 Wolfgang Buckel
Wolfgang Buckel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 13 September 2021
Sec. Microbial Physiology and Metabolism
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.703525
This article is part of the Research TopicMicrobial BioenergeticsView all 14 articles
Anaerobic bacteria ferment carbohydrates and amino acids to obtain energy for growth. Due to the absence of oxygen and other inorganic electron acceptors, the substrate of a fermentation has to serve as electron donor as well as acceptor, which results in low free energies as compared to that of aerobic oxidations. Until about 10 years ago, anaerobes were thought to exclusively use substrate level phosphorylation (SLP), by which only part of the available energy could be conserved. Therefore, anaerobes were regarded as unproductive and inefficient energy conservers. The discovery of electrochemical Na+ gradients generated by biotin-dependent decarboxylations or by reduction of NAD+ with ferredoxin changed this view. Reduced ferredoxin is provided by oxidative decarboxylation of 2-oxoacids and the recently discovered flavin based electron bifurcation (FBEB). In this review, the two different fermentation pathways of glutamate to ammonia, CO2, acetate, butyrate and H2 via 3-methylaspartate or via 2-hydroxyglutarate by members of the Firmicutes are discussed as prototypical examples in which all processes characteristic for fermentations occur. Though the fermentations proceed on two entirely different pathways, the maximum theoretical amount of ATP is conserved in each pathway. The occurrence of the 3-methylaspartate pathway in clostridia from soil and the 2-hydroxyglutarate pathway in the human microbiome of the large intestine is traced back to the oxygen-sensitivity of the radical enzymes. The coenzyme B12-dependent glutamate mutase in the 3-methylaspartate pathway tolerates oxygen, whereas 2-hydroxyglutaryl-CoA dehydratase is extremely oxygen-sensitive and can only survive in the gut, where the combustion of butyrate produced by the microbiome consumes the oxygen and provides a strict anaerobic environment. Examples of coenzyme B12-dependent eliminases are given, which in the gut are replaced by simpler extremely oxygen sensitive glycyl radical enzymes.
Fermentation is a chemical reaction catalyzed by living cells with two different meanings. In biotechnology the bacterial production of a certain compound mainly from glucose under aerobic conditions is called fermentation, e.g., L-lysine fermentation. In this review, however, fermentation is defined as an anaerobic bacterial redox process of an organic substrate leading to different products, e.g., fermentation of glutamate to ammonia, CO2, acetate, butyrate and hydrogen. Thereby glutamate is oxidized to CO2 and acetate; the resulting reducing equivalents are used to synthesize butyrate and H2 to establish a balanced chemical equation. Inorganic electron acceptors such as nitrate, sulfate or Fe(III) are not involved; otherwise the process would be called respiration rather than fermentation. Thermodynamically, a fermentation is an exergonic process with a free energy of ΔG°′ <−20 kJ/mol. ΔG° is defined for 1 M substrates and products, 105 Pa for gasses and at a temperature of 298 K. Under biological conditions, in dilute aqueous solutions (55.5 M H2O) and at pH 7.0, ΔG°′ is used. In this review, the ΔG°′ values have been calculated from the data given by Thauer et al. (1977).
A well-established fermentation is the conversion of glucose by lactic acid bacteria to lactate at pH 7.0 with ΔG°′ = −185 kJ/mol (eq. 1). In contrast, the aerobic oxidation of glucose in mammalian cells with O2 to CO2 and H2O yields ΔG°′ = −2,872 kJ/mol (eq. 2).
Glucose oxidation via glycolysis, pyruvate dehydrogenase, Krebs’ (citric acid) cycle and mitochondrial respiratory chain are very well analyzed processes leading to about 38 mol ATP/mol glucose; (−2,872 kJ/mol glucose): (38 mol ATP/mol glucose) = −76 kJ/mol ATP. Hence in a catabolic reaction with one or more irreversible steps, a free energy change of −76 kJ is required to generate 1 mol ATP, whereas under equilibrium conditions only about −50 kJ are necessary (Thauer et al., 1977; Buckel and Thauer, 2013). If this value is applied to the fermentation of glucose to 2 lactate, the ATP yield should be (−185 kJ/mol): (−76 kJ/mol ATP) = 2.4 ATP, which is 20% higher than 2.0 ATP via substrate level phosphorylation in glycolysis. This examples tells why energy conservation in fermentative anaerobic bacteria has been considered as inefficient. The discovery that anaerobic bacteria are able use in addition electrochemical ion gradients for ATP synthesis, catalyzed by ubiquitous H+ or Na+ dependent F1F0-ATP-synthases (von Ballmoos et al., 2009), changed this view. Work by Konings and colleagues showed that in the glucose fermenting Streptococcus cremoris, the export of a formed lactic acid molecule is accompanied by a proton, which establishes an electrochemical proton gradient (Otto et al., 1980). Since 2 lactic acids are produced from glucose, two additional protons are exported which can give rise to 0.5 ATP catalyzed by ATP synthase. Thus the involvement of electrochemical ion gradients allows bacteria to use energy increments that are smaller than that of 1 ATP, which makes anaerobes even more efficient than aerobes. Therefore, the amount of energy to synthesize 1 ATP in anaerobes is now considered as about −66 kJ rather than −76 kJ in aerobes; see section “Fermentation of Glutamate: 2-Hydroxyglutarate or 3-Methylaspartate Pathway?”
In this review, I will show that also anaerobes have enzymes which are able to generate a Na+/H+ motive force, the biotin-containing decarboxylases (see section “Biotin-Containing Decarboxylases as Sodium Ion Pumps”) and the NAD:ferredoxin oxidoreductase (Rnf, see section “The Na+/H+ Pump Ferredoxin:NAD Oxidoreductase Also Called Rnf”). Sections “Flavin Based Electron Bifurcation” and “On the Mechanism of Electron Bifurcation Catalyzed by EtfAB-Bcd” will give an introduction to flavin based electron bifurcation, by which “energy rich” reduced ferredoxin and flavodoxin are obtained. In section “Fermentation of Glutamate: 2-Hydroxyglutarate or 3-Methylaspartate Pathway?” the 2-hydroxyglutarate and the 3-methylaspartate pathway of the fermentation of glutamate are introduced, which apply the biotin-containing decarboxylases and Rnf together with substrate level phosphorylation (SLP) to obtain the maximum theoretical yield of ATP. Section “Oxygen-Tolerant and -Intolerant Radical Enzymes” tackles the question why nature evolved two different pathways of glutamate fermentation. Finally, section “Butyrate Provides Anaerobiosis in the Gut” shows how butyrate converts the human gut to an extremely oxygen-free environment and an ideal environment for strict anaerobic bacteria.
Concomitant with the discovery of Konings, bacterial Na+ pumps were detected and isolated, driven by decarboxylation (Buckel, 2001a). The first decarboxylase of this type was detected by Dimroth (1980) in Klebsiella pneumoniae and characterized in the following years as biotin containing integral membrane enzyme catalyzing the decarboxylation of oxaloacetate to pyruvate, ΔG°′ ≈ −30 kJ/mol coupled to the translocation of 2 Na+. This breakthrough was followed by methylmalonyl-CoA decarboxylase from Veillonella aerogenes (Hilpert and Dimroth, 1982) and glutaconyl-CoA decarboxylases from Acidaminococcus fermentans, Peptostreptococcus asaccharolyticus (formerly called Peptococcus aerogenes; Ezaki et al., 1983) and Clostridium symbiosum (Buckel and Semmler, 1982, 1983). These enzymes share a common two step mechanism: the carboxyl group of the substrate is transferred to enzyme bound biotin. Addition of a proton to the carbonyl group of the formed N-carboxybiotin causes decarboxylation, which drives the translocation of Na+ ions from the cytoplasm through the membrane to the outside of the bacterium. These biotin containing decarboxylases are composed of 3–5 subunits. The largest and hydrophilic α-subunit catalyzes the carboxy transfer from substrate to biotin and the extremely hydrophobic β-subunit is responsible for the decarboxylation of carboxybiotin and Na+ transport. Biotin is covalently attached to a conserved lysine residue of the γ-subunit and the small δ-subunit connects the α-subunit in the cytoplasm with the β-subunit in the membrane (Vitt et al., 2020). Variations are observed with the γ-subunit. In oxaloacetate decarboxylase from K. aerogenes, a small domain of the α-subunit serves as γ-subunit (Schwarz et al., 1988; Xu et al., 2020), whereas glutaconyl-CoA decarboxylase from C. symbiosum contains two slightly different γ-subunits (Kress et al., 2009). The amount of Na+ ions transported by these pumps could be determined for oxaloacetate decarboxylase from K. pneumoniae as up to 2 Na+ per decarboxylation (Dimroth and Thomer, 1993). This is supported by the recent cryo-EM structure (4.5 Å resolution) of the trimeric βγ-subunit complex of oxaloacetate decarboxylase from Salmonella typhimurium, where two Na+ binding sites in one β-subunit were detected. Unfortunately, the coupling between decarboxylation of carboxybiotin and Na+-transport could not be elucidated (Xu et al., 2020).
Oxaloacetate decarboxylase from K. pneumoniae is involved in the fermentation of citrate to 2 acetates, 1.2 CO2 and 0.5 formate. The pathway involves citrate cleavage to acetate and oxaloacetate, decarboxylation of the latter to CO2, pyruvate and ΔμNa+, cleavage of pyruvate with CoA to acetyl-CoA and formate, substrate level phosphorylation (SLP) with acetyl-CoA to acetate, CoA and ATP. Generation of ΔμNa+ is used for citrate transport and for driving the endergonic reduction of NAD+ for anabolic purposes by formate via ubiquinol (Pfenninger-Li and Dimroth, 1992). A balanced stoichiometry of citrate fermentation would give ΔG°′ = −78.7 kJ/mol, which is the amount of energy required for the synthesis of about 1.2 ATP (eq. 3).
Propionigenium modestum thrives from an apparently very simple chemical reaction, the decarboxylation of succinate to propionate (eq. 4) (Hilpert et al., 1984). Propionate CoA-transferase converts succinate to succinyl-CoA, which is rearranged to (R)-methylmalonyl-CoA in a coenzyme B12-dependent manner. Epimerization affords (S)-methylmalonyl-CoA, which is decarboxylated to propionyl-CoA, whereby ΔμNa+ is generated (Hilpert and Dimroth, 1982). At the low succinate concentrations in the Canale Grande in Venice, Italy (ca. 1 mM), from which the organism was isolated (Schink, 1982), the free energy is sufficient for about ½ ATP (eq. 4). ΔμNa+ directly drives the Na+-dependent F1Fo-ATP synthase (Laubinger and Dimroth, 1988).
The genes coding for Rnf have been known from Rhodobacter capsulatus, where it is involved in nitrogen fixation and named Rnf (for Rhodobacter nitrogen fixation) (Schmehl et al., 1993). Similar genes were detected in the genome of C. tetani, a close relative of C. tetanomorphum, whose membranes catalyzed the oxidation of NADH with ferricyanide [hexacyanoferrate(III)] (Brüggemann et al., 2003; Imkamp et al., 2007). The enzyme complex in membranes from C. tetanomorphum, which contained the highest activity, could be solubilized with dodecylmaltoside and purified (Boiangiu et al., 2005; Jayamani, 2008). SDS-PAGE indicated that the complex was composed of six different subunits RnfCDGEAB (Figure 1), RnfCDGE are related to four subunits of the sodium pumping NADH:quinone oxidoreductase (Nqr) from Vibrio cholerae (Steuber et al., 2014a,b). The yellow-brownish native enzyme complex contained covalently bound flavin, non-covalently bound FMN, riboflavin and 23 ± 1 mol Fe per mol protein, most likely in six iron-sulfur clusters (Boiangiu et al., 2005).
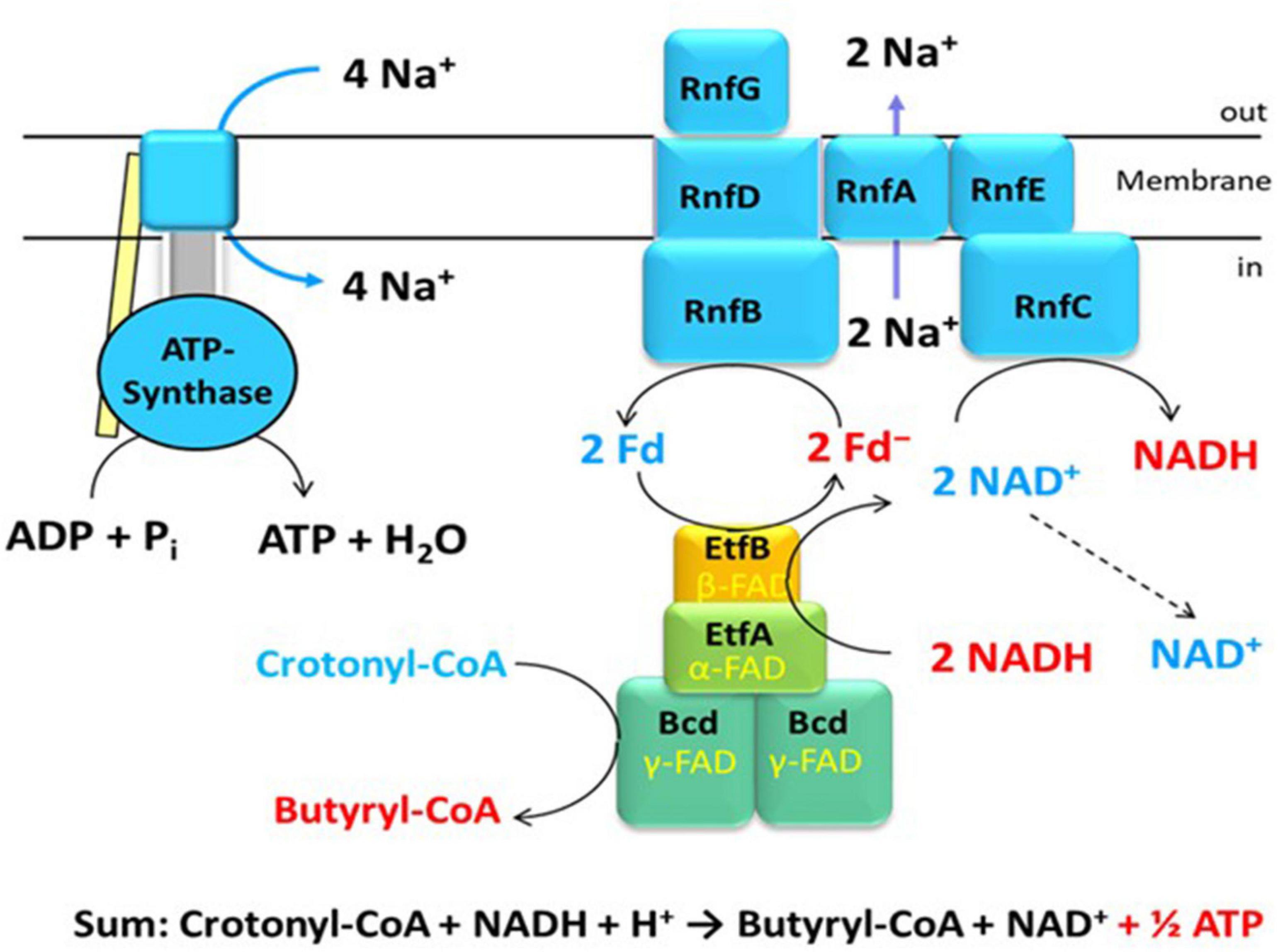
Figure 1. An example of energy conservation in butyrate producing anaerobic bacteria via the generation of an ion motif force. The bifurcating EtfAB-Bcd complex reduces 2 Fd with NADH driven by the exergonic reduction of crotonyl-CoA to butyryl-CoA with a second NADH. The Rnf complex in the membrane produces 2 ΔμNa+ from the exergonic reduction of NAD+ by 2 Fd–. The ATPase generates 1 ATP from 4 ΔμNa+.
The oxidation of NADH with ferricyanide catalyzed by Rnf in membrane vesicles from C. tetanomorphum showed no Na+-dependence; probably the electrons did not pass through all six subunits due to a short cut. Therefore, the reaction was performed in reverse. The reduction of NAD+ was measured in an assay, in which 1 mM Ti(III)citrate was used to keep ferredoxin in the reduced state. Controls showed that Ti(III)citrate reduced NAD+ without ferredoxin and membranes at pH ≥ 7.0. But at pH 6.8 again no Na+-dependence could be observed (Imkamp et al., 2007; Jayamani, 2008). The first Na+-transport with an Rnf was demonstrated with membrane vesicles from Acetobacterium woodii, NAD+, Ti(III)citrate and 22Na+ by Biegel and Müller (2010). The electrogenic transport was stimulated by valinomycin in the presence of K+ and inhibited by 100 μM Na+-ionophore N,N,N’,N’-tetracyclohexyl-1,2-phenylendioxydiacetamide (ETH 2120). Later Ti(III)citrate was replaced by purified carbon monoxide dehydrogenase/acetyl-CoA synthase and CO as reductant for ferredoxin, which resulted in much more reliable dependencies on Na+ (Hess et al., 2013). In a similar manner reduced ferredoxin and flavodoxin quinol were generated with the electron bifurcating electron transfer flavoprotein (EtfAB) and butyryl-CoA dehydrogenase (Bcd) both from A. fermentans (see also next chapter, eq. 5). With membranes from A. fermentans it was shown that Rnf activity was Na+-dependent with apparent Km = 120 ± 20 μM Na+ and Km = 280 ± 50 μM Li+ at pH 6.8; in addition, Km = 2.0 ± 0.4 μM flavodoxin and 1.4 ± 0.1 μM ferredoxin were measured (Chowdhury et al., 2016). The values for Rnf from A. woodii were: Km = 201 ± 30 μM Na+ at pH 6 and Km = 155 ± 39 μM Na+ at pH 7.7 (Hess et al., 2013). Surprisingly, the Km values for Na+ and Li+ are much lower than those of the biotin-containing Na+ pumps glutaconyl-CoA decarboxylase from A. fermentans (Buckel and Semmler, 1982) or oxaloacetate decarboxylase from K. pneumoniae (Dimroth and Thomer, 1986), which exhibit apparent Km values of 1.0–1.5 mM Na+ and 25–100 mM Li+. Recently a survey showed that not all anaerobic bacteria contain Rnfs and not all Rnfs are Na+-dependent. Thus the Rnf activity is absent in R. capsulatus and Escherichia coli, but present also in Bacteroides fragilis, Clostridium ljungdahlii, V. cholerae, and Clostridium kluyveri. Besides C. tetanomorphum, A. fermentans, and A. woodii, a Na+-dependence could be only detected in Rnf from B. fragilis (Hess et al., 2016).
Ferredoxin-dependent electron bifurcation represents an important additional process to generate the energy rich molecule. Only a few enzymes, such as pyruvate ferredoxin: oxidoreductases (PFOR), reduce ferredoxin, the substrate of Rnf. A search for a reductant other than PFOR, took the butyryl-CoA dehydrogenases (Bcd) into consideration, because the difference between the reduction potentials of NAD/NADH, E0’ = −320 mV, and crotonyl-CoA/butyryl-CoA, E0’ = −10 mV, amounts to +310 mV, equal to ΔG°′ = −n × F × ΔEo’ = −2 × 96.5 × 0.310 Volt = −59.8 kJ mol–1. Probably Bcd also reduces ferredoxin or pumps Na+ driven by this large ΔG°’. The hypothesis of a membrane bound butyryl-CoA dehydrogenase (Bcd) could be refuted by the already known isolation of a soluble green homotetrameric Bcd from A. fermentans which could be separated from the yellow heterodimeric electron transferring flavoprotein (EtfAB) (Buckel, 1990). A very similar green soluble Bcd has been known from Megasphaera elsdenii (formerly called Peptostreptococcus elsdenii) (Engel and Massey, 1971), which like A. fermentans belongs to the Negativicutes, a class with a Gram-negative cell wall of the, predominantly, Gram-positive phylum Firmicutes (Latin: cutis firma = strong outer skin) (Marchandin et al., 2010). In contrast, butyryl-CoA dehydrogenase from the Gram-positive C. tetanomorphum forms a tight complex with the heterodimeric EtfAB (EtfAB-Bcd). But immunogold labeling and electron microscopy clearly showed that this EtfAB-Bcd complex was also not associated with the membrane (Herrmann, 2008). Nevertheless, a hypothesis arose from these negative results that the reduction of crotonyl-CoA with NADH catalyzed by Bcd from C. tetanomorphum was somehow involved in the reduction of ferredoxin (Eo’ = −420 mV). Furthermore, since 1969 it was known that cell extracts of Clostridium kluyveri catalyzed an acetyl-CoA dependent reduction of ferredoxin with NADH (Thauer et al., 1969). Since the cell extract contained all enzymes for the synthesis of butyrate (Seedorf et al., 2008) as did C. tetanomorphum cells, it was ferredoxin that indeed should be reduced by NADH. Similar to complex III of the mitochondrial respiratory chain, it was postulated that NADH reduced EtfAB, which bifurcated one electron exergonically to δ-FAD of Bcd and the other endergonically to ferredoxin (eq. 5) (Herrmann et al., 2008). To reduce crotonyl-CoA with two electrons, repetition of the reaction is required:
This hypothesis was verified first with the purified Etf-Bcd complex from C. kluyveri (Li et al., 2008). The catalytic amounts of purified ferredoxin from Clostridium pasteurianum were regenerated with purified [FeFe]hydrogenase also from C. pasteurianum. If one of the components of this assay (NADH, crotonyl-CoA, Fd, hydrogenase or EtfAB-Bcd) was omitted, no reaction could be observed. Hence the electron bifurcation system is tightly coupled. The purified Etf-Bcd complex from C. tetanomorphum catalyzed exactly the same reaction as in eq. 5, as did the combination of Bcd and EtfAB from A. fermentans or M. elsdenii (Chowdhury et al., 2014, 2015).
To study the mechanism of electron bifurcation, EtfAB-Bcd was chosen, because it contains no iron-sulfur cluster and in the resting state, it is state stable under air; for other bifurcating systems see: Buckel and Thauer (2018a, b) and Kayastha et al. (2021). EtfAB-Bcd harbors three differently bound FAD molecules, α- and β-FAD in EtfAB, and δ-FAD, one in each subunit of the tetrameric Bcd (Figure 1). As source for EtfABs, A. fermentans (Chowdhury et al., 2014) and M. elsdenii (Chowdhury et al., 2015; Vigil et al., 2021) are the most suitable organisms, because their EtfABs form no tight complexes with their Bcds and can be studied separately. The crystal structure EtfAB from A. fermentans (Af-EtfAB) revealed its composition of 3 domains, similar to those of Etfs involved in fatty acid and anaerobic toluene oxidations (Figure 2, Bifurcation-like state) (Chowdhury et al., 2014; Vogt et al., 2019). Domains I and II form subunit A and domain III subunit B. α-FAD is located in domain II (subunit A) with its isoalloxazine ring at the interface to domain III (subunit B). β-FAD sits on domain III with its isoalloxazine ring at the interface between domains III and I. β-FAD is only present in bifurcating Etfs, whereas in non-bifurcating Etfs, e.g., those acting as electron acceptors of acyl-CoA dehydrogenases, AMP alone occupies the place of the AMP part of β-FAD; the place for the isoalloxazine part is empty (Roberts et al., 1996). In Af-EtfAB, the distance between α- and β-FAD is 18 Å; the possible rotation of α-FAD on the flexible domain II toward β-FAD by about 10° shortens the distance between the two FADs to 14 Å, the longest distance of fast electron tunneling (Moser et al., 1992). NADH is located close to β-FAD. Since the already known structure of Bcd from M. elsdenii did not tell anything about the interaction with Etf (Djordjevic et al., 1995), the structure of the tight EtfAB-Bcd complex from Clostridium difficile was solved (Figure 2, Dehydrogenase state) (Demmer et al., 2017). The structure revealed the homotetrameric Bcd with one EtfAB at each subunit, (EtfAB)4(Bcd)4. Furthermore, α-FAD on the flexible domain II has rotated by about 80° in the opposite direction toward δ-FAD of Bcd, until 8 Å distance. Thus the two structures represent two states of the bifurcating system. The structure of EtfAB alone is close to the bifurcating state with a distance between α-FAD and β-FAD of 18 Å. In complex with Bcd, α-FAD swings into the dehydratase D-state, which brings it 34 Å apart from β-FAD (Figure 2).
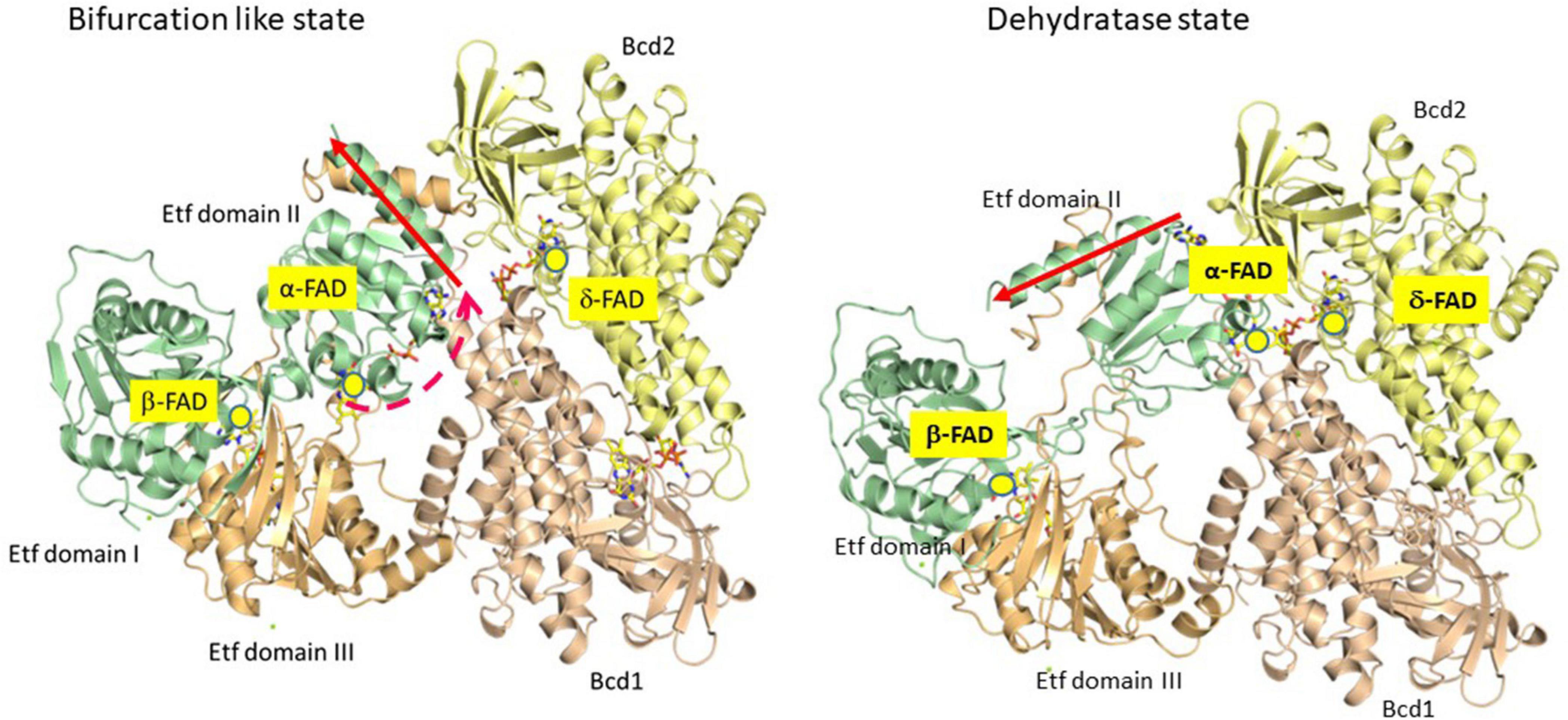
Figure 2. Partial crystal structures of the recombinant Etf-Bcd complex from C. difficile produced in E. coli; EtfAB and 2 subunits of the tetrameric Bcd are displayed. The dehydratase state shows the structure as solved. In the bifurcation state domain II of Etf rotated CW by 80° as found in Etf from A. fermentans (see red arrow); Etf, domains I + II (subunit A) in green, domain III (subunit B) in light brown, Bcd1 in yellow and Bcd2 in pinkish-brown. In the dehydratase state, α-FAD and δ-FAD are located close together (8 Å distance), ready for ET. In the bifurcation-like state, α-FAD and β-FAD are 18 Å apart; further rotation by 10° CW would bring them closer together by 4 Å, ready for electron bifurcation (taken from Buckel and Thauer, 2018a).
Potentiometric titrations with dithionite under anaerobic conditions followed by UV/vis spectroscopy revealed the reduction potentials E0’ for EtfAB, α-FAD/α-FAD•– = +134 mV, α-FAD•–/α-FADH– = −36 mV, β-FAD/β-FADH•– = −271 mV; for Bcd, δ-FAD/δ-FAD•– = −42 mV, δ-FAD–/δ-FADH– = −64 mV (Figure 3; Sucharitakul et al., 2021b). The one-electron reduction potentials of β-FAD could not be measured by the applied method, because the half-life of the semiquinone is expected to be extremely short, T1/2 ca. 10 ps (Lubner et al., 2017). In analogy to the long-known Q-cycle in the mitochondrial complex III, a reactive β-FAD•– with an extremely low stability constant (log Ks<<0) is necessary for electron bifurcation (eq. 6) (Bergdoll et al., 2016). Therefore, the β-FAD/β-FAD•– reduction potential must be much more negative than that of β-FAD•–/β-FADH–, whereas in flavoproteins with stable semiquinones the opposite is the case (log Ks > 0), e.g., flavodoxin from A. fermentans with reduction potentials for FAD/FAD– = −60 mV are much higher than that for FAD–/FADH– = −420 mV (Hans et al., 2002).
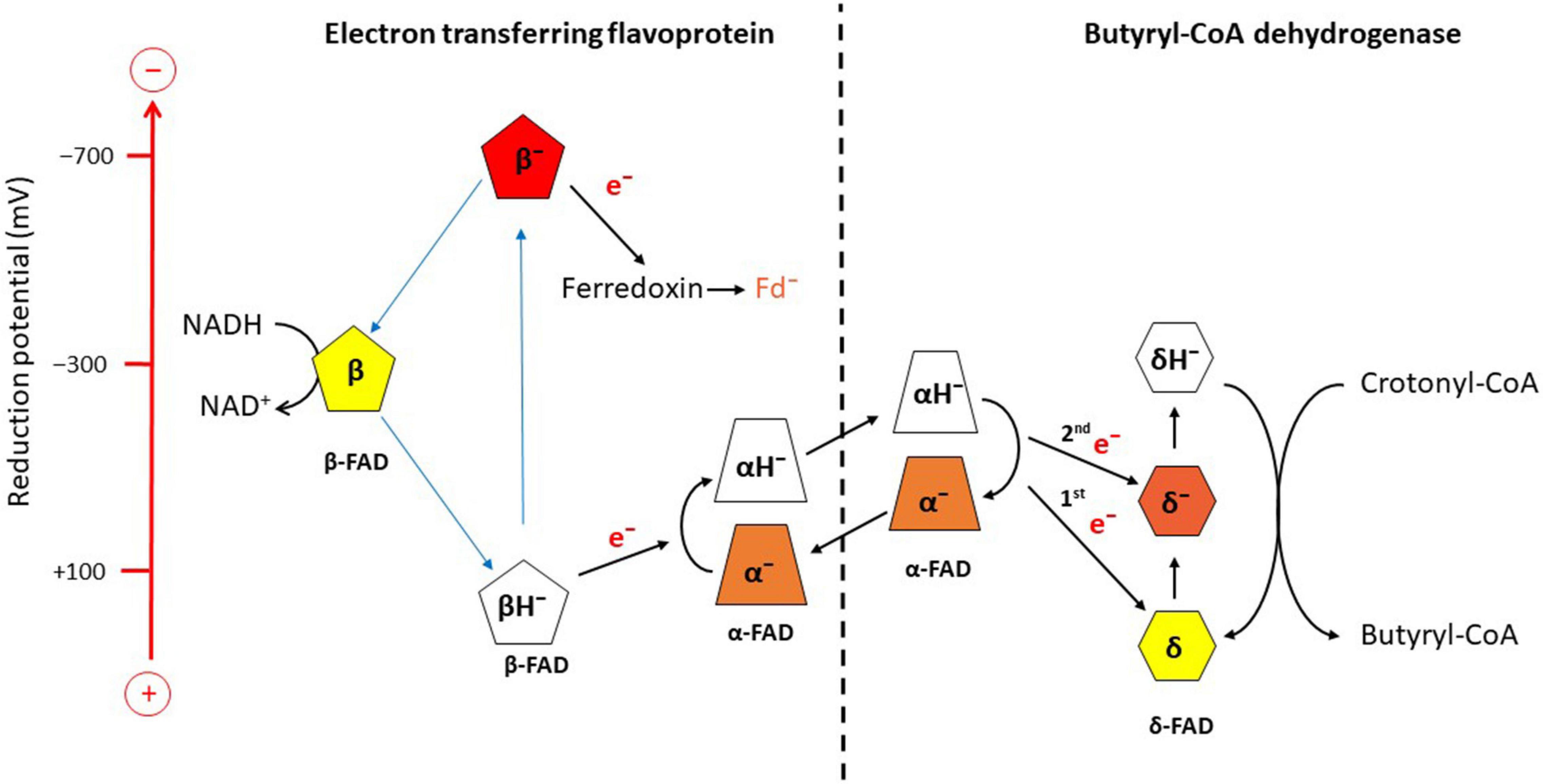
Figure 3. Scheme of electron bifurcation in the EtfAB-Bcd complex. The different FADs are placed according to their approximate reduction potential. NADH reduces β-FAD to β-FADH–, which bifurcates. One electron goes to α-FAD– and the formed semiquinone, β-FAD•−, reduces ferredoxin (Fd) to Fd–. The formed hydroquinone (α-FADH–) swings over to Bcd and transfers one electron to δ-FAD. Note the decrase of the reduction potential of α-FAD•− due to the change in location from Etf to Bcd. Repetition of this process yields δ-FADH–, which reduces crotonyl-CoA to butyryl-CoA. The FAD semiquinones are red, the quinones yellow and the hydroquinones colorless (Sucharitakul et al., 2021a,b).
To convert Ks to the one-electron reduction potentials of the bifurcating cofactor with a reduction potential of E0’, the relationship of eq. 7 is used. A graphical plot of E’ versus log Ks gives two straight lines which cross at E’ = Eo’ and log Ks = 0. At log Ks > 0, the potentials are normal and at log Ks < 0 the potentials are inverse or called “crossed” (Nitschke and Russell, 2012).
Two values of Ks were determined experimentally, 10–14 to 10–15 with the cytochrome bc1 complex III (Zhang et al., 2007) and 10–21 with the bifurcating archaeal enzyme NAD+ ferredoxin-NADP reductase (Nfn), which reversibly bifurcates the electrons from NADPH to ferredoxin and NAD+ (eq. 8) (Lubner et al., 2017).
Besides from the Archaeon Pyrococcus furiosus (Lubner et al., 2017), Nfn has also been characterized from the bacteria C. kluyveri (Wang et al., 2010), Thermotoga maritima (Demmer et al., 2015) and Sporomusa ovata (Kremp et al., 2020). The enzyme acts as transhydrogenase and in the reverse direction ensures with the “energy rich” Fd– that the NAD+/NADPH ratio is kept low at E’ = −380 mV and the NAD+/NADH ratio high at E’ = −280 mV. Hence in vivo NADPH is a better reductant and NAD+ a better oxidant.
According to Eq. 7, the one-electron reduction potentials of β-FADH– are calculated with an assumed log Ks = −15.2 to ± 0.0295 × (−15.2) + (−0.271) = ± 0.448−0.271 = +0.177 and −0.719 Volt (Figure 3; Buckel and Thauer, 2018a). The electron with the higher potential goes endergonically from +177 mV to α-FAD– (−36 mV) and the lower potential electron goes exergonically from −719 mV to ferredoxin (−390 mV) sitting about 6 Å apart from β-FAD. Thus the two one-electron transfers are coupled; the first electron moves uphill only if the second electron falls downhill to ferredoxin and vice versa (Figure 3). Now the formed α-FADH– rotates by 90° toward Bcd and transfers one electron to δ-FAD of Bcd and the regenerated α-FAD– returns to β-FAD. Surprisingly, in the presence of Bcd, the reduction potential of the formed α-FADH– changes to close to the two-electron reduction potential of α-FAD/α-FADH– = −228 mV, which enables much better electron transfer to the potential of δ-FAD/δ-FAD– = +162 mV. The next electron bifurcation transfers one electron via α-FADH– to δ-FAD– (−63 mV) and the formed δ-FADH– changes to the two-electron reduction potential of −100 mV, which easily reduces crotonyl-CoA to butyryl-CoA (−10 mV; Figure 3).
Probably the reader wonders why during electron bifurcation, α-FAD•– rather than α-FAD acts as high potential acceptor. There are three reasons, firstly the reduction potential of α-FAD with +134 mV is too high, whereas that of α-FAD•– is more in the right range of −36 mV. In electron bifurcation, the potential difference between β-FADH– and α-FAD•– with + 179−(−36) = +215 mV should be similar to the negative difference between the potentials of β-FADH– and Fd with − 719−(−390) = −329 mV, to ensure tight coupling. With α-FAD as acceptor, the difference of + 179−(+134) = +45 mV would be much too low. Secondly, the change of the one-electron reduction potential of α-FADH– to the two-electron reduction potential cannot occur at the state of α-FAD•–. Thirdly, as soon as EtfAB encounters NADH, either in vitro or in vivo, α-FAD is reduced to the semiquinone (Sucharitakul et al., 2021a). Finally, stopped flow measurements by Jeerus Sucharitakul directly showed that Etf with α-FADH– indeed transferred one electron to Bcd, whereas with α-FAD•– no electron transfer was observed (Sucharitakul et al., 2021b, unpublished).
The previous chapters described enzymatic systems which are able to establish electrochemical Na+ or H+ gradients for energy conservation in anaerobic bacteria. In addition, electron bifurcation as a major source of reduced ferredoxin has been presented. This chapter shows how the two pathways of glutamate fermentation apply these systems to obtain the theoretical yield of ATP/glutamate (eq. 9). −314 kJ/5 glutamate: −66 kJ/ATP = 4.8 ATP/5 glutamate; 0.96 ATP/glutamate.
In the 2-hydroxyglutarate pathway (Figure 4), which has been found in A. fermentans, C. symbiosum, P. asaccharolyticus, and F. nucleatum (Buckel and Barker, 1974), NAD+ oxidizes glutamate to 2-oxoglutarate and ammonia (Hornby and Engel, 1984). The formed NADH reduces 2-oxoglutarate to (R)-2-hydroxyglutarate (Yu et al., 2012), which is converted to the thioester by acetyl-CoA (Buckel et al., 1981) and dehydrated to glutaconyl-CoA; for a review see Buckel (2019). Glutaconyl-CoA is the substrate of the third group of biotin-containing and Na+-dependent decarboxylases (Buckel and Semmler, 1983). Glutaconyl-CoA is a vinylogous malonyl-CoA, in which the δ-carboxylate is equally activated as the β-carboxylates in malonyl-CoA and methylmalonyl-CoA. Decarboxylation of 5 glutaconyl-CoA yields 5 × 2 ΔμNa+ and 5 crotonyl-CoA (2-butenoyl-CoA), three of which disproportionate oxidatively to 6 acetyl-CoA and 3 NADH (Figure 5). Four NADH reduce 2 crotonyl-CoA to 2 butyryl-CoA and 4 ferredoxin (Fd) to 4 Fd– via electron bifurcation. Two Fd– give rise to H2 catalyzed by a [FeFe]hydrogenase and the other two Fd– to 2 ΔμNa+ + 1 NADH via Rnf. In summary, the 5 ΔμNa+ used for the transport of 5 glutamates into the cell have to be considered (Brüggemann et al., 2003). The net conserved energy amounts to 5 × 2 + 2 – 5 = 7 ΔμNa+ and 2 butyryl-CoA + 6 acetyl-CoA, 5 of which are required for the activation of 5 × 2-hydroxyglutarates. The remaining 3 thioesters are equivalent to 3 ATP by SLP (Table 1). If 4 ΔμNa+ give 1 ATP, then the yield is 7/4 + 3 = 4.75 (≈ 4.8) ATP/5 glutamate, as expected.
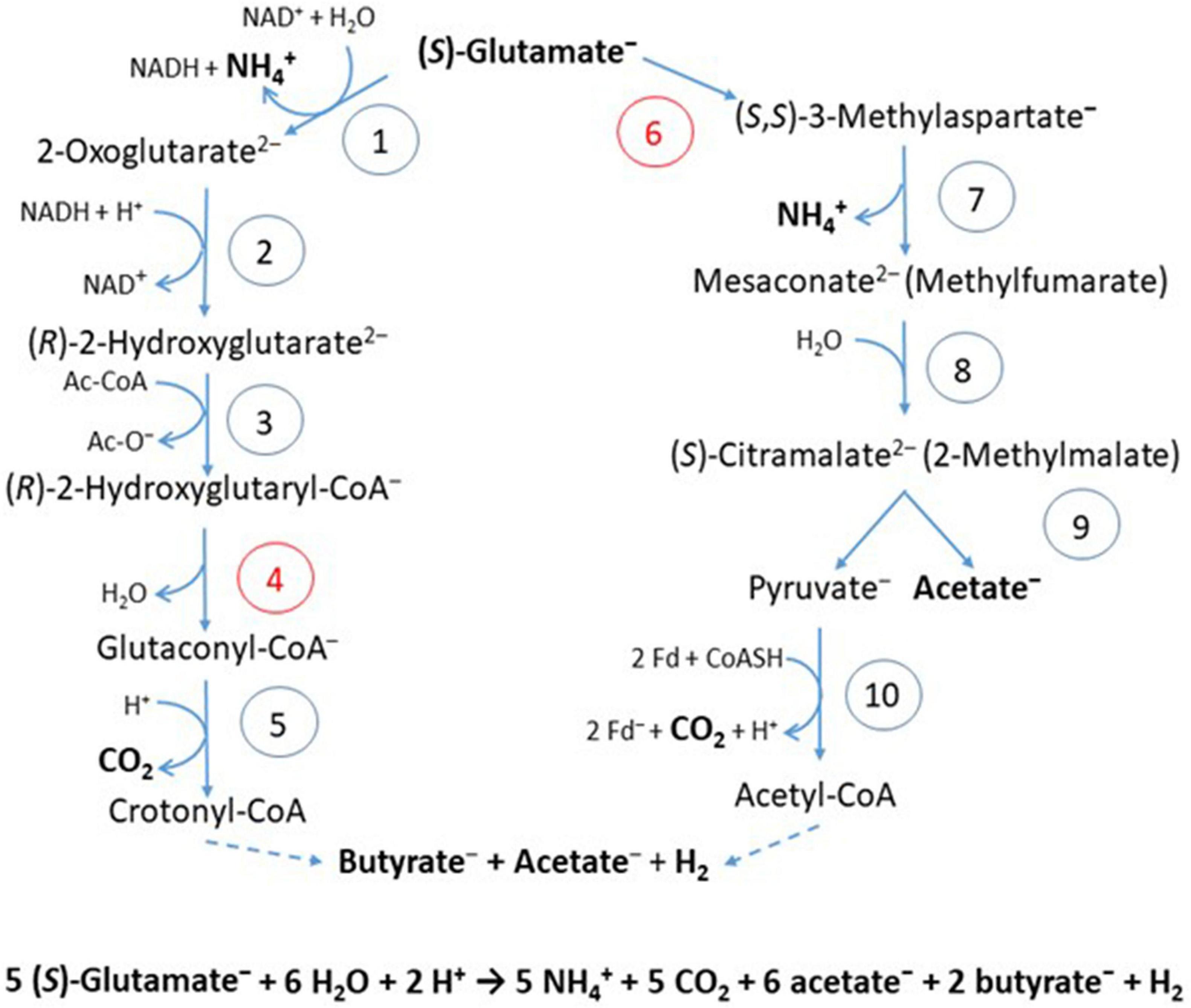
Figure 4. Fermentation of glutamate via 2-hydroxyglutarate or via 3-methylaspartate. The numbers in the circles represent the corresponding enzymes; red numbers denote radical enzymes: 1, (S)-glutamate dehydrogenase; 2, (R)-2-hydroxyglutarate dehydrogenase; 3, glutaconate CoA-transferase; 4, (R)-2-hydroxyglutaryl-CoA dehydratase; 5, glutaconyl-CoA decarboxylase, Na+-pumping; 6, glutamate mutase, coenzyme B12-dependent; 7, methylaspartase; 8, mesaconase; 9, (S)-citramalate lyase; 10, pyruvate:ferredoxin oxidoreductase (PFOR). Ac-CoA, acetyl or glutaconyl-CoA; Fd, ferredoxin; Fd–, reduced ferredoxin. The formation of butyrate and acetate are shown in Figure 5. Hydrogen, H2 is formed from 2 H+ and 2 Fd–, catalyzed by a [FeFe]-hydrogenase.
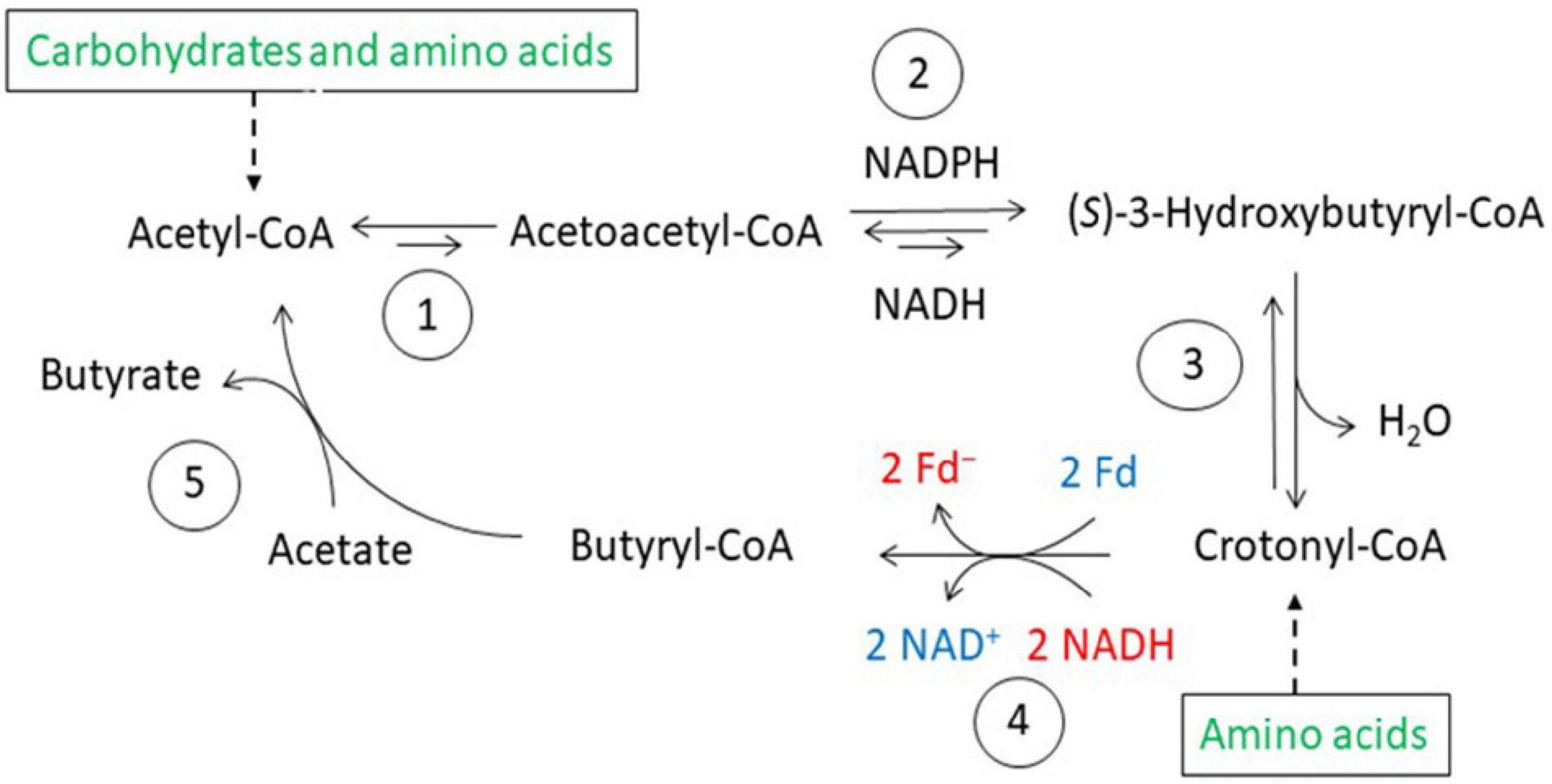
Figure 5. Butyrate synthesis in anaerobic bacteria. 1, Thiolase; 2, (S)-3-hydroxybutyryl-CoA dehydrogenases, NADH and NADPH specific; 3, (S)-3-hydroxybutyryl-CoA dehydratase; 4, electron bifurcating EtfAB-butyryl-CoA dehydrogease complex; 5, butyrate CoA-transferase.
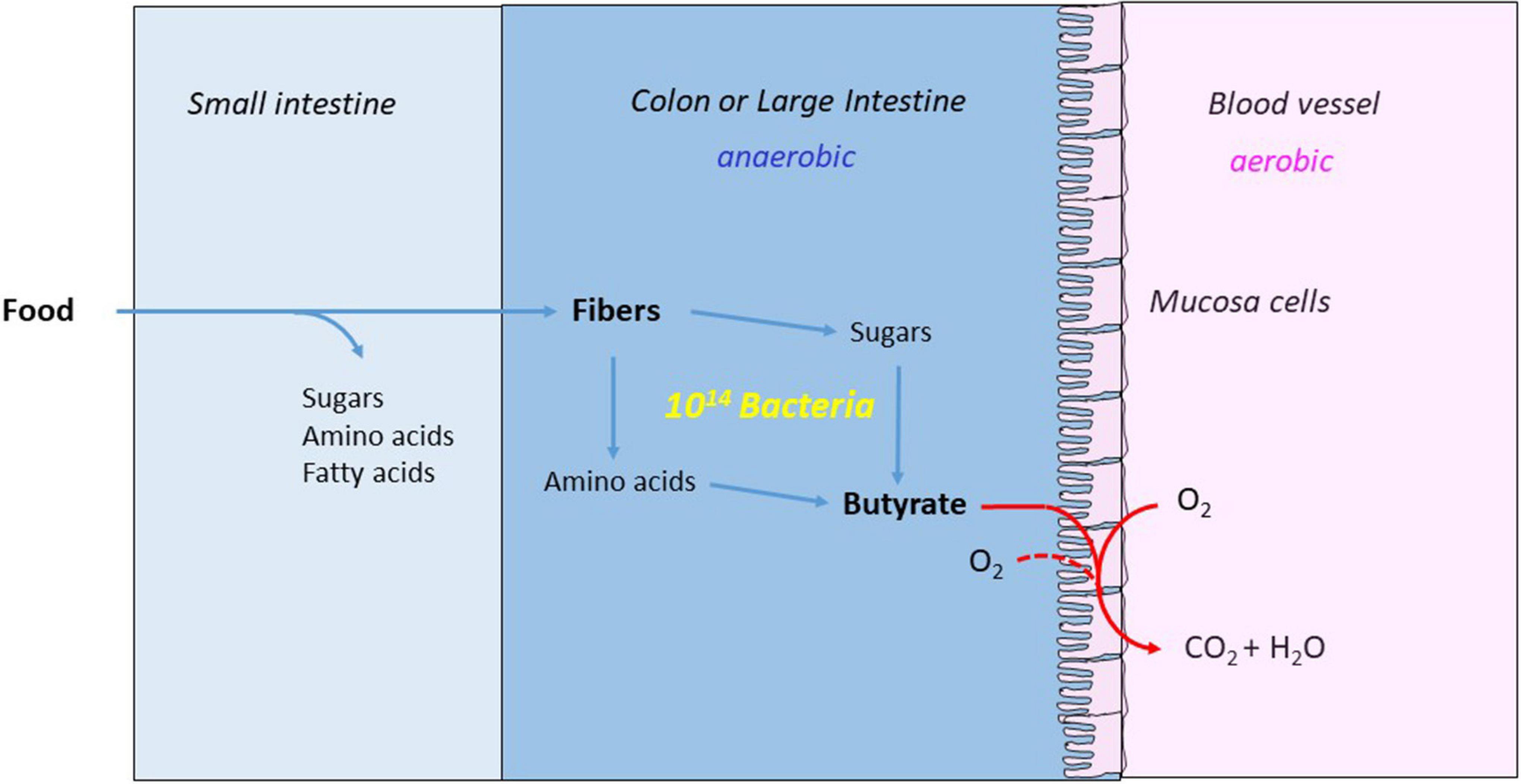
Figure 6. Butyrate nourishes the mucosa cells and makes the colon anaerobic. The major part of the food is digested and absorbed in the small intestine. The remaining fibers are hydrolyzed to monosaccharides and amino acids by the anaerobic bacteria and fermented the large intestine or colon to acetate, propionate and butyrate. The mucosa cells combust the produced butyrate with oxygen from blood and colon, whereby the colon becomes strictly anaerobic.
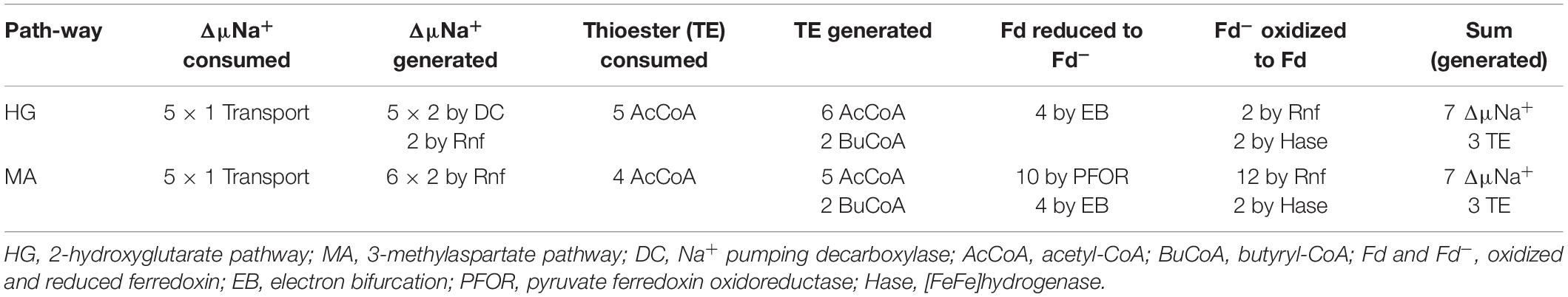
Table 1. Energy conservation in the two pathways of glutamate fermentation.
Several clostridia, Clostridium tetanomorphum, C. cochlearium, and C. tetani, use a completely different pathway of glutamate fermentation, but with the same stoichiometry as that via 2-hydroxyglutarate (eq. 9) (Buckel and Barker, 1974). This 3-methylaspartate pathway is initiated by the coenzyme B12-dependent carbon skeleton rearrangement of (S)-glutamate to (2S,3S)-3-methylaspartate (Barker et al., 1964), from which ammonia is readily eliminated affording mesaconate (methylfumarate) (Barker et al., 1959). Addition of water yields (S)-citramalate (2-methylmalate) (Blair and Barker, 1966), which is cleaved to acetate and pyruvate (Buckel and Bobi, 1976). Oxidation of 5 pyruvate to 5 acetyl-CoA, 5 CO2 and 10 Fd– is catalyzed by pyruvate:ferredoxin oxidoreductase (PFOR) (Figure 4). 4 Acetyl-CoA condense to 2 acetoacetyl-CoA, which are reduced with 2 NADH to 2 (S)-3-hydroxybutyryl-CoA, dehydrated to 2 crotonyl-CoA and further reduced with 4 NADH to 2 butyryl-CoA and 4 reduced ferredoxin via electron bifurcation (Figure 5). For the reduction of 6 NAD+ by Rnf, 12 Fd– are consumed and give rise to 12 ΔμNa+. The remaining 2 Fd– are used to produce 1 H2. The import of 5 glutamate consumes 5 ΔμNa+. In summary, 3 ATP are obtained via SLP from 1 acetyl-CoA and 2 butyryl-CoA and 7/4 ATP from the remaining 12–5 = 7 ΔμNa+, altogether 4.75 ATP/5 glutamate, the same as in the 2-hydroxyglutarate pathway (Table 1).
The fermentations of glutamate via methylaspartate or 2-hydroxyglutarate lead to identical products and to identical amounts of conserved ATP. Therefore, the question arises why bacteria exclusively use one of these pathways. One would expect that bacteria prefer the 2-hydroxyglutarate pathway because the 3-methylaspartate pathway requires about 30 additional enzymes for the anaerobic biosynthesis of the coenzyme B12 (Moore and Warren, 2012), whereas in the 2-hydroxyglutarate pathway only the ubiquitous cofactors NAD, FAD, CoA, pyridoxal-5′-phosphate and biotin are involved. These cofactors are not pathway specific, because they are also necessary for anabolism. In absence of vitamin B12, Fusobacterium varium uses the 2-hydroxyglutarate pathway, whereas after addition of 1 μM vitamin B12 or just 1 μM CoCl2 to the medium, the organism ferments glutamate via 3-methylaspartate (Ramezani et al., 2011). Hence, F. varium is able to synthesize coenzyme B12 when Co2+ is present and the argument that the expression of 30 additional genes for B12 synthesis favors the 2-hydroxyglutarate pathway does not apply. Therefore, the reason for the existence of this pathway must be found somewhere else.
The organisms using the 2-hydroxyglutarate pathway, A. fermentans, P. asaccharolyticus, C. symbiosum, F. nucleatum, and F. varium were isolated from anaerobic niches of the human host, preferentially from the human large intestine. In contrast, the members of the 3-methylaspartate pathway C. tetanomorphum, C. tetani, C. cochlearium, C. lentoputrescens, C. limosum, C. malenominatum have been detected outside the gut on places like soil, where organic matter decomposes anaerobically (Buckel, 2001b). The employment of the 2-hydroxyglutarate pathway in the human gut could be due to the much lower oxygen concentration as compared to soil which encounters often exposures to air through plant roots, earthworms, moles, etc. Probably, the oxygen sensitivity of enzymes of an important pathway might be responsible for the ecology of the organism.
Both glutamate fermenting pathways contain oxygen sensitive radical enzymes. Whereas coenzyme B12-dependent glutamate mutase in the 3-methylaspartate pathway is only moderately oxygen sensitive, 2-hydroxyglutaryl-CoA dehydratase in the 2-hydroxyglutarate pathway immediately becomes inactive after exposure to air. Coenzyme B12-dependent mutases catalyze the reversible radical rearrangement of a methine radical to a methylene radical. The process is initiated by homolysis of the carbon-cobalt bond of coenzyme B12. The formed 5′-deoxyadenosyl radical abstracts the Si-hydrogen atom from the methylene group at C3 of (S)-glutamate and after rearrangement adds it back to the formed methylene radical yielding the methyl group of (2S,3S)-3-methylaspartate. After each turnover, the radical disappears by reformation of the carbon-cobalt bond of the coenzyme, for a review see Buckel and Golding (1996). Thus, in the presence of air, the radicals are only transiently exposed to oxygen as the coenzyme B12-dependent methylmalonyl-CoA mutase in human mitochondria.
In contrast, 2-hydroxyglutaryl-CoA dehydratase acts with a completely different radical mechanism (Buckel and Keese, 1995), for a review see Buckel (2019). The enzyme system is composed of two proteins. The homodimeric activator contains one ADP in each subunit and one [4Fe-4S] cluster coordinated by four cysteines, two from each subunit, similar to the iron protein (NifH) of nitrogenase (Locher et al., 2001). The heterodimeric dehydratase holds one [4Fe-4S] cluster in each subunit. Each cluster is coordinated by three cysteines; the fourth coordination is occupied in subunit A by a sulfur atom and in subunit B by water, which can be replaced by the thioester carbonyl of the substrate. The dehydration is initiated by ATP/ADP exchange of the activator and reduction of its [4Fe-4S]2+ cluster by reduced ferredoxin (Fd–). Driven by ATP hydrolysis, the extra electron is transferred to or “shot into” the [4Fe-4S] cluster of subunit A and further shifted to cluster B, where 2-hydroxyglutaryl-CoA is bound. The electron in cluster B reduces the thioester carbonyl to a ketyl radical, which due to its lower basicity is replaced from the iron by the hydroxyl group at C-2, a process called ligand swapping. Aided by the iron of cluster B, the nucleophilic ketyl eliminates the hydroxyl group to form an enoxy radical. The hydroxyl group at the iron of the cluster acts as base to remove the now acidic β-proton at C-3. The resulting allylic ketyl replaces the formed water by a second ligand swapping and returns the electron via cluster B to cluster A. The formed product glutaconyl-CoA is released and cluster B is able to accept the next substrate together with the electron form cluster A. The dehydratase is able to catalyze at least 1000 turnovers before another ATP molecule has to be hydrolyzed to continue catalysis (Kim et al., 2005). Proof of this mechanism was obtained by detection of the corresponding allylic ketyl radical of (R)-2-hydroxyisocaproyl-CoA dehydratase from C. difficile (Kim et al., 2008).
It has been shown experimentally that (R)-2-hydroxyacyl-CoA dehydratases are indeed much more oxygen sensitive than coenzyme B12-dependent carbon skeleton mutases. Whereas 2-hydroxyglutarate dehydratases had to be purified and assayed under strict exclusion of oxygen (Kim et al., 2005), glutamate mutase could be purified under air. During catalysis, however, after about 1 min a significant effect of air on the rate of 3-methylaspartate formation from glutamate was observed (Leutbecher et al., 1992). In the dehydratases, the electron never disappears during the catalytic cycle and the iron-sulfur cluster of the activator is solvent accessible, which makes it extremely oxygen sensitive. The question arises why the gut, where (R)-2-hydroxyacyl-CoA dehydratases are active, contains such low oxygen concentrations despite its ample supply with oxygen-rich blood (see section “Butyrate Provides Anaerobiosis in the Gut”).
In the previous chapters it has been shown that the 3-methylaspartate pathway tolerates oxygen because after each turnover of glutamate mutase, the cobalt-carbon bond between the 5′-deoxyadenosyl radical and cob(II)alamin is reformed. Thus, only during catalysis the radical is exposed to oxygen. In contrast, in (R)-2-hydroxyglutaryl-CoA dehydratase of the alternative glutamate fermenting pathway, the radical is always present and exposed to the medium. In addition, the iron-sulfur clusters of the dehydratase and its activator are very oxygen-sensitive. Most likely, this is the reason, why organisms of the 2-hydroxyglutarate pathway are only found in the gut or in strictly anaerobic marine sediments. Furthermore, B12-dependent carbon skeleton mutases and eliminases as well as thiamin diphosphate dependent enzymes are substituted in the gut by glycyl radical enzymes (Table 2). Similar to (R)-2-hydroxyacyl-CoA dehydratases, glycyl radical enzymes also require specific activating enzymes. These activases belong to the large family of radical SAM enzymes, which catalyze the one electron-reduction of S-adenosylmethionine (SAM) to methionine and the 5′-deoxyadenosyl radical. This radical irreversibly abstracts one hydrogen from a conserved glycine residue of the enzyme to give a stable protein-bound radical. Upon binding of substrate, the glycyl radical abstracts the sulfhydryl hydrogen form a nearby cysteine residue, which in turn removes the hydrogen atom from the substrate. After the rearrangement, the radical returns to the glycine residue and remains stable until the next turnover, unless it is attacked by oxygen (Knappe and Sawers, 1990; Shisler and Broderick, 2014).

Table 2. Pairs of oxygen-tolerant and intolerant enzymes which catalyze the same reaction or are key enzymes of alternative pathways leading to the same products.
Many organisms including humans are able to degrade propionate via carboxylation of propionyl-CoA to (S)-methylmalonyl-CoA and racemization to (R)-methylmalonyl-CoA, which is rearranged to succinyl-CoA mediated by coenzyme B12. The Krebs cycle converts succinyl-CoA to oxaloacetate, which enters gluconeogenesis or is degraded via pyruvate to acetyl-CoA. Propionibacteria use the reverse pathway to produce propionate from succinate. Veillonella alcalescens and P. modestum conserve energy with the biotin-dependent Na+-pump (S)-methylmalonyl-CoA decarboxylase (Hilpert and Dimroth, 1982; Schink, 1982). In contrast, Clostridium propionicum, isolated from marine mud (Cardon and Barker, 1946), converts alanine, cysteine and serine via pyruvate (Hofmeister et al., 1993) to (R)-lactate, which is dehydrated via (R)-lactyl-CoA to acrylyl-CoA (Schweiger and Buckel, 1984; Hofmeister and Buckel, 1992) and reduced to propionyl-CoA (Hetzel et al., 2003). Like 2-hydroxyglutaryl-CoA dehydratase, lactyl-CoA dehydratase is an extremely oxygen sensitive radical enzyme (Parthasarathy et al., 2010). The rumen micro-organism Megasphaera elsdenii reduces about half of the consumed lactate in the same way as C. propionicum; the other half is oxidized to acetyl-CoA, from which butyrate (C2 + C2), valerate (C2 + C3) and caproate (3 × C2) are formed (Lewis and Elsden, 1955).
The pyridoxal-5′-phosphate dependent elimination of water from threonine leads to 2-oxobutyrate and ammonia (Hofmeister et al., 1993). Homocysteine, derived from methionine, suffers a similar elimination to 2-oxobutyrate, ammonia and sulfide. Some anaerobic bacteria oxidize 2-oxobutyrate with ferredoxin to propionyl-CoA catalyzed by a thiamin diphosphate (TDP)-dependent enzyme and excrete propionate. C. propionicum reduces 2-oxobutyrate to (R)-2-hydroxybutyrate and dehydrates (R)-2-hydroxybutyryl-CoA to crotonyl-CoA, catalyzed by (R)-lactyl-CoA dehydratase (Hofmeister and Buckel, 1992).
Three eliminations of water from 1,2-diols are known, each of which is catalyzed by two different enzymes, either coenzyme B12-dependent or by the much more oxygen-sensitive glycyl radical enzymes (Table 2). The coenzyme B12-dependent glycerol dehydratase catalyzes the removal of the central hydroxyl group of glycerol yielding 3-hydroxypropanal (Forage and Foster, 1982), which can be reduced to 1,3-propanediol, a building block for polyesters. The discovery of the glycyl radical dehydratase from Clostridium butyricum (O’Brien et al., 2004) though extremely oxygen-sensitive, paved the road to a more economic fermentation of 1,3-propanediol from glycerol, because the already established procedure with the coenzyme B12-dependent enzyme required feeding with the precious vitamin B12. The mechanism of coenzyme B12-dependent eliminations was first studied with propane-1,2-diol dehydratase from Aerobacter aerogenes by Janós Rétey and Dulio Arigoni in Switzerland (Rétey et al., 1966) as well as Robert H. Abeles (Zagalak et al., 1966) in the United States. In the human gut, degradation of L-fucose (6-deoxygalactose) and L-rhamnose (6-deoxymannose) affords propane-1,2-diol, which is dehydrated to propanal, catalyzed by the glycyl radical enzyme propane-1,2-diol dehydratase (Levin and Balskus, 2018). The third pair of coenzyme B12 and glycyl radical enzymes catalyzing the same reaction are classes II and III of ribonucleotide reductase (Greene et al., 2020). The formation of the conserved thiyl radical in this enzyme is performed in class II by the 5′-deoxyadenosyl radical from coenzyme B12 and in class III by the glycyl radical. Abstraction of the 3′-hydrogen of the ribonucleotide by the thiyl radical and deprotonation of the 3′-hydroxyl group affords a nucleophilic ketyl (radical anion), which eliminates the adjacent 2′-hydroxyl group (Buckel, 2019). The reduction of the formed enoxy radical differs between the classes. In class II the reductants are two cysteine residues which form a disulfide, whereas class III uses formate for this purpose.
In the eliminations of ammonia from ethanolamine or trimethylamine from choline, carbon nitrogen bonds are broken. Interestingly, for the deamination of ethanolamine only a coenzyme B12-dependent enzyme is known (Shibata et al., 2010), whereas the elimination of trimethylamine from choline is exclusively catalyzed by a glycyl radical enzyme (Craciun et al., 2014). Perhaps the different elimination mechanisms, but not the oxygen sensitivity, are responsible for the kind of radical enzyme applied. It has been proposed theoretically (Buckel, 1996; Feliks and Ullmann, 2012; Kovacevic et al., 2018) and demonstrated experimentally (Levin and Balskus, 2018) that in the dehydration of propane-1,2-diol catalyzed by the glycyl radical enzyme, the hydroxyl group at C2 is eliminated. Coenzyme B12-dependent propane-1,2-diol dehydratase and glycerol dehydratase, however, shift the hydroxyl group at C2 to C1, forming propane-1,1-diol or 3-hydroxypropane-1,1-diol which dehydrate to the aldehydes. Because trimethylamine is a better leaving group than ammonia, chemistry might compel choline lyase to use the glycyl radical mechanism. Trimethylamine (TMA) is of medical importance, because in the liver it is oxidized to the N-oxide (TMAO), which may cause cancer and other diseases (Gatarek and Kaluzna-Czaplinska, 2021).
The oxidation of pyruvate to acetyl-CoA is a complex reaction for which nature has developed thiamin diphosphate, which causes Umpolung of the carbonyl group of the 2-oxo acid to enable decarboxylation. Most likely a concealed radical is involved in the reaction (Chen et al., 2018). The oxidant is either NAD+ or ferredoxin. In aerobes with NAD+ the reaction is irreversible, but reversible with ferredoxin in anaerobes (Eqs. 10, 11). Thus this reaction is an example of anaerobes being more efficient energy converters than aerobes.
Under strict anaerobic conditions, pyruvate dehydrogenase of E. coli (Eq. 10) is replaced by the glycyl radical enzyme, pyruvate formate lyase (PFL), which catalyzes the reversible cleavage of pyruvate to acetyl-CoA and formate (Eq. 12). The enzyme is also present in other enterobacteria and several clostridia.
The degradation of taurine (2-aminoethylsulfonate) in aerobes and facultative anaerobes affords sulfoacetaldehyde by amino transfer to pyruvate. The aldehyde is converted by the TDP-dependent sulfoacetaldehyde acetyltransferase Xsc to acetyl phosphate and sulfite (Ruff et al., 2003). Under the strict anaerobic conditions in the gut, the Bacillus Bilophila wadsworthia bypasses Xcs with an aldehyde reductase and the glycyl radical enzyme isethionate lyase. This enzyme catalyzes the radical diol dehydratase-like elimination of isethionate (2-sulfoethanol) yielding acetaldehyde and sulfite which is reduced to H2S (Peck et al., 2019). AdhE catalyzes the oxidation of acetaldehyde to acetyl-CoA, which as well as acetyl phosphate yields ATP by SLP. The genes iseG and iseH coding for isethionate lyase and its activating enzyme were also detected in several well-known sulfate reducing organisms (Dawson et al., 2021). The genes from Desulfovibrio vulgaris str. Hildenborough were expressed in E. coli and the produced proteins characterized as active isethionate lyase (Xing et al., 2019; Wei and Zhang, 2021).
In this review it has become apparent that radical reactions play an important part in the metabolism of anaerobes and most likely already in prebiotic chemistry (Dragicevic et al., 2015). Table 2 summarizes the pairs of oxygen-tolerant and intolerant enzymes which catalyze radical reactions leading to the same product or are key enzymes of alternative pathways. Whereas the oxygen-intolerant 2-hydroxyacyl-CoA dehydratases and glycyl radical enzymes have simple cofactors, the oxygen-tolerant coenzyme B12 and TDP-dependent enzymes use the most complex cofactors known in biochemistry (Jurgenson et al., 2009; Moore and Warren, 2012). Apparently, the oxygen-intolerant radical enzymes evolved first and prevailed under the primeval atmosphere until the oxygen concentration raised, which forced nature to develop the oxygen tolerant and much more complex cofactors coenzyme B12 and TDP. The oxygen-intolerant radical enzymes survived only in strictly anaerobic places such as the human gut and marine sediments. Probably, these coenzyme B12 and TDP dependent fermentations were not present at the origin of life, but emerged just before or during the Great Oxidation event at 2.1 billion years ago (Anbar et al., 2007).
Many obligate anaerobic bacteria in the human gut produce butyrate. The most common pathway is the condensation of two acetyl-CoA to acetoacetyl-CoA, followed by the first reduction to (S)-3-hydroxybutyryl-CoA, dehydration to crotonyl-CoA and the second reduction to butyryl-CoA as shown in the fermentation of glutamate via 3-methylaspartate. The first reductants are NADH or at low acetyl-CoA concentrations NADPH as found in Clostridium kluyveri. The stronger reductant NADPH helps to shift the unfavorable equilibrium of acetyl-CoA condensation to acetoacetyl-CoA toward (S)-3-hydroxybutyryl-CoA (Figure 5). The second reduction by NADH is coupled to the reduction of ferredoxin via electron bifurcation (Buckel and Thauer, 2013). Butyrate is released either by a CoA-transferase with acetate or by SLP via butyryl-phosphate (Louis and Flint, 2017). This pathway is used by most organisms fermenting carbohydrates, as are bacteria related to Roseburia intestinalis, Faecalibacterium prausnitzii, and Eubacterium hallii (all belong to Firmicutes). They are among the most abundant known butyrate-producing bacteria in human feces, comprising about 8% of the total flora (Hold et al., 2003). The majority of the bacteria in the human gut belong to Bacteroidetes (Gevers et al., 2012; Huttenhower and Gevers, 2012), which produce acetate and succinate rather than butyrate. As mentioned above A. fermentans, M. elsdenii, and C. difficile also use electron bifurcation for butyrate synthesis.
To the author’s knowledge, electron bifurcation with crotonyl-CoA appears to be the only pathway to butyrate in anaerobes. Probably no anaerobe wants to abandon the large energy released during butyrate synthesis by taking another route. Some acetate assimilating aerobes, however, reduce crotonyl-CoA with NADH directly to butyryl-CoA without proceeding via Etf. Actually the enzyme acts as crotonyl-CoA carboxylase yielding ethylmalonyl-CoA. Only at low CO2 concentrations the product is butyryl-CoA (Erb et al., 2007).
Fermentations of amino acids, which give rise to butyrate, are degraded to acetyl-CoA or directly to crotonyl-CoA as shown by the fermentation of glutamate via 2-hydroxyglutarate. Clostridium propionicum oxidizes threonine and methionine to (R)-2-hydroxybutyrate which is converted to crotonyl-CoA by a mechanism similar to that shown for (R)-2-hydroxyglutarate (Barker and Wiken, 1948). The complex anaerobic degradation of lysine in Clostridium subterminale SB4 (Barker, 1981) and Fusobacterium nucleatum (Barker et al., 1982) involves the radical SAM-dependent 2,3-shift and the coenzyme B12-dependent 6,5-shift of the amino groups to 3,5-diaminohexanoate (Ruzicka et al., 2000). The latter is deaminated to 3-oxo-5-aminohexanoate and cleaved with acetyl-CoA to acetoacetate and 3-aminobutyryl-CoA followed by deamination to crotonyl-CoA (Jeng and Barker, 1974) and reduction to butyryl-CoA, which forms butyrate and acetoacetyl-CoA with acetoacetate. Histidine is degraded to butyrate via glutamate either by C. tetanomorphum via the 3-methylaspartate pathway or by C. symbiosum via the 2-hydroxyglutarate pathway (Barker, 1981). In a Stickland type fermentation, proline is reduced to 5-aminovalerate by Clostridium sporogenes (Stickland, 1935), which is deaminated to 5-hydroxyvalerate and further converted via homocrotonyl-CoA to valerate (homobutyrate) by Clostridium viride (Eikmanns and Buckel, 1991; Buckel et al., 1994).
In the human gut, butyrate is the preferred nutrient of the mucosa cells, which form the inner wall of the large intestine, also called colonic epithelia cells. Butyrate activates the nuclear peroxisome proliferator-activated receptor-γ (PPAR-γ), a transcription factor, which in turn stimulates mitochondrial β-oxidation. The very active β-oxidation of butyrate is fueled by oxygen from the blood and consumes any remaining oxygen inside of the intestine resulting in an oxygen-free medium, where anaerobes optimally thrive. PPAR-γ also inhibits expression of the NO2 gene, which codes for an enzyme that catalyzes the oxidation of arginine with oxygen to nitric oxide (NO). NO is further oxidized to nitrate, an electron acceptor for potentially pathogenic enterobacteria. For example, E. coli, the best known bacterium of the human gut, is well adapted to the strict anaerobic environment as demonstrated by its glycyl radical enzymes class III ribonucleotide reductase and pyruvate:formate lyase (PFL, eq. 11). However, in the presence of oxygen or nitrate the facultative anaerobe forms pyruvate dehydrogenase (eq. 10) and multiplies very fast, from about 1% of the anaerobic microbiome to a major inhabitant of the gut. In case, the quickly spreading E. coli strain is pathogenic, severe diseases can occur. To prevent such events, butyrate nourishes colonic epithelia cells, which provides an oxygen-free medium for the microbiome in the large intestine and protects the host from harmful bacteria (Cani, 2017). For a constant butyrate production, the microbiome must be well fed with carbohydrates and amino acids. However, glucose, lactose, starch, glycogen and most proteins are digested and resorbed already in the small intestine, and nothing is left for the microbiome in the gut. Therefore, the human diet must contain indigestible carbohydrate and protein fibers for the mircrobiome, which is able to hydrolyze almost every glycosidic and peptide bond. Hence, fibers for the human diet are necessary for a healthy gut. The microbiologists Erica and Justin Sonnenburg remind their children to eat enough fibers by saying “Feed your bugs!” (Sonnenburg and Sonnenburg, 2015).
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The author thank Tobias Erb, Max Planck Institute for Terrestrial Microbiology, Marburg, Germany for generous support, Bernard T. Golding, University of Newcastle upon Tyne, United Kingdom, and Rudolf K. Thauer, Max Planck Institute for Terrestrial Microbiology, for helpful discussions.
FAD•–, FAD anionic semiquinone; FADH–, FAD hydroquinone; Fd, ferredoxin; Fd– reduced ferredoxin.
Anbar, A. D., Duan, Y., Lyons, T. W., Arnold, G. L., Kendall, B., Creaser, R. A., et al. (2007). A whiff of oxygen before the great oxidation event? Science 317, 1903–1906. doi: 10.1126/science.1140325
Barker, H. A. (1981). Amino acid degradation by anaerobic bacteria. Annu. Rev. Biochem. 50, 23–40. doi: 10.1146/annurev.bi.50.070181.000323
Barker, H. A., Kahn, J. M., and Hedrick, L. (1982). Pathway of lysine degradation in Fusobacterium nucleatum. J. Bacteriol. 152, 201–207. doi: 10.1128/jb.152.1.201-207.1982
Barker, H. A., Smyth, R. D., Wilson, R. M., and Weissbach, H. (1959). The purification and properties of beta-methylaspartase. J. Biol. Chem. 234, 320–328. doi: 10.1016/s0021-9258(18)70297-4
Barker, H. A., Suzuki, F., Iodice, A., and Rooze, V. (1964). Glutamate mutase reaction. Ann. N. Y. Acad. Sci. 112, 644–654. doi: 10.1111/j.1749-6632.1964.tb45041.x
Barker, H. A., and Wiken, T. (1948). The origin of butyric acid in the fermentation of threonine by Clostridium propionicum. Arch. Biochem. 17, 149–151.
Bergdoll, L., Ten Brink, F., Nitschke, W., Picot, D., and Baymann, F. (2016). From low- to high-potential bioenergetic chains: thermodynamic constraints of Q-cycle function. Biochim. Biophys. Acta 1857, 1569–1579. doi: 10.1016/j.bbabio.2016.06.006
Biegel, E., and Müller, V. (2010). Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142. doi: 10.1073/pnas.1010318107
Blair, A. H., and Barker, H. A. (1966). Assay and purification of (+)-citramalate hydro-lyase components from Clostridium tetanomorphum. J. Biol. Chem. 241, 400–408. doi: 10.1016/s0021-9258(18)96931-0
Boiangiu, C. D., Jayamani, E., Brügel, D., Herrmann, G., Kim, J., Forzi, L., et al. (2005). Sodium ion pumps and hydrogen production in glutamate fermenting anaerobic bacteria. J. Mol. Microbiol. Biotechnol. 10, 105–119. doi: 10.1159/000091558
Brüggemann, H., Baumer, S., Fricke, W. F., Wiezer, A., Liesegang, H., Decker, I., et al. (2003). The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc. Natl. Acad. Sci. U.S.A. 100, 1316–1321. doi: 10.1073/pnas.0335853100
Buckel, W. (1990). “Amino acid fermentation: coenzyme B12-dependent and -independent pathways,” in The Molecular Basis of Bacterial Metabolism 41. Colloquium - Mosbach 1990, eds G. Hauska and R. Thauer (Heidelberg: Springer Verlag), 21–30. doi: 10.1007/978-3-642-75969-7_3
Buckel, W. (1996). Unusual dehydrations in anaerobic bacteria: considering ketyls (radical anions) as reactive intermediates in enzymatic reactions. FEBS Lett. 389, 20–24. doi: 10.1016/0014-5793(96)00530-3
Buckel, W. (2001a). Sodium ion-translocating decarboxylases. Biochim. Biophys. Acta 1505, 15–27. doi: 10.1016/s0005-2728(00)00273-5
Buckel, W. (2001b). Unusual enzymes involved in five pathways of glutamate fermentation. Appl. Microbiol. Biotechnol. 57, 263–273. doi: 10.1007/s002530100773
Buckel, W. (2019). Enzymatic reactions involving ketyls: from a chemical curiosity to a general biochemical mechanism. Biochemistry 58, 5221–5233. doi: 10.1021/acs.biochem.9b00171
Buckel, W., and Barker, H. A. (1974). Two pathways of glutamate fermentation by anaerobic bacteria. J. Bacteriol. 117, 1248–1260. doi: 10.1128/jb.117.3.1248-1260.1974
Buckel, W., and Bobi, A. (1976). The enzyme complex citramalate lyase from Clostridium tetanomorphum. Eur. J. Biochem. 64, 255–262. doi: 10.1111/j.1432-1033.1976.tb10295.x
Buckel, W., Dorn, U., and Semmler, R. (1981). Glutaconate CoA-transferase from Acidaminococcus fermentans. Eur. J. Biochem. 118, 315–321. doi: 10.1111/j.1432-1033.1981.tb06404.x
Buckel, W., and Golding, B. (1996). Glutamate and 2-methyleneglutarate mutase: from microbial curiosities to paradigms for coenzyme B12-dependent enzymes. Chem. Soc. Rev. 25, 329–337. doi: 10.1039/cs9962500329
Buckel, W., Janssen, P. H., Schuhmann, A., Eikmanns, U., Messner, P., Sleytr, U., et al. (1994). Clostridium viride sp. nov., a strictly anaerobic bacterium using 5-aminovalerate as growth substrate, previously assigned to Clostridium aminovalericum. Arch. Microbiol. 162, 387–394.
Buckel, W., and Keese, R. (1995). One-electron redox reactions of CoASH esters in anaerobic bacteria – a mechanistic proposal. Angew. Chem. 34, 1502–1506. doi: 10.1002/anie.199515021
Buckel, W., and Semmler, R. (1982). A biotin-dependent sodium pump: glutaconyl-CoA decarboxylase from Acidaminococcus fermentans. FEBS Lett. 148, 35–38. doi: 10.1016/0014-5793(82)81237-4
Buckel, W., and Semmler, R. (1983). Purification, characterisation and reconstitution of glutaconyl-CoA decarboxylase, a biotin-dependent sodium pump from anaerobic bacteria. Eur. J. Biochem. 136, 427–434. doi: 10.1111/j.1432-1033.1983.tb07760.x
Buckel, W., and Thauer, R. K. (2013). Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113. doi: 10.1016/j.bbabio.2012.07.002
Buckel, W., and Thauer, R. K. (2018a). Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem. Rev. 118, 3862–3886. doi: 10.1021/acs.chemrev.7b00707
Buckel, W., and Thauer, R. K. (2018b). Flavin-based electron bifurcation, ferredoxin, flavodoxin, and anaerobic respiration with protons (Ech) or NAD+ (Rnf) as electron acceptors: a historical review. Front. Microbiol. 9:401. doi: 10.3389/fmicb.2018.00401
Cani, P. D. (2017). Gut cell metabolism shapes the microbiome. Science 357, 548–549. doi: 10.1126/science.aao2202
Cardon, B. P., and Barker, H. A. (1946). Two new amino-acid-fermenting bacteria, Clostridium propionicum and Diplococcus glycinophilus. J. Bacteriol. 52, 629–634. doi: 10.1128/jb.52.6.629-634.1946
Chen, P. Y., Aman, H., Can, M., Ragsdale, S. W., and Drennan, C. L. (2018). Binding site for coenzyme a revealed in the structure of pyruvate:ferredoxin oxidoreductase from Moorella thermoacetica. Proc. Natl. Acad. Sci. U.S.A. 115, 3846–3851. doi: 10.1073/pnas.1722329115
Chowdhury, N. P., Kahnt, J., and Buckel, W. (2015). Reduction of ferredoxin or oxygen by flavin-based electron bifurcation in Megasphaera elsdenii. FEBS J. 282, 3149–3160. doi: 10.1111/febs.13308
Chowdhury, N. P., Klomann, K., Seubert, A., and Buckel, W. (2016). Reduction of flavodoxin by electron bifurcation and sodium ion-dependent reoxidation by NAD+ catalyzed by ferredoxin-NAD+ reductase (Rnf). J. Biol. Chem. 291, 11993–12002. doi: 10.1074/jbc.m116.726299
Chowdhury, N. P., Mowafy, A. M., Demmer, J. K., Upadhyay, V., Koelzer, S., Jayamani, E., et al. (2014). Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J. Biol. Chem. 289, 5145–5157. doi: 10.1074/jbc.m113.521013
Craciun, S., Marks, J. A., and Balskus, E. P. (2014). Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem. Biol. 9, 1408–1413. doi: 10.1021/cb500113p
Dawson, C. D., Irwin, S. M., Backman, L. R. F., Le, C., Wang, J. X., Vennelakanti, V., et al. (2021). Molecular basis of C-S bond cleavage in the glycyl radical enzyme isethionate sulfite-lyase. Cell Chem. Biol. 28, 1–14. doi: 10.1016/j.chembiol.2021.03.001
Demmer, J. K., Huang, H., Wang, S., Demmer, U., Thauer, R. K., and Ermler, U. (2015). Insights into flavin-based electron bifurcation via the NADH-dependent reduced ferredoxin:NADP oxidoreductase structure. J. Biol. Chem. 290, 21985–21995. doi: 10.1074/jbc.m115.656520
Demmer, J. K., Pal Chowdhury, N., Selmer, T., Ermler, U., and Buckel, W. (2017). The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-CoA dehydrogenase complex from Clostridium difficile. Nat. Commun. 8:1577.
Dimroth, P. (1980). A new sodium-transport system energized by the decarboxylation of oxaloacetate. FEBS Lett. 122, 234–236. doi: 10.1016/0014-5793(80)80446-7
Dimroth, P., and Thomer, A. (1986). Kinetic analysis of the reaction mechanism of oxaloacetate decarboxylase from Klebsiella aerogenes. Eur. J. Biochem. 156, 157–162. doi: 10.1111/j.1432-1033.1986.tb09561.x
Dimroth, P., and Thomer, A. (1993). On the mechanism of sodium ion translocation by oxaloacetate decarboxylase of Klebsiella pneumoniae. Biochemistry 32, 1734–1739. doi: 10.1021/bi00058a006
Djordjevic, S., Pace, C. P., Stankovich, M. T., and Kim, J. J. (1995). Three-dimensional structure of butyryl-CoA dehydrogenase from Megasphaera elsdenii. Biochemistry 34, 2163–2171. doi: 10.1021/bi00007a009
Dragicevic, I., Baric, D., Kovacevic, B., Golding, B. T., and Smith, D. M. (2015). Non-enzymatic ribonucleotide reduction in the prebiotic context. Chemistry 21, 6132–6143. doi: 10.1002/chem.201405741
Eikmanns, U., and Buckel, W. (1991). Crystalline green 5-hydroxyvaleryl-CoA dehydratase from Clostridium aminovalericum. Eur. J. Biochem. 197, 661–668. doi: 10.1111/j.1432-1033.1991.tb15956.x
Engel, P. C., and Massey, V. (1971). Green butyryl-coenzyme a dehydrogenase. an enzyme-acyl-coenzyme a complex. Biochem. J. 125, 889–902. doi: 10.1042/bj1250889
Erb, T. J., Berg, I. A., Brecht, V., Müller, M., Fuchs, G., and Alber, B. E. (2007). Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. U.S.A. 44, 31496–31502.
Ezaki, T., Yamamoto, N., Ninomiya, K., Suzuki, S., and Yabuuchi, E. (1983). Transfer of Peptococcus indolicus, Peptococcus asaccharolyticus, Peptococeus prevotii and Peptococcus magnus to the genus Peptostreptococcus and proposal of Peptostreptococcus tetardius sp. nov. Int. J. Syst. Bacteriol. 33, 683–689. doi: 10.1099/00207713-33-4-683
Feliks, M., and Ullmann, G. M. (2012). Glycerol dehydratation by the B12-independent enzyme may not involve the migration of a hydroxyl group: a computational study. J. Phys. Chem. B 116, 7076–7087. doi: 10.1021/jp301165b
Forage, R. G., and Foster, M. A. (1982). Glycerol fermentation in Klebsiella pneumoniae: functions of the coenzyme B12-dependent glycerol and diol dehydratases. J. Bacteriol. 149, 413–419. doi: 10.1128/jb.149.2.413-419.1982
Gatarek, P., and Kaluzna-Czaplinska, J. (2021). Trimethylamine N-oxide (TMAO) in human health. EXCLI J. 20, 301–319.
Gevers, D., Pop, M., Schloss, P. D., and Huttenhower, C. (2012). Bioinformatics for the human microbiome project. PLoS Comput. Biol. 8:e1002779. doi: 10.1371/journal.pcbi.1002779
Greene, B. L., Kang, G., Cui, C., Bennati, M., Nocera, D. G., Drennan, C. L., et al. (2020). Ribonucleotide reductases: structure, chemistry, and metabolism suggest new therapeutic targets. Annu. Rev. Biochem. 98, 45–75. doi: 10.1146/annurev-biochem-013118-111843
Hans, M., Bill, E., Cirpus, I., Pierik, A. J., Hetzel, M., Alber, D., et al. (2002). Adenosine triphosphate-induced electron transfer in 2-hydroxyglutaryl-CoA dehydratase from Acidaminococcus fermentans. Biochemistry 41, 5873–5882. doi: 10.1021/bi020033m
Herrmann, G. (2008). Enzymes of Two Clostridial Amino-Acid Fermentation Pathways. Ph. D. thesis. Germany: Philipps Universität Marburg.
Herrmann, G., Jayamani, E., Mai, G., and Buckel, W. (2008). Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791. doi: 10.1128/jb.01422-07
Hess, V., Gallegos, R., Jones, J. A., Barquera, B., Malamy, M. H., and Müller, V. (2016). Occurrence of ferredoxin:NAD(+) oxidoreductase activity and its ion specificity in several Gram-positive and Gram-negative bacteria. PeerJ 4:e1515. doi: 10.7717/peerj.1515
Hess, V., Schuchmann, K., and Müller, V. (2013). The ferredoxin:NAD+ oxidoreductase (Rnf) from the acetogen Acetobacterium woodii requires Na+ and is reversibly coupled to the membrane potential. J. Biol. Chem. 288, 31496–31502. doi: 10.1074/jbc.m113.510255
Hetzel, M., Brock, M., Selmer, T., Pierik, A. J., Golding, B. T., and Buckel, W. (2003). Acryloyl-CoA reductase from Clostridium propionicum. an enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur. J. Biochem. 270, 902–910. doi: 10.1046/j.1432-1033.2003.03450.x
Hilpert, W., and Dimroth, P. (1982). Conversion of the chemical energy of methylmalonyl-CoA decarboxylation into a Na+ gradient. Nature 296, 584–585. doi: 10.1038/296584a0
Hilpert, W., Schink, B., and Dimroth, P. (1984). Life by a new decarboxylation-dependent energy conservation mechanism with Na+ as coupling ion. EMBO J. 3, 1665–1670. doi: 10.1002/j.1460-2075.1984.tb02030.x
Hofmeister, A. E., and Buckel, W. (1992). (R)-lactyl-CoA dehydratase from Clostridium propionicum. Stereochemistry of the dehydration of (R)-2-hydroxybutyryl-CoA to crotonyl-CoA. Eur. J. Biochem. 206, 547–552. doi: 10.1111/j.1432-1033.1992.tb16958.x
Hofmeister, A. E., Grabowski, R., Linder, D., and Buckel, W. (1993). L-serine and L-threonine dehydratase from Clostridium propionicum. two enzymes with different prosthetic groups. Eur. J. Biochem. 215, 341–349. doi: 10.1111/j.1432-1033.1993.tb18040.x
Hold, G. L., Schwiertz, A., Aminov, R. I., Blaut, M., and Flint, H. J. (2003). Oligonucleotide probes that detect quantitatively significant groups of butyrate-producing bacteria in human feces. Appl. Environ. Microbiol. 69, 4320–4324. doi: 10.1128/aem.69.7.4320-4324.2003
Hornby, D. P., and Engel, P. C. (1984). Characterization of Peptostreptococcus asaccharolyticus glutamate dehydrogenase purified by dye-ligand chromatography. J. Gen. Microbiol. 130, 2385–2394. doi: 10.1099/00221287-130-9-2385
Huttenhower, C., and Gevers, D. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Imkamp, F., Biegel, E., Jayamani, E., Buckel, W., and Müller, V. (2007). Dissection of the caffeate respiratory chain in the acetogen Acetobacterium woodii: Identification of an Rnf-type NADH dehydrogenase as a potential coupling site. J. Bacteriol. 189, 8145–8153. doi: 10.1128/jb.01017-07
Jayamani, E. (2008). A Unique Way of Energy Conservation in Glutamate Fermenting Clostridia Ph D dissertation. Germany: Philipps-University Marburg.
Jeng, I., and Barker, H. A. (1974). Purification and properties of l-3-aminobutyryl coenzyme a deaminase from a lysine-fermenting Clostridium. J. Biol. Chem. 249, 6578–6584. doi: 10.1016/s0021-9258(19)42195-9
Jurgenson, C. T., Begley, T. P., and Ealick, S. E. (2009). The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78, 569–603. doi: 10.1146/annurev.biochem.78.072407.102340
Kayastha, K., Vitt, S., Buckel, W., and Ermler, U. (2021). Flavins in the electron bifurcation process. Arch. Biochem. Biophys. 701:108796. doi: 10.1016/j.abb.2021.108796
Kim, J., Darley, D., and Buckel, W. (2005). 2-Hydroxyisocaproyl-CoA dehydratase and its activator from Clostridium difficile. FEBS J. 272, 550–561. doi: 10.1111/j.1742-4658.2004.04498.x
Kim, J., Darley, D. J., Buckel, W., and Pierik, A. J. (2008). An allylic ketyl radical intermediate in clostridial amino-acid fermentation. Nature 452, 239–242. doi: 10.1038/nature06637
Knappe, J., and Sawers, G. (1990). A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol. Rev. 6, 383–398. doi: 10.1111/j.1574-6968.1990.tb04108.x
Kovacevic, B., Baric, D., Babic, D., Bilic, L., Hanzevacki, M., Sandala, G. M., et al. (2018). Computational tale of two enzymes: glycerol dehydration with or without B12. J. Am. Chem. Soc. 140, 8487–8496. doi: 10.1021/jacs.8b03109
Kremp, F., Roth, J., and Müller, V. (2020). The Sporomusa type Nfn is a novel type of electron-bifurcating transhydrogenase that links the redox pools in acetogenic bacteria. Sci. Rep. 10:14872.
Kress, D., Brügel, D., Schall, I., Linder, D., Buckel, W., and Essen, L. O. (2009). An asymmetric model for Na+-translocating glutaconyl-CoA decarboxylases. J. Biol. Chem. 284, 28401–28409. doi: 10.1074/jbc.m109.037762
Laubinger, W., and Dimroth, P. (1988). Characterization of the ATP synthase of Propionigenium modestum as a primary sodium pump. Biochemistry 27, 7531–7537. doi: 10.1021/bi00419a053
Leutbecher, U., Böcher, R., Linder, D., and Buckel, W. (1992). Glutamate mutase from Clostridium cochlearium. purification, cobamide content and stereospecific inhibitors. Eur. J. Biochem. 205, 759–765. doi: 10.1111/j.1432-1033.1992.tb16840.x
Levin, B. J., and Balskus, E. P. (2018). Characterization of 1,2-propanediol dehydratases reveals distinct mechanisms for B12-dependent and glycyl radical enzymes. Biochemistry 57, 3222–3226. doi: 10.1021/acs.biochem.8b00164
Lewis, D., and Elsden, S. R. (1955). The fermentation of L-threonine, L-serine, L-cysteine and acrylic acid by a gram-negative coccus. Biochem. J. 60, 683–692. doi: 10.1042/bj0600683
Li, F., Hinderberger, J., Seedorf, H., Zhang, J., Buckel, W., and Thauer, R. K. (2008). Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850. doi: 10.1128/jb.01417-07
Locher, K. P., Hans, M., Yeh, A. P., Schmid, B., Buckel, W., and Rees, D. C. (2001). Crystal structure of the Acidaminococcus fermentans 2-hydroxyglutaryl-CoA dehydratase component A. J. Mol. Biol. 307, 297–308. doi: 10.1006/jmbi.2000.4496
Louis, P., and Flint, H. J. (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41. doi: 10.1111/1462-2920.13589
Lubner, C. E., Jennings, D. P., Mulder, D. W., Schut, G. J., Zadvornyy, O. A., Hoben, J. P., et al. (2017). Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat. Chem. Biol. 13, 655–659. doi: 10.1038/nchembio.2348
Marchandin, H., Teyssier, C., Campos, J., Jean-Pierre, H., Roger, F., Gay, B., et al. (2010). Negativicoccus succinicivorans gen. nov., sp. nov., isolated from human clinical samples, emended description of the family Veillonellaceae and description of Negativicutes classis nov., Selenomonadales ord. nov. and fam. nov. in the bacterial phylum Firmicutes. Int. J. Syst. Evol. Microbiol. 60, 1271–1279. doi: 10.1099/ijs.0.013102-0
Moore, S. J., and Warren, M. J. (2012). The anaerobic biosynthesis of vitamin B12. Biochem. Soc. Trans. 40, 581–586. doi: 10.1042/bst20120066
Moser, C. C., Keske, J. M., Warncke, K., Farid, R. S., and Dutton, P. L. (1992). Nature of biological electron transfer. Nature 355, 796–802. doi: 10.1038/355796a0
Nitschke, W., and Russell, M. J. (2012). Redox bifurcations: mechanisms and importance to life now, and at its origin: a widespread means of energy conversion in biology unfolds. Bioessays 34, 106–109. doi: 10.1002/bies.201100134
O’Brien, J. R., Raynaud, C., Croux, C., Girbal, L., Soucaille, P., and Lanzilotta, W. N. (2004). Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemisty 43, 4635–4645. doi: 10.1021/bi035930k
Otto, R., Sonnenberg, A. S. M., Veldkamp, H., and Konings, W. N. (1980). Generation of an electrochemical proton gradient in Streptococcus cremoris by lactate efflux. Proc. Natl. Acad. Sci. U.S.A. 77, 5502–5506. doi: 10.1073/pnas.77.9.5502
Parthasarathy, A., Buckel, W., and Smith, D. M. (2010). On the thermodynamic equilibrium between (R)-2-hydroxyacyl-CoA and 2-enoyl-CoA. FEBS J. 277, 1738–1746. doi: 10.1111/j.1742-4658.2010.07597.x
Peck, S. C., Denger, K., Burrichter, A., Irwin, S. M., Balskus, E. P., and Schleheck, D. (2019). A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc. Natl. Acad. Sci U.S.A. 116, 3171–3176. doi: 10.1073/pnas.1815661116
Pfenninger-Li, X. D., and Dimroth, P. (1992). NADH formation by Na+-coupled reversed electron transfer in Klebsiella pneumoniae. Mol. Microbiol. 6, 1943–1948. doi: 10.1111/j.1365-2958.1992.tb01367.x
Ramezani, M., Resmer, K. L., and White, R. L. (2011). Glutamate racemization and catabolism in Fusobacterium varium. FEBS J. 278, 2540–2551. doi: 10.1111/j.1742-4658.2011.08179.x
Rétey, J., Umani-Ronchi, A., Seibl, J., and Arigoni, D. (1966). Zum mechanismus der propandioldehydrase-reaktion. Experientia 22, 502–503. doi: 10.1007/bf01898652
Roberts, D. L., Frerman, F. E., and Kim, J. J. (1996). Three-dimensional structure of human electron transfer flavoprotein to 2.1-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 93, 14355–14360. doi: 10.1073/pnas.93.25.14355
Ruff, J., Denger, K., and Cook, A. M. (2003). Sulphoacetaldehyde acetyltransferase yields acetyl phosphate: purification from Alcaligenes defragrans and gene clusters in taurine degradation. Biochem. J. 369, 275–285. doi: 10.1042/bj20021455
Ruzicka, F. J., Lieder, K. W., and Frey, P. A. (2000). Lysine 2,3-aminomutase from Clostridium subterminale SB4: mass spectral characterization of cyanogen bromide-treated peptides and cloning, sequencing, and expression of the gene kamA in Escherichia coli. J. Bacteriol. 182, 469–476. doi: 10.1128/jb.182.2.469-476.2000
Schink, B. (1982). Propionigenium modestum gen. nov. sp. nov. a new strictly anaerobic, nonsporing bacterium growing on succinate. Arch. Microbiol. 133, 209–216. doi: 10.1007/bf00415003
Schmehl, M., Jahn, A., Meyer zu Vilsendorf, A., Hennecke, S., Masepohl, B., Schuppler, M., et al. (1993). Identification of a new class of nitrogen fixation genes in Rhodobacter capsulatus: a putative membrane complex involved in electron transport to nitrogenase. Mol. Gen. Genet. 241, 602–615. doi: 10.1007/bf00279903
Schwarz, E., Oesterhelt, D., Reinke, H., Beyreuther, K., and Dimroth, P. (1988). The sodium ion translocating oxalacetate decarboxylase of Klebsiella pneumoniae. sequence of the biotin-containing alpha-subunit and relationship to other biotin-containing enzymes. J. Biol. Chem. 263, 9640–9645. doi: 10.1016/s0021-9258(19)81564-8
Schweiger, G., and Buckel, W. (1984). On the dehydration of (R)-lactate in the fermentation of alanine to propionate by Clostridium propionicum. FEBS Lett. 171, 79–84. doi: 10.1016/0014-5793(84)80463-9
Seedorf, H., Fricke, W. F., Veith, B., Brüggemann, H., Liesegang, H., Strittmatter, A., et al. (2008). The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc. Natl. Acad. Sci. U.S.A. 105, 2128–2133. doi: 10.1073/pnas.0711093105
Shibata, N., Tamagaki, H., Hieda, N., Akita, K., Komori, H., Shomura, Y., et al. (2010). Crystal structures of ethanolamine ammonia-lyase complexed with coenzyme B12 analogs and substrates. J. Biol. Chem. 285, 26484–26493. doi: 10.1074/jbc.m110.125112
Shisler, K. A., and Broderick, J. B. (2014). Glycyl radical activating enzymes: structure, mechanism, and substrate interactions. Arch. Biochem. Biophys. 546, 64–71. doi: 10.1016/j.abb.2014.01.020
Steuber, J., Halang, P., Vorburger, T., Steffen, W., Vohl, G., and Fritz, G. (2014a). Central role of the Na(+)-translocating NADH:quinone oxidoreductase (Na(+)-NQR) in sodium bioenergetics of Vibrio cholerae. Biol. Chem. 295, 1389–1399. doi: 10.1515/hsz-2014-0204
Steuber, J., Vohl, G., Casutt, M. S., Vorburger, T., Diederichs, K., and Fritz, G. (2014b). Structure of the V. cholerae Na+-pumping NADH:quinone oxidoreductase. Nature 516, 62–67. doi: 10.1038/nature14003
Stickland, L. H. (1935). Studies in the metabolism of the strict anaerobes (Genus Clostridium): the reduction of proline by Cl. sporogenes. Biochem. J. 29, 288–290. doi: 10.1042/bj0290288
Sucharitakul, J., Buckel, W., and Chaiyen, P. (2021a). Rapid kinetics reveal surprising flavin chemistry in the bifurcating electron transfer flavoprotein from Acidaminococcus fermentans. J. Biol. Chem. 296:100124. doi: 10.1074/jbc.RA120.016017
Sucharitakul, J., Buttranon, S., Wongnate, T., Chowdhury, N. P., Prongjit, M., Buckel, W., et al. (2021b). Modulations of the reduction potentials of flavin-based electron bifurcation complexes and semiquinone stabilities are key to control directional electron flow. FEBS J. 288, 1008–1026. doi: 10.1111/febs.15343
Thauer, R. K., Jungermann, K., Rupprecht, E., and Decker, K. (1969). Hydrogen formation from NADH in cell-free extracts of Clostridium kluyveri. acetyl coenzyme a requirement and ferredoxin dependence. FEBS Lett. 4, 108–112. doi: 10.1016/0014-5793(69)80208-5
Thauer, R. K., Jungermann, R., and Decker, K. (1977). Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41, 100–180. doi: 10.1128/br.41.1.100-180.1977
Vigil, W. Jr., Niks, D., Franz-Badur, S., Chowdhury, N., Buckel, W., and Hille, R. (2021). Spectral deconvolution of redox species in the crotonyl-CoA-dependent NADH:ferredoxin oxidoreductase from Megasphaera elsdenii. a flavin-dependent bifurcating enzyme. Arch. Biochem. Biophys. 701:108793. doi: 10.1016/j.abb.2021.108793
Vitt, S., Prinz, S., Hellwig, N., Morgner, N., Ermler, U., and Buckel, W. (2020). Molecular and low-resolution structural characterization of the Na+-translocating glutaconyl-CoA decarboxylase from Clostridium symbiosum. Front. Microbiol. 11:48011. doi: 10.3389/fmicb.2020.00480
Vogt, M. S., Schühle, K., Koelzer, S., Peschke, P., Chowdhury, N. P., Kleinsorge, D., et al. (2019). Structural and functional characterization of an electron transfer flavoprotein involved in toluene degradation in strictly anaerobic bacteria. J. Bacteriol. 201, e00326. doi: 10.1128/JB.00326-19
von Ballmoos, C., Wiedenmann, A., and Dimroth, P. (2009). Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Rev. Biochem. 78, 649–672. doi: 10.1146/annurev.biochem.78.081307.104803
Wang, S., Huang, H., Moll, J., and Thauer, R. K. (2010). NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192, 5115–5123. doi: 10.1128/jb.00612-10
Wei, Y., and Zhang, Y. (2021). Glycyl radical enzymes and sulfonate metabolism in the microbiome. Annu. Rev. Biochem. 90, 817–846. doi: 10.1146/annurev-biochem-080120-024103
Xing, M., Wei, Y., Zhou, Y., Zhang, J., Lin, L., Hu, Y., et al. (2019). Radical-mediated C-S bond cleavage in C2 sulfonate degradation by anaerobic bacteria. Nat. Commun. 10:1609.
Xu, X., Shi, H., Gong, X., Chen, P., Gao, Y., Zhang, X., et al. (2020). Structural insights into sodium transport by the oxaloacetate decarboxylase sodium pump. Elife 9:e53853. doi: 10.7554/eLife.53853
Yu, X., Bresser, J., Schall, I., Djurdjevic, I., Buckel, W., Wang, X., et al. (2012). Development of a satisfactory and general continuous assay for aminotransferases by coupling with (R)-2-hydroxyglutarate dehydrogenase. Anal. Biochem. 432, 127–131. doi: 10.1016/j.ab.2012.09.009
Zagalak, B., Frey, P. A., Karabatsos, G. L., and Abeles, R. H. (1966). The stereochemistry of the conversion of D and L 1,2-propanediols to propionaldehyde. J. Biol. Chem. 241, 3028–3035. doi: 10.1016/s0021-9258(18)96492-6
Keywords: ΔμNa+, decarboxylation, ferredoxin, Rnf, electron bifurcation, coenzyme B12, glycyl radical enzymes, oxygen sensitivity
Citation: Buckel W (2021) Energy Conservation in Fermentations of Anaerobic Bacteria. Front. Microbiol. 12:703525. doi: 10.3389/fmicb.2021.703525
Received: 30 April 2021; Accepted: 30 July 2021;
Published: 13 September 2021.
Edited by:
Wolfgang Nitschke, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
William Martin, University of Düsseldorf Medical School, GermanyCopyright © 2021 Buckel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Buckel, YnVja2VsQHN0YWZmLnVuaS1tYXJidXJnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.