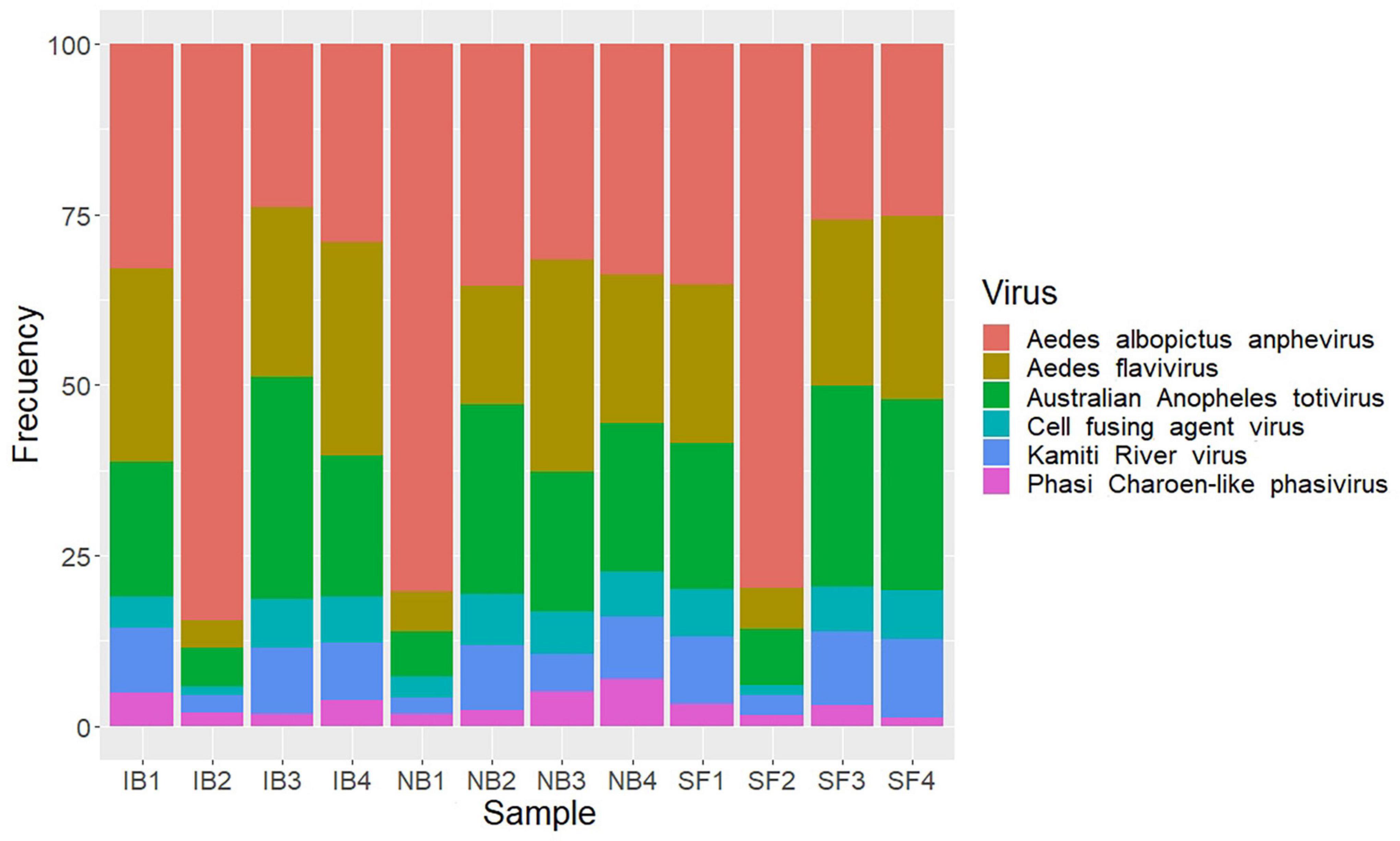
Figure 9. Stacked bar plots showing relative abundance of viral species identified after feed treatments.
Discussion
Aedes albopictus mosquitoes are important vectors of arboviruses worldwide. Given the effect of blood meal and its active components on the mosquito transcriptome, we decided to evaluate the effect of complement on Ae. albopictus physiology and microbial composition. Specifically, this work is a continuation of our previous study in which we found that human complement proteins modify transcription in numerous Ae. aegypti genes, affecting multiple metabolic pathways, including the immune system (Giraldo-Calderón et al., 2020). RNA-seq has served as a robust method to identify the gene expression profile of organisms and has been used to meta-taxonomy analysis (Cottier et al., 2018). Ae. aegypti mosquitoes, unlike Ae. albopictus, have a strong feeding preference for humans while Ae. albopictus shows an opportunistic feeding behavior on a wide range of hosts (from cold-blooded to warm-blooded animals) so we aim to study the effect of human complement factors in the later species (Richards et al., 2006; Delatte et al., 2010). This study provided insights into the effects of human complement protein activity over the gene expression, and the microbiome of Ae. albopictus.
We observed that human complement proteins influence the expression of at least 600 genes in Ae. albopictus. Because many Ae. albopictus genes have unknown function, full interpretation of the effect of human complement on gene expression could not be attained. Nevertheless, we found an alteration in the expression of 47 genes related to the immune system and may influence the pathogen survival ingested in the blood. One of the most interesting pathways found downregulated in the IB group was the autophagy pathway. This is a tightly regulated catabolic process whereby cells degrade intracellular components via the lysosomal machinery and it plays an important role in homeostasis maintenance, cell development, growth, and immunity (Chang and Neufeld, 2010). In humans, autophagy promotes cell survival under these stress conditions by recycling the organism own materials (proteins and organelles) (Kwon et al., 2019). This suggest that feeding with normal blood is important in maintaining the autophagy pathway functional. Interestingly, we observed that genes associated with apoptosis were downregulated in mosquitoes from the NB group. Gathering all this together suggest that the presence of active complement in human blood induces proliferation of gut cells and protects against apoptosis.
RNA interference is one of the most important anti-viral mechanism in mosquitoes (Blair, 2011). Several components of this pathway were found upregulated in the NB group. Specifically, we observed an increased transcript accumulation for Dicer 1 and PIWI genes. Dcr1 interacts with dsRNA-binding proteins (i.e., R3D1/Loqs) to process and load miRNA duplex into the RISC complex. Dcr1 displays some role in antiviral RNAi in mosquitoes through miRNA synthesis (Bernhardt et al., 2012), for example, an increase in miRNAs production in C6/36 cells has been detected in response to West Nile virus (Slonchak et al., 2014) and DENV infections (Yan et al., 2014). PIWI is a member of the PIWI small RNA (piRNA) pathway proposed to be involved in anti-viral defense (Marconcini et al., 2019), which has been identified in Aedes cultured cells and adult mosquitoes after infection by arthropod-borne alpha-, bunya-, and flaviviruses (Blair, 2019).
In our current study, we found the upregulated expression of ten genes of the CYP450 family suggesting that complement proteins contained in NB group may increase the expression of the oxidative stress response. Although, oxidative stress may damage host tissues if not controlled, it plays an important role in defense against pathogen replication and survival (Oliveira et al., 2011). In this regard, mechanisms protecting mosquito tissues from oxidative stress may have an impact on pathogen prevalence (Oliveira et al., 2017). Heme and iron contained in the blood meal may induce oxidative damage in the midgut (Whiten et al., 2017); as protection, hematophagous arthropods have evolved different mechanisms, including the synthesis of antioxidants such as catalase. In fact, a study showed that catalase levels increase at 24 and 36 h after a blood meal to recover to sugar fed mosquito levels once digestion is finished (Oliveira et al., 2017). Another study demonstrated that a blood meal decreases the production of reactive oxygen species through a heme-mediated activation of the protein kinase C (Oliveira et al., 2011).
Arboviruses are normally acquired through feeding and the midgut epithelial correspond to the first line of defense against infection (Franz et al., 2015). The midgut is a complex tissue, composed by several types of cells helping in the digestion and absorption of nutrients. In concordance with our observed upregulation of oxidative stress at 6 h post feeding, a recent study demonstrated that silencing of the Ae. aegypti factor erythroid-derived factor 2 (AeNrf2) increases reactive oxygen species levels and stimulates intestinal stem cell proliferation (Bottino-Rojas et al., 2018). The presence of mitotic cells in the midgut of Ae. albopictus (Janeh et al., 2017) and drastic changes in midgut cells have also been observed in Culex mosquitoes, which suggest that blood feeding induces midgut cells proliferation (Houk, 1977). The JAK/STAT and the EGFR pathways, and more specifically the Socs36E and Keren genes, respectively, are activated in Ae. albopictus midgut following feeding with human blood (Janeh et al., 2017). In our study, significant differences in the expression Keren were found when comparing midguts of mosquitoes fed with IB or NB, suggesting significant activation of the EGFR pathways regenerative signaling in midguts of mosquitoes fed with NB where the complement factors are still active. Although no significant differences among treatments were found in the expression of Socs36E, we found that the Hop ortholog in Ae. albopictus of a negative regulator of the JAK/STAT pathway in Drosophila, the JAK/STAT pathway signaling Janus Kinase Hopscotch (Hop) gene (AALF028230) associated with proliferation of diploid imaginal cells (Binari and Perrimon, 1994) was significantly upregulated in mosquitoes fed with NB, suggesting that both EGFR and JAK/STAT signaling pathways may be activated in NB mosquitoes with low suppression effect from SOCS36E (Baeg et al., 2005; Muller et al., 2005).
We found several genes involved in lipid metabolism and oogenesis upregulated in mosquitoes fed with NB. Although studying the specific blood components regulating oogenesis was out of the scope of this work, other studies suggest that lipid transport from the fat body to oocytes occurs within the first 30 h post blood feeding (Briegel et al., 2003) and that lipid storage and metabolism in the fat body after blood feeding are closely related to vitellogenesis (Pinch et al., 2021). Another study also suggests that blood feeding stimulates the release of two hormones, insulin-like peptide 3 and ovary ecdysteroidogenic hormone in Ae. aegypti, leading to the activation of the insulin/insulin growth factor signaling pathway in ovaries inducing oogenesis (Vogel et al., 2015). Taken all this information together, we believe that components present in non-heat inactivated serum have an effect on egg production signaling, but further research is needed to determine whether active human complement in the blood meal has a direct effect on oviposition or hatching rates.
On the other hand, multiple methods have been used to describe the host microbiome composition, like 16S and DNA-seq, and an accurate taxonomy classification based on RNA-seq reads can now be used to gather taxonomic information about the microbiota present in a biological sample without conducting additional sequencing, allowing efficiently and robustly identify bacteria, fungi, and viruses in the same sample (Cox et al., 2017). Herein, the meta-taxonomic analysis of the microbiome of Ae. albopictus showed the presence of at least 31 OTUs in the mosquito population analyzed. It has previously been known that the composition of Ae. albopictus microbiome varies between individuals, populations, even between field and colony mosquitoes (Seabournid et al., 2020); some studies have shown that 24–48 h after a blood meal, bacterial abundance increases, but diversity decreases (Wang et al., 2011; Terenius et al., 2012). In this study, we found that 6 h after blood-feeding the activity of Actinobacteria, Rhodospirillaceae, and Anaplasmataceae increase significantly. In beetles and hemipterans, Actinobacteria have been found as important nutrient processors aiding in the digestion of sugar and blood meals (Zucchi et al., 2012), and producing antibiotic barriers against pathogens (Seipke et al., 2011; Zucchi et al., 2011). Rhodospirillaceae is part of the phylum Proteobacteria, the dominant group in the Ae. albopictus microbiome (Juma et al., 2020). The results observed in this study suggest that the Rhodospirillaceae family may play a role in blood metabolism in mosquitoes. Interestingly, the increase in Anaplasmataceae was observed only in the NB treatment, suggesting that the observed change was caused by human complement proteins. We also found that, within Anaplasmataceae, the increase OTU corresponded to Wolbachia, a commonly found group in mosquitoes including Ae. albopictus. Interestingly, Ae. albopictus is naturally found super-infected with two Wolbachia strains, wAlbA and wAlbB (Sinkins et al., 1995; Zhou et al., 1998) as we found in this study. This genus of bacteria is a focus of interest because some strains have been shown to influence the vector competition of Ae. aegypti (Moreira et al., 2009). However, more studies are needed to elucidate the effect of blood meal on native strains of Wolbachia found in Ae. albopictus since previous studies suggest a minimal effect on the infection with human arboviruses (Mousson et al., 2012; Raquin et al., 2015; Joanne et al., 2017) in contrast to what is observed in the transinfected strains from Drosophila melanogaster used in Ae. aegypti (Fraser et al., 2017).
Our analysis revealed that the presence of the active complement in NB induced a significant increase in the number of Wolbachia sequences. Since Wolbachia is found in multiples mosquito tissues but is particularly abundant in the reproductive tissues of mosquitoes (Zouache et al., 2009; Joubert et al., 2016), we consider that changes in the Ae. albopictus transcriptome can serve as the triggering signal for the increase in Wolbachia activity. Possible signals linked to this could be an increase in the transcription of genes related to oogenesis and lipid metabolism. In this study we found an increase in the expression of genes related to lipid metabolism when the complement system is active. Mainly involving synthesis, transport, and storage of lipids. Wolbachia are unable to synthesize certain vital biomolecules, including lipids and amino acids, relying instead on the host metabolism for these compounds (Cho et al., 2011; Caragata et al., 2014) in vitro studies suggested a significant impact of Wolbachia replication and the depletion of lipids groups (sphingolipid, diacylglycerols, and phosphatidylcholines) that has been demonstrated increased in DENV-infected cells and that are required for optimal replication (Perera et al., 2012; Molloy et al., 2016). Consequently, further test with additional timepoints is necessary to determine if Wolbachia replication affects lipid metabolism upon feeding with active complement factors leading to changes in lipid metabolism or vice versa.
As we discussed before, previous studies have shown that the human complement system can remain active for up to 6 h post blood meal before its inactivation by blood digestion proteases (Margos et al., 2001; Simon et al., 2018; Pereira-Filho et al., 2020), and may interact with the mosquito cells and its associated gut microbiome. Also, since the presence of active human complement in mosquito midgut can decrease pathogen populations, we decided to investigate the impact of heat inactivation of human serum on the microbiome/virome of Ae. albopictus mosquitoes. To date, most of the conducted mosquito microbiota studies have focused on the bacterial component in mosquitoes and their possible intrinsic roles in mosquito biology. However, numerous insect-specific viruses (ISV) have been reported in culicids, and increasing evidence suggest that ISVs might influence the mosquito physiology as well as its ability to transmit important arboviruses (Öhlund et al., 2019). Some studies, mainly with Ae. albopictus from Asia, have begun to reveal information about the existence of numerous viruses that remain stable in mosquito populations, apparently transmitted vertically (Zhao et al., 2019; Kubacki et al., 2020; Shi et al., 2020). For instance, a study reports that although Ae. albopictus cell lines are permissive to the Phasi Charoen-like virus, it was only found in Ae. aegypti cells. Yet the presence of such virus interrupts dengue and Zika virus replication during co-infection with Cell-fusing agent virus (Schultz et al., 2018). Still, more studies are needed to elucidate how ingestion of blood can affect the structure of the viral community in mosquitoes and whether these communities can influence their ability to prevent or facilitate pathogen colonization.
Through RNA sequencing it has been possible to know the presence of numerous viruses in mosquitoes, however, obtaining this information is limited by the sequencing technology and depth of sequencing, for which some methods have been suggested to carry out viral enrichment of libraries (Sadeghi et al., 2018; Shi et al., 2020). Diversity of insect viruses found in this study was low, compared to previous studies, in which they have proposed the presence between 1 and 23 viral species in Ae. albopictus populations, however, many of them unclassified (Kubacki et al., 2020; Shi et al., 2020). The number of viruses identified in this work could be affected by the astringent identification criteria (contigs identity and cover >60%), sequencing depth, and having used F1 colonized mosquitoes. In this regard, it is possible that the conditions used to raise the F1 in the laboratory, such as the use of distilled water and larvae food, influenced the microbial composition (Martinson and Strand, 2021). A recent study compared the virome of Ae. albopictus derived from field and from colonies and concluded that both in populations, the virome seem to be stable through the life cycle with each group of mosquitoes displayed the respective “core virome” (Shi et al., 2020). Still, it has been reported that the virome diversity in lab Ae. albopictus is lower than in field mosquitoes, probably due to the less complex environment and the availability of clean water and food resources (Shi et al., 2020). In this study, we found the presence of six viruses associated with insects that are present in this population of Ae. albopictus studied, which were not affected after the blood meals. Therefore, we consider that this set of viruses found is a representation of the natural core virome of Ae. albopictus, which seems to be mainly made up of flaviviruses, and is kept in circulation through vertical transmission (Shi et al., 2020). The Ae. albopictus anphevirus was the virus with the highest number of average sequences, however, in most samples it presented a proportion like Aedes flavivirus and Australian Anopheles totivirus, therefore, its apparent dominance could be caused as an artifact of the sequencing of a batch of samples. The effect of anphevirus on the biology of Ae. albopictus mosquitoes is largely unknown, however, it has been reported that another Anphevirus (Aedes Anphevirus) has complex interplays with Wolbachia and can interfere with DENV replication (Parry and Asgari, 2018). In addition, an Ae. albopictus anphevirus has been reported as globally distributed and may interfere with vector competence through modulation of the RNAi pathway (Manni and Zdobnov, 2020).
In conclusion, our results show that after a blood meal, human complement proteins influence the transcription of multiple Ae. albopictus genes, modifying functions such as the immune system, lipid metabolism, oxidative stress, and oogenesis. Also, the presence of Wolbachia increases after the ingestion of a normal blood meal, which suggests a direct or indirect effect of human complement in the activity of these bacteria (a figure summarizing the main findings of this study can be found on Figure 10). Therefore, it will be necessary to consider the characteristics of the blood meal as a factor that can influence vector competence in mosquito infection studies. This study evaluates the changes that occur 6 h post-feeding, however, physiological changes can manifest up to 36 h, therefore, future studies should evaluate different post-feeding times to fully identify the derived changes in gene expression, the microbiome and the virome, caused by the blood feeding, and specially by human complement proteins.
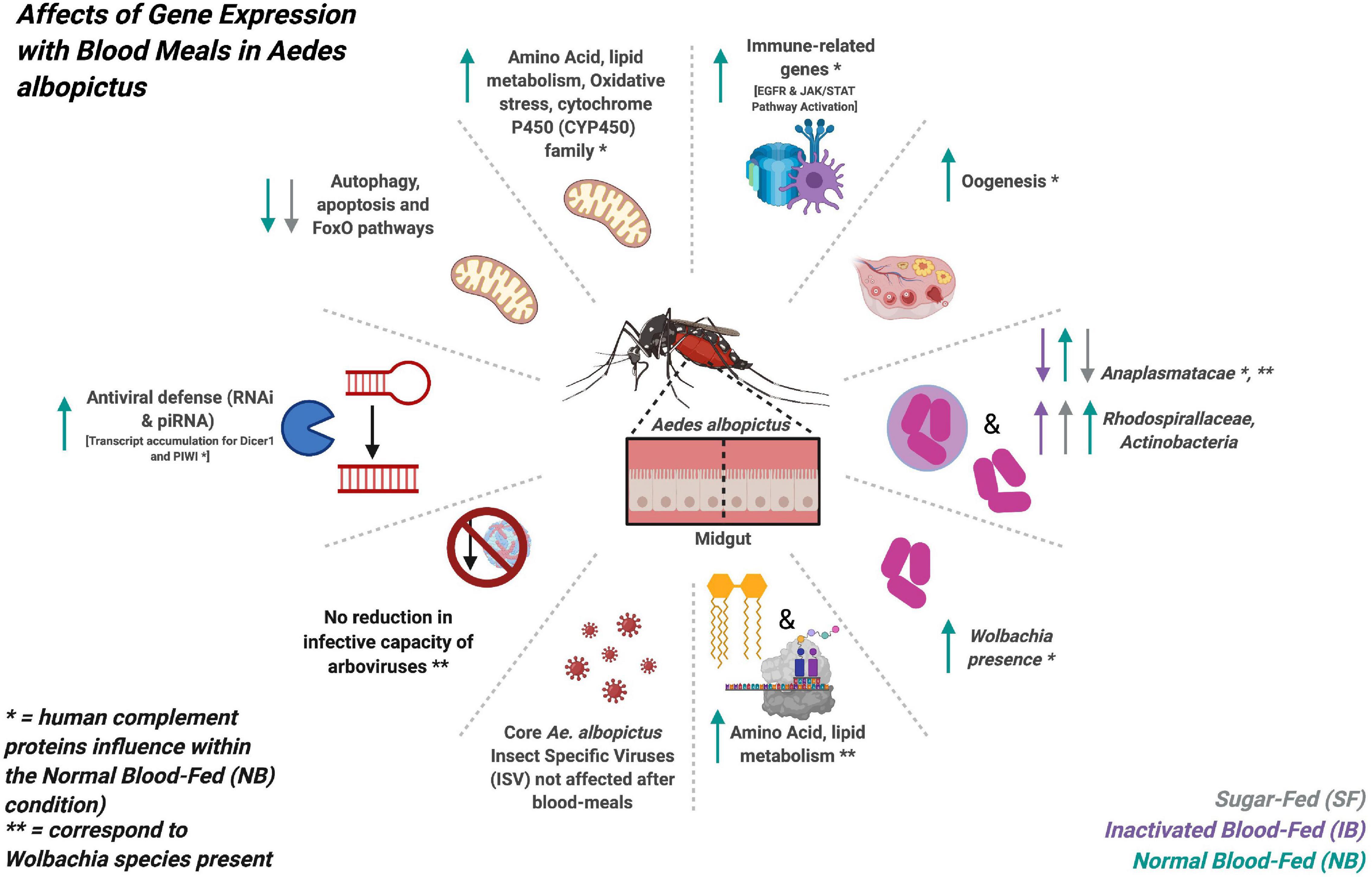
Figure 10. Graphic summary of the main findings obtained in the transcriptome expression and microbiome changes of Ae. albopictus after feeding with sugar, heat-inactivated, and normal blood.
Materials and Methods
Mosquitoes Sampling and Maintenance
Mosquitoes were collected using ovitraps set in Manhattan, KS, United States. Each trap consisted of a 10 cm diameter black plastic cup, a rectangle (8 cm × 15 cm) of 3MM CHR filter paper (Whatman, Maidstone, United Kingdom), and 350 ml of rainwater. Six traps were set weekly from June 10th to August 26th, 2019, and from June 2nd to July 28th, 2020. All collections were made in urban settings. The traps were left in the field for 7 days and then transported to the lab. Filter papers were checked under a microscope for the presence of Aedes-like eggs, and the content of each trap was emptied into a transparent tray and checked for the presence of mosquito larvae.
Mosquito eggs were allowed to hatch in the breeding site water. F0 larvae and pupae were maintained in distilled water with dog chow as a food source. Mosquitoes were reared at 27 ± 1°C, 75 ± 5% RH, with a photoperiod of 16:8 h (L:D). Adults were maintained on 10% sucrose ad libitum. Randomly picked fourth-instar larvae were selected for morphological identification, and newly emerged adults were identified by visual inspection. F0 adults were allowed to mate for 2–3 days following emergence, then were offered blood meals twice a week, and allowed to lay eggs. Mosquitoes were fed for 60 min, at room temperature, using a Hemotek feeder system warmed at 37°C and 1 ml of bovine blood (Lampire Biological Laboratories, Pipersville, PA, United States). A 50 ml plastic cup with distilled water and a 42.5 mm circle of Whatman Grade 1 filter paper was placed inside F0-adult cages to collect F1 eggs. All F1 eggs were pooled and hatched in distilled water. F1 Female mosquitoes (n = 20 per experiment) 5–10 days-old were feed with a mixture of human red blood cells in a 1:1 ratio with either heat-inactivated or normal non-inactivated autologous plasma.
Blood Source
The protocols included in this study were approved by the Kansas State University Review Board (IBC# 1205, IRB #8811). Human peripheral blood was collected in tubes containing ethylenediaminetetraacetic acid (EDTA), then, plasma was separated from the red blood cells (RBCs) by centrifugation. Red blood cells (RBCs) were washed three times in 1X PBS and kept at 4°C until use. Plasma was separated in two categories, normal non-inactivated and heat-inactivated 30 min at 56°C. Aliquots were stored at −20°C until use.
Mosquito Blood-Feeding
Female adult mosquitoes were fed with either heat-inactivated blood plasma (500 μL inactivated plasma + 500 μL of homologous packed RBC), or normal non-inactivated plasma (500 μL normal plasma + 500 μL homologous packed RBC). Mosquitoes were fed during 30 min at room temperature using 1 mL of blood mixture in a Hemotek feeder maintained at 37°C. Blood-fed females were placed in a separate cage. Six hours post-feeding, whole abdomens or midguts were dissected and transferred to 1.5-mL tubes with 200 μL of DNA/RNA shield (Zymo Research, Irvine, CA, United States), in pools of five abdomens or midguts. Tissue of females fed sugar solution 10% were used as control. Mosquito tissues was homogenized in ZR Bashing-Bead lysis tubes.
Mosquito RNA Isolation and Sequencing
Mosquito total RNA was extracted from the homogenizated per the manufacturer’s instructions by Quick-RNA miniprep kit (Zymo Research, Irvine, CA, United States). Each treatment was conducted in four replicates (n = 5 mosquitoes per replicate). The Ribo-Zero Gold Kit (Human/Mouse/Rat) was used to deplete ribosomal RNA (New England Biolabs, Ipswich, MA, United States). A total of 100 ng of depleted RNA per sample was sent for sequencing to The Genome Sequencing Laboratory at The University of Kansas (Lawrence, KS, United States), where the sample QC, libraries preparation, and were sequenced as 150 pb single reads in the Illumina HiSeq [NextSeq Mid-Output (MO)].
Transcriptome Analysis
Raw files were processed for removal of Illumina adaptor sequences, trimmed and quality-based filtered using Fastp software v.0.20.0 (Chen et al., 2018). The remaining high-quality reads were mapped onto the reference genome of Ae. albopictus assembly AaloF1.2 using STAR v2.7 (Dobin et al., 2013). The unmapped reads were separated to perform the microbiome analysis. Mapped reads were counted using RNA-Seq by Expectation-Maximization (RSEM) v1.3.3 (Li and Dewey, 2011). The expression of genes in different libraries was normalized by the Trimmed Mean of M-values (TMM), and differential expressed genes (DEG) (Log2FC ≥ | 1| and Benjamini–Hochberg adjusted p-value ≤ 0.05) were identified using edgeR v3.32.1 (Robinson et al., 2010), which assumes a Negative Binomial Distribution model.
The gene description, gene ontology (GO) terms and orthologs of Ae. aegypti of DEG were obtained using the BiomaRt package v.2.44.1 (Durinck et al., 2005). To identify DEG-enriched metabolic pathways, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis was performed in the Search Tool for the Retrieval of Interacting Genes (STRING) v.11.0 platform1, using the Ae. aegypti orthologs.
Verification of DEG and Gut Damage Detection by RT-qPCR
To confirm the results of the RNA-seq analyses, relative expression levels of eight genes (Supplementary Table 3) differentially expressed between sugar fed and blood fed mosquitoes were chosen. For real-time quantitative PCR analysis a total of 100 ng of RNA were used for cDNA synthesis with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacture instructions. Real-time quantitative PCR reactions of 10 μl were performed in triplicate with SYBR Green Supermix (Bio-Rad). Real-time quantitative PCR reactions were run on a real Applied Biosystems QuantStudio 3. No primer dimer was detected when inspecting the melting curves.
Fold-changes in gene expression between IB and NB mosquitoes were derived by the 2(-Delta Delta C(T)) method (Livak and Schmittgen, 2001), using the constitutive ribosomal protein 49 (RP49) as housekeeping gene, and sugar fed treatment as control. Samples were considered negative if the cycle threshold (Ct) value was greater than 30.
For the detection of cell damage, RNA from dissected guts. Four biological replicates were used for each treatment, each one with three dissected midguts. The RT-qPCR assays were performed as described elsewhere (Janeh et al., 2017). Ct values for target genes were normalized to Rp49 and compared to controls using the delta Ct method.
Virome and Bacteriome Analysis
The unmapped sequences in the genome of Ae. albopictus were used to identify the set of viruses and bacteria present. First, a custom database of ribosomal RNA sequences was generated using SILVA v132 LSU, SSU and 5S rRNA (RF00001), 5.8S rRNA (RF00002), tRNA (RF00005), Ribonuclease P (RF00010, RF00011, and RF00373). And the software SortMeRNA v.2.1 (Kopylova et al., 2012) was used to identify ribosomal sequences in the transcriptome data using an e-value cut-off of 10–5 (Zhao et al., 2019). Reads with an identity >60% and 60% of the read length covered were marked as ribosomal and excluded from further analysis.
The remaining sequences were assembled using SPAdes v.3.12.0 (Bankevich et al., 2012), and contings greater than 450 nt were classified using DIAMOND v.2.0.8 (Buchfink et al., 2014) against the nr viral and bacterial databases (updated February 2021) using the sensitive mode for taxonomic annotation with an e-value cut-off of 10–5 and bit score cut-off of 100 (Zhao et al., 2019; Kubacki et al., 2020). KronaTools v.2.7.1 (Ondov et al., 2011) were used to parse the output file of DIAMOND, which found the least common ancestor of the best 25 DIAMOND hits (based on BLASTx score). The number of reads for each contig present in the detected taxonomic units was normalized using the RPM method with respect to the reads mapped in the Ae. albopictus genome per library and GraphPad Prism software v.9.0.1 was used to determine modifications in viral and bacterial abundance.
Wolbachia RT-qPCR Detection
We used an RT-qPCR to verify the presence of Wolbachia. Specific primers were designed for the groEL gene (Supplementary Table 3), which is a constitutive gene. RT-qPCR was performed in the same way as described above. The relative abundance of Wolbachia was estimated with respect to the mosquito housekeeping RP49 expression.
Phylogenetic Analysis
The contigs mapped on the Wolbachia surface protein (Wsp) gene were extracted and aligned with respect to 21 reference sequences. Alignments of the sequences were performed with MAFFT v.7.475 (Katoh and Standley, 2013) using the most accurate algorithm, L-INS-I, with 1,000 cycles of iterative refinement. The phylogenetic tree was reconstructed from 1,000 ultrafast bootstrap ML tree replicates using IQ-TREE v1.6.12 (Nguyen et al., 2015) with best-fit model selection by ModelFinder (Kalyaanamoorthy et al., 2017). FigTree v.1.4.42 was used for phylogenetic tree visualization. A figure summarizing the methods used and the main finding of this study can be found on Figure 11.
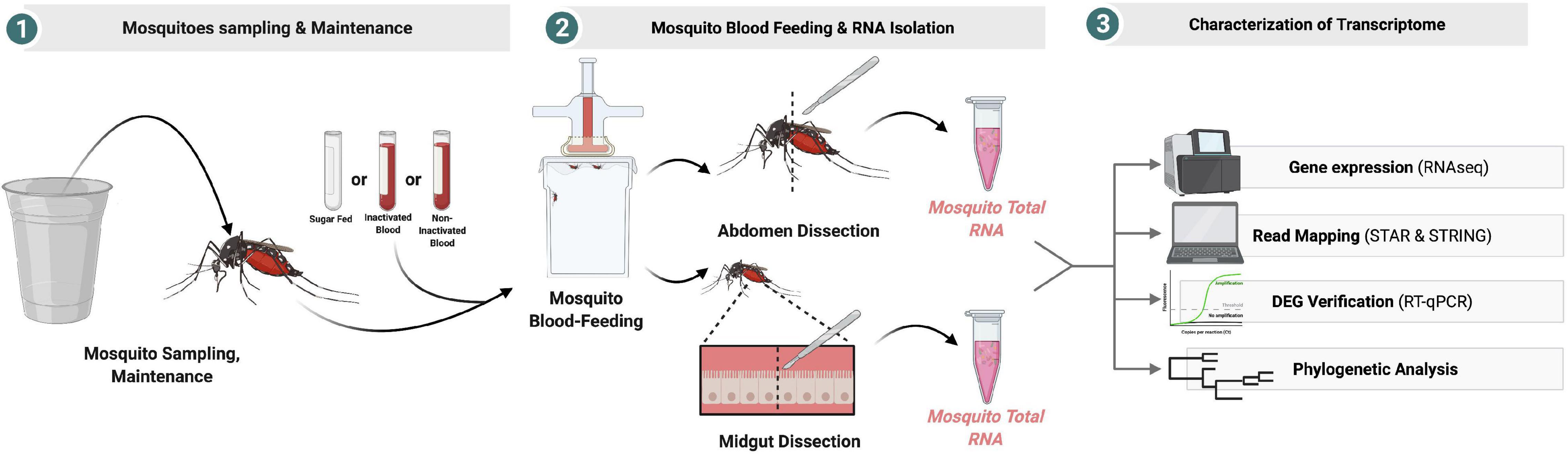
Figure 11. Graphic summary of the experimental design.
Data Availability Statement
The data presented in the study are deposited in the SRA repository, accession number PRJNA722323. The data are publicly available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA722323.
Ethics Statement
The studies involving human participants were reviewed and approved by the Kansas State University (IRB#8811). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
BL-R: conceptualization and funding acquisition. BL-R, PR-L, SF, and AC-T: methodology. BL-R, AC-T, PR-L, AH-R, CM, MR-B, and JH: experiments. AC-T: bioinformatics analysis and original draft preparation. BL-R, AC-T, YP, and GR-U: writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the USDA National Institute of Food and Agriculture, Hatch-Multistate, project 1021430 and the COBRE–NIH 5P20GM103638-08, BL-R. The 2019 Kansas State Summer Research Fellowship program for Undergraduate, JH. AC-T thanks to the financial support provided by COLCIENCIAS to project no. 111574455690, contract no. 634-2017, and by the Research Development Committee (CODI, by its Spanish acronym) of Universidad de Antioquia to project no. 2017-16393.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Department of Entomology at Kansas State University for the support in this project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.724345/full#supplementary-material
Supplementary Figure 1 | Virome classification of Ae. albopictus samples using DIAMOND and the lowest common ancestor (LCA) approach, with results visualized by Krona. Normalized abundance of viral reads aligning to the indicated virus species.
Supplementary Table 1 | KEGG metabolic pathways differentially expressed between the feeding treatments in Aedes albopictus, using the orthologs of Aedes aegypti.
Supplementary Table 2 | Bacteriome and virome detected in Aedes albopictus. Number of contigs and reads per OUT, identity percentage, and Average of Log (e-value).
Supplementary Table 3 | List of primers used in the RT-qPCR assays.
Footnotes
References
Adams, L. E., Martin, S. W., Lindsey, N. P., Lehman, J. A., Rivera, A., Kolsin, J., et al. (2019). Epidemiology of dengue, chikungunya, and zika virus disease in U.S. States and Territories 2017. Am. J. Trop. Med. Hyg. 101, 884–890. doi: 10.4269/ajtmh.19-0309
Baeg, G. H., Zhou, R., and Perrimon, N. (2005). Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 19, 1861–1870. doi: 10.1101/gad.1320705
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barzon, L. (2018). Ongoing and emerging arbovirus threats in Europe. J. Clin. Virol. 107, 38–47. doi: 10.1016/j.jcv.2018.08.007
Benedict, M. Q., Levine, R. S., Hawley, W. A., and Lounibos, L. P. (2007). Spread of the tiger: global risk of invasion by the mosquito Aedes albopictus. Vector-Borne Zoonotic Dis. 7, 76–85.
Bernhardt, S. A., Simmons, M. P., Olson, K. E., Beaty, B. J., Blair, C. D., and Black, W. C. (2012). Rapid intraspecific evolution of miRNA and siRNA genes in the mosquito Aedes aegypti. PLoS One 7:e44198. doi: 10.1371/journal.pone.0044198
Bhattacharya, T., Newton, I. L. G., and Hardy, R. W. (2017). Wolbachia elevates host methyltransferase expression to block an RNA virus early during infection. PLoS Pathog 13:e1006427. doi: 10.1371/journal.ppat.1006427
Binari, R., and Perrimon, N. (1994). Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. Genes Dev. 8, 300–312. doi: 10.1101/gad.8.3.300
Blair, C. D. (2011). Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol. 6, 265–277. doi: 10.2217/fmb.11.11
Blair, C. D. (2019). Deducing the role of virus genome-derived PIWI-Associated RNAs in the mosquito-arbovirus arms race. Front. Genet. 10:1114. doi: 10.3389/fgene.2019.01114
Bottino-Rojas, V., Talyuli, O. A. C., Carrara, L., Martins, A. J., James, A. A., Oliveira, P. L., et al. (2018). The redox-sensing gene Nrf2 affects intestinal homeostasis, insecticide resistance, and Zika virus susceptibility in the mosquito Aedes aegypti. J. Biol. Chem. 293, 9053–9063. doi: 10.1074/jbc.RA117.001589
Briegel, H., Gut, T., and Lea, A. O. (2003). Sequential deposition of yolk components during oogenesis in an insect, Aedes aegypti (Diptera: Culicidae). J. Insect Physiol. 49, 249–260. doi: 10.1016/s0022-1910(02)00272-x
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Caragata, E. P., Dutra, H. L., and Moreira, L. A. (2016). Inhibition of Zika virus by wolbachia in Aedes aegypti. Microb Cell 3, 293–295. doi: 10.15698/mic2016.07.513
Caragata, E. P., Rances, E., O’Neill, S. L., and McGraw, E. A. (2014). Competition for amino acids between Wolbachia and the mosquito host, Aedes aegypti. Microb. Ecol. 67, 205–218. doi: 10.1007/s00248-013-0339-4
Chang, Y.-Y., and Neufeld, T. P. (2010). Autophagy takes flight in Drosophila. FEBS Lett. 584, 1342–1349. doi: 10.1016/j.febslet.2010.01.006
Chen, Q., Wu, G., Chen, H., Li, H., Li, S., Zhang, C., et al. (2019). Quantification of human oral and fecal Streptococcus parasanguinis by use of quantitative real-time PCR targeting the groEL Gene. Front. Microbiol. 10:2910. doi: 10.3389/fmicb.2019.02910
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890.
Cho, K. O., Kim, G. W., and Lee, O. K. (2011). Wolbachia bacteria reside in host Golgi-related vesicles whose position is regulated by polarity proteins. PLoS One 6:e22703. doi: 10.1371/journal.pone.0022703
Cottier, F., Srinivasan, K. G., Yurieva, M., Liao, W., Poidinger, M., Zolezzi, F., et al. (2018). Advantages of meta-total RNA sequencing (MeTRS) over shotgun metagenomics and amplicon-based sequencing in the profiling of complex microbial communities. NPJ Biofilms Microbiomes 4:2.
Cox, J. W., Ballweg, R. A., Taft, D. H., Velayutham, P., Haslam, D. B., and Porollo, A. (2017). A fast and robust protocol for metataxonomic analysis using RNAseq data. Microbiome 5:7. doi: 10.1186/s40168-016-0219-5
Delatte, H., Desvars, A., Bouétard, A., Bord, S., Gimonneau, G., Vourc’h, G., et al. (2010). Blood-feeding behavior of Aedes albopictus, a vector of chikungunya on la réunion. Vector-Borne Zoonotic Dis. 10, 249–258. doi: 10.1089/vbz.2009.0026
Ding, F., Fu, J., Jiang, D., Hao, M., and Lin, G. (2018). Mapping the spatial distribution of Aedes aegypti and Aedes albopictus. Acta Trop. 178, 155–162. doi: 10.1016/j.actatropica.2017.11.020
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Durinck, S., Moreau, Y., Kasprzyk, A., Davis, S., De Moor, B., Brazma, A., et al. (2005). BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinforma Appl. 21, 3439–3440. doi: 10.1093/bioinformatics/bti525
Franz, A. W., Kantor, A. M., Passarelli, A. L., and Clem, R. J. (2015). Tissue barriers to arbovirus infection in mosquitoes. Viruses 7, 3741–3767. doi: 10.3390/v7072795
Fraser, J. E., De Bruyne, J. T., Iturbe-Ormaetxe, I., Stepnell, J., Burns, R. L., Flores, H. A., et al. (2017). Novel Wolbachia-transinfected Aedes aegypti mosquitoes possess diverse fitness and vector competence phenotypes. PLoS Pathog 13:e1006751. doi: 10.1371/journal.ppat.1006751
Gao, H., Cui, C., Wang, L., Jacobs-Lorena, M., and Wang, S. (2020). Mosquito microbiota and implications for disease control. Trends Parasitol. 36, 98–111. doi: 10.1016/j.pt.2019.12.001
Geoghegan, V., Stainton, K., Rainey, S. M., Ant, T. H., Dowle, A. A., Larson, T., et al. (2017). Perturbed cholesterol and vesicular trafficking associated with dengue blocking in Wolbachia-infected Aedes aegypti cells. Nat. Commun. 8:526. doi: 10.1038/s41467-017-00610-8
Giraldo-Calderón, G. I., Calle-Tobón, A., Rozo-López, P., Colpitts, T. M., Park, Y., Rua-Uribe, G. L., et al. (2020). Transcriptome of the Aedes aegypti mosquito in response to human complement proteins. Int. J. Mol. Sci. 21:6584. doi: 10.3390/ijms21186584
Hahn, M. B., Eisen, L., McAllister, J., Savage, H. M., Mutebi, J.-P., and Eisen, R. J. (2017). Updated reported distribution of Aedes (Stegomyia) aegypti and Aedes (Stegomyia) albopictus (Diptera: Culicidae) in the United States, 1995–2016. J. Med. Entomol. 54, 1420–1424. doi: 10.1093/jme/tjx088
Hegde, S., Khanipov, K., Albayrak, L., Golovko, G., Pimenova, M., Saldaña, M. A., et al. (2018). Microbiome interaction networks and community structure from laboratory-reared and field-collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus mosquito vectors. Front. Microbiol. 9:2160. doi: 10.3389/fmicb.2018.02160
Hixson, B., Taracena, M. L., and Buchon, N. (2021). Midgut epithelial dynamics are central to mosquitoes’ physiology and fitness, and to the transmission of vector-borne disease. Front. Cell Infect. Microbiol. 11:653156. doi: 10.3389/fcimb.2021.653156
Houk, E. J. (1977). Midgut ultrastructure of Culex tarsalis (Diptera: Culcidae) before and after a bloodmeal. Tissue Cell 9, 103–118. doi: 10.1016/0040-8166(77)90052-0
Ippolito, M. M., Denny, J. E., Langelier, C., Sears, C. L., and Schmidt, N. W. (2018). Malaria and the microbiome: a systematic review. Clin. Infect. Dis. 67, 1831–1839. doi: 10.1093/cid/ciy374
Janeh, M., Osman, D., and Kambris, Z. (2017). Damage-Induced cell regeneration in the midgut of Aedes albopictus mosquitoes. Sci. Rep. 7:44594. doi: 10.1038/srep44594
Joanne, S., Vythilingam, I., Teoh, B.-T., Leong, C.-S., Tan, K.-K., Wong, M. L., et al. (2017). Vector competence of Malaysian Aedes albopictus with and without Wolbachia to four dengue virus serotypes. Trop Med. Int. Heal. 22, 1154–1165. doi: 10.1111/tmi.12918
Joubert, D. A., Walker, T., Carrington, L. B., De Bruyne, J. T., Kien, D. H. T., Hoang, N. L. T., et al. (2016). Establishment of a Wolbachia superinfection in Aedes aegypti mosquitoes as a potential approach for future resistance management. PLoS Pathog. 12:e1005434. doi: 10.1371/journal.ppat.1005434
Juma, E. O., Allan, B. F., Kim, C. H., Stone, C., Dunlap, C., and Muturi, E. J. (2020). Effect of life stage and pesticide exposure on the gut microbiota of Aedes albopictus and Culex pipiens. Sci. Rep. 10:9489.
Kalappa, D. M., Subramani, P. A., Basavanna, S. K., Ghosh, S. K., Sundaramurthy, V., Uragayala, S., et al. (2018). Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections. Malar. J. 17:385. doi: 10.1186/s12936-018-2535-7
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kamgang, B., Nchoutpouen, E., Simard, F., and Paupy, C. (2012). Notes on the blood-feeding behavior of Aedes albopictus (Diptera: Culicidae) in Cameroon. Parasites Vectors 5:57. doi: 10.1186/1756-3305-5-57
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khattab, A., Barroso, M., Miettinen, T., and Meri, S. (2015). Anopheles midgut epithelium evades human complement activity by capturing factor H from the blood meal. PLoS Negl. Trop. Dis. 9:e0003513. doi: 10.1371/journal.pntd.0003513
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kraemer, M. U. G., Reiner, R. C., Brady, O. J., Messina, J. P., Gilbert, M., Pigott, D. M., et al. (2019). Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 4, 854–863. doi: 10.1038/s41564-019-0376-y
Kubacki, J., Flacio, E., Qi, W., Guidi, V., Tonolla, M., and Fraefel, C. (2020). Viral metagenomic analysis of Aedes albopictus mosquitos from Southern Switzerland. Viruses 12:929. doi: 10.3390/v12090929
Kwon, Y., Kim, M., Jung, H. S., Kim, Y., and Jeoung, D. (2019). Targeting autophagy for overcoming resistance to Anti-EGFR treatments. Cancers (Basel) 11:1374. doi: 10.3390/cancers11091374
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12:323. doi: 10.1186/1471-2105-12-323
Lindh, E., Argentini, C., Remoli, M. E., Fortuna, C., Faggioni, G., Benedetti, E., et al. (2019). The Italian 2017 outbreak chikungunya virus belongs to an emerging aedes albopictus–adapted virus cluster introduced from the indian subcontinent. Open Forum Infect Dis. 6:ofy321. doi: 10.1093/ofid/ofy321/5239584
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, L., Jiang, L. Y., Xiao, X. C., Di, B., Jing, Q. L., Wang, S. Y., et al. (2017). The dengue preface to endemic in mainland China: the historical largest outbreak by Aedes albopictus in Guangzhou, 2014. Infect. Dis. Poverty. 6:148. doi: 10.1186/s40249-017-0352-9
Mahmud, M. A. F., Abdul Mutalip, M. H., Lodz, N. A., Muhammad, E. N., Yoep, N., Hashim, M. H., et al. (2019). Environmental management for dengue control: a systematic review protocol. BMJ Open 9:e026101. doi: 10.1136/bmjopen-2018-026101
Manni, M., and Zdobnov, E. M. (2020). A novel anphevirus in aedes albopictus mosquitoes is distributed worldwide and interacts with the host RNA interference pathway. Viruses 12:1264. doi: 10.3390/v12111264
Marconcini, M., Hernandez, L., Iovino, G., Houé, V., Valerio, F., Palatini, U., et al. (2019). Polymorphism analyses and protein modelling inform on functional specialization of Piwi clade genes in the arboviral vector Aedes albopictus. PLoS Negl. Trop. Dis. 13:e0007919. doi: 10.1371/journal.pntd.0007919
Margos, G., Navarette, S., Butcher, G., Davies, A., Willers, C., Sinden, R. E., et al. (2001). Interaction between host complement and mosquito-midgut-stage Plasmodium berghei. Infect. Immun. 69, 5064–5071. doi: 10.1128/IAI.69.8.5064-5071.2001
Martinson, V. G., and Strand, M. R. (2021). Diet-Microbiota interactions alter mosquito development. Front. Microbiol. 12:650743. doi: 10.3389/fmicb.2021.650743
Molloy, J. C., Sommer, U., Viant, M. R., and Sinkins, S. P. (2016). Wolbachia modulates lipid metabolism in Aedes albopictus mosquito Cells. Appl. Environ. Microbiol. 82, 3109–3120. doi: 10.1128/AEM.00275-16
Moore, C. G., and Mitchell, C. J. (1997). Aedes albopictus in the United States: ten-year presence and public health implications. Emerg. Infect. Dis. 3, 329–334. doi: 10.3201/eid0303.970309
Moreira, L. A., Iturbe-Ormaetxe, I., Jeffery, J. A., Lu, G., Pyke, A. T., Hedges, L. M., et al. (2009). A wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and plasmodium. Cell 139, 1268–1278. doi: 10.1016/j.cell.2009.11.042
Moreno-Madriñán, M. J., and Turell, M. (2017). Factors of concern regarding zika and other aedes aegypti-transmitted viruses in the United States. J. Med. Entomol. 54, 251–257. doi: 10.1093/jme/tjw212
Moreno-Madriñán, M. J., and Turell, M. (2018). History of mosquitoborne diseases in the United States and implications for new pathogens. Emerg. Infect. Dis. 24, 821–826. doi: 10.3201/eid2405.171609
Mousson, L., Zouache, K., Arias-Goeta, C., Raquin, V., Mavingui, P., and Failloux, A. B. (2012). The native wolbachia symbionts limit transmission of dengue virus in Aedes albopictus. PLoS Negl. Trop. Dis. 6:e1989. doi: 10.1371/journal.pntd.0001989
Muller, P., Kuttenkeuler, D., Gesellchen, V., Zeidler, M. P., and Boutros, M. (2005). Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 436, 871–875. doi: 10.1038/nature03869
Nguyen, L. T., Schmidt, H. A., Von, Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Öhlund, P., Lundén, H., and Blomström, A. L. (2019). Insect-specific virus evolution and potential effects on vector competence. Virus Genes. 55, 127–137. doi: 10.1007/s11262-018-01629-9
Oliveira, J. H., Goncalves, R. L., Lara, F. A., Dias, F. A., Gandara, A. C., Menna-Barreto, R. F., et al. (2011). Blood meal-derived heme decreases ROS levels in the midgut of Aedes aegypti and allows proliferation of intestinal microbiota. PLoS Pathog 7:e1001320. doi: 10.1371/journal.ppat.1001320
Oliveira, J. H. M., Talyuli, O. A. C., Goncalves, R. L. S., Paiva-Silva, G. O., Sorgine, M. H. F., Alvarenga, P. H., et al. (2017). Catalase protects Aedes aegypti from oxidative stress and increases midgut infection prevalence of Dengue but not Zika. PLoS Negl. Trop. Dis. 11:e0005525. doi: 10.1371/journal.pntd.0005525
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
O’Neill, S. L. (2018). The use of wolbachia by the world mosquito program to interrupt transmission of Aedes aegypti transmitted viruses. Adv. Exp. Med. Biol. 1062, 355–360. doi: 10.1007/978-981-10-8727-1_24
Pakpour, N., Riehle, M. A., and Luckhart, S. (2014). Effects of ingested vertebrate-derived factors on insect immune responses. Curr. Opin. Insect Sci. 3, 1–5. doi: 10.1016/j.cois.2014.07.001
Parry, R., and Asgari, S. (2018). Aedes anphevirus: an insect-specific virus distributed worldwide in Aedes aegypti mosquitoes that has complex interplays with wolbachia and dengue virus infection in cells. J. Virol. 92:e00224-18. doi: 10.1128/JVI.00224-18
Pereira-Filho, A. A., Mateus Pereira, R. H., da Silva, N. C. S., Ferreira Malta, L. G., Serravite, A. M., and Carvalho, et al. (2020). The gut anti-complement activity of Aedes aegypti: investigating new ways to control the major human arboviruses vector in the Americas. Insect Biochem. Mol. Biol. 120:103338. doi: 10.1016/j.ibmb.2020.103338
Perera, R., Riley, C., Isaac, G., Hopf-Jannasch, A. S., Moore, R. J., Weitz, K. W., et al. (2012). Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog 8:e1002584. doi: 10.1371/journal.ppat.1002584
Pinch, M., Mitra, S., Rodriguez, S. D., Li, Y., Kandel, Y., Dungan, B., et al. (2021). Fat and happy: profiling mosquito fat body lipid storage and composition post-blood meal. Front. Insect Sci. 1:168. doi: 10.3389/finsc.2021.693168
Raquin, V., Valiente Moro, C., Saucereau, Y., Tran, F.-H., Potier, P., and Mavingui, P. (2015). Native Wolbachia from Aedes albopictus blocks chikungunya virus infection in cellulo. PLoS One 10:e0125066. doi: 10.1371/journal.pone.0125066
Rezza, G. (2012). Aedes albopictus and the reemergence of Dengue. BMC Public Health 12:72. doi: 10.1186/1471-2458-12-72
Richards, S. L., Ponnusamy, L., Unnasch, T. R., Hassan, H. K., and Apperson, C. S. (2006). Host-Feeding patterns of Aedes albopictus (Diptera: Culicidae) in relation to availability of human and domestic animals in suburban landscapes of Central North Carolina. J. Med. Entomol. 43, 543–551. doi: 10.1603/0022-2585(2006)43[543:hpoaad]2.0.co;2
Robert, M. A., Stewart-Ibarra, A. M., and Estallo, E. L. (2020). Climate change and viral emergence: evidence from Aedes-borne arboviruses. Curr. Opin. Virol. 40, 41–47. doi: 10.1016/j.coviro.2020.05.001
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Sadeghi, M., Altan, E., Deng, X., Barker, C. M., Fang, Y., Coffey, L. L., et al. (2018). Virome of 12 thousand Culex mosquitoes from throughout California. Virology 523, 74–88. doi: 10.1016/j.virol.2018.07.029
Schultz, M. J., Frydman, H. M., and Connor, J. H. (2018). Dual Insect specific virus infection limits arbovirus replication in Aedes mosquito cells. Virology 518, 406–413. doi: 10.1016/j.virol.2018.03.022
Scolari, F., Casiraghi, M., and Bonizzoni, M. (2019). Aedes spp. and their microbiota: a review. Front. Microbiol. 10:2036. doi: 10.3389/fmicb.2019.02036
Seabournid, P., Spafford, H., Yoneishi, N., and Medeiros, M. (2020). The Aedes albopictus (Diptera: Culicidae) microbiome varies spatially and with ascogregarine infection. PLoS Negl. Trop. Dis. 14:e0008615. doi: 10.1371/journal.pntd.0008615
Seipke, R. F., Barke, J., Brearley, C., Hill, L., Yu, D. W., Goss, R. J. M., et al. (2011). A single streptomyces symbiont makes multiple antifungals to support the fungus farming ant acromyrmex octospinosus. PLoS One 6:e22028. doi: 10.1371/journal.pone.0022028
Shi, C., Zhao, L., Atoni, E., Zeng, W., Hu, X., Matthijnssens, J., et al. (2020). Stability of the virome in lab-and field-collected aedes albopictus mosquitoes across different developmental stages and possible core viruses in the publicly available virome data of Aedes mosquitoes. mSystems 5:e00640-20. doi: 10.1128/mSystems.00640-20
Simon, N., Friedrich, O., and Kappes, B. (2018). Quantification of human complement factor H binding to asexual malaria blood stages by an enzyme-linked immunosorbent assay. Vaccine 36, 1545–1547. doi: 10.1016/j.vaccine.2018.01.080
Sinkins, S. P., Braig, H. R., and O’Neill, S. L. (1995). Wolbachia superinfections and the expression of cytoplasmic incompatibility. Proc. R. Soc. B Biol. Sci. 261, 325–330.
Slonchak, A., Hussain, M., Torres, S., Asgari, S., and Khromykh, A. A. (2014). Expression of mosquito microRNA Aae-miR-2940-5p is downregulated in response to West Nile virus infection to restrict viral replication. J. Virol. 88, 8457–8467. doi: 10.1128/JVI.00317-14
Smith, R. C., Vega-Rodriguez, J., and Jacobs-Lorena, M. (2014). The Plasmodium bottleneck: malaria parasite losses in the mosquito vector. Mem. Inst. Oswaldo Cruz 109, 644–661. doi: 10.1590/0074-0276130597
Taracena, M. L., Bottino-Rojas, V., Talyuli, O. A. C., Walter-Nuno, A. B., Oliveira, J. H. M., Anglero-Rodriguez, Y. I., et al. (2018). Regulation of midgut cell proliferation impacts Aedes aegypti susceptibility to dengue virus. PLoS Negl. Trop. Dis. 12:e0006498. doi: 10.1371/journal.pntd.0006498
Terenius, O., Lindh, J. M., Eriksson-Gonzales, K., Bussière, L., Laugen, A. T., Bergquist, H., et al. (2012). Midgut bacterial dynamics in Aedes aegypti. FEMS Microbiol Ecol. 80, 556–565. doi: 10.1111/j.1574-6941.2012.01317.x
Vogel, K. J., Brown, M. R., and Strand, M. R. (2015). Ovary ecdysteroidogenic hormone requires a receptor tyrosine kinase to activate egg formation in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. U S A. 112, 5057–5062. doi: 10.1073/pnas.1501814112
Wang, Y., Gilbreath, T. M., Kukutla, P., Yan, G., and Xu, J. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. doi: 10.1371/journal.pone.0024767
Whiten, S. R., Eggleston, H., and Adelman, Z. N. (2017). Ironing out the details: exploring the role of iron and heme in blood-sucking arthropods. Front. Physiol. 8:1134. doi: 10.3389/fphys.2017.01134
Yan, H., Zhou, Y., Liu, Y., Deng, Y., and Chen, X. (2014). miR-252 of the Asian tiger mosquito Aedes albopictus regulates dengue virus replication by suppressing the expression of the dengue virus envelope protein. J. Med. Virol. 86, 1428–1436. doi: 10.1002/jmv.23815
Zakrzewski, M., Rasic, G., Darbro, J., Krause, L., Poo, Y. S., Filipovic, I., et al. (2018). Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 8:4690. doi: 10.1038/s41598-018-22945-y
Zhang, X., Huang, S., Jin, T., Lin, P., Huang, Y., Wu, C., et al. (2018). Discovery and high prevalence of Phasi Charoen-like virus in field-captured Aedes aegypti in South China. Virology 523, 35–40. doi: 10.1016/j.virol.2018.07.021
Zhao, L., Atoni, E., Shi, C., Yuan, Z., and Xia, H. (2019). Mapping the virome in lab-reared and wild-caught Aedes albopictus mosquitoes. Access Microbiol. 1:4.
Zhou, W., Rousset, F., and O’Neil, S. (1998). Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc. R Soc. B Biol. Sci. 265, 509–515. doi: 10.1098/rspb.1998.0324
Zouache, K., Voronin, D., Tran-Van, V., Mousson, L., Failloux, A.-B., and Mavingui, P. (2009). Persistent Wolbachia and cultivable bacteria infection in the reproductive and somatic tissues of the mosquito vector Aedes albopictus. PLoS One 4:e6388. doi: 10.1371/journal.pone.0006388
Zucchi, T. D., Guidolin, A. S., and Cônsoli, F. L. (2011). Isolation and characterization of actinobacteria ectosymbionts from Acromyrmex subterraneus brunneus (Hymenoptera, Formicidae). Microbiol Res. 166, 68–76. doi: 10.1016/j.micres.2010.01.009
Keywords: RNA-sequencing, tiger-mosquito, Aedes-mosquitoes, RNA viruses, Wolbachia, complement-system, immunity
Citation: Calle-Tobón A, Holguin-Rocha AF, Moore C, Rippee-Brooks M, Rozo-Lopez P, Harrod J, Fatehi S, Rua-Uribe GL, Park Y and Londoño-Rentería B (2021) Blood Meals With Active and Heat-Inactivated Serum Modifies the Gene Expression and Microbiome of Aedes albopictus. Front. Microbiol. 12:724345. doi: 10.3389/fmicb.2021.724345
Received: 12 June 2021; Accepted: 12 August 2021;
Published: 09 September 2021.
Edited by:
Camila Carlos-Shanley, Texas State University, United StatesReviewed by:
Stefanie Christine Becker, University of Veterinary Medicine Hannover, GermanyNathalie Chazal, Université de Montpellier, France
Copyright © 2021 Calle-Tobón, Holguin-Rocha, Moore, Rippee-Brooks, Rozo-Lopez, Harrod, Fatehi, Rua-Uribe, Park and Londoño-Rentería. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berlin Londoño-Rentería, YmVybGlubG9uZG9ub0BnbWFpbC5jb20=