
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 August 2021
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.718487
 Christopher J. Waite*†
Christopher J. Waite*† Anya Lindström Battle
Anya Lindström Battle Mark H. Bennett
Mark H. Bennett Matthew R. Carey
Matthew R. Carey Chun K. Hong
Chun K. Hong Ioly Kotta-Loizou
Ioly Kotta-Loizou Martin Buck
Martin Buck Jörg Schumacher*†
Jörg Schumacher*†Free-living nitrogen-fixing bacteria can improve growth yields of some non-leguminous plants and, if enhanced through bioengineering approaches, have the potential to address major nutrient imbalances in global crop production by supplementing inorganic nitrogen fertilisers. However, nitrogen fixation is a highly resource-costly adaptation and is de-repressed only in environments in which sources of reduced nitrogen are scarce. Here we investigate nitrogen fixation (nif) gene expression and nitrogen starvation response signaling in the model diazotroph Klebsiella oxytoca (Ko) M5a1 during ammonium depletion and the transition to growth on atmospheric N2. Exploratory RNA-sequencing revealed that over 50% of genes were differentially expressed under diazotrophic conditions, among which the nif genes are among the most highly expressed and highly upregulated. Isotopically labelled QconCAT standards were designed for multiplexed, absolute quantification of Nif and nitrogen-stress proteins via multiple reaction monitoring mass spectrometry (MRM-MS). Time-resolved Nif protein concentrations were indicative of bifurcation in the accumulation rates of nitrogenase subunits (NifHDK) and accessory proteins. We estimate that the nitrogenase may account for more than 40% of cell protein during diazotrophic growth and occupy approximately half the active ribosome complement. The concentrations of free amino acids in nitrogen-starved cells were insufficient to support the observed rates of Nif protein expression. Total Nif protein accumulation was reduced 10-fold when the NifK protein was truncated and nitrogenase catalysis lost (nifK1–1203), implying that reinvestment of de novo fixed nitrogen is essential for further nif expression and a complete diazotrophy transition. Several amino acids accumulated in non-fixing ΔnifLA and nifK1–1203 mutants, while the rest remained highly stable despite prolonged N starvation. Monitoring post-translational uridylylation of the PII-type signaling proteins GlnB and GlnK revealed distinct nitrogen regulatory roles in Ko M5a1. GlnK uridylylation was persistent throughout the diazotrophy transition while a ΔglnK mutant exhibited significantly reduced Nif expression and nitrogen fixation activity. Altogether, these findings highlight quantitatively the scale of resource allocation required to enable the nitrogen fixation adaptation to take place once underlying signaling processes are fulfilled. Our work also provides an omics-level framework with which to model nitrogen fixation in free-living diazotrophs and inform rational engineering strategies.
Most organisms are unable to directly assimilate inert atmospheric dinitrogen (N2) and rely instead on the bio-availability of chemically reduced, or “fixed,” forms of nitrogen (N), such as ammonia, nitrate or amino acids. Food security is fundamentally dependent on the uptake of fixed N by agricultural crops. Biological N fixation – the reduction of N2 to NH3 by specialised microorganisms – replenishes most of the fixed N in the biosphere and supports approximately 60% of the N required for human nutrition (Smil, 2001). However, as crop production must increase by up to 70% to support the estimated global food demand in 2050 (Hunter et al., 2017), agriculture is growing ever more reliant on the synthetically fixed and environmentally damaging N fertilisers produced by the industrial Haber–Bosch process. Addressing the severe anthropogenic imbalances in the global N cycle is critical to managing the triple global challenges of food security, climate change, and environmental degradation (Steffen et al., 2015; Zhang et al., 2015). Modern biotechnological approaches offer great promise for expanding the capacity of biological N fixation and the development of more sustainable solutions for the delivery of N to plants (e.g., Rogers and Oldroyd, 2014; Batista and Dixon, 2019).
Biological N fixation, widely distributed among bacterial lineages and compatible with a range of physiologies, is catalysed by the structurally and mechanistically conserved nitrogenase enzyme (Dixon and Kahn, 2004). The universal molybdenum-iron (MoFe) nitrogenase consists of two oxygen-sensitive metalloproteins. The MoFe protein is a heterotetramer of NifD and NifK subunits and contains an iron-molybdenum cofactor in each catalytic site for N2 reduction. The iron (Fe) protein, which acts as an electron donor for MoFe, is a homodimer of NifH. N2 reduction occurs with a slow catalytic turnover rate (∼5 s–1) (Hageman and Burris, 1978) and an optimal stoichiometry of: N2 + 8 H+ + 8 e– + 16 MgATP → 2 NH3 + H2 + 6 MgADP + 16 Pi (Thorneley and Lowe, 1985; Seefeldt et al., 2009). As such, diazotrophic growth on N2 as a sole N source necessitates both a high nitrogenase concentration (up to 20% of total cell protein) and considerable energetic (ATP) input (Dixon and Kahn, 2004).
The 24-kb nitrogen fixation (nif) gene cluster of the free-living diazotroph Klebsiella oxytoca (Ko) has been the model system for molecular biology studies of biological nitrogen fixation (Arnold et al., 1988). Consisting of 20 genes arranged into 8 operons, this cluster is sufficient for heterologous N fixation in non-diazotrophs such as Escherichia coli (Dixon and Postgate, 1972). Besides the structural proteins NifHDK, many other nif gene products function coordinately in the assembly of the mature nitrogenase complex: synthesis and insertion of the FeMo-cofactor (NifBEQNVXY); synthesis of Fe–S and P metalloclusters (NifSUZ); maturation of MoFe protein (NifWYZ); maturation of (NifMW); and electron transport to (NifFJ) the Fe protein (Jimenez-Vicente et al., 2018; Mus et al., 2018). The products of the nifLA operon regulate the transcription of the other nif operons via conserved promoter elements. Given the significant energetic costs associated with nitrogenase expression and function, diazotrophs utilise common regulatory principles to restrict nif gene expression to the optimal environmental conditions. Among proteobacteria, the σ54-type bacterial enhancer binding protein (bEBP) NifA stringently couples nif transcription to the availability of fixed N, energy status, and microaerobic oxygen concentration (Dixon and Kahn, 2004). In Ko, NifA is sequestered in an inactive form in non-optimal conditions by the anti-activator flavoprotein NifL (Henderson et al., 1989; Martinez-Argudo et al., 2004). Upon anaerobiosis, NifL is reduced and sequestered to the cytoplasmic membrane, contributing to the release of active NifA (Klopprogge et al., 2002).
NifA is embedded in the global N regulation (ntr) signaling cascade at two levels, with both nifLA transcription and the stability of the NifLA complex independently coupled to the availability of fixed N (Drummond et al., 1983). During N limitation stress, the two-component system NtrB–NtrC is responsible for expression of the large ntr regulon (∼50 genes) associated with N assimilation, scavenging, and growth on alternative N sources (Zimmer et al., 2000). Like NifA, the NtrC response regulator is a σ54-type bEBP, which is activated via phosphorylation in N limiting conditions. The paralogous PII-type signal transduction proteins GlnB and GlnK are subject to reversible covalent modification by the uridylyltransferase/uridylylremovase (UT/UR) enzyme in direct response to the intracellular glutamine concentration, a key indicator of cellular N status (Merrick, 2015). The PII proteins also report on cellular carbon status via allosteric interactions with 2-oxoglutarate (α-KG) and ATP. When intracellular glutamine is low, the trimeric PII-type proteins are uridylylated and stimulate phosphorylation of NtrC by NtrB. Activated NtrC initiates transcription of the nifLA operon, in addition to glnA [encoding the glutamine synthetase (GS) enzyme essential for ATP-dependent assimilation of ammonium at low concentrations], amtB (ammonium transporter channel), and glnK. Conversely, the sensing of high glutamine levels in the presence of replete ammonium, the preferred N source for rapid growth, results in de-phosphorylation of NtrC and stringent repression of the Ntr regulon (Schumacher et al., 2013). GlnK expression is induced by N limitation whilst GlnB is constitutively expressed, with the result that the PII proteins are believed to fulfill distinct roles in N regulation (Dixon and Kahn, 2004; Gosztolai et al., 2017). GlnK specifically exerts the second layer of N-mediated control over nif gene expression through its interaction with the NifLA complex. Evidence suggests that in Ko, GlnK is required to relieve NifA of inhibition by NifL (Jack et al., 1999), independently of its uridylylation state (He et al., 1998). NifA is constitutively active in the absence of NifL (Kim et al., 1989), highlighting the importance of the anti-activator in integrating physiological signals of N limitation (GlnK) and anaerobiosis (redox state) in the control of nif expression (Martinez-Argudo et al., 2004).
Re-wiring the metabolism of free-living soil diazotrophs to achieve excretion of surplus fixed N in the plant root environment represents a short-term alternative to the two ambitious synthetic biology strategies being explored for the development of N-fixing crops (Batista and Dixon, 2019): engineering N-fixing symbioses in non-leguminous crops (Rogers and Oldroyd, 2014) and expression of nitrogenase function directly in plant cells (Burén et al., 2018). Mutant diazotroph strains defective in the regulation of N metabolism have proved effective in plant growth promotion in N limited soils (Iniguez et al., 2007; Setten et al., 2013; Barney et al., 2015; Pankievicz et al., 2015; Ambrosio et al., 2017) and are now a key constituent of newly emerging commercial agricultural inoculants (Bloch et al., 2020; Temme et al., 2020).
Essential to these ongoing biotechnological approaches is the understanding and repurposing of the Ko nif regulon, which has previously been refactored using standardised synthetic parts for robust heterologous control (Temme et al., 2012; Smanski et al., 2014), reorganised into large cleavable polyproteins for stoichiometric expression in eukaryotic hosts (Yang et al., 2018) and partially expressed in the plant mitochondrial matrix (Allen et al., 2017). However, there are fundamental aspects of the free-living diazotrophy adaptation still to be understood, such as how cells can afford the significant protein investment in nitrogenase under N limitation stress. A long evolved and complex regulatory network exists to maximise cell growth over a wide range of N sources, likely preventing adaptive resource investment in non-optimal conditions and minimizing leakage of fixed N into the environment. This implies that perturbing N metabolism has the potential to yield significant fitness penalties in competitive soil environments. A mechanism by which both fixed N excretion and competitive fitness can be achieved has yet to be revealed (Batista and Dixon, 2019).
To provide a basis to rationally engineer diazotrophy in bacteria, here we present a multi-omics framework, exploiting quantitative proteomics, targeted metabolite estimates, and transcriptome analyses, with which to model N economy in Ko. By focusing on the transition from N-replete to diazotrophic growth states, we explore how the N regulatory network enables adaptation to transient fluctuations in extracellular N similar to those experienced in natural environments.
The bacterial strains, plasmids, and oligonucleotides used in this study are described in Supplementary Table 1. E. coli and Ko strains were grown in Lysogeny Broth (LB) at 37 or 30°C, respectively, for general propagation, cloning, and transformations. The Ko M5a1 strain was obtained from Ruth Schmitz (Christian Albrechts University, Kiel). The ΔnifLA, ΔglnK, and ΔglnB knockout mutants and nifK1–1203 truncation mutant were derived by Lambda Red recombineering (Datsenko and Wanner, 2000). Oligonucleotides were designed in order to amplify a kanamycin resistance cassette (nptII) from the pGEM-T-KanFRT plasmid (Zumaquero et al., 2010) with 60 nt overhangs homologous to the flanking regions of the Ko M5a1 genomic region targeted for knockout mutagenesis (pGEM-T-KanFRT binding region in upper case in Supplementary Table 1). PCR was performed using Phusion polymerase (Thermo Scientific) with 25 ng template plasmid and an annealing temperature of 55°C. Template DNA was removed by DpnI digestion prior to gel extraction of products. Ko M5a1 was transformed with the pKD46 plasmid, expressing the Lambda Red genes required for homologous recombination under the control of an arabinose-inducible promoter. Competent Red-recombinase expressing cells were prepared in super optimal broth (SOB) [0.5% (w/v) yeast extract, 2% (w/v) tryptone, 10 mM NaCl, 2.5 mM KCl, 20 mM MgSO4], to which L-arabinose was added to a final concentration of 10 mM once OD600 reached 0.1. Cells were grown to an OD600 of 0.4–0.6 before being washed three times in sterile cold 10% glycerol solution. Approximately 1 μg of purified PCR product was incubated with 100 μL of competent cells on ice for 30 min prior to electroporation. Transformed cells were recovered at 30°C for 3 h before being plated out on LB agar plates supplemented with kanamycin. Kanamycin-resistant colonies were used to generate seed cultures from which genomic DNA was extracted. Locus-specific homologous recombination was confirmed by diagnostic PCR, combining primers specific to regions flanking the knockout locus with those specific to the nptII cassette, and Sanger sequencing.
NH4+ run-out was used to de-repress gln/nif gene expression and achieve a reproducible transition into diazotrophic growth. K. oxytoca strains were cultured in Nitrogen-Free David and Mingioli (NFDM) medium [69 mM K2HPO4, 25 mM KH2PO4, 0.1 mM Na2MoO4, 90 μM FeSO4, 0.8 mM MgSO4, 2% (w/v) glucose] supplemented with NH4Cl as a nitrogen source. To ensure replete cellular N status, seed cultures were supplemented with 20 mM NH4Cl and grown to an OD600 of approximately 2–3. Following centrifugation at 4000 × g and 4°C, cell pellets were washed once and resuspended in NFDM supplemented with 0.5 mM NH4Cl to an OD600 of 0.1. Where required, N-replete controls were prepared similarly but supplemented with an additional 10 mM NH4Cl. For small-scale growth assays, 200-μL cultures were incubated in triplicate in sterile 96-well culture plates and OD600 was measured at 10-min intervals using a CLARIOstar plate reader (BMG LABTECH), in which a microaerobic atmosphere (0.2% O2, 1% CO2, and 98.8% N2) was maintained using an atmospheric control unit and gas cylinders. For batch culture experiments, 100 mL cultures were sealed in 150-mL Erlenmeyer flasks using Suba-Seal septa (Sigma-Aldrich) and chilled on ice to slow cell growth and NH4Cl depletion whilst flushed with N2 gas for 45 min. Colorimetric O2xyDot sensors (OxySense) fixed inside the flasks were used to verify micro-aerobic conditions. Cultures were warmed to 25°C and shaken at 200 rpm for up to 24 h. To sample cell culture, an equivalent volume of oxygen free N2 gas was injected into the headspace via gas-tight syringe and the flask inverted in order to extract cells and medium. All experiments were performed with three independent biological replicate cultures for each strain or condition.
One milliliter pure, O2-free acetylene (BOC) at room temperature and pressure was injected into air-tight culture vessels at the beginning of run-out experiments (t = 0 h). The partial pressure of acetylene used was sub-saturating and approximately equivalent to the Km of the nitrogenase (Harris et al., 2018). Five hundred microliters of culture headspace was sampled via gas-tight syringe at 2.5-h intervals, immediately prior to cell culture sampling, and subject to gas chromatography through a HayeSep N column (Agilent) at 90°C in N2 carrier gas. Acetylene and ethylene were detected by flame ionisation (FID) at 300°C and ChemStation software (Agilent) was used to integrate signal peak areas. Pure standards of acetylene and ethylene were injected prior to samples to confirm retention times and a ΔnifLA culture was used as a negative control. The rate of ethylene production per unit cell density (nmol ethylene OD600–1 mL–1 h–1) was estimated using a standard calibration curve (data not shown). Dilution of ethylene as a result of culture sampling was corrected for by measuring the concomitant dilution of acetylene.
The concentration of NH4+ ions in 200 μL centrifuged culture supernatant was determined semi-quantitatively using the colorimetric MColorTest kit (Merck). A color change from yellow to blue was measured by absorbance at 690 nm and NH4+ concentration calculated against a standard calibration curve (78–5000 μM).
Micro-aerobic Ko M5a1 cell cultures were prepared as described above (see section “Diazotrophic Growth”). Briefly, duplicate cultures with an OD600 of 0.1 were prepared from an N-replete seed culture and supplemented with 0.5 mM (N run-out) or 10 mM (N-replete) NH4Cl, respectively. Once N-fixation was confirmed by acetylene reduction (t = 8 h), diazotrophic cell samples were fixed with a 1/10 volume of 5% phenol/95% ethanol (v/v), harvested by centrifugation and stored on dry ice. A single sample of N-replete cells was harvested early during exponential phase (t = 1.5 h). Whole-cell RNA extraction, library preparation and quality control, next-generation sequencing, and read alignment was performed commercially by Vertis Biotechnologie AG (Freising, Germany). Cell samples were treated with lysozyme for 15 min in order to extract total RNA, which was then isolated using the mirVana microRNA isolation kit (Ambion). DNase was used to specifically remove contaminant DNA and ribosomal RNA was depleted using the Ribo-Zero rRNA Removal Kit for bacteria (Illumina). The primary RNA transcripts were fragmented using ultrasound (four pulses of 30 s each at 4°C) prior to ligation of 3′ oligonucleotide adapters. First-strand cDNA synthesis via Moloney murine leukemia virus (M-MLV) reverse transcriptase was primed using the 3′ adapter. Following purification of first-strand cDNA, a 5′ Illumina TruSeq sequencing adapter (Illumina), inclusive of an 8 nt barcode sequence, was ligated to the 3′ end of the antisense cDNA prior to PCR-amplification of double-stranded cDNA to about 10–20 ng/μL (12–13 thermocycles) using a high-fidelity DNA polymerase. cDNA was purified, pooled in equimolar quantities, and size-fractionated (200–500 bp) using the Agencourt AMPure XP kit (Beckman Coulter Genomics) prior to analysis by capillary electrophoresis. The cDNA pool was sequenced strand-specifically on an Illumina NextSeq 500 system, utilising a read length of 75 nt and a depth of 100 million reads. The output reads were trimmed to remove adapter sequences and filtered to remove duplicates, prior to alignment to the Ko M5a1 genome (GenBank WGS accession JAFHKG010000000; BioSample SAMN17288411). Data was provided by Vertis Biotechnologie in the form of raw reads (fastq), binary alignment map (bam) files, and normalised gene counts (reads per kilobase per million, RPKM) (Mortazavi et al., 2008). Raw and processed datasets are accessible via the NCBI Gene Expression Omnibus (GEO Series accession GSE164668). Across all samples, between 22.0 and 25.1 million reads (81.4–87.7%) were mapped to the genome, including 56.7–68.5% mapped to annotated genes. Alignments were visualised manually using Integrative Genomics Viewer (IGV) (Robinson et al., 2011). Differential gene expression analysis was performed using the R/Bioconductor statistical package DEseq2 (Love et al., 2014), utilising settings of regularised logarithm (rlog) normalisation of read counts, Wald hypothesis testing, and p-value adjustment using a False Discovery Rate threshold of 0.05 (Benjamini and Hochberg, 1995). Log2 fold changes between conditions were calculated for differentially expressed genes (DEGs) with an adjusted p-value of <0.05. DEG lists were mapped to the KEGG Orthology database using the KEGG Automatic Annotation Server (Moriya et al., 2007), using translated coding sequences as queries for the bi-directional best-hit (BBH) method of ortholog assignment.
A methanol-chloroform extraction procedure (Sapcariu et al., 2014) was used for simultaneous, reliable, and medium-throughput extraction of total cell protein and polar metabolites. Fifteen milliliters cell culture (at variable OD600) was harvested by centrifugation at 4000 × g and 4°C and the pellet rapidly frozen in liquid nitrogen. Cell sample was thawed and resuspended in 800 μL ice-cold 50% (v/v) methanol, followed by the addition of 400 μL ice-cold chloroform. The mixture was agitated at 1000 rpm for 10 min in a cooled TissueLyser II cell disruptor (Qiagen) prior to phase separation by centrifugation at 15,000 × g. Seven-fifty microliters of the upper polar phase was extracted and evaporated in a rotary vacuum evaporator until dry, prior to storage at −80°C. The protein interphase was washed in 50% methanol and vortex solubilised in 500 μL urea-phosphate buffer (7 M urea, 69 mM K2HPO4, 25 mM KH2PO4). Total protein concentration was determined using the colorimetric DC assay (Biorad) according to manufacturer instructions.
Analyte peptides were prepared by incubating 30 μL total cell protein sample and a mix of protein standards (see below) with 2 μg sequencing grade modified trypsin (Promega) for 5 h at 37°C followed by 20 h at 25°C in a final volume of 300 μL digestion buffer [100 mM Tris–HCl (pH 8), 50 mM NH4HCO3, 1 mM TCEP]. Ten microliters control samples taken before and after incubation with trypsin were run on SDS-PAGE to verify complete digestion prior to inactivation of trypsin with 2% formic acid. Dried polar metabolites were resuspended in 100 μL 50% methanol prior to a 10 μL aliquot being derivatised using the 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate AccQTag Ultra reagent (Waters), as described previously (Gray et al., 2017). Both peptide and amino acid samples were separated by reverse-phase chromatography on a Prominence HPLC system (Shimadzu) and analyzed using a linear ion trap triple-quadrupole 6500 Qtrap mass spectrometer (AB Sciex). Forty microliters peptide sample injections were separated isothermally at 50°C through a Luna C18 silica column (Phenomenex, 100 Å pore size, 3 μm particle size, 100 mm × 4.6 mm) with a multi-step solvent gradient: 100% solvent A (94.9% H2O, 5% acetonitrile, 0.1% formic acid), equilibration; 0–25% solvent B (94.9% acetonitrile, 5% H2O, 0.1% formic acid), 30 min; 25–50% B, 5 min; 50–100% B; 100% B, 5 min. Five microliters derivatised metabolite sample injections were separated isothermally at 45°C and 300 μL per minute through an Acquity T3 C18 high-strength silica UPLC column (Waters, 100 Å pore size, 1.8 μm particle size, 100 mm × 2.1 mm) with gradient: 4% B, 0.1 min; 28% B, 10 min; 80% B, 1.5 min; 4% B, 1.5 min. The MS was operated in positive ion mode with the following general parameters: curtain gas, 40 psi; ion source nebulising gas, 40 psi; ion source turbo gas, 60 psi; curtain gas, 40 psi; ion spray voltage, 5500 V; source gas temperature, 500°C; entrance potential, 10 V. The collision cell exit potential was set at a constant 15 V and the retention time detection window at 180–360 s. For MS/MS of amino acids, the following parameters were set as constants: collision energy, 30 V; declustering potential 30 V, collision cell exit potential, 10 V; retention time detection window, 90 s. Transitions lists detailing further analytical parameters specific to peptide and amino acid targets (e.g., Q1/Q3 mass, retention time) are provided in Supplementary Data 1, 3, respectively. Signal and standard peak areas were manually validated and integrated using Skyline software (MacLean et al., 2010) prior to calculation of analyte concentrations and statistical analysis in R. Peptide quantification was achieved by summing the integrated peak areas of two or more validated transitions. Where possible, the abundances of at least two correlative peptides for each protein were averaged to assign protein copy number. Protein copy number per cell values assume an OD-specific cell density of 109 CFU mL–1 OD–1. Post-translational modification (PTM) values are expressed as the ratio of modified/total (unmodified + modified) states. Amino acid concentrations were calculated relative to external standard curves of a standard mix of 20 amino acids (Cell Free Amino Acid Mixture-15N, Sigma-Aldrich) diluted to 0.7–10.0 μM and assuming a similar cell volume of Ko M5a1 to that characteristic of E. coli (10–15 L; Milo, 2013) and an OD-specific cell density of 109 CFU mL–1 OD–1.
Skyline software (MacLean et al., 2010) was used for peptide screening, peak selection, method development, analysis, and data processing purposes. Candidate analytical peptides were selected according to the following criteria: unique to Ko M5a1 proteome (extracted from GenBank WGS accession JAFHKG010000000), 5–25 residues in length, 50–1800 m/z ratio, and doubly charged with more than three fragment y-ions. Peptides containing His, Met, or Cys residues, proline-adjacent tryptic sites or KK/RR adjacent cut sites were excluded where possible. Unscheduled selected reaction monitoring (SRM) methods were assigned a dwell time of 3 ms per transition and peptide-specific collision energy and declustering potential parameters predicted by Skyline. A total 12–17 candidate peptides verified in test diazotrophic samples (see section “Results”) were included in each QconCAT design, flanked by the three amino acid residues upstream and downstream of the native trypsin cleavage site in order to maintain digestion efficiencies equivalent to the native target proteins. Furthermore, unique quantitative bovine serum albumin (BSA) peptides were incorporated at both termini to act as internal controls against potential degradation by exoproteolysis. Each QconCAT sequence was codon optimised for expression in E. coli, synthesised, and inserted into the pET100/D-TOPO expression vector (GeneArt, Life Technologies). For in vivo double-labelling at arginine and lysine residues, the His-tagged QconCATs were expressed in a modified ΔargA ΔlysA BL21 strain (Matic et al., 2011) cultured in Gutnick minimal salts medium (33.79 mM KH2PO4, 77.51 mM K2HPO4, 5.74 mM K2SO4, 0.41 mM MgSO4) supplemented with 0.4% glucose, 10 mM NH4Cl, 1 mM heavy-labelled L-[13C6,15N2]arginine and L-[13C6,15N2]lysine (Sigma-Aldrich), and 18 unlabelled amino acids at 1 mM concentrations each. Following sonication, cell protein was extracted from inclusion bodies using 7 M urea. Bench-top column purification of His-tagged protein was performed using Ni-NTA agarose resin with a binding buffer consisting of 95 mM Na2HPO4, 5.2 mM NaH2PO4, 50 mM NaCl, and 3 M urea. QconCAT purity was determined by SDS-PAGE. Protein samples were mixed 1:1 with 2 × Laemmli buffer, boiled for 5 min and separated through a 15% polyacrylamide mini-gel over 60 min at 200 mA. Gels were stained with SYPRO Ruby® fluorescent stain and % purity estimated via lane-specific densitometry. QconCAT concentration, corrected for purity, was determined via DC assay. Following validation of standard response linearity (see section “Results”), QconCATs were added to sample digests at the following concentrations (μg/mL): QNif1, 0.7; QNif2, 0.124; QNif3, 0.081; QNtr, 0.97.
The bacterial nitrogen stress response was induced experimentally upon metabolic consumption of a limited concentration of ammonium in the growth medium (here termed run-out) (Schumacher et al., 2013; Gosztolai et al., 2017). We supplemented Ko M5a1 with an initial ammonium salt concentration (0.5 mM NH4Cl) that enabled both nitrogen starvation and, subsequently, diazotrophic growth to be observed over the course of a day culture (approximately 12 h). Cells were pre-cultured overnight in excess NH4Cl (20 mM) to ensure a replete cellular N status prior to run-out. The limiting NH4+ concentration in the culture supernatant was largely depleted (<3% of starting concentration) after 5 h, coinciding with a plateauing of cell growth (Figure 1A). Nitrogenase activity was first detected after 7.5 h via the acetylene reduction assay (Figure 1B). An increase in OD600 suggests that cell growth slowly resumed between 10 and 12.5 h, during which period nitrogenase activity also increased. An endpoint OD600 measurement was taken after 24 h, at which point the cell density of wild-type Ko M5a1 had increased by more than threefold with respect to the onset of nitrogen starvation. However, this time point was not considered for further analyses given that the cell state may be strongly affected by secondary variables such as carbon limitation and changing pH of the growth medium. Growth did not resume following nitrogen starvation in a ΔnifLA mutant control, confirming that wild-type growth under these conditions is diazotrophic and dependent on nitrogenase expression. The supernatant NH4+ concentration remained depleted during diazotrophic growth, suggesting that NH4+ derived from fixation of N2 by the nitrogenase is rapidly assimilated and its net excretion (accounting for AmtB-mediated re-uptake) is negligible in the growth conditions used.
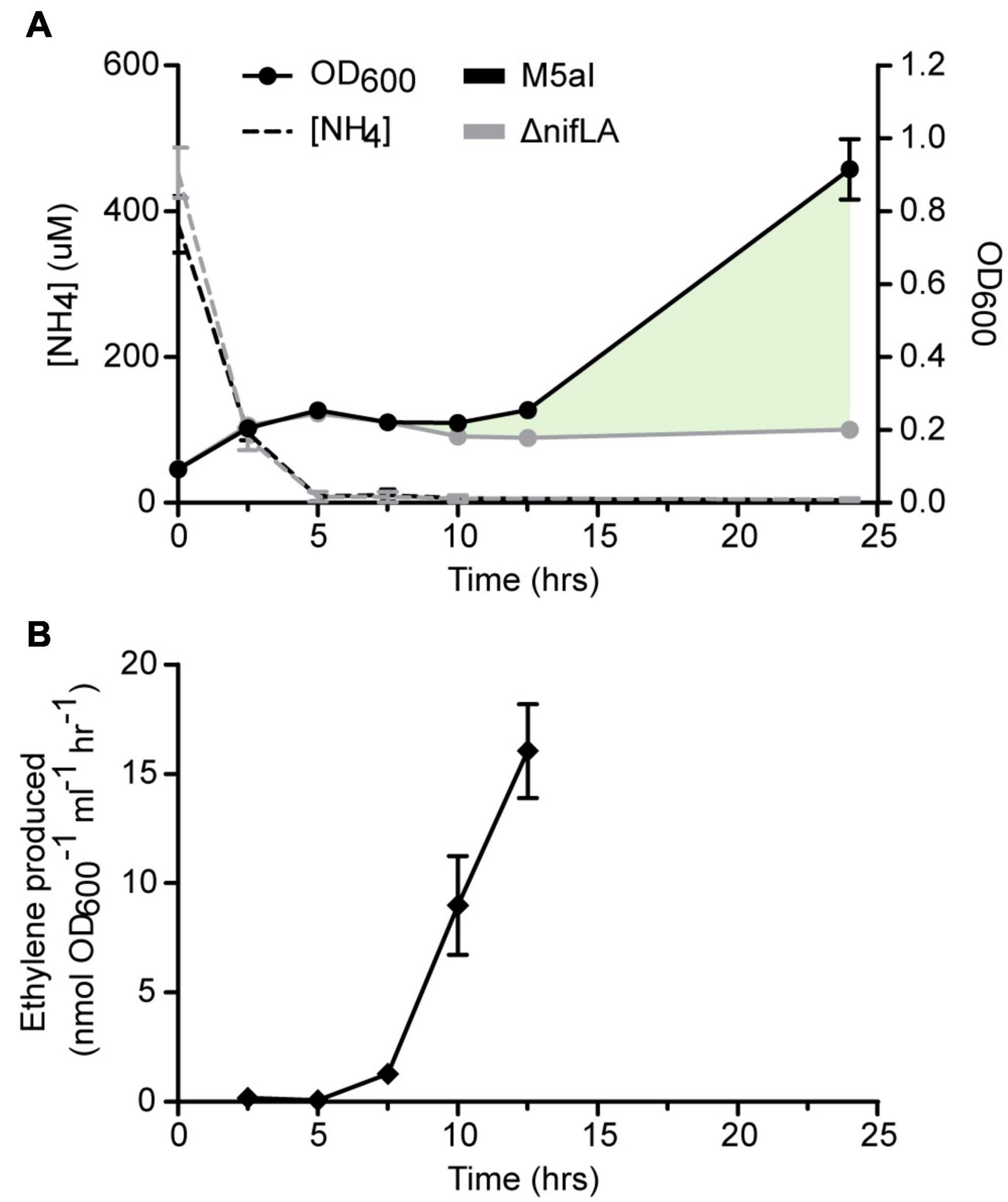
Figure 1. Physiological parameters of NH4+ run-out and diazotrophy. Ko M5a1 wild-type (black) and ΔnifLA (gray) strains were cultured for 24 h in NFDM medium supplemented with 0.5 mM NH4Cl under micro-aerobic conditions. (A) Cell density (OD600, filled circles) and supernatant NH4+ concentration (dashed lines) were measured at 2.5 h intervals and again after 24 h. Cell growth attributable to NifLA expression and diazotrophy is represented by shaded fill (pale green). (B) Culture headspace was sampled at 2.5-h intervals (between 2.5 and 12.5 h) for detection of nitrogenase activity via the acetylene reduction assay. Acetylene was injected into the head space at t = 0 h. Activity values correspond to ethylene evolved per hour and per unit cell density at 25°C. Error bars represent SEM of three biological replicates. Acetylene reduction was not assayed at 24 h.
In order to assess the global gene expression profile associated with diazotrophic growth, the Ko M5a1 transcriptome in N-fixing (t = 8 h; OD600 = 0.21, 0.19) and N-replete (exponential growth in 10 mM NH4Cl; OD600 = 0.24) conditions was compared by RNA-seq. As only one N-replete replicate was sequenced, requiring variance for this condition to be estimated and resulting in reduced statistical power (Anders and Huber, 2010), the analysis presented here is exploratory in nature and focused on genes exhibiting large fold changes. More than 50% of the transcriptome (2694/5279 genes) was highlighted as differentially expressed (adjusted p-value < 0.05) under diazotrophic conditions (Figure 2 and Supplementary Data 2). Given that glucose concentration and micro-aerobiosis were constant across both conditions, adaptation to nitrogen stress and its indirect global effects on metabolism and growth was most likely responsible for this concerted reprogramming of transcription. The nif genes were both highly upregulated and highly expressed in the diazotrophic condition. Normalizing for transcript length (RPKM), nifH was the most highly expressed gene and all 20 nif genes were among the 200 most highly expressed genes by rank. Nine hundred and twenty-five genes were upregulated and 1117 downregulated by more than twofold. Of these, 1515 correspond to annotated proteins in the KEGG functional orthology (KO) database, including metabolic enzymes, gene expression machinery, and transporters (Supplementary Figure 1). In addition to the nif genes, numerous NtrC-dependent genes associated with the bacterial nitrogen stress response were upregulated, including glnA (GS), glnK (PII-type signaling protein), the rut (pyrimidine scavenging), and ast (arginine catabolism) operons and several operons encoding amino acid transporters. Operons encoding enzymes and transporters required for metabolism of nitrate/nitrite (KO: NrtABC-NirB-NasA) and urea (KO: UreABC-UrtABCDE) and purine scavenging (KO: HpxAB-HpxO-HpxWXYZ) were also highly upregulated. Whilst these operons are not associated with the canonical NtrC regulon described in E. coli (Zimmer et al., 2000), analysis of the promoter sequences upstream of their apparent transcriptional start sites suggests that NtrC and σ54 may play a role in their expression in Ko M5a1 (data not shown). This contrasts with the operon encoding a branched-chain amino acid transport system (KO: LivKHMGF) which is highly upregulated but exhibits no obvious promoter NtrC binding sites. Whilst glnA is highly expressed, the downregulation of gdh by more than 10-fold is consistent with the periphery role of glutamate dehydrogenase (with a far lower affinity for ammonium than GS) in nitrogen assimilation during N limitation stress conditions. The array of other downregulated genes observed may reflect the wider impacts of nitrogen starvation on metabolism, growth, and phenotype. Downregulation of genes associated with DNA replication, RNA transcription, and purine (pur) and pyrimidine (pyr) nucleotide biosynthesis implies a decreased resource allocation into cell division and gene expression. Similarly, downregulation of 48 ribosomal proteins, 35 ribosome biogenesis genes and 55 tRNA biosynthesis genes may reflect a partial hibernation of the protein translation machinery. This hypothesis is supported by the upregulation of raiA and sra, encoding stationary phase-associated ribosome inhibitors, by more than 15- and 4-fold, respectively. General reprogramming of carbohydrate metabolism, amino acid metabolism, and membrane transport (ABC) is reflected by similar numbers of upregulated and downregulated genes in these functional groups. However, it is noted that many genes may be differentially expressed due to indirect effects of N starvation on metabolism and growth rather than active regulation in response to starvation.
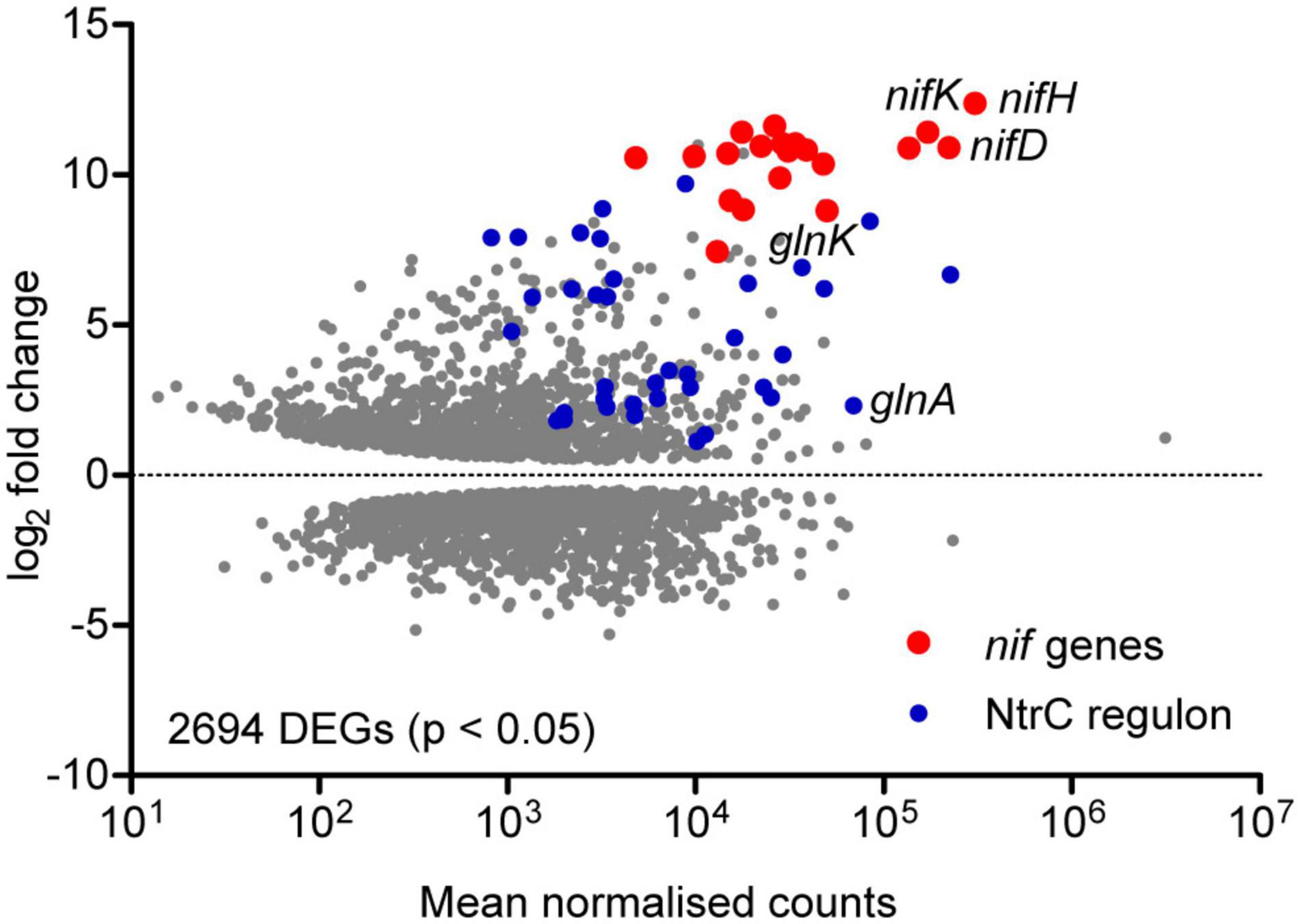
Figure 2. Exploratory analysis of the diazotrophic transcriptome. MA plot of the log2 fold change and mean RNA-seq read count for all Ko M5a1 genes. Gray points represent differentially expressed genes (DEGs) in N-fixing conditions (0.5 mM NH4Cl run-out) with respect to N-replete (10 mM NH4Cl) conditions, with adjusted p-values of <0.05 as determined by DESeq2. Read counts were averaged (mean) across all N-fixing and N-replete samples and normalised using the regularised logarithm (rlog) transformation. DEGs belonging to the known nif and NtrC-dependent regulons are highlighted in red and blue, respectively.
In order to assess the extent of post-transcriptional regulation in nif gene expression, the sequencing read mapping profile of the nif cluster was examined at nucleotide resolution for signatures of antisense transcription or variation in mRNA stability (Supplementary Figure 2). No evidence of antisense transcription was observed within any of the eight known nif transcriptional units. Each transcriptional unit was defined by a 5′ peak in read density commencing 12 bp downstream of a putative sigma-54 recognition sequence (consensus TGGC-N7-TTGC). The notable spike in read density observed at the end of the nifHDKTY operon raises the possibility of an internal secondary promoter for nifY.
Selected reaction monitoring-MS was utilised for targeted quantification of the Ko M5a1 proteome compartment associated with N metabolism and its regulation during diazotrophic growth. Furthermore, the QconCAT approach, highly suited to SRM workflows, was adopted for development of multiplexed standards for absolute quantification (Supplementary Figure 3). Protein sequences of interest were subjected to in silico trypsin digest and candidate analytical peptides were selected for empirical validation. At least three parent–daughter ion transitions for each candidate peptide were assimilated into a series of unscheduled SRM methods for screening against biological samples. Total cell protein was isolated from three sample cell cultures representing different expression states of the N proteome: (i) Ko M5a1, 5 h after NH4+ run-out (N stress and Nif expression); (ii) ΔnifLA, 5 h after NH4+ run-out (N stress but no Nif expression); (iii) Ko M5a1, supplemented with 10 mM NH4Cl (N-replete). Trypsin digested samples were analyzed by SRM-MS and peptides exhibiting multiple co-eluting transition peaks in the diazotrophic sample (i) but a low background in the corresponding negative control sample (ii and iii for Nif peptides; iii for Ntr peptides] were selected for further QconCAT standard development. The nifLA-dependent and N stress-dependent detection of SRM transitions specific to exemplar NifA (GAFTGAVR) and NifD (GVVFGPIK) peptides is outlined in Figure 3A. An enhanced product ion (EPI) scan was performed to confirm the identity of peptides with low peak intensity (data not shown). Empirical screening identified signature proteotypic peptides for 19 of the 20 Nif proteins. All four candidate peptides for NifV were undetectable in all samples.
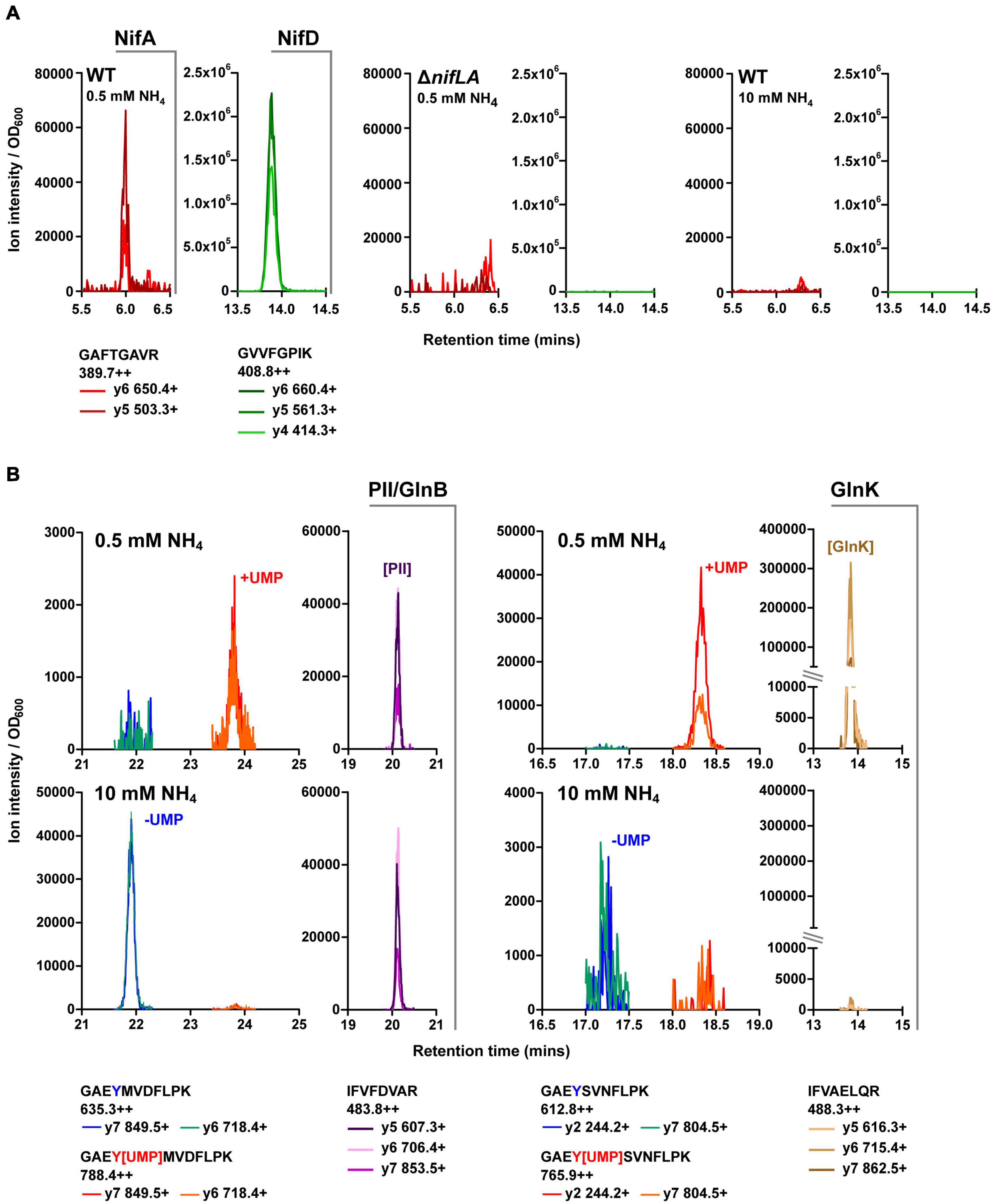
Figure 3. Validation of key Nif and Ntr peptides. Retention time specific LC–MS peaks corresponding to candidate peptides (see Supplementary Data 1) in representative biological test samples: diazotrophic (wild-type, 0.5 mM NH4Cl), non-diazotrophic/N limiting (ΔnifLA, 0.5 mM NH4Cl) and N-replete (wild-type, 10 mM NH4Cl). Multiple daughter y-ion fragments (+) are shown for each parent ion (++), along with the corresponding m/z values. Peak ion intensity is normalised to sample OD600 for relative quantification between samples. (A) NifD and NifA. (B) GlnB and GlnK. Non-modifiable peptides are indicative of relative PII-protein abundance between samples. Peaks at discrete retention times represent uridylylated (+UMP; red/orange) and non-uridylylated (–UMP; blue/green) forms of modifiable peptides. Note variable y-axis scales.
Peptides we have previously shown to be quantotypic for the Ntr signaling proteins NtrC, GlnB, GlnK, and the GS enzyme in E. coli (Schumacher et al., 2013; Gosztolai et al., 2017) exhibit high sequence identity with their Ko M5a1 homologues. Pairs of post-translationally modifiable (containing the uridylylated Y51 residue) and non-modifiable peptides of the PII-type proteins GlnB and GlnK were analyzed for their utility in reporting on both N stress-dependent protein expression level and uridylylation state. The modifiable peptides were detected predominantly in their uridylylated forms in N-limiting conditions and their non-uridylylated forms in NH4Cl-replete conditions (Figure 3B). The covalent addition of the uridine monophosphate (UMP) moiety resulted in an increase in peptide retention time of approximately 2 min for both GlnB and GlnK. Whilst the non-modifiable peptide of GlnB was observed at similar intensities in both cellular N states, the equivalent GlnK peptide was only detected in the N-limiting condition. This is consistent with the expression patterns of the PII-type proteins in E. coli, whereby glnK transcription is governed by NtrC activity while glnB expression is constitutive.
We designed four QconCAT sequences (QNif1, QNif2, QNif3, QNtr; see Supplementary Data 1) encompassing a total of 55 validated signature peptides, with at least two per target protein. Purified and heavy-labelled (13C15N-arginine and lysine) QconCATs were purified (Supplementary Figure 4) and mixed with cell protein samples (i, ii, iii), prior to trypsin digest and SRM-MS for sample peptide validation and SRM method scheduling. The enzymatic cleavage of all signature peptides from the QconCAT standards was confirmed (Supplementary Figure 5) and the absence of unlabelled peptide peaks in standard-only control samples confirmed completeness of in vivo labelling. The linearity and limit of detection for each peptide was assayed by titrating a fixed concentration of QconCATs with a dilution series (100–10–5) of cell protein extract (Supplementary Figure 6). SRM transitions exhibiting non-linearity or a poor limit of detection were excluded during method optimisation. The absolute quantification of Nif protein copy numbers in diazotrophic cells, inferred from the unlabelled/labelled (sample/QconCAT) ratio of SRM signal peak area and QconCAT standard concentration, was largely consistent across independent signature peptides (Supplementary Figure 7). Besides NifQ and NifZ, which exhibited no linear peptides, each Nif protein exhibited at least one peptide with a limit of detection below 1 nM in the digested sample.
Figure 4 shows the cellular copy number of the 19 quantifiable Nif proteins at two time points during the adaptation to diazotrophy, corresponding to (a) onset of nitrogen starvation (5 h following supplementation with 0.5 mM NH4) and (b) onset of slow diazotrophic growth (10 h). Negligible Nif protein concentrations, below the limits of detection, were observed in the two control conditions (ΔnifLA; wild-type supplemented with 10 mM NH4Cl), highlighting the strong dependence of nif promoter activation on both NifA and nitrogen limitation. Although absolute quantification cannot reveal the fraction of any given protein that is either functional or in a specific complex, the observed NifH/NifDK ratio of approximately 2 closely reflects the known in vivo catalytic stoichiometry of the nitrogenase enzyme (Peters et al., 1995). Likewise, the abundances of the NifL and NifA regulators were broadly similar, with a modest excess of NifL suggesting that all NifA activator proteins are able to be sequestered by NifL acting as an inhibitor. Whilst the stoichiometry of the nitrogenase subunits remained constant over time, the ratio of the nitrogenase complex to the accessory Nif proteins (involved in co-factor biosynthesis, assembly, and regulation) increased between 5 and 10 h (Supplementary Figure 8). This pattern of accumulation may reflect both the order in which the Nif protein functions are required for diazotrophy to be established and the relative rates of different steps/reactions required. For instance, the accessory protein NifU was the third most abundant Nif protein at 5 h, consistent with its role in the synthesis of Fe–S clusters for both nitrogenase structural (NifH) and co-factor biosynthesis (NifB) proteins (Jimenez-Vicente et al., 2018), but does not accumulate further at a similar rate to NifHDK.
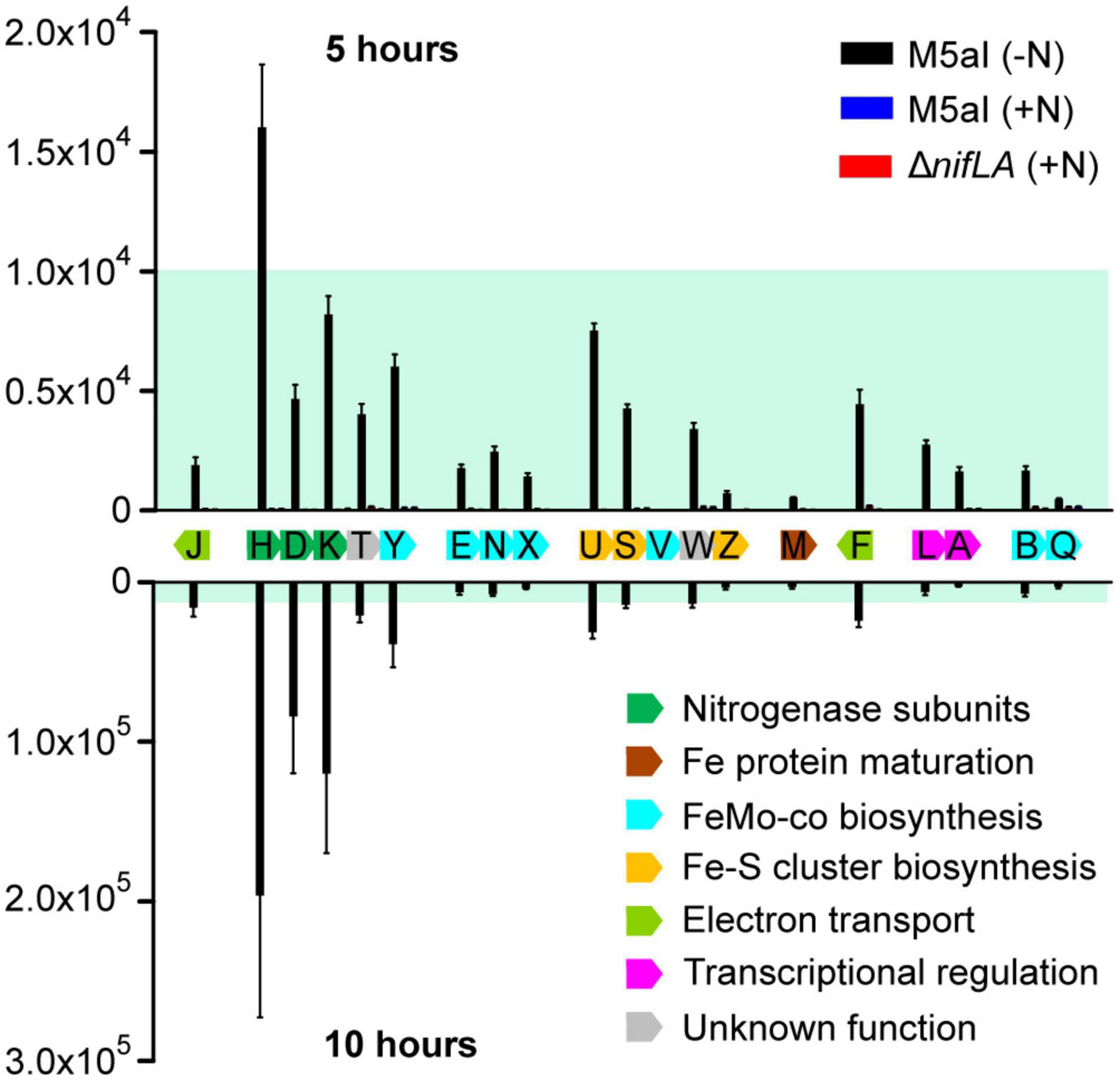
Figure 4. Stoichiometry of Nif proteins during the onset of diazotrophy. Absolute copy number of 19 detectable Nif proteins at the onset of nitrogen starvation (5 h following supplementation with 0.5 mM NH4; above the x-axis) and at the onset of slow diazotrophic growth (10 h; below the x-axis) in Ko M5a1 wild-type cells (black bars). N-replete conditions (wild-type plus 10 mM NH4Cl; blue bars) and Nif– (ΔnifLA; red bars) cells were used as negative controls, which are barely visible in the figure. Protein samples were supplemented with heavy-labelled QconCAT standards (QNif1, QNif2, QNif3) prior to tryptic digestion. Multiple peptide transitions per Nif protein (see Supplementary Data 1) were integrated for calculation of absolute protein copy number per cell. Estimates inferred from known QconCAT concentrations and the mean sample/QconCAT LC–MS peak area ratio, assuming an OD600 of 1.0 = 109 CFU mL–1 and a cell volume of 10–15 L. Error bars represent SEM of three biological replicates. NifV was undetectable in all samples. Nif proteins are displayed according to genomic organization and colored by function. Blue shading representative of ≤10,000 copies and highlights variable y-axis scale.
Absolute Nif protein expression was correlated with cellular nitrogen status throughout a complete time course encompassing NH4+ run-out and subsequent diazotrophic growth, as in Figure 1. In order to relate the benefit of N fixation activity to the cost of Nif expression, protein samples were extracted in parallel from two non-diazotrophic strains representative, at the level of protein expression, of Nif+ (nifK1–1203) and Nif− (ΔnifLA) states alongside wild-type (diazotrophic and Nif+). The nifK1–1203 mutant was constructed such that a C-terminal domain of NifK necessary for catalytic activity of the nitrogenase is lacking but nif expression otherwise unimpaired. Complete protein copy number data are provided in Supplementary Data 1. Protein signatures of N limitation stress were evident prior to complete NH4+ starvation in all three strains, with increases in both NtrC-dependent protein expression (GlnK and GS) (Figure 5A) and the percentage of GlnB and GlnK protein uridylylation (Figure 5B) between 0 and 2.5 h. This response coincided with a supernatant NH4+ concentration of approximately 100 μM at 2.5 h, prior to depletion to a level representative of complete starvation (<10 μM) by 5 h (see Figure 1). GlnK levels increased more than 40-fold over the course of the diazotrophy adaptation whilst GlnB protein levels remained constant, consistent with glnB being constitutive and regulated independently of NtrC activity. At the onset of N starvation (t = 5 h), the wild-type copy numbers of GlnK and GlnB were 8200 and 350 per cell, respectively. GS increased approximately threefold during NH4+ run-out, peaking at approximately 17,000 copies per cell (t = 2.5 h).
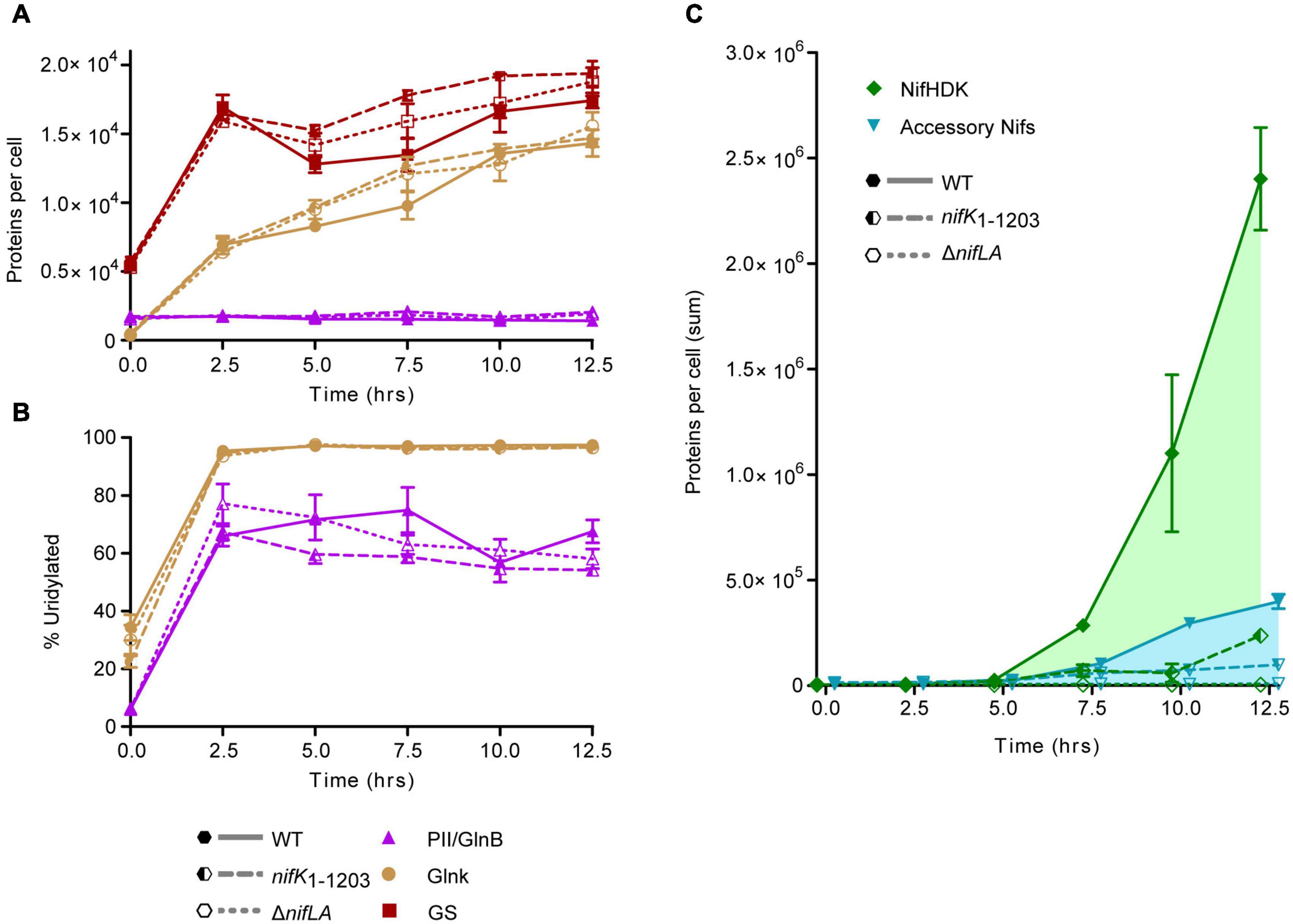
Figure 5. Time-course of Nif and Ntr protein expression and post-translational N stress signaling. Ko M5a1 wild-type (full lines, full shapes) nifK1–1203 (dashed lines, half shapes) and ΔnifLA (dotted lines, empty shapes) strains were cultured for 12.5 h in NFDM medium supplemented with 0.5 mM NH4Cl under micro-aerobic conditions. Cell protein was extracted at 2.5 h intervals, supplemented with a mix of four QconCAT standards and trypsin digested. Absolute copy number was inferred from the sample/QconCAT LC–MS peak area ratio for quantotypic non-modifiable peptides and QconCAT concentration, assuming an OD600 of 1.0 = 109 CFU mL–1 and a cell volume of 10–15 L. Error bars represent SEM of three biological replicates. (A) Copy number of Ntr proteins: GlnB (purple), GlnK (brown), and glutamine synthetase (GS) (orange). (B) Post-translational modification of PII-type proteins GlnB and GlnK. Percentage uridylylation was inferred from the modified/total LC–MS peak area ratio for modifiable peptides. (C) Summed absolute copy numbers of nitrogenase subunits NifHDK (green) and accessory proteins associated with co-factor biosynthesis and assembly (blue). Overlapping data points offset for clarity.
Both PII-type signaling proteins were highly uridylylated during NH4+ runout and remained in this PTM state for the duration of the diazotrophy adaptation. Furthermore, there was no notable difference in PTM status between the wild-type and the non-fixing ΔnifLA and nifK1–1203 strains. These findings imply that diazotrophy does little to relieve the cells perceived N limitation stress, which remains a persistent characteristic of the diazotrophic growth state. However, there was a marked difference between the absolute levels of GlnK and GlnB uridylylation, both in N starved and N-replete conditions. Whilst >95% of GlnK was uridylylated following N starvation, the maximal level of GlnB uridylylation observed was around 75%, peaking around the onset of detectable nitrogenase activity in the wild-type and prior to complete NH4+ run-out in ΔnifLA and nifK1–1203. In N-replete conditions (t = 0 h) where GlnB outnumbers GlnK approximately fourfold, their uridylylation levels were approximately 5 and 35%, respectively. It is also notable that the level of GlnB uridylylation decreased following the onset of diazotrophy in the wild-type (t = 7.5–10 h) and steadily throughout prolonged N starvation in the non-diazotrophic strains. Taken together, these observations suggest that uridylylated GlnK is the by far the most abundant PII isoform during diazotrophy, whilst GlnB is the more sensitive of the two proteins to finer shifts in N status.
The detection of Nif proteins at levels exceeding limits of detection coincided with the onset of N starvation (t = 5 h). The time-resolved expression of the individual Nif proteins is shown in Supplementary Figure 9. As observed in previous experiments (see Figure 4), the nitrogenase subunits NifHDK accumulated more rapidly than the accessory Nif proteins (Figure 5C). Between 10 and 12.5 h, the concentration of nitrogenase subunits more than doubled, whilst the sum of the other Nif proteins increased by only 35%. Assuming that the concentration of fully assembled nitrogenase complex is limited by the least abundant of the individual NifHDK proteins, it can be inferred that the nitrogenase increased more than 50-fold (800–50,000 enzymes per cell) between the onset of N starvation (5 h) and the resumption of slow diazotrophic growth (12.5 h). Nitrogenase far exceeded the concentration of the GS complex, which does not exceed 1500 molecules per cell (taking into account its 12 subunit assembly) throughout N starvation. The apparent disparity in concentration may compensate somewhat for the significantly different turnover rates for nitrogenase (∼5 s–1) and GS (>500 s–1; van Heeswijk et al., 2013). It is notable that the NtrC-dependent gene products (GlnK, GS) accumulated several hours prior to the NifA-dependent nif gene products, suggesting that the two regulons are induced at different thresholds of N stress.
The total abundance of NifHDK and accessory Nif proteins in the non-diazotrophic nifK1–1203 mutant fell to 10 and 25% of wild-type levels, respectively, by 12.5 h into the time course (Figure 5C). This disparity between the strains was not evident at the 5 h time point (Supplementary Figure 9F), implying that it was not the result of differences in the kinetics of nif gene de-repression in response to N starvation. Rather, the exaggeration of the nifK1–1203 phenotype throughout the diazotrophy transition suggests that N fixation function is required to sustain further Nif protein expression and, by implication, that stored cellular biomass derived from nutrients in the growth medium alone is insufficient to support the expression levels observed in the wild-type. As expected, no evidence of Nif protein expression was observed in the ΔnifLA mutant throughout the time course. Despite the difference in Nif protein burden, however, no significant difference in cell density between ΔnifLA and nifK1–1203 was evident (Supplementary Figure 9G). This may reflect the fact that neither strain can undergo diazotrophic growth following N starvation and therefore any fitness penalty due to futile Nif expression in nifK1–1203 is not manifest in this respect.
To explore whether the burden of Nif protein expression and N stress more broadly are manifest at the level of substrate amino acids, we quantified the intracellular pools, extracted simultaneously with total protein, in the wild-type, ΔnifLA, and nifK1–1203 strains via LC–MS. The limit of quantitation of each amino acid was determined via a dilution series of amino acid standards of known concentration (0.1–100 μM). MS signal response was linear for all amino acids, with robust quantitation at 0.1 μM for all except asparagine, aspartate, and glutamine, for which signal response was linear at 2 μM and above (Supplementary Figure 10A). The calculated amino acid pool concentrations for NH4+ replete cells sampled immediately following transfer to 0.5 mM NH4+ medium were consistent with those observed in E. coli cells grown in similar N-replete conditions (Supplementary Figure 10B; Bennett et al., 2009), indicative of effective metabolite extraction and quantification.
Absolute amino acid concentrations varied by several orders of magnitude during the wild-type transition to diazotrophy, from a high of 70 mM glutamate in N-replete conditions to a low of 5 μM histidine following NH4+ run-out. Total amino acid concentrations are shown in Supplementary Figure 11 and listed individually in Supplementary Data 3. To provide a comparative overview of how amino acid pool sizes respond to N starvation and the onset of diazotrophy, absolute concentrations were normalised against the base mean for each amino acid (Figure 6A). Thirteen amino acids were characterised by a decrease in abundance consistent with extracellular NH4+ run-out. This trend was most striking for glutamine, asparagine, aspartic acid, isoleucine, and valine, which were all more than 10-fold depleted following N starvation, relative to N-replete conditions (0 h). In contrast, the abundance of four amino acids (arginine, lysine, proline, tyrosine) peaked following NH4+ starvation (5–7.5 h). This suggests that whilst N starvation is characterised by limitation in many amino acids, some pools are not only robust to the effects of starvation but may accumulate in this condition. This hypothesis is supported by the phenotype of the non-diazotrophic strains ΔnifLA and nifK1–1203, in which the same four amino acids accumulate in spite of the inability to recover from prolonged N starvation (Figure 6B). At 12.5 h, proline and lysine were approximately 10- and 5-fold more abundant, respectively, in ΔnifLA and nifK1–1203 cells relative to wild-type. In terms of absolute concentration, alanine, glutamate, and proline are in >5 mM excess in the non-diazotrophic strains. It is remarkable that most amino acids were at similar or moderately increased concentrations in both ΔnifLA and nifK1–1203, implying that prolonged N starvation is not characterised by a critical depletion of amino acids, even when there is the additional burden of Nif expression.
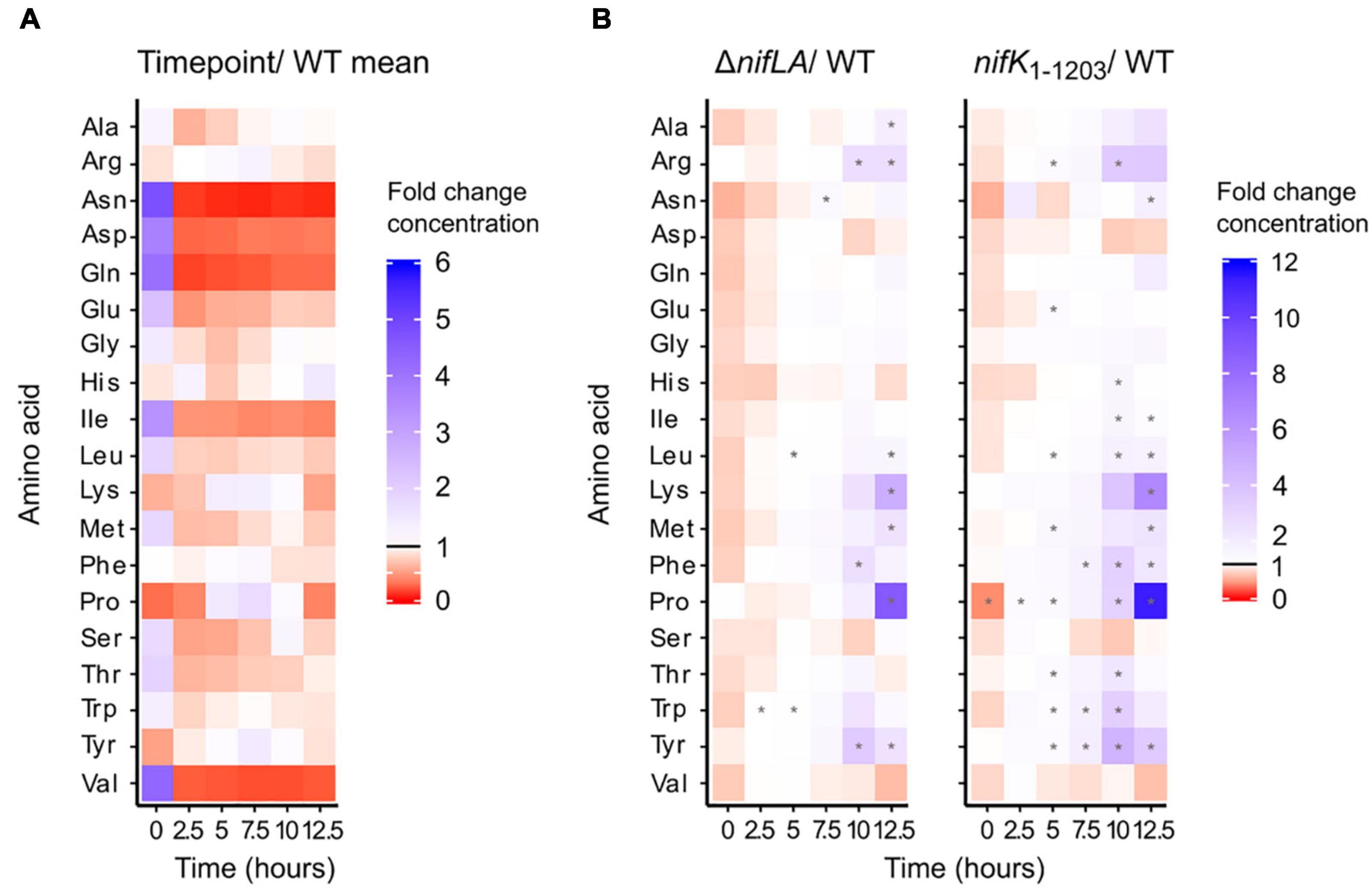
Figure 6. Dynamics of the intracellular amino acid pools during the diazotrophy transition. Ko M5a1 wild-type, nifK1–1203, and ΔnifLA strains were cultured for 12.5 h in NFDM medium supplemented with 0.5 mM NH4Cl. Polar metabolites were extracted at 2.5 h intervals, derivatised with the AccQTag reagent (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate) and analyzed via LC–MS. Absolute amino acid concentrations were calculated by referencing external standard curves, assuming a characteristic cell volume of 10–15 L. (A) Time course of wild-type amino acid pools. Heat map density representative of fold-change between time-specific concentrations and the base mean amino acid concentration across all time points. Asterisks denote statistically significant differences from base mean (one sample unpaired Student’s t-test, N = 3, p < 0.05). (B) Time course of ΔnifLA and nifK1–1203 amino acid pools. Heat map density representative of fold-change between ΔnifLA and nifK1–1203 amino acid concentrations and wild-type. Asterisks denote statistically significant differences between mutant and wild-type means (two sample unpaired Student’s t-test, N = 3, adjusted p-value (False Discovery Rate; Benjamini and Hochberg, 1995; p < 0.05). Note different heat map density scales between panels (A,B). Absolute amino acid concentrations are detailed in Supplementary Data 3.
We calculated amino acid supply and demand in order to estimate the relative burden of Nif protein expression on the total substrate pool. Estimates for the production rate of each Nif protein (Supplementary Figure 12A) and the apparent demand for each amino acid substrate (Supplementary Figure 12B) were derived from the rate of change in measured protein concentration between 7.5 and 12.5 h in the wild-type and nifK1–1203. The observed rate of nitrogenase (NifHDK) synthesis during this period implies incorporation of >17,000 amino acid residues per second on average in the wild-type and approximately 950 per second in nifK1–1203. We do not account for Nif protein degradation in this model, such that amino acid demand may be underestimated. Furthermore, we approximated for how long the N starved amino acid pools (quantified at 5 h) could support Nif translation if they were finite in concentration and not replenished (Supplementary Figure 12C). Given the relative abundance and amino acid composition of the Nif proteins, alanine, glycine, glutamine, and leucine were in highest demand (>1000 molecules per second), and histidine and tryptophan in lowest demand (<250 molecules per second) in the wild-type. Taking into account pool concentrations, the least limiting amino acids were glutamate, proline, and alanine, which are sufficient to support >1 h of Nif protein expression in the wild-type and >10 h in nifK1–1203. However, 10 amino acid pools were insufficient to satisfy the theoretical demands of wild-type Nif protein translation for more than 5 min. These estimates support a model in which N fixation is necessary to support further Nif expression (see Figure 5C) and suggest that fixed N is rapidly re-distributed through amino acid metabolism via de novo biosynthesis and/or metabolism reflecting transamination. In the case of nifK1–1203, the approximately 10-fold lower levels of Nif protein accumulation may stem from rate limitation in a few amino acids, such as asparagine and histidine, which must be replenished by transamination alone.
To better understand the temporal signaling roles of the PII-type proteins GlnB and GlnK during diazotrophy, in particular the reported role of GlnK in activation of NifA, the effects of ΔglnB and ΔglnK knockouts on N fixation activity, Nif and Ntr protein expression and post-translational modification were assayed 3 h after NH4+ run-out. The ΔglnK mutant exhibited a threefold reduced acetylene reduction activity relative to wild-type, while ΔglnB had no notable effect (Figure 7A). Similar phenotypes were observed in the presence of 0.25 mM extracellular glutamine (equivalent N atoms to 0.5 mM NH4), which has been used interchangeably with NH4+ as a limiting N source (Supplementary Figure 13A). Consistent with acetylene reduction, NifHDK protein abundance was reduced threefold in ΔglnK cells but not in ΔglnB (Figure 7B). Albeit in a microtiter context rather than batch flask, which may lead to subtle differences in O2 concentration and redox state, diazotrophic growth was severely delayed in the ΔglnK mutant, whilst ΔglnB grows at the same rate as the wild-type (Supplementary Figure 13B). The knockout mutations were validated by the absence of independent signature peptides for GlnB and GlnK, respectively (Figures 7C,D). Given that non-zero levels of Nif abundance and nitrogenase activity were measured in both mutants, neither GlnK nor GlnB is strictly necessary for diazotrophy to occur in the presence of the remaining homologue. However, GlnK is more strongly influential in full de-repression of nif expression, consistent with nuanced roles for the partially redundant PII proteins. GlnB may exhibit some function in the dissociation of NifLA and de-repression.
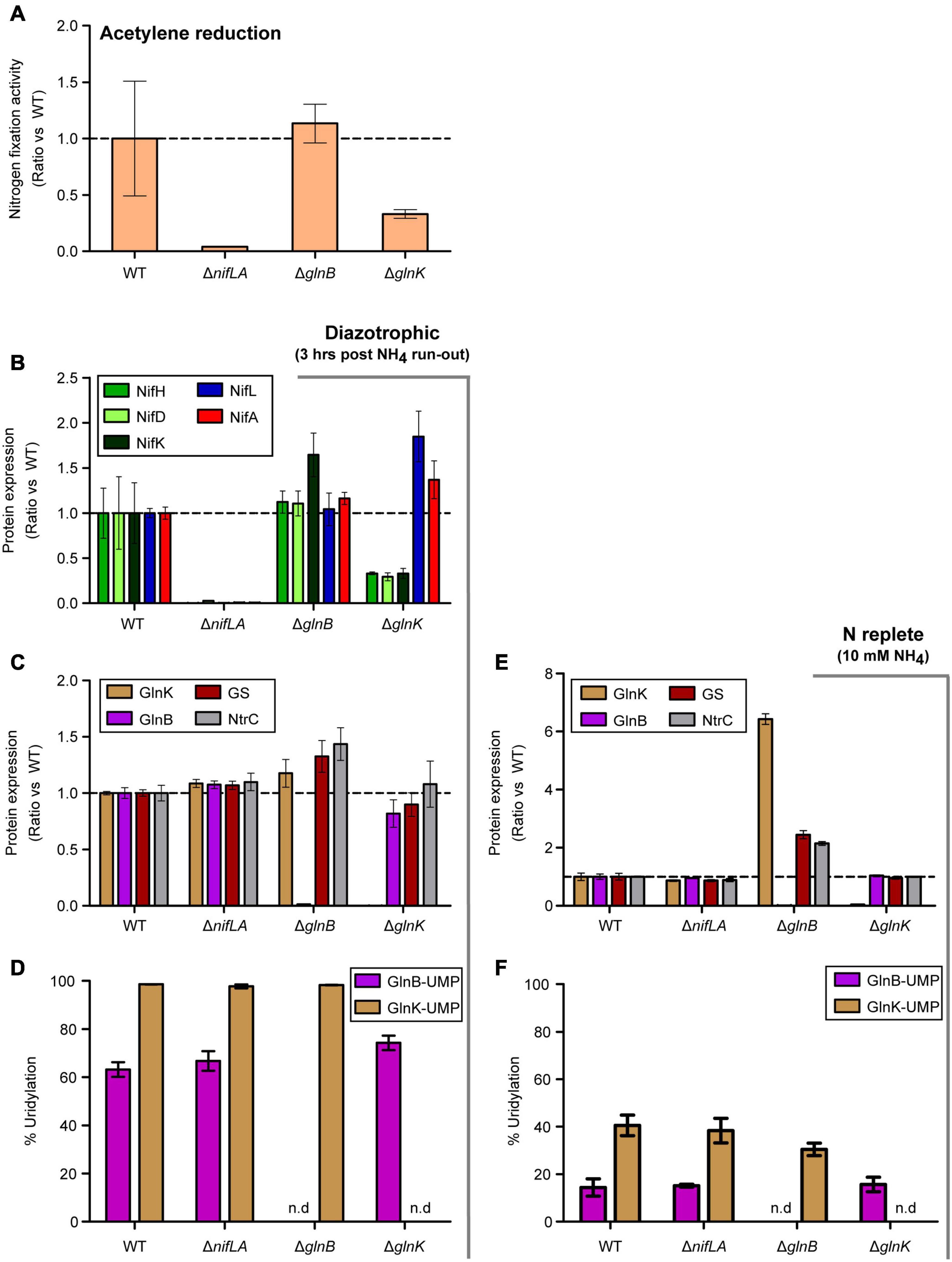
Figure 7. Effects of ΔglnB and ΔglnK knockout mutations on diazotrophy. Ko M5a1 wild-type, ΔnifLA, ΔglnB, and ΔglnK strains were cultured for 3 h following run-out of 0.5 mM NH4Cl in NFDM medium. Fold-change in various parameters with respect to wild-type: (A) Nitrogen fixation activity. Culture headspace was analyzed for ethylene evolution via the acetylene reduction assay. (B) Nif protein abundance. Extracted cell protein was supplemented with a mix of four QconCAT standards, trypsin digested, and the derived peptides analyzed via LC–MS. The absolute copy numbers of NifHDK (green shades) and NifLA (blue, red) were inferred from the sample/QconCAT peak area ratio for quantotypic peptides and QconCAT concentration, assuming an OD600 of 1.0 = 109 CFU mL–1 and a cell volume of 10–15 L. (C,E) Ntr protein abundance. The absolute copy numbers of GlnK (brown), GlnB (pink), GS (maroon), and NtrC (gray). Dashed lines correspond to no fold change relative to wild-type. (D,F) PTM uridylylation status of PII-type proteins. Percentage uridylylation of GlnK (brown) and GlnB (pink) was inferred from the modified/total LC–MS peak area ratio for modifiable peptides. Note variable y-axis scales. Error bars represent SEM of three biological replicates. n.d. refers to no data shown.
Both NifLA abundance (Figure 7B) and the fraction of uridylylated GlnB (Figure 7D) were moderately increased in ΔglnK, suggesting that both N stress and NtrC activity were elevated. However, the fact that NifHDK were downregulated despite an increase in abundance of NifLA (Figure 7B) implies that the dominant role of GlnK is in dissociation of the NifLA complex and its role in perception of N stress for the activation of NtrC is of secondary importance. Whilst ΔglnB had no apparent effect on Nif expression, increased levels of GlnK, GS, and NtrC were observed in both diazotrophic and N-replete conditions (Figure 7C,E). Given that glnK expression is understood to be induced specifically under N limitation stress (as observed in Figure 5), it is notable that GlnK levels were elevated by more than sixfold in the ΔglnB strain, relative to wild-type, in the presence of excess NH4Cl (Figure 7E). This implies the absence of GlnB leads to perceived N limitation signaling despite actual N-replete conditions. In the context of GlnK expression as a marker for NtrC activity, the loss of the repressive effect of non-uridylylated GlnB on NtrB kinase activity may explain why NtrC phosphorylation, and therefore GlnK expression, is elevated in ΔglnB.
N run-out and upshift experiments enable faithful and non-intrusive study of the physiological adaptations induced in response to changes in environmental and cellular N status (Schumacher et al., 2013; Gosztolai et al., 2017). In contrast to other approaches used to establish N stress, such as rapid cell washing followed by medium replacement (Poza-Carrión et al., 2014) and continuous culture (Schreiber et al., 2016), N run-out enables both the pairwise comparison of distinct N statuses (N-replete and N starved) and the consumption-dependent kinetics of the intervening adaptive transition to be revealed. We performed NH4+ run-out experiments in which Ko M5a1 cells were sampled over a time course for multi-omics analyses of N-replete, N starved, and diazotrophic growth states. Nitrogen fixation activity was first detected within 2.5 h of complete NH4+ consumption, with evidence for diazotrophic growth observed at the subsequent time point (Figure 1). A notable hallmark of diazotrophy in these conditions was the continuation of N stress signaling despite the resumption of growth, with maximal levels of PII protein uridylylation reached prior to complete NH4+ starvation (at an extracellular NH4+ concentration approximately 100 μM) and remaining elevated for the remainder of the time course (Figure 5). This is perhaps not surprising given that glutamine, the key metabolic indicator of cellular N status, is consumed as the amino donor in the cyclical GS-GOGAT pathway necessary for assimilation of fixed NH4+ and decreases in concentration with NH4+ starvation (Figure 7).
The fact that expression of both nitrogenase and GS is dependent on NtrC activity implies that diazotrophic growth is inherently dependent on some level of N stress signaling. However, the accumulation of NtrC-dependent gene products (GlnK, GS) several hours prior to NifA-dependent nif gene products (Figure 5) suggests that the two regulons may be induced at different thresholds of N stress. The apparent delay in Nif expression may be due to differences in either NtrC-dependent promoter activities or the N stress thresholds at which post-translational events yield active NtrC and NifA. It is known that the nifLA promoter has a lower affinity for NtrC than the glnA promoter (Austin et al., 1987; Minchin et al., 1988), whilst the reported role for GlnK in liberating NifA from its inhibitive complex with NifL (Jack et al., 1999) may in principle occur at a different threshold N stress than GlnB/GlnK-dependent phosphorylative activation of NtrC.
Nitrogen fixation protein expression was induced rapidly alongside onset of N stress (Figure 5). Whilst the regulation of the nif genes is well understood (Dixon and Kahn, 2004), in particular their dependence on N stress and microaerobiosis (signalled via NifLA), this study reveals the scale with which gene expression capacity is redirected toward synthesis of nitrogenase during the diazotrophy transition, highlighted by the estimated fractions of the transcriptome and proteome accounting for nif transcripts and Nif proteins, respectively. According to our comparison of the Ko M5a1 transcriptome between N-replete and N-fixing conditions, the nif genes are among both the most highly upregulated and highly expressed sets of genes during the transition to diazotrophy (Figure 2). Here, the use of RNA-seq has the advantage over previous studies based on qRT-PCR techniques (e.g., Poza-Carrión et al., 2014) in that the relative expression of the nif genes as a function of the whole transcriptome can be determined qualitatively. Highly stringent regulation of the nif genes by NifA is reflected both by negligible transcription activation in N-replete conditions and the apparent absence of additional internal promoters or signatures of antisense regulation (Supplementary Figure 2). Putative σ54-dependent transcription start sites were identified for seven of the eight described nif operons, with the exception of nifM, although these remain hypothetical until followed up using a primary transcript selective methodology, such as differential RNA-seq (dRNA-seq).
The most notable feature of Nif protein expression dynamics was the accumulation of the structural nitrogenase subunits (NifHDK) at a higher rate than the accessory Nif proteins involved in biosynthesis and maturation (Figure 5C), mirroring the pattern observed in Azotobacter vinelandii (Poza-Carrión et al., 2014). This bifurcation likely reflects the optimal order and reaction rate of different protein functions during diazotrophy, with biosynthesis and maturation especially critical for early establishment while the concentration of nitrogenase may be more closely correlated to subsequent growth rate and fitness. Assuming a degree of subunit stability, once an optimal nitrogenase concentration is established, only minimal expression of the accessory Nif proteins that catalytically support maturation may be necessary. In A. vinelandii, despite being actively degraded during diazotrophic growth, NifB and NifEN provide sufficient FeMo-co for an excess of NifDK molecules as a result of multiple enzymatic turnovers (Martínez-Noël et al., 2011; Poza-Carrión et al., 2014). The pattern of Nif protein accumulation observed is also supportive of a simple model of polycistronic transcription of the nif operons, in line with our understanding of nif promoter architecture, with minimal post-transcriptional regulation. The stoichiometry of the proteins expressed from each operon remained relatively constant throughout the time course studied (Supplementary Figure 9) and the most upstream gene in each operon tended to be expressed at the highest protein copy, with the exception of NifE, which was consistently exceeded by the NifN concentration.
The concurrent measurements of absolute protein and intracellular amino acid concentrations performed in this study enable coarse grained predictions as to how intracellular N, both pre-existing and de novo fixed, is re-allocated to permit high levels of Nif expression in spite of N starvation conditions. The concentrations of most amino acids following N run-out would be insufficient as a finite substrate pool for the subsequent surge in Nif protein expression (Supplementary Figure 12), implying that their replenishment via a combination of de novo N fixation and scavenging of existing biomass is necessary. The fact that Nif proteins accumulated to only 10% of wild-type levels in the presence of a non-functional nitrogenase (nifK1–1203) provides strong evidence that fixed N is rapidly re-invested into further Nif expression. Focusing on the time window following the first detected N fixation activity (7.5–12.5 h), 900,000 excess Nif proteins were synthesised in wild-type relative to the non-diazotrophic nifK1–1203, requiring the supply of an additional 16,900 amino acids per second. The NifHDK concentrations at 10 h imply that there may be up to 35,000 nitrogenase enzymes per cell (70,745 NifK copies). Assuming a theoretical di-nitrogen reduction rate of 5 s–1 (Hageman and Burris, 1978) this equates to a total fixation rate in the range of 350,000 N atoms s–1, implying that it is indeed feasible for fixed N to account for the excess Nif protein expression, in addition to other housekeeping processes such as the onset of slow cell division between 10 and 12.5 h.
Whilst it is difficult to make robust comparisons across species boundaries, the general parameters of protein synthesis during diazotrophy can be inferred from earlier studies that have subjected bacterial models to growth under N limiting conditions. Li et al. (2018) observed a growth rate of E. coli on 1.9 mM NH4+ (0.1 doublings per hour) similar to that of diazotrophic Ko M5a1 in this study (0.098/h between 10 and 12.5 h). Assuming that total protein content in such a slowly growing cell equates to approximately 7.6 × 107 polymerised amino acids, as measured by Bremer and Dennis (2008), it follows that Nif proteins account for around half of total cell protein following the onset of diazotrophic growth (52.2%; 12.5 h), with the nitrogenase largely responsible for this protein burden (46.3%). These estimates are somewhat higher than, but broadly in line with, previous measurements of Nif protein content (Dixon and Kahn, 2004). Under N-limiting conditions, active E. coli ribosomes are both relatively few in number (approximately 8000 copies per cell) and sparsely distributed along translating mRNA molecules (142 nt/ribosome) (Bremer and Dennis, 2008; Li et al., 2018). Considering the effects of both N limitation and growth at 25°C, it is probable that the translation elongation rate of Ko M5a1 ribosomes is constrained to five amino acids per second or less (Zhu et al., 2016; Li et al., 2018). If that assumption were to hold true then almost half (47.5%) of the active ribosome complement would need to be engaged in Nif protein translation to account for the accumulation rates observed between 7.5 and 12.5 h. Taken together, these rudimentary estimates imply that the proteome of early diazotrophy can be modelled as two compartments of broadly similar size, one consisting of the Nif proteins and one comprising all other proteins, with a reduced pool of active ribosomes divided between them. However, further studies involving both an accurate measurement of the total proteome size and ribosome profiling are necessary to test this hypothesis further.
The reduced availability of functional ribosome general to N limited growth (Li et al., 2018) has been directly related to a decrease in the pool of amino acids used as substrates for translation. Amino acids are the common currency that couple metabolism, protein synthesis, and growth rate, with auto-regulatory mechanisms in place that tune ribosome concentration and translation rate to the amino acid supply and nutrient conditions (Scott et al., 2014). Sub-saturating levels of tRNA charging stimulates downregulation of ribosomal gene expression via the stringent response (Condon et al., 1995) along with ribosome hibernation (Sanchuki et al., 2017). Therefore, the decrease in abundance of many amino acids that occurs concurrently with extracellular NH4+ run-out (Figure 6 and Supplementary Data 3) may not only place a direct substrate limitation on translation rate but further constrain it indirectly via down-regulation of ribosome availability. Furthermore, these same regulatory mechanisms that couple amino acid supply to translation rate may function to explain why the N starved amino acid pools in non-diazotrophic mutants (ΔnifLA and nifK1–1203) remained so similar to wild-type despite no influx of fixed N. In effect, the active slowdown of translation rate acts to maintain a steady-state of amino acids and prevent complete run-out (Scott et al., 2014). Real-time measurement of translation elongation speed, as demonstrated by Zhu et al. (2016), in N starved and either diazotrophic or non-diazotrophic cell samples could be utilised in the future to test the hypothesis that translation rate increases in response to N fixation specifically.
The primary signal of cellular N status, glutamine, was depleted more than 25-fold during experimental NH4+ run-out (0–5 h). This is in line with previous reports in E. coli (Schumacher et al., 2013) and Salmonella typhimurium (Ikeda et al., 1996) and points to a model in which the perception of external nitrogen limitation via internal glutamine limitation is general to the Enterobacteriaceae family. Conversely, the fivefold decrease in glutamate concentration seen during the same time period contrasts with previous observations that this amino acid is relatively insensitive to NH4+ limitation (Brauer et al., 2006; Schumacher et al., 2013), even in Klebsiella pneumoniae specifically (Schmitz, 2000). Glutamate levels were partially restored by the 10 h time point, twofold higher that at 2.5 h. It is possible that variation in cell size throughout the diazotrophy transition may modestly skew calculations of amino acid concentrations which rely on an assumption of constant cell volume (1 fL). We have shown previously that the median cell volume of E. coli may decrease by approximately 30% following NH4+ run out (F. Heydenreich unpublished). Cell volume measurements were not performed in this study, however, a decrease in volume due to N starvation would effectively equate to an underestimation of intracellular metabolite concentrations.
Studying the non-diazotrophic ΔnifLA and nifK1–1203 mutants revealed several unexpected characteristics of amino acid metabolism during N starved conditions. As discussed above, prolonged N starvation did not lead to complete depletion of any one or more amino acids. Rather, in the absence of N fixation, subsets of amino acids accumulate to concentrations in excess of those observed in N-replete conditions (proline, lysine, and arginine) or in excess of time point-equivalent wild-type pools (glutamate, alanine, proline, lysine, and arginine). The accumulation of specific amino acids in non-diazotrophic mutants may signify bottlenecks in N metabolism when Nif expression is absent and/or nitrogenase function is lost. Low GS activity due to NH4+ limitation may explain the marginal excess of glutamate. Furthermore, both proline and arginine can be synthesised de novo from glutamate, which may explain their accumulation. Proline has also been shown to serve as an osmoprotectant during diazotrophy in Ko M5a1 (Le Rudulier et al., 1982). Alterations in glutamate metabolism cannot explain the accumulation of lysine, whose biosynthesis is unrelated. An alternative hypothesis is that some accumulating amino acids are derived preferentially from protein degradation and other N scavenging pathways.
Several studies have attempted to discern the distinct roles of the paralogous PII-type proteins GlnB and GlnK in the regulation of N metabolism. Functional redundancy between the pair is most apparent in vitro, whereby, for instance, uridylylated GlnK can activate both NtrB and the adenylation of GS and non-uridylylated GlnB can interact with the AmtB channel, albeit with lower affinity than the other paralogue in each case (Van Heeswijk et al., 1996; Atkinson and Ninfa, 1999; Javelle et al., 2004). Their distinct functional associations in vivo appear to relate as much to their condition-specific copy numbers as their structural differences, considering that GlnK exceeds constitutive GlnB many fold when upregulated during N limiting conditions. It has been proposed that GlnB represents the primary glutamine sensor in E. coli, with GlnK acting as a dynamic buffer to prevent excess N shock following starvation (van Heeswijk et al., 2013; Gosztolai et al., 2017).
The de-repression of NifA in a subset of diazotrophic bacteria such as Ko M5a1, however, is a PII-type function that has been attributed exclusively to GlnK (He et al., 1998; Jack et al., 1999), with GlnB only able to substitute for GlnK at excessively high and non-physiological concentrations (Arcondéguy et al., 1999). We observed that a ΔglnK knockout mutation reduced both the de-repressed level of nitrogenase activity and NifHDK protein levels by around two thirds when either NH4+ or glutamine were used as a limiting N source but still allowed N fixation at a reduced rate (Figure 7 and Supplementary Figure 13). This phenotype is in line with the 75% reduction in nif promoter activation seen previously in K. pneumoniae, albeit when using a strain in which nifLA were expressed independently of cellular N status from a low copy plasmid (Jack et al., 1999). However, it contrasts with the study by He et al. (1998), which reported complete repression of nif promoter activation in the absence of GlnK in a heterologous E. coli model system and founded the long-held view of an essential regulatory role. We propose that our study of GlnK in the native Ko M5a1 background is more representative of its true function during diazotrophy. Whilst the knockout strains generated in this report contain a nptII resistance cassette at the mutated loci, which may impose polar effects on neighbouring genes, it is important to note that previous studies have confirmed that the phenotypes of similarly derived ΔglnK strains are manifest independently of polar effects on the downstream amtB gene (Atkinson and Ninfa, 1998; He et al., 1998).
Although the ΔglnB mutation had no effect on N fixation or growth rate, our results suggest that GlnB is the more sensitive of the PII-type paralogues to changes in cellular N status. Whilst the GlnK uridylylation level remained at >90% following NH4+ runout, GlnB was marginally de-uridylylated following the onset of diazotrophic growth (Figure 5). Furthermore, the level of GlnB uridylylation varied in the range of 60–75% during N limiting conditions, suggesting that a trimer consisting of 2/3 uridylylated subunits is the dominant functional state. The contrasting uridylylation states of the two PII-type proteins may reflect different affinities for the UT/UR enzyme (GlnD) and allosteric signaling molecule 2-ketoglutarate (α-KG). It has been shown that GlnB is de-uridylylated around 10-fold faster than GlnK in the presence of moderate glutamine concentrations in E. coli (Atkinson and Ninfa, 1999), supporting the hypothesis that GlnB plays a rapid and fine-tuning role in cellular N signaling. The lack of a notable ΔglnB phenotype in the N limiting conditions studied may simply reflect the fact that GlnK is in excess, which enables it to substitute for GlnB but not to the same extent vice versa. In contrast, the ΔglnB mutant had a clear impact on NtrC activity in N-replete conditions when it represents the most abundant PII-type protein (Figure 7). Future studies might explore this hypothesis further by utilising a double ΔglnBglnK knockout as a baseline genotype in which to vary GlnB and GlnK expression levels independently. The fact that uncomplemented double ΔglnBglnK mutants render some diazotrophs unable to fix N (e.g., Michel-Reydellet and Kaminski, 1999) implies that whilst there may be redundancy between GlnB and GlnK, as observed here, some PII-type protein function is likely necessary for de-repression of nif gene expression. Our data would suggest that GlnB can function in place of GlnK, but at low constitutive copy number only minimal de-repression via NifLA is achieved.
Our comparison of the Ko M5a1 transcriptome between N-replete and N-fixing conditions revealed differential expression in more than 50% of genes (Figure 2 and Supplementary Figure 1), highlighting the breadth of cellular functions associated with both regulation of and adaptation to diazotrophic growth. As expected, many of the operons upregulated in diazotrophic conditions are associated with the conserved bacterial N stress response. Eighteen of the 25 canonical NtrC/Nac-regulated operons described in E. coli via microarray (Zimmer et al., 2000) were apparently upregulated here also. However, we also describe several putative NtrC-dependent operons relating to the metabolism of N-containing compounds (urea, nitrate/nitrite, purines) which may enable valuable scavenging functions during starvation conditions. There is potential value in performing a complete characterisation of the NtrC-dependent regulon in Ko M5a1, which may differ significantly to that of E. coli and provide a clearer picture of how N fixation is associated to other modules of N metabolism. Whilst comparative genomic analyses indicate a high degree of similarity between K. pneumoniae and E. coli, almost 800 genes are unique between the two species, many of which are thought to correspond to novel metabolic phenotypes and nutrient growth capabilities (Liao et al., 2011).
The gene families apparently downregulated during diazotrophy include several associated with gene expression and cell division, suggesting that the slow growth rate observed during N starvation is in part regulated. The alarmone of the stringent response, ppGpp, is released by the ribosome-associated ppGpp synthetase (RelA) and in E. coli the expression of relA is induced by NtrC during N stress (Brown et al., 2014). This study saw upregulation of relA in diazotrophic cells, suggesting that the stringent response may be responsible for a large component of differential global gene expression, downstream of NtrC, under N starved conditions. The stringent response has also been strongly implicated in the global deceleration and hibernation of ribosomes (Li et al., 2018).
Given the longstanding biotechnological interest in enhancing biological nitrogen fixation, it is remarkable that our understanding of diazotrophic bacteria has seldom progressed beyond a description of the various gene regulatory mechanisms governing nif gene expression to complete systems-level studies that integrate omics datasets and their interactions. Various refactored and synthetically inducible Ko M5a1 nif gene clusters reproduce only a fraction of wild-type nitrogenase activity in heterologous hosts (Temme et al., 2012; Ryu et al., 2020), highlighting the fact that even the most cutting edge of synthetic biology tools cannot compensate for an incomplete predictive model of native physiology. The genetic organization of nif genes would suggest that the N fixation function is sufficiently modular to be readily transferred between chassis microorganisms (Dixon and Postgate, 1972). However, it is deeply embedded in the context of multiple aspects of physiology beyond N status, including redox state, energy level, oxygen status, and metallochemistry.
This study provides a methodological framework within which predictive models of free-living diazotrophs can be developed and we have generated several hypotheses to be further explored. Analyses of Nif expression at the transcript and protein levels reveal a switch-like transition from tightly repressed to highly upregulated in response to N status. Given the scale of cellular burden associated with Nif expression, it is critical that metabolic engineering approaches applied to diazotrophy incorporate elements of temporal control in response to multiple stimuli.
Targeted mass spectrometry for absolute protein quantification represents an improvement in accuracy and sensitivity over previous methods used to estimate Nif protein levels, such as immunoblotting alongside external purified standards for semi-quantitative estimation of Nif expression in A. vinelandii (Poza-Carrión et al., 2014). Mass spectrometry capability is becoming ever more accessible and the QconCAT approach to method development, in particular, enables multiplexed, high-throughput, and reproducible protein quantification. Digested peptide samples can be further assessed via untargeted proteomics. In the context of our observation that Nif proteins make up a significant fraction of the diazotrophic proteome, a critical objective of further studies should be to assess the most significant, and perhaps compensatory, changes that occur in the general non-Nif compartment of the proteome. Likewise, global proteomics may shed light on the ΔnifLA and nifK1–1203 phenotypes, in particular with regard to whether the excess of free amino acids is reflected by compensatory changes in non-Nif protein levels.
We also show that co-extraction of proteins and amino acids can shed light on the availability and allocation rate of N resources prior to and during starvation. However, a limitation of this study is that it is not possible to distinguish directly which N atoms in cellular resource pools, such as amino acids, are derived de novo from N2 fixation or from pre-existing biomass via degradation and scavenging. Utilizing stable N isotopes (15N) to differentially label atmospheric N2 and N-containing biomass would address this question while being well suited to a mass spectrometry approach.
Finally, in light of growing evidence that phenotypic heterogeneity is a pervasive fitness strategy for many biotechnologically relevant microbes, even within controlled, artificial environments (Binder et al., 2017), it is important to note that many of the methods employed to study the regulation of diazotrophy, as exemplified here, assume homogenous cell behavior and that bulk measurements therefore represent the average cell in the population. Indeed, single-cell studies performed by Schreiber et al. (2016) determined that Ko M5a1 exhibits mixed-substrate growth with distinct N2-fixing and NH4-feeding sub-populations when grown continuously in states of intermediate N stress. However, the fact that diazotrophy was the homogenous phenotype under complete NH4+ starvation would imply that this behavior can be assumed following complete NH4+ run-out in this study.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
CW, JS, and MBu designed the study. CW, AL, and CH performed the experiments. MBe contributed to analytical chemistry (LC–MS). IK-L and MC contributed to the analysis of transcriptome data. CW analyzed the data and prepared the manuscript. All authors contributed to critical review and editing of the manuscript.
This work was supported by a BBSRC Strategic LOLA grant (BBSRC, BB/N003608/1) awarded to MBu. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Ruth Schmitz (Christian Albrechts University, Kiel) for the Ko M5a1 strain and Ron Hay (University of Dundee) for the BL21(DE3) ΔlysΔarg strain.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.718487/full#supplementary-material
Allen, R. S., Tilbrook, K., Warden, A. C., Campbell, P. C., Rolland, V., Singh, S. P., et al. (2017). Expression of 16 nitrogenase proteins within the plant mitochondrial matrix. Front. Plant Sci. 8:287. doi: 10.3389/fpls.2017.00287
Ambrosio, R., Ortiz-Marquez, J. C. F., and Curatti, L. (2017). Metabolic engineering of a diazotrophic bacterium improves ammonium release and biofertilization of plants and microalgae. Metab. Eng. 40, 59–68. doi: 10.1016/j.ymben.2017.01.002
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Arcondéguy, T., Heeswijk, W. C., and Merrick, M. (1999). Studies on the roles of GlnK and GlnB in regulating Klebsiella pneumoniae NifL-dependent nitrogen control. FEMS Microbiol. Lett. 180, 263–270. doi: 10.1111/j.1574-6968.1999.tb08805.x
Arnold, W., Rump, A., Klipp, W., Priefer, U. B., and Pühler, A. (1988). Nucleotide sequence of a 24,206-base-pair DNA fragment carrying the entire nitrogen fixation gene cluster of Klebsiella pneumoniae. J. Mol. Biol. 203, 715–738. doi: 10.1016/0022-2836(88)90205-7
Atkinson, M. R., and Ninfa, A. J. (1998). Role of the GlnK signal transduction protein in the regulation of nitrogen assimilation in Escherichia coli. Mol. Microbiol. 29, 431–447. doi: 10.1046/j.1365-2958.1998.00932.x
Atkinson, M. R., and Ninfa, A. J. (1999). Characterization of the GlnK protein of Escherichia coli. Mol. Microbiol. 32, 301–313. doi: 10.1046/j.1365-2958.1999.01349.x
Austin, S., Henderson, N., and Dixon, R. (1987). Requirements for transcriptional activation in vitro of the nitrogen-regulated gInA and nifLA promoters from Klebsiella pneumoniae: dependence on activator concentration. Mol. Microbiol. 1, 92–100. doi: 10.1111/j.1365-2958.1987.tb00532.x
Barney, B. M., Eberhart, L. J., Ohlert, J. M., Knutson, C. M., and Plunkett, M. H. (2015). Gene deletions resulting in increased nitrogen release by azotobacter vinelandii: application of a novel nitrogen biosensor. Appl. Environ. Microbiol. 81, 4316–4328. doi: 10.1128/AEM.00554-15
Batista, M. B., and Dixon, R. (2019). Manipulating nitrogen regulation in diazotrophic bacteria for agronomic benefit. Biochem. Soc. Trans. 47, 603–614. doi: 10.1042/BST20180342
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery ratea practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodological 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bennett, B. D., Kimball, E. H., Gao, M., Osterhout, R., Van Dien, S. J., and Rabinowitz, J. D. (2009). Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599. doi: 10.1038/nchembio.186
Binder, D., Drepper, T., Jaeger, K.-E., Delvigne, F., Wiechert, W., Kohlheyer, D., et al. (2017). Homogenizing bacterial cell factories: analysis and engineering of phenotypic heterogeneity. Metab. Eng. 42, 145–156. doi: 10.1016/J.YMBEN.2017.06.009
Bloch, S. E., Ryu, M. H., Ozaydin, B., and Broglie, R. (2020). Harnessing atmospheric nitrogen for cereal crop production. Curr. Opin. Biotechnol. 62, 181–188. doi: 10.1016/j.copbio.2019.09.024
Brauer, M. J., Yuan, J., Bennett, B. D., Lu, W., Kimball, E., Botstein, D., et al. (2006). Conservation of the metabolomic response to starvation across two divergent microbes. Proc. Natl. Acad. Sci. U.S.A. 103, 19302–19307. doi: 10.1073/pnas.0609508103
Bremer, H., and Dennis, P. P. (2008). Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 3. doi: 10.1128/ecosal.5.2.3
Brown, D. R., Barton, G., Pan, Z., Buck, M., and Wigneshweraraj, S. (2014). Nitrogen stress response and stringent response are coupled in Escherichia coli. Nat. Commun. 5:4115. doi: 10.1038/ncomms5115
Burén, S., López-Torrejón, G., and Rubio, L. M. (2018). Extreme bioengineering to meet the nitrogen challenge. Proc. Natl. Acad. Sci. U.S.A. 115, 8849–8851. doi: 10.1073/pnas.1812247115
Condon, C., Squires, C., and Squires, C. L. (1995). Control of rRNA transcription in Escherichia coli. Microbiol. Mol. Biol. Rev. 59, 623–645. doi: 10.1128/mr.59.4.623-645.1995
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Dixon, R. A., and Postgate, J. R. (1972). Genetic transfer of nitrogen fixation from klebsiella pneumoniae to escherichia coli. Nature 237, 102–103. doi: 10.1038/237102a0
Dixon, R., and Kahn, D. (2004). Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631. doi: 10.1038/nrmicro954
Drummond, M., Clements, J., Merrick, M., and Dixon, R. (1983). Positive control and autogenous regulation of the nifLA promoter in Klebsiella pneumoniae. Nature 301, 302–307. doi: 10.1038/301302a0
Gosztolai, A., Schumacher, J., Behrends, V., Bundy, J. G., Heydenreich, F., Bennett, M. H., et al. (2017). GlnK facilitates the dynamic regulation of bacterial nitrogen assimilation. Biophys. J. 112, 2219–2230. doi: 10.1016/J.BPJ.2017.04.012
Gray, N., Zia, R., King, A., Patel, V. C., Wendon, J., McPhail, M. J. W., et al. (2017). High-speed quantitative UPLC-MS analysis of multiple amines in human plasma and serum via precolumn derivatization with 6-aminoquinolyl- N -hydroxysuccinimidyl Carbamate: application to acetaminophen-induced liver failure. Anal. Chem. 89, 2478–2487. doi: 10.1021/acs.analchem.6b04623
Hageman, R. V., and Burris, R. H. (1978). Nitrogenase and nitrogenase reductase associate and dissociate with each catalytic cycle. Proc. Natl. Acad. Sci. U.S.A. 75, 2699–2702. doi: 10.1073/pnas.75.6.2699
Harris, D. F., Lukoyanov, D. A., Shaw, S., Compton, P., Tokmina-Lukaszewska, M., Bothner, B., et al. (2018). Mechanism of N2 reduction catalyzed by fe-nitrogenase involves reductive elimination of H2. Biochemistry 57, 701–710. doi: 10.1021/acs.biochem.7b01142
He, L., Soupene, E., Ninfa, A., and Kustu, S. (1998). Physiological role for the GlnK protein of enteric bacteria: relief of NifL inhibition under nitrogen-limiting conditions. J. Bacteriol. 180, 6661–6667. doi: 10.1128/jb.180.24.6661-6667.1998
Henderson, N., Austin, S., and Dixon, R. (1989). Role of metal ions in negative regulation of nitrogen fixation by the nitL gene product from Klebsiella pneumoniae. Mol. General Genet. MGG 216, 484–491. doi: 10.1007/bf00334394
Hunter, M. C., Smith, R. G., Schipanski, M. E., Atwood, L. W., and Mortensen, D. A. (2017). Agriculture in 2050: recalibrating targets for sustainable intensification. Bioscience 67, 386–391. doi: 10.1093/biosci/bix010
Ikeda, T. P., Shauger, A. E., and Kustu, S. (1996). Salmonella typhimurium apparently perceives external nitrogen limitation as internal glutamine limitation. J. Mol. Biol. 259, 589–607. doi: 10.1006/jmbi.1996.0342
Iniguez, A. L., Dong, Y., and Triplett, E. W. (2007). Nitrogen fixation in wheat provided by Klebsiella pneumoniae 342. Mol. Plant Microbe. Interact. 17, 1078–1085. doi: 10.1094/MPMI.2004.17.10.1078
Jack, R., De Zamaroczy, M., and Merrick, M. (1999). The signal transduction protein GlnK is required for NifL-dependent nitrogen control of Nif gene expression in Klebsiella pneumoniae. J. Bacteriol. 181, 1156–1162. doi: 10.1128/jb.181.4.1156-1162.1999
Javelle, A., Severi, E., Thornton, J., and Merrick, M. (2004). Ammonium sensing in Escherichia coli: role of the ammonium transporter Amtb and AmtB-GlnK complex formation. J. Biol. Chem. 279, 8530–8538. doi: 10.1074/jbc.M312399200
Jimenez-Vicente, E., Yang, Z.-Y., Ray, W. K., Echavarri-Erasun, C., Cash, V. L., Rubio, L. M., et al. (2018). Sequential and differential interaction of assembly factors during nitrogenase MoFe protein maturation. J. Biol. Chem. 293, 9812–9823. doi: 10.1074/jbc.RA118.002994
Kim, Y.-M., Hidaka, M., Masaki, H., Beppu, T., and Uozumi, T. (1989). Constitutive expression of nitrogenase system in Klebsiella oxytoca by gene targeting mutation to the chromosomal nifLA operon. J. Biotechnol. 10, 293–301. doi: 10.1016/0168-1656(89)90073-4
Klopprogge, K., Grabbe, R., Hoppert, M., and Schmitz, R. A. (2002). Membrane association of Klebsiella pneumoniae NifL is affected by molecular oxygen and combined nitrogen. Arch. Microbiol. 177, 223–234. doi: 10.1007/s00203-001-0379-x
Le Rudulier, D., Yang, S. S., and Csonka, L. N. (1982). Nitrogen fixaton in Klebsiella pneumoniae during osmotic stress effect of exogenous proline or a proline overproducing plasmid. BBA Gen. Subj. 719, 273–283. doi: 10.1016/0304-4165(82)90099-X
Li, S. H.-J., Li, Z., Park, J. O., King, C. G., Rabinowitz, J. D., Wingreen, N. S., et al. (2018). Escherichia coli translation strategies differ across carbon, nitrogen and phosphorus limitation conditions. Nat. Microbiol. 3, 939–947. doi: 10.1038/s41564-018-0199-2
Liao, Y. C., Huang, T. W., Chen, F. C., Charusanti, P., Hong, J. S. J., Chang, H. Y., et al. (2011). An experimentally validated genome-scale metabolic reconstruction of Klebsiella pneumoniae MGH 78578, iYL1228. J. Bacteriol. 193, 1710–1717. doi: 10.1128/JB.01218-10
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
MacLean, B., Tomazela, D. M., Shulman, N., Chambers, M., Finney, G. L., Frewen, B., et al. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. doi: 10.1093/bioinformatics/btq054
Martinez-Argudo, I., Little, R., Shearer, N., Johnson, P., and Dixon, R. (2004). The NifL-NifA system: a multidomain transcriptional regulatory complex that integrates environmental signals. J. Bacteriol. 186, 601–610. doi: 10.1128/JB.186.3.601-610.2004
Martínez-Noël, G., Curatti, L., Hernandez, J. A., and Rubio, L. M. (2011). NifB and NifEN protein levels are regulated by ClpX2 under nitrogen fixation conditions in Azotobacter vinelandii. Mol. Microbiol. 79, 1182–1193. doi: 10.1111/j.1365-2958.2011.07540.x
Matic, I., Jaffray, E. G., Oxenham, S. K., Groves, M. J., Barratt, C. L., Tauro, S., et al. (2011). Absolute SILAC-compatible expression strain allows Sumo-2 copy number determination in clinical samples. J. Proteome. Res. 10, 4869–4875. doi: 10.1021/pr2004715
Merrick, M. (2015). Post-translational modification of PII signal transduction proteins. Front. Microbiol. 5:763. doi: 10.3389/fmicb.2014.00763
Michel-Reydellet, N., and Kaminski, P. A. (1999). Azorhizobium caulinodans PII and GlnK proteins control nitrogen fixation and ammonia assimilation. J. Bacteriol. 181, 2655–2658. doi: 10.1128/jb.181.8.2655-2658.1999
Milo, R. (2013). What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 35, 1050–1055. doi: 10.1002/bies.201300066
Minchin, S. D., Austin, S., and Dixon, R. A. (1988). The role of activator binding sites in transcriptional control of the divergently transcribed nifF and nif LA promoters from Klebsiella pneumoniae. Mol. Microbiol. 2, 433–442. doi: 10.1111/j.1365-2958.1988.tb00049.x
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. doi: 10.1038/nmeth.1226.1038/nmeth.1226
Mus, F., Alleman, A. B., Pence, N., Seefeldt, L. C., and Peters, J. W. (2018). Exploring the alternatives of biological nitrogen fixation. Metallomics 10, 523–538. doi: 10.1039/C8MT00038G
Pankievicz, V. C. S., Do Amaral, F. P., Santos, K. F. D. N., Agtuca, B., Xu, Y., Schueller, M. J., et al. (2015). Robust biological nitrogen fixation in a model grass-bacterial association. Plant J. 81, 907–919. doi: 10.1111/tpj.12777
Peters, J. W., Fisher, K., and Dean, D. R. (1995). Nitrogenase Structure and Function: A Biochemical-Genetic Perspective. Available online at: www.annualreviews.org (accessed October 1, 2020)
Poza-Carrión, C., Jiménez-Vicente, E., Navarro-Rodríguez, M., Echavarri-Erasun, C., and Rubio, L. M. (2014). Kinetics of nif gene expression in a nitrogen-fixing bacterium. J. Bacteriol. 196, 595–603. doi: 10.1128/JB.00942-13
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Rogers, C., and Oldroyd, G. E. D. (2014). Synthetic biology approaches to engineering the nitrogen symbiosis in cereals. J. Exp. Bot. 65, 1939–1946. doi: 10.1093/jxb/eru098
Ryu, M. H., Zhang, J., Toth, T., Khokhani, D., Geddes, B. A., Mus, F., et al. (2020). Control of nitrogen fixation in bacteria that associate with cereals. Nat. Microbiol. 5, 314–330. doi: 10.1038/s41564-019-0631-2
Sanchuki, H. B. S., Gravina, F., Rodrigues, T. E., Gerhardt, E. C. M., Pedrosa, F. O., Souza, E. M., et al. (2017). Dynamics of the Escherichia coli proteome in response to nitrogen starvation and entry into the stationary phase. Biochim. Biophys. Acta Proteins Proteomics 1865, 344–352. doi: 10.1016/j.bbapap.2016.12.002
Sapcariu, S. C., Kanashova, T., Weindl, D., Ghelfi, J., Dittmar, G., and Hiller, K. (2014). Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 1, 74–80. doi: 10.1016/j.mex.2014.07.002
Schmitz, R. A. (2000). Internal glutamine and glutamate pools in Klebsiella pneumoniae grown under different conditions of nitrogen availability. Curr. Microbiol. 41, 357–362. doi: 10.1007/s002840010149
Schreiber, F., Littmann, S., Lavik, G., Escrig, S., Meibom, A., Kuypers, M. M. M., et al. (2016). Phenotypic heterogeneity driven by nutrient limitation promotes growth in fluctuating environments. Nat. Microbiol. 1:16055. doi: 10.1038/nmicrobiol.2016.55
Schumacher, J., Behrends, V., Pan, Z., Brown, D. R., Heydenreich, F., Lewis, M. R., et al. (2013). Nitrogen and carbon status are integrated at the transcriptional level by the nitrogen regulator NtrC in vivo. MBio 4:e00881-13. doi: 10.1128/mBio.00881-13
Scott, M., Klumpp, S., Mateescu, E. M., and Hwa, T. (2014). Emergence of robust growth laws from optimal regulation of ribosome synthesis. Mol. Syst. Biol. 10, 747–747. doi: 10.15252/msb.20145379
Seefeldt, L. C., Hoffman, B. M., and Dean, D. R. (2009). Mechanism of mo-dependent nitrogenase. Annu. Rev. Biochem. 78, 701–722. doi: 10.1146/annurev.biochem.78.070907.103812
Setten, L., Soto, G., Mozzicafreddo, M., Fox, A. R., Lisi, C., Cuccioloni, M., et al. (2013). Engineering Pseudomonas protegens Pf-5 for nitrogen fixation and its application to improve plant growth under nitrogen-deficient conditions. PLoS One 8:e63666. doi: 10.1371/journal.pone.0063666
Smanski, M. J., Bhatia, S., Zhao, D., Park, Y., Woodruff, L. B. A., Giannoukos, G., et al. (2014). Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol. 32, 1241–1249. doi: 10.1038/nbt.3063
Smil, V. (2001). Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. Cambridge, MA: MIT Press.
Steffen, W., Richardson, K., Rockström, J., Cornell, S. E., Fetzer, I., Bennett, E. M., et al. (2015). Sustainability. planetary boundaries: guiding human development on a changing planet. Science 347:1259855. doi: 10.1126/science.1259855
Temme, K., Tamsir, A., and Bloch, S. (2020). Methods And Compositions For Improving Engineered Microbes. Available online at: https://lens.org/112-138-962-836-769 (accessed May 31, 2021).
Temme, K., Zhao, D., and Voigt, C. A. (2012). Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. U.S.A. 109, 7085–7090. doi: 10.1073/pnas.1120788109
Van Heeswijk, W. C., Hoving, S., Molenaar, D., Stegeman, B., Kahn, D., and Westerhoff, H. V. (1996). An alternative P(II) protein in the regulation of glutamine synthetase in Escherichia coli. Mol. Microbiol. 21, 133–146. doi: 10.1046/j.1365-2958.1996.6281349.x
van Heeswijk, W. C., Westerhoff, H. V., and Boogerd, F. C. (2013). Nitrogen assimilation in Escherichia coli: putting molecular data into a systems perspective. Microbiol. Mol. Biol. Rev. 77, 628–695. doi: 10.1128/MMBR.00025-13
Yang, J., Xie, X., Xiang, N., Tian, Z.-X., Dixon, R., and Wang, Y.-P. (2018). Polyprotein strategy for stoichiometric assembly of nitrogen fixation components for synthetic biology. Proc. Natl. Acad. Sci. U.S.A. 115, E8509–E8517. doi: 10.1073/pnas.1804992115
Zhang, X., Davidson, E. A., Mauzerall, D. L., Searchinger, T. D., Dumas, P., and Shen, Y. (2015). Managing nitrogen for sustainable development. Nature 528, 51–59. doi: 10.1038/nature15743
Zhu, M., Dai, X., and Wang, Y. P. (2016). Real time determination of bacterial in vivo ribosome translation elongation speed based on LacZα complementation system. Nucleic Acids Res. 44:e155. doi: 10.1093/nar/gkw698
Zimmer, D. P., Soupene, E., Lee, H. L., Wendisch, V. F., Khodursky, A. B., Peter, B. J., et al. (2000). Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc. Natl. Acad. Sci. U.S.A. 97, 14674–14679. doi: 10.1073/pnas.97.26.14674
Keywords: nitrogen stress, absolute protein quantification, Klebsiella oxytoca, nif gene expression, PII proteins, nitrogenase, diazotrophic bacteria: biological fixation of nitrogen, resource allocation
Citation: Waite CJ, Lindström Battle A, Bennett MH, Carey MR, Hong CK, Kotta-Loizou I, Buck M and Schumacher J (2021) Resource Allocation During the Transition to Diazotrophy in Klebsiella oxytoca. Front. Microbiol. 12:718487. doi: 10.3389/fmicb.2021.718487
Received: 01 June 2021; Accepted: 12 July 2021;
Published: 09 August 2021.
Edited by:
Harry Beller, U.S. Environmental Protection Agency (EPA), United StatesReviewed by:
Marcelo Bueno Batista, Department of Molecular Microbiology, John Innes Centre, United KingdomCopyright © 2021 Waite, Lindström Battle, Bennett, Carey, Hong, Kotta-Loizou, Buck and Schumacher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher J. Waite, Y2hyaXN0b3BoZXIud2FpdGUxMEBpbXBlcmlhbC5hYy51aw==; Jörg Schumacher, ai5zY2h1bWFjaGVyQGltcGVyaWFsLmFjLnVr
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.