
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 August 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.711577
 Mauricio Flores-Valdez1,2†
Mauricio Flores-Valdez1,2† Miguel A. Ares1,3†
Miguel A. Ares1,3† Roberto Rosales-Reyes4
Roberto Rosales-Reyes4 Javier Torres1
Javier Torres1 Jorge A. Girón5
Jorge A. Girón5 Bart C. Weimer6
Bart C. Weimer6 Alfonso Mendez-Tenorio2*
Alfonso Mendez-Tenorio2* Miguel A. De la Cruz1*
Miguel A. De la Cruz1*Klebsiella pneumoniae is recognized as a common cause of nosocomial infections and outbreaks causing pneumonia, septicemia, and urinary tract infections. This opportunistic bacterium shows an increasing acquisition of antibiotic-resistance genes, which complicates treatment of infections. Hence, fast reliable strain typing methods are paramount for the study of this opportunistic pathogen’s multi-drug resistance genetic profiles. In this study, thirty-eight strains of K. pneumoniae isolated from the blood of pediatric patients were characterized by whole-genome sequencing and genomic clustering methods. Genes encoding β-lactamase were found in all the bacterial isolates, among which the blaSHV variant was the most prevalent (53%). Moreover, genes encoding virulence factors such as fimbriae, capsule, outer membrane proteins, T4SS and siderophores were investigated. Additionally, a multi-locus sequence typing (MLST) analysis revealed 24 distinct sequence types identified within the isolates, among which the most frequently represented were ST76 (16%) and ST70 (11%). Based on LPS structure, serotypes O1 and O3 were the most prevalent, accounting for approximately 63% of all infections. The virulence capsular types K10, K136, and K2 were present in 16, 13, and 8% of the isolates, respectively. Phylogenomic analysis based on virtual genome fingerprints correlated with the MLST data. The phylogenomic reconstruction also denoted association between strains with a higher abundance of virulence genes and virulent serotypes compared to strains that do not possess these traits. This study highlights the value of whole-genomic sequencing in the surveillance of virulence attributes among clinical K. pneumoniae strains.
Klebsiella pneumoniae is a ubiquitous Gram-negative encapsulated bacterium that is commonly found in the mucosal surfaces of mammals and the environment. In humans, these bacteria can colonize the gastrointestinal and nasopharynx tracts, from where they gain entry to blood circulation and other tissues causing infection (Piperaki et al., 2017). K. pneumoniae is well-known as one of the major causes of community- and hospital-acquired Gram-negative bloodstream infections (Meatherall et al., 2009). In immunocompromised patients it can cause biliary infection, peritonitis, meningitis, septicemia, and liver abscesses (Ares et al., 2013). Also, K. pneumoniae nosocomial isolates often display high rates of antimicrobial multi-drug resistance (Chong et al., 2018). This opportunistic microorganism causes infection through several virulence factors such as surface antigens, fimbriae, capsule, outer membrane proteins, and siderophores; all of which allow the bacteria to enter and multiply within the host (Podschun and Ullmann, 1998; Ares et al., 2016, 2019). The genus Klebsiella expresses two types of surface antigens: a lipopolysaccharide (O antigen) and a capsular polysaccharide (K antigen). The K. pneumoniae capsule is a complex structure that measures up to 160 nm wide (Clements et al., 2008). The number of genes that code for the capsule of K. pneumoniae vary from 16 to 20 genes according to the capsular serotype. The capsular polysaccharide synthesis (cps) locus comprises genes that are organized into three consecutive transcriptional units that give rise to a large number of proteins involved in biosynthesis and export of capsular components. The capsule is the most thoroughly studied virulence factor of K. pneumoniae. The CPS confers resistance against the bactericidal activity of antimicrobial peptides, complement, and phagocytes (Paczosa and Mecsas, 2016). In addition, the capsule also protects the bacterium from opsonization and suppresses the early inflammatory response (Ko, 2017; Ares et al., 2019). The LPS is essential for the bacterial cell envelope structure, protects against antibiotics, and promotes adhesion. The LPS consists of three domains: the lipid A, an oligosaccharide core and the O antigen. The polysaccharide chains of the O antigen facilitate the initial adhesion process and confer resistance to serum bactericidal activity (Podschun and Ullmann, 1998; Stojkovic et al., 2017). Thus, both capsule and LPS antigens significantly contribute to the pathogenicity of the bacteria (Evrard et al., 2010). Seventy-eight K antigens and eight O antigens have been described in K. pneumoniae strains. The structural variability of these antigens has been used to classify strains into several serotypes, which in turn have elucidated differences between the degree of virulence among different strains (Follador et al., 2016). Strains carrying the K1 and K2 capsular serotypes, which mainly cause liver abscess, are known to be hypermucoviscous (Chung et al., 2012) and the serotype O1 are the most prevalent in clinical infections (Hsieh et al., 2014). Host cell adherence and biofilm formation of K. pneumoniae isolates are mediated by type 1, type 3, and ECP fimbriae whose major fimbrial subunits are encoded by the fimA, mrkA, and ecpA genes, respectively (Alcántar-Curiel et al., 2013). OmpA is one of the major outer membrane proteins of Gram-negative bacteria, and it is highly conserved among the Enterobacteriaceae. K. pneumoniae’s OmpA contributes to the attenuation of the airway epithelial cell-mediated inflammatory response and the absence of OmpA makes the microorganism more susceptible to antimicrobial peptides (Li et al., 2014). Additionally, K. pneumoniae produces two major porins OmpK35 and OmpK36, which are homologs of OmpF and OmpC of Escherichia coli, respectively, functioning as channels for export and import of hydrophilic molecules (e.g., nutrients and cephalosporins/carbapenems). The loss of outer membrane porins in K. pneumoniae leads to increased resistance to cephalosporins/carbapenems and reduced virulence (March et al., 2013; Li et al., 2014). The production of siderophores confers to bacteria the ability to sequester and uptake iron from infected tissues. K. pneumoniae produces different siderophores among which the most relevant are enterobactin, yersiniabactin, aerobactin, and salmochelin. In this study, we characterized the most relevant phenotypic and genomic virulence traits occurring among a set of 38 clinical K. pneumoniae strains isolated from a pediatric population in Mexico City. The outcome of this study highlights the importance whole genome sequence analysis in the surveillance of virulence attributes of clinical K. pneumoniae strains.
Klebsiella pneumoniae strains were isolated (September 2010 to August 2013) from individuals diagnosed with sepsis from the Laboratory of Clinical Microbiology, Pediatric Hospital, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social (IMSS), Mexico City, Mexico (Ares et al., 2013). All pediatric patients included in this study were hospitalized for more than 48 h. A clinical examination was conducted by the medical staff in order to rule out sepsis acquired outside the hospital. Microbiological methods were performed as described (Ares et al., 2013). To isolate and identify the microbial species, positive blood cultures were selected. An aliquot of 50 μL was inoculated onto MacConkey agar plates and incubated in aerobic conditions for 24–48 h at 37°C. After incubation, colonies grown on agar plates were suspended in 3 mL of sterile saline solution (0.45% NaCl, pH 7.0) to reach a McFarland turbidity of 0.5–0.63. Bacterial identification was carried out using the VITEK-2 system (bioMérieux) and genomic DNA was extracted using the DNeasy Mini Kit (Qiagen, Hilden, Germany). The clinical data are shown in Table 1.
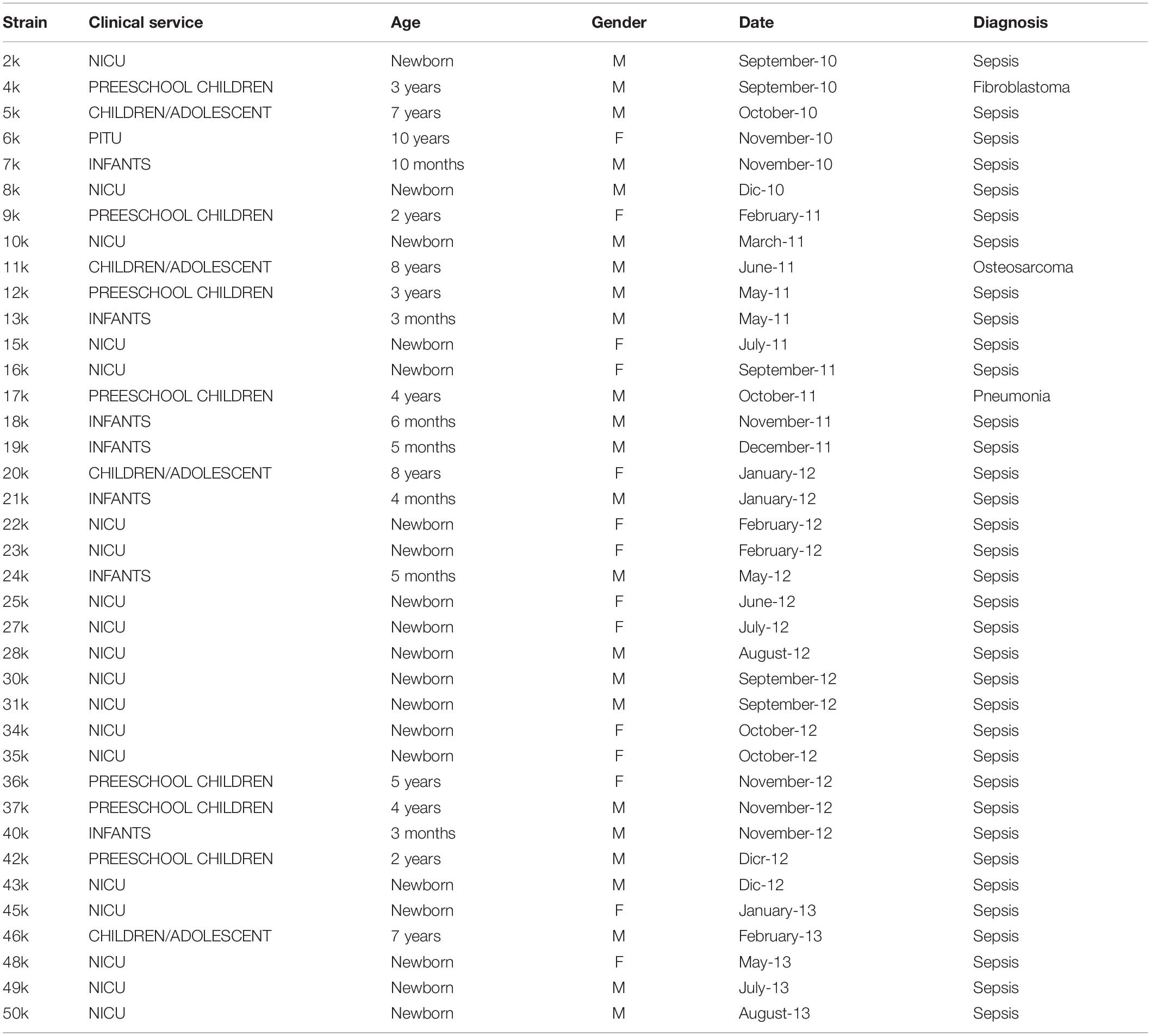
Table 1. Clinical data of K. pneumoniae strains.
Antimicrobial susceptibility testing for K. pneumoniae strains was carried out using Gram-negative bacilli cards (VITEK-2, AST- GN04) as was previously reported (Ares et al., 2013).
Genomic DNA was obtained from 50 random strains of the clinical K. pneumoniae strain collection and subjected to whole genome sequencing with the HiSeq 2000 platform (Illumina) producing 150 bp paired end reads at the laboratory of Dr. Bart Weimer, University of California, Davis. De novo genome assembly was accomplished using SPAdes v.3.13.0 (Bankevich et al., 2012) using a range of k-mer sizes of 21, 33, 55, 77-mers for the assembly. The assembled draft genomes were annotated by Prokka v.1.13 (Seemann, 2014). Finally, 38 K. pneumoniae genomes showed good data quality and were deposited at NCBI. Five Klebsiella quasipneumoniae (29k, 33k, 39k, 41k, and 44k) and three Klebsiella variicola (3k, 26k, and 32k) strains were added to the bioproject. In addition, strain 1k, which is the genome of wild-type K. pneumoniae used in our group, was also included (Ares et al., 2016).
Sequence data were submitted to the NCBI’s Gen Bank under BioProject Accession Number PRJNA605025. Each of the strains is stored with the Accession Numbers SAMN14008666-SAMN14008712 and the genomic sequences can be accessed with the Accession Numbers JAAGZT000000000-JAAHBN000000000.
Genome sequences of both K. pneumoniae isolates and NTUH K-2044 reference strain (Wu et al., 2009) were provided in FASTA format to calculate a phylogenomic tree based on Virtual Genome Fingerprints (VGF) using the VAMPhyRE software1. This method consists of carrying out a virtual hybridization analysis in which the recognition sites of a collection of 15,264 13-mer probe sequences previously designed to maximize their diversity, are searched in both strands of each genome and allowing a single-base mismatch during hybridization. The collection of hybridization sites obtained by this technique is known as the Virtual Genomic Fingerprint (VGF) and consists of a sample of sites regularly distributed throughout the genome. The VGFs are compared pairwise to estimate the fraction of shared homologous sites. A site comparison optimization analysis is previously performed, including a symmetric extension to both sites of genome alignment at each hybridization site and applying threshold match value that guarantees that only those sites not shared by chance are counted. From the fraction of homologous sites shared between each pair of genomes, a measure of their genomic distance, equivalent to the number of nucleotide substitutions between each pair, is estimated according to a method described by Nei and Li (1979). The distance is used in the MEGAX program to calculate phylogenetic trees using the Neighbor-Joining method (Kumar et al., 2018). Phylogenomic trees were edited with iTol (Interactive Tree of Life). Likewise, all assembled genomes in the dataset study were annotated using the Prokka prokaryotic annotation pipeline (Seemann, 2014) and the pan-genome was calculated with Roary: The Pan Genome Pipeline (Page et al., 2015) negative bacilli cards (VITEK-2, AST- GN04), as was previously reported (Ares et al., 2013).
Multilocus sequence typing (MLST) analysis was performed using seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) (Diancourt et al., 2005). The allele sequences and sequence types (STs) were determined by the BIGS database of the Institute Pasteur2 (Jolley and Maiden, 2010). Only exact matches of the allelic profile were reported.
The bacterial resistome was obtained by assessing the occurrence of antimicrobial resistance genes and plasmid replicons using both ResFinder 3.1 (Zankari et al., 2012) and PlasmidFinder 2.0 (Carattoli et al., 2014) from the Center for Genomic Epidemiology3. The analysis was performed with an identity threshold of 98% and a minimum match length of 80%. The families of the antibiotics assessed were fosfomycin, quinolones, phenicoles, aminoglycosides, sulphonamides, trimethoprim, tetracycline, and β-lactam.
Known virulence genes were searched on the K. pneumoniae genomes obtained above with BLASTN (Altschul et al., 1990; Camacho et al., 2009). For type 1 and type 3 fimbriae the clusters fim (fimB, fimE, fimA, fimI, fimC, fimD, fimF, fimG, fimH, and fimK) and mrk (mrkA, mrkB, mrkC, mrkD, and mrkF) were used, respectively. For ECP, the ecpA, ecpB, ecpC, ecpD, and ecpE gene cluster was analyzed. Likewise, the genes that code for outer membrane proteins (ompA, ompK35, and ompK36), the cluster virB (virB1, virB2, virB3-B4, virB5, virB6, virB7, virB8, virB9, virB10, and virB11) of the T4SS, and the hypermucoviscous associated genes magA and rmpA were screened. Additionally, fes, iroN, fyuA, and iutA genes, which correspond to enterobactin, salmochelin, yersiniabactin, and aerobactin siderophores respectively, were also searched. All the genes used for querying were obtained from the NTUH K-2044 reference strain. All analyses were carried out with an identity threshold of 96% and a minimum match length of 80%.
The genes wzi and wzc were searched in the draft genomes of K. pneumoniae using BLASTN (Altschul et al., 1990; Camacho et al., 2009), a database was assembled utilizing the corresponding locus of K serotypes published by Wyres et al. (2016). In the same manner the locus wb was searched and compared with the data published by Fang et al. (2016) to identify O serotypes. All analyses were carried out with an identity threshold of 96% and a minimum length of 80%. Additionally, the software Kaptive4 (Wick et al., 2018) was used to determine the K and O serotypes. Results that matched in both methodologies were reported as positive for the searched serotype.
Capsular polysaccharides were extracted and quantified as previously described (Ares et al., 2016). The glucuronic acid concentration in each bacterium was expressed in micrograms/109 CFU.
The phagocytosis assay was performed using THP-1 (ATCC TIB-202) human monocytes as previously described (Ares et al., 2016). Phagocytosis resistance was expressed in intracellular colony-forming-units per mL (CFUs/mL).
All experiments were performed with an initial inoculum of ∼106 CFU/mL in 20 mL of LB medium in 50 mL pyrogen-free and sterile polypropylene tubes. Polymyxin B activity was examined at 4 mg/mL. Serial strains in 20 mL were collected at 4 h for viable counting on LB agar plates after 24 h of incubation at 37°C as described Nang et al., 2019.
Genomic sequences of 38 bacterial strains positively identified as K. pneumoniae were obtained and they were named with a number followed by a letter “k” (e.g., 1k; Table 1). The draft genomes had an average assembly length of 5.55 Mb ± 0.14; a GC content of 57.23% ± 0.16%; N50 of 169,445 ± 41,243; and CDS of 5,135 ± 144 (assembly data and accession are shown in Table 2). The K. pneumoniae isolates shared a core genome consisting of 1,192 genes (present in ≥99% isolates and with 95% identity). The genomes analysis showed that with exception of strain 46k, all K. pneumoniae presented at least one plasmid sequence (Table 3).
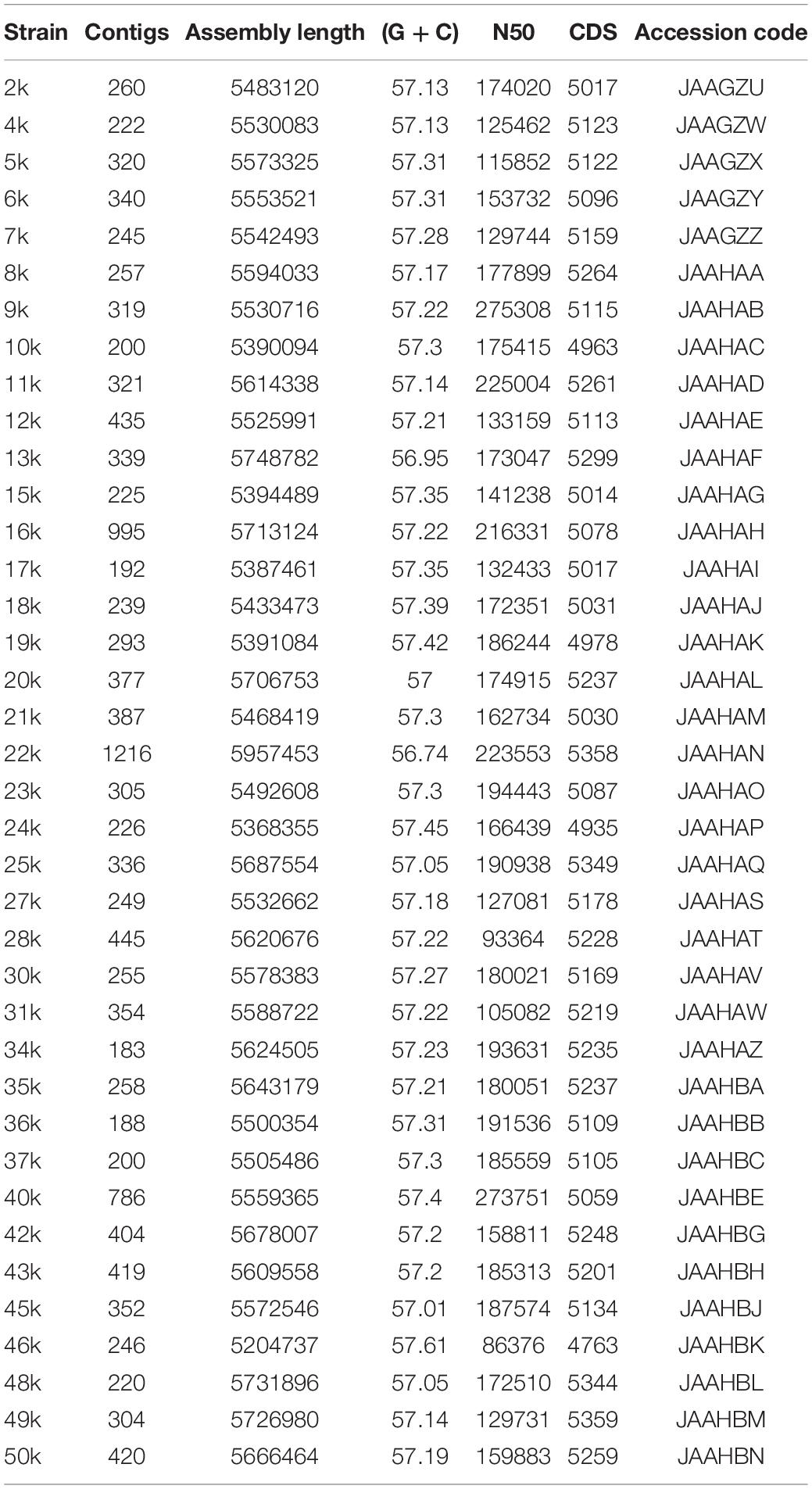
Table 2. Assembly data of K. pneumoniae genomes.

Table 3. Plasmid replicons found in K. pneumoniae strains.
Twenty-four different sequence types (ST) were identified among the K. pneumoniae isolates in this study. The most prevalent STs were ST-76 in six strains followed by ST-70 with four occurrences (Table 4). Interestingly, two K. pneumoniae strains, 46k and 43k, presented two new sequence types ST-4872 and ST-4873, respectively. Our analysis showed that ST-76 and ST-70 are the most prevalent ST in the strains collection and that new STs such as ST-4872 and ST-4873, appeared in the bacterial population of K. pneumoniae. The phylogenetic analysis was carried out based on the virtual genomic fingerprint technique (VAMPhyRE) using 13 k-mer probes and the Nearest-Neighbor method in MEGAX with default settings. The final phylogenomic representation (Figure 1) denotes consistent correlation with MLST findings.
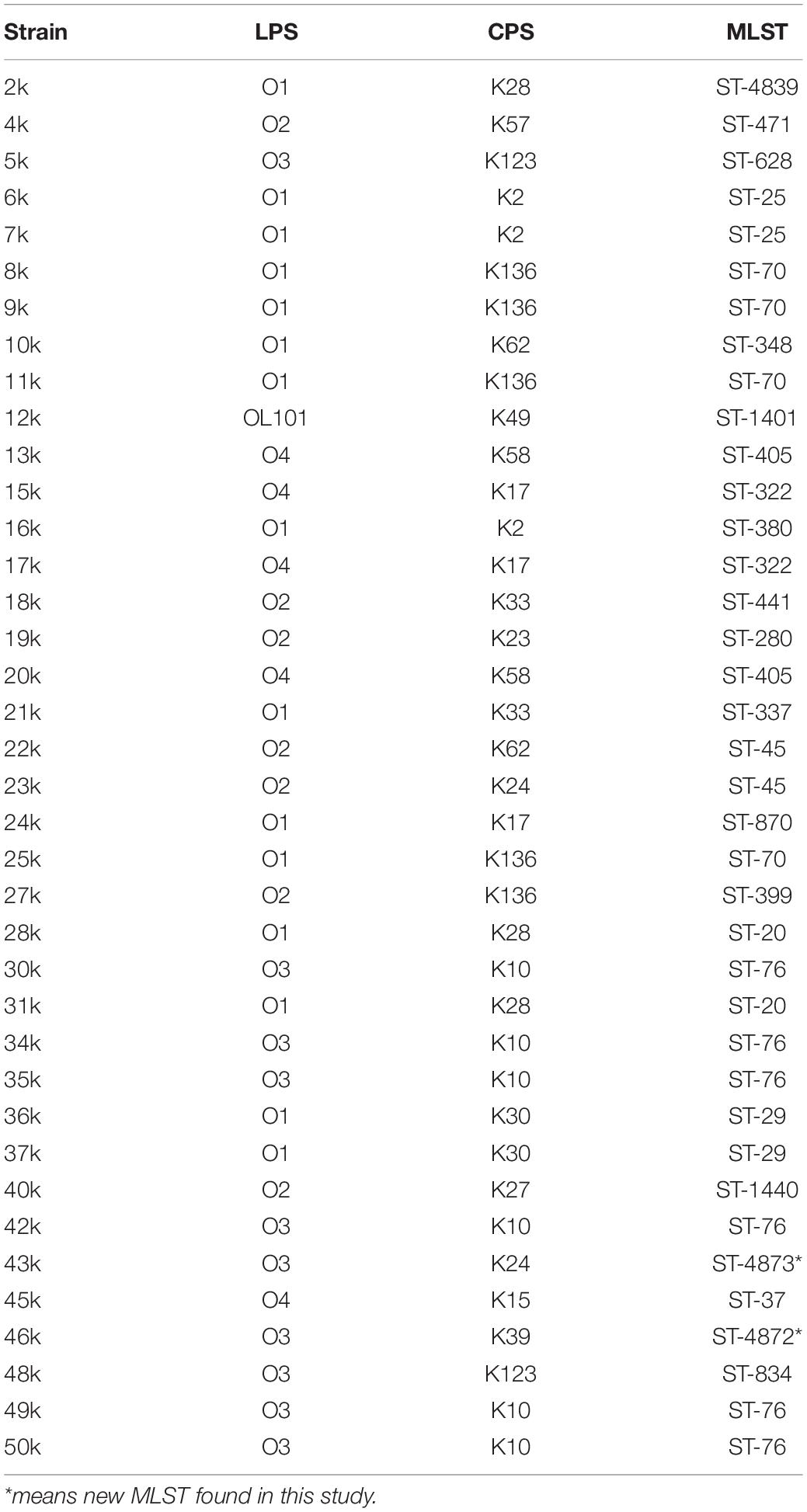
Table 4. Capsular polysaccharide synthesis (CPS), LPS, and MLST in K. pneumoniae strains.
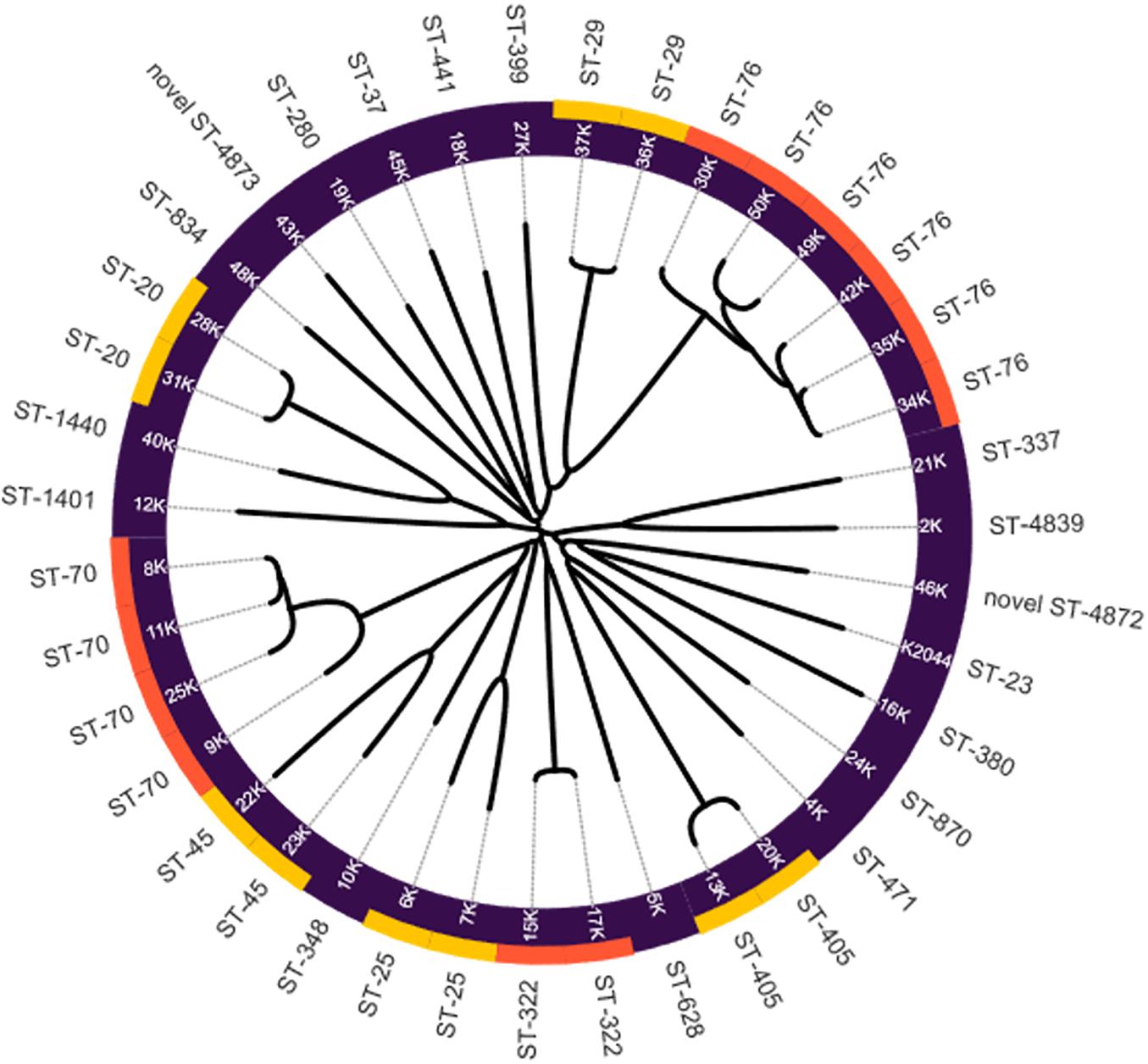
Figure 1. Phylogenomic reconstruction of Klebsiella pneumoniae strains by VGF. NTUH-K2044 strain was added as reference. Yellow and orange colors denote the clustering of strains by MLST.
The results for the search of resistance genes in the K. pneumoniae strains studied is presented in Table 5. The prevalence of genes encoding resistance to fosfomycin, quinolones, phenicols, aminoglycosides, sulphonamides, trimethoprim, tetracycline was 100, 97, 5, 79, 68, 39, and 45%, respectively. Moreover, β-lactamase genes (Table 5) such as blaSHV, blaOXA, blaTEM, and blaCTX–M were present in 53, 53, 5, and 26%, of the strains, respectively. No blaOKP, blaLEN genes were found in this bacterial collection. All the strains carried at least two different types of resistance genes. Antimicrobial susceptibility testing of K. pneumoniae strains (Table 6) showed a similar profile correspondent to the resistance genes encoded in the bacterial genome.
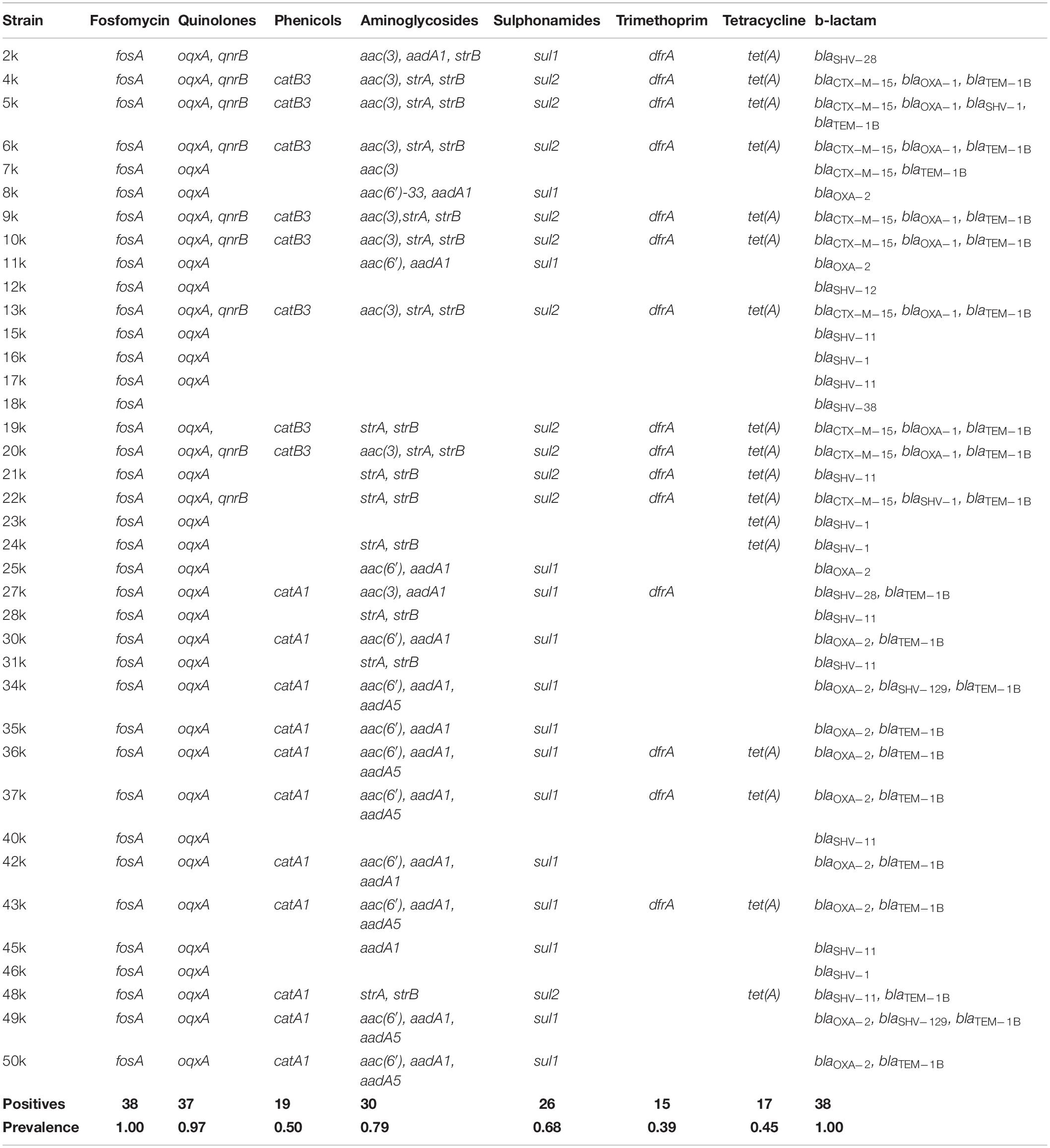
Table 5. Antibiotic resistance genes in K. pneumoniae strains.
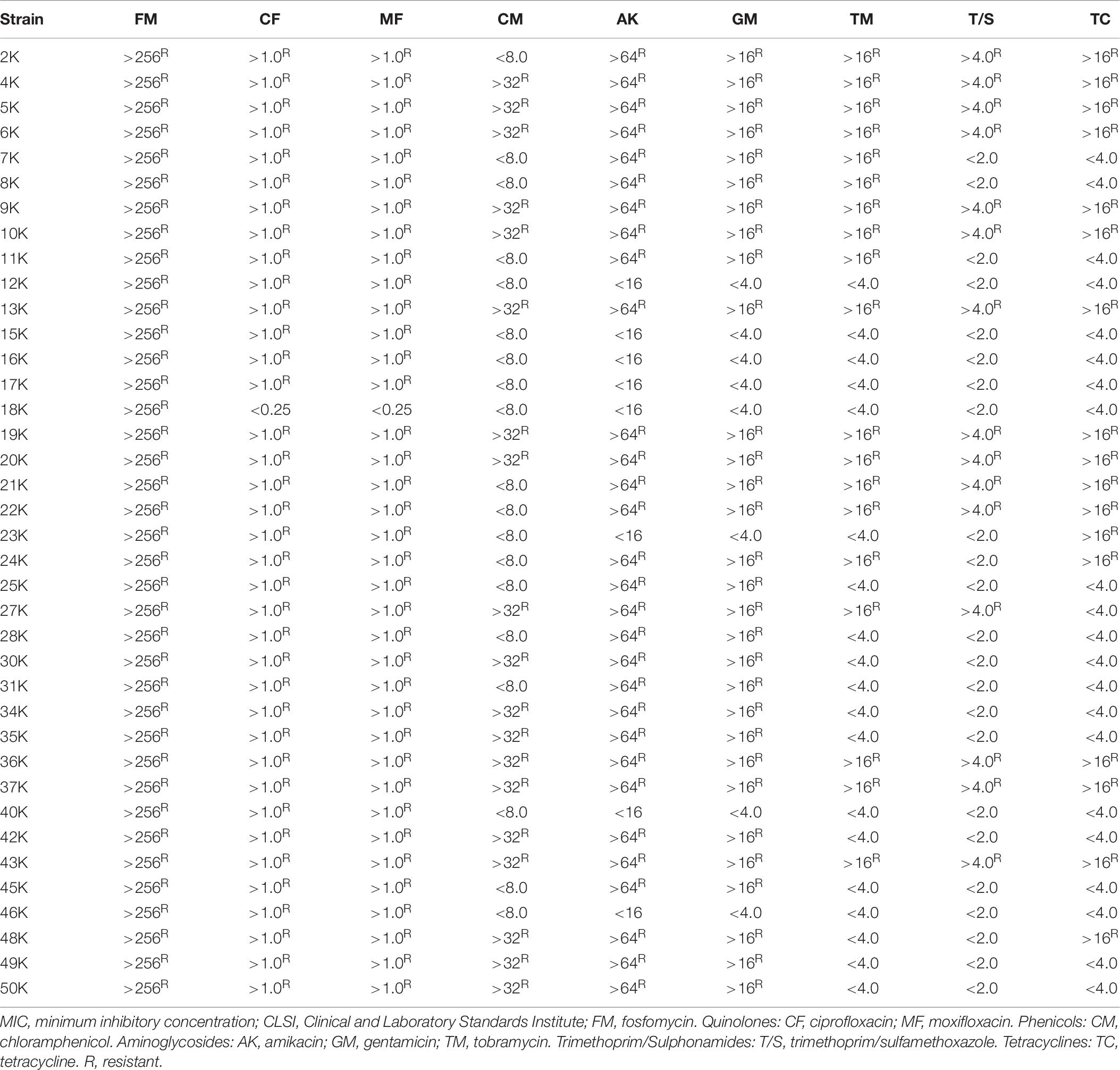
Table 6. MIC (μg/mL) of the 38 clinical isolates of K. pneumoniae according to CLSI.
The prevalence of virulence genes in the genomes of K. pneumoniae strains is shown in Figure 2. For fimbrial genes, the prevalence was very high showing that, with exception of strain 27k that lacks the fim cluster, all the strains possess the fim (Type 1 fimbriae), mrk (Type 3 fimbriae) and ecp (E. coli common pilus) genes. In terms of genes that code for outer membrane proteins, ompK35 and ompA were present in all K. pneumoniae genomes, while ompK36 gene presented in 63% of the strains. Interestingly, seven (18%) K. pneumoniae strains were positive for vir genes, which code for a putative T4SS. Regarding siderophores, fes (enterobactin) and fyuA (yersiniabactin) genes were present in 100 and 18% of the K. pneumoniae strains, respectively. Of note, only strain 16k carried genes that code for the four siderophores: enterobactin, yersiniabactin, aerobactin, and salmochelin. In addition, this strain was the only one to possess the hypermucoviscous associated gene rmpA. None of the strains were positive for magA gene. Furthermore, a correlation is depicted between the presence of virulence factors and the clustering by the VGF in Figure 2. Strains 16k and the NTUH K-2044, which possess the higher number of the virulence factors, were grouped into a common clade. Strains 6k, 22k, 23k, and 9k that contain the presence of fyuA and vir genes, formed an independent clade. Interestingly, strains 13k and 20k that belong to the O4:K58 serotype were allocated into a different clade although they shared the fyuA and vir genes.
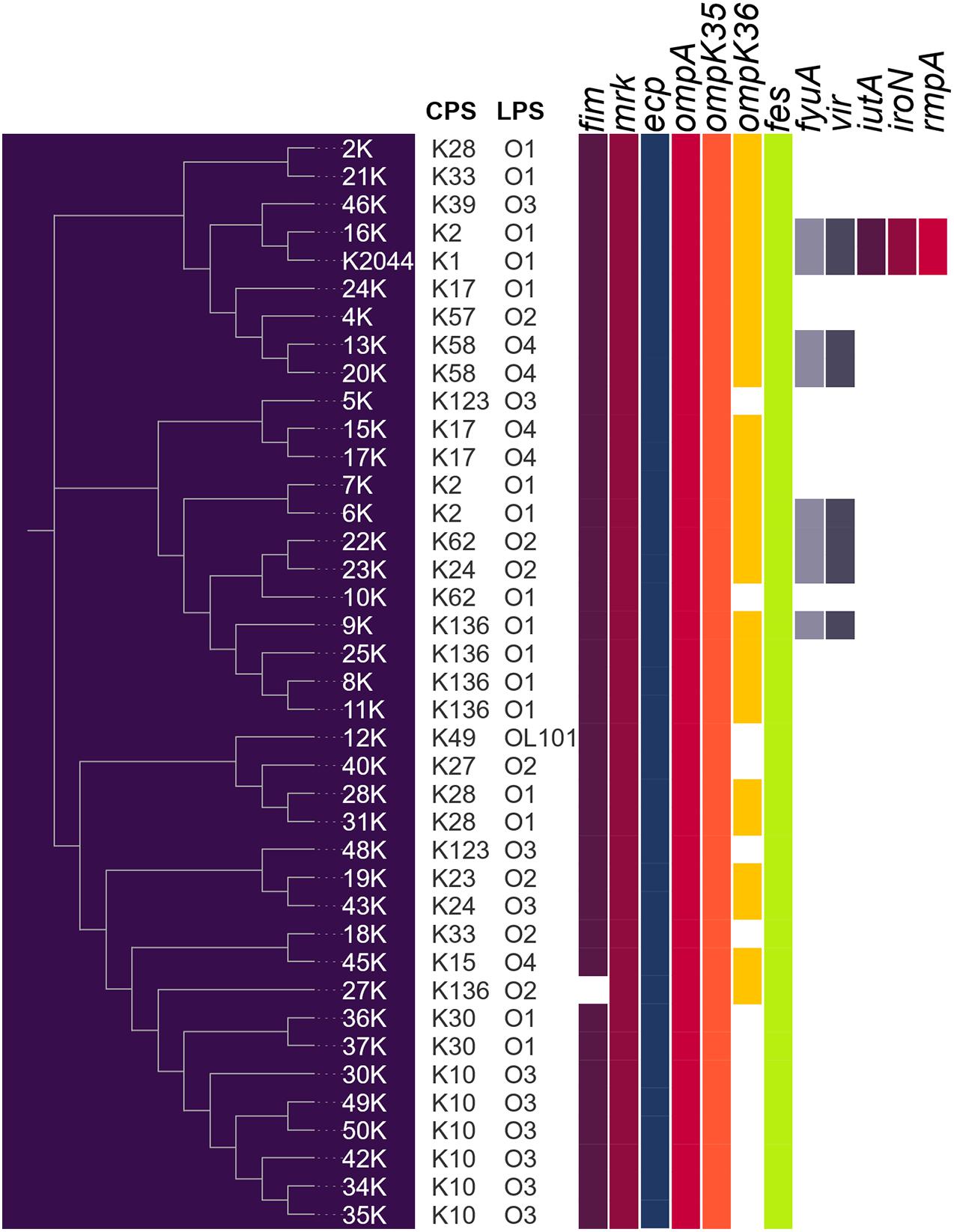
Figure 2. Virulome of K. pneumoniae strains. Color filled box mean presence of virulence factor. NTUH-K2044 was added as reference strain.
The analysis of the O groups among the strains showed that the most prevalent serogroup was O1 (39%), followed by O3 (26%) (Table 4). Seventeen different K-serotypes were identified in the all strains, being serotype K10 the most prevalent (16%), followed by K136 (13%) (Table 4). Three strains were positive for serotype K2 (6k, 7k, and 16k), which is associated to hypervirulence phenotype due to capsule hyperproduction (Yeh et al., 2007; Yu et al., 2008). We analyzed the capsule production and phagocytosis resistance in K. pneumoniae K2 serotype strains and compared them to the K10 and K136 serotypes, which were the most prevalent (Figure 3). In addition, polymyxin B susceptibility assays were performed to evaluate the protective capacity of the capsule against this antimicrobial cationic peptide (Campos et al., 2004). As controls, we used the wild-type K. pneumoniae 123/01 strain (K39 serotype) and an Δhns-derivative mutant, which show low and high level of capsule production, respectively (Ares et al., 2016). Indeed, the K2 serotype strains produced more capsule than K10/K136 serotypes to similar levels as the Δhns mutant, which is a hypercapsulated K. pneumoniae. Therefore, K. pneumoniae K2 serotype strains were less phagocytized by THP-1 macrophages than K10 and K136 serotypes strains. In addition, the action of polymyxin B was lower in the three K2 serotype K. pneumoniae.
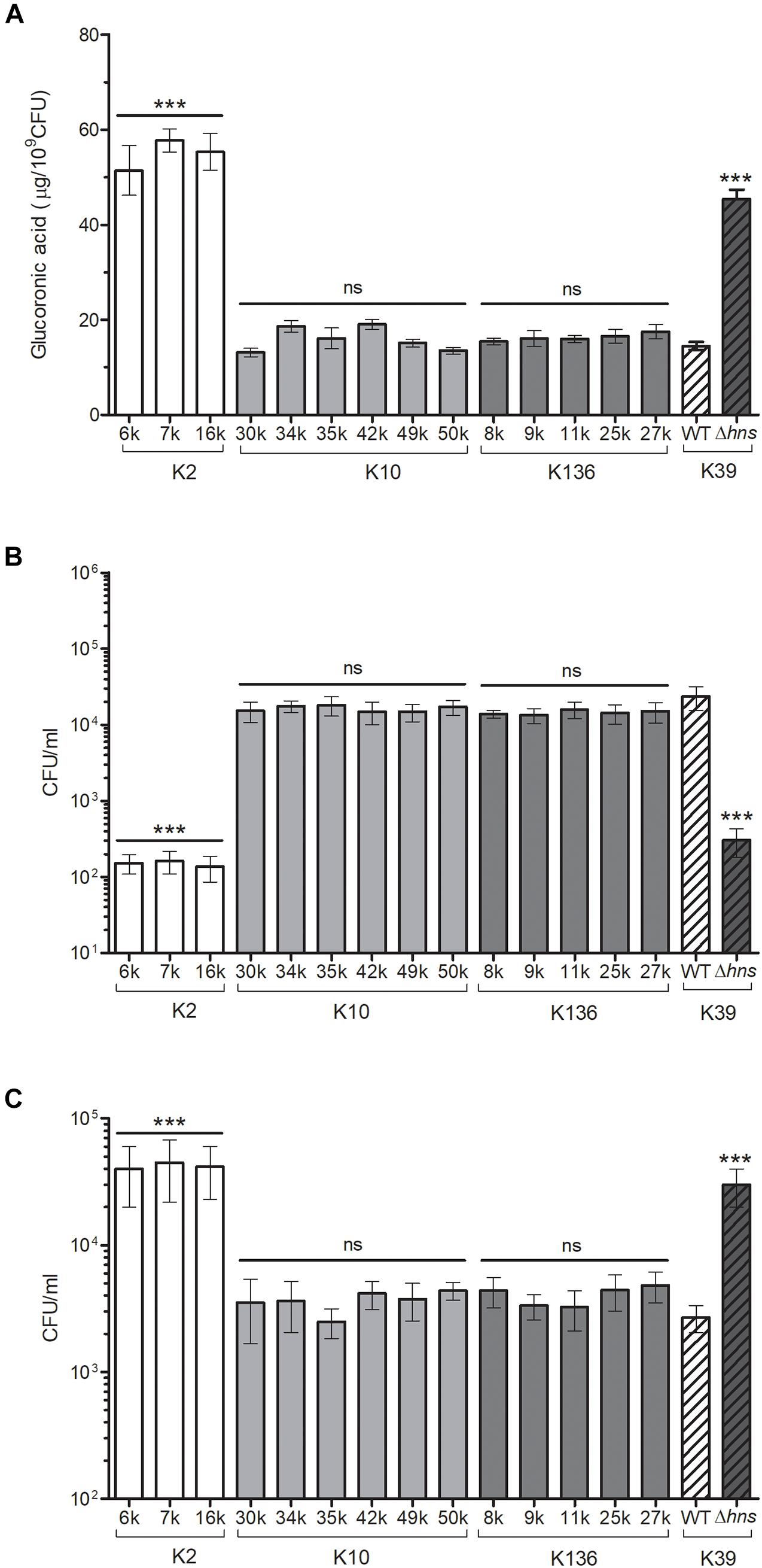
Figure 3. K2 serotype K. pneumoniae is highly resistant to phagocytosis and polymyxin B. (A) Capsule quantification by determination of the glucuronic acid concentration from capsular polysaccharides extracted from 0.5 ml bacterial cultures. (B) Comparison of phagocytic uptake of K. pneumoniae grown under the indicated different conditions by THP-1 macrophages. (C) Polymyxin B activity (4 mg/ml) against K. pneumoniae strains. An Δhns mutant was evaluated as control of phagocytosis and polymyxin B. ns, not significant; statistically significant with respect to bacteria grown in LB medium: ***p < 0.001.
Gram-negative bacilli infections are the most serious and potentially life-threatening causes of infectious disease in hospitalized patients (Joram et al., 2012; Ares et al., 2013). K. pneumoniae is an opportunistic pathogen associated with both community-acquired and nosocomial infections, causing pneumonia, abscess, bacteremia, and urinary tract infections (Gassama-Sow et al., 2010). Due to its ability to rapidly acquire antimicrobial resistance, K. pneumoniae has become a global concern calling for actions to prevent the spread of multidrug resistant bacteria (Jansen et al., 2018). Over the last few years, whole-genome sequencing (WGS) has become more accessible and affordable, resulting in its increased use in many fields, including clinical microbiology (Mathers et al., 2015). One of the main benefits of using WGS is to characterize the genomic content of clinically relevant bacteria and relate it to virulence-associated phenotypes to allow the understanding of their transmission within the hospital and to provide accurate diagnostics and to apply prompt therapies. The present study aimed to characterize K. pneumoniae isolates from pediatric patients hospitalized between 2010 and 2013 in Mexico City. This is the first study in Mexico that uses WGS to deeply characterize this opportunistic pathogen. In this work, 24 STs were identified in the bacterial genomes analyzed, showing in general, a heterogeneous distribution among which ST-76 (16%) and ST-70 (11%) were the most prevalent STs. Of note, the ST-29 represented by strains 36k and 37k, has previously been associated with hypervirulence (Shon et al., 2013; Lev et al., 2018). This same ST-29 was previously reported in isolates from community-acquired infections in the State of Guerrero, México, presenting a hypermucoviscosity phenotype (Garza-Ramos et al., 2018).
Regarding the prevalence of antibiotic resistance, K. pneumoniae genomes showed high prevalence (97%) for oqxA or qnrB genes, which conferred resistance to quinolones. Only one strain (18k) was sensitive to ciprofloxacin and moxifloxacin, which corresponded to the lack of oqxA/qnrB genes in the bacterial genome. This high resistance to this class of antibiotics has been reported by other researchers in both Mexico and worldwide (Rodríguez-Zulueta et al., 2013; Osei Sekyere and Amoako, 2017; Salah et al., 2019; Geetha et al., 2020). Sulfonamide resistance genes such as sul1 or sul2 were highly prevalent (68%) in the K. pneumoniae strains analyzed in this study, which is in agree with other reports (Khamesipour and Tajbakhsh, 2016; Taitt et al., 2017). Genes that confer tetracycline [tet(A), tet(B), tet(C), tet(D), or tet(E)] were present in 45% of the strains, which is low in comparison to other studies (Bokaeian et al., 2015; Lev et al., 2018). All strains presented at least one type of gene coding for β-lactamases and the SHV-type was the most prevalent in the sequenced genomes (53%). However, for the blaTEM-type, only blaTEM–1B gene was identified in 20 strains (53%), which has been reported as the most prevalent β-lactamases genes in K. pneumoniae (Chang et al., 2001; Feizabadi et al., 2010; Shaikh et al., 2015). The β-lactamases genes blaOXA–1 and blaOXA–2 were found in 20 strains (53%), while for the blaCTX–M–15 gene was found in 10 bacterial genomes analyzed (26%), which has been reported by different authors as common in nosocomial outbreaks (Zhou et al., 2015; Bevan et al., 2017; Gautam et al., 2019). β-lactamases genes blaOKP and blaLEN were not found in the strains.
Seventeen different types of CPS were found among the clinical K. pneumoniae collection. Three strains (6k, 7k, and 16k) were identified as belonging to the K2 capsular serotype, which is the main antigen associated with hypervirulence in K. pneumoniae (Brisse et al., 2013; Lin et al., 2014; Lev et al., 2018). These three strains produced more capsule and consequently they were more resistant to both macrophage-mediated phagocytosis and polymyxin, than the K10 and K136 capsular types, which were the most prevalent serotypes in our study. Our results are consistent with previous studies confirming that the K2 serotype confer K. pneumoniae higher resistance to both macrophage-mediated phagocytosis and polymyxin B due to increased capsule production (Yu et al., 2015). In the case of the LPS O-antigen, five different serotypes were found. In agreement with other studies (Shankar-Sinha et al., 2004; Hsieh et al., 2014; Follador et al., 2016), the O1 antigen, which confers greater resistance to the serum’s bactericidal activity, was the most prevalent O1 serogroup (39%) in the K. pneumoniae strains isolated from blood. Whilst O2 serogroup is more sensitive to human serum killing than O1 (Pennini et al., 2017), there are not reports about the O3 antigen immunogenicity, which was the second serogroup most prevalent (24%) in the K. pneumoniae strains collection.
In terms of adhesive structures, type 3 fimbria is the most important organelle of K. pneumoniae, playing an important role in adherence to both abiotic and biotic surfaces (Langstraat et al., 2001; Di Martino et al., 2003; Struve et al., 2009; Schroll et al., 2010; Ares et al., 2016). Virulence gene analysis revealed that the mrk cluster was present in all the strains, supporting the notion that type 3 fimbriae are a hallmark of K. pneumoniae virulence. Porins have been shown to be important for bacterial survival due to their role in the exchange of substances, including nutrients and toxic metabolites (Tsai et al., 2011). All the strains presented both the ompA and ompK35 genes. In contrast, only 25 (66%) K. pneumoniae strains were positive for ompK36 gene. The high prevalence of ompK35 versus ompK36, supports the structural role of OmpK35 as a porin related to the outer membrane integrity, whilst OmpK36 provides increased antibiotic resistance with no significant loss of fitness in the host (Fajardo-Lubián et al., 2019). Interestingly, K. pneumoniae strains lacking ompK36 presented over 70% prevalence of blaOXA carbapenemase gene, which support the notion that the resistance to carbapenem antibiotics is related to the absence of ompK36 porin gene (Landman et al., 2009; Wong et al., 2019). The combination of siderophores secreted by K. pneumoniae during infection affect tissue localization, systemic dissemination, and host survival (Holden et al., 2016). Enterobactin genes were present in 100% of the K. pneumoniae genomes, which is understandable since the catecholate is typically encoded in the core genome of K. pneumoniae (Martin and Bachman, 2018). Genes that encodes yersiniabactin siderophore were present in seven (18%) K. pneumoniae, and two of these genomes presented the K2 capsular serotype, reflecting the virulence properties of these strains, since yersiniabactin is significantly associated with invasive infections in humans (Holt et al., 2015).
With exception of the strain 46k, at least one plasmid sequence was detected in 37 of the 38 genomes analyzed. IncFIB plasmid was present in 36 of 37 of the bacterial isolates. This conjugative plasmid has been associated with the dissemination of the blaNDM–1, blaSHV–12, blaCTXM–15, and blaOXA–1 genes in K. pneumoniae (Oliveira et al., 2020). Methods of circularization and annotation of the plasmid DNA using sequencing techniques that generate long-read sequences will offer insights into the content of those plasmids and its evolution and spread (Ramsamy et al., 2020).
Traditional phylogenetic approaches require the calculation of multiple sequence alignment of homologous sequences for finding informative characters or for the calculation of sequence differences that are used as overall measures to estimate evolutionary distances. Thus, the time required for a phylogenetic study is distributed into that which is needed for calculating the alignment and that which is needed for the phylogenetic method itself (Yang and Rannala, 2012). The VAMPhyRE method is an alignment free tool based in the calculation of Virtual Genomic Fingerprints by a virtual hybridization with predesigned probe sets. Additionally, SNPs methods usually require a collection of SNPs relative to a reference sequence which is not required by the VAMPhyRE approach. The phylogenomic reconstructions obtained by VAMPhyRE correlated to the MLST determination. These results demonstrated a strong relation between MLST classification scheme and the phylogenomic analysis in the bacterial strains. This same behavior has been reported by Muñoz-Ramírez et al. (2017) in Helicobacter pylori strains. Furthermore, the VAMPhyRE methodology was able to cluster together strains with high number of virulence factors and hypervirulent serotypes from those lacking them. Interestingly, strain 16k presented a strong phylogenetic relation with NTUH-K2044 reference strain, carrying all virulence factors analyzed in our study, including the four siderophores and the T4SS genes. Ongoing work in our laboratory aims to characterize the pathogenic features of this strain. In summary, the search of virulence factors by using whole genome sequencing and virtual genome fingerprinting is an alternative method to classify clinical K. pneumoniae strains.
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MD and AM-T conceived and designed the experiments. MA and RR-R performed the experiments. MF-V, MA, JT, JG, BW, AM-T, and MD analyzed the data. MF-V, JG, and MD wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants FIS/IMSS/PROT/G16/1617 (to MD) from the Fondo de Investigación en Salud (FIS)-Instituto Mexicano del Seguro Social (IMSS), Mexico. MF-V was supported by a pre-doctoral fellowship from CONACYT (586925).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The genomes were sequenced using the 100K Pathogen Genome Project resources. We thank the team of curators of the Institut Pasteur MLST and whole genome MLST databases for curating the data and making them publicly available at http://bigsdb.pasteur.fr.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.711577/full#supplementary-material
Alcántar-Curiel, M. D., Blackburn, D., Saldaña, Z., Gayosso-Vázquez, C., Iovine, N. M., De la Cruz, M. A., et al. (2013). Multi-functional analysis of Klebsiella pneumoniae fimbrial types in adherence and biofilm formation. Virulence 4, 129–138. doi: 10.4161/viru.22974
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-80362
Ares, M. A., Fernández-Vázquez, J. L., Rosales-Reyes, R., Jarillo-Quijada, M. D., von Bargen, K., Torres, J., et al. (2016). H-NS nucleoid protein controls virulence features of Klebsiella pneumoniae by regulating the expression of Type 3 pili and the capsule polysaccharide. Front. Cell. Infect. Microbiol. 6:13. doi: 10.3389/fcimb.2016.00013
Ares, M. A., Sansabas, A., Rodríguez-Valverde, D., Siqueiros-Cendón, T., Rascón-Cruz, Q., Rosales-Reyes, R., et al. (2019). The interaction of Klebsiella pneumoniae with lipid rafts-associated cholesterol increases macrophage-mediated phagocytosis due to down regulation of the capsule polysaccharide. Front. Cell. Infect. Microbiol. 9:255. doi: 10.3389/fcimb.2019.00255
Ares, M. Á, Alcántar-Curiel, M. D., Jiménez-Galicia, C., Rios-Sarabia, N., Pacheco, S., and De la Cruz, M. Á (2013). Antibiotic resistance of gram-negative bacilli isolated from pediatric patients with nosocomial bloodstream infections in a Mexican tertiary care hospital. Chemotherapy 59, 361–368. doi: 10.1159/000362085
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bevan, E. R., Jones, A. M., and Hawkey, P. M. (2017). Global epidemiology of CTX-M β-lactamases: temporal and geographical shifts in genotype. J. Antimicrob. Chemother. 72, 2145–2155. doi: 10.1093/jac/dkx146
Bokaeian, M., Shahraki Zahedani, S., Soltanian Bajgiran, M., and Ansari Moghaddam, A. (2015). Frequency of PER, VEB, SHV, TEM and CTX-M genes in resistant strains of Pseudomonas aeruginosa producing extended spectrum β-Lactamases. Jundishapur J Microbiol. 8:e13783. doi: 10.5812/jjm.13783
Brisse, S., Passet, V., Haugaard, A. B., Babosan, A., Kassis-Chikhani, N., Struve, C., et al. (2013). wzi gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J. Clin. Microbiol. 51, 4073–4078. doi: 10.1128/JCM.01924-1913
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Campos, M. A., Vargas, M. A., Regueiro, V., Llompart, C. M., Albertí, S., and Bengoechea, J. A. (2004). Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 72, 7107–7114. doi: 10.1128/iai.72.12.7107-7114.2004
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chang, F.-Y., Siu, L. K., Fung, C.-P., Huang, M.-H., and Ho, M. (2001). Diversity of SHV and TEM β-Lactamases in Klebsiella pneumoniae: gene evolution in northern taiwan and two novel β-Lactamases, SHV-25 and SHV-26. Antimicrob. Agents Chemother. 45, 2407–2413. doi: 10.1128/AAC.45.9.2407-2413.2001
Chong, Y., Shimoda, S., and Shimono, N. (2018). Current epidemiology, genetic evolution and clinical impact of extended-spectrum β-lactamase-producing Escherichia coli and Klebsiella pneumoniae. Infect. Genet. Evol. 61, 185–188. doi: 10.1016/j.meegid.2018.04.005
Chung, D. R., Park, M. H., Kim, S. H., Ko, K. S., Kang, C.-I., Peck, K. R., et al. (2012). Prevalence and molecular characterization of serotype K1 Klebsiella pneumoniae strains from various clinical specimen sources in 11 Asian countries. J. Infect. 64, 622–625. doi: 10.1016/j.jinf.2012.02.007
Clements, A., Gaboriaud, F., Duval, J. F. L., Farn, J. L., Jenney, A. W., Lithgow, T., et al. (2008). The major surface-associated saccharides of Klebsiella pneumoniae contribute to host cell association. PLoS One 3:e3817. doi: 10.1371/journal.pone.0003817
Di Martino, P., Cafferini, N., Joly, B., and Darfeuille-Michaud, A. (2003). Klebsiella pneumoniae type 3 pili facilitate adherence and biofilm formation on abiotic surfaces. Res. Microbiol. 154, 9–16. doi: 10.1016/S0923-2508(02)00004-9
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A. D., and Brisse, S. (2005). Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J. Clin. Microbiol. 43, 4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005
Evrard, B., Balestrino, D., Dosgilbert, A., Bouya-Gachancard, J.-L. J., Charbonnel, N., Forestier, C., et al. (2010). Roles of capsule and lipopolysaccharide O antigen in interactions of human monocyte-derived dendritic cells and Klebsiella pneumoniae. Infect. Immun. 78, 210–219. doi: 10.1128/IAI.00864-869
Fajardo-Lubián, A., Ben Zakour, N. L., Agyekum, A., Qi, Q., and Iredell, J. R. (2019). Host adaptation and convergent evolution increases antibiotic resistance without loss of virulence in a major human pathogen. PLoS Pathog. 15:e1007218. doi: 10.1371/journal.ppat.1007218
Fang, C.-T., Shih, Y.-J., Cheong, C.-M., and Yi, W.-C. (2016). Rapid and accurate determination of lipopolysaccharide O-Antigen types in Klebsiella pneumoniae with a novel PCR-Based O-Genotyping method. J. Clin. Microbiol. 54, 666–675. doi: 10.1128/JCM.02494-2415
Feizabadi, M. M., Mahamadi-Yeganeh, S., Mirsalehian, A., Mirafshar, S.-M., Mahboobi, M., Nili, F., et al. (2010). Genetic characterization of ESBL producing strains of Klebsiella pneumoniae from Tehran hospitals. J. Infect. Dev. Ctries. 4, 609–615. doi: 10.3855/jidc.1059
Follador, R., Heinz, E., Wyres, K. L., Ellington, M. J., Kowarik, M., Holt, K. E., et al. (2016). The diversity of Klebsiella pneumoniae surface polysaccharides. Microb. Genom. 2:e000073. doi: 10.1099/mgen.0.000073
Garza-Ramos, U., Barrios-Camacho, H., Moreno-Domínguez, S., and Toribio-Jiménez, J. (2018). Phenotypic and molecular characterization of Klebsiella spp. isolates causing community-acquired infections. New Microbes New Infect. 23, 17–27. doi: 10.1016/j.nmni.2018.02.002
Gassama-Sow, A., Diallo, M. H., Wane, A. A., Seck, A., Samb-Ba, B., Sow, P. S., et al. (2010). Genetic determinants of antibiotic resistance in diarrheagenic Klebsiella pneumoniae subspecies ozaenae: an emerging enteropathogen in Senegal. Clin. Infect. Dis. 50, 453–455. doi: 10.1086/649892
Gautam, V., Thakur, A., Sharma, M., Singh, A., Bansal, S., Sharma, A., et al. (2019). Molecular characterization of extended-spectrum β-lactamases among clinical isolates of Escherichia coli & Klebsiella pneumoniae: a multi-centric study from tertiary care hospitals in India. Indian J. Med. Res. 149, 208–215. doi: 10.4103/ijmr.IJMR_172_18
Geetha, P. V., Aishwarya, K. V. L., Mariappan, S., and Sekar, U. (2020). Fluoroquinolone resistance in clinical isolates of e. J. Lab. Phys. 12, 121–125.
Holden, V. I., Breen, P., Houle, S., Dozois, C. M., and Bachman, M. A. (2016). Klebsiella pneumoniae siderophores induce inflammation, bacterial dissemination, and HIF-1α stabilization during pneumonia. mBio 7:e01397-16. doi: 10.1128/mBio.01397-1316
Holt, K. E., Wertheim, H., Zadoks, R. N., Baker, S., Whitehouse, C. A., Dance, D., et al. (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. U S A. 112, E3574–E3581.
Hsieh, P.-F., Wu, M.-C., Yang, F.-L., Chen, C.-T., Lou, T.-C., Chen, Y.-Y., et al. (2014). D-galactan II is an immunodominant antigen in O1 lipopolysaccharide and affects virulence in Klebsiella pneumoniae: implication in vaccine design. Front. Microbiol. 5:608. doi: 10.3389/fmicb.2014.00608
Jansen, K. U., Knirsch, C., and Anderson, A. S. (2018). The role of vaccines in preventing bacterial antimicrobial resistance. Nat. Med. 24, 10–19. doi: 10.1038/nm.4465
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Joram, N., de Saint Blanquat, L., Stamm, D., Launay, E., and Gras-Le Guen, C. (2012). Healthcare-associated infection prevention in pediatric intensive care units: a review. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2481–2490. doi: 10.1007/s10096-012-1611-1610
Khamesipour, F., and Tajbakhsh, E. (2016). Analyzed the genotypic and phenotypic antibiotic resistance patterns of Klebsiella pneumoniae isolated from clinical samples in Iran. Biomedical Research 27, 0970–938X.
Ko, K. S. (2017). The contribution of capsule polysaccharide genes to virulence of Klebsiella pneumoniae. Virulence 8, 485–486. doi: 10.1080/21505594.2016.1240862
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Landman, D., Bratu, S., and Quale, J. (2009). Contribution of OmpK36 to carbapenem susceptibility in KPC-producing Klebsiella pneumoniae. J. Med. Microbiol. 58(Pt 10), 1303–1308. doi: 10.1099/jmm.0.012575-0
Langstraat, J., Bohse, M., and Clegg, S. (2001). Type 3 fimbrial shaft (MrkA) of Klebsiella pneumoniae, but not the fimbrial adhesin (MrkD), facilitates biofilm formation. Infect. Immun. 69, 5805–5812. doi: 10.1128/iai.69.9.5805-5812.2001
Lev, A. I., Astashkin, E. I., Kislichkina, A. A., Solovieva, E. V., Kombarova, T. I., Korobova, O. V., et al. (2018). Comparative analysis of Klebsiella pneumoniae strains isolated in 2012-2016 that differ by antibiotic resistance genes and virulence genes profiles. Pathog. Glob. Health 112, 142–151. doi: 10.1080/20477724.2018.1460949
Li, B., Zhao, Y., Liu, C., Chen, Z., and Zhou, D. (2014). Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 9, 1071–1081. doi: 10.2217/fmb.14.48
Lin, J.-C., Koh, T. H., Lee, N., Fung, C.-P., Chang, F.-Y., Tsai, Y.-K., et al. (2014). Genotypes and virulence in serotype K2 Klebsiella pneumoniae from liver abscess and non-infectious carriers in Hong Kong, Singapore and Taiwan. Gut Pathog. 6:21. doi: 10.1186/1757-4749-6-21
March, C., Cano, V., Moranta, D., Llobet, E., Pérez-Gutiérrez, C., Tomás, J. M., et al. (2013). Role of bacterial surface structures on the interaction of Klebsiella pneumoniae with phagocytes. PLoS One 8:e56847. doi: 10.1371/journal.pone.0056847
Martin, R. M., and Bachman, M. A. (2018). Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 8:4. doi: 10.3389/fcimb.2018.00004
Mathers, A. J., Stoesser, N., Sheppard, A. E., Pankhurst, L., Giess, A., Yeh, A. J., et al. (2015). Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: insights into endemicity from whole-genome sequencing. Antimicrob. Agents Chemother. 59, 1656–1663. doi: 10.1128/AAC.04292-4214
Meatherall, B. L., Gregson, D., Ross, T., Pitout, J. D. D., and Laupland, K. B. (2009). Incidence, risk factors, and outcomes of Klebsiella pneumoniae bacteremia. Am. J. Med. 122, 866–873. doi: 10.1016/j.amjmed.2009.03.034
Muñoz-Ramírez, Z. Y., Mendez-Tenorio, A., Kato, I., Bravo, M. M., Rizzato, C., Thorell, K., et al. (2017). Whole genome sequence and phylogenetic analysis show strains from latin america have followed a unique evolution pathway. Front. Cell. Infect. Microbiol. 7:50. doi: 10.3389/fcimb.2017.00050
Nang, S. C., Han, M.-L., Yu, H. H., Wang, J., Torres, V. V. L., Dai, C., et al. (2019). Polymyxin resistance in Klebsiella pneumoniae: multifaceted mechanisms utilized in the presence and absence of the plasmid-encoded phosphoethanolamine transferase gene mcr-1. J. Antimicrob. Chemother. 74, 3190–3198. doi: 10.1093/jac/dkz314
Nei, M., and Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U S A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Oliveira, É. M., Beltrão, E., Scavuzzi, A., Barros, J. F., and Lopes, A. (2020). High plasmid variability, and the presence of IncFIB, IncQ, IncA/C, IncHI1B, and IncL/M in clinical isolates of Klebsiella pneumoniae with bla KPC and bla NDM from patients at a public hospital in Brazil. Revista da Sociedade Brasileira de Medicina Tropical 53: e20200397. doi: 10.1590/0037-8682-0397-2020
Osei Sekyere, J., and Amoako, D. G. (2017). Genomic and phenotypic characterisation of fluoroquinolone resistance mechanisms in Enterobacteriaceae in Durban. South Africa. PLoS One 12:e0178888. doi: 10.1371/journal.pone.0178888
Paczosa, M. K., and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/MMBR.00078-15
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pennini, M. E., De Marco, A., Pelletier, M., Bonnell, J., Cvitkovic, R., Beltramello, M., et al. (2017). Immune stealth-driven O2 serotype prevalence and potential for therapeutic antibodies against multidrug resistant Klebsiella pneumoniae. Nat. Commun. 8:1991. doi: 10.1038/s41467-017-02223-2227
Piperaki, E.-T., Syrogiannopoulos, G. A., Tzouvelekis, L. S., and Daikos, G. L. (2017). Klebsiella pneumoniae: virulence, biofilm and antimicrobial resistance. Pediatr. Infect. Dis. J. 36, 1002–1005. doi: 10.1097/INF.0000000000001675
Podschun, R., and Ullmann, U. (1998). Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603. doi: 10.1128/CMR.11.4.589
Ramsamy, Y., Mlisana, K. P., Allam, M., Amoako, D. G., Abia, A. L. K., Ismail, A., et al. (2020). Genomic analysis of carbapenemase-producing extensively drug-resistant Klebsiella pneumoniae isolates reveals the horizontal spread of p18-43_01 plasmid encoding blaNDM-1 in South Africa. Microorganisms 8:137. doi: 10.3390/microorganisms8010137
Rodríguez-Zulueta, P., Silva-Sánchez, J., Barrios, H., Reyes-Mar, J., Vélez-Pérez, F., Arroyo-Escalante, S., et al. (2013). First outbreak of KPC-3-producing Klebsiella pneumoniae (ST258) clinical isolates in a Mexican Medical Center. Antimicrob. Agents Chemother. 57, 4086–4088. doi: 10.1128/AAC.02530-2512
Salah, F. D., Soubeiga, S. T., Ouattara, A. K., Sadji, A. Y., Metuor-Dabire, A., Obiri-Yeboah, D., et al. (2019). Distribution of quinolone resistance gene (qnr) in producing and in Lomé. Togo. Antimicrob. Resist. Infect. Control 8:104.
Schroll, C., Barken, K. B., Krogfelt, K. A., and Struve, C. (2010). Role of type 1 and type 3 fimbriae in Klebsiella pneumoniae biofilm formation. BMC Microbiol. 10:179. doi: 10.1186/1471-2180-10-179
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shaikh, S., Fatima, J., Shakil, S., Rizvi, S. M., and Kamal, M. A. (2015). Risk factors for acquisition of extended spectrum beta lactamase producing Escherichia coli and Klebsiella pneumoniae in North-Indian hospitals. Saudi J. Biol. Sci. 22, 37–41. doi: 10.1016/j.sjbs.2014.05.006
Shankar-Sinha, S., Valencia, G. A., Janes, B. K., Rosenberg, J. K., Whitfield, C., Bender, R. A., et al. (2004). The Klebsiella pneumoniae O antigen contributes to bacteremia and lethality during murine pneumonia. Infect. Immun. 72, 1423–1430. doi: 10.1128/iai.72.3.1423-1430.2004
Shon, A. S., Bajwa, R. P. S., and Russo, T. A. (2013). Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence 4, 107–118. doi: 10.4161/viru.22718
Stojkovic, K., Szijártó, V., Kaszowska, M., Niedziela, T., Hartl, K., Nagy, G., et al. (2017). Identification of d-Galactan-III as part of the lipopolysaccharide of serotype O1. Front. Microbiol. 8:684. doi: 10.3389/fmicb.2017.00684
Struve, C., Bojer, M., and Krogfelt, K. A. (2009). Identification of a conserved chromosomal region encoding Klebsiella pneumoniae type 1 and type 3 fimbriae and assessment of the role of fimbriae in pathogenicity. Infect. Immun. 77, 5016–5024. doi: 10.1128/IAI.00585-589
Taitt, C. R., Leski, T. A., Erwin, D. P., Odundo, E. A., Kipkemoi, N. C., Ndonye, J. N., et al. (2017). Antimicrobial resistance of Klebsiella pneumoniae stool isolates circulating in Kenya. PLoS One 12:e0178880. doi: 10.1371/journal.pone.0178880
Tsai, Y.-K., Fung, C.-P., Lin, J.-C., Chen, J.-H., Chang, F.-Y., Chen, T.-L., et al. (2011). Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 55, 1485–1493. doi: 10.1128/AAC.01275-1210
Wick, R. R., Heinz, E., Holt, K. E., and Wyres, K. L. (2018). Kaptive web: user-friendly capsule and lipopolysaccharide serotype prediction for klebsiella genomes. J. Clin. Microbiol. 56:e00197-18. doi: 10.1128/JCM.00197-118
Wong, J. L. C., Romano, M., Kerry, L. E., Kwong, H.-S., Low, W.-W., Brett, S. J., et al. (2019). OmpK36-mediated carbapenem resistance attenuates ST258 Klebsiella pneumoniae in vivo. Nat. Commun. 10:3957.
Wu, K.-M., Li, L.-H., Yan, J.-J., Tsao, N., Liao, T.-L., Tsai, H.-C., et al. (2009). Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J. Bacteriol. 191, 4492–4501. doi: 10.1128/jb.00315-09
Wyres, K. L., Wick, R. R., Gorrie, C., Jenney, A., Follador, R., Thomson, N. R., et al. (2016). Identification of capsule synthesis loci from whole genome data. Microb. Genom. 2:e000102. doi: 10.1099/mgen.0.000102
Yang, Z., and Rannala, B. (2012). Molecular phylogenetics: principles and practice. Nat. Rev. Genet. 13, 303–314. doi: 10.1038/nrg3186
Yeh, K.-M., Kurup, A., Siu, L. K., Koh, Y. L., Fung, C.-P., Lin, J.-C., et al. (2007). Capsular serotype K1 or K2, rather than magA and rmpA, is a major virulence determinant for Klebsiella pneumoniae liver abscess in Singapore and Taiwan. J. Clin. Microbiol. 45, 466–471. doi: 10.1128/JCM.01150-1156
Yu, W.-L., Ko, W.-C., Cheng, K.-C., Lee, C.-C., Lai, C.-C., and Chuang, Y.-C. (2008). Comparison of prevalence of virulence factors for Klebsiella pneumoniae liver abscesses between isolates with capsular K1/K2 and non-K1/K2 serotypes. Diagn. Microbiol. Infect. Dis. 62, 1–6. doi: 10.1016/j.diagmicrobio.2008.04.007
Yu, Z., Qin, W., Lin, J., Fang, S., and Qiu, J. (2015). Antibacterial mechanisms of polymyxin and bacterial resistance. BioMed. Res. Int. 2015:679109. doi: 10.1155/2015/679109
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Klebsiella pneumoniae, whole-genome sequence, Mexico, blood samples, phylogenomic
Citation: Flores-Valdez M, Ares MA, Rosales-Reyes R, Torres J, Girón JA, Weimer BC, Mendez-Tenorio A and De la Cruz MA (2021) Whole Genome Sequencing of Pediatric Klebsiella pneumoniae Strains Reveals Important Insights Into Their Virulence-Associated Traits. Front. Microbiol. 12:711577. doi: 10.3389/fmicb.2021.711577
Received: 18 May 2021; Accepted: 27 July 2021;
Published: 13 August 2021.
Edited by:
Leonard Peruski, Centers for Disease Control and Prevention (CDC), United StatesReviewed by:
Jason Sahl, Northern Arizona University, United StatesCopyright © 2021 Flores-Valdez, Ares, Rosales-Reyes, Torres, Girón, Weimer, Mendez-Tenorio and De la Cruz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. De la Cruz, bWlndWVsX2FuZ2VsXzgxQGxpdmUuY29t; Alfonso Mendez-Tenorio, YW1lbmRlenRAaXBuLm14
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.