
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 29 July 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.694489
 David Krones1†
David Krones1† Marcel Rühling1†
Marcel Rühling1† Katrin Anne Becker2
Katrin Anne Becker2 Tobias C. Kunz1
Tobias C. Kunz1 Carolin Sehl2
Carolin Sehl2 Kerstin Paprotka1
Kerstin Paprotka1 Erich Gulbins2,3
Erich Gulbins2,3 Martin Fraunholz1*
Martin Fraunholz1*Staphylococcus aureus (S. aureus) is well known to express a plethora of toxins of which the pore-forming hemolysin A (α-toxin) is the best-studied cytolysin. Pore-forming toxins (PFT) permeabilize host membranes during infection thereby causing concentration-dependent effects in host cell membranes ranging from disordered ion fluxes to cytolysis. Host cells possess defense mechanisms against PFT attack, resulting in endocytosis of the breached membrane area and delivery of repair vesicles to the insulted plasma membrane as well as a concurrent release of membrane repair enzymes. Since PFTs from several pathogens have been shown to recruit membrane repair components, we here investigated whether staphylococcal α-toxin is able to induce these mechanisms in endothelial cells. We show that S. aureus α-toxin induced increase in cytosolic Ca2+ in endothelial cells, which was accompanied by p38 MAPK phosphorylation. Toxin challenge led to increased endocytosis of an extracellular fluid phase marker as well as increased externalization of LAMP1-positive membranes suggesting that peripheral lysosomes are recruited to the insulted plasma membrane. We further observed that thereby the lysosomal protein acid sphingomyelinase (ASM) was released into the cell culture medium. Thus, our results show that staphylococcal α-toxin triggers mechanisms in endothelial cells, which have been implicated in membrane repair after damage of other cell types by different toxins.
The opportunistic pathogen Staphylococcus aureus (S. aureus) produces a plethora of toxins of which the pore-forming α-toxin is most investigated (e.g., reviewed in Bhakdi and Tranum-Jensen, 1991; von Hoven et al., 2019). However, the bi-component PFTs γ-hemolysin and the so called leukocidins also form pores in target membranes, but are specific for certain cell types or even species-specific (Alonzo and Torres, 2014), whereas α-toxin is able to permeabilize different cell types (Berube and Bubeck Wardenburg, 2013). Further, the so-called phenol-soluble modulins (PSM) are secreted by S. aureus in copious amounts (Wang et al., 2007). PSMs, membrane active peptides, are strongly cytolytic in absence of serum lipoproteins (Surewaard et al., 2012) and thus are thought to be required for the intracellular virulence of S. aureus upon internalization by epithelial or endothelial cells, where they can cause the translocation of the pathogen from phago-endosomes to the host cytoplasm (Grosz et al., 2014).
Staphylococcus aureus α-toxin induces cell death in mammalian host cells (Menzies and Kourteva, 2000; Haslinger-Loffler et al., 2005). In sensitive target cell membranes, α-toxin monomers assemble to a heptameric pre-pore (Bhakdi and Tranum-Jensen, 1991) and subsequent pore formation can lead to calcium influx and efflux of potassium (Fink et al., 1989; Walev et al., 1993; Kwak et al., 2012), or even larger molecules such as ATP (Gierok et al., 2014), eventually leading to host cell death (Baaske et al., 2016). Specific binding of α-toxin to A Disintegrin And Metalloproteinase Domain-containing protein 10 (ADAM10) facilitates pre-pore assembly on target membranes (Popov et al., 2015). Furthermore, the activated metalloprotease leads to degradation of cell barrier proteins in epithelial and endothelial cells, allowing for S. aureus deep tissue penetration (Inoshima et al., 2011; Becker et al., 2018). α-toxin sensitivity of target membranes is further dependent on sphingomyelin, since mutants in sphingomyelin synthetase SGMS1 were identified as α-toxin resistant in a genome-wide CRISPR screen (Virreira Winter et al., 2016) and treatment with the bacterial sphingomyelinase (bSMase) staphylococcal β-toxin abrogated α-toxin sensitivity in epithelial cells HBE16o– (Ziesemer et al., 2019).
Mammalian cells also express sphingomyelinases, which serve crucial cellular functions in lipid signaling, membrane fusion, apoptosis induction (Santana et al., 1996; Zhao et al., 2016), infection (Hauck et al., 2000; Riethmüller et al., 2006; Grassmé et al., 2008; Faulstich et al., 2015; Seitz et al., 2015; Peters et al., 2019) or membrane repair mechanisms after plasma membrane insult, e.g., by pore forming toxins such as streptolysin O (SLO) or listeriolysin O (LLO) (Andrews et al., 2014). Bacteria (Grassmé et al., 1997; Grassmé et al., 2003; Simonis and Schubert-Unkmeir, 2018), viruses (Grassmé et al., 2005; Miller et al., 2012; Shivanna et al., 2015) as well as fungi (Bryan et al., 2015) have been shown to exploit enzymes of the sphingolipid metabolism. Upon S. aureus infections, a rapid activation of acid sphingomyelinase (ASM) occurs and this activation is triggered by staphylococcal α-hemolysin (Ma et al., 2017; Becker et al., 2018).
Plasma membrane damage by PFT may trigger membrane repair mechanisms, which depend on the pore size, type of toxin and the toxin receptors involved in the process (Blazek et al., 2015; von Hoven et al., 2019). The rapid change of intracellular ion concentration upon plasma membrane insult leads to an activation of different signaling pathways. In some cases, mitogen activated kinases (MAPK) are activated after membrane insult, which initiate the production of interleukins (IL) such as IL-4, IL-6 or tumor necrosis factor α (TNFα) for host cell protection as well as signaling to cells of the immune system (Chow et al., 2010; Fedele et al., 2010). At the same time, activation of MAPK is connected to the initiation of death pathways such as apoptosis, whereby in human endothelial cells a cleavage of procaspases 6, and 9, and cleavage of procaspase 3 was detected, leading to DNA fragmentation (N’Guessan et al., 2005). Ca2+ influx also leads to the recruitment of peripheral lysosomes to the plasma membrane, which in turn causes exocytosis of membrane repair enzymes such as lysosomal ASM. This process is accompanied by short-term externalization of lysosomal-associated membrane protein 1 (LAMP1) (Reddy et al., 2001). Externalized ASM cleaves sphingomyelin in the outer leaflet of the plasma membrane thereby generating ceramide-enriched platforms (Grassmé et al., 2001a), which serve to cluster receptors (Grassmé et al., 2001b), but also cause an ATP-independent invagination of the plasma membrane (Babiychuk et al., 2008) and a subsequent endocytosis of the pore (Idone et al., 2008). Endocytosis of α-toxin pores had been identified previously to reseal the plasma membranes of cells intoxicated with staphylococcal α-toxin (Husmann et al., 2009) and thus represents one additional pathway of plasma membrane repair next to the removal of pores by vesicle budding and the clogging by annexins (reviewed, e.g., in Bischofberger et al., 2012; Brito et al., 2019).
We here show that treatment with staphylococcal α-toxin induces the recruitment of LAMP1 positive vesicles to the extracellular environment of endothelial cells, which thereby release the membrane repair enzyme ASM into the extracellular environment. This recruitment coincides with elevation of cytosolic calcium levels suggesting that α-toxin triggers membrane repair mechanisms in host cells as they were observed for other pore-forming toxins.
Recombinant S. aureus toxins were purchased from Gentaur GmbH (Aachen, Germany). α-toxin was purchased from Gentaur GmbH or Sigma (Taufkirchen, Germany).
Staphylococcus aureus strains (Table 1) were grown on trypticase soy agar (TSA) plates or TSA containing 5% defibrinated sheep blood. Appropriate antibiotics were added when selection was required. For infection experiments, S. aureus strains were grown over night in brain heart infusion (BHI) medium with appropriate antibiotics. Overnight cultures were incubated aerobically under constant shaking (200 rpm) at 37°C. For preparation of bacterial supernatants, overnight cultures were centrifuged for 2 min at 20,800 g and the supernatant was sterile-filtered through a syringe filter with a pore size of 0.2 μm.

Table 1. Strains used in this study.
S. aureus strains JE2 Δhla (Becker et al., 2018) were transduced with ϕ11 lysates of S. aureus RN4220 carrying either pCerulean or pHla-Cerulean (Giese et al., 2009) in order to allow for genetic complementation of α-toxin production. In order to induce toxin production 200 ng/ml anhydrous tetracycline was added to the bacterial culture and culture supernatants were harvested as mentioned above.
All cell lines were grown mycoplasma-free in media supplemented with 10% fetal calf serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). The murine endothelioma cell line bEnd.3 (ATCC® CRL-2299) was grown in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 2% L-Glutamine. Human monocytic THP-1 cells were seeded with a density of 2.5 × 105 cells/well in a 24 well plate and challenged with sterile culture supernatants or purified alpha-toxin for 30 min at 37°C.
The human brain microvascular endothelial cell line HBMEC (kindly provided by A. Schubert-Unkmeir, Würzburg) and the human lung microvascular endothelial line HuLEC-5a (ATCC® CRL-3244TM) were grown in MCDB 131 Medium with a final concentration of 10% FBS and supplemented with 2.76 μM hydrocortisol, 0.01 ng/ml human epidermal growth factor (hEGF), 2 mM L-glutamine and microvascular growth supplement (25 ml of a 20x stock solution per 500 ml medium). All media were purchased from Thermo Scientific.
ASM activity assays were based on a published protocol (Mühle and Kornhuber, 2017). Briefly, supernatants (SNT) of eukaryotic cells were collected in 1.5 ml centrifugation tubes and immediately stored on ice. The adherent cells were lysed with ASM lysis buffer (250 mM sodium acetate, pH 5.0, 0.1% Nonidet P-40, 1.3 mM EDTA, and 1x Roche Complete protease inhibitor cocktail) for at least 15 min and were scraped off with a pipette tip and collected. Protein concentrations were determined by Bicinchoninic Acid (BCA)-Assay (PierceTM BCA Protein Assay Kit) according to the manufacturer’s instructions. ASM activity in the cell culture SNT was measured by addition of 100 μl SNT to 100 μl ASM SNT assay buffer (200 mM sodium acetate buffer pH 5.0, 500 mM NaCl, 0.2% Nonidet P-40, 500 μM ZnCl2). ASM activity in the whole cell lysate (LYS) was measured in 100 μl lysate volume, equivalent to 1 μg total protein, and addition of 100 μl ASM LYS assay buffer (200 mM sodium acetate buffer (pH 5.0), 500 mM NaCl, 0.2% Nonidet P-40). To each assay buffer solution, 58 pmol of BODIPY-FL-C12 Sphingomyelin (Invitrogen) was added (1:2000 dilution) and samples were either incubated for 24 h (supernatant samples) or for 4 h (lysate samples) at 37°C under constant shaking (300 rpm). Assays were terminated by addition of 250 μl chloroform:methanol (2:1) and brief mixing of the samples. Samples were centrifuged (3 min, 13,000 g), 100 μl of the lower phase was collected and evaporated in a SpeedVac at 45°C for 20 min. Samples were resuspended in 10 μl chloroform:methanol (2:1) and were separated by thin-layer chromatography (TLC) on silica-gel plates (ALUGRAM® Xtra SIL G 5 × 10 cm). Separation was performed in sealed glass chambers containing chloroform:methanol (80:20). The fluorescence of lipids was detected with a Typhoon 9200 Scanner (Amersham) using excitation with the 532 nm Laser.
After incubation of bEnd.3 cells with several different S. aureus toxins, ASM activity was determined using a radioactive assay. Cells were lysed in ASM lysis buffer II (250 mM sodium acetate, pH 5.0, 1% Nonidet P-40 and 100 μM ZnCl2) for 10 min on ice and then tip-sonicated for 10 s. Samples were diluted to 0.1% Nonidet P-40 and incubated with [14C]sphingomyelin (0.05 mCi per sample; 52 mCi/mmol; Perkin Elmer) for 30–60 min at 37°C. Sphingomyelinase reaction was terminated by addition of chloroform:methanol (2:1, v/v). Phases were separated and the upper, [14C]choline chloride containing phase was collected and radioactivity was determined by liquid scintillation counting.
Mammalian cells were challenged with sterile bacterial supernatant at multiplicities of infection (MOI) or concentrations as indicated. Host cell supernatants were collected at indicated time points and centrifuged at 14,000 g for 2 min. For each sample, triplicates were transferred to 96 well cell culture plates and measurement of lactate dehydrogenase (LDH) release into growth medium (as direct indicator of cell membrane integrity loss) was performed with the Cytotoxicity Detection KitPLUS (Roche) according to the supplier’s manual.
Hemolysis of red blood cells (RBC) was performed with sheep and rabbit blood samples (Fiebig Nährstofftechnik, Germany). Briefly, 1 ml whole blood was centrifuged at 150 g for 5 min. Supernatant was carefully removed and cell pellet washed with 0.9% sodium chloride solution. This step was repeated two times. The washed cell pellet was then diluted to yield a 1% cell dilution. Washed, diluted blood was then incubated with diluted α-toxin or bacterial supernatants by shaking for 1 h at 500 rpm and 37°C. Samples then were centrifuged for 1 min at 20,800 g in a tabletop centrifuge to yield cell free supernatants. Triplicates of the sample supernatants were transferred to 96-well microplates and were analyzed in a Tecan InfinitePro200 plate reader at 506 nm ± 9 nm. Relative hemolysis was calculated with reference to maximal lysis (0.1% Triton-X 100) and a negative control (0.9% NaCl).
HuLEC were seeded 2–7 days prior experiment in a density of 2 × 105 cells/ml on glass cover slips. After treatment, cells were fixed for 15 min on RT with 4% PFA and subsequently permeabilized with PBS/5% FBS/0.1% Triton X-100. Cells were then incubated with primary antibody (ZO-1 - Cell signaling #13663 1:400; VE-Cadherin – R&D MAB9381 1:500) overnight in a humid chamber. The following day, glass cover slips were rinsed thrice with PBS, and incubated with secondary antibody for 1 h at RT. Cells were finally rinsed thrice with PBS and mounted on microscopy slides in Mowiol and dried overnight.
HuLECs were seeded in a density of 3 × 104 cells/well one day prior use in a μ-Slide 8 well live-cell chamber (Ibidi). The following day, cells were rinsed thrice in PBS and individually incubated with MCDB131 growth medium supplemented with 4 μM Fluo 3-AM (Invitrogen) for 25 min at 37°C. Cells were again rinsed with PBS and RPMI1640 without phenol red (ThermoFisher, #32404014) complemented with 10% FBS was added. Imaging was performed on a TCS SP5 confocal microscope (Leica Biosystems, Wetzlar, Germany) using a 63x oil-immersion UV objective with a numerical aperture of 1.4. Images were taken at 37°C with a resolution of 1024 × 1024 every 7.5 s for up to 25 min. Treatment with ionomycin, toxin or supernatants was performed during image acquisition after 10 images of untreated cells were recorded. Time series of the treated samples were analyzed with Fiji/ImageJ (Schindelin et al., 2012). Rectangular regions of interest (ROI) for each Fluo 3-AM labeled cell were defined. The average intensity over time was determined by the ImageJ plugin Time Series Analyzer (version 3). Fluorescence intensities were calculated relative to the initial time points (before addition of toxins, controls or bacterial supernatants) and were plotted using GraphPad Prism.
HuLECs were seeded in a density of 1 × 105 cells/well 4 days prior the experiment. The cells were washed thrice with PBS and then challenged for 30 min at 37°C, 5% CO2. At the start of each challenge, LAMP1 monoclonal antibody (Santa Cruz, H4A3) in a concentration of 1 μg per 1 × 106 cells was added. The cells were then rinsed five times with PBS and fixed by addition of 4% PFA. Cells were again rinsed thrice with PBS, permeabilized and blocked with PBS containing 0.1% saponin and 5% FBS for 1 h at RT. Secondary antibody (Alexa Fluor 488-coupled anti-mouse IgG, Santa Cruz) was diluted 1:1000 in PBS/0.1% saponin/5% FBS/Hoechst 33258 and was left on samples for 1 h at 37°C. Cells were finally washed five times with PBS, mounted in Mowiol and dried overnight at RT. Fluorescence was detected with a Leica TCS SP5 confocal laser scanning microscope (Leica Biosystems, Wetzlar, Germany) using a 63x oil-immersion objective (Numerical Aperture 1.4).
bEnd.3 cells were seeded 48 h before the experiment in 24 well (1 × 105 cells/well). Before α-toxin challenge, cells were washed thrice with HEPES buffered saline (H/S: 132 mM NaCl, 20 mM HEPES pH 7.3, 5.4 mM KCl, 1 mM CaCl2, 0.7 mM MgCl2, 0.8 mM MgSO4) or with calcium free phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 1.15 mM KH2PO4, 6,5 mM Na2HPO4). After indicated times of toxin incubation, cells were lysed for 5 min in SDS lysis buffer (25 mM HEPES, 0.1 % SDS, 0,5 % deoxycholate, 1 % triton, 10 mM EDTA, 10 mM NaPP, 10 mM NaF, 125 mM NaCl, 10 mM NaVO4, 10 μg/ml aprotinin/leupeptin) on ice. Samples were centrifuged at 20,800 g for 5 min at 4°C and supernatants were added to 5x SDS Laemmli buffer and boiled. An equivalent of about 6,000 cells of each sample was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were blotted onto nitrocellulose membranes at 4°C overnight. Membranes were blocked in Starting Block Tris-buffered saline (TBS) blocking buffer (Thermo-Fisher Scientific, Darmstadt, Germany) for 1 h and incubated with antibodies specific for p38 (Cell Signaling Technology, #9212) or phospho-p38 (Cell Signaling Technology, #9211), respectively. All antibodies were diluted 1:1000 in blocking buffer and were incubated for 1 h at room temperature. Blots were washed five times with TBS/0.05% Tween 20 (TBS/T), incubated with alkaline phosphatase coupled anti-rabbit antibody (Abcam, Berlin, Germany) for 1 h, washed again five times with TBS/T and afterward two times in alkaline wash buffer, and developed with CDP-STAR with Nitro-Block II Enhancer system (Perkin Elmer, Rodgau, Germany).
HuLEC were seeded with a density of 2 × 105 cells/well in a 12 well-plate and cultivated for 3 days. Cells were washed thrice in PBS and treated with 1 μg/ml α-toxin, or 10% sterile-filtered S. aureus JE2 SNT for 15 min in cell culture medium (MCDB131 medium containing 2 mM L-glutamine and 10% FBS inactivated at 70°C). After treatment, cells were washed once with PBS on ice and lysed by adding per well 80 μL 2x Laemmli buffer (100 mM Tris–HCL pH 6.8, 4% SDS, 20% glycerol, 2% beta-mercaptoethanol, 25 mM EDTA, 0.04% bromophenol blue.). Lysates were incubated for 10 min on 95°C and separated by 10% SDS-PAGE. Semi-dry Western transfer on a PVDF membrane (Sigma) was performed for 2 h at RT with 60 mA/10 V per blot. Membranes were blocked for 1 h in 5% bovine serum albumin (BSA; Albumin Fraktion V, Roth) in TBS/T at room temperature. Incubation with primary antibody (p38 [CST #9212] and phospho-p38 [CST #9215], 1:500 dilution) was performed overnight at 4°C. Membranes were washed thrice in TBS/T and incubated with secondary horseradish peroxidase-conjugated antibody (Dianova 111-035-144, 1:2500 in 5% BSA in TBS/T) for 1 h at room temperature. Subsequently, blots were washed thrice with TBS/T, developed using the ECL system (Pierce) and imaged on an Intas HR 16-3200 chemiluminescence reader.
One way analysis of variance (ANOVA) or Kruskal-Wallis rank sum tests determined significant differences in the analyzed data, followed by Sidka’s multiple comparison test or two-tailed Student’s t-tests as post-hoc tests using GraphPad Prism.
Murine endothelia have been shown to degrade tight junctions upon challenge with staphylococcal α-toxin thereby leading to lung edema in vivo (Seeger et al., 1990; Bartlett et al., 2008; Becker et al., 2018). We here treated bEnd.3 endothelial cells with staphylococcal α-toxin and investigated phosphorylation of p38 MAPK, which has been previously shown to be activated by α-toxin in epithelial cells (Eiffler et al., 2016). 10–20 min after addition of the toxin we observed an increase in phospho-p38 (Supplementary Figure 1A). This increase lasted for about 60 min, after which phosphorylation of p38 decreased. Phosphorylation of p38 was abolished when host cells were treated with the ADAM10 inhibitor GI254023X (Selleck Chemicals, Biozol, Germany) prior to toxin addition (Supplementary Figure 1B).
We next investigated, if p38 phosphorylation depended on the extracellular divalent cations Ca2+ and Mg2+. We therefore added α-toxin to host cells in H/S buffer or Ca2+/Mg2+-free PBS. Cells incubated in medium without divalent cations showed no change in α-toxin-dependent p38 phosphorylation (Supplementary Figure 1C), demonstrating that the α-toxin-dependent increase in phospho-p38 at the 15 min time point required divalent cations in the medium.
We then tested α-toxin dependent activation of p38 in a human microvascular endothelial cell line (HuLEC) and observed an increase of relative phospho-p38 levels in HuLEC treated with 1 μg/ml purified α-toxin (1.81 ± 0.46 fold relative to the medium control), wild-type (WT) S. aureus culture supernatant (SNT; 2.12 ± 0.45 fold), and SNT of the Δhla mutant complemented with 1 μg/ml α-toxin (2.21 ± 0.51). Δhla SNT was leading to significantly less p38 activation (1.46 ± 0.31 fold relative to control; p = 0.0068) when compared to WT SNT or purified α-toxin (p = 0.033), whereas differences in normalized fold-changes between either purified α-toxin (p = 0.064) or WT SNT (0.61) and complemented Δhla SNT were not significant (n.s.; Figure 1A). This demonstrated that α-toxin activated p38 MAPK in the HuLEC in a similar fashion as observed for the murine endothelial cell line bEnd.3 (Supplementary Figure 1).
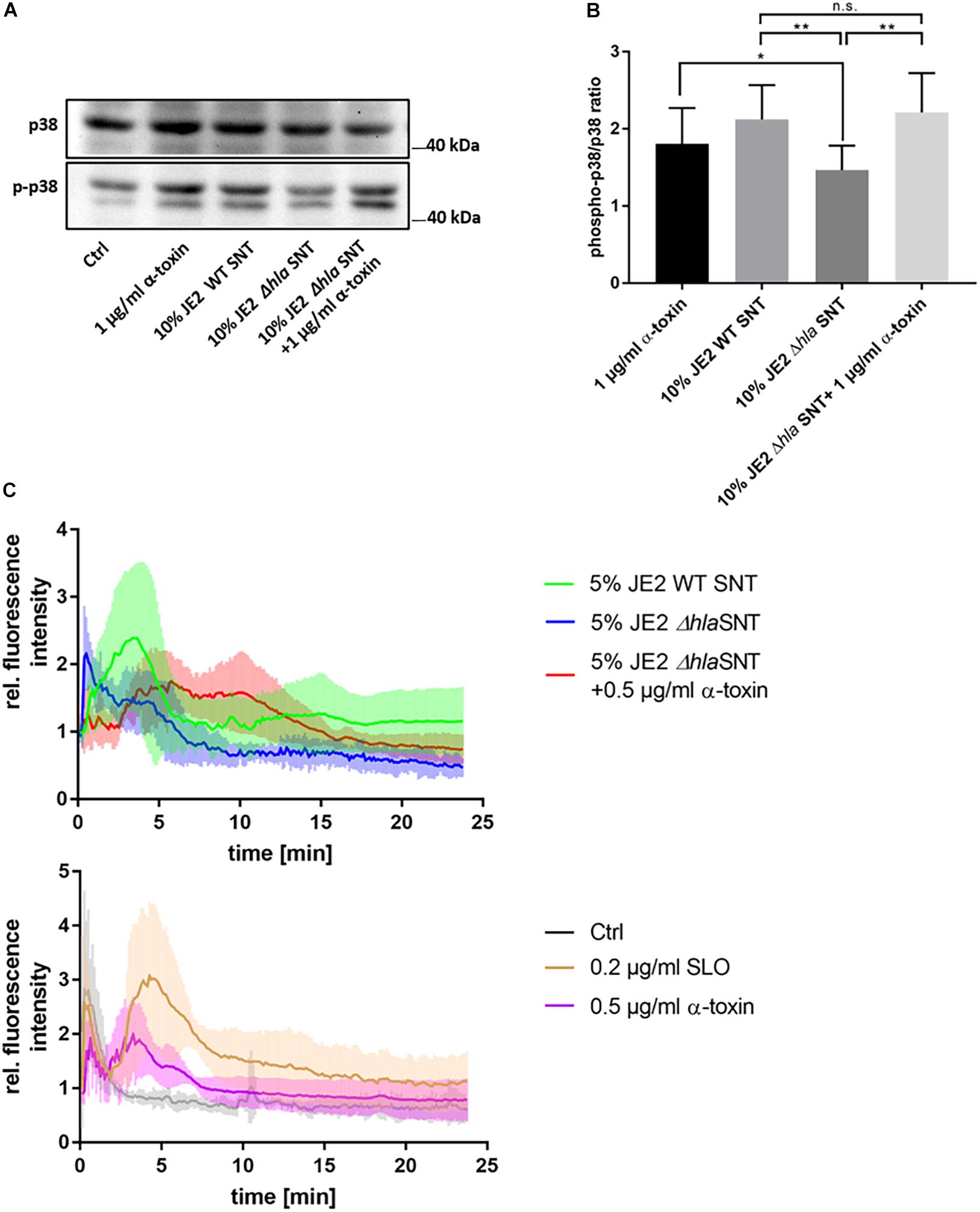
Figure 1. Human microvascular endothelial cells (HuLEC) treated with α-toxin induce phosphorylation of p38 MAPK. (A,B) HuLEC were treated with 1 μg/ml α-toxin, or SNT of S. aureus WT, a Δhla mutant or Δhla SNT complemented with 1 μg/ml α-toxin and phosphorylation of p38 was determined densitometrically by Western blot. Ratios of phospho-p38/p38 ratios were calculated and normalized to the untreated control, whose ratio was set to 1. Significance was established by ANOVA followed by paired Student’s t-test as post-hoc test. *: p < 0.05; **: p < 0.01. (C) Calcium-influx is detected after α-toxin challenge. HuLEC cells were incubated with 4 μM Fluo 3-AM for 25 min at 37°C/5% CO2 and subsequently analyzed for calcium-specific increase of fluorescence by time-lapse imaging. After 75 s (10 time frames), cells were treated and the Fluo-3 fluorescence was recorded in 7.5 s intervals. The top panel shows the fluorescence of cells treated with 5% supernatant of JE2 WT (JE2 SNT) or mutant JE2 lacking α-toxin (JE2 Δhla SNT), or JE2 Δhla SNT complemented with 0.5 μg/ml α-toxin. The bottom panel shows the fluorescence recorded for 0.2 μg/ml SLO (3.32 nM), 0.5 μg α-toxin (15 nM) or the negative control (bacterial culture medium only). Shown are mean relative fluorescence intensities of four independent experiments +/- standard deviation.
Next, we investigated by time lapse imaging α-toxin-dependent changes in cytosolic Ca2+ levels in HuLEC (Figure 1C and Supplementary Video 1). Cells reproducibly reacted to bacterial supernatants of wild-type S. aureus and purified α-toxin with an increase in intracellular Ca2+ levels (Figure 1C and Supplementary Video 1). These elevations in cytosolic Ca2+depended on the presence of α-toxin, although the time points of the Ca2+ peaks varied between the experiments. Treatment with bacterial supernatants lacking α-toxin (JE2 Δhla SNT) prevented the strong Ca2+ signal in the endothelial cells, which was reinstated by adding 0.5 μg/ml α-toxin to culture SNT of the Δhla strain. We observed a similar Ca2+ signal when Fluo-3 loaded cells were treated with SLO, another proteinaceous PFT (Figure 1C). This SLO amount is equivalent to one fifth of the concentration of α-toxin monomers, demonstrating the differential sensitivity of HuLEC cells for both toxins.
For complementation of Δhla SNT with purified α-toxin we determined the cytotoxic and hemolytic potential of bacterial supernatants and compared it with a commercially available preparation of α-toxin. Cytotoxicity was tested with the monocytic cell line THP-1. Cytotoxicity of bacterial SNT from the WT S. aureus strain JE2 was reduced in the Δhla mutant (p = 0.014) which was reflected by a similar increase in cytotoxicity upon addition of 1 μg/ml α-toxin to medium. Supernatants of the non-cytotoxic strain Cowan I (p = 0.0001) and a JE2 mutant in the two-component system saeR (ΔsaeR, p = 0.0002) were drastically reduced (Supplementary Figure 2A). Our data thus suggests that the residual cytotoxicity observed for the Δhla mutant is due to Sae-dependently produced leukocidins, which have been shown to target monocytic cells such as THP-1. Hemolysis was tested on washed blood from rabbit (Supplementary Figures 2B,C) and sheep (Supplementary Figure 2D). A titration of hemolytic activity of different toxin concentrations on rabbit RBC determined half-maximal hemolysis at 0.375 μg/ml for our preparation of the PFT. 0.5 μg/ml of toxin were approximately equivalent to a 5% dilution of WT SNT (Supplementary Figure 2C). Similar results were obtained for RBC derived from sheep blood. As expected rabbit RBC were more sensitive for α-toxin hemolysis when compared to sheep RBC (Supplementary Figure 2D).
Since one Ca2+-dependent pathway of membrane repair after PFT insult involves exocytosis of peripheral lysosomes (reviewed in Bischofberger et al., 2012; Etxaniz et al., 2018; Brito et al., 2019), we tested whether the supernatant of S. aureus cultures would transiently recruit lysosomal vesicles to the cell surface. In order to test this, we added antibodies directed against the lysosomal membrane protein LAMP-1 to the culture medium and left the cells untreated (Figure 2A) or treated the cells with purified α-toxin (Figure 2B), ionomycin (Figure 2C), or bacterial supernatants of wild-type bacteria or an isogenic α-toxin mutant. After 30 min, we fixed and permeabilized the cells, and detected anti-LAMP-1 antibodies with fluorescently labeled secondary antibodies. We thereby specifically detected LAMP-1 proteins that were surface-exposed during the course of the challenge (Figure 2D). By fluorescence microscopy we enumerated LAMP1 signals in the cells and observed significantly increased numbers of LAMP-1 positive vesicles in cells treated with ionomycin and supernatants from α-toxin-proficient bacteria when compared to supernatant from the Δhla mutant (Figure 2E).
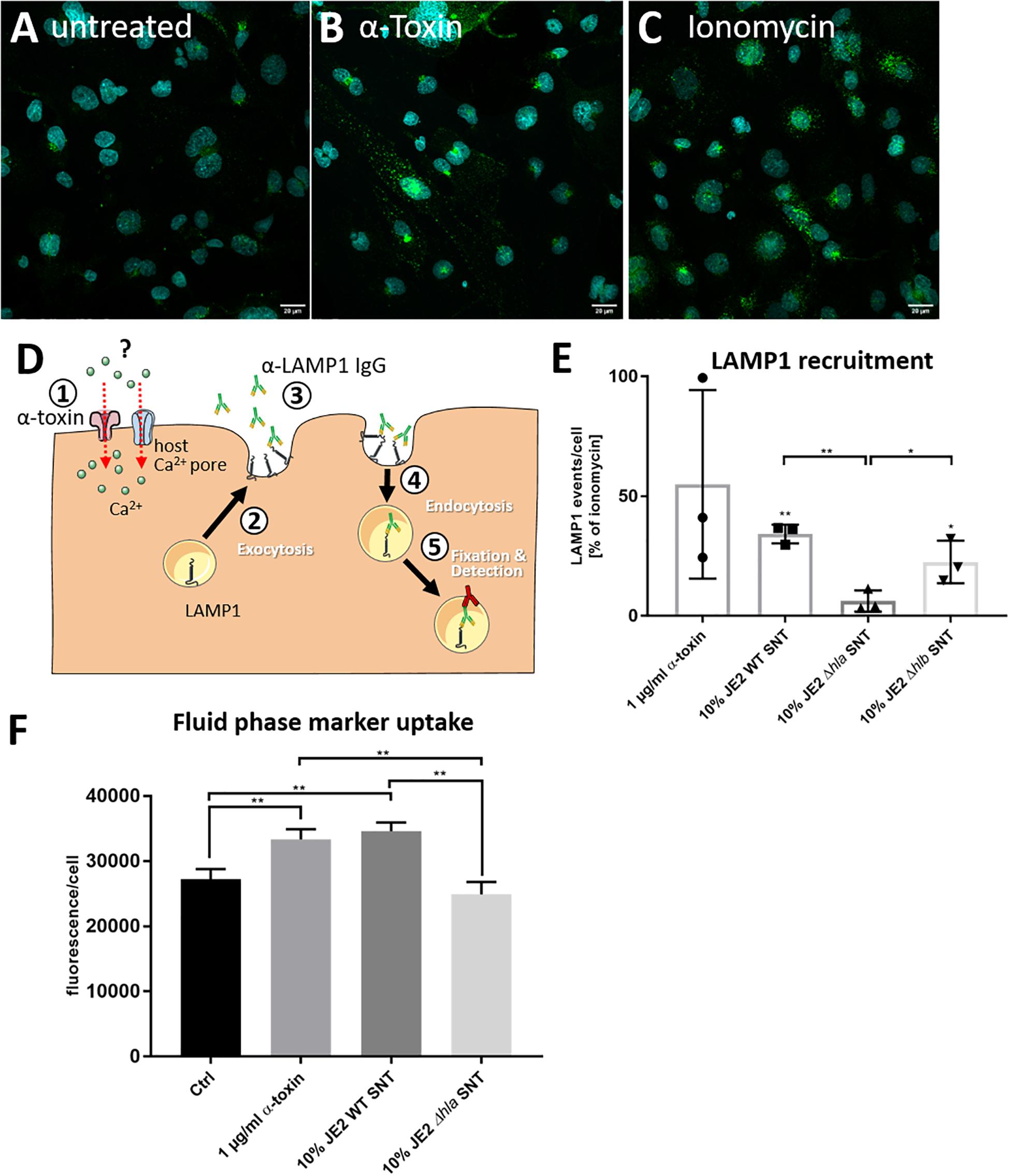
Figure 2. S. aureus induces recruitment of LAMP-1 positive vesicles to the plasma membrane. HuLEC cells were left untreated (A) or were treated for 30 min with 1 μg/ml purified α-toxin (B) or 1 μM of the Ca2+ ionophore ionomycin (C). Fluorescence microscopy in untreated cells and cells treated with α-toxin or ionomycin demonstrates more and stronger LAMP1 signals in the treated cells. (D) Assay schematic: 1 μg LAMP1 antibody per 1 × 106 cells was added together with α-toxin ①. Either by directly establishing a Ca2+ permissive pore or indirectly by activating endogenous host Ca2+ channels, the cytosolic Ca2+ concentration rises. Thereby, the antibodies can recognize exocytosed lysosomal membranes ② currently displayed at the host cell surface ③ as well as LAMP1-molecules which were taken up again during the course of the treatment ④. After fixation and permeabilization, an Alexa488-labeled secondary antibody was added and LAMP1 was detected by fluorescence microscopy ⑤. (E) Microscopic quantification of LAMP1-positive vesicles left untreated, or treated with α-toxin- and ionomycin, or cells treated with 10% S. aureus culture supernatants (SNT) of wild-type bacteria (JE2) or its isogenic α-toxin mutant (JE2 Δhla) demonstrates that the LAMP1 surface recruitment is abrogated in the Δhla strain. (F) Epithelial cells increase uptake of a membrane impermeable fluid phase dye after challenge with α-toxin. HBMEC were incubated in cell culture medium containing Alexa633-hydrazide and either 1 μg/ml purified S. aureus α-toxin or 10% SNT from JE2 WT or its isogenic Δhla mutant was added. After 30 min cells were detached by trypsinization and fluorescence was analyzed by flow cytometry. For statistical analysis, experiments in E and F were independently conducted three times (n = 3). A Kruskal-Wallis rank sum test determined significant differences in the analyzed data, which were further tested with pairwise Student’s t-tests. *: p < 0.05, **: p < 0.01; ***: p < 0.001. For more details see text.
We next added the membrane impermeable fluorescent dye Alexa633 hydrazide to the cell culture medium. The dye thus served as fluid phase marker. We then added either 1 μg/ml purified S. aureus α-toxin or 10% bacterial culture supernatant from either S. aureus JE2 wild-type (WT) or its isogenic Δhla mutant for 15 min. After the incubation, cells were washed, detached by trypsinization and analyzed for Alexa633 fluorescence by flow cytometry. We observed an increased uptake of the membrane impermeable fluid phase dye after challenge with purified α-toxin or S. aureus WT SNT, whereas fluorescence of cells treated with SNT from the Δhla mutant were at the level of the negative control (Figure 2F).
Thus our data suggest that upon treatment with α-toxin or bacterial SNT containing the toxin, lysosomes are transiently recruited to the cell surface.
We have previously shown that the acid sphingomyelinase is activated rapidly after staphylococcal α-toxin challenge in macrophages and endothelial cells (Becker et al., 2018). Therefore, we investigated ASM activity in host cells by treatment with sterile bacterial supernatants. For that purpose we adapted a previously published ASM assay (Mühle and Kornhuber, 2017) which is based on separation of fluorescently labeled C12 sphingomyelin and ceramide by thin-layer chromatography. However, we observed a prominent ASM activity in our cell culture medium, which was caused by fetal bovine serum (FBS) (Supplementary Figure 3). We here used culture supernatants of S. aureus, which are known to contain copious amounts of short amphiphilic cytolytic peptides, the so called phenol-soluble modulins (PSM) (Surewaard et al., 2012). We therefore had to include FBS in the cell culture media, since serum lipoproteins inhibit host cytolysis by PSM. Heat-inactivation of FBS at 55°C for 30 min (Spence et al., 1989) reduced ASM activity, but only heat-treatment of FBS for 1 h at 70°C resulted in complete loss of ASM activity, whereas PSM-dependent cytolysis was still abrogated under these conditions (Supplementary Figure 4).
Using the activity assays described above, we identified an additional background sphingomyelinase activity. The S. aureus genome harbors the structural gene for a bacterial sphingomyelinase (bSMase) β-toxin (β-hemolysin, hlb). We found sphingomyelinase activity not only in culture supernatants from the strongly β-toxigenic strain 6850, but also in the MRSA strain JE2, suggesting that the latter strain had an excised bacteriophage. bSMase activity was only completely abolished in JE2 mutants, which carried an additional transposon insertion within the structural hlb gene. By contrast, the bacterial phospholipase C (plc) activity did not contribute to sphingomyelin degradation (Supplementary Figure 5). Compared to supernatants from S. aureus strains 6850 and RN4220, sphingomyelinase activity of the clinically relevant S. aureus strain JE2 was smaller but still detectable.
With the adapted assay conditions we next compared effects of bacterial culture supernatants from S. aureus or purified toxin on endothelial cells and challenged the cells with 10% bacterial culture supernatant of wild-type S. aureus JE2, or its isogenic Δhla or Δhlb mutants, or treated the cells with 1 μg/ml purified α-toxin. At different time points during a 30-min incubation we collected both cell culture medium and cell lysates. Both fractions were assayed for ASM activity (Figures 3A–D). We thereby observed an α-toxin-dependent increase in host ASM activity in the culture medium over time, whereas ASM activity in cell lysates decreased (Figures 3A–D). The absence of lactate dehydrogenase (LDH) in the media indicated that the endothelial cell membranes remained intact during the treatment (Supplementary Figure 6) suggesting that ASM is released by peripheral lysosomes that fuse with the plasma membrane. ASM release by HuLEC was also induced by treatment with streptolysin O (SLO) as well as the calcium ionophore ionomycin (Figure 3E), which again is reminiscent of membrane repair processes leading to ASM release (Andrews et al., 2014). The residual sphingomyelinase activity observed in SNT of the Δhla strain may be derived from the bacterial SMase β-toxin, which can be expressed by S. aureus (Supplementary Figure 5, see also above). We also tested an hlb insertional mutant of S. aureus JE2, which was β-toxin deficient (Supplementary Figure 5). Challenging endothelial cells with supernatant of this mutant strain showed similar dynamics of ASM activation when compared to cells challenged with supernatants of the wild-type strain. This demonstrated that the bSMase does not contribute to the observed effects (Figure 3C). We next treated HuLEC with bacterial culture supernatant of the α-toxin mutant genetically complemented with an expression plasmid for α-toxin (pHla-Cerulean) or a vector control (pCerulean; Giese et al., 2009). We observed that bacterial supernatants from α-toxin-expressing S. aureus JE2 Δhla demonstrated significantly more ASM release when compared to the medium or vector controls (Figure 3F).
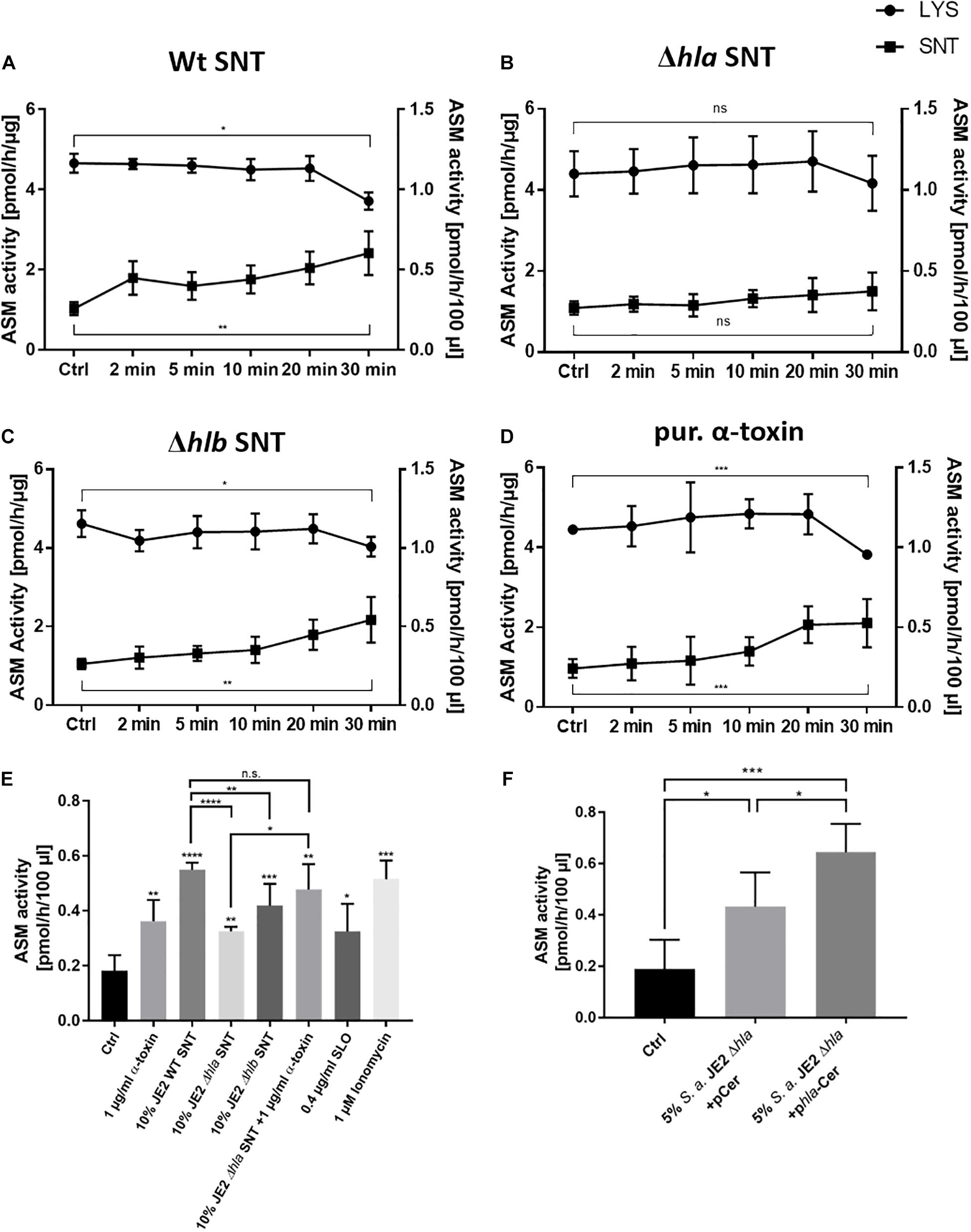
Figure 3. Staphylococcal α-toxin induces ASM release into the supernatant of endothelial cells: HuLEC were either challenged with 10% bacterial culture supernatant of (A) S. aureus JE2 WT (SNT WT), (B) JE2 lacking α-toxin (Δhla SNT) or (C) JE2 lacking β-toxin (Δhlb SNT), or were treated with 1 μg/ml of purified α-toxin (D) or were left untreated (Ctrl). ASM activity was measured at the indicated time points in cell culture medium (“SNT”, closed squares) as well as in endothelial cell lysate (“LYS”, closed circles). ASM activity of lysates are given in pmol/h/μg (left y-axis) and were normalized to the total protein amount in cell lysates. ASM activities in culture media are given as pmol/h/100 μl (right y-axis). (E) LDH assays of the culture supernatants in (A–D) demonstrate that the endothelial cells remained intact over the course of the experiment. (F) HuLEC were challenged for 30 min with the indicated samples. The reduced ASM release in the Δhla SNT with regard to WT SNT is complemented by external addition of 1 μg/ml α-toxin. ASM release is also induced by 0.4 μg/ml SLO and 1 μM ionomycin. (E,F) n = 5 for Ctrl, 1 μg/ml α-toxin, WT SNT and Δhlb SNT; n = 3 for the remaining samples. Significance in the dataset was determined by ANOVA and further analyzed by unpaired Student’s t-test. *: p < 0.05, **: p < 0.01; ***: p < 0.001; ns: not significant.
Thus, our results demonstrate that peripheral lysosomes are recruited to the endothelial cell surface upon addition of staphylococcal α-toxin suggesting induction of a membrane repair mechanism that involves the release of lysosomal ASM. In line with this, the challenge of endothelial cells with supernatants lacking α-toxin failed to recruit ASM to the extracellular space. This indicated that α-toxin is the major toxin involved in this process in endothelial cells. Supporting this hypothesis, we observed that HuLEC cells, which were grown for 7 days on a collagen matrix and displayed intact tight junctions, were not lysed by bacterial culture supernatants of the S. aureus JE2 Δhla mutant, whereas supernatant collected from the wild type readily lead to tight junction breakdown and endothelial cell lysis (Supplementary Figure 7). Since our previous study determined that α-toxin induced ASM activity in murine bEnd.3 cells, we also tested, if other staphylococcal pore-forming toxins would lead to ASM activation in bEnd.3 cells. We thus treated the cells with combinations of purified subunits of the bi-component pore-forming toxins hemolysin γ (HlgA + HlgB, or HlgC + HlgB, respectively) or the leukocidins LukDE or LukSF (Supplementary Figure 8). None of these combinations led to ASM activation over the course of a 60-min treatment. This again demonstrates that α-toxin is the main staphylococcal toxin leading to ASM release by endothelial cells.
α-toxin is secreted by S. aureus as a monomer and is assembled as a heptameric protein complex after binding to non-specific lipid receptors (Schwiering et al., 2013; Ziesemer et al., 2019) or the metalloprotease ADAM10 (Wilke and Bubeck Wardenburg, 2010) on target membranes. Due to its small pore diameter, it was suggested that S. aureus α-toxin pores are only permeable for small cations, predominantly potassium (Harshman et al., 1989; Jonas et al., 1994), although the permeability of the plasma membrane for Ca2+ and larger molecules such as ATP was also demonstrated (Jonas et al., 1994; Gierok et al., 2014). A hallmark consequence of cell intoxication with pore-forming toxins is the uncontrolled exchange of ions between the extra- and intracellular space (Dal Peraro and van der Goot, 2016). Intracellular concentration changes of specific ions induce tailored cell responses in order to re-establish homeostasis (Cooper and McNeil, 2015). When α-toxin elicits Ca2+ influx, activation of kinases critical for cell autonomous defense mechanisms ensues (Rosen et al., 1994; Liu et al., 2016). Central to this mechanism is the phosphorylation of the mitogen-activated protein kinase p38 (p38 MAPK), which is activated upon exposure of mammalian cells to staphylococcal α-toxin (Husmann et al., 2006; Kloft et al., 2009; Eiffler et al., 2016) and which is also important for target cell recovery from the toxin insult (Husmann et al., 2006). Accordingly, we detected phosphorylation of p38 MAPK after incubation with purified staphylococcal α-toxin in human (Figure 1) and murine endothelial cells (Supplementary Figure 1). p38 phosphorylation was reduced upon removal of extracellular divalentions or upon inhibition of the metalloprotease ADAM10 (Supplementary Figure 1), the receptor for α-toxin on target membranes (Wilke and Bubeck Wardenburg, 2010). Further, it has been demonstrated by others that α-toxin heptamerization is inhibited upon addition of the ADAM10 antagonist GI254023X (Inoshima et al., 2011), whereas the protease activity of ADAM10 is not required for α-toxin pore formation (Ezekwe et al., 2016). We here observed that endothelial cell p38 phosphorylation in presence of α-toxin was abrogated by pharmacological inhibition of ADAM10 with GI254023X (Supplementary Figure 1B).
Upon α-toxin treatment we observed a rise in intracellular Ca2+ concentrations, which peaked in presence of purified α-toxin or bacterial SNT containing the PFT (Figure 1C and Supplementary Video 1). We observed similar peaks of Fluo-3 fluorescence when applying the PFT streptolysin O (Figure 1C). By contrast, treatment with the Ca2+ ionophore ionomycin led to a quickly appearing and strong Ca2+ peak (Supplementary Video 1). This strong Ca2+ influx after ionomycin treatment has been detected over a variety of cell types (Yoshida and Plant, 1992; Mason and Grinstein, 1993; Gutierrez et al., 1999; Vasilev et al., 2012), whereas oscillating Ca2+ fluxes are often observed after mechanical cell injury (Reddy et al., 2001) and eventually result in calcium spikes (Justet et al., 2019). Thus our data suggests that α-toxin treatment of human and murine endothelial cells leads to p38 activation, which depends on divalent cations in the medium and ADAM10. It is currently unclear, if α-toxin itself thereby forms a Calcium-conductive channel or if challenge with the toxin results in activation of endogenous host Ca2+ channels, endogenous pores or even ruptures as has been discussed previously (von Hoven et al., 2019).
It has been observed that phosphorylation of p38 MAPK already occurs at low doses of α-toxin, whereas ADAM10 dependent breakdown of E-cadherin in toxin-challenged keratinocytes required higher concentrations of α-toxin (von Hoven et al., 2016). This may correspond with alternate states of membrane permeability, which might be caused by different α-toxin concentrations or the different cell types (von Hoven et al., 2019). This is further supported by the induction of Ca2+-independent membrane repair after α-toxin insult of fibroblasts (Valeva et al., 2000).
Plasma membrane disruption e.g., by PFT results in changes in intracellular ion-concentrations, which in turn trigger cell responses leading to membrane repair or cell death (reviewed in Bischofberger et al., 2012; Etxaniz et al., 2018; Brito et al., 2019). p38 is implicated in survival pathways such as autophagy as well as plasma membrane repair after challenge with staphylococcal α-toxin (Husmann et al., 2006; Kloft et al., 2009; Kao et al., 2011) and interference with p38 MAPK-dependent membrane recovery pathways caused host cell death (Husmann et al., 2006). Calcium-dependent membrane repair pathways for instance include recruitment of annexins to damaged membrane areas where they assemble to clog pores. The survival mechanism against mechanical membrane injury (Reddy et al., 2001) as well as insult by some PFT involves the Ca2+-dependent exocytosis of peripheral lysosomes to the cell surface thereby releasing membrane repair enzymes such as lysosomal ASM into the extracellular environment. ASM-dependent conversion of outer leaflet sphingomyelin to ceramide facilitates endocytosis of the toxin pores (reviewed in Andrews, 2019; e.g., Brito et al., 2019).
In line with the latter repair strategy, our ASM activity assays revealed that intoxication with S. aureus α-toxin leads to a re-partitioning of ASM to the extracellular space (Figure 3). These findings are supported by the increased exposure of the lysosomal membrane protein LAMP-1 on the plasma membrane after α-toxin challenge. This suggested that the ASM activity present in the cell culture media originated from peripheral lysosomes that fuse with the plasma membrane to deliver membrane repair enzymes.
LAMP1 exposure to the extracellular environment is described to be rather short, since these externalized lysosomal membranes are quickly re-endocytosed (Andrews, 2017). Accordingly, we observed increased rates of endocytosis of a fluorescent fluid phase marker, which was α-toxin-dependent in our observations (Figure 2F). Since host cells also have been shown to recover from α-toxin injury by endocytosis of the affected membrane patches (Husmann et al., 2009), which is also observed for other PFTs (Idone et al., 2008), host cell plasma membrane permeabilized by staphylococcal α-toxin may dispose of the pores by recruitment of peripheral lysosomes, the accompanied release of repair enzymes such as ASM, as well as re-endocytosis of the additional membrane fractions.
ASM is a crucial factor for the conversion of sphingomyelin to ceramide in the extracellular leaflet of the injured plasma membrane (Andrews et al., 2014). Since this removal of plasma membrane sphingomyelin mediates resistance to α-toxin (Ziesemer et al., 2019) it is tempting to speculate that ASM release of injured mammalian cells do not only initiate plasma membrane repair mechanisms, but also may decrease membrane sensitivity against the toxin.
In addition, α-toxin pores were found to be excreted, if cells were unable to remove the pore by proteolysis (Husmann et al., 2009). Thus, the processes of pore endocytosis, excretion as well as release of ASM to the culture medium may be interlinked in certain cell types.
In order to assess ASM activity in the culture supernatants of endothelial cells treated with staphylococcal culture supernatants, we had to include fetal bovine serum in order to inactivate phenol-soluble modulins, but were confronted with a strong background ASM activity in the serum. This activity was reliably inactivated by treatment at 70°C for 1 h (Supplementary Figure 3), while the serum was still inhibiting cytolysis by PSMs (Supplementary Figure 4). Further, we found that the commonly used laboratory strain RN4220 and the strongly cytotoxic strain 6850 could not be used for our ASM analyses due to their strong production of bacterial SMase. Although β-toxin is a neutral SMase (Doery et al., 1963), its activity is still detectable under acidic assay conditions. Clinical strains are often positive for so-called β-converting phages, which inactivate the bSMase by lysogeny (Goerke et al., 2006; Katayama et al., 2013). However, reactivation of the phage results in β-toxin expression (Salgado-Pabon et al., 2014; Tran et al., 2019) which was recently corroborated by bacterial single cell RNAseq in S. aureus culture (Blattman et al., 2020). We found sphingomyelinase activity in S. aureus JE2 was abrogated only by an additional insertional mutation in the hlb gene. Although bSMase activity in strain JE2 was rather low and only measurable in concentrated bacterial supernatants, we recommend testing each genetic background of S. aureus for a potential confounding SMase activity.
In summary, we here show – to our knowledge for the first time – that treatment with S. aureus α-toxin recruits acid sphingomyelinase to the extracellular space of endothelial cells. Our data further suggest that α-toxin is the major staphylococcal toxin causing the observed effects in these cells, since supernatant of a S. aureus α-toxin mutant did not induce ASM release and tight-junction disruption in endothelial cells. In addition, murine endothelial cells treated with other purified staphylococcal toxins did not show increase in ASM activity.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
DK, MR, TK, KB, EG, and MF designed the experiments and wrote the manuscript. DK, MR, TK, CS, KP, and KB performed the experiments. DK, MR, TK, KB, and MF analyzed the data. All authors contributed to the article and approved the submitted version.
We thank the Deutsche Forschungsgemeinschaft for funding this project within the research group FOR2123 (to EG and MF) and the training group GRK2581 (to MF). This publication was supported by the Open Access Publication Fund of the University of Wuerzburg.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are indebted to Simon Peters and Alexandra Schubert-Unkmeir (Institute for Hygiene and Medical Microbiology, Universitätsklinikum Würzburg) as well as Kathrin Stelzner and Nadine Vollmuth (Chair of Microbiology, University Würzburg) for materials or valuable help with techniques.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.694489/full#supplementary-material
Supplementary Figure 1 | α-toxin induces p38 MAPK phosphorylation which is dependent on divalent cations in the medium and ADMA10: bEnd.3 were incubated with 10 μg/ml α-toxin for 5–120 min and phosphorylation of p38 was determined by Western blot (A–C). Pre-treatment of cells with the ADAM10 inhibitor GI254023X inhibited p38 phosphorylation (B). Ca2+ is required for p38 activation, since a buffer lacking Ca2+ reduced α-toxin induced p38 phosphorylation. Shown are representative Western blots of three independent experiments. Phosphorylation of p38 was determined densitometrically and was normalized by signals measured for total p38. p38 is activated significantly during a period ranging from 10 to 60 min post-intoxication (A). (B) Pre-treatment of cells with the ADAM10 inhibitor GI254023X inhibited p38 phosphorylation significantly at all investigated time points. (C) Ca2+ is required for p38 activation, which is observed at 15 and 30 min after intoxication. Activation is not observed in buffers lacking Ca2+. Statistical analysis was performed with GraphPad Prism using One way ANOVA followed by Sidka’ multiple comparison test. ∗: p < 0.05; ∗∗: p < 0.001; ****: p < 0.0001.
Supplementary Figure 2 | THP-1 monocyte cytotoxicity and hemolysis of bacterial culture supernatants and α-toxin. (A) Cytotoxicity of S. aureus JE2 SNT is reduced when compared to SNT of its isogenic α-toxin mutant (Δhla), Cowan I, or a saeR mutant (ΔsaeR), whereas 1 μg/ml α-toxin lysed host cells. Statistical analysis was performed with GraphPad Prism using One-way ANOVA and a post-hoc unpaired Student’s t-tests. ∗: p < 0.05; ∗∗: p < 0.001; ∗∗∗: p < 0.0001. Hemolysis assays were performed using washed rabbit (B,C) and sheep blood (D). For details see main manuscript text.
Supplementary Figure 3 | FBS exhibits a strong ASM activity which is inactivated upon treatment at elevated temperatures. (A) MCDB131 medium supplemented with untreated of heat inactivated (55°C for 30 min) FBS was tested for its ASM activity. Heat-inactivation for 30 min at 55°C reduced ASM activity. (B) Heat inactivation for 1 h at 70°C (Medium 70°C) abolishes ASM activity of FBS completely. Thin-layer results are representative images of multiple experiments.
Supplementary Figure 4 | S. aureus phenol soluble modulins are inhibited by heat-treated FBS. Epithelial cells were challenged with increasing concentrations of culture supernatants of S. aureus overnight cultures diluted in cell culture media. Additionally 70°C-treated FBS was added or the assays were left FBS-free. The heat-treated FBS shows the same inhibition of cytotoxicity as normal FBS. n = 3. Significance was calculated with one-way ANOVA followed by post-hoc Student’s t-tests. ∗∗∗: p < 0.001; ns: not significant.
Supplementary Figure 5 | S. aureus exhibits toxin specific sphingomyelinase activity. Equal amounts of supernatants (SNT) from S. aureus overnight cultures were tested for sphingomyelinase activity. S. aureus exhibited a strain-specific conversion of sphingomyelin to ceramide with strains 6850 and RN4220 showing strongest bSMase activities. Even the clinical strain JE2 demonstrates measurable activities, although the responsible ORF hlb is inactivated by phage lysogeny in this strain suggesting phage reactivation. Sphingomyelinase activity is absent from insertional mutants within hlb (termed “Δhlb”) in strain JE2 and 6850, as well as in strain Cowan I. Thin-layer results are representative images of three independent experiments.
Supplementary Figure 6 | Lactate dehydrogenase is not released from HuLEC treated with bacterial supernatants. HuLEC were either challenged with 10% bacterial culture supernatant of S. aureus JE2 WT (SNT WT), JE2 lacking α-toxin (Δhla SNT) or JE2 lacking β-toxin (Δhlb SNT), were treated with 1 μg/ml of purified α-toxin, or were left untreated (Ctrl). Cell supernatants were collected and were tested for LDH release. Shown is LDH release as per cent of the lysis control. LDH assays of the culture supernatants demonstrate that the endothelial cells remained intact over the course of the experiment.
Supplementary Figure 7 | S. aureus JE2 Δhla supernatant does not induce tight junction degradation and endothelial cell death. HuLEC were seeded 7 days prior to toxin challenge on glass cover slips coated with collagen. Cells were either treated with 10% bacterial culture supernatants of wild-type S. aureus JE2 (A,B) or its isogenic Δhla mutant (C,D). Phase contrast microscopy images (A,C) were taken at 10× magnification. Immunofluorescence images (B,D) taken at 40× magnification. Blue – Hoechst 33258; Red – ZO-1; Yellow – VE-Cadherin.
Supplementary Figure 8 | Purified S. aureus toxins other than α-toxin fail to induce ASM activation in murine endothelial cells. bEnd.3 cells were challenged with various combinations of hemolysin γ (Hlg) or leukocidin (Luk) subunits for 5–60 min and ASM activity was measured in cell lysate. n = 3.
Supplementary Video 1 | Eleveated Calcium levels in host cytosol are α-toxin-dependent. Human lung microvascular endothelial cells were loaded with the calcium-sensor Fluo 3-AM (4 μM) for 25 min at 37°C/5% CO2 and subsequently imaged every 7.5 s. After three time frames, cells were treated with 500 nM ionomycin, 0.4 μg/ml SLO, 0.5 μg α-toxin or 5% of bacterial culture supernatants (SNT) of wild-type S. aureus JE2, its isogenic α-toxin mutant JE2 Δhla, or JE2 Δhla SNT complemented with 0.5 μg/ml α-toxin.
Alonzo, F. III, and Torres, V. J. (2014). The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 78, 199–230. doi: 10.1128/MMBR.00055-13
Andrews, N. W. (2017). “Detection of lysosomal exocytosis by surface exposure of lamp1 luminal epitopes,” in Lysosomes: Methods and Protocols, eds K. Öllinger and H. Appelqvist (New York, NY: Springer New York), 205–211. doi: 10.1007/978-1-4939-6934-0_13
Andrews, N. W. (2019). Solving the secretory acid sphingomyelinase puzzle: insights from lysosome-mediated parasite invasion and plasma membrane repair. Cell. Microbiol. 21:e13065. doi: 10.1111/cmi.13065
Andrews, N. W., Almeida, P. E., and Corrotte, M. (2014). Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 24, 734–742. doi: 10.1016/j.tcb.2014.07.008
Baaske, R., Richter, M., Moller, N., Ziesemer, S., Eiffler, I., Muller, C., et al. (2016). ATP release from human airway epithelial cells exposed to Staphylococcus aureus alpha-toxin. Toxins 8:365. doi: 10.3390/toxins8120365
Babiychuk, E. B., Monastyrskaya, K., and Draeger, A. (2008). Fluorescent annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic 9, 1757–1775. doi: 10.1111/j.1600-0854.2008.00800.x
Bartlett, A. H., Foster, T. J., Hayashida, A., and Park, P. W. (2008). Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 198, 1529–1535. doi: 10.1086/592758
Becker, K. A., Fahsel, B., Kemper, H., Mayeres, J., Li, C., Wilker, B., et al. (2018). Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect. Immun. 86:e00606-17. doi: 10.1128/iai.00606-17
Berube, B. J., and Bubeck Wardenburg, J. (2013). Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins 5, 1140–1166. doi: 10.3390/toxins5061140
Bhakdi, S., and Tranum-Jensen, J. (1991). Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 55, 733–751. doi: 10.1128/mmbr.55.4.733-751.1991
Bischofberger, M., Iacovache, I., and van der Goot, F. G. (2012). Pathogenic pore-forming proteins: function and host response. Cell Host Microbe 12, 266–275. doi: 10.1016/j.chom.2012.08.005
Blattman, S. B., Jiang, W., Oikonomou, P., and Tavazoie, S. (2020). Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat. Microbiol. 5, 1192–1201. doi: 10.1038/s41564-020-0729-6
Blazek, A. D., Paleo, B. J., and Weisleder, N. (2015). Plasma membrane repair: a central process for maintaining cellular homeostasis. Physiology 30, 438–448. doi: 10.1152/physiol.00019.2015
Brito, C., Cabanes, D., Sarmento Mesquita, F., and Sousa, S. (2019). Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 76, 1319–1339. doi: 10.1007/s00018-018-2992-8
Bryan, A. M., Del Poeta, M., and Luberto, C. (2015). Sphingolipids as regulators of the phagocytic response to fungal infections. Mediators Inflamm. 2015:640540. doi: 10.1155/2015/640540
Chow, E. M., Batty, S., and Mogridge, J. (2010). Anthrax lethal toxin promotes dephosphorylation of TTP and formation of processing bodies. Cell. Microbiol. 12, 557–568. doi: 10.1111/j.1462-5822.2009.01418.x
Cooper, S. T., and McNeil, P. L. (2015). Membrane repair: mechanisms and pathophysiology. Physiol. Rev. 95, 1205–1240. doi: 10.1152/physrev.00037.2014
Dal Peraro, M., and van der Goot, F. G. (2016). Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92. doi: 10.1038/nrmicro.2015.3
Doery, H. M., Magnusson, B. J., Cheyne, I. M., and Gulasekharam, J. (1963). A phospholipase in staphylococcal toxin which hydrolyses sphingomyelin. Nature 198, 1091–1092. doi: 10.1038/1981091a0
Eiffler, I., Behnke, J., Ziesemer, S., Muller, C., and Hildebrandt, J. P. (2016). Staphylococcus aureus alpha-toxin-mediated cation entry depolarizes membrane potential and activates p38 MAP kinase in airway epithelial cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 311, L676–L685. doi: 10.1152/ajplung.00090.2016
Etxaniz, A., Gonzalez-Bullon, D., Martin, C., and Ostolaza, H. (2018). Membrane repair mechanisms against permeabilization by pore-forming toxins. Toxins 10:234. doi: 10.3390/toxins10060234
Ezekwe, E. A. Jr., Weng, C., and Duncan, J. A. (2016). ADAM10 cell surface expression but not activity is critical for Staphylococcus aureus alpha-hemolysin-mediated activation of the NLRP3 inflammasome in human monocytes. Toxins 8:95. doi: 10.3390/toxins8040095
Faulstich, M., Hagen, F., Avota, E., Kozjak-Pavlovic, V., Winkler, A. C., Xian, Y., et al. (2015). Neutral sphingomyelinase 2 is a key factor for PorB-dependent invasion of Neisseria gonorrhoeae. Cell. Microbiol. 17, 241–253. doi: 10.1111/cmi.12361
Fedele, G., Spensieri, F., Palazzo, R., Nasso, M., Cheung, G. Y., Coote, J. G., et al. (2010). Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS One 5:e8734. doi: 10.1371/journal.pone.0008734
Fey, P. D., Endres, J. L., Yajjala, V. K., Widhelm, T. J., Boissy, R. J., Bose, J. L., et al. (2013). A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12
Fink, D., Contreras, M. L., Lelkes, P. I., and Lazarovici, P. (1989). Staphylococcus aureus alpha-toxin activates phospholipases and induces a Ca2+ influx in PC12 cells. Cell. Signal. 1, 387–393. doi: 10.1016/0898-6568(89)90057-0
Gierok, P., Harms, M., Richter, E., Hildebrandt, J. P., Lalk, M., Mostertz, J., et al. (2014). Staphylococcus aureus alpha-toxin mediates general and cell type-specific changes in metabolite concentrations of immortalized human airway epithelial cells. PLoS One 9:e94818. doi: 10.1371/journal.pone.0094818
Giese, B., Dittmann, S., Paprotka, K., Levin, K., Weltrowski, A., Biehler, D., et al. (2009). Staphylococcal alpha-toxin is not sufficient to mediate escape from phagolysosomes in upper-airway epithelial cells. Infect. Immun. 77, 3611–3625. doi: 10.1128/iai.01478-08
Goerke, C., Wirtz, C., Fluckiger, U., and Wolz, C. (2006). Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 61, 1673–1685. doi: 10.1111/j.1365-2958.2006.05354.x
Grassmé, H., Becker, K. A., Zhang, Y., and Gulbins, E. (2008). Ceramide in bacterial infections and cystic fibrosis. Biol. Chem. 389, 1371–1379. doi: 10.1515/BC.2008.162
Grassmé, H., Gulbins, E., Brenner, B., Ferlinz, K., Sandhoff, K., Harzer, K., et al. (1997). Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91, 605–615. doi: 10.1016/s0092-8674(00)80448-1
Grassmé, H., Jekle, A., Riehle, A., Schwarz, H., Berger, J., Sandhoff, K., et al. (2001a). CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 276, 20589–20596. doi: 10.1074/jbc.M101207200
Grassmé, H., Jendrossek, V., Riehle, A., von Kurthy, G., Berger, J., Schwarz, H., et al. (2003). Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9, 322–330. doi: 10.1038/nm823
Grassmé, H., Riehle, A., Wilker, B., and Gulbins, E. (2005). Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 280, 26256–26262. doi: 10.1074/jbc.M500835200
Grassmé, H., Schwarz, H., and Gulbins, E. (2001b). Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem. Biophys. Res. Commun. 284, 1016–1030. doi: 10.1006/bbrc.2001.5045
Grosz, M., Kolter, J., Paprotka, K., Winkler, A. C., Schafer, D., Chatterjee, S. S., et al. (2014). Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell. Microbiol. 16, 451–465. doi: 10.1111/cmi.12233
Gutierrez, A. A., Arias, J. M., Garcia, L., Mas-Oliva, J., and Guerrero-Hernandez, A. (1999). Activation of a Ca2+-permeable cation channel by two different inducers of apoptosis in a human prostatic cancer cell line. J. Physiol. 517(Pt 1), 95–107. doi: 10.1111/j.1469-7793.1999.0095z.x
Harshman, S., Boquet, P., Duflot, E., Alouf, J. E., Montecucco, C., and Papini, E. (1989). Staphylococcal alpha-toxin: a study of membrane penetration and pore formation. J. Biol. Chem. 264, 14978–14984. doi: 10.1016/s0021-9258(18)63799-8
Haslinger-Loffler, B., Kahl, B. C., Grundmeier, M., Strangfeld, K., Wagner, B., Fischer, U., et al. (2005). Multiple virulence factors are required for Staphylococcus aureus-induced apoptosis in endothelial cells. Cell. Microbiol. 7, 1087–1097. doi: 10.1111/j.1462-5822.2005.00533.x
Hauck, C. R., Grassmé, H., Bock, J., Jendrossek, V., Ferlinz, K., Meyer, T. F., et al. (2000). Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of Neisseria gonorrhoeae. FEBS Lett. 478, 260–266. doi: 10.1016/s0014-5793(00)01851-2
Husmann, M., Beckmann, E., Boller, K., Kloft, N., Tenzer, S., Bobkiewicz, W., et al. (2009). Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 583, 337–344. doi: 10.1016/j.febslet.2008.12.028
Husmann, M., Dersch, K., Bobkiewicz, W., Beckmann, E., Veerachato, G., and Bhakdi, S. (2006). Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 344, 1128–1134. doi: 10.1016/j.bbrc.2006.03.241
Idone, V., Tam, C., Goss, J. W., Toomre, D., Pypaert, M., and Andrews, N. W. (2008). Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180, 905–914. doi: 10.1083/jcb.200708010
Inoshima, I., Inoshima, N., Wilke, G. A., Powers, M. E., Frank, K. M., Wang, Y., et al. (2011). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17, 1310–1314. doi: 10.1038/nm.2451
Jonas, D., Walev, I., Berger, T., Liebetrau, M., Palmer, M., and Bhakdi, S. (1994). Novel path to apoptosis: small transmembrane pores created by staphylococcal alpha-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect. Immun. 62, 1304–1312. doi: 10.1128/iai.62.4.1304-1312.1994
Justet, C., Chifflet, S., and Hernandez, J. A. (2019). Calcium oscillatory behavior and its possible role during wound healing in bovine corneal endothelial cells in culture. Biomed Res. Int. 2019:8647121. doi: 10.1155/2019/8647121
Kao, C. Y., Los, F. C., Huffman, D. L., Wachi, S., Kloft, N., Husmann, M., et al. (2011). Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 7:e1001314. doi: 10.1371/journal.ppat.1001314
Katayama, Y., Baba, T., Sekine, M., Fukuda, M., and Hiramatsu, K. (2013). Beta-hemolysin promotes skin colonization by Staphylococcus aureus. J. Bacteriol. 195, 1194–1203. doi: 10.1128/jb.01786-12
Kloft, N., Busch, T., Neukirch, C., Weis, S., Boukhallouk, F., Bobkiewicz, W., et al. (2009). Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 385, 503–506. doi: 10.1016/j.bbrc.2009.05.121
Kreiswirth, B. N., Lofdahl, S., Betley, M. J., O’Reilly, M., Schlievert, P. M., Bergdoll, M. S., et al. (1983). The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. doi: 10.1038/305709a0
Kwak, Y. K., Vikstrom, E., Magnusson, K. E., Vecsey-Semjen, B., Colque-Navarro, P., and Mollby, R. (2012). The Staphylococcus aureus alpha-toxin perturbs the barrier function in Caco-2 epithelial cell monolayers by altering junctional integrity. Infect. Immun. 80, 1670–1680. doi: 10.1128/iai.00001-12
Liu, X., Wang, N., Zhu, Y., Yang, Y., Chen, X., Chen, Q., et al. (2016). Extracellular calcium influx promotes antibacterial autophagy in Escherichia coli infected murine macrophages via CaMKKbeta dependent activation of ERK1/2, AMPK and FoxO1. Biochem. Biophys. Res. Commun. 469, 639–645. doi: 10.1016/j.bbrc.2015.12.052
Ma, J., Gulbins, E., Edwards, M. J., Caldwell, C. C., Fraunholz, M., and Becker, K. A. (2017). Staphylococcus aureus alpha-toxin induces inflammatory cytokines via lysosomal acid sphingomyelinase and ceramides. Cell. Physiol. Biochem. 43, 2170–2184. doi: 10.1159/000484296
Mason, M. J., and Grinstein, S. (1993). Ionomycin activates electrogenic Ca2+ influx in rat thymic lymphocytes. Biochem. J. 296(Pt 1), 33–39. doi: 10.1042/bj2960033
Menzies, B. E., and Kourteva, I. (2000). Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol. Med. Microbiol. 29, 39–45. doi: 10.1111/j.1574-695X.2000.tb01503.x
Miller, M. E., Adhikary, S., Kolokoltsov, A. A., and Davey, R. A. (2012). Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J. Virol. 86, 7473–7483. doi: 10.1128/jvi.00136-12
Mühle, C., and Kornhuber, J. (2017). Assay to measure sphingomyelinase and ceramidase activities efficiently and safely. J. Chromatogr. A 1481, 137–144. doi: 10.1016/j.chroma.2016.12.033
N’Guessan, P. D., Schmeck, B., Ayim, A., Hocke, A. C., Brell, B., Hammerschmidt, S., et al. (2005). Streptococcus pneumoniae R6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb. Haemost. 94, 295–303. doi: 10.1160/TH04-12-0822
Peters, S., Schlegel, J., Becam, J., Avota, E., Sauer, M., and Schubert-Unkmeir, A. (2019). Neisseria meningitidis type IV pili trigger Ca(2+)-dependent lysosomal trafficking of the acid sphingomyelinase to enhance surface ceramide levels. Infect. Immun. 87:e00410-19. doi: 10.1128/iai.00410-19
Popov, L. M., Marceau, C. D., Starkl, P. M., Lumb, J. H., Shah, J., Guerrera, D., et al. (2015). The adherens junctions control susceptibility to Staphylococcus aureus alpha-toxin. Proc. Natl. Acad. Sci. U.S.A. 112, 14337–14342. doi: 10.1073/pnas.1510265112
Reddy, A., Caler, E. V., and Andrews, N. W. (2001). Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106, 157–169. doi: 10.1016/s0092-8674(01)00421-4
Riethmüller, J., Riehle, A., Grassmé, H., and Gulbins, E. (2006). Membrane rafts in host-pathogen interactions. Biochim. Biophys. Acta 1758, 2139–2147. doi: 10.1016/j.bbamem.2006.07.017
Rosen, L. B., Ginty, D. D., Weber, M. J., and Greenberg, M. E. (1994). Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12, 1207–1221. doi: 10.1016/0896-6273(94)90438-3
Salgado-Pabon, W., Herrera, A., Vu, B. G., Stach, C. S., Merriman, J. A., Spaulding, A. R., et al. (2014). Staphylococcus aureus beta-toxin production is common in strains with the beta-toxin gene inactivated by bacteriophage. J. Infect. Dis. 210, 784–792. doi: 10.1093/infdis/jiu146
Santana, P., Pena, L. A., Haimovitz-Friedman, A., Martin, S., Green, D., McLoughlin, M., et al. (1996). Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86, 189–199. doi: 10.1016/s0092-8674(00)80091-4
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schwiering, M., Brack, A., Stork, R., and Hellmann, N. (2013). Lipid and phase specificity of alpha-toxin from S. aureus. Biochim. Biophys. Acta 1828, 1962–1972. doi: 10.1016/j.bbamem.2013.04.005
Seeger, W., Birkemeyer, R. G., Ermert, L., Suttorp, N., Bhakdi, S., and Duncker, H. R. (1990). Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab. Invest. 63, 341–349.
Seitz, A. P., Grassmé, H., Edwards, M. J., Pewzner-Jung, Y., and Gulbins, E. (2015). Ceramide and sphingosine in pulmonary infections. Biol. Chem. 396, 611–620. doi: 10.1515/hsz-2014-0285
Shivanna, V., Kim, Y., and Chang, K. O. (2015). Ceramide formation mediated by acid sphingomyelinase facilitates endosomal escape of caliciviruses. Virology 483, 218–228. doi: 10.1016/j.virol.2015.04.022
Simonis, A., and Schubert-Unkmeir, A. (2018). The role of acid sphingomyelinase and modulation of sphingolipid metabolism in bacterial infection. Biol. Chem. 399, 1135–1146. doi: 10.1515/hsz-2018-0200
Spence, M. W., Byers, D. M., Palmer, F. B., and Cook, H. W. (1989). A new Zn2+-stimulated sphingomyelinase in fetal bovine serum. J. Biol. Chem. 264, 5358–5363. doi: 10.1016/s0021-9258(18)83553-0
Surewaard, B. G., Nijland, R., Spaan, A. N., Kruijtzer, J. A., de Haas, C. J., and van Strijp, J. A. (2012). Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 8:e1002606. doi: 10.1371/journal.ppat.1002606
Tran, P. M., Feiss, M., Kinney, K. J., and Salgado-Pabon, W. (2019). varphiSa3mw prophage as a molecular regulatory switch of Staphylococcus aureus beta-toxin production. J. Bacteriol. 201:e00766-18. doi: 10.1128/JB.00766-18
Valeva, A., Walev, I., Gerber, A., Klein, J., Palmer, M., and Bhakdi, S. (2000). Staphylococcal alpha-toxin: repair of a calcium-impermeable pore in the target cell membrane. Mol. Microbiol. 36, 467–476. doi: 10.1046/j.1365-2958.2000.01865.x
Vann, J. M., and Proctor, R. A. (1987). Ingestion of Staphylococcus aureus by bovine endothelial cells results in time- and inoculum-dependent damage to endothelial cell monolayers. Infect. Immun. 55, 2155–2163. doi: 10.1128/iai.55.9.2155-2163.1987
Vasilev, F., Chun, J. T., Gragnaniello, G., Garante, E., and Santella, L. (2012). Effects of ionomycin on egg activation and early development in starfish. PLoS One 7:e39231. doi: 10.1371/journal.pone.0039231
Virreira Winter, S., Zychlinsky, A., and Bardoel, B. W. (2016). Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus alpha-hemolysin-mediated toxicity. Sci. Rep. 6:24242. doi: 10.1038/srep24242
von Hoven, G., Qin, Q., Neukirch, C., Husmann, M., and Hellmann, N. (2019). Staphylococcus aureus alpha-toxin: small pore, large consequences. Biol. Chem. 400, 1261–1276. doi: 10.1515/hsz-2018-0472
von Hoven, G., Rivas, A. J., Neukirch, C., Klein, S., Hamm, C., Qin, Q., et al. (2016). Dissecting the role of ADAM10 as a mediator of Staphylococcus aureus alpha-toxin action. Biochem. J. 473, 1929–1940. doi: 10.1042/bcj20160062
Walev, I., Martin, E., Jonas, D., Mohamadzadeh, M., Muller-Klieser, W., Kunz, L., et al. (1993). Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun. 61, 4972–4979. doi: 10.1128/iai.61.12.4972-4979.1993
Wang, R., Braughton, K. R., Kretschmer, D., Bach, T. H., Queck, S. Y., Li, M., et al. (2007). Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13, 1510–1514. doi: 10.1038/nm1656
Wilke, G. A., and Bubeck Wardenburg, J. (2010). Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. U.S.A. 107, 13473–13478. doi: 10.1073/pnas.1001815107
Yoshida, S., and Plant, S. (1992). Mechanism of release of Ca2+ from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. J. Physiol. 458, 307–318. doi: 10.1113/jphysiol.1992.sp019419
Zhao, M., Pan, W., Shi, R. Z., Bai, Y. P., You, B. Y., Zhang, K., et al. (2016). Acid sphingomyelinase mediates oxidized-LDL induced apoptosis in macrophage via endoplasmic reticulum stress. J. Atheroscler. Thromb. 23, 1111–1125. doi: 10.5551/jat.32383
Keywords: Staphylococcus aureus, acid sphingomyelinase, staphylococcal alpha-toxin, sphingomyelinase release, lysosomal recruitment
Citation: Krones D, Rühling M, Becker KA, Kunz TC, Sehl C, Paprotka K, Gulbins E and Fraunholz M (2021) Staphylococcus aureus α-Toxin Induces Acid Sphingomyelinase Release From a Human Endothelial Cell Line. Front. Microbiol. 12:694489. doi: 10.3389/fmicb.2021.694489
Received: 13 April 2021; Accepted: 05 July 2021;
Published: 29 July 2021.
Edited by:
Christine Josenhans, Ludwig Maximilian University of Munich, GermanyReviewed by:
Volker Winstel, Twincore Center for Experimental and Clinical Infection Research GmbH, GermanyCopyright © 2021 Krones, Rühling, Becker, Kunz, Sehl, Paprotka, Gulbins and Fraunholz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Fraunholz, bWFydGluLmZyYXVuaG9sekB1bmktd3VlcnpidXJnLmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.