 Xin Wang1,2Gengan Du1,2Hong Chen1,2Xuejun Zeng1,2
Xin Wang1,2Gengan Du1,2Hong Chen1,2Xuejun Zeng1,2 Bin Liu1,2
Bin Liu1,2 Chunfeng Guo1,2Qinglin Sheng3
Chunfeng Guo1,2Qinglin Sheng3 Yahong Yuan1,2*Tianli Yue1,2,3*
Yahong Yuan1,2*Tianli Yue1,2,3*- 1College of Food Science and Engineering, Northwest A&F University, Xianyang, China
- 2Laboratory of Quality & Safety Risk Assessment for Agro-Products (Yangling), Ministry of Agriculture, Xianyang, China
- 3College of Food Science and Technology, Northwest University, Xi’an, China
Fuzhuan brick tea (FBT) is a unique post-fermented tea product, naturally co-fermented by microorganisms, and has gained global popularity due to its potential health benefits for humans. Considerable efforts have been made toward elucidating the microbial diversity within FBT, but an understanding of the underlying FBT community interactions and functions remains poorly studied. Consequently, the microbial communities of two types of FBT, originating from Hunan and Shaanxi provinces, were investigated using comparative shotgun metagenomic sequencing and functional annotations. Metagenomic analysis indicated that two communities shared similar taxonomic and functional attributes. Two samples shared 486 genera, in which Pseudomonas contributed most to the abundant functions within the two samples. The carbohydrate active enzyme functions of the communities primarily comprised GH (32.92%), GT (26.8%), CEs (20.43%), and AAs (18.04%). Furthermore, the overall metabolic pathways encoded by the metagenomes were largely associated with carbohydrate and amino acid metabolism, with nine metabolic pathways that were differential between two groups including penicillin and cephalosporin biosynthesis. Significantly, a total of 35 potential probiotics were inferred, with Pseudomonas putida being the most abundant inferred probiotic (80%) within the FBT communities. This study provides new insights into FBT microbial communities on their potential functions and roles in FBT characteristics.
Introduction
Fuzhuan brick tea (FBT) is a fermented tea product that has been consumed in China over 3,000 years and has since become popular in many countries (Li et al., 2018). The manufacturing of FBT comprises primary processing (proceeds from fresh tea leaves to fixing, rolling, piling fermentation, and drying) and press processing steps (screening, blending, steaming, piling fermentation, pressing, fungal fermentation, and drying) (Li et al., 2017), which requires numerous biochemical activities and microbial community successional processes that consequently produce many positive effects on human health (Liu et al., 2019). Increasing numbers of studies have confirmed the health benefits of FBT including anti-hyperlipidemia (Wang and Ho, 2009), anti-dysentery (Zhang et al., 2013), anti-obesity (Li et al., 2013), anti-hyperglycemia (Yamashita et al., 2012), and anti-oxidation (Cheng et al., 2015) effects. These health effects are closely related to the bacterial and fungal communities present within FBT that have been recently studied with culture-based or cultivation-independent methods (Tamang et al., 2016). Indeed, investigations of bacterial and fungal diversity have been intensively conducted in recent years. Nevertheless, the effects of microbial community changes on the functional diversity in FBT remain poorly understood.
The development of metagenomic methods has allowed new insights into the functions of natural microbial communities (Knight et al., 2018). Consequently, increasing attention has been paid to the integration of the microbial community genetic taxonomic and functional diversity. Concomitantly, developments over the last few decades in next-generation DNA sequencing and high-throughput computational methods have greatly enhanced our understanding of fermented food microbial communities (Arikan et al., 2020). Culture-independent taxonomic methods primarily based on high-throughput sequencing have been frequently applied to analyze fermented food microbial communities, for example, studies of wine (Johansson et al., 2021), kimchi (Kim et al., 2021), and fermented Chinese xiaoqu (Su et al., 2020). Importantly, metagenomic sequencing can identify novel species with higher resolution and better accuracy compared to traditional rRNA gene sequencing (Chen et al., 2017). Nevertheless, few studies have investigated the overall microbial community structure of FBT. Thus, efforts are needed to generate deeper insights into the underlying interactions within FBT microbial communities and their associated functions.
Hunan and Shaanxi have a long history of producing FBT in China. Thus, the objective of the present study was to comprehensively evaluate the response of microbial communities and their functions in two different types of FBT_H and FBT_S. The distribution of FBT microbial communities and their functional potentials was thus comparatively investigated by shotgun metagenomic sequencing for the first time. Detailed taxonomic and functional characteristics of the FBT communities were determined through genome assembly and subsequent comparative genomic analysis. The novel insights identified herein provide new understanding of the complex microbiomes in FBT and highlight the utility of integrated meta-omics approaches for understanding microbiomes within fermented tea product ecosystems.
Materials and Methods
Sample Collection
Individual FBT samples were purchased from the Yiyang Fu Cha Industry Development Co., Ltd. (Yiyang, Hunan Province, China) and the Xianyang Jingwei Fu Tea Industry Co., Ltd. (Xianyang, Shaanxi Province, China). Samples were weighed after grinding with sterile instruments. One gram of FBT was added to 10 ml of 0.1 M PBS (pH 8.0), subjected to ultrasonic extraction for 1 min and vortexed for 10 s. The above steps were repeated twice to ensure complete extraction and filtrates were collected. Subsequently, sediments were collected by centrifugating at 13,000 × g for 10 min, washed twice with 70% ethanol, and stored in a refrigerator at −80°C for subsequent analysis. Three replicates were set for each type of sample.
DNA Extraction, Sequencing Library Construction, and Metagenomics Sequencing
To generate metagenomic datasets for both FBT samples, genomic DNA was extracted from 0.5 g of prepared samples using the Fast DNA SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, United States) according to the manufacturer’s protocol. The concentration and purity of DNA were quantified with a TBS-380 mini-fluorometer and NanoDrop 2000 spectrometer, respectively (Li et al., 2020). DNA was then sheared into approximately 400-bp fragments using a Covaris M220 instrument. The metagenomic libraries were subsequently prepared using the NEXTFLEXTM Rapid DNA-Seq Kit (Illumina, San Diego, CA, United States). Paired-end sequencing was performed on the Illumina HiSeq 4000 platform at Majorbio Bio-Pharm Technology Co., Ltd., using a NovaSeq and HiSeq X Reagent Kits according to the manufacturer’s instructions.
Sequence Quality Control, Contig Assembly, Gene Prediction, Taxonomic Identification, and Functional Annotation
Fastp1 was used to remove the adapter sequences from the 3′ and 5′ ends of paired-end Illumina reads. Low-quality reads (quality <20 or containing N bases) and short reads (<50 bp) were also removed to generate clean reads. A total of 369,511,938 high-quality sequences were obtained from two samples. Reads were then assembled de novo using the IDBA-UD assembler with settings of -mink 40, -maxk 97, -min_contig 300 and also mapped back to assembled contigs using Bowtie2 (Langmead and Salzberg, 2012). Unmapped reads were also assembled with MEGAHIT. The IDBA-UD and MEGAHIT assemblies were then merged and sorted into pools with fragments longer or shorter than 1,000 bp. To generate longer contigs, those <1,000 bp were assembled using Newbler (Mason et al., 2012). Contigs longer than 1,000 bp and those generated with Newbler (≥300 bp) were combined for gene prediction. Open reading frames (ORFs) were predicted using MetaGene. ORFs longer than 100 bp were translated to amino acid sequences by NCBI translation table. After sequence assembly and gene prediction, a total of 217,271 final contigs were obtained that comprised 544,374 ORFs. All protein coding genes exhibiting >90% sequence identity and 90% coverage to reference proteins were clustered using CD-HIT, and the longest sequences were chosen as representatives to construct a non-redundant gene catalog (Fu et al., 2012). The quality-filtered reads were then mapped to the representative sequences using SOAPaligner2 to determine gene abundances. Relative gene expression within each genome was then calculated by relativizing the expression of each ORF by the median reads per kilobase million (RPKM) value calculated across the genome (Lawson et al., 2017).
BLASTP comparison against the NR database was then used to taxonomically annotate the representative sequences using DIAMOND3 with an e-value cutoff of 1e––5. All unique ORFs were also functionally annotated using DIAMOND based on kyoto encyclopedia of genes and genomes (KEGG)4 and CAZyme5 databases in order to evaluate potential functions encoded by communities. KEGG modules were also used for annotating biological pathways of sequenced genomes. Overall, a non-redundant gene catalog of 121,737 genes was constructed to describe the information for genes identified in FBT samples. The SOAPaligner was used to align the high-quality reads to a non-redundant gene catalog and enumerate gene abundances.
Statistical Analysis
Relative abundance of taxonomic groups and functional profiles was visualized using STAMP (v2.1.3). Differences in the microbial diversity were compared using ANOVA tests, with statistical significance determined by Tukey’s test (p < 0.05). Principal component analysis (PCA) was used to visualize the variation among the FBT microbial communities. Linear discriminant analysis effect size (LEfSe) statistical analysis was performed using the Galaxy interface.6 Metabolic pathways were evaluated using the online ipath2.0 platform7 based on KEGG annotations.
Sequence Data Availability
The six FBT samples are available from the NCBI Sequence Read Archive. The BioProject has an accession ID of PRJNA729248. The SRA of six BioSamples is SRR14520614, SRR14520615, SRR14520616, SRR14520617, SRR14520618, and SRR14520619.8
Results and Discussion
Taxonomic Composition of the Fuzhuan Brick Tea Communities
Taxonomic classification was successfully assigned to 70% of the total sequences. Approximately 99.95% of metagenomic sequences were classified as Bacteria and Archaea, with only 0.03% being classified as eukaryotic and the remaining 0.02% not being classified. The results are consistent with a previous study indicating that bacteria play indispensable roles in FBT quality with additional significant correlations observed between fungal and bacterial community compositions (Rui et al., 2019). Nearly all of the communities (98%) analyzed here were classified as Proteobacteria, with an additional small subset of community classified as Actinobacteria (<1%). Furthermore, the two FBT communities were dominated by the genus Pseudomonas (73.97%, 78.74%), followed by Enterobacteriaceae (4.83%, 7.13%), and Citrobacter (4.59%, 7.10%); Comamonas (4.35%, 0.26%), Stenotrophomonas (2.05%, 0.19%), Leclercia (1.08%, 0), and Rhizobiaceae (0.87%, 0) were more abundant in FBT_H than FBT_S (Figure 1A), suggesting that the difference might be due to the unique processing of FBT of the two brands not due to different regions. Moreover, it is noticed that there is greater variation in FBT_H while FBT_S replicates share more similarity. Strictly speaking, this difference is allowed in the experiment and may be caused by random error. Pseudomonas was clearly the most abundant taxa within FBT microbial communities, and Pseudomonas putida (60%) was the most abundant species within the genus (Supplementary Figure 1A). A previous study demonstrated that P. putida can degrade caffeine to xanthine via a purine-dependent metabolic pathway (i.e., N-demethylation pathway) and further alleviate the bitter and astringent taste of fermented tea (Jiang et al., 2019). Thus, P. putida may be an important regulator of flavor and taste profiles of fermented tea. Further metagenomic analysis indicated that the two communities shared similar taxonomic and functional attributes (Figure 1B). A total of 486 genera were shared in two samples, accounting for 78% of the total 623 genera. An additional 126 unique genera were identified in FBT_H, while 11 unique genera were only identified in FBT_S, indicating a higher level of microbial diversity in FBT_H than FBT_S.
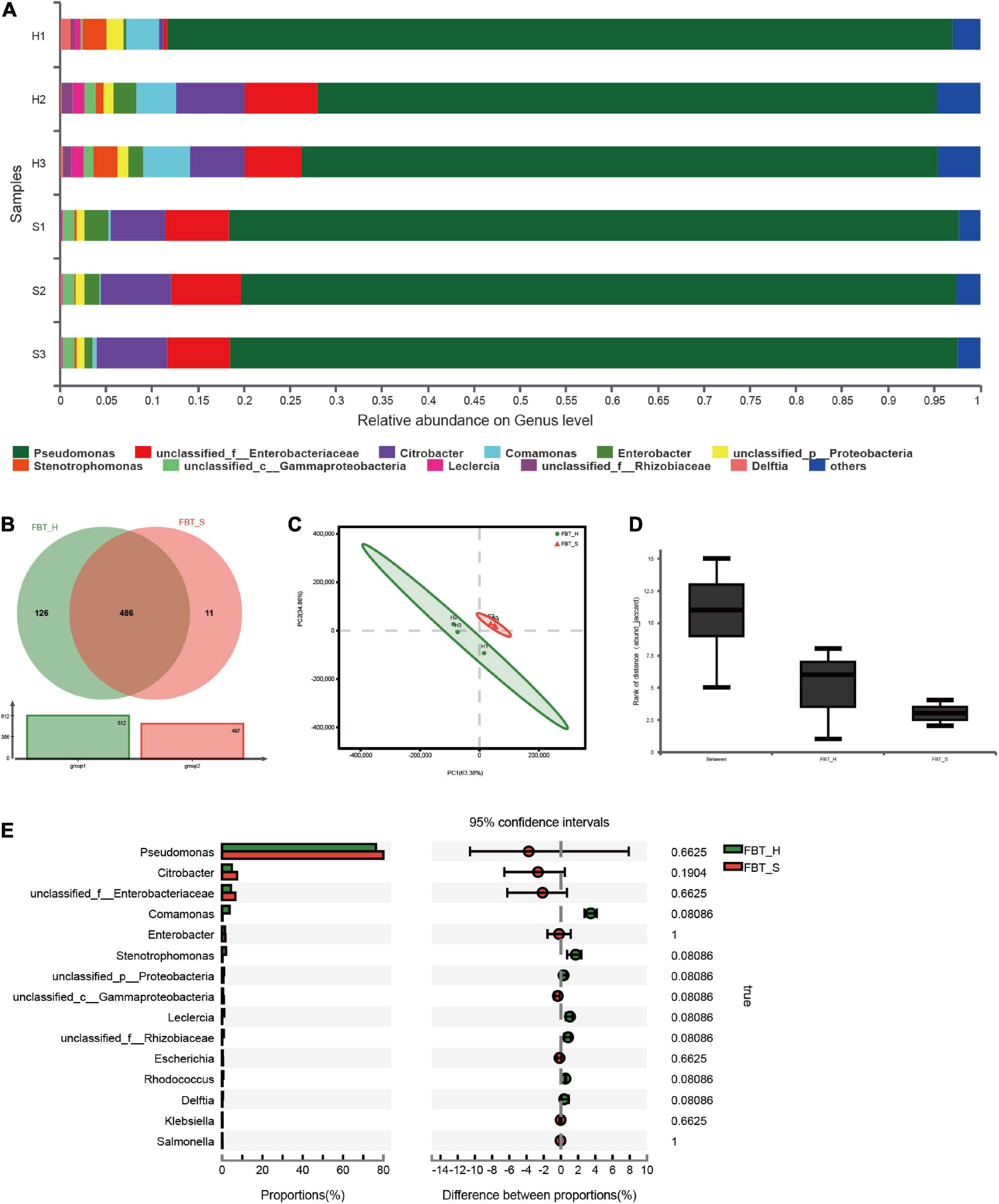
Figure 1. Microbial community composition at the genus level among two FBT groups. (A) The relative abundances of microbial populations. (B) Venn diagram depicting shared taxonomic groups between FBT_H and FBT_S samples. (C) Variation in taxonomic composition among FBT types based on PCA ordination. Different colors indicate different FBT types. The proportion of variation explained by PCA1 and 2 was 63.38 and 34.86%, respectively. FBT_H: H1–H3. FBT_S: S1–S3. (D) ANOSIM analysis of variation based on FBT communities. Inter-group differences are shown by the “Between” box. The “FBT_H and FBT_S” boxes show intra-group differences. (E) STAMP differential analysis of microbial communities between FBT_H and FBT_S samples (error bars indicate Wilcoxon rank-sum tests).
PCA was used to evaluate variation in microbial community composition between the FBT_H and FBT_S at the genus and species level (Figure 1C and Supplementary Figure 1B). PCA clearly separated FBT_H and FBT_S with the first two components comprising 63.38 and 34.86% of the total variation, respectively. Analysis of similarities (ANOSIM) results also suggested that the intergroup differences were greater than intragroup differences, although this result did not show statistical differences (Figure 1D). The ORFs annotated against the NR database were further analyzed and the 15 most abundant genera are shown in Figure 1E. Among these, Pseudomonas, Citrobacter, and unclassified Enterobacteriaceae were the main taxa in two sample types, with no statistical differences in their taxonomic compositions. Previous studies showed that Pseudomonas sp., a potential biocontrol strain, provided beneficial protection against branch canker diseases in tea plants (Mareeswaran and Premkumar, 2020). Pseudomonas fluorescens also demonstrated notable antagonistic activity against fungal species, including Fusarium oxysporum and Alternaria alternata (Chakrabarti, 2009). Fu et al. (2020) suggested that Citrobacter sp. can degrade tea saponin (exhibits strong hemolytic and toxic properties to cold-blooded animals upon overdose) in tea seed cake, implicating a development potential within feed. In contrast, the Enterobacteriaceae group of Proteobacteria has been little studied, and their potential activities or roles in FBT require further analysis. Nevertheless, we hypothesized that the Enterobacteriaceae may originate from raw tea plants rather than via fermentation, based on previous studies (Williams et al., 2010).
Pseudomonas putida, unclassified Pseudomonas, unclassified Enterobacteriaceae, unclassified Citrobacter, and Citrobacter freundii were more abundant in FBT_S samples than FBT_H, while Pseudomonas sp. FH4, Comamonas testosteroni, and P. fluorescens were less abundant in the FBT_S samples than in FBT_H, although significant differences in abundances were again not observed (Supplementary Figure 1E). Thus, there was no significant difference in microbial community composition of FBT_H and FBT_S samples. Combined with a previous literature, it may be concluded that composition of microbial community in FBTs was mainly affected by the different fermentation stage, rather than other factors (Xu et al., 2011).
LEfSe analysis was then performed to confirm the differential species (species level) abundance that contributed significantly to the two FBT samples (Figure 2). A total of 76 bacteria and 21 fungi exhibited significant differences in abundances based on LEfSe analysis. Pseudomonas sp. FH4 and C. testosteroni (LDA scores > 4) were the taxa with the highest contribution scores for FBT_H enrichment. In contrast, P. putida and unclassified Pseudomonas (LDA scores > 4) were the taxa that contributed most to the FBT_S sample distinctiveness. For the eukaryotic community members, Pantholops hodgsonii, Spinacia oleracea, Ascochyta rabiei, Puccinia triticina, and Monomorium pharao (LDA score > 4) contributed most to FBT_H community enrichment. Lipotes vexillifer and Plasmopara halstedii (LDA scores > 4) were all identified as characteristic eukaryote species in the FBT_S samples. Numerous eukaryotic species were common to both samples and did not exhibit significant differences, including the phyla Nematoda, Ascomycota, and Basidiomycota (Supplementary Figures 2, 3). A previous study also reported that Ascomycota and Basidaiomycota comprised the dominant microorganisms within FBT (Zhang et al., 2013).
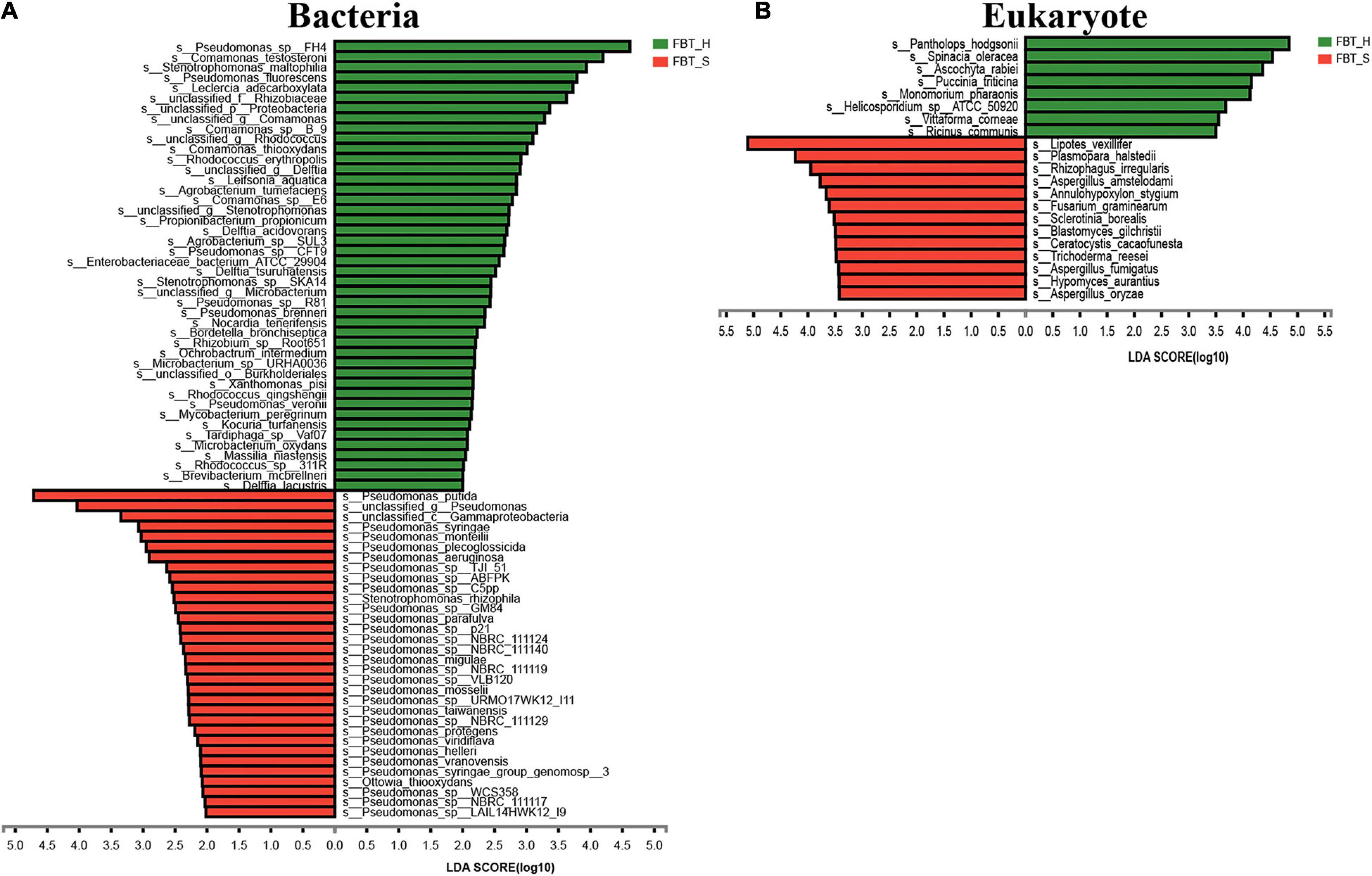
Figure 2. LEfSe analysis of (A) bacterial and (B) eukaryal populations that differed between FBT_H and FBT_S samples based on linear discriminant analysis (LDA) scores. Higher LDA scores indicate greater differential effects of samples on species abundances.
KEGG Functional Analysis
The correlation coefficients of species diversity (on genus level) and enzyme functional profiles were highly consistent at 0.94 and 0.92, respectively (Supplementary Figure 5). To evaluate the functional contributions of FBT microbial populations and metabolic pathways, the functional profiles of the communities were investigated (Figure 3). Clear metabolic contributions were identified for the 10 most abundant microbial populations and specific metabolic functions. The 10 most abundant enzyme functions were contributed from Pseudomonas including F1: histidine kinase, F2: DNA polymerase, F3: peptidylprolyl isomerase, F4: DNA helicase, F5: NADH:ubiquinone reductase (H+-translocating), F6: glutamine synthetase, F7: glutathione transferase, F8: DNA topoisomerase, F9: acetyl-CoA C-acetyltransferase, and F10: shikimate dehydrogenase in both FBT_H and FBT_S, especially for F6 and F10 functions. Shikimate dehydrogenase mainly promoted the biosynthesis of phenylalanine, tyrosine, and tryptophan, which can synthesize important neurotransmitters and hormones involved in glucose metabolism and lipid metabolism, and then regulated the blood sugar serum lipids of the human body (Suzuki et al., 2019). It is known that glutamine synthetase can control the production of glutamine; interestingly, a literature reported that glutamine limitation can mitigate cancer (Jiang et al., 2018). The relative contributions of Comamonas and Stenotrophomonas to FBT_H functional profiles (mean = 5.08%) was much higher than FBT_S (mean = 0.43%). Conversely, the relative contributions of unclassified Enterobacteriaceae were lower in FBT_H (mean = 4.99%) than in FBT_S (mean = 7.72%) samples (Figure 3A). Bacterial–bacterial interactions are ubiquitous in natural environments, including in FBT. For example, the F5 function comprised genes from different bacterial taxa including unclassified Proteobacteria, Stenotrophomonas, Enterobacter, unclassified Gammaproteobacteria, Comamonas, unclassified Enterobacteriaceae, Citrobacter, and Pseudomonas. As is known to all, F5 is associated with NADH:ubiquinone reductase, a large enzyme that is the first segment of respiratory chain in most eukaryotes and many bacteria including Gammaproteobacteria (Volker et al., 2000). NADH:ubiquinone reductase is largely associated with respiration, wherein NADH can be produced during nutrient breakdown and is subsequently used to generate a proton motive force for ATP synthesis (Spero et al., 2015).
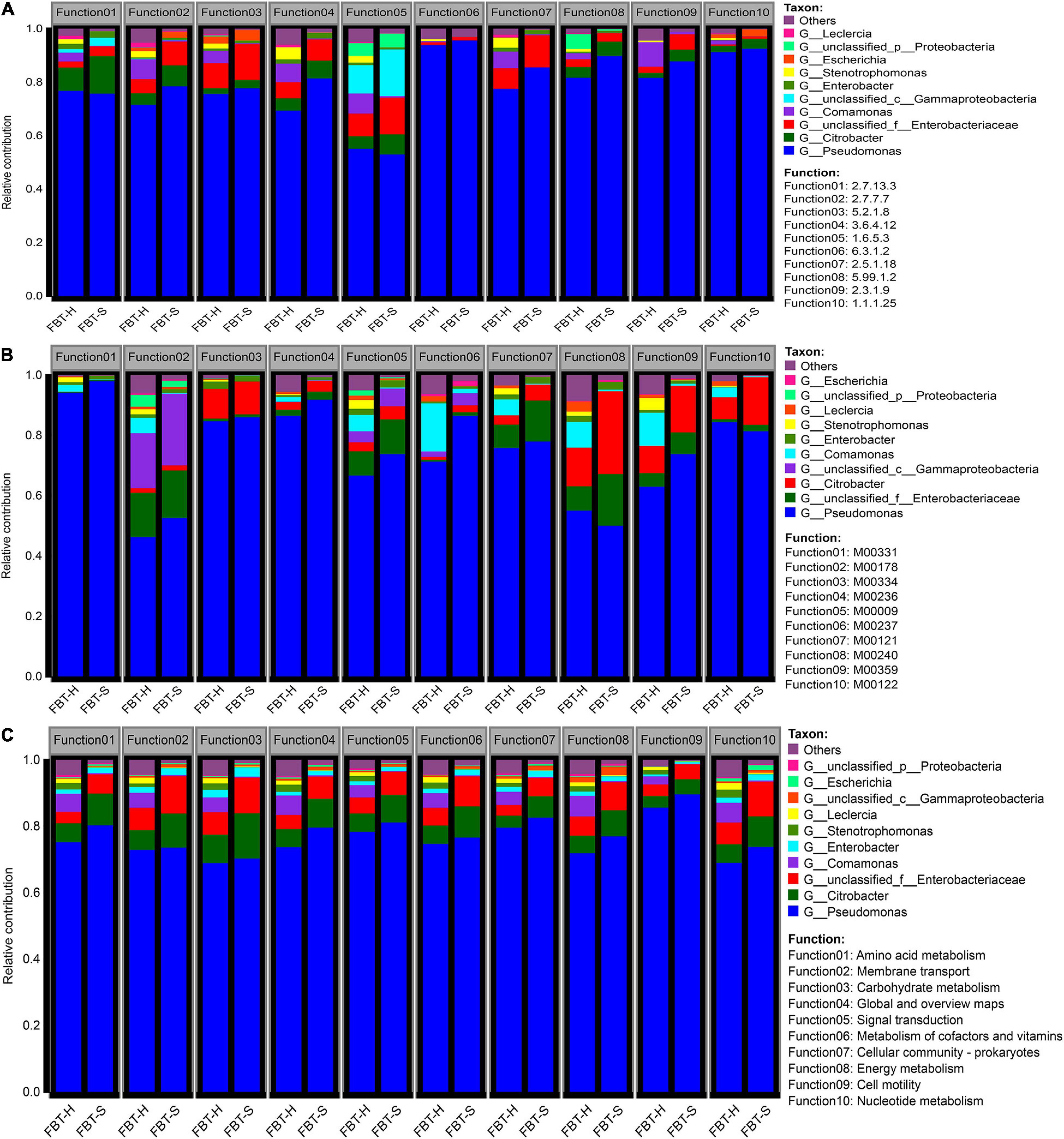
Figure 3. Contribution of microbial populations to (A) enzyme functions, (B) functional modules, and (C) KEGG pathways. Colors indicate relative abundances of different taxa. The abundance contributed by taxa = (reads of the specific taxa associated with function)/(total reads of all taxa associated with function).
Also, microbial community and module function diversity exhibited high correlation with R2 value of 0.92 and 0.95, respectively (Supplementary Figure 5). Pseudomonas contributed the 10 most abundant module functions for both FBT_H and FBT_S communities (Figure 3B). In addition, considerable contributions of Citrobacter to the module functions of F3 (type VI secretion system), F8 (iron complex transport system), F9 (aminoacyl-tRNA biosynthesis, eukaryotes), and F10 (cobalamin biosynthesis) were also observed. The type VI secretion system is a versatile secretion mechanism involved in various functions including antibacterial activity and antagonistic inter-population competition in Gram-negative bacteria like Citrobacter (Gueguen and Cascales, 2013). Chen et al. (2000) reported that Citrobacter is a microorganism found in a compost participated in iron complex transport by producing aerobactin (a siderophore) to improve Fe acquisition. Besides, another previous study demonstrated the production of cobalamin (Vitamin B12) was associated with microbial fermentation in Pseudomonas denitrificans, Salmonella typhimurium, Propionibacterium freudenreichii, and Propionibacterium shermanii (Guo and Chen, 2018), suggesting that Vitamin B12 may exist in FBT metabolites. Thus, our results may provide new insights into cobalamin-producing bacteria taxa. Moreover, unclassified Gammaproteobacteria contributed the most to the abundance of function F2 (ribosome, bacteria), F5 (central carbohydrate metabolism–citrate cycle), and F6 (branched-chain amino acid transport system). Interestingly, a gene encoding a ribosome modulation factor was widely observed throughout the Gammaproteobacteria class organisms, but was not present in any other bacteria taxa (Ueta et al., 2013). Similarly, many Gammaproteobacteria utilize citric acid cycle to conserve energy in their metabolism (Assie et al., 2020). Furthermore, Escherichia coli belongs to the Gammaproteobacteria class and has been observed to use amino acid transport system to secrete threonine (Liu et al., 2012).
Considering metabolism pathway level 2 (Figure 3C), the 10 most prevalent functions were all contributed by numerous microorganisms, with more diverse species contributions for FBT_H samples compared to FBT_S. Nevertheless, Pseudomonas again contributed the most to functional abundances, followed by unclassified Enterobacteriaceae and Citrobacter. Interestingly, the functional contribution of Comamonas to FBT_H samples was much higher than FBT_S. Moreover, microbial taxa associated with the core-housekeeping functions like signal transduction (F1: two-component system), membrane transport (F2: ABC transporters), global and overview maps (F3: biosynthesis of amino acids, F4: carbon metabolism), nucleotide metabolism (F7: purine metabolism), cellular community prokaryotes (F5: bacterial secretion system, F8: biofilm formation-Vibrio cholerae, F9: quorum sensing), and cell motility (F6: flagellar assembly, F10: bacterial chemotaxis) were abundant metabolic categories represented in both samples (Supplementary Figure 4). The contribution of unclassified Proteobacteria to flagellar assembly and bacterial chemotaxis functional abundance in FBT_H samples was higher than in FBT_S samples, while the contribution of Escherichia to the bacterial secretion system abundance in FBT_H was much lower than in FBT_S, indicating that the functional contributions of species were related to the specific microbial environments of FBT types. Specific organisms can be associated with different metabolic functions, while the collective metabolic activities of communities can substantially modify the environment (Mazumdar et al., 2013).
A significant positive correlation was observed between Pseudomonas abundances and the enzyme function of 2.1.1.107 (uroporphyrinogen methyltransferase, p < 0.01), while Enterobacter abundances were positively correlated with 2.7.7.65 (diguanylate cyclase), but negatively correlated with 1.2.1.26 (2,5-dioxovalerate dehydrogenase, p < 0.01) (Figure 4A). The specific contributions of Citrobacter and unclassified Enterobacteriaceae were associated with the M00122 (cobalamin biosynthesis) (Figure 4B), consistent with the module function analysis above. Similarly, Pseudomonas abundances exhibited a significant positive correlation with the functional category arginine biosynthesis and porphyrin and chlorophyll metabolism (p < 0.01, p < 0.05). Chlorophyll catabolites are an indispensable part of phytochemicals and human nutrition, as Karg et al. (2021) reported that a phylloxanthobilin (a yellow chlorophyll catabolite) possesses antioxidative and anti-inflammatory activities. Citrobacter and unclassified Enterobacteriaceae abundances were also positively correlated with fructose and mannose metabolism, which may be related to the reduction of hyperglycemia by drinking FBT (p < 0.05) (Zhu et al., 2021); Comamonas abundances were also closely associated with the functions of homologous recombination, pyrimidine metabolism, and quorum sensing. In contrast, Enterobacter abundances were more associated with the metabolism pathways of glycerophospholipid metabolism (Figure 4C), which may participate in inhibiting fat accumulation and avoiding fatty liver produced (Wu et al., 2016).
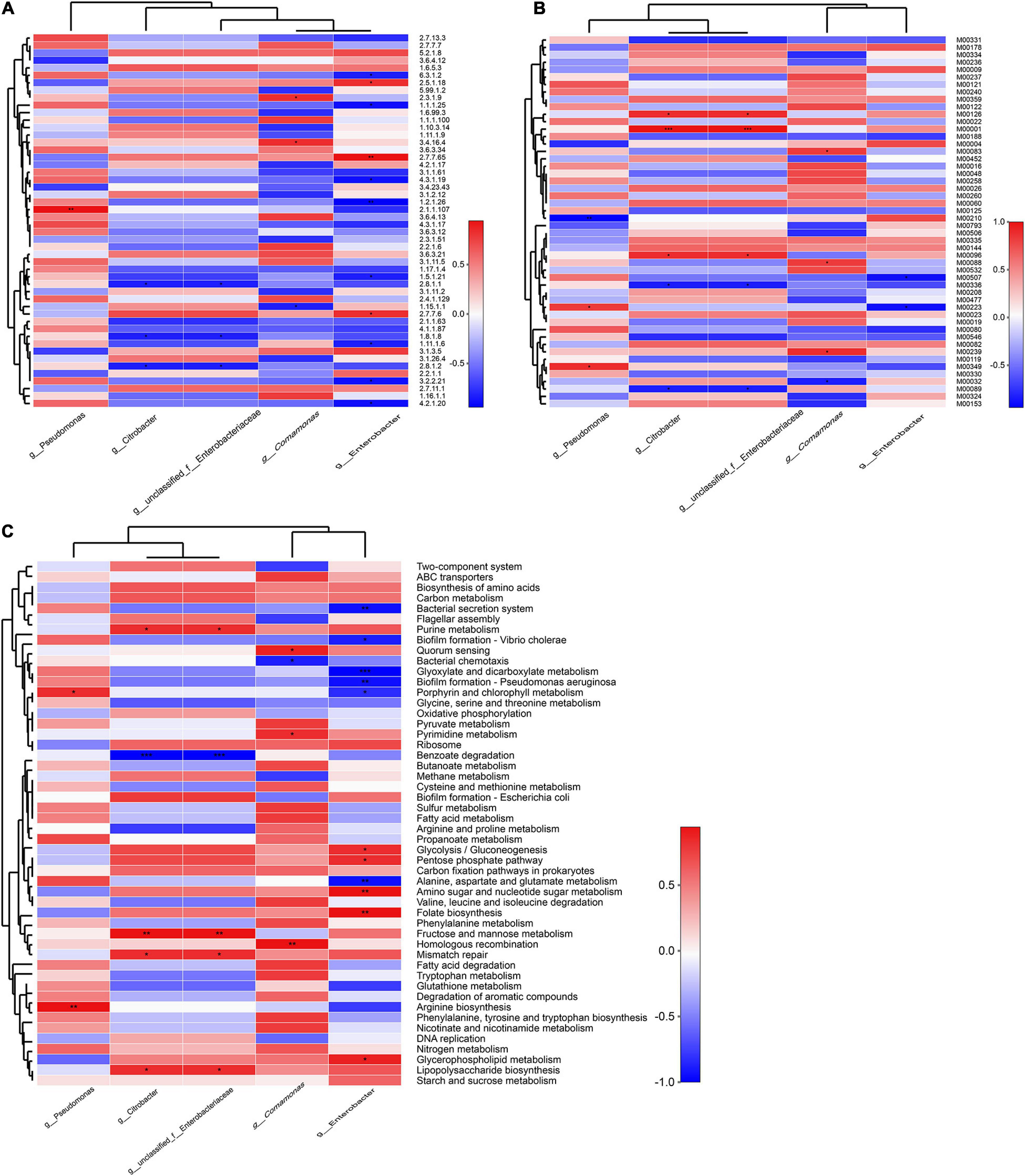
Figure 4. Correlation of encoded functions and the most abundant microbial taxa. Cell colors denote correlation coefficient values based on the scales to the right of the heatmaps (*p < 0.05, **p < 0.01, and ***p < 0.001). Profiles are shown for (A) enzyme profiles, (B) module profiles, and (C) KEGG pathway level 3.
CAZy Functional Analysis
A total of 6,339,740 reads were mapped to 203 CAZymes among all metagenomes (Figure 5). The carbohydrate active enzymes (CAZy) functional distributions were shown as follows: class glycoside hydrolases (GH) exhibited a functional abundance of 32.92%; glycosyl transferases (GT), 26.8%; carbohydrate esterases (CE), 20.43%; and auxiliary activities (AA), 18.04% (Supplementary Figure 5). Among these, the abundance of function CE1 (the abundance > 11%) was highest, followed by AA3, CE10, GH23, GT4, GT2, and GT41, and no significant differences were observed among the two samples (Figures 5A,B). Thus, the species contributions to CAZy functional profiles were consistent across both sample groups. Notably, GH are associated with the oxidation, conversion, or degradation of phenolic compounds (Zhao et al., 2019). A previous study also reported that a thermo-stable GH, α-rhamnosidase, may be present in FBT communities that contributed to produce glycosidically bound volatiles such as benzyl alcohol, 2-phenylethanol, methyl salicylate, linalool, geraniol, coumarin, and damascenone (Fang et al., 2019). In addition, increases in GT expression levels have been proven, which is beneficial for metabolizing starch and cellulose to glucose, thereby providing energy to populations tolerating fluoride stress (Yang et al., 2020). Moreover, Habrylo et al. (2018) reported that some CEs exhibit optimal activity at pH 6–8 and 40°C, which renders them suitable for both acidic and alkaline applications, such as coffee and tea fermentations. Similarly, most AA in FBT reflected the adaptations of microbial populations to tea substrate (Kanokratana et al., 2018).
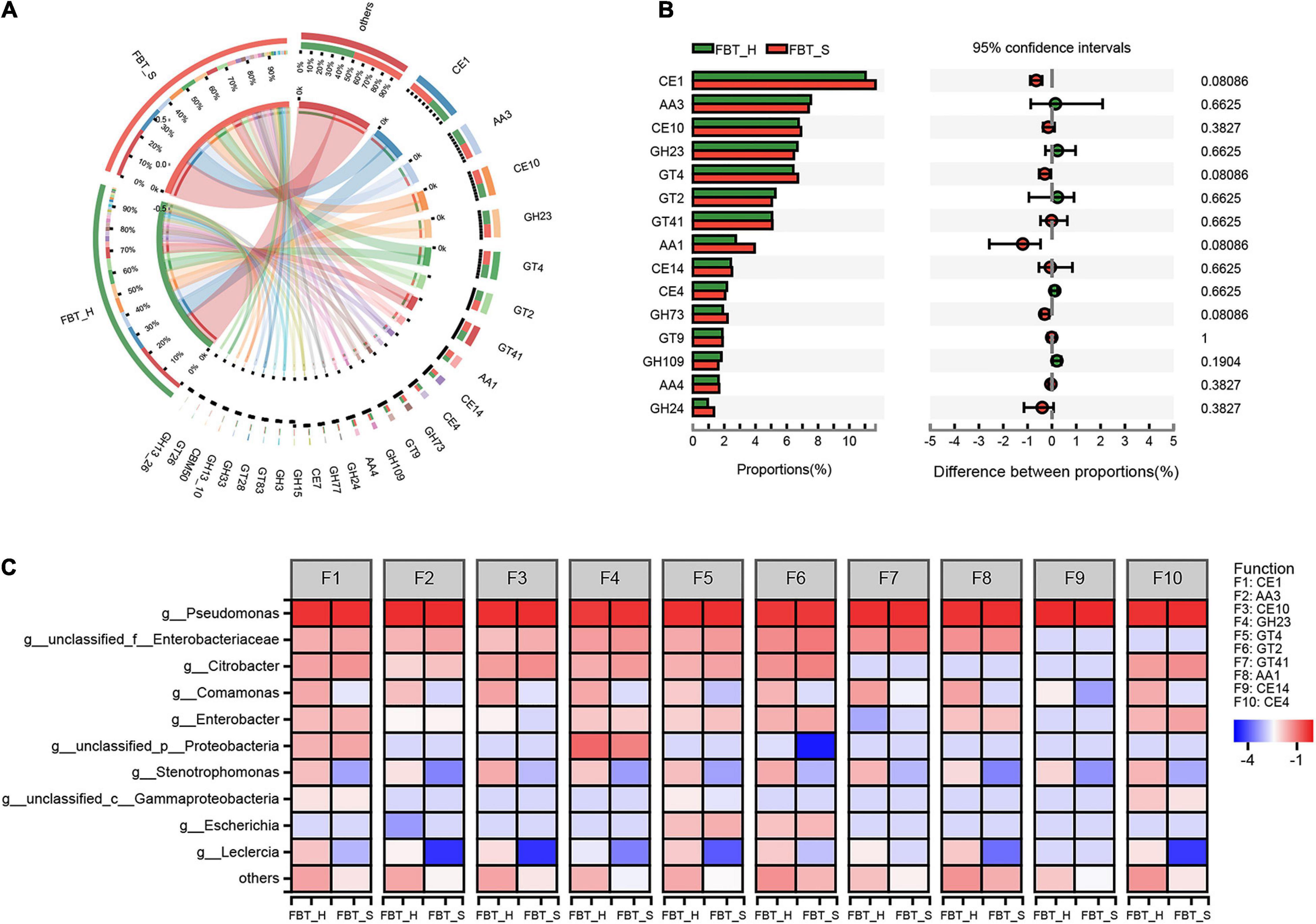
Figure 5. Distribution of CAZyme database functions. (A) The overall distribution of CAZyme among FBT samples. The length of bars for each FBT on the outer ring represents the percentage of CAZymes within each FBT sample. (B) Comparison of CAZyme functions between FBT_H and FBT_S samples. The data were visualized using STAMP (error bars represent Wilcoxon rank-sum test). (C) The contribution of species to CAZyme functional profiles. AA, auxiliary activities; CBM, carbohydrate-binding modules; CE, carbohydrate esterases; GH, glycoside hydrolases; GT, glycosyl transferases; PL, polysaccharide lyases.
The contributions of Comamonas, Stenotrophomonas, and Leclercia to each of the CAZy functions were higher in FBT_H samples than FBT_S (Figure 5C). Additionally, AA3_2 and GH103 were key characteristic CAZy functions in FBT_H samples, while AA1 was a key CAZy function for FBT_S samples based on LDA scores (Supplementary Figure 5). An obvious positive correlation was observed between Comamonas and the abundances of function CE4, reflecting that Comamonas harbors and contributes to the abundances of CE4 family enzymes including chitin deacetylase (EC 3.5.1.41), chitooligosaccharide deacetylase (EC 3.5.1.-), peptidoglycan GlcNAc deacetylase (EC 3.5.1.-) and peptidoglycan N-acetylmuramic acid deacetylase (EC 3.5.1.-). However, some significant negative correlations were observed including the CE1 function and Comamonas abundances (Spearman R = −0.94, p < 0.01), and CE10 and GT41 functions and Enterobacter abundances (Spearman R = 0.94, p < 0.01).
Differential Metabolic Pathways
Seven level 2 KEGG pathways were overrepresented in FBT_H samples relative to FBT_S, while six were less represented in FBT_H samples compared to FBT_S. The most represented metabolic pathways comprised carbohydrate and amino acid metabolism for both samples and their abundances were not significantly difference (Figure 6A). In addition, the two-component system (ko02020) and ABC transporters (ko02010) were the most abundant level 3 KEGG metabolism pathways. A total of 255 and 287 enzymes were primarily affiliated with the ko02020 and ko02010 pathways, respectively, all of which did not exhibit significant differences between FBT_H or FBT_S samples (Figure 6B and Supplementary Tables 4, 5). Comparative analysis of secondary metabolite biosynthesis pathways between FBT_H and FBT_S was conducted using the ipath platform, with one differential pathway (penicillin and cephalosporin biosynthesis) being observed only in FBT_H samples. This pathway is primarily involved in the coordinated transformation of two metabolites: isopenicillin N (C05557) and penicillin N (C06564) (Figure 7A). Cephalosporins and penicillins are the most frequently used β-lactam antibiotics for treating human infections globally. The primary organisms used for industrial production of these antibiotics are Acremonium chrysogenum, Penicillium chrysogenum, Aspergillus nidulans, and the Gram-positive bacterium Streptomyces lipmanii (Terfehr et al., 2017). Isopenicillin N is the bioactive intermediate of the penicillin biosynthesis pathway (Ziemons et al., 2017). Furthermore, the gene pa4191 of Pseudomonas aeruginosa has provisionally been annotated as a member of the isopenicillin N synthase family that mediates a key step in penicillin biosynthesis (Zhang et al., 2019). In brief, penicillin is biosynthesized by several eukaryotic and bacterial species. Metagenomic analyses showed here that several FBT community strains were involved in penicillin and cephalosporin biosynthesis including Agrobacterium, Comamonas, Delftia, Leclercia, Microbacterium, Ochrobactrum, Paraburkholderia, Pseudomonas, Stenotrophomonas, and unclassified Rhizobiaceae.
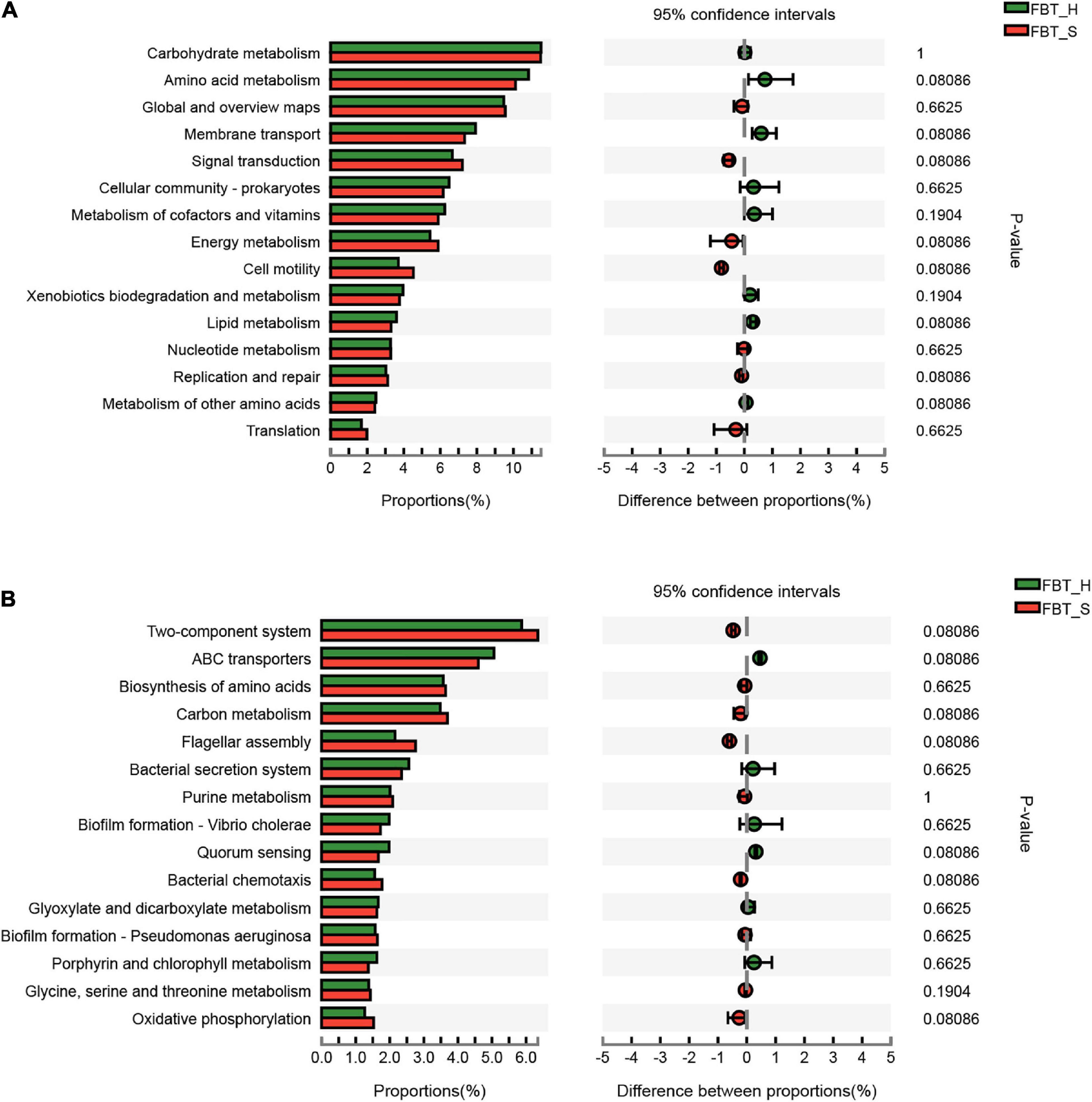
Figure 6. Comparison of KEGG pathway level functions between FBT type communities. (A) KEGG pathway level 2 and (B) KEGG pathway level 3.
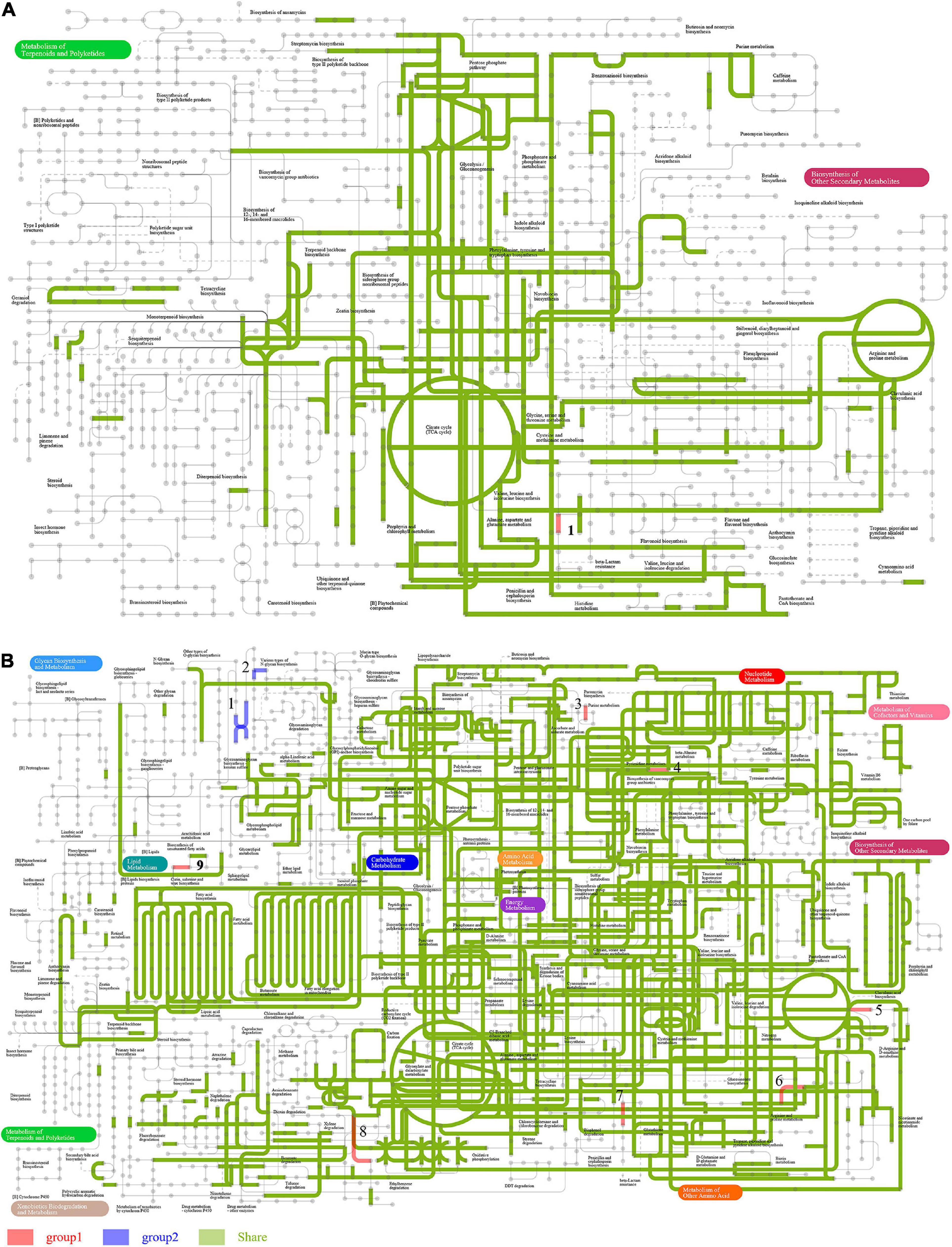
Figure 7. Comparison of ipath metabolic pathways between FBT-type communities. Red and blue lines indicate unique metabolic pathways for FBT_H and FBT_S communities, respectively, while green lines indicate shared metabolic pathways among the two samples. (A) Biosynthesis of secondary metabolites and overall (B) metabolic pathways.
Further analysis of the global overview map of metabolic pathways provided additional insights into the central metabolisms of two FBT community populations (Figure 7B). A total of nine metabolic pathways were observed that were differentially abundant among two FBT types. Pathways I and II were related to glycan biosynthesis and only observed in the FBT_S community, while other seven central metabolic pathways (III–IX) were only present in FBT_H samples, namely, III, ascorbate and aldarate metabolism; IV, purine metabolism and pyrimidine metabolism; V, glycine, serine, and threonine metabolism, and arginine and proline metabolism; VI, cysteine and methionine metabolism; VII, penicillin and cephalosporin biosynthesis; VIII, ethylbenzene degradation; and IX, sphingolipid metabolism. Based on the above results, it is possible that greater abundances of tea polysaccharides may be produced in FBT_S type teas that contribute to reducing the risk of type 2 diabetes and obesity (Chen et al., 2019). In addition, some abundant microbial populations participated in amino acid metabolism activity (e.g., with glycine, serine, threonine, arginine, proline, cysteine, and methionine substrates) that can improve the taste of tea (Chen et al., 2020). The metabolic pathway of ethylbenzene degradation in FBT_H samples contributed to the regulation of fermented tea aroma (Zhu et al., 2018). Moreover, several microbial populations within FBT samples were also associated with the regulation of ascorbate and aldarate metabolism, nucleotide, and sphingolipid metabolism.
Probiotic Analysis
A total of 35 probiotics were predicted (annotated against probio database) within two FBT sample groups. Among these, nine were only annotated in FBT_H samples, namely, Vibrio alginolyticus, Lactobacillus helveticus, Serratia liquefaciens, Lysinibacillus fusiformis, Lactobacillus brevis, Lactobacillus crispatus, Lactobacillus kefiranofaciens, Bifidobacterium catenulatum, and Pantoea agglomerans; and four were only annotated in FBT_S communities, namely, Bacillus thuringiensis, Clostridium butyricum, Bacillus licheniformis, and Acinetobacter calcoaceticus (Table 1). Obviously, P. putida was the most abundant inferred probiotic with 80% proportions in both sample groups, which can use caffeine as its sole source of carbon and nitrogen, rendering it a potential candidate for bio-decaffeination, the production of caffeine derivatives, and environmental remediation of caffeine (Ma et al., 2018). It also has been used to biodegrade agrochemicals like the triazole fungicide propiconazole (Sarkar et al., 2010). Thus, P. putida played a key role in ensuring the safety of tea products.
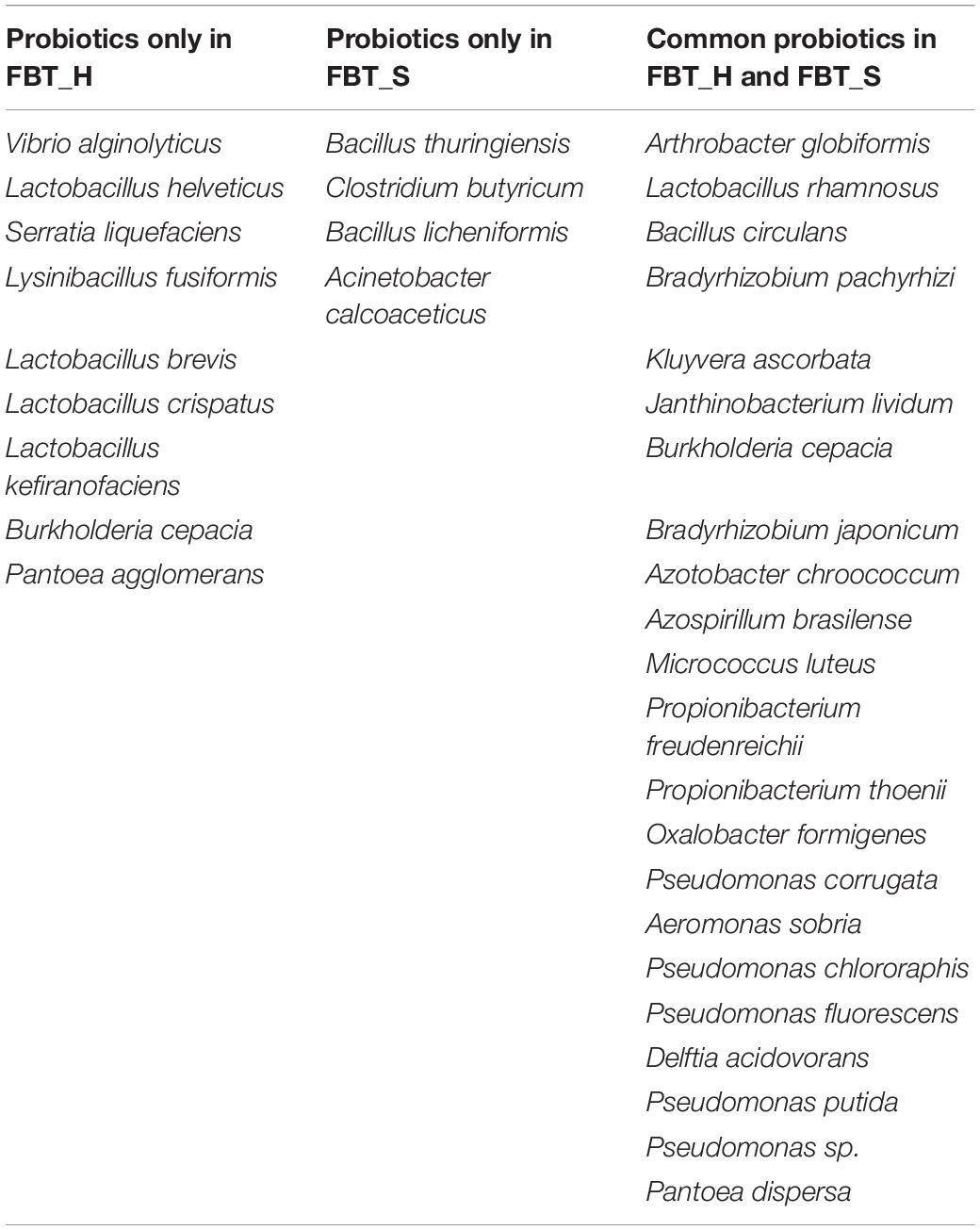
Table 1. Probiotic composition between FBT_H and FBT_S based on probio database.
Additionally, the abundances of other potential probiotics in FBT_H were higher than FBT_S, except for P. putida and P. fluorescens. Thus, potential probiotic effects may be higher in FBT_H (Figure 8A). Furthermore, some significant positive correlations were observed between annotated probiotics and functions (p < 0.001), including the fact that P. putida abundances are associated with the effects of plant nutrient supplies, microbial infections in plants, and plant product yields, while the abundances of P. fluorescens are closely related to the normal absorption and assimilation of nutrients in intestines, the maintenance of healthy gastrointestinal tract microfloral ecologies, the promotion of normal bowel functions, assistance in stabilizing gut mucosal barrier, and supporting gut immune system. Furthermore, Delftia acidovorans is highly associated with plant growth effects, Aeromonas sobria is highly associated with bacterial infections in animals, Pseudomonas chlororaphis is closely related to the effects of fungal infections in plants and plant nitrogen supplies, and Oxalobacter formigenes is highly associated with hyperoxaluria effects (Figure 8B).
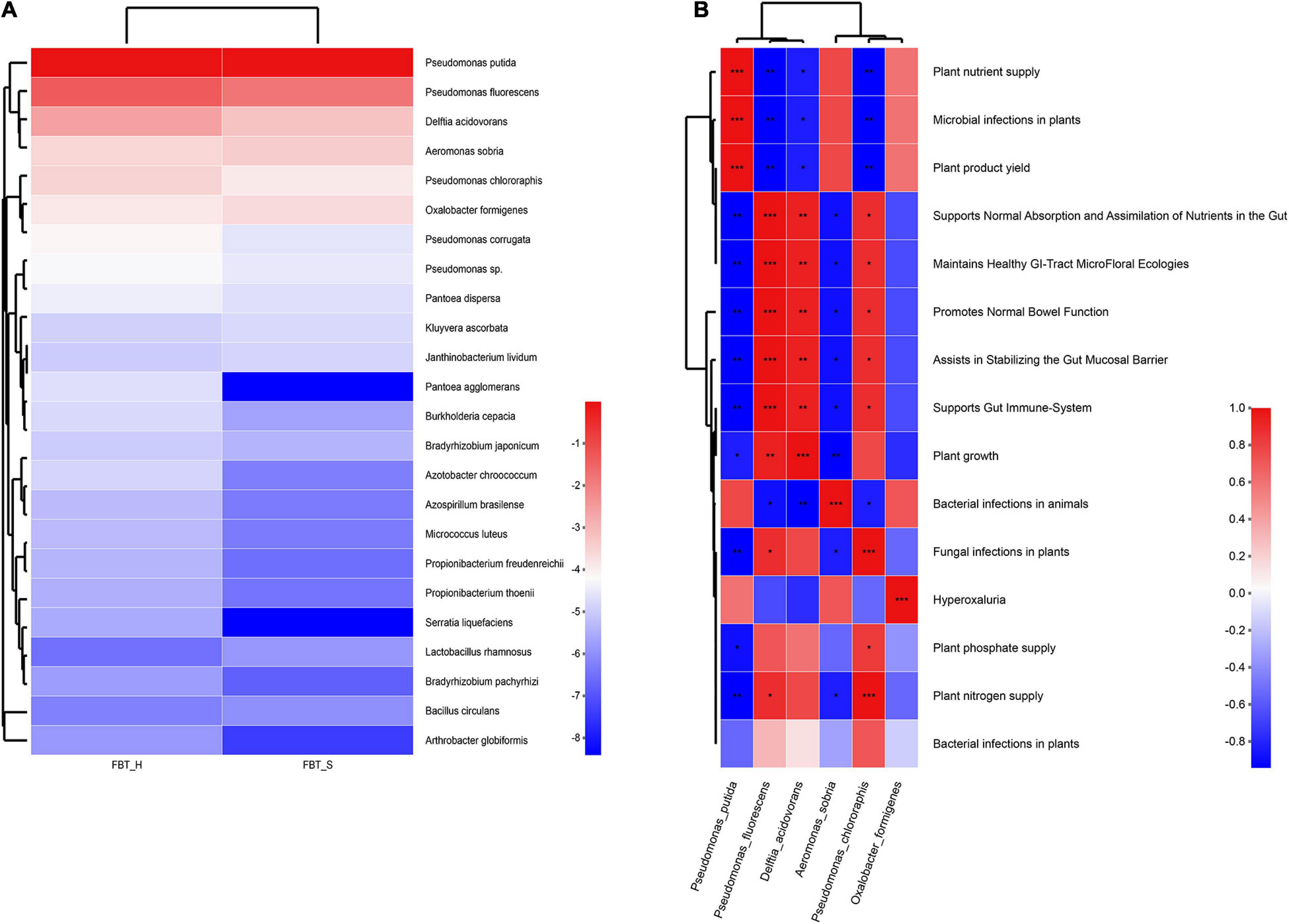
Figure 8. Probiotic analysis for FBT communities against probio database. (A) The relative abundances of inferred probiotic in FBT communities. (B) Correlation between probiotic and functional abundances (*p < 0.05, **p < 0.01, and ***p < 0.001). Cell colors indicate the correlation coefficients based on the scale to the right.
Metagenomic analysis of FBT microbial communities here provided novel insights into the huge probiotic flora of FBT. Overall, numerous previously reported probiotic fungi including Eurotium cristatum and Aspergillus cristatus (Kang et al., 2019; Rui et al., 2019) were identified from FBT, while several predicted bacteria were also identified in FBT using culture-dependent methods and 16s RNA sequencing including P. fluorescens, P. putida, Lactobacillus rhamnosus, L. kefiranofaciens, and B. licheniformis (Supplementary Figure 6 and Supplementary Table 6).
Conclusion
The potential interactions of microbial populations and their associated functions were evaluated in two types of FBT using metagenomics for the first time. Our results showed that microbial community composition was largely similar among two types of FBT samples, with no significant differences. However, diversity in FBT_H (612 genera) was higher than FBT_S (497 genera). Pseudomonas contributed most to the abundant functional groups in both FBT_H and FBT_S. Moreover, the carbohydrate enzyme functional distribution of microbial communities in FBT exhibited highly abundant functional potential for GHs (32.92%), GTs (26.8%), CEs (20.43%), and AAs (18.04%). Carbohydrate and amino acid metabolism were the primary and key metabolic pathways in two FBT samples. Notably, Pseudomonas contributed the most to enzyme function, module function, and pathway level 2. A significant positive correlation was observed between Pseudomonas abundances and the enzyme function of 2.1.1.107 (uroporphyrinogen methyltransferase, p < 0.01). In addition, considerable contributions of Citrobacter to the module functions of F3 (type VI secretion system), F8 (iron complex transport system), F9 (aminoacyl-tRNA biosynthesis, eukaryotes), and F10 (cobalamin biosynthesis) were also observed. The specific contributions of Citrobacter and unclassified Enterobacteriaceae were associated with the M00122 (cobalamin biosynthesis). Differently, nine metabolic pathways exhibited differential abundances between two types of communities. A total of 35 probiotics were present in FBT samples, with nine being unique to FBT_H and four being unique to FBT_S communities. Furthermore, P. putida was the most abundant inferred probiotic (80%) among two samples. This study provided new and detailed insights into the taxonomic composition and potential function of FBT communities (including carbohydrate and metabolic functions), especially for the potential probiotics in FBT communities.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: www.ncbi.nlm.nih.gov/, PRJNA729248.
Author Contributions
XW prepared the manuscript, did the literature survey, and designed the figures. YY, TY, and GD revised the manuscript. HC and XZ performed some of the experiments. BL, CG, and QS revised some of the mistakes in grammar. All authors read and approved the manuscript.
Funding
This work was supported by the Shaanxi Special Project of China (2018ZDXM-NY-084).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the online platform of Majorbio Cloud Platform for data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.705681/full#supplementary-material
Supplementary Figure 1 | Microbial community compositions among FBT samples. (A) Relative abundances of microbial community. (B) Variation in genus-level microbial composition within FBT samples. The proportion of variation explained by PCA1 and 2 was 80.08 and 19.15%, respectively. FBT_H: H1–H3. FBT_S: S1–S3. (C) Hierarchical clustering of genus-level taxonomic profiles. (D) Hierarchical clustering analysis of species-level profiles. (E) Comparison of species groups between FBT_H and FBT_S samples.
Supplementary Figure 2 | LEfSe analysis of bacterial communities among FBT samples. Red nodes indicate taxa are significantly enriched in FBT_S samples, while green nodes indicate taxa are significantly enriched in FBT_H samples. Yellow nodes indicate the lack of significant effect when comparing FBT_H and FBT_S samples.
Supplementary Figure 3 | LEfSe analysis of eukaryotic communities among FBT samples. Red nodes indicate taxa are significantly enriched in FBT_S samples, while green nodes indicate taxa are significantly enriched in FBT_H samples. Yellow nodes indicated the lack of significant effect when comparing FBT_H and FBT_S samples.
Supplementary Figure 4 | Species and KEGG pathway level 3 contribution analysis.
Supplementary Figure 5 | Distribution of CAZyme functions among FBT samples. (A) α diversity between microbial genus-levels and CAZyme functions. (B) β diversity between genus-levels and CAZyme functions. (C) Differences in CAZyme function between FBT_H and FBT_S samples. Higher LDA scores indicate greater effects of sample type on functional abundances. (D) Correlational analysis between species and CAZyme functions (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (E) Distribution of CAZyme functions.
Supplementary Figure 6 | The colony morphology of 5 potential probiotics.
Supplementary Table 1 | Biosynthesis of secondary metabolite pathways enriched in FBT_H samples.
Supplementary Table 2 | Metabolic pathways enriched in FBT_S samples.
Supplementary Table 3 | Metabolic pathways enriched in FBT_H samples.
Supplementary Table 4 | ko02010 ABC transporter annotations in FBT samples.
Supplementary Table 5 | ko02020 two-component system annotations in FBT samples.
Footnotes
- ^ https://github.com/OpenGene/fastp
- ^ https://github.com/aquaskyline/SOAPdenovo2
- ^ http://ab.inf.uni-tuebingen.de/software/diamond/
- ^ http://kobas.cbi.pku.edu.cn/home.do
- ^ http://www.cazy.org/
- ^ http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload, α<0.05, LDA score>2.
- ^ http://pathways.embl.de
- ^ https://dataview.ncbi.nlm.nih.gov/?search=SUB9613785&archive=bioproject
References
Arikan, M., Mitchell, A. L., Finn, R. D., and Gurel, F. (2020). Microbial composition of Kombucha determined using amplicon sequencing and shotgun metagenomics. J. Food Sci. 85, 455–464. doi: 10.1111/1750-3841.14992
Assie, A., Leisch, N., Meier, D. V., Gruber-Vodicka, H., Tegetmeyer, H. E., Meyerdierks, A., et al. (2020). Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria). ISME J. 14, 104–122. doi: 10.1038/s41396-019-0508-7
Chakrabarti, S. (2009). “Diversity of rhizosphere microflora in Himalayan tea gardens of Darjeeling, India,” in Proceedings of the 3rd International Symposium on Biodiversity and Natural Heritage of the Himalaya, 31–33.
Chen, G., Chen, C., and Lei, Z. (2017). Meta-omics insights in the microbial community profiling and functional characterization of fermented foods. Trends Food Sci. Tech. 65, 23–31. doi: 10.1016/j.tifs.2017.05.002
Chen, G. J., Chen, R. S., Chen, D., Ye, H., Hu, B., Zeng, X. X., et al. (2019). Tea polysaccharides as potential therapeutic options for metabolic diseases. J. Agric. Food Chem. 67, 5350–5360. doi: 10.1021/acs.jafc.8b05338
Chen, L., Dick, W. A., and Streeter, J. G. (2000). Production of aerobactin by microorganisms from a compost enrichment culture and soybean utilization. J. Plant Nutr. 23, 2047–2060. doi: 10.1080/01904160009382164
Chen, Z., Kang, J., Zhang, Y., Yi, X., Pang, X., Li-Byarlay, H., et al. (2020). Differences in the bacterial profiles and physicochemical between natural and inoculated fermentation of vegetables from Shanxi Province. Ann. Microbiol. 70:66.
Cheng, Q., Cai, S., Ni, D., Wang, R., Zhou, F., Ji, B., et al. (2015). In vitro antioxidant and pancreatic alpha-amylase inhibitory activity of isolated fractions from water extract of Qingzhuan tea. J. Food Sci. Tech. Mys. 52, 928–935. doi: 10.1007/s13197-013-1059-y
Fang, X., Dong, Y., Xie, Y., Wang, L., Wang, J., Liu, Y., et al. (2019). Effects of beta-glucosidase and alpha-rhamnosidase on the contents of flavonoids, ginkgolides, and aroma components in Ginkgo tea drink. Molecules 24:2009. doi: 10.3390/molecules24102009
Fu, G. M., Chen, K. D., Wang, J. T., Wang, M., Li, R. Y., Wu, X. J., et al. (2020). Screening of tea saponin-degrading strain to degrade the residual tea saponin in tea seed cake. Prep. Biochem. Biotech. 50, 697–707. doi: 10.1080/10826068.2020.1731827
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gueguen, E., and Cascales, E. (2013). Promoter swapping unveils the role of the Citrobacter rodentium CTS1 type VI secretion system in interbacterial competition. Appl. Environ. Microbiol. 79, 32–38. doi: 10.1128/aem.02504-12
Guo, M., and Chen, Y. (2018). Coenzyme cobalamin: biosynthesis, overproduction and its application in dehalogenation—a review. Rev. Environ. Sci. Biol. 17, 259–284. doi: 10.1007/s11157-018-9461-6
Habrylo, O., Evangelista, D. E., Castilho, P. V., Pelloux, J., and Henrique-Silva, F. (2018). The pectinases from Sphenophorus levis: potential for biotechnological applications. Int. J. Biol. Macromol. 112, 499–508. doi: 10.1016/j.ijbiomac.2018.01.172
Jiang, J., Pavlova, N. N., and Zhang, J. (2018). Asparagine, a critical limiting metabolite during glutamine starvation. Mol. Cell. Oncol. 5:e1441633. doi: 10.1080/23723556.2018.1441633
Jiang, Y., Lu, Y., Huang, Y., Chen, S., and Ji, Z. (2019). Bacillus amyloliquefaciens HZ-12 heterologously expressing NdmABCDE with higher ability of caffeine degradation. LWT 109, 387–394. doi: 10.1016/j.lwt.2019.04.033
Johansson, L., Nikulin, J., Juvonen, R., Krogerus, K., Magalhaes, F., Mikkelson, A., et al. (2021). Sourdough cultures as reservoirs of maltose-negative yeasts for low-alcohol beer brewing. Food Microbiol. 94:103629. doi: 10.1016/j.fm.2020.103629
Kang, D., Su, M., Duan, Y., and Huang, Y. (2019). Eurotium cristatum, a potential probiotic fungus from fuzhuan brick tea, alleviated obesity in mice by modulating gut microbiota. Food Funct. 10, 5032–5045. doi: 10.1039/c9fo00604d
Kanokratana, P., Wongwilaiwalin, S., Mhuantong, W., Tangphatsornruang, S., Eurwilaichitr, L., Champreda, V., et al. (2018). Characterization of cellulolytic microbial consortium enriched on Napier grass using metagenomic approaches. J. Biosci. Bioeng. 125, 439–447. doi: 10.1016/j.jbiosc.2017.10.014
Karg, C. A., Doppler, C., Schilling, C., Jakobs, F., Dal Colle, M. C. S., Frey, N., et al. (2021). A yellow chlorophyll catabolite in leaves of Urtica dioica L.: an overlooked phytochemical that contributes to health benefits of stinging nettle. Food Chem. 359:129906. doi: 10.1016/j.foodchem.2021.129906
Kim, E., Cho, E.-J., Yang, S.-M., Kim, M.-J., and Kim, H.-Y. (2021). Novel approaches for the identification of microbial communities in kimchi: MALDI-TOF MS analysis and high-throughput sequencing. Food Microbiol. 94:103641. doi: 10.1016/j.fm.2020.103641
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lawson, C. E., Wu, S., Bhattacharjee, A. S., Hamilton, J. J., McMahon, K. D., Goel, R., et al. (2017). Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun. 8:15416.
Li, J.-Y., Jin, X.-Y., Zhang, X.-C., Chen, L., Liu, J.-L., Zhang, H.-M., et al. (2020). Comparative metagenomics of two distinct biological soil crusts in the Tengger Desert, China. Soil Biol. Biochem. 140:107637. doi: 10.1016/j.soilbio.2019.107637
Li, Q., Chai, S., Li, Y., Huang, J., Luo, Y., Xiao, L., et al. (2018). Biochemical components associated with microbial community shift during the pile-fermentation of primary dark tea. Front. Microbiol. 9:1509. doi: 10.3389/fmicb.2018.01509
Li, Q., Huang, J., Li, Y., Zhang, Y., Luo, Y., Chen, Y., et al. (2017). Fungal community succession and major components change during manufacturing process of Fu brick tea. Sci. Rep. 7:6947.
Li, Q., Liu, Z., Huang, J., Luo, G., Liang, Q., Wang, D., et al. (2013). Anti-obesity and hypolipidemic effects of Fuzhuan brick tea water extract in high-fat diet-induced obese rats. J. Sci. Food Agric. 93, 1310–1316. doi: 10.1002/jsfa.5887
Liu, Q., Cheng, Y., Xie, X., Xu, Q., and Chen, N. (2012). Modification of tryptophan transport system and its impact on production of L-tryptophan in Escherichia coli. Bioresour. Technol. 114, 549–554. doi: 10.1016/j.biortech.2012.02.088
Liu, Z., Gao, L., Huang, J. A., Cai, S., Zhang, S., Xiong, L., et al. (2019). Leading progress on genomics, health benefits and utilization of tea resources in China. Nature 566, S15–S19.
Ma, Y. X., Wu, X. H., Wu, H. S., Dong, Z. B., Ye, J. H., Zheng, X. Q., et al. (2018). Different catabolism pathways triggered by various methylxanthines in caffeine-tolerant bacterium Pseudomonas putida CT25 isolated from tea garden soil. J. Microbiol. Biotechnol. 28, 1147–1155. doi: 10.4014/jmb.1801.01043
Mareeswaran, J., and Premkumar, R. (2020). Effect of chemicals and biological agents on branch canker disease in tea. Indian J. Exp. Biol. 58, 271–275.
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to deepwater horizon oil spill. ISME J. 6, 1715–1727. doi: 10.1038/ismej.2012.59
Mazumdar, V., Amar, S., and Segre, D. (2013). Metabolic proximity in the order of colonization of a microbial community. PLoS One 8:e77617. doi: 10.1371/journal.pone.0077617
Rui, Y., Wan, P., Chen, G., Xie, M., Sun, Y., Zeng, X., et al. (2019). Analysis of bacterial and fungal communities by Illumina MiSeq platforms and characterization of Aspergillus cristatus in Fuzhuan brick tea. LWT 110, 168–174. doi: 10.1016/j.lwt.2019.04.092
Sarkar, S., Seenivasan, S., and Asir, R. P. S. (2010). Biodegradation of propargite by Pseudomonas putida, isolated from tea rhizosphere. J. Hazard. Mater. 174, 295–298. doi: 10.1016/j.jhazmat.2009.09.050
Spero, M. A., Aylward, F. O., Currie, C. R., and Donohue, T. J. (2015). Phylogenomic analysis and predicted physiological role of the proton-translocating NADH:quinone oxidoreductase (complex I) across bacteria. mBio 6:e00389-15.
Su, C., Zhang, K.-Z., Cao, X.-Z., and Yang, J.-G. (2020). Effects of Saccharomycopsis fibuligera and Saccharomyces cerevisiae inoculation on small fermentation starters in Sichuan-style Xiaoqu liquor. Food Res. Int. 137:109425. doi: 10.1016/j.foodres.2020.109425
Suzuki, Y., Kido, J., Matsumoto, S., Shimizu, K., and Nakamura, K. (2019). Associations among amino acid, lipid, and glucose metabolic profiles in childhood obesity. BMC Pediatr. 19:273. doi: 10.1186/s12887-019-1647-8
Tamang, J. P., Watanabe, K., and Holzapfel, W. H. (2016). Review: diversity of microorganisms in global fermented foods and beverages. Front. Microbiol. 7:377. doi: 10.3389/fmicb.2016.00377
Terfehr, D., Dahlmann, T. A., and Kuck, U. (2017). Transcriptome analysis of the two unrelated fungal beta-lactam producers Acremonium chrysogenum and Penicillium chrysogenum: velvet-regulated genes are major targets during conventional strain improvement programs. BMC Genom. 18:272. doi: 10.1186/s12864-017-3663-0
Ueta, M., Wada, C., Daifuku, T., Sako, Y., Bessho, Y., Kitamura, A., et al. (2013). Conservation of two distinct types of 100S ribosome in bacteria. Genes Cells 18, 554–574. doi: 10.1111/gtc.12057
Volker, Z., Sari, K., Marko, K., Ilmo, H., and Moshe, F. (2000). The NADH oxidation domain of complex i: do bacterial and mitochondrial enzymes catalyze ferricyanide reduction similarly? BBA Bioenerget. 1459, 61–68.
Wang, Y., and Ho, C. T. (2009). Polyphenolic chemistry of tea and coffee: a century of progress. J. Agric. Food Chem. 57, 8109–8114. doi: 10.1021/jf804025c
Williams, K. P., Gillespie, J. J., Sobral, B. W., Nordberg, E. K., Snyder, E. E., Shallom, J. M., et al. (2010). Phylogeny of gammaproteobacteria. J. Bacteriol. 192, 2305–2314. doi: 10.1128/jb.01480-09
Wu, T., Guo, Y., Liu, R., Wang, K., and Zhang, M. (2016). Black tea polyphenols and polysaccharides improve body composition, increase fecal fatty acid, and regulate fat metabolism in high-fat diet-induced obese rats. Food Funct. 7, 2469–2478. doi: 10.1039/c6fo00401f
Xu, A., Wang, Y., Wen, J., Liu, P., Liu, Z., and Li, Z. (2011). Fungal community associated with fermentation and storage of Fuzhuan brick-tea. Int. J. Food Microbiol. 146, 14–22. doi: 10.1016/j.ijfoodmicro.2011.01.024
Yamashita, Y., Wang, L., Tinshun, Z., Nakamura, T., and Ashida, H. (2012). Fermented tea improves glucose intolerance in mice by enhancing translocation of glucose transporter 4 in skeletal muscle. J. Agric. Food Chem. 60, 11366–11371.
Yang, P., Liu, Z., Zhao, Y., Cheng, Y., Li, J., Ning, J., et al. (2020). Comparative study of vegetative and reproductive growth of different tea varieties response to different fluoride concentrations stress. Plant Physiol. Biochem. 154, 419–428.
Zhang, H., Che, S., Wang, R., Liu, R., Zhang, Q., and Bartlam, M. (2019). Structural characterization of an isopenicillin N synthase family oxygenase from Pseudomonas aeruginosa PAO1. Biochem. Biophys. Res. Commun. 514, 1031–1036.
Zhang, L., Zhang, Z.-Z., Zhou, Y.-B., Ling, T.-J., and Wan, X.-C. (2013). Chinese dark teas: postfermentation, chemistry and biological activities. Food Res. Int. 53, 600–607.
Zhao, M., Su, X. Q., Nian, B., Chen, L. J., Zhang, D. L., Duan, S. M., et al. (2019). Integrated meta-omics approaches to understand the microbiome of spontaneous fermentation of traditional Chinese pu-erh tea. mSystems 4:e00680-19.
Zhu, J. X., Yu, C., Zhou, H., Wei, X. L., and Wang, Y. F. (2021). Comparative evaluation for phytochemical composition and regulation of blood glucose, hepatic oxidative stress and insulin resistance in mice and HepG2 models of four typical Chinese dark teas. J. Sci. Food Agric. 15. doi: 10.1002/jsfa.11328
Zhu, Y., Lv, H. P., Shao, C. Y., Kang, S. Y., Zhang, Y., Guo, L., et al. (2018). Identification of key odorants responsible for chestnut-like aroma quality of green teas. Food Res. Int. 108, 74–82.
Keywords: Fuzhuan brick tea (FBT), microbial community, potential function, metabolic pathway, shotgun metagenomic sequencing
Citation: Wang X, Du G, Chen H, Zeng X, Liu B, Guo C, Sheng Q, Yuan Y and Yue T (2021) Comparative Metagenomics Reveals Microbial Communities and Their Associated Functions in Two Types of Fuzhuan Brick Tea. Front. Microbiol. 12:705681. doi: 10.3389/fmicb.2021.705681
Received: 06 May 2021; Accepted: 19 August 2021;
Published: 16 September 2021.
Edited by:
Jyoti Prakash Tamang, Sikkim University, IndiaReviewed by:
Dhananjaya Pratap Singh, National Bureau of Agriculturally Important Microorganisms (ICAR), IndiaSenthil Kumar Nachimuthu, Mizoram University, India
Mandira Kochar, The Energy and Resources Institute (TERI), India
Copyright © 2021 Wang, Du, Chen, Zeng, Liu, Guo, Sheng, Yuan and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yahong Yuan, eXloMzI0QDEyNi5jb20=; Tianli Yue, eXVldGwzMDVAbndzdWFmLmVkdS5jbg==