 Min Hu
Min Hu Fangbai Li1,2
Fangbai Li1,2 Li Zhuang
Li Zhuang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol. , 06 October 2021
Sec. Microbiotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.691913
Gene encoding the large subunit of As(III) oxidase (AioA), an important component of the microbial As(III) oxidation system, is a widely used biomarker to characterize As(III)-oxidizing communities in the environment. However, many studies were restricted to a few sequences generated by clone libraries and Sanger sequencing, which may have underestimated the diversity of As(III)-oxidizers in natural environments. In this study, we designed a primer pair, 1109F (5′-ATC TGG GGB AAY RAC AAY TA−3′) and 1548R (5′-TTC ATB GAS GTS AGR TTC AT−3′), targeting gene sequence encoding for the conserved molybdopterin center of the AioA protein, yielding amplicons approximately 450 bp in size that are feasible for highly parallel amplicon sequencing. By utilizing in silico analyses and the experimental construction of clone libraries using Sanger sequencing, the specificity and resolution of 1109F/1548R are approximated with two other previously published and commonly used primers, i.e., M1-2F/M3-2R and deg1F/deg1R. With the use of the 1109F/1548R primer pair, the taxonomic composition of the aioA genes was similar both according to the Sanger and next-generation sequencing (NGS) platforms. Furthermore, high-throughput amplicon sequencing using the primer pair, 1109F/1548R, successfully identified the well-known As(III)-oxidizers in paddy soils and sediments, and they also revealed the differences in the community structure and composition of As(III)-oxidizers in above two biotopes. The random forest analysis showed that the dissolved As(III) had the highest relative influence on the Chao1 index of the aioA genes. These observations demonstrate that the newly designed PCR primers enhanced the ability to detect the diversity of aioA-encoding microorganisms in environments using highly parallel short amplicon sequencing.
Arsenic (As) is a ubiquitous element and an extremely toxic metalloid in the environment. It is either released via natural processes (weathering or hydrothermal and volcanic emissions) or anthropogenic activities (use of arsenical pesticides/herbicides, the release of industrial waste, incineration, and the combustion of fossil fuels) (Oremland and Stolz, 2005). The toxicity and mobility of inorganic As species depend on the redox state. Arsenite [As(III)] is more toxic than arsenate [As(V)] to an active cell. As(V) is much more strongly adsorbed by commonly occurring surface minerals, such as aluminum oxyhydroxides or iron (hydro)oxides, than As(III) (Oremland and Stolz, 2005).
As(III) oxidizing microorganisms exist widely in nature as both heterotrophic and chemo/photosynthetic autotrophic microorganisms (Hamamura et al., 2009). As(III) oxidation in nature is primarily mediated by microorganisms and catalyzed by the Aio enzyme system. The Aio enzyme contains two subunits: a large catalytic subunit (AioA, approximately 90 kDa) that contains a molybdopterin center bound to the pyranopterin cofactor and a [3Fe–4S] cluster and an associated small subunit (AioB, approximately 14 kDa) containing a single Rieske-type [2Fe–2S] cluster (Mukhopadhyay et al., 2002). Homologous genes encoding these two subunits were formerly assigned different names (aoxB and aoxA, aroA and aroB, and asoA and asoB, accordingly). The nomenclature for genes involved in prokaryotic As(III) oxidation has been unified as the name aio, and the two subunits are now denoted as aioA and aioB, respectively (Yamamura and Amachi, 2014). To date, homologs of genes encoding AioAB have been found in phylogenetically diverse strains including members of α-,β-,γ-Proteobacteria, Actinobacteria, Aquificae, Bacteroidetes, Chlorobi, Chloroflexi, Crenarchaeota, Deinococcus-Thermus, Firmicutes, and Nitrospira (Yamamura and Amachi, 2014). The aioA gene has been successfully used as a molecular marker to capture the diversity and taxonomic information for As(III)-oxidizing microbial communities in environments (Zhang et al., 2017; Xue et al., 2020; Zhai et al., 2020). Primers targeting the aioA genes, such as M1-2F/M3-2R (approximate amplicon size of 1,100 bp) (Quéméneur et al., 2008) and deg1F/deg1R (530 bp) (Inskeep et al., 2007) have been developed. The aioA gene amplicon size obtained using some recently reported primer pairs ranged from 530 to 1,100 bp (Inskeep et al., 2007; Quéméneur et al., 2008; Hamamura et al., 2009; Sultana et al., 2012), which are not feasible for application in highly parallel amplicon sequencing for which an amplicon size of less than 500 bp is recommended. However, classical surveys of aioA diversity are performed by constructing clone libraries and Sanger sequencing, which have limitations of the sample number and sequencing depth (normally dozens of clones per sample), which possibly underestimate the aioA diversity in natural environments.
Simultaneous comparative analysis of the bacterial community structure across many samples is currently made possible by the advent of next-generation sequencing (NGS) technologies for highly multiplexed amplicon sequencing of 16S rRNA genes (Klindworth et al., 2012). Amplicon NGS surveys are now also increasingly applied to target functional genes that are indicative of a particular microbial guild, such as mcrA for methane oxidizers (Cai et al., 2018), amoA for ammonia oxidizers (Roots et al., 2019), pmoA for methanotrophs (Chiri et al., 2020), and dsrAB for sulfate reducers (Zeleke et al., 2013; Pelikan et al., 2016). To date, investigations targeting metal biotransformation using NGS have only involved the arrA gene for dissimilatory arsenate reducers (Mirza et al., 2017) and the arsM gene for arsenate methylated microorganisms (Zhang S. Y. et al., 2015). However, no available PCR primer is suitable for applications in high-throughput sequencing for genes that encode As(III)-oxidases.
The aim of the current study is to design degenerate primers that target the aioA gene and have the subsequent amplicon length suitable for highly multiplexed amplicon sequencing by NGS. The specificity and coverage of the newly designed primers are then compared with the primer sets previously published in silico and experimentally by constructing clone libraries. Finally, the utility of the newly designed primer pair for highly parallel amplicon sequencing is verified with As-contaminated paddy soil and sediment samples using the Illumina Hiseq platform. The newly designed primer pair constitutes a useful tool for the further study of microbial communities to investigate the diversity of As(III) oxidizers in environmental samples.
A total of 67 full-length bacterial AioA proteins (orthology number: K08355) and aioA gene sequences were downloaded from the KEGG database (Kanehisa and Goto, 2000) (version 91.0, released on July 1, 2019) (Supplementary Table 1). The taxonomic profiles of these AioA sequences were obtained using a Perl script utilizing a tax dump file from the NCBI FTP (ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/). Proteobacteria-related sequences were in the greatest abundance (accounting for 87.67% of all sequences), followed by Deinococcus-Thermus (5.48%). At the order level, the AioA sequences originated from Burkholderiales (39.73%) and Rhizobiales (20.55%), which were predominant, followed by Rhodobacterales (10.96%) and Thermales (5.48%) (Supplementary Figure 1). Phylogenetic analysis of the AioA sequences was conducted using MEGA version 7 (Kumar et al., 2018). Their evolutionary history was inferred using the Neighbor-Joining method and bootstrapping of 1,000. All of the positions containing gaps and missing data were eliminated. The aioA gene sequences, all with their complete coding regions, were aligned using MUSCLE software v3.8.31 (Edgar, 2004) with the default parameters. Then, the aligned sequences encoding the conserved molybdopterin center and the [3Fe-4S] binding motif of the aioA protein were extracted and used for the downstream primer designs. These two alignments were then imported into DegePrime (Hugerth et al., 2014) to generate degenerate primers for conserved regions unique to the aioA gene at the following parameters: oligomer length, 20; maximum oligomer degeneracy, 96; the number of bases at the sequence termini that would not be considered in the analysis, 30. Only the sequence alignments of the conserved molybdopterin center of the AioA successfully produced primers using DegePrimer were namely 1109F (5′-ATC TGG GGB AAY RAC AAY TA−3′) and 1548R (5′-TTC ATB GAS GTS AGR TTC AT−3′) (corresponding to the nucleotides 1,168–1,184 and 1,628–1,609 of the aioA gene of Alcaligenes (A.) faecalis) (Table 1). The 1109F/1548R primer sets were in silico predicted and generated a PCR product size of approximately 450 bp, and these are suitable for parallel amplicon sequencing using NGS platforms such as Illumina Hiseq PE250 or the ThermoFisher Ion S5 XL system.
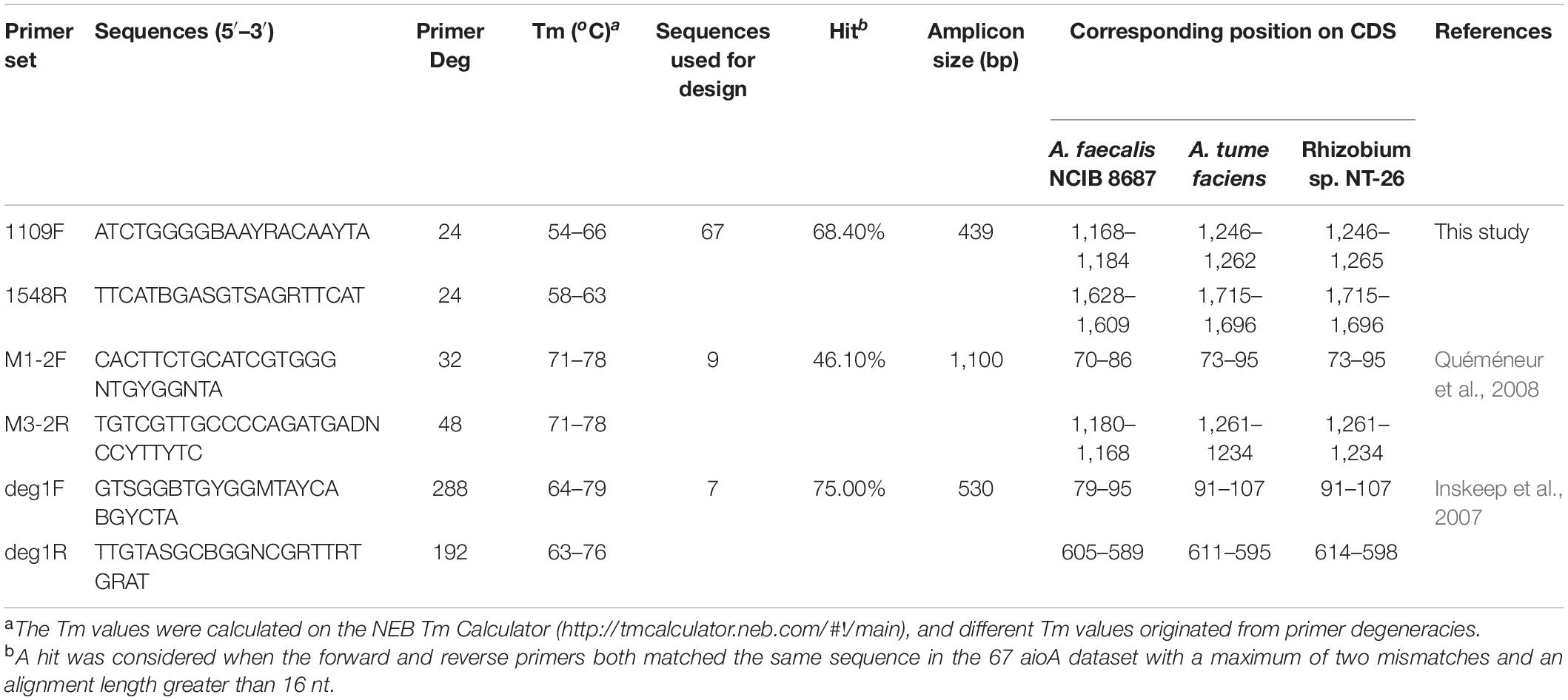
Table 1. Primers for the amplification of the aioA genes.
Then the performance of the newly designed primer set was compared with the other two previously published and commonly used primers, i.e., M1-2F/M3-2R (Inskeep et al., 2007) and deg1F/deg1R (Quéméneur et al., 2008). Direct amplification of the aioA genes from pure culture isolates were not evaluated because the isolates were not available in our laboratory. An in silico evaluation of the primers against the 67 aioA gene sequences from the KEGG database using the BLASTN algorithm was performed. A hit was considered when the forward and reverse primers both matched the same sequence, with a maximum of two mismatches and an alignment length larger than 16 nt. Coverage was displayed as the ratio of sequence hits in all the constructed 67 aioA gene dataset. Furthermore, a taxonomic analysis of the hit aioA gene sequences was also performed to estimate the phylogenetic coverage of each primer pair.
As-contaminated paddy soil and sediment samples were selected to estimate the amplification efficiency and performance of the newly designed primer pair, 1109F/1548R, together with another two published aioA-targeting primer pairs (M1-2F/M3-2R and deg1F/1R). The soil sample was collected from an As-contaminated paddy field (23°37′28″′N, 116°48′43″E) that was surrounded by the Lianhuashan tungsten mine located in Shantou City, Guangdong Province, China. The severe impact of mining and metal processing activities on the paddy field has been documented (Liu et al., 2010, 2015). Soil samples were randomly collected from the arable layer (0–20 cm deep) and combined into one compost sample in 50-ml polypropylene centrifuge tubes. The sediment sample was collected from the river nearby the As-contaminated paddy soil field. Samples were then transported on ice to the laboratory and stored at −80°C prior to DNA extraction. For the soil physico-chemical properties analysis, the soil samples were air-dried at room temperature and crushed using a wooden roller to pass through a 2 mm sieve. The soil was characterized, and the results were a total organic C content of 35.12 g kg–1, total N of 2.72 g kg–1, total P of 0.86 g kg–1, total K of 18.2 mg kg–1, and total As of 102.5 mg kg–1, with a pH of 5.75. The physico-chemical properties of the sediment are shown in Supplementary Table 1 (listed as S1 sample). Genomic DNA from the 0.5 g paddy soil and sediment samples was extracted using the PowerSoilTM DNA isolation kit (MoBio, Carlsbad, CA). All extractions were performed in triplicate according to the manufacturer’s instructions, and the extracted DNA was stored at −80°C until analysis.
The DNA extracted from the paddy soil and sediment was amplified using the 1109F/1548R primer pair and the two published primer pairs, M1-2F/M3-2R and deg1F/deg1R. Each PCR reaction contained 25 μl of a PCR mixture consisting of 5 μL 5 × PCR ExTaq buffer, 100 μmol dNTPs, 0.5 μmol each primer, 50 ng template, and 1 U of ExTaq DNA polymerase (TaKaRa, Dalian, China). Following verification of the amplification reaction on a 1.5% agarose gel, the PCR product was excised and purified using a QIAquick gel extraction kit (Qiagen, Valencia, CA). The purified PCR products were cloned into a pGEM-T vector (Promega, Madison, WI) and used to transform Escherichia coli DH5α competent cells (TaKaRa, Dalian, China) according to the protocol provided by the manufacturer. Approximately 100 clones from each library were sequenced using the ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA). The cloned sequences were trimmed and assembled using SeqMan (DNAstar, Madison, WI) after verification using a BLASTX similarity search against the NCBI-nr database at e < e–10 and an alignment length > 150 nt. The specificity of the primer sets was calculated using the aioA-annotated sequence against the total number of cloned sequences. The BLASTX analysis was also used for the taxonomic assignment of the aioA sequences.
Samples from the As-contaminated paddy soil and sediment were collected from the region surrounding the Lianhuashan tungsten mine, as described elsewhere (Liu et al., 2010, 2015; Supplementary Table 2). Arsenic species (including dissolved, phosphate-extractable and oxalate-extractable As(III) and As(V)) were detected as described (Yu et al., 2017; Qiao et al., 2019). In brief, the dissolved As was determined by adding 20 ml of ultrapure water (pH = 7.0) to 1 g of soil and shaking in a rotary shaker (200 rpm) for 15 m at room temperature. This was followed by centrifugal separation at 4,500 r/min for 15 m. The supernatant then was then filtered through a sterile 0.22-μm filter for determination of the dissolved As(III) and As(V) concentrations using atomic fluorescence spectroscopy (SA-20, Jitian, Beijing, China). Soil pellets were used to sequentially fractionate arsenic in the soil using 1M KH2PO4 + 0.1 M ascorbic acid and 0.2 M NH4+-oxalate that primarily target As adsorbed on iron (oxyhydr)oxides and As incorporated into iron (oxyhydr)-oxides, respectively. The mixtures were shaken and centrifuged. Then, each supernatant was passed through a 0.22 μm filter, and the As concentrations were determined using atomic fluorescence spectroscopy as described above.
For the parallel amplicon sequencing of the aioA gene, DNA from the As-contaminated paddy soil and sediment was extracted using the PowerSoilTM DNA isolation kit (MoBio) and amplified using the 1109F/1548R primer pair with Illumina adapters and a unique 12bp-Golay barcode sequence attached to the reverse PCR primer (Supplementary Table 3). The PCR conditions were as follows: 3 m at 95°C; followed by 28 cycles of denaturation at 95°C for 30 s, primer annealing at 55°C for 30 s, an extension at 72°C for 30 s, and a final extension for 5 m at 72°C. The PCR products were checked by resolving an aliquot on a 2% agarose gel. Triplicate reactions were combined and the amplicons were purified using a QIAquick gel extraction kit (Qiagen). The concentration of purified PCR products was determined by using a Qubit 3.0 Fluorometer and the Qubit dsDNA BR Assay kit (Life Technologies, Grand Island, NY) following the manufacturer’s instructions. The purified PCR products were pooled in equimolar concentrations and sequenced using an Illumina Hiseq platform at Novogene Bioinformatics Technology (Beijing, China). The HiSeq analysis was performed in the 2 × 250 cycle combination, i.e., producing 2 × 250 nt paired-end reads.
The paired-end reads were merged using Flash (Magoč and Salzberg, 2011), followed by quality control using the QIIME pipeline (Caporaso et al., 2010b). The quality control included retaining sequences with minimum merged sequence lengths of 250 bp and maximum homopolymers of eight bases. In addition, sequences with unidentified bases (N) and sequences with more than one inexact match with a unique barcode identifier and perfect primer matches were also removed. The remaining sequences were aligned using PyNAST (Caporaso et al., 2010a) and then uploaded into the UPARSE pipeline implementing USEARCH (Edgar, 2010) for the dereplication and de novo clustering to a 90% similarity level after the removal of singletons and chimeras. Denoising was performed using the PyroClean software (Ramirez-Gonzalez et al., 2013) with default parameters. The conservation in the sequences of gene candidates in element biogeochemical cycles is much lower than that of 16S rRNA and ITS genes, which are phylogenetic markers used for microbial community analyses. The standard threshold of operational taxonomic unit (OTU) clustering has not been unified for arsenic metabolism genes. In this study, the aioA sequences were clustered into OTUs at a 90% identity threshold. Representative OTU sequences were then confirmed as the aioA gene sequences using the Diamond software (Buchfink et al., 2014) against the NCBI-nr database at e < e–10 and an alignment length > 200 nt. Phylogenetic trees were generated using FastTree and representative OTU sequences (Price et al., 2010). To assess the OTU abundance, UPARSE was used to map the aioA sequences to the representative OTU sequences at a 90% identity threshold (as a cutoff of the subgenus level). A distance matrix was then constructed based on the OTU abundance. To estimate the alpha diversity, a random subsampling method for each sequence library was used for the microbial community diversity index calculations to control for the effects of the library size. The alpha diversity indices (observed OTUs, Chao1, phylogenic diversity [PD], Shannon, and Simpson) were calculated for all the samples with 1,000 repetitions using a size of 33,637 sequences per sample. Furthermore, the beta (unweighted Unifrac) diversity was calculated in QIIME software (Caporaso et al., 2010b). The taxonomic composition of the aioA sequences in each sample was determined using the MEGAN software (Huson et al., 2007), with the results searched against the NCBI-nr using Diamond software (Buchfink et al., 2014). The phylogeny of the abundant representative OTU sequences whose average relative abundance exceeded 1% was determined using MEGAX (Kumar et al., 2018).
Statistical analyses were conducted using SPSS 17.0 (SPSS Inc.). Statistical significance was estimated using two-way analyses of variance (ANOVA) to determine the significance in the differences of alpha diversity between the paddy soil and the sediment samples. The Spearman’s correlation coefficient (rho) was used as a measure of the correlation between the taxonomic compositions of aioA genes sequenced by Sanger and NGS platforms, giving a value between + 1 and -1, where 1 stands for the total positive correlation, 0 stands for no correlation, and –1 stands for the total negative correlation. The random forest (RF) analysis was conducted using the R package randomForest to investigate the impact of environmental parameters on the aioA gene diversity (Liaw and Wiener, 2002).
The amplicon sequences of the aioA gene from paddy soil and sediment samples have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SRR9915017-SRR9915024.
The specificity of 1109F/1548R together with another two previously published aioA-targeted primers, M1-2F/M3-2R and deg1F/deg1R (Table 1), was compared to the 67 aioA gene sequences from the KEGG database. The sequences matched by the three primer pairs primarily originated from Proteobacteria, especially from the orders of Burkholderiales, Rhizobiales, and Rhodobacterales (Supplementary Figure 1). Primer pairs 1109F/1548R exactly matched the aioA gene sequences from the well-studied As(III)-oxidizers, such as A. faecalis subsp. faecalis NCIB 8687, Rhizobium sp. NT-26, Xanthobacter autotrophicus Py2, Achromobacter xylosoxidans NCTC10807, Herminiimonas arsenicoxydans, Thiomonas arsenitoxydans, and Chloroflexus aurantiacus. The phylogenetic coverage of the different primer pairs was different. Primer pairs 1109F/1548R, M1-2F/M3-2R, and deg1F/deg1R targeted sequences from 31, 31, and 40 genera, respectively (the 67 aioA sequences used for the primer design covered 43 genera) (Supplementary Figure 1). No primer pairs tested in the current study matched all the aioA gene sequences retrieved from the KEGG database.
The phylogenetic distribution of the amplified sequences and specificity of the primer sets were compared by constructing clone libraries using DNA isolated from As-contaminated paddy soil and sediment samples (100 clones for each primer pair). The taxonomic information for the aioA gene sequences in the three clone libraries was also compared. At the genus level, most of the aioA sequences from the deg1F/deg1R libraries were unclassified both in the paddy soil and sediment samples, with relative abundances of 68.61 and 99.65%, respectively (Figure 1). For the paddy soil sample, the 1109F/1548R library data were similar to the deg1F/deg1R data in that Ralstonia sequences accounting for 25.16 and 22.03%, respectively, of all the aioA sequences compared with 42.72% using the M1-2F/M3-2R primer pair. However, Devosia and Kaistia predominantly been identified using the 1109F/1548R and M1-2F/M3-2R primer pair (Figure 1A). A sediment sample was also selected to compare the differences in the taxonomic distribution among these three aioA gene primer pairs (Figure 1B). The Curvibacter-associated aioA gene sequences consisted of 57.38 and 84.04% in the M1-2F/M3-2R and 1109F/1548R libraries, respectively, and was a rare biome in the deg1F/deg1R library. Furthermore, the dominant Methylobacterium-affiliated sequence (19.67%) has only been detected using the M1-2F/M3-2R primer pair (Figure 1B).
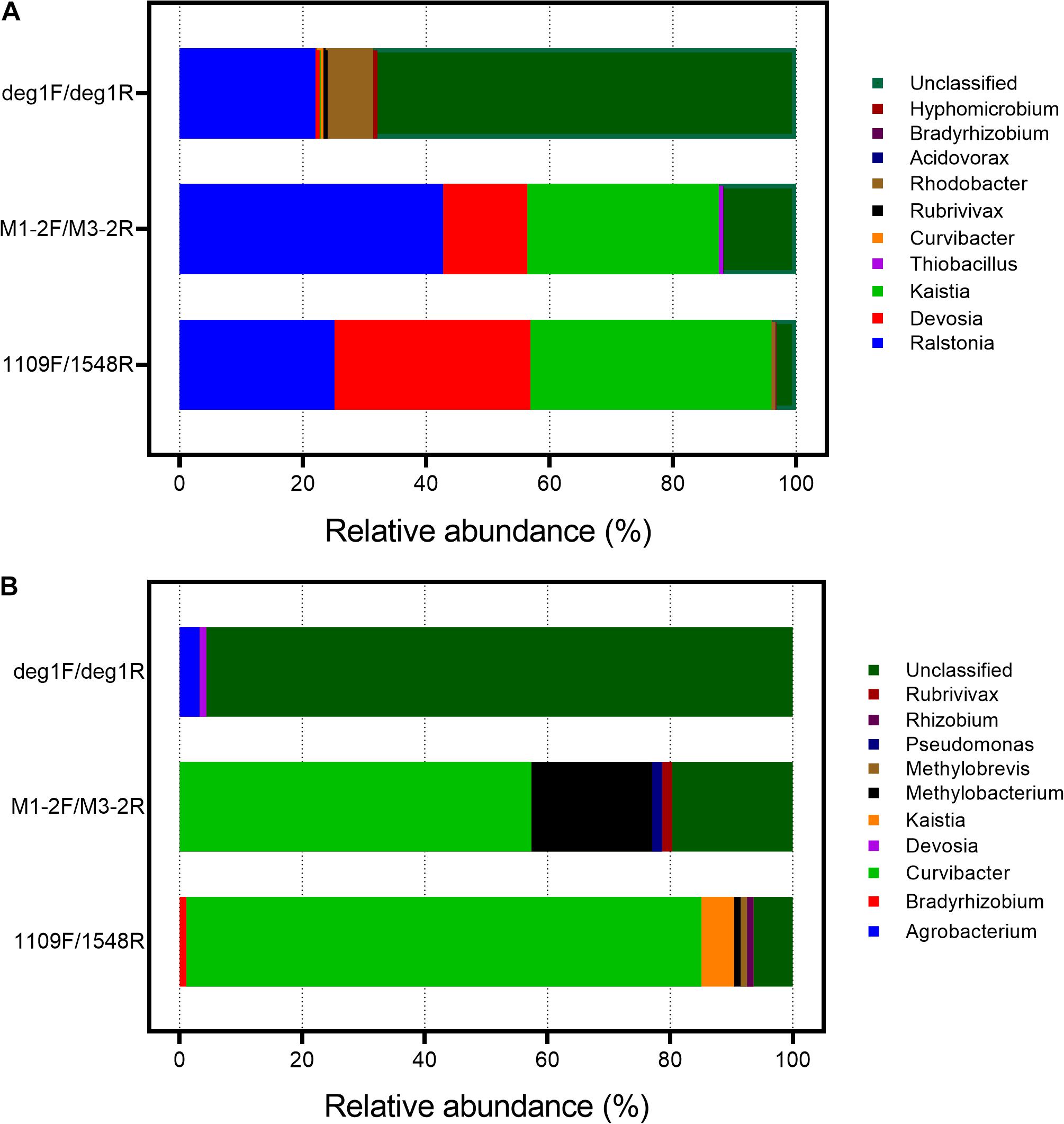
Figure 1. Taxonomic information of the clone sequences generated using three primer pairs and DNA from the As-contaminated paddy soil (A) and sediment (B). The aioA-targeted clone sequences were verified using the BLASTX similarity search against the NCBI-nr database at e < e– 10 and alignment length > 150 nt. Specificities of the primer sets were calculated as the aioA-target sequence hits to the total number of clone sequences. The taxonomic information for the aioA clone sequences is shown at the genus level after disarming non-aioA sequences.
A comparison of 1109F/1548R on the Sanger and NGS sequencing platforms was also conducted to verify the effects of sequencing methods on the detected community of As(III)-oxidizing bacteria using the 1109F/1548R primer pair. It was shown that the taxonomic compositions of the aioA genes were similar using these two sequencing strategies. Although, the taxonomic composition of the aioA genes had slight differences between the Sanger and NGS sequencing, the most abundant aioA genes from the paddy soil sample were affiliated with Kaisita (with a relative abundance of 28.94% in the Sanger sequencing and 39.05% in the NGS sequencing). This was followed by Devosia (Sanger: 22.53%; NGS: 31.77%) and Ralstonia (Sanger: 32.30%; NGS: 25.16%). The predominant genera in the sediment examined using the 1109F/1548R primer pair were also similar both on the Sanger and NGS platforms, and Curvibacter (Sanger: 84.04%; NGS: 76.80%) was the most identified abundant genera (Figure 2). Spearman’s rho tests showed a significant correlation between taxonomic compositions of aioA genes sequenced by Sanger and NGS platforms, using paddy soil (Spearman rho coefficient = 0.838, p-value = 0.009) and sediment (Spearman rho coefficient = 0.829, p-value = 0.002) samples. The above results suggested the taxonomic profiles are similar between these two sequencing approaches.
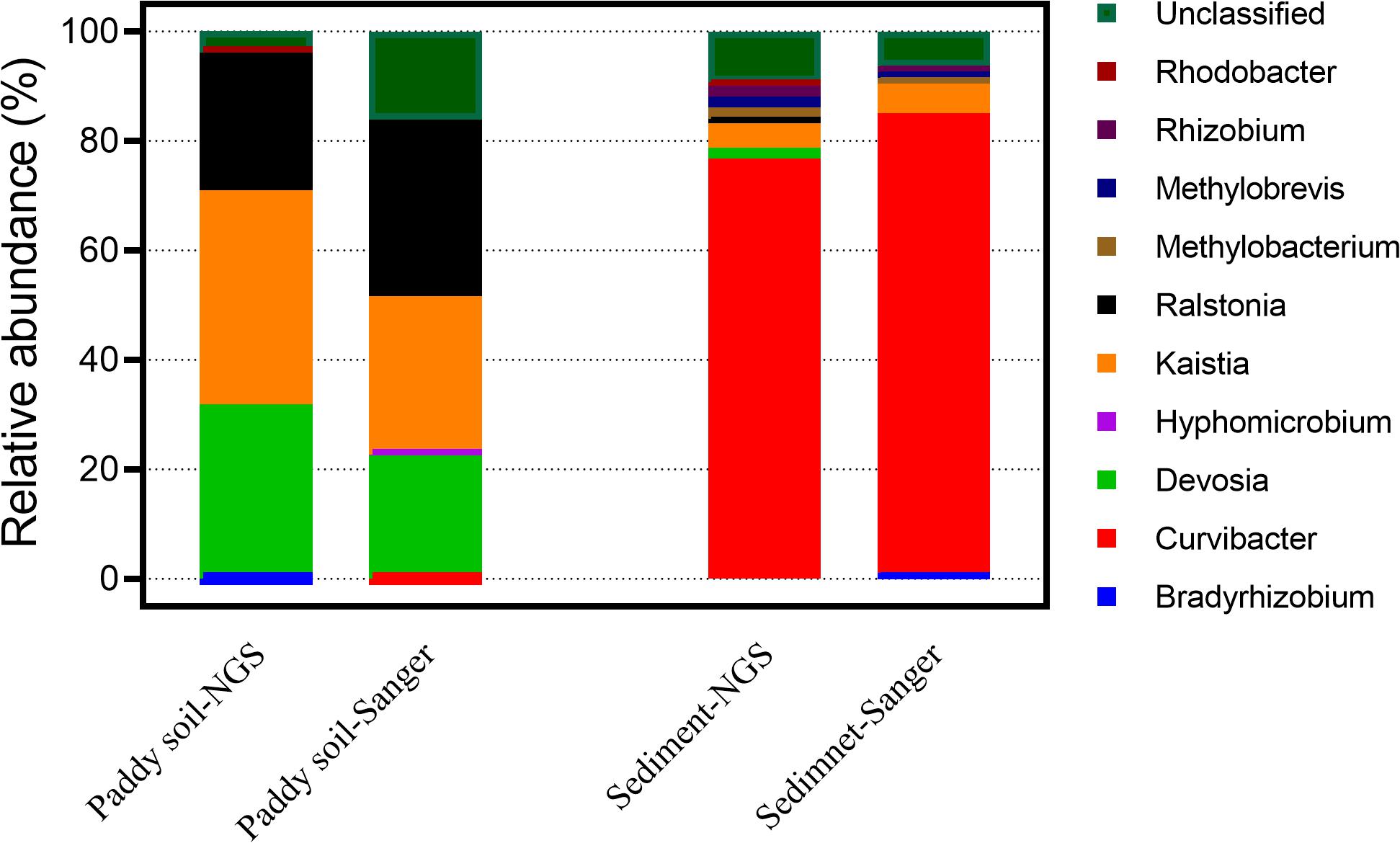
Figure 2. The taxonomic comparison of Miseq (NGS) and clone (Sanger) sequencing of aioA genes in paddy soil and sediment samples using 1109F/1548R primers.
The availability of the newly developed primer pair, 1109F/1548R, for the amplification of aioA genes from five samples of As-contaminated paddy soil and five samples of sediment was tested using high-throughput sequencing (Supplementary Table 4). In addition, the community diversity and structure of the As(III) oxidizers from these two habitats were compared. After quality screening, chimera removal, and the BLASTX analysis against the NCBI-nr database, 151,287 aioA sequences were extracted from the 1109F/1548R libraries (Supplementary Table 4). After resampling, 10,313 sequences per sample were used for further analysis. The aioA amplicon sequences were clustered into 280 OTUs at a 90% sequence identity. The Good coverage ranged from 0.996 to 0.998, indicating that the sequencing had sampled the majority of the diversity present (Supplementary Table 4). The alpha diversity of aioA genes, as measured by the observed OTUs, Chao1, Shannon, Simpson, and Faith PD indexes, was estimated using the QIIME2 platform. Briefly, the observed OTUs were 102.6 ± 18.022 and 81.4 ± 21.22 for the paddy soil and sediment samples, respectively. The Chao1 indexes were 128.97 ± 19.21 and 104.64 ± 22.39 for the paddy soil and sediment samples, respectively. The Shannon indexes were 3.47 ± 0.61 and 3.20 ± 0.47 for the paddy soil and sediment samples, respectively. The Simpson indexes were 0.81 ± 0.07 and 0.79 ± 0.06 for the paddy soil and sediment samples, respectively. The Faith PD indices were 12.63 ± 1.36 and 10.90 ± 1.64 for the paddy soil and sediment samples, respectively. The Chao1 and Faith PD diversity indices were significantly higher in the paddy soil than in the sediment (both p = 0.076, Supplementary Table 5). However, the number of observed OTUs and the values of the Chao1, Shannon, and Simpson indexes did not differ significantly between the paddy soil and sediment samples (all p > 0.05, Supplementary Table 5). The results of the unweighted Unifrac PCoA showed that the sediment samples clearly clustered together and separately from the paddy soil samples, and the Unifrac distance among the paddy soil samples was higher than that among the sediment samples. These results indicated that the As(III)-oxidizing community structure was strongly associated with the sampled habitats (Figure 3A).
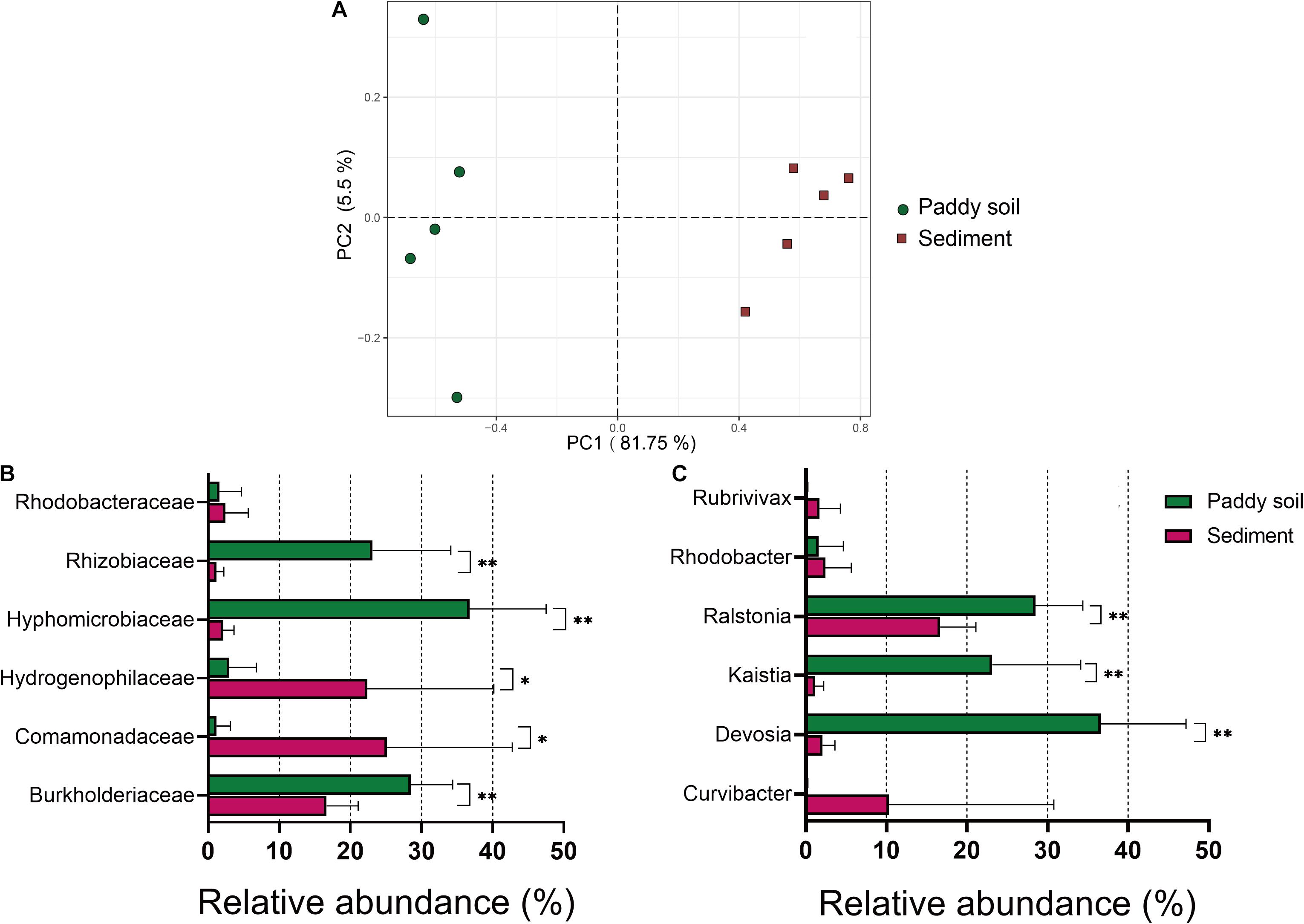
Figure 3. Differences in the As(III)-oxidizing bacteria between the paddy soil and sediment samples. (A) Unweighted PCoA clustering of the As(III)-oxidizing community in the paddy soil (red circles) and sediment (blue squares) samples generated using the 1109F/1548R primer sets. The percentage variation in the plotted principal component is indicated on the axes. (B) Taxonomic composition of the As(III)-oxidizing bacteria at the family level. (C) Taxonomic composition of the As(III)-oxidizing bacteria at the genus level. The taxonomic composition of the aioA sequences in each sample was determined using the MEGAN software against the NCBI-nr with Diamond software at e < e–10 and an alignment length > 200 nt. The average ± SD values of the samples in each group are expressed in each column. The significance cutoff for the corrected P-value using the Benjamini–Hochberg false discovery rate (FDR) procedure is shown. Significant differences between the sampled groups are indicated as ∗∗p < 0.01 and ∗p < 0.05.
The phylogenetic distribution of the As(III)-oxidizing communities was also determined using the BLASTX data. Among the aioA sequences retrieved from the paddy soil, the largest fraction of these sequences was affiliated with the families Hyphomicrobiaceae (36.79 ± 10.75%), Rhizobiaceae (23.14 ± 10.97%), and Burkholderiaceae (28.50 ± 5.88%) (Figure 3B). Only 2.96 ± 3.84%, 1.60 ± 3.07%, and 1.19 ± 1.94% of the sequences aioA were distributed among the families Hydrogenophilaceae, Rhodobacteraceae, and Comamonadaceae, respectively. For the As-contaminated sediments, the average, 25.16 ± 17.6% sequences fell into the family Comamonadaceae, and 22.39 ± 17.78% was affiliated with Hydrogenophilaceae, followed by Burkholderiaceae (16.65 ± 4.45%). Multiple t-test analyses indicated significant differences in the abundance (p < 0.05) between the dominant families in the paddy soils and the sediments (with relative abundances greater than 1% including Burkholderiaceae, Comamonadaceae, Hydrogenophilaceae, Hyphomicrobiaceae, and Rhizobiaceae) (Figure 3B). At the genus level, Ralstonia, Kaistia, and Devosia exhibited statistically significant differences in abundances between the paddy soil and the sediment samples (P < 0.01) (Figure 3C).
To investigate the phylogenetic affiliation of the most abundant OTUs in detail, the 20 most abundant OTUs were aligned with reference sequences and a Neighbor-Joining tree was constructed. As shown in Supplementary Figure 2 and Supplementary Table 6, the majority of the abundant OTUs clustered with uncultured bacteria. The 20 most abundant OTUs were grouped into seven clades that belonged to Alphaproteobacteria (in the orders of Hyphomicrobiales and Rhodobacterales), Betaproteobacteria (in the orders of Burkholderiales and Hydrogenophilales), and uncultured bacteria (three clusters). Clade I consisted of two OTU (OTU 11 and OTU 106) and showed high similarity with bacterium HR39 and bacterium HR40) with additive abundances of 23.65 and 1.12% in the paddy soil and sediment samples, respectively. In addition, two OTUs (OTU 13 and OTU 170 with total abundances of 33.15 and 1.53% in the paddy soil and sediment samples, respectively) clustered with Devosia sp. 67–54 (OJX19890) (with an average 82.75% amino acid identity). Furthermore, OTU 40 (average abundance in the paddy soil: 1.57%; sediment: 2.36%) clustered closely with Rhodobacter sp. CACIA14H1 (ESW59939) (93.00% identity). The abundance of OTUs (OTU3 and OTU 162) of clade III in the paddy soil (with total abundances of 1.00%) was lower than that in the sediment (25.79%), which clustered with Curvibacter sp. GWA2 64 110 (OGP03110) (89.44% identity). Three OTUs (OTU 1, OTU 42, and OTU 16, with total abundances of 1.18 and 20.15% in the paddy soil and sediment samples, respectively) clustered with sequences from a reference Hydrogenophilaceae bacterium CG1_02_62_3903 (OIO79863) (with an average 79.58% amino acid identity). Six OTUs (OTU 20, OTU 258, OTU 105, OTU 251, OTU 38, and OTU 12, and with total abundances of 16.39 and 8.75% in the paddy soil and sediment samples, respectively) and with an uncultured microorganism (BAM75656) (average 76.21% amino acid identity). Finally, OTU 2, OTU 5, OTU 32, and OTU 68 (with a total abundance in the paddy soil: 1.7%; sediment: 13.93%) clustered with Betaproteobacteria bacterium RIFCSPLOWO2_02_FULL_62_17 (OFZ99153) (average 86.64% identity). The remaining OTUs were placed closely together with a number of uncultured aioA sequences obtained in previous studies that were not affiliated with any cultured isolate (Supplementary Figure 2 and Supplementary Table 6).
Samples from the As-contaminated paddy soil and sediment were collected from the same region that was used to examine the performance of the 109F/1548R primer pairs using high-throughput amplicon sequencing. The paddy samples exhibited a pH range from 6.44 to 6.75 compared to a range of 6.15–6.49 in the sediment samples. The pH values in the paddy soil samples were significantly higher than that in the sediment samples (Student’s t-tests, p < 0.01). The paddy soil and sediment samples contained organic matter concentrations of 27.86 ± 2.50 g kg–1 and 27.11 ± 8.55 g kg–1, respectively. The total As concentration in the paddy soil samples varied from 37.19 to 94.42 mg kg–1, and all exceeded the maximum allowable concentration (MAC) of As for agricultural soils in China (30mg kg–1, National Environmental Protection Agency of China GB 15618, 1995). The total concentration of As in the sediment samples was 57.50 ± 21.30 mg kg–1, which did not exhibit a significant difference to the paddy soil samples. Phosphate-extractable and oxalate-extractable As(III)/As(V) in the paddy soil and sediment samples, which represented the As species adsorbed on iron (oxyhydr) oxides and the As species incorporated into iron (oxyhydr)-oxides, respectively. The concentration of phosphate-extractable As(III)/As(V) and oxalate-extractable As(III)/As(V) in the sediment samples (PO4- As(III) with an average ± SD value of 4.13 ± 4.00 mg kg–1; PO4- As(V) value of 20.34 ± 13.67 mg kg–1; oxalate-As(III) value of 1.26 ± 0.68 mg kg–1; and oxalate-As(V) value of 9.84 ± 4.78 mg kg–1) were significantly higher than that in the paddy soil sample (PO4-As(III) value of 1.30 ± 0.95 mg kg–1; PO4-As(V) value of 6.08 ± 2.93 mg kg–1; oxalate-As(III) value of 0.75 ± 0.22 mg kg–1; and oxalate-As(V) value of 3.52 ± 0.86 mg kg-1) (all p < 0.01). RF predictions were employed to explore how the environmental parameters affected the diversity of aioA (Figure 4). The results showed that dissolved As(III) had the highest relative influence (8.14%) on the Chao1 index of the aioA genes followed by oxalate-As(V) (5.82%), PO4-As(V) (4.28%), and PO4-As(III) (1.98%). These findings implied that dissolved As(III) may be a key factor driving the species richness of As(III)-oxidizing bacteria in natural environments.
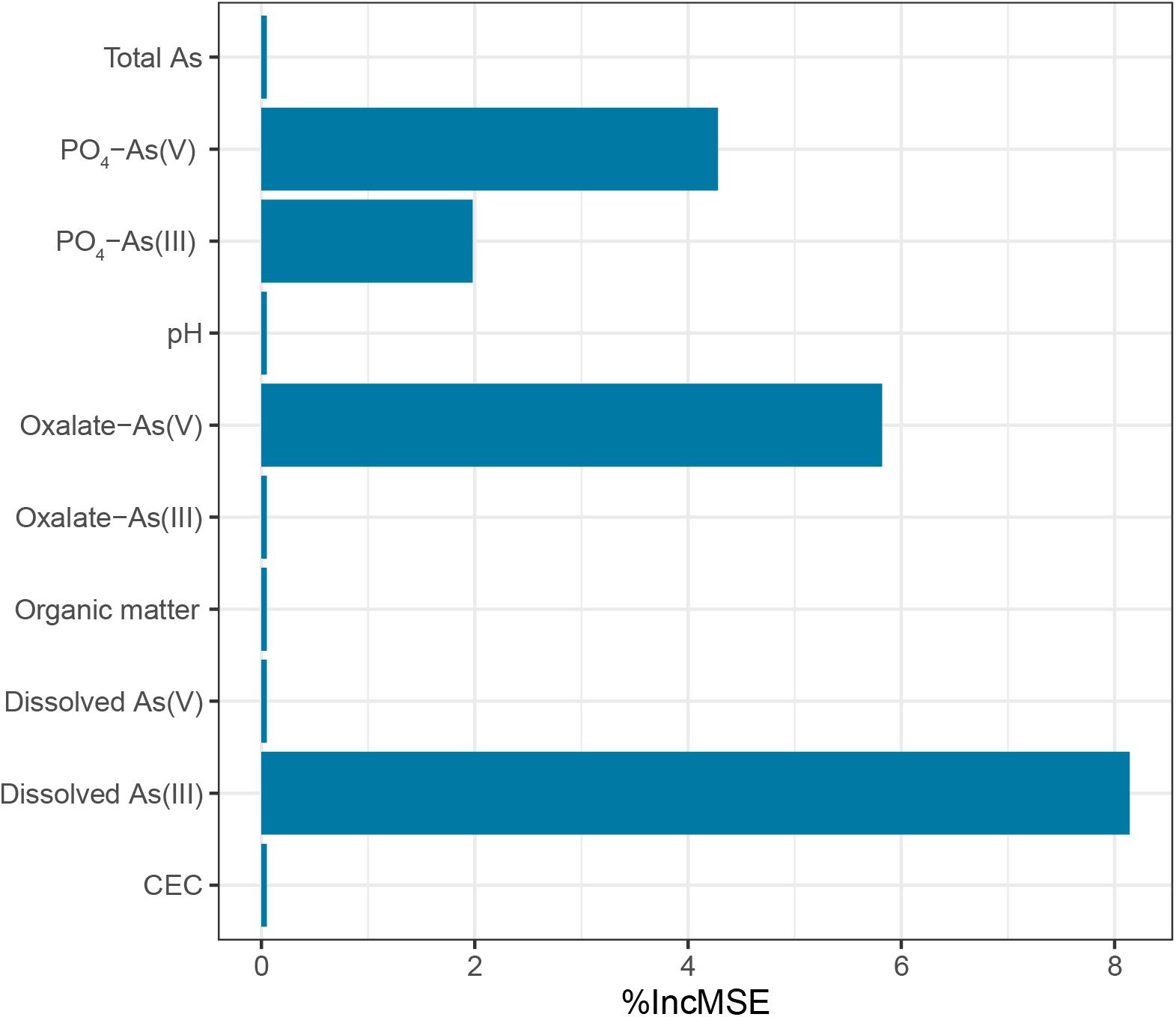
Figure 4. Random forest (RF) predictions of the most influential environmental parameters on the Chao1 index of the aioA genes.
Based on the in silico evaluation, the best performing primer pair was deg1F/deg1R, with an overall coverage of 85.09% of all the aioA gene sequences analyzed (Supplementary Figure 1). That was because of primer degeneracy of deg1F and deg1R was much higher than that of the other two primer pairs (Table 1). The high degeneracy of primers increased the probability of matching aioA gene sequences. However, it should be noted that primer degeneracy can also lead to a biased picture of microbial diversity (Baker et al., 2003; Huse et al., 2008), and preferential amplification might lead to under-representation of important members of a community (Huse et al., 2008). In addition, the primers used for high-throughput amplicon sequencing normally consisted of the Illumina adapter, barcoder, pad, and linker sequences, followed by the gene targeted primers (Caporaso et al., 2010b). Then, the high degeneracy of the aioA primers may also increase the probability of secondary structure generation, such as dimers and hairpins. Such a secondary structure of the primers adversely affect primer template annealing and reduce the availability of primers to participate in the reaction. This can lead to poor or no yields of the product. Thus, the high degeneracy of the aioA primers are not suitable for high-throughput amplicon sequencing.
Although the phylogenetic coverage of deg1F/deg1R was best in the in silico analysis, the taxonomic resolution was not high in the other reports that used this primer pair. In an investigation of the impact of nitrate additions on As species dynamics in the porewater of four As-contaminated paddy soils, the results of the aioA clone libraries using deg1F/deg1R showed that 35.42–70.83% of the clone sequences were classified as uncultured organisms (Zhang et al., 2017). In a similar study on the diversity of bacterial As(III) oxidase and reductase genes in the rhizosphere of the As-hyperaccumulator, the result of the aioA gene clone libraries using the deg1F/deg1R primer pair also showed that most of the clone sequences were affiliated with uncultured bacterium (Han et al., 2017). Hence, the phylogenetic coverage of deg1F/deg1R should be carefully examined using natural samples in the future. In addition, the isolated As(III)-oxidizing bacteria primarily belonged to Proteobacteria (Supplementary Figure 1), and this may lead to an over-estimation of the relative abundance of Proteobacteria in identifying As(III)-oxidizers in nature. Indeed, various primer pair targeted aioA genes could not cover all the lineages of As(III)-oxidizers, and a gene diversity analysis may produce different taxonomic information. We think this bias could be settled with high-throughput strain isolation methods and the recently rising metagenome binning technology (to obtain the nearly complete genome without cultivation).
The conserved regions of the aioA gene used for primer design may be an important factor influencing the taxonomic resolution of the aioA clone sequences. The deg1F/deg1R primers targeted the sequence encoding the conserved [3Fe–4S]-binding motif of the AioA protein compared with the 1109F/1548R primers targeting the sequence for the molybdopterin center of the AioA protein and the M1-2F/M3-2R primers targeting these two conserved regions (Table 1). Combined with the taxonomic information obtained from the analyses involving these three primer pairs, the taxonomic resolution of the primer pairs targeting the molybdopterin center sequence was much better than that of the primers targeting the [3Fe–4S]-binding motif sequence.
Traditional molecular fingerprint methods, such as T-RFLP, DGGE, and clone library, have been used to investigate the distribution and diversity of aioA genes (Zhang J. et al., 2015; Han et al., 2017; Pipattanajaroenkul et al., 2021). These studies have reported that the community of As(III)-oxidizing bacteria in paddy soil was dominated by Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria (Dong et al., 2014; Jia et al., 2014), most of which are affiliated with Rhizobiales, Burkholderiales, Comamonadaceae, Phyllobacteriaceae, Bradyrhizobiaceae, and Methylobacteriaceae (Zhang S. Y. et al., 2015). In this study, the bacterial diversity and compositions of the aioA gene in paddy soil and sediment were first explored using NGS. Compared with the sediment, the paddy soil samples were significantly enriched in the aioA gene affiliated with the genus of Devosia (in the family of Hyphomicrobiaceae), Kaistia (in the family of Rhizobiaceae), and Ralstonia (in the family of Burkholderiaceae) (Figure 3). Devosia dominated in the community of the heterotrophic enrichment using As(III) as the only electron donor, suggesting its important role in As(III) oxidation in soil (Sultana et al., 2012). Kaistia is typically a rhizospheric genera, and the aioA gene has been identified in the genome of Kaistia sp. SCN 65-12 (Pal and Sengupta, 2021). Ralstonia sp. 22 was isolated from mine soil and shown to be capable of As(III) oxidation (Lieutaud et al., 2010). In addition, Ralstonia sp. b3 was the predominant species of an autotrophic arsenic(III)-oxidizing population in reactors (Battaglia-Brunet et al., 2002). Additionally, the relative abundance of Curvibacter (in the family of Comamonadaceae) was much higher in sediment than that in paddy soil samples (Figure 3). In a study of metagenomic insights into microbial arsenic metabolism in shallow groundwater, aioA genes primarily contributed to Curvibacter (Wang et al., 2020). In addition, the ferrous-oxidizing Curvibacter sp. CD03 has been retrieved from alluvial sediment (Hassan et al., 2016), indicating the bacteria in the genus Curvibacter have potential for As(III) and Fe(II) oxidation. This simultaneously suggests that it could be utilized as a bioremediation agent for reducing As toxicity and immobilization in sediments.
In this study, the majority of abundant OTUs in the paddy soil was placed to uncultured aioA sequences reported previously that are not affiliated with any cultured isolates. However, the OTUs from the sediment samples always clustered with reference aioA sequences of As(III)-oxidizing strains, such as Thiobacillus sp. SCN 64-35, Comamonadaceae bacterium SCN 68-20, and Curvibacter sp. GWA2 64 110 (Supplementary Figure 2). Although very few As(III) oxidizers have been successfully isolated from the paddy soil environment (only Paracoccus sp. SY and Acidovorax sp. ST3 have been reported) (Zhang J. et al., 2015; Zhang et al., 2017), the predicted AioA protein sequences of some OTUs from the paddy soil shared a high amino acid identity with well-known As(III) oxidizers, such as Rhizobium sp. NT-26, H. arsenicoxydans, and Agrobacterium sp. C13 (Supplementary Figure 2). These microbes have been shown to contribute significantly to the oxidation of As(III) under both aerobic and anaerobic conditions. The above findings indicated that the As(III)-oxidizing community structure and composition in the paddy soil and sediment environments were remarkably different. Therefore, additional efforts should be dedicated toward delineation of the diversity and distribution pattern of the As(III) oxidizers from diverse environments globally, and this would involve high-throughput parallel sequencing of a large number of samples.
Microbial As(III) oxidation contributes to efficient As immobilization in environments. Thus, an understanding of the environmental factors that influence aioA genes is important for the bioremediation of arsenic-contaminated environments in the future. We found that the Chao1 and Faith PD diversity indices of the aioA genes were significantly higher in paddy soil than in sediment (Supplementary Table 5). In a previous study, a redundancy analysis indicated that soil pH, available Ca and P, and As(V) concentration were key factors driving diverse compositions in the aioA gene community, and As(V) amendment elevated aioA gene diversity in the rhizosphere of the As-hyperaccumulator Pteris vittata (Han et al., 2017). Zhang S. Y. et al. (2015) also found pH, SO42–-S, and the total C/N ratio significantly explained the variation in the microbial community compositions based on aioA genes. However, the effects of arsenic species on the As(III)-oxidizing communities have been less reported. To explore how environmental parameters affected the diversity of aioA genes, an RF analysis was conducted. The results showed that dissolved As(III) had the highest relative influence on the Chao1 index of the aioA genes (Figure 4). In arsenic contaminated soils, phosphate-extractable As was one of the most important factors in shaping the As transformation functional genes (Gu et al., 2017). Furthermore, As(III) was identified as the major environmental factor influencing the community and abundance of As(III)-oxidizing bacteria in groundwater (Pipattanajaroenkul et al., 2021). Normally, As fractionation in soil and sediment includes dissolved, poorly crystalline Fe (oxyhydr)oxide-bound (extracted by NH4+-oxalate) and Fe (oxyhydr)oxides-bound (extracted by KH2PO4 and ascorbic acid) forms that represent the different degrees of bioavailability (Liu et al., 2015). It has been shown that dissolved As(III) is easily utilized as electron donor by bacteria as Fe (oxyhydr)oxide-bound As(III) (Jeong et al., 2010). Thus, it is not strange that the diversity of the aioA genes were very highly influenced by dissolved As(III) than by PO4-As(III) and oxalate- As(III).
In summary, in the current study, it was demonstrated that the newly developed primer pair, 1109F/1548R, was sufficiently long to capture genetic variation, allowing for the discrimination of phylogenetic groups of the aioA genes. The obtained data encourages further use of the aioA gene as a functional marker specific to As(III) oxidizers in environmental diversity surveys. The newly developed aioA primers tested in the current study will aid other researchers wishing to detect As(III) oxidases (or their expression) in environmental systems, especially for the identification of the As(III)-oxidizing community composition and diversity, which requires multiple-sample analysis and deep sequencing. Future studies should focus on evaluating the link between As speciation and concentrations and aioA gene diversity and abundances in the environment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MH and FL conceived and designed the experiments. MH and JQ performed most of the experiments. CY, HY, and LZ supervised the execution of the experiments. MH wrote the manuscript. All authors read and approved the final manuscript.
This work was supported by grants from the National Science Foundation of China (Grant No. 41977138), the Natural Science Foundation of Guangdong Province (Grant No. 2020A1515011577), the Outstanding Young Talent for the National “Ten Thousand Talents Program,” and the GDAS Special Project of Science and Technology Development (Grant No. 2020GDASYL-20200104017).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.691913/full#supplementary-material
Supplementary Figure 1 | Phylogenetics of the 67 aioA genes used for the primer design, and the hits (a pair matched the same sequence with a maximum of two mismatches and alignment length larger than 16 nt) of the two primer pair matches) of the three primer pairs to the 67 aioA gene sequences. The solid square represents a hit of the primer pair to the corresponding aioA gene sequences, whereas the hollow square represents the primer pair could not match to the aioA gene sequences. The order of the squares in each aioA sequence are shown as1109F/1548R, M1-2F/M3-2R, and deg1F/deg1R. Sequences of arrA from Shewanella sp. ANA and Bacillus selenitireducens MLS10 functioned as outgroups in building the phylogenetic tree.
Supplementary Figure 2 | Neighbor-joining tree of the 20 most abundant OTUs in the library of the aioA-1109F/1548R. The average relative abundances (%) for the paddy soil and sediment samples are given to the right of each OTU. The scale bar indicates the sequence dissimilarity between the nodes.
Supplementary Table 1 | Information of arsenite oxidase and the corresponding genes used in the primer design.
Supplementary Table 2 | Geochemistry of the paddy soils and sediments used for the aioA genes high-throughput amplicon sequencing.
Supplementary Table 3 | Barcoding primer sequences for the amplicon sequencing on the Illumina Hiseq platform.
Supplementary Table 4 | The amplicon sequencing information and alpha diversity index of the aioA genes on the Illumina Hiseq platform using the two newly designed primer pairs.
Supplementary Table 5 | The difference in α-diversity of the paddy soil and sediment samples in the libraries of 1109F/1548R.
Supplementary Table 6 | Phylogenetics of the aioA OTUs (with relative abundance > 1%) with their closest relatives.
Baker, G. C., Smith, J. J., and Cowan, D. A. (2003). Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 55, 541–555. doi: 10.1016/j.mimet.2003.08.009
Battaglia-Brunet, F., Dictor, M.-C., Garrido, F., Crouzet, C., Morin, D., Dekeyser, K., et al. (2002). An arsenic(III)-oxidizing bacterial population: selection, characterization, and performance in reactors. J. Appl. Microbiol. 93, 656–667. doi: 10.1046/j.1365-2672.2002.01726.x
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59. doi: 10.1038/nmeth.3176
Cai, W. W., Liu, W. Z., Zhang, Z. J., Feng, K., Ren, G., Pu, C. L., et al. (2018). mcrA sequencing reveals the role of basophilic methanogens in a cathodic methanogenic community. Water Res. 136, 192–199. doi: 10.1016/j.watres.2018.02.062
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Bittinger, K., Bushman, F. D., Desantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Chiri, E., Greening, C., Lappan, R., Waite, D. W., Jirapanjawat, T., Dong, X. Y., et al. (2020). Termite mounds contain soil-derived methanotroph communities kinetically adapted to elevated methane concentrations. ISME J. 14, 2715–2731. doi: 10.1038/s41396-020-0722-3
Dong, D. T., Yamaguchi, N., Makino, T., and Amachi, S. (2014). Effect of soil microorganisms on arsenite oxidation in paddy soils under oxic conditions. Soil Sci. Plant Nutrit. 60, 377–383. doi: 10.1080/00380768.2014.897924
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Gu, Y. F., Van Nostrand, J., Wu, L. Y., He, Z. L., Qin, Y. J., Zhao, F. J., et al. (2017). Bacterial community and arsenic functional genes diversity in arsenic contaminated soils from different geographic locations. PLoS One 12:e0176696. doi: 10.1371/journal.pone.0176696
Hamamura, N., Macur, R. E., Korf, S., Ackerman, G., Taylor, W. P., Kozubal, M., et al. (2009). Linking microbial oxidation of arsenic with detection and phylogenetic analysis of arsenite oxidase genes in diverse geothermal environments. Environ. Microbiol. 11, 421–431. doi: 10.1111/j.1462-2920.2008.01781.x
Han, Y. H., Fu, J. W., Xiang, P., Cao, Y., Rathinasabapathi, B., Chen, Y., et al. (2017). Arsenic and phosphate rock impacted the abundance and diversity of bacterial arsenic oxidase and reductase genes in rhizosphere of As-hyperaccumulator Pteris vittata. J. Hazard. Mater. 321, 146–153. doi: 10.1016/j.jhazmat.2016.08.079
Hassan, Z., Sultana, M., Westerhoff, H. V., Khan, S. I., and Röling, W. F. M. (2016). Iron cycling potentials of arsenic contaminated groundwater in Bangladesh as revealed by enrichment cultivation. Geomicrobiol. J. 33, 779–792. doi: 10.1080/01490451.2015.1111471
Hugerth, L. W., Wefer, H. A., Lundin, S., Jakobsson, H. E., Lindberg, M., Rodin, S., et al. (2014). DegePrime, a program for degenerate primer design for broad-taxonomic-range PCR in microbial ecology studies. Appl. Environ. Microbiol. 80, 5116–5123. doi: 10.1128/aem.01403-14
Huse, S. M., Dethlefsen, L., Huber, J. A., Welch, D. M., Relman, D. A., and Sogin, M. L. (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. doi: 10.1371/journal.pgen.1000255
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Inskeep, W. P., Macur, R. E., Hamamura, N., Warelow, T. P., Ward, S. A., and Santini, J. M. (2007). Detection, diversity and expression of aerobic bacterial arsenite oxidase genes. Environ. Microbiol. 9, 934–943. doi: 10.1111/j.1462-2920.2006.01215.x
Jeong, H. Y., Han, Y. S., Park, S. W., and Hayes, K. F. (2010). Aerobic oxidation of mackinawite (FeS) and its environmental implication for arsenic mobilization. Geochim. Cosmochim. Acta 74, 3182–3198. doi: 10.1016/j.gca.2010.03.012
Jia, Y., Huang, H., Chen, Z., and Zhu, Y. G. (2014). Arsenic uptake by rice is influenced by microbe-mediated arsenic redox changes in the rhizosphere. Environ. Sci. Technol. 48, 1001–1007. doi: 10.1021/es403877s
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1. doi: 10.1093/nar/gks808
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evolut. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lieutaud, A., Van Lis, R., Duval, S., Capowiez, L., Muller, D., Lebrun, R., et al. (2010). Arsenite oxidase from Ralstonia sp. 22: Characterization of the enzyme and its interaction with soluble cytochromes. J. Biol. Chemistry 2010:113761. doi: 10.1074/jbc.M110.113761
Liu, C. P., Luo, C. L., Gao, Y., Li, F. B., Lin, L. W., Wu, C. A., et al. (2010). Arsenic contamination and potential health risk implications at an abandoned tungsten mine, southern China. Environ. Pollut. 158, 820–826. doi: 10.1016/j.envpol.2009.09.029
Liu, C. P., Yu, H. Y., Liu, C. S., Li, F. B., Xu, X. H., and Wang, Q. (2015). Arsenic availability in rice from a mining area: Is amorphous iron oxide-bound arsenic a source or sink? Environ. Pollut. 199, 95–101. doi: 10.1016/j.envpol.2015.01.025
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mirza, B. S., Sorensen, D. L., Dupont, R. R., and Mclean, J. E. (2017). New arsenate reductase gene (arrA) PCR primers for diversity assessment and quantification in environmental samples. Appl. Environ. Microbiol. 83, e2725–e2716. doi: 10.1128/aem.02725-16
Mukhopadhyay, R., Rosen, B. P., Phung, L. T., and Silver, S. (2002). Microbial arsenic: From geocycles to genes and enzymes. FEMS Microbiol. Rev. 26, 311–325. doi: 10.1111/j.1574-6976.2002.tb00617.x
Oremland, R. S., and Stolz, J. F. (2005). Arsenic, microbes and contaminated aquifers. Trends Microbiol. 13, 45–49. doi: 10.1016/j.tim.2004.12.002
Pal, S., and Sengupta, K. (2021). In silico analysis of phylogeny, structure, and function of arsenite oxidase from unculturable microbiome of arsenic contaminated soil. J. Genet. Enginee. Biotechnol. 19:47. doi: 10.1186/s43141-021-00146-x
Pelikan, C., Herbold, C. W., Hausmann, B., Müller, A. L., Pester, M., and Loy, A. (2016). Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ. Microbiol. 18, 2994–3009. doi: 10.1111/1462-2920.13139
Pipattanajaroenkul, P., Chotpantarat, S., Termsaithong, T., and Sonthiphand, P. (2021). Effects of arsenic and iron on the community and abundance of arsenite-oxidizing bacteria in an arsenic-affected groundwater aquifer. Curr. Microbiol. 78, 1324–1334. doi: 10.1007/s00284-021-02418-8
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Qiao, J. T., Li, X. M., Li, F. B., Liu, T. X., Young, L. Y., Huang, W. L., et al. (2019). Humic substances facilitate arsenic reduction and release in flooded paddy soil. Environ. Sci. Technol. 53, 5034–5042. doi: 10.1021/acs.est.8b06333
Quéméneur, M., Heinrich-Salmeron, A., Muller, D., Lièvremont, D., Jauzein, M., Bertin, P. N., et al. (2008). Diversity surveys and evolutionary relationships of aoxB genes in aerobic arsenite-oxidizing bacteria. Appl. Environ. Microbiol. 74, 4567–4573. doi: 10.1128/aem.02851-07
Ramirez-Gonzalez, R., Yu, D. W., Bruce, C., Heavens, D., Caccamo, M., and Emerson, B. C. (2013). PyroClean: Denoising pyrosequences from protein-coding amplicons for the recovery of interspecific and intraspecific genetic variation. PLoS One 8:e57615. doi: 10.1371/journal.pone.0057615
Roots, P., Wang, Y. B., Rosenthal, A. F., Griffin, J. S., Sabba, F., Petrovich, M., et al. (2019). Comammox Nitrospira are the dominant ammonia oxidizers in a mainstream low dissolved oxygen nitrification reactor. Water Res. 157, 396–405. doi: 10.1016/j.watres.2019.03.060
Sultana, M., Vogler, S., Zargar, K., Schmidt, A.-C., Saltikov, C., Seifert, J., et al. (2012). New clusters of arsenite oxidase and unusual bacterial groups in enrichments from arsenic-contaminated soil. Arch. Microbiol. 194, 623–635. doi: 10.1007/s00203-011-0777-7
Wang, L. Y., Yin, Z. P., and Jing, C. Y. (2020). Metagenomic insights into microbial arsenic metabolism in shallow groundwater of Datong basin, China. Chemosphere 245:125603. doi: 10.1016/j.chemosphere.2019.125603
Xue, S. G., Jiang, X. X., Wu, C., Hartley, W., Qian, Z. Y., Luo, X. H., et al. (2020). Microbial driven iron reduction affects arsenic transformation and transportation in soil-rice system. Environ. Pollut. 260:114010. doi: 10.1016/j.envpol.2020.114010
Yamamura, S., and Amachi, S. (2014). Microbiology of inorganic arsenic: from metabolism to bioremediation. J. Biosci. Bioengine. 118, 1–9. doi: 10.1016/j.jbiosc.2013.12.011
Yu, H. Y., Wang, X. Q., Li, F. B., Li, B., Liu, C. P., Wang, Q., et al. (2017). Arsenic mobility and bioavailability in paddy soil under iron compound amendments at different growth stages of rice. Environ. Pollut. 224, 136–147. doi: 10.1016/j.envpol.2017.01.072
Zeleke, J., Sheng, Q., Wang, J. G., Huang, M. Y., Xia, F., Wu, J. H., et al. (2013). Effects of Spartina alterniflora invasion on the communities of methanogens and sulfate-reducing bacteria in estuarine marsh sediments. Front. Microbiol. 4:243. doi: 10.3389/fmicb.2013.00243
Zhai, W. W., Qin, T. Y., Li, L. G., Guo, T., Yin, X. L., Khan, M. I., et al. (2020). Abundance and diversity of microbial arsenic biotransformation genes in the sludge of full-scale anaerobic digesters from a municipal wastewater treatment plant. Environ. Int. 138:105535. doi: 10.1016/j.envint.2020.105535
Zhang, J., Zhao, S. C., Xu, Y., Zhou, W. X., Huang, K., Tang, Z., et al. (2017). Nitrate stimulates anaerobic microbial arsenite oxidation in paddy soils. Environ. Sci. Technol. 51, 4377–4386. doi: 10.1021/acs.est.6b06255
Zhang, S. Y., Zhao, F. J., Sun, G. X., Su, J. Q., Yang, X. R., Li, H., et al. (2015). Diversity and abundance of arsenic biotransformation genes in paddy soils from Southern China. Environ. Sci. Technol. 49, 4138–4146. doi: 10.1021/acs.est.5b00028
Keywords: aioA gene, arsenite oxidation, primer design, As-contaminated paddy soil and sediment, highly-parallel amplicon sequencing
Citation: Hu M, Li F, Qiao J, Yuan C, Yu H and Zhuang L (2021) New Arsenite Oxidase Gene (aioA) PCR Primers for Assessing Arsenite-Oxidizer Diversity in the Environment Using High-Throughput Sequencing. Front. Microbiol. 12:691913. doi: 10.3389/fmicb.2021.691913
Received: 22 May 2021; Accepted: 16 September 2021;
Published: 06 October 2021.
Edited by:
José E. Barboza-Corona, University of Guanajuato, MexicoReviewed by:
Alfonso Benítez-Páez, Principe Felipe Research Center (CIPF), SpainCopyright © 2021 Hu, Li, Qiao, Yuan, Yu and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Hu, aHVtaW5Ac29pbC5nZC5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.