 Yao Yang
Yao Yang Wenjun Zhang1
Wenjun Zhang1 Wenxu Xia
Wenxu Xia- 1School of Food Science and Pharmaceutical Engineering, Nanjing Normal University, Nanjing, China
- 2Jiangsu Academy of Agricultural Sciences, Nanjing, China
- 3Geneception (Shanghai) Bio-technology Co., Ltd., Shanghai, China
- 4Nanjing Institute of Product Quality Inspection, Nanjing, China
Thorough intestinal adhesion and colonization greatly promote the probiotic properties of lactic acid bacteria (LAB). Labeling and tracing with fluorescent proteins are effective and reliable for studying the in vivo physiological activities of LAB including localization, adhesion, and colonization. Lactiplantibacillus plantarum WCFS1 was successfully traced with a red fluorescent protein (RFP), which was expressed by the bacteria-carrying recombinant plasmids. In this study, we aimed to construct a stable RFP mCherry expression system, whose encoding gene was integrated into the bacterial chromosome via double-crossed homologous recombination, and use it for labeling WCFS1 with the goal of avoiding the potential loss of non-chromosomal plasmids along with intestinal growth. First, the constitutive expression of the mCherry protein was improved after adjusting the length of the spacer between the promoter and the gene start codon. Then, the optimized mCherry gene expression cassette was integrated into the chromosome of WCFS1. The resulting strain had normal unimpaired growth and strong fluorescent signals, even after 100 generations, indicating its stability. Furthermore, quantitative polymerase chain reaction (PCR) results revealed a strong positive correlation between the fluorescence intensity of the strain and the number of viable cells, demonstrating its potential usage for the quantification of in vivo WCFS1 cells. Finally, the increased adhesion ability of WCFS1 due to the recombinant expression of the bsh gene was visualized and evaluated using fluorescence intensity, the results of which were consistent with those obtained using the previously established quantification methods. These results suggest that the chromosomal-integrated mCherry labeling system can be extensively used to examine the distribution, colonization, and survival of LAB in vivo in order to determine the mechanism of its probiotic function.
Introduction
Lactic acid bacteria (LAB) are safe for human consumption and beneficial to human health when used as probiotics. Clinically, they can improve digestive health and have various medical applications (Holzapfel and Wood, 2014). For instance, Limosilactobacillus reuteri DSM 17938 helps to prevent and alleviate antibiotic-associated diarrhea (Kołodziej and Szajewska, 2019). Lactobacillus sakei CRL1862 can control biofilms from Listeria monocytogenes by producing bacteriocin (Pérez-Ibarreche et al., 2016). As common inhabitants of the gastrointestinal tract (Salas-Jara et al., 2016), Lactobacillus spp. have been used in genetic and molecular biology studies for heterologous expression, demonstrating their important roles in investigating the properties of probiotics and their potential as live in vivo carriers (Wyszyńska et al., 2015). A previous study showed that recombinant invasive Lactiplantibacillus plantarum expressing fibronectin binding protein A specifically modulated humoral immune responses (Liu et al., 2019). Alimolaei et al. (2017) found that Lcb. casei harboring the α-toxoid from Clostridium perfringens, as an oral vaccine candidate, was able to protect the host from gas gangrene and necrotic enteritis. However, the specific interaction mechanisms between LAB and their hosts remain unknown. In our previous study, we suggested that bile salt hydrolase (BSH), as a functional enzyme in LAB, is related to the intestinal colonization of LAB (Yang et al., 2019). To further study the probiotic mechanism of LAB carrying bsh in vivo, a stable and specific microbial tracer technology is necessary for monitoring the in vivo distribution, colonization, and survival of LAB.
Fluorescent proteins (FPs) are stable and species spanning and thus have served as intracellular biochemical markers for more than two decades (Zhang and Xing, 2010). Labeling probiotics with FPs is an effective strategy for tracing target cells in complex in vivo ecosystems and evaluating in vitro gene expression (Landete et al., 2016). Among them, mTagBFP2, mCherry, and green fluorescent protein (GFP) have been expressed in LAB (Landete et al., 2016; Spacova et al., 2018). The adhesion of Lacticaseibacillus rhamnosus to Caco-2 cells was visualized with mTagBFP2 and mCherry (Spacova et al., 2018). The activities of the Lpb. plantarum strain B35 labeled with GFP were monitored in the digestive tract of honey bees (Javorský et al., 2017). In our previous study, the fluorescence emitted from eGFP protein in Lpb. plantarum WCFS1 did not distinguish the background and host cells, whereas mCherry protein displayed prominent fluorescence activity without interference (Chen et al., 2019), indicating that mCherry is more suitable for tracing Lpb. plantarum WCFS1. Moreover, mCherry protein does not exert toxic or additional physiological effects on host organisms (Carroll et al., 2010).
Most fluorescent markers rely on plasmid expression systems, in which antibiotic selection is required (Landete et al., 2016). In other words, when the selection pressure is lost, the number of cells maintaining the plasmids tends to decrease during subculture. Segregational stability studies estimated that only 3% of recombinant pSIP-series vectors are retained after 84 generations without antibiotic selection (Nguyen et al., 2011). Therefore, integrating the genes encoding FPs into probiotic genomes is a better strategy for ensuring the expression stability of exotic FPs in the absence of antibiotics, especially for tracking targets in vivo.
In our previous study aimed at establishing an appropriate FP labeling technology, we discovered that the constitutive promoter PldhL was optimal for mCherry expression in WCFS1 (Chen et al., 2019), as it does not require additional inducers and makes the recombinant bacteria useful for in vivo tracing research. In the same study, we labeled Lpb. plantarum with FPs by constructing and introducing a plasmid constitutively expressing mCherry (Chen et al., 2019).
In the present study, we describe the construction of a novel mCherry labeling system, the encoding gene of which was integrated into the bacterial chromosome via double-crossed homologous recombination. The stability of the new mCherry labeling system was observed and compared to the previous one harboring a plasmid. Furthermore, the research application of the integrated mCherry labeling system in studying LAB in vivo adhesion and colonization was tested by visualizing and quantifying the in vivo colonies of the strain carrying bsh, which encodes the enzyme that promotes LAB adhesion to Caco-2 cells (Yang et al., 2019).
Materials and Methods
Bacterial Strains and Culture Conditions
The bacterial strains used in this study are listed in Table 1. All the Lpb. plantarum WCFS1 and derivative strains used in this study were cultivated in MRS broth (Haibo Ltd., Qingdao, China) at 37°C with erythromycin (Em), chloramphenicol (Cm), and rifampicin (Rm) at a final concentration of 10, 10, and 50 μg ml–1 when required, respectively. Escherichia coli DH10B was used for cloning purposes, and it was cultivated at 37°C in Luria-Bertani (LB) medium (Oxoid Ltd., Basingstoke Hampshire, United Kingdom) with Em, Cm, and ampicillin (Amp) at a final concentration of 250, 10, and 100 μg ml–1 when required, respectively.

Table 1. Strains and plasmids used in this study.
General DNA Techniques
Genomic DNAs of WCFS1 were extracted according to the instructions provided in the Rapid Bacterial Genomic DNA Isolation Kit (Sangon, Shanghai, China). All plasmids used in this study were extracted using AxyPrep Plasmid MiniPrep Kit (Axygen, CA, United States) according to the manufacturer’s protocol. Enzymes were purchased from Takara (Dalian, China). PCRs for cloning purposes were performed with ExTaq PCR DNA polymerase (Takara, Dalian, China) following the manufacturer’s instructions. All amplified sequences were verified and the nucleotide sequencing in this study was performed at Sangon (Shanghai, China).
Optimization of the Promoter Spacer for mCherry Expression
To optimize the expression of the mcherry gene, three different plasmids, pLDHL673, pLDHL674, and pLDHL675, were constructed, respectively. All these three plasmids were, respectively, constructed by cloning a PldhL-mcherry gene cassette into the BglII–HindIII site of plasmid pSIPH460 (Figure 1), in which the different PldhL-mcherry gene cassette was constructed based on the previous literature method (Kahala and Palva, 1999; Anbazhagan et al., 2013; van Zyl et al., 2015). In pLDHL673, the promoter PldhL of the ldhL gene was retained and the original start codon of mcherry was replaced from TTG to ATG. The spacer of pLDHL673 between the Shine-Dalgarno (SD) sequence of the promoter PldhL and the start codon of mcherry was 12 bases. In pLDHL674 (van Zyl et al., 2015), the spacer between the SD region of the promoter PldhL and the ATG start codon of mcherry was also 12 bases; however, six bases CTTATT upstream of the ATG start codon of mcherry was replaced by a BamHI site. Similarly, in pLDHL675 (Kahala and Palva, 1999), the spacer between the SD region and the start codon of the mcherry gene contained the 15 nucleotides coding for the first five amino acids of ldhL but lacking the ATG start codon of mcherry. All these three PldhL-mcherry fragments were oligonucleotides synthesized by GenScript (Nanjing, China) with the BglII and HindIII sites at its 5′ and 3′ ends, respectively, cloned and introduced to E. coli DH10B. Positive colonies were selected on plate assays. After confirming the correctness of the plasmid constructs, the resulting expression vectors were designated as pLDHL673, pLDHL674, and pLDHL675, respectively. Subsequently, plasmids pLDHL673, pLDHL674, and pLDHL675 were, respectively, electroporated in WCFS1, and transformants were screened by selecting for 250 μg ml–1 of Em resistant. Resulting strains were designated as WCFS1:pLDHLH673, WCFS1:pLDHLH674, and WCFS1:pLDHLH675, respectively.
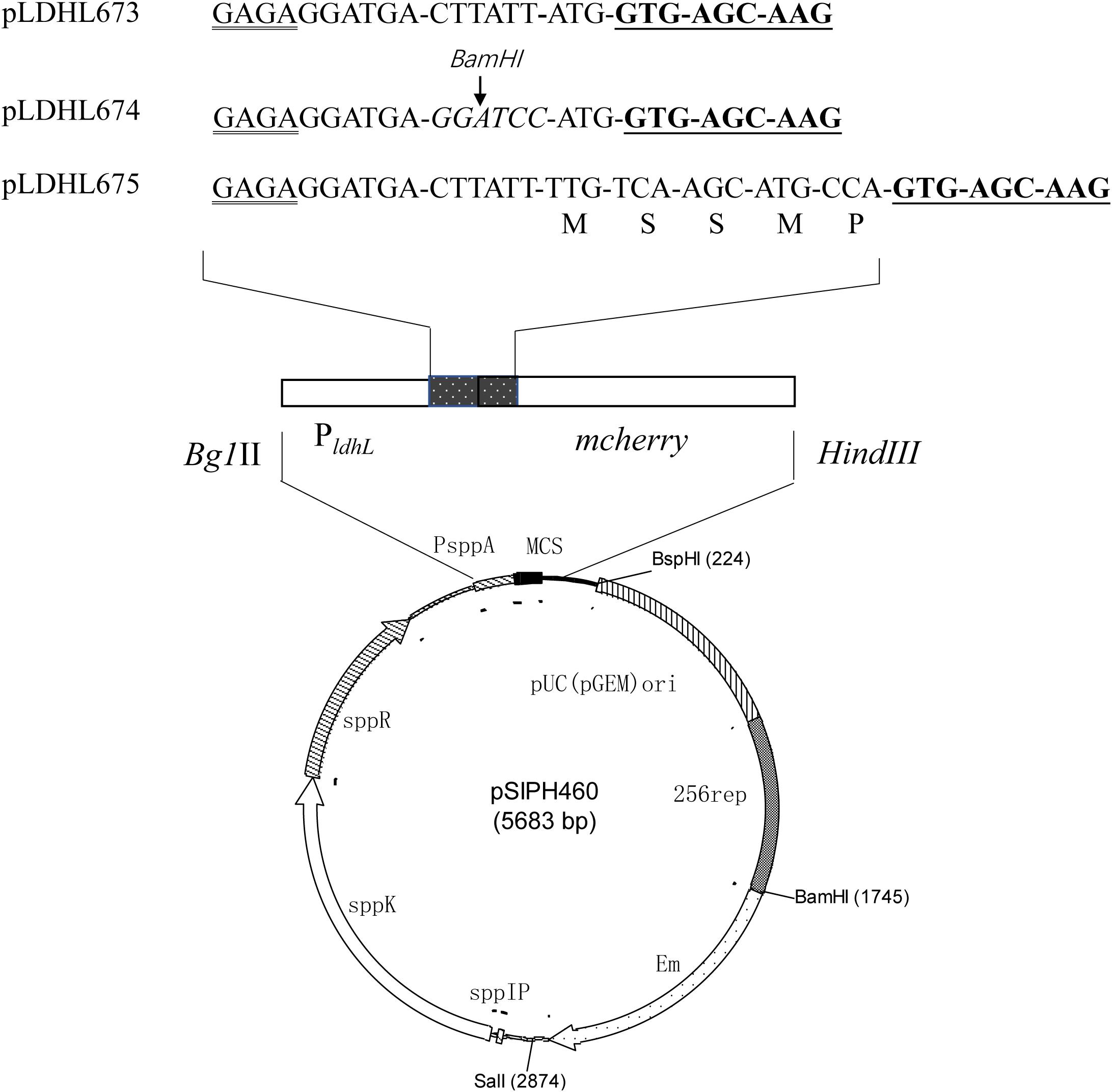
Figure 1. Optimization of the promoter spacer for mCherry expression. The Shine–Dalgarno (SD) sequence is doubly underlined and the mcherry gene is underlined. The spacer, which is between the SD sequence of the promoter PldhL and the start codon in mcherry-containing expression vectors, was designed variously. The amino acid residues, as the restriction enzyme recognition sites, are marked with italics.
Fluorescence Detection of mCherry in Recombinant WCFS1
Recombinant WCFS1 carrying different expression vectors were, respectively, grown in MRS broth for 12 h with 10 μg ml–1 of Em. It is worth noting that WCFS1:pSIPH472 needs to be induced by adding SppIP as an inducer peptide at a final concentration of 50 ng L–1 and incubated at 30°C as previously described (Chen et al., 2019). The 0.5-ml cultures were then harvested (8,000 × g, 3 min), and the cells were washed twice with PBS (pH 7.0) to remove culture medium and then resuspended in 0.5 ml of PBS (pH 7.0). The fluorescent intensity of the cells was detected with Multiscan Spectrum (Tecan, Swiss) at an excitation of 587 nm and an emission of 612 nm, respectively. In each assay, the strain carrying pSIPH460 was used as the control.
Construction of the Genomic Integrated mCherry Mutants
Expression and integration plasmids constructed during this study are based on the pNZ5319 LAB/E. coli shuttle vector (Rolain et al., 2012). The vector contains an EmR gene for Em resistance, the promoter P32, and a cat gene for Cm resistance. All primers are listed in Table 2. The targets used for homologous recombination were the restriction modification type 1 (Rm1) gene for WCFS1 (Serrano et al., 2007). The double-crossover integration vector was constructed in such a way that the lox66-P32-cat-lox71-PldhL-mcherry gene cassette (Figure 2) was flanked with the target genes’ sequences including some of their upstream and downstream regions, to facilitate homologous recombination.

Table 2. PCR and quantitative PCR primers used in this study.
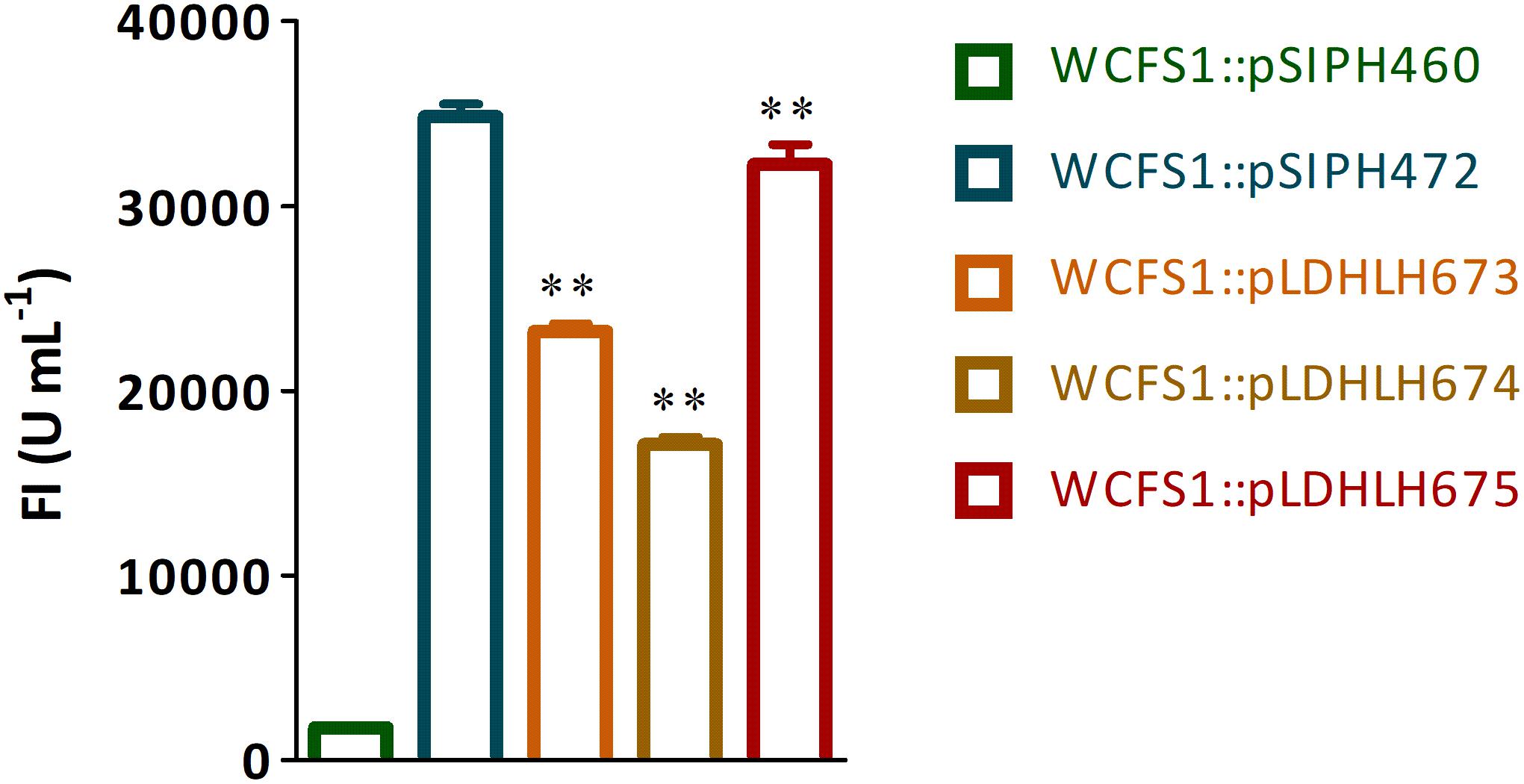
Figure 2. Fluorescence detection of mCherry protein expressed in recombinant WCFS1. The fluorescence intensity of recombinants WCFS1 containing the different mCherry expression vectors were detected at the corresponding wavelength, respectively. The pLDHLH-series plasmids and pSIPH472 plasmid all carry the target mcherry gene, while the pSIPH460 plasmid is set as the control. ** Represents a significant (p < 0.01) difference between groups.
The upstream sequence (primers 211 and 212, 900 bp, Table 2) and the downstream sequence (primers 221 and 222, 900 bp, Table 2) of the Rm1 gene were amplified from the WCFS1 genome DNAs, respectively. The downstream fragment was digested with HindIII and then ligated to the LAB/E. coli shuttle vector pLDHLH675 that contains the PldhL-mcherry gene and was digested with the same restriction enzyme. The presence of target genes in the resulting recombinant plasmid pLDHLH675-Rmdown isolated from Em-resistant transformants was confirmed by sequencing with primers 221/222. Plasmid pLDHLH675-Rmdown was digested with Bg1II and SacI, and a 2.2-kb DNA fragment, containing the mcherry gene, flanked by the PldhL promoter and the downstream regions, was purified from a 1% (w/v) agarose gel. The upstream fragment was digested with XhoI and SwaI and then ligated to plasmid pNZ5319, carrying the lox66-P32-cat-lox71 gene cassette, which was digested with the same restriction enzyme. Resulting recombinant plasmid pNZ5319-Rmup was confirmed by sequencing with primers 211/212. Plasmid pNZ5319-Rmup was linearized following digestion with Bg1II and SacI, purified, and then ligated with T4 DNA ligase to the 2.2-kb PldhL-mcherry-Rmdown gene fragment. E. coli DH10B electro-cells were then transformed for propagating the resulting plasmid, named pNZ5328. Transformants were screened by selecting for Cm and Em resistant and a red colony color phenotype. The integrity of the plasmid was verified by restriction digests and PCR with primers 211 and 222. Following sequence confirmation of the integration vectors, pNZ5328 was extracted and transformed into WCFS1 by electroporation as described previously (Lambert et al., 2007). The Cm marker was associated with the mcherry gene to enable selection of integrants that have lost the EmR-associated vector. The lox66-P32-cat-lox71-PldhL-mcherry gene was verified by PCR with primers SW/85 and PF/XW, respectively (Table 2). The resulting mutant was named NZ5328.
Stability of the Integrated mCherry Expression
The integrated mCherry mutant NZ5328, with WCFS1:pLDHLH675 as a control, were continuously subcultured in MRS medium without antibiotics for 100 generations, respectively. Subsequently, the cell numbers of these two strains were counted in the corresponding MRS agar, with or without antibiotic, after 100 generations by replica plating, respectively (Nguyen et al., 2011). All colonies in MRS agar with antibiotics were re-verified through high-throughput PCR to detection of the mcherry gene using primers Qm1 and Qm2 (Table 2).
Growth Curve of the Integrated mCherry Mutant
The integrated mCherry mutant NZ5328, with WCFS1:pLDHLH675 as control, were cultivated in MRS broth overnight at 37°C, respectively. The growth curve of the strain was performed by measuring OD600 nm value of bacterial solution, and the corresponding fluorescent intensity was also measured as previously described in this study. Meanwhile, the wild-type WCFS1 was set as a negative control.
Enumeration for Viable Cells of the Integrated mCherry Mutant
The integrated mCherry mutant NZ5328 was cultivated in MRS broth for 10 h at 37°C. After then, the culture was serially diluted, and the fluorescence of each cell suspension was measured as previously described in this study. Cell suspensions were plated onto MRS agar, and the plates were incubated at 37°C for 48 h. Fluorescence emitted was expressed as fluorescent intensity against viable cell numbers.
In addition, cell suspensions (1 ml) of the 10-h-old cultures NZ5328 were harvested. The cell suspensions were disrupted by glass beads (0.1 mm, biospec). Genome DNAs of viable cells were then extracted and serially diluted 10-fold, and 1 μl of each dilution was subjected to the qPCR assay on an ABI 7500 Fast real-time PCR system with 2.0.1 version software. The reaction mix contained 1.0 μl of DNA, 0.3 μM each of mcherry-specific primers (Qm1 and Qm2, Table 2), and 10 μl of 2×TaqMan Universal PCR Master Mix (TaKaRa Ltd., Dalian, China). The thermal cycle program was as follows: 95°C for 30 s, 95°C for 5 s, followed by 40 cycles of 58°C for 30 s and 72°C for 30 s. Meanwhile, the rpoB gene from the WCFS1 genome was used as a reference gene as well as the control (Marco et al., 2007).
Fluorescence Detection of WCFS1 Mutants in Mice Gut
The integrated mCherry mutant NZ5328 and the wild-type WCFS1 were cultivated in MRS medium overnight, and harvested by centrifugation (8,000 × g, 3 min), respectively. These two kinds of cells were washed twice and re-suspended with 1 ml of PBS (pH 7.0). In total, 14 healthy mice (C57BL/6, 8 weeks old) were raised for tracing experiment and divided into two groups (seven mice per group). Each group was fed with 0.2 ml of bacterial suspensions (NZ5328 or WCFS1, 109 cells) daily for two consecutive days, respectively. Moreover, one mouse in each group was randomly selected and euthanized with ketamine injection, after feeding the bacterial suspensions for 6 h. The intestinal tissues of these two mice were used for fluorescence imaging and observed by the in vivo imaging system (IVIS, PerkinElmer, Massachusetts, United States). Ethical approval for animal experiments was obtained from the Animal Research Ethics Committee of Nanjing Normal University (Permit No. AREC 2018-10-26). All animal procedures were performed according to European Community guidelines (Directive 2010/63/EU).
In vitro Bacterial Adhesion Assay
Previously, the results of the plate colony counting assay proved that BSH protein promotes the adhesion of LAB to Caco-2 cells (Yang et al., 2019). In this study, the same expression vector pSIPH462 was cloned into mCherry labeled NZ5328 to study the change of its adhesion characteristics. After electrotransformation of the plasmid pSIPH462 carrying the bsh gene into NZ5328, the strain NZ5328 carrying pSIPH462 (named NZ5328-bsh) was cultivated in MRS medium with 10 μg ml–1 of Em and confirmed by PCR using primer cexu4 and cexu5 (Table 2). The recombinant strain was cultured in MRS medium supplemented with SppIP and until the OD600 nm reached about 1.8, the BSH activities of NZ5328-bsh or WCFS1:pSIPH462 (named WCFS1-bsh) were detected as described previously, respectively (Yang et al., 2019). The recombinant strain NZ5328:pSIPH460 or WCFS1:pSIPH460 was set as an experimental control. Subsequently, the adhesion of NZ5328-bsh strain to Caco-2 cells was performed as described by Yang et al. (2019). The total number of adhered BSH recombinant cells was estimated by plate count in MRS agar, fluorescent assay, and mcherry-qPCR, respectively. The adhesion ability (CFU/cell) was based on the ratio of adhered BSH recombinant cells to total Caco-2 cells, and the adhered BSH recombinant cells were observed under the confocal laser scanning microscope (CLSM, Olympus, Tokyo, Japan). The recombinant strain NZ5328:pSIPH460 was also set as an empty control, and each sample was repeated at least three times.
Statistical Analysis
The values in this study were presented as mean ± standard deviation (SD). Statistical analyses are conducted by GraphPad prism 6.0 (GraphPad Software Inc., San Diego, CA, United States). Statistical comparisons are analyzed by Student’s t-test, and multiple comparisons are performed with one-way ANOVA analysis, followed by Turkey’s post-hoc test. A p-value of less than 0.05 was considered as statistically significant, and ∗p < 0.05, ∗∗p < 0.01.
Results
Optimization Promoter Spacer for mCherry Expression in WCFS1
To optimize the expression of mCherry controlled by the promoter of ldhL in WCFS1, a series of mCherry expression vectors was constructed by adjusting the spacer between the SD region and start codon of mcherry (Figure 1). Three different recombinant plasmids (pLDHLH674, pLDHLH675, and pLDHLH673) were, respectively, constructed and electrotransformed into the host strain WCFS1. The expression of mcherry was evaluated by detecting its fluorescence intensity. The results showed that the fluorescent mCherry proteins were successfully expressed in the three recombinant strains (Figure 2). The fluorescence intensity of strain WCFS1:pLDHLH675 (32254.25 U ml–1) was higher than that of strains WCFS1:pLDHLH673 (23217.5 U ml–1) and WCFS1:pLDHLH674 (17085.25 U ml–1) (Figure 2), indicating that the promoter spacer of pLDHLH675 was more suitable for expressing the fluorescent mCherry protein. Finally, we determined the optimized spacer structure of pLDHLH675, which was used for subsequent mCherry expression in this study.
Construction of Genomic-Integrated mCherry Mutants of WCFS1
The double-crossover integrated mCherry expression plasmid pNZ5328 was constructed as described previously (Figure 3). Escherichia coli DH10B cells transformed with pNZ5328 produced fluorescent colonies that were both Em- and Cm-resistant. NZ5328 retained resistance to Cm, but not to Em, indicating that the lox66-P32-cat-lox71-PldhL-mcherry cassette was successfully integrated by double-crossover into the host chromosome without the plasmid DNA encoding EmR (Figure 3). The presence of cat and mcherry in NZ5328 was confirmed using polymerase chain reaction (PCR) with the primers SW/85 and PF/XW, respectively. No 4.2-kb target PCR fragment was amplified, confirming that Rm1 was successfully disrupted by the insertion of the lox66-P32-cat-lox71-PldhL-mcherry cassette. The results of fluorescence detection showed that NZ5328 was fluorescent with an intensity of 26,394 U ml–1, indicating that the genomic-integrated strain for expressing mCherry was successfully constructed.
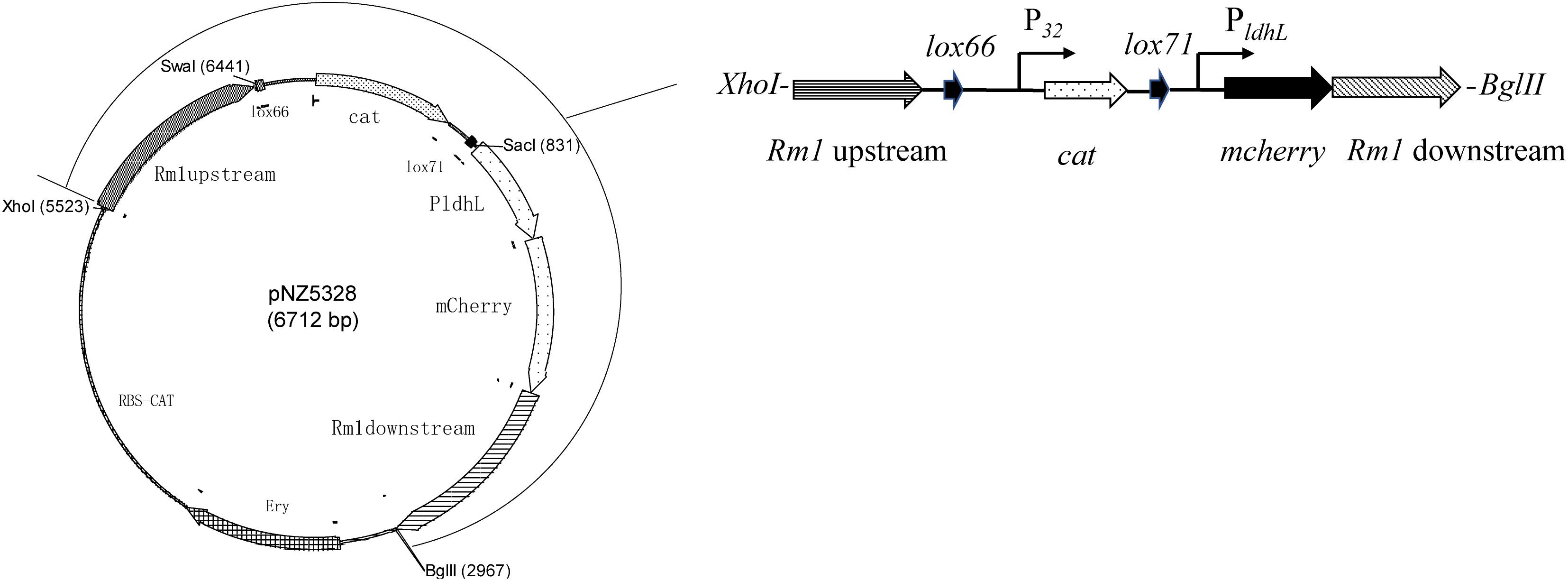
Figure 3. Schematic overview of exogenous gene insertion in pNZ5328 vector. Vector pNZ5328 contains Rm1 gene upstream, a lox66 gene, the promoter P32, a cat gene, a lox71 gene, the promoter PldhL, the mcherry gene, and Rm1 gene downstream.
Stability of the Integrated mCherry Expression System in WCFS1
To verify the stability of the integrated mCherry expression system in NZ5328, the target mcherry and fluorescence intensity of the system were detected and evaluated. First, the growth curve of NZ5328 was measured, and its corresponding fluorescence intensity was detected (Figure 4). Compared to those of the control strain WCFS1:pLDHLH675 and wild-type WCFS1, there was no difference in the growth curves of the three strains (Figure 4A). Compared to that of the control strain WCFS1:pLDHLH675, the fluorescence intensity of the integrated strain NZ5328 decreased slightly (p < 0.05) but tended to be stable at the stationary stage (maximum fluorescence: 30,447.33 ± 959.37 U ml–1; Figure 4B).
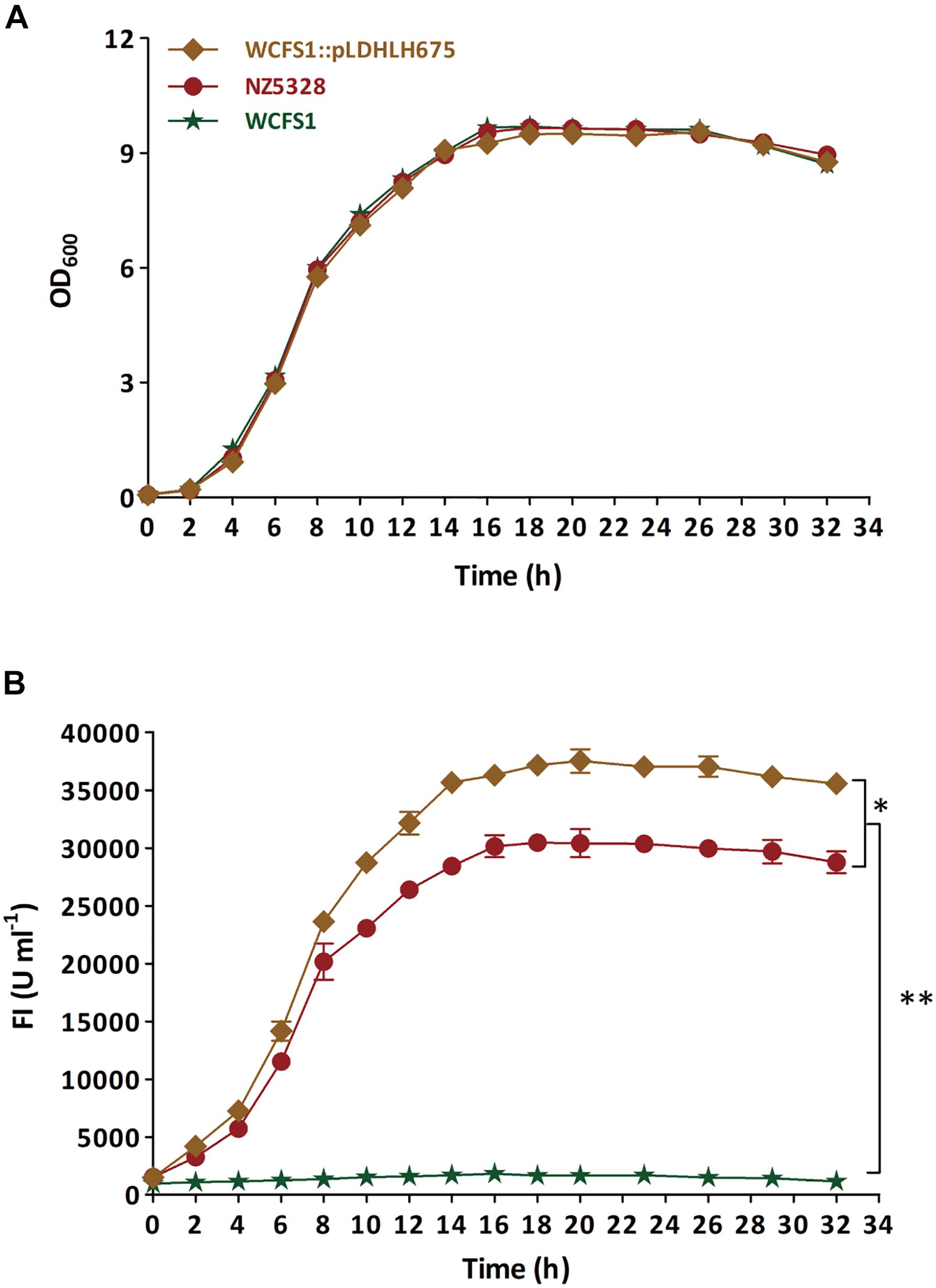
Figure 4. The growth curve (A) of recombinants carrying mcherry gene and detection of corresponding fluorescence values (B). The WCFS1:Pldhlh675 is plasmid-based recombinant, and the NZ5328 is genomic-integrated recombinant, while the wild-type WCFS1 is set as the control. * Represents a significant (p < 0.05) difference between groups. ** Represents a significant (p < 0.01) difference between groups.
After 100 generations, both replica plating and PCR verification results showed that the integrated strain NZ5328 stably carried mcherry without loss (Figure 5, PCR validation data not shown), and the fluorescence intensity of the NZ5328 derivatives was maintained at a high level (32,164.25 ± 128.12 U ml–1). However, the WCFS1:pLDHLH675 derivatives exhibited significant mcherry gene loss. Almost 95% of WCFS1:pLDHLH675 derivatives could not grow on plates containing antibiotics (Figure 5). The results showed that plasmids were easily lost owing to the lack of resistant stress selection.
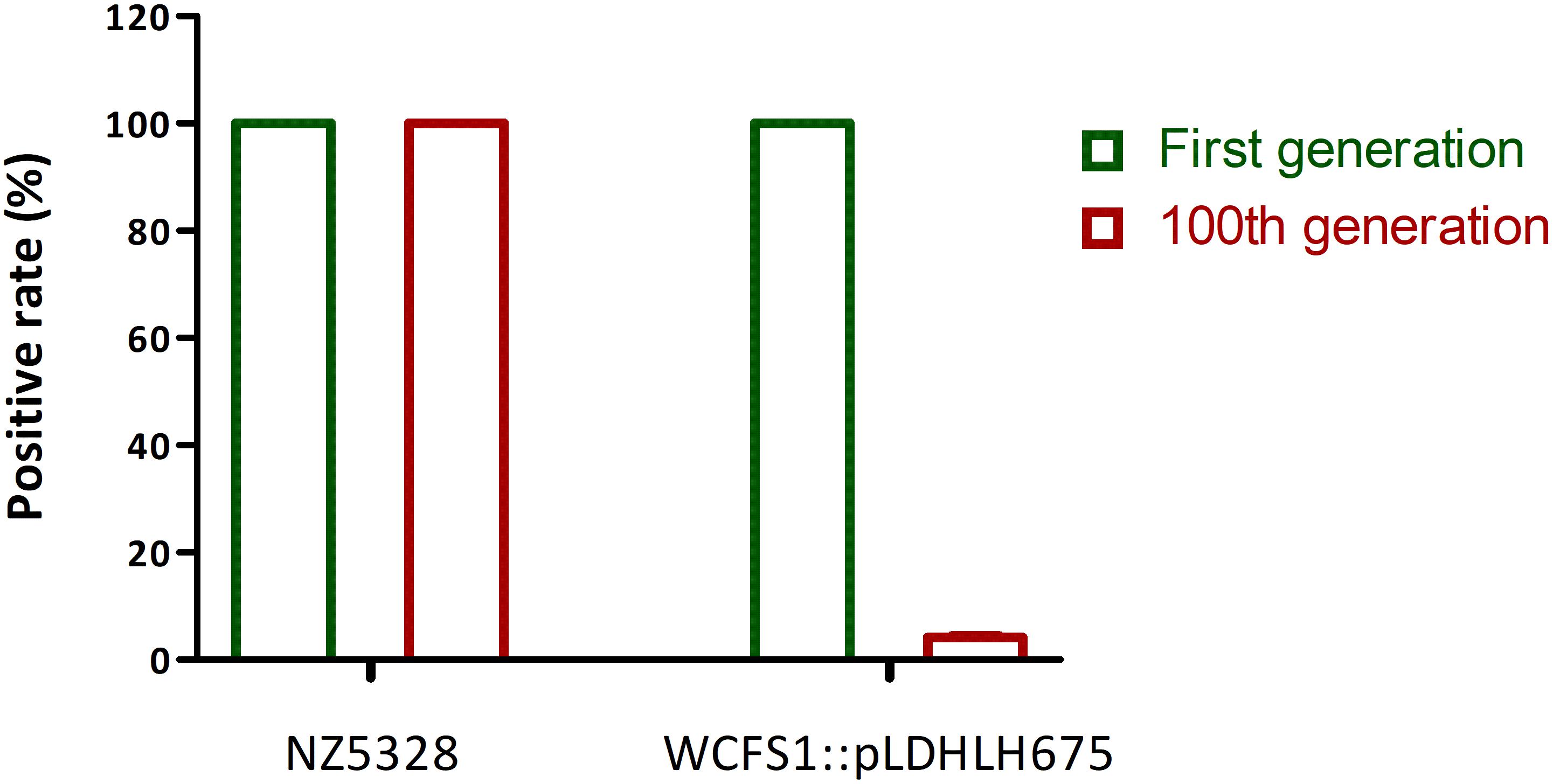
Figure 5. Stability of mcherry gene in genomic-integrated and plasmid-based recombinants (after 100 generations). The left Y-axis is the percentage of mcherry-containing (positive) colonies in the MRS agar with antibiotic.
Enumeration of Viable Cells With the Integrated mCherry Mutant
To verify whether the integrated mCherry expression system can be used for the quantitative detection of the target bacteria, the correlation between fluorescence intensity and cell numbers of the integrated mCherry mutant was investigated. As expected, the change in the trend of cell numbers was similar to that of the fluorescence intensity of mcherry-containing strains over time. There was a linear relationship between the fluorescence intensity and number of cells in NZ5328 (R2 = 0.998; Figure 6A), indicating that fluorescence detection can be used for enumeration of cells.
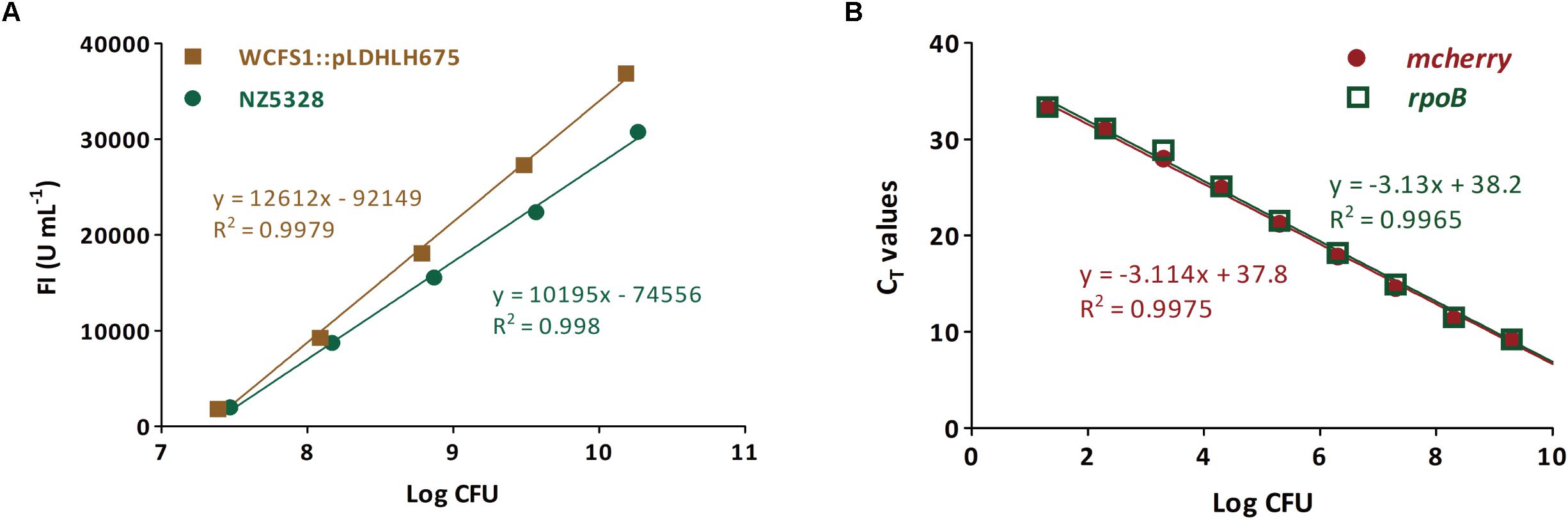
Figure 6. Enumeration for viable cells of the mcherry-containing mutants (genomic-integrated and plasmid-based) based on the fluorescence intensity (A) and mcherry-qPCR (B). The rpoB gene from NZ5328 genome was used as an internal gene.
Subsequently, to confirm that mcherry in the integrated mutant can be used as the target gene for quantification, a pair of specific primers for mcherry was developed for enumerating cells. The quantitative (q)PCR assay was positive for mcherry, and no cross-reaction with non-target genes was observed. Moreover, mcherry quantification was stable and highly reproducible (data not shown). To further verify the quantitative accuracy, mcherry in NZ5328 was evaluated using the reference gene rpoB (Marco et al., 2007). The standard curve based on rpoB almost overlapped with that of mcherry, with a correlation coefficient of 0.9965 (Figure 6B), suggesting that quantification of mcherry in NZ5328 was consistent with that of rpoB. Thus, the qPCR assay based on mcherry detection can be applied to measure viable counts over a dynamic range from 10 to 1010 colony-forming units (CFU).
Fluorescence Localization and Detection of Target Bacteria in Mouse Intestines
To evaluate the fluorescence of the integrated mCherry mutant in the mouse gut without interference from the host, mice were fed integrated mCherry mutants, and red fluorescence was monitored. Fluorescence was detected in the gastrointestinal tract of the mice after removing the abdominal skin and fur (Figure 7), and only the limbs and tails of the mice presented red fluorescence at the emission wavelength of mCherry (data not shown). At 6 h after NZ5328 administration, the persistence of fluorescent cells in the small intestine was perceived when the mice were imaged (Figure 7). Fluorescence was not emitted by WCFS1 or the intestinal tissues, indicating that the background without mCherry protein did not interfere with fluorescence determination at the corresponding wavelength.
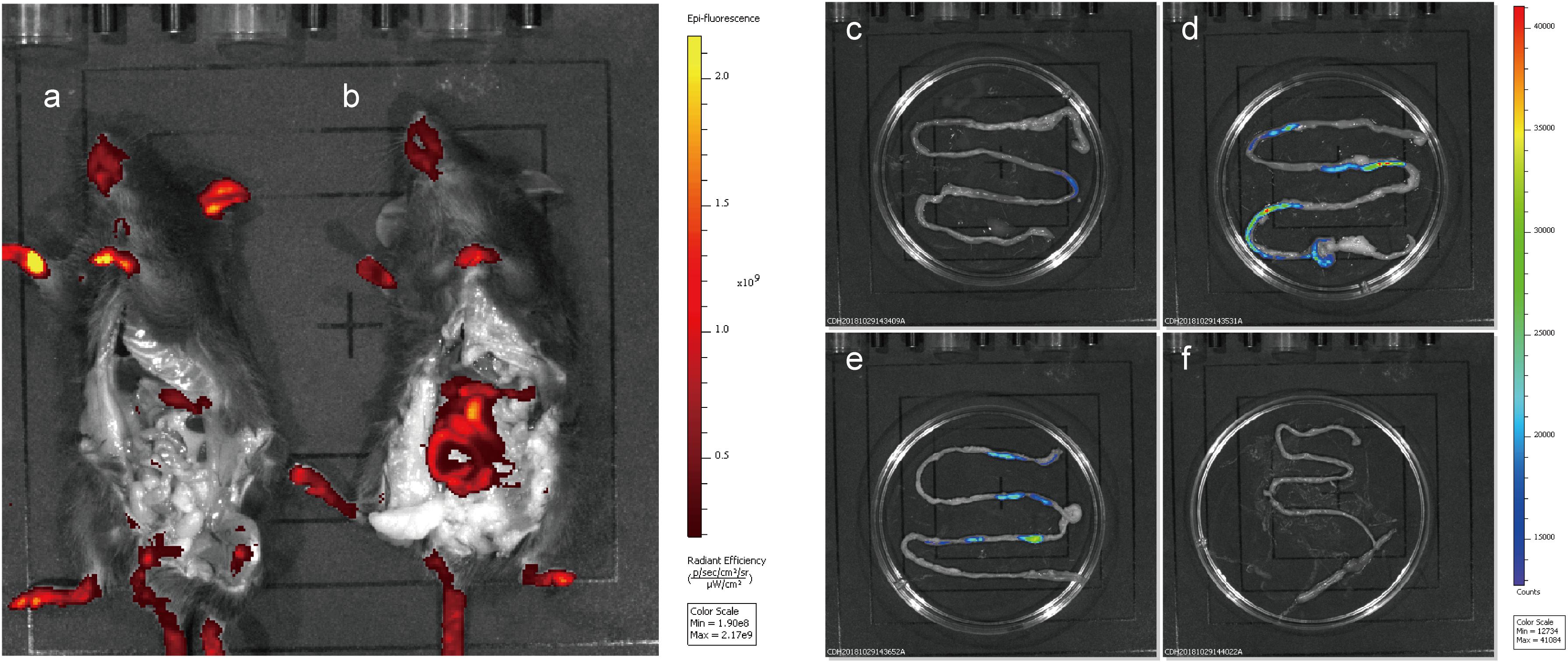
Figure 7. Fluorescence imaging and tracing of WCFS1 and derivative cells in the abdomen and small intestine of the C57BL/6 mice. After 6 h of feeding, euthanized mice were treated with the removal of ventral skin when fluorescence imaging was collected. Fluorescence determination of the C57BL/6 mouse fed WCFS1 (a) and NZ5328 (b) by the in vivo imaging system, and fluorescence detection of WCFS1 (f) and NZ5328 (c–e) in the small intestine of the C57BL/6 mouse.
Verification of Integrated Bacteria in Cell Adhesion, Quantification, and Protein Expression
BSH activity determined in BSH recombinants showed that NZ5328-bsh expressing heterogenous bsh was not distinct from that of WCFS1 harboring pSIPH462 (Figure 8A), suggesting that the chromosomal-integrated FP gene did not interfere with the heterologous expression of BSH protein in the WCFS1 strain. These results provide a theoretical basis for NZ5328 to be used as an expression host for other target genes. In this study, with the assistance of mCherry labeling, changes in the adhesion of LAB promoted by BSH proteins could be directly observed through microscopes. After co-culture with Caco-2 cells, BSH recombinant cells were visualized using confocal laser scanning microscopy as red fluorescence emitted by mCherry (Figure 8B). Furthermore, to complement the approach using fluorescent signals to quantify the LAB cells, qPCR analysis of the mcherry gene was performed to quantify LAB cells. The results obtained using mcherry-qPCR and plate counts were similar (Figure 8C). The approach used in this study has fewer steps and higher efficiency than the plate colony counting assay, and the results were comparable between them.
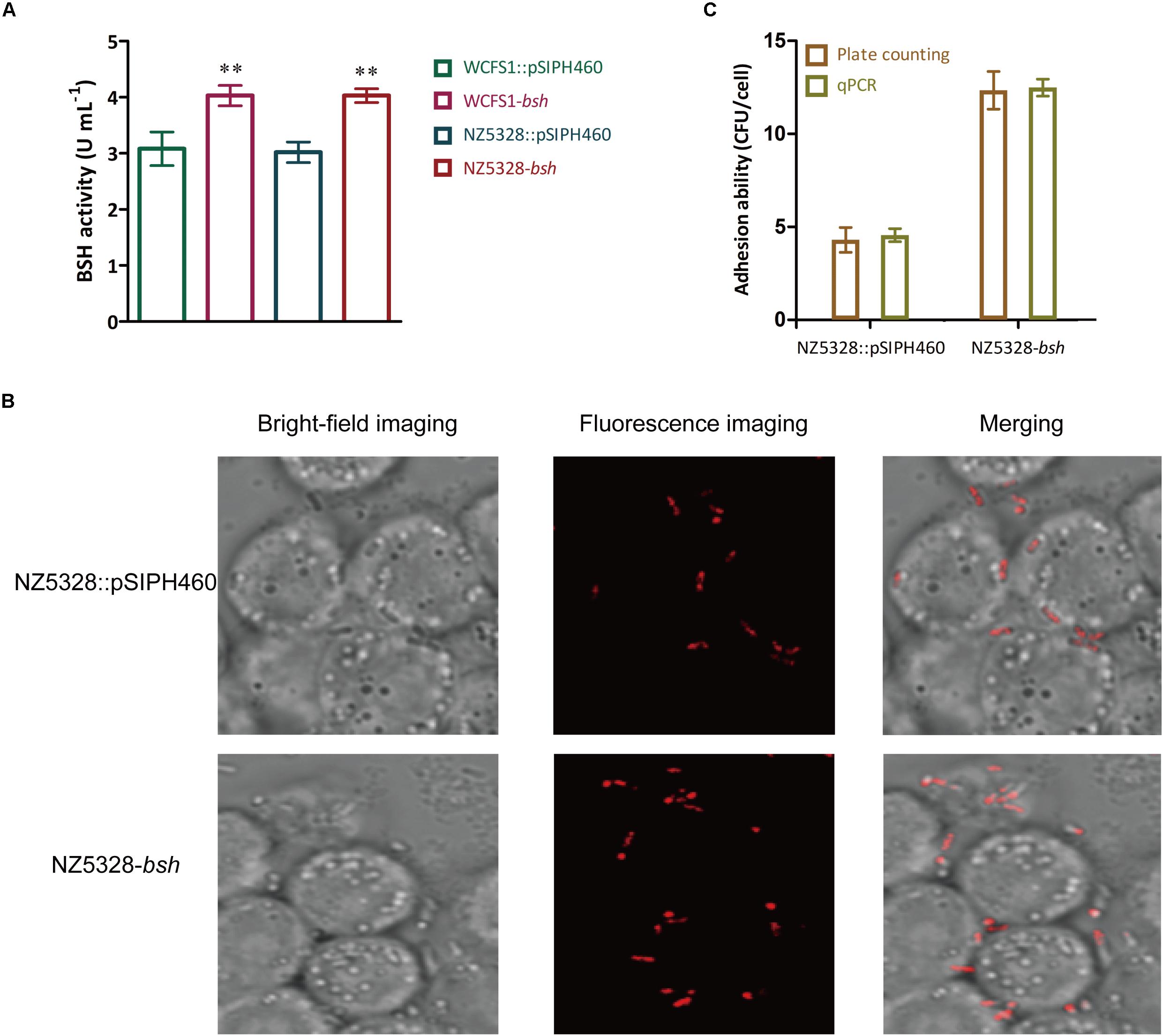
Figure 8. BSH activity detection (A), fluorescence imaging (B), and quantitative detection (C) of the bacterial adhesion assay in vitro. ** Represents a significant (p < 0.01) difference from the control.
Discussion
In this study, we aimed to construct a stable red fluorescent protein (RFP) mCherry expression system in WCFS1 and found that the chromosomal-integrated mCherry labeling system can be extensively used to examine the distribution, colonization, and survival of LAB in vivo.
Previous studies have suggested that photons emitted by the mCherry protein reflect the in vivo metabolic activity of L. sakei and Lpb. plantarum 423 (van Zyl et al., 2015). Our previous studies also suggested that the RFP mCherry has potential applications in the labeling of WCFS1 (Chen et al., 2019). The mCherry protein can be constitutively expressed under regulation of the promoter PldhL. In our previous study, the fluorescence intensity emitted by mCherry regulated by the promoter PldhL in WCFS1:pLDHLH673 was visible but significantly lower than the induced expression (Chen et al., 2019). The spacer region modified by embedding simple sequence repeats affected the expression of target genes (Egbert and Klavins, 2012). In this study, we first optimized the mCherry expression level by changing the spacer size. Compared to that of the inducible strain WCFS1:pSIPH472, the fluorescence intensity of WCFS1 harboring pLDHLH674 and pLDHLH673 was markedly lower than that of WCFS1:pSIPH472 (Figure 2), which stimulated intense fluorescence activity as described previously (Chen et al., 2019). In contrast, we found that the insertion of restriction enzyme recognition sequences into the space between the SD region and start codon altered the mCherry expression level. Fluorescence emission by mCherry in WCFS1:pLDHLH675 reached a maximum (Figure 2), indicating that the 9-bp spacer was optimal for higher protein expression.
Among the fluorescent markers used in LAB, GFP, RFP, and their derivatives are commonly employed (Landete et al., 2016; Javorský et al., 2017; Spacova et al., 2018). Cellular auto-fluorescence emission of WCFS1 has an intensive wavelength overlap with that of eGFP (Chen et al., 2019). In contrast, mCherry produced clear red fluorescence without background interference in WCFS1, which is consistent with RFP performance in other Lactobacillus spp. (van Zyl et al., 2015; Spacova et al., 2018). Notably, the Lpb. plantarum strain labeled with mCherry was not pink under natural light, unlike the color of other mCherry-labeled bacteria in previous research (Castro-Bravo et al., 2017; Martinez-Jaramillo et al., 2017; Mohedano et al., 2019); we speculate that the unique cell wall structure of WCFS1 may be the main reason.
Most fluorescence reporter systems are dependent on expression plasmids carrying a fluorescence gene. The segregational stability of plasmids varies with selection pressure. Without antibiotics, expression vectors can be easily lost from the host cells during continuous proliferation, particularly in vivo. Integration of mcherry into the genome of a probiotic strain not only excludes the need for antibiotic selection but also provides an expression system that is genetically stable. When mcherry was integrated into WCFS1, the level of fluorescence emitted from labeled cells decreased compared to that observed in plasmids (Figure 4B). This may have resulted from the low transcriptional abundance of the insertion locus. Genetic stability was high, although the fluorescence intensity of mcherry-integrated cells was low (Figure 4B). After 100 generations, the integrated strain NZ5328 stably carried mcherry without loss (Figure 5) and fluorescence emitted from the labeled strains remained constant (data not shown), reflecting the superiority of the fluorescence integration strategy. Thus, fluorescence genes integrated into chromosomes can be genetically stable and used to track each step of cell movement in real time, while eliminating the potential risk of plasmid loss.
In addition to tracing, the integrated mCherry expression system shows potential for enumeration. mcherry-labeled cells were determined based on standard fluorescence curve intensity and threshold cycle (CT) values for mcherry. In our study, a positive linear correlation between fluorescence intensity and CFU was successfully established (Figure 6). The number of Lpb. plantarum cells carrying integrated mcherry positively correlated with fluorescence intensity, which is in agreement with the findings of a colonization study (van Zyl et al., 2015). However, considering the limited detection sensitivity, the intensity of fluorescent signals cannot reflect the number of cells when <107 CFU. Therefore, we used qPCR to quantify mcherry in the NZ5328 genome as an alternative. The relationship between the cell numbers and CT values of amplified mcherry was linear over a broad dynamic range, and starting material as low as 10 CFU could still be detected. The two quantitative methods facilitated rapid detection of target strains in vivo and in vitro, and considerably improved work efficiency.
In addition to the extensive studies on LAB as probiotics, they have also been intensively utilized as the host strains for in vivo heterologous expression of many important proteins, along with the improvement of LAB expression systems. In this study, bsh gene was expressed in the mCherry-labeled WCFS1 strain. The results suggested that the chromosomal integrated FP gene did not interfere with the heterologous expression of BSH protein in the WCFS1 strain. Previously, the results of the plate colony counting assay proved that BSH protein promotes the adhesion of LAB to Caco-2 cells (Yang et al., 2019). In this study, with the assistance of mCherry labeling, changes in the adhesion of LAB promoted by BSH proteins could be directly observed through microscopes (Figure 8B). In order to complement the approach using fluorescence signals to quantify LAB cells, qPCR analysis of the mcherry gene was performed to further quantify the LAB cells.
Nowadays, Lactobacillus spp. have been extensively studied not only for their probiotic properties (Holzapfel and Wood, 2014), but also for their roles as producers for expressing various recombinant proteins of importance. Thus, FP labeling systems have potentially broader applications than plasmid systems. Overall, this FP labeling system with the integration of mcherry is an important, non-invasive, and effective strategy for tracking and quantifying probiotics in various in vivo tissues as well as monitoring heterologous protein expression in vitro. Our results exemplify the research application potential of the integrated FP-labeled LAB strains as probiotics and bioengineering tools. Further investigation of the interaction between hosts and intestinal microbes using the FP labeling system and similar labeling techniques is underway.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by the Animal Research Ethics Committee of Nanjing Normal University (Permit No. AREC 2018-10-26).
Author Contributions
YY, WZ, WX, YC, HH, PW, and YL participated in the design and discussion of the study. WZ and YC carried out the experiments. WX and YC wrote the manuscript. YY and WX discussed, revised, and edited the manuscript. All authors read and approved the final version to be published.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31501507).
Conflict of Interest
WX was employed by company Geneception (Shanghai) Bio-technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alimolaei, M., Golchin, M., Abshenas, J., Ezatkhah, M., and Bafti, M. S. (2017). A Recombinant Probiotic, Lactobacillus casei, Expressing the Clostridium perfringens α-toxoid, as an Orally Vaccine Candidate Against Gas Gangrene and Necrotic Enteritis. Probiot. Antim. Prot. 10, 251–257. doi: 10.1007/s12602-017-9276-8
Anbazhagan, K., Sasikumar, P., Gomathi, S., Priya, H. P., and Selvam, G. S. (2013). In vitro degradation of oxalate by recombinant Lactobacillus plantarum expressing heterologous oxalate decarboxylase. J. Appl. Microbiol. 115, 880–887. doi: 10.1111/jam.12269
Carroll, P., Schreuder, L. J., Muwanguzikarugaba, J., Wiles, S., Robertson, B. D., Ripoll, J., et al. (2010). Sensitive Detection of Gene Expression in Mycobacteria under Replicating and Non-Replicating Conditions Using Optimized Far-Red Reporters. PLoS One 5:e9823. doi: 10.1371/journal.pone.0009823
Castro-Bravo, N., Hidalgo-Cantabrana, C., Rodriguez-Carvajal, M. A., Ruas-Madiedo, P., and Margolles, A. (2017). Gene Replacement and Fluorescent Labeling to Study the Functional Role of Exopolysaccharides in Bifidobacterium animalis subsp. lactis. Front. Microbiol. 8:1405. doi: 10.3389/fmicb.2017.01405
Chen, Y., Wang, P., Zhang, W., Yang, Q., and Yang, Y. (2019). Construction and application of mCherry red fluorescent protein fusion expression system in lactic acid bacteria. Sheng wu gong cheng xue bao = Chinese J. Biotech. 35, 492–504. doi: 10.13345/j.cjb.180306
Chen, Y., Qi, M., Xu, M., Huan, H., Shao, W., and Yang, Y. (2018). Food-grade gene transformation system constructed in Lactobacillus plantarum using a GlmS-encoding selection marker. FEMS Microbiol. Lett. 365:254. doi: 10.1093/femsle/fny254
Durfee, T., Nelson, R., Baldwin, S. F., Plunkett, G., Burland, V., Mau, B., et al. (2008). The Complete Genome Sequence of Escherichia coli DH10B: Insights into the Biology of a Laboratory Workhorse. J. Bacteriol. 190, 2597–2606. doi: 10.1128/JB.01695-07
Egbert, R. G., and Klavins, E. (2012). Fine-tuning gene networks using simple sequence repeats. Proc. Natl. Acad. Sci. U S A. 109, 16817–16822. doi: 10.1073/pnas.1205693109
Holzapfel, W. H., and Wood, B. (2014). Overview of the ecology and biodiversity of the LAB. Hoboken, NJ: John Wiley & Sons, Ltd.
Javorský, P., Fecskeová, L. K., Hrehová, L., Sabo, R., Legáth, J., and Pristas, P. (2017). Establishment of Lactobacillus plantarum strain in honey bee digestive tract monitored using gfp fluorescence. Benef Microbes. 8, 291–297. doi: 10.3920/BM2016.0022
Kahala, M., and Palva, A. (1999). The expression signals of the Lactobacillus brevis slpA gene direct efficient heterologous protein production in lactic acid bacteria. Appl. Microbiol. Biotechnol. 51, 71–78. doi: 10.1007/s002530051365
Kleerebezem, M., Boekhorst, J., Van Kranenburg, R., Molenaar, D., Kuipers, O. P., Leer, R., et al. (2003). Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. U S A 100, 1990–1995. doi: 10.1073/pnas.0337704100
Kołodziej, M., and Szajewska, H. (2019). Lactobacillus reuteri DSM 17938 in the prevention of antibiotic-associated diarrhoea in children: a randomized clinical trial. Clin. Microbiol. Infect. 25, 699–704. doi: 10.1016/j.cmi.2018.08.017
Lambert, J. M., Bongers, R. S., and Kleerebezem, M. (2007). Cre-lox-Based System for Multiple Gene Deletions and Selectable-Marker Removal in Lactobacillus plantarum. Appl. Environ. Microbiol. 73, 1126–1135. doi: 10.1128/AEM.01473-06
Landete, J. M., Medina, M., and Arqués, J. L. (2016). Fluorescent reporter systems for tracking probiotic lactic acid bacteria and bifidobacteria. World J. Microbiol. Biotechnol. 32:119. doi: 10.1007/s11274-016-2077-5
Liu, J., Yang, G., Gao, X., Zhang, Z., Liu, Y., Liu, Q., et al. (2019). Recombinant invasive Lactobacillus plantarum expressing fibronectin binding protein A induce specific humoral immune response by stimulating differentiation of dendritic cells. Benef Microbes. 10, 589–604. doi: 10.3920/BM2018.0157
Marco, M. L., Bongers, R. S., De Vos, W. M., and Kleerebezem, M. (2007). Spatial and Temporal Expression of Lactobacillus plantarum Genes in the Gastrointestinal Tracts of Mice. Appl. Environ. Microbiol. 73, 124–132. doi: 10.1128/AEM.01475-06
Martinez-Jaramillo, E., Garza-Morales, R., Loera-Arias, M. J., Saucedo-Cardenas, O., Montes-de-Oca-Luna, R., McNally, L. R., et al. (2017). Development of Lactococcus lactis encoding fluorescent proteins, GFP, mCherry and iRFP regulated by the nisin-controlled gene expression system. Biotech. Histochem. 92, 167–174. doi: 10.1080/10520295.2017.1289554
Mohedano, M. L., Hernández-Recio, S., Yépez, A., Requena, T., Martínez-Cuesta, M. C., Peláez, C., et al. (2019). Real-Time Detection of Riboflavin Production by Lactobacillus plantarum Strains and Tracking of Their Gastrointestinal Survival and Functionality in vitro and in vivo Using mCherry Labeling. Front. Microbiol. 10:1748. doi: 10.3389/fmicb.2019.01748
Nguyen, T. T., Mathiesen, G., Fredriksen, L., Kittl, R., Nguyen, T. H., Eijsink, V. G., et al. (2011). A Food-Grade System for Inducible Gene Expression in Lactobacillus plantarum Using an Alanine Racemase-Encoding Selection Marker. J. Agric Food Chem. 59, 5617–5624. doi: 10.1021/jf104755r
Pérez-Ibarreche, M., Castellano, P., Leclercq, A., and Vignolo, G. (2016). Control of Listeria monocytogenes biofilms on industrial surfaces by the bacteriocin-producing Lactobacillus sakei CRL1862. FEMS Microbiol. Lett. 363:fnw118. doi: 10.1093/femsle/fnw118
Rolain, T., Bernard, E., Courtin, P., Bron, P. A., Kleerebezem, M., Chapot-Chartier, M. P., et al. (2012). Identification of key peptidoglycan hydrolases for morphogenesis, autolysis, and peptidoglycan composition of Lactobacillus plantarum WCFS1. Microb. Cell Fact. 11:137. doi: 10.1186/1475-2859-11-137
Salas-Jara, M. J., Ilabaca, A., Vega, M., and García, A. (2016). Biofilm Forming Lactobacillus: New Challenges for the Development of Probiotics. Microorganisms 4:35. doi: 10.3390/microorganisms4030035
Serrano, L. M., Molenaar, D., Wels, M., Teusink, B., Bron, P. A., de Vos, W. M., et al. (2007). Thioredoxin reductase is a key factor in the oxidative stress response of Lactobacillus plantarum WCFS1. Microb. Cell Fact. 6:29. doi: 10.1186/1475-2859-6-29
Spacova, I., Lievens, E., Verhoeven, T., Steenackers, H., Vanderleyden, J., Lebeer, S., et al. (2018). Expression of fluorescent proteins in Lactobacillus rhamnosus to study host-microbe and microbe-microbe interactions. Microb. Biotechnol. 11, 317–331. doi: 10.1111/1751-7915.12872
Taylor, M. J., Perrais, D., and Merrifield, C. J. (2011). A High Precision Survey of the Molecular Dynamics of Mammalian Clathrin-Mediated Endocytosis. PLoS Biol. 9:e1000604. doi: 10.1371/journal.pbio.1000604
van Zyl, W. F., Deane, S. M., and Dicks, L. M. (2015). Use of the mCherry Fluorescent Protein To Study Intestinal Colonization by Enterococcus mundtii ST4SA and Lactobacillus plantarum 423 in Mice. Appl. Environ. Microbiol. 81, 5993–6002. doi: 10.1128/AEM.01247-15
Wyszyńska, A., Kobierecka, P., Bardowski, J., and Jagusztyn-Krynicka, E. K. (2015). Lactic acid bacteria-20 years exploring their potential as live vectors for mucosal vaccination. Appl. Microbiol. Biotechnol. 99, 2967–2977. doi: 10.1007/s00253-015-6498-0
Yang, Y., Liu, Y., Zhou, S., Huang, L., Chen, Y., and Huan, H. (2019). Bile salt hydrolase can improve Lactobacillus plantarum survival in gastrointestinal tract by enhancing their adhesion ability. FEMS Microbiol. Lett. 366:fnz100. doi: 10.1093/femsle/fnz100
Keywords: mCherry fluorescent protein, probiotic tracking, probiotic labeling, Lactiplantibacillus plantarum WCFS1, fluorescent reporter system
Citation: Yang Y, Zhang W, Huan H, Xia W, Chen Y, Wang P and Liu Y (2021) Construction of an Integrated mCherry Red Fluorescent Protein Expression System for Labeling and Tracing in Lactiplantibacillus plantarum WCFS1. Front. Microbiol. 12:690270. doi: 10.3389/fmicb.2021.690270
Received: 02 April 2021; Accepted: 20 May 2021;
Published: 22 June 2021.
Edited by:
Xiao-Jun Ji, Nanjing Tech University, ChinaReviewed by:
Yanling Hao, China Agricultural University, ChinaJosé María Landete, Instituto Nacional de Investigación y Tecnología Agroalimentaria (INIA), Spain
Copyright © 2021 Yang, Zhang, Huan, Xia, Chen, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yao Yang, Y3Jhbm55X3lhbmdAaG90bWFpbC5jb20=