 Haixiu Yang
Haixiu Yang Fan Tong
Fan Tong Changlu Qi
Changlu Qi Ping Wang
Ping Wang Jiangyu Li
Jiangyu Li Liang Cheng
Liang Cheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 July 2021
Sec. Systems Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.685549
This article is part of the Research Topic Multi-omics Study on Gut Microbiota Related to Faecal Microbiota Transplantation View all 13 articles
Many microbes are parasitic within the human body, engaging in various physiological processes and playing an important role in human diseases. The discovery of new microbe–disease associations aids our understanding of disease pathogenesis. Computational methods can be applied in such investigations, thereby avoiding the time-consuming and laborious nature of experimental methods. In this study, we constructed a comprehensive microbe–disease network by integrating known microbe–disease associations from three large-scale databases (Peryton, Disbiome, and gutMDisorder), and extended the random walk with restart to the network for prioritizing unknown microbe–disease associations. The area under the curve values of the leave-one-out cross-validation and the fivefold cross-validation exceeded 0.9370 and 0.9366, respectively, indicating the high performance of this method. Despite being widely studied diseases, in case studies of inflammatory bowel disease, asthma, and obesity, some prioritized disease-related microbes were validated by recent literature. This suggested that our method is effective at prioritizing novel disease-related microbes and may offer further insight into disease pathogenesis.
Microbial communities, including fungi, archaea, protozoa, bacteria, and viruses, are distributed across various organs of the human body, such as the skin, oral cavity, respiratory tract, and intestine (Cheng et al., 2020; Qi et al., 2021; Sommer and Backhed, 2013). It is reported that about 1014 microbial cells reside in the adult intestine, nearly 10 times the number of human cells. Therefore, microbes play an important role in the human body, engaging in various physiological processes, including metabolism regulation and immune defense (Das and Nair, 2019), and disorders relating to microbial communities within the human body have been linked to various human diseases (Huang et al., 2020; Yang et al., 2016). For example, Qin et al. (2010) found that inflammatory bowel disease (IBD), mainly in the forms of ulcerative colitis and Crohn’s disease, was usually caused by low microbial diversity. The diversity of the gut microbiota has also been associated with obesity, and the microbial-community composition can be intentionally manipulated to regulate the energy balance of obese individuals (Ley et al., 2005). Chen and Blaser (2007) found that colonization with Helicobacter pylori was inversely associated with asthma and allergy occurrence, and childhood acquisition of H. pylori can reduce these risks. The imbalance of microbial communities has also been associated with various types of cancer, including oral cancer (Zhang L. et al., 2019), colorectal cancer (Kim D.J. et al., 2020), and lung cancer (Zheng et al., 2020). Microbe-based disease pathogenesis is complex and can be influenced by environmental factors such as diet, smoking, and antibiotics therapy (Human Microbiome Project Consortium, 2012; Althani et al., 2016; Chen H. et al., 2017; Liu W. et al., 2020). Exploring and understanding microbe-disease associations, therefore, presents a significant challenge (Cheng et al., 2019; Cheng, 2019).
With the development of high-throughput sequencing technologies, such as 16S ribosomal RNA (16S rRNA), an increasing number of microbes have been identified, accelerating human disease research. Furthermore, projects such as the Human Microbiome Project (HMP) (Gevers et al., 2012; Nadia, and Ramana, 2020) and the Metagenomics of the Human Intestinal Tract (MetaHIT) Project1 were initiated to reveal the relationships between microbes and human diseases. However, traditional experimental methods for investigating microorganism-based pathogenesis are laborious and time-consuming, hindering progress in this field. In recent years, many computational methods have been successfully applied to the prediction of new associations, for example, miRNA–target association prediction (Deng et al., 2019; Yousef et al., 2007), lncRNA–target association prediction (Wang et al., 2019a; Zhang J. et al., 2019; Zhang Z. et al., 2019; Zhao et al., 2020), drug–target association prediction (Liu H. et al., 2020; Luo et al., 2017; Munir et al., 2019; Wang et al., 2020), drug–ncRNA association prediction (Yang et al., 2020), and association prediction between physical examination indicators with diabetes (Yang et al., 2021). However, these computational methods were only extended to the field of microbe–disease association prediction when the Human Microbe–Disease Association Database (HMDAD) became available (Ma et al., 2017). The HMDAD is the first resource that collects human microbe–disease associations through manual curation from 61 microbiota publications before July 2014. HMDAD documents 483 microbe–disease entries, including 39 diseases and 292 microbes, providing the foundation for subsequent computational–based microbe–disease association predictions.
Based on HMDAD, Chen X. et al. (2017) constructed a microbe–disease network and developed the KATZHMDA model for microbe–disease association prediction using the KATZ measurement and Gaussian interaction profile kernel similarity for microbes and diseases. Then, a series of computational methods were proposed to infer potential microbe–disease associations (Qu et al., 2019; Yang and Zou, 2020; Zhou et al., 2020). For example, Shen et al. (2017) extended the random walk to the microbe–disease heterogeneous network to compute the possibilities of microbe–disease associations. Huang et al. (2017) proposed NGRHMDA, which adopted neighbor-based collaborative filtering and a graph-based scoring method, to infer potential microbe–disease associations. Wang et al. developed a prediction model, NBLPIHMDA, to predict new microbe–disease associations. This model applied bidirectional label propagation on the disease similarity network and the microbe similarity network (Wang et al., 2019b). Liu Y. et al. (2020) proposed a deep matrix factorization microbe–disease association (DMFMDA) model, which combined the linear modeling ability of matrix factorization and the non-linear modeling ability of multi-layer perceptron to infer potential microbe–disease associations. To our knowledge, current computational methods for potential microbe–disease association predictions are all based on known microbe–disease associations from HMDAD. However, HMDAD documents the microbe–disease entries of only 61 publications before July 2014 and has not been updated. In recent years, research into microbe–disease associations have increased exponentially. Accordingly, some online repositories have been developed to record highly credible microbe–disease associations, such as Peryton (Skoufos et al., 2021), Disbiome (Janssens et al., 2018), and gutMDisorder (Cheng et al., 2020), which include thousands of curated microbe–disease associations.
In this study, we constructed a two-layer heterogeneous network by integrating large-scale known microbe–disease associations from the Peryton, Disbiome, and gutMDisorder databases, then extending the random walk with restart (RWR) to the network to prioritize candidate microbe–disease associations. The method fully considered the topological properties of the comprehensive network and achieved reasonable efficacy. Exploring microbe–disease relationships may not only help to reveal the mechanisms of disease pathogenesis but also provide insights to aid the prevention, diagnosis, and prognosis of various diseases.
The known microbe–disease associations used in this study were downloaded from the Peryton database2 (Skoufos et al., 2021), the Disbiome database3 (Janssens et al., 2018), and the gutMDisorder database4 (Cheng et al., 2020). Peryton is a novel resource that hosts more than 7,900 experimentally supported microbe–disease associations through manual curation of 314 publications. The database incorporates 43 diseases and 1,396 microorganisms, which are standardized via Medical Subject Headings (MeSH) and the NCBI Taxonomy database, respectively. Disbiome is a comprehensive database that collects microbe–disease associations from nearly 1,200 publications. Disbiome records 372 diseases and 1,622 organisms. The diseases are classified using the Medical Dictionary for Regulatory Activities (MedDRA) classification system and the microorganisms are normalized using NCBI and SILVA taxonomies. The gutMDisorder database provides a comprehensive resource for dysbiosis of the gut microbiota in disorders and interventions. gutMDisorder documents 2,263 experimentally supported microbe–disease associations between 579 gut microbes and 123 disorders or 77 intervention measures in humans. The microbes and diseases are standardized via the NCBI Taxonomy database and Disease Ontology (DO), respectively. The human microbe–disease associations were collected from the databases mentioned above to construct the composite heterogeneous network.
The human microbe–disease associations were collected from the three databases mentioned above. Since the identifiers of diseases and microbes were inconsistent between different databases, we standardized the diseases and microbes via MeSH and the NCBI Taxonomy database, respectively. Finally, we obtained 7,810 microbe–disease associations (1,389 microbes and 41 diseases) from the Peryton database, 7,378 microbe–disease associations (1,439 microbes and 251 diseases) from the Disbiome database, and 1,249 microbe–disease associations (412 microbes and 84 diseases) from the gutMDisorder database (see Figure 1). We removed any repeated microbe–disease associations from different resources, and finally obtained 11,037 distinct microbe–disease associations involving 287 human diseases and 2,106 microbes, which were used to construct the microbe–disease network.
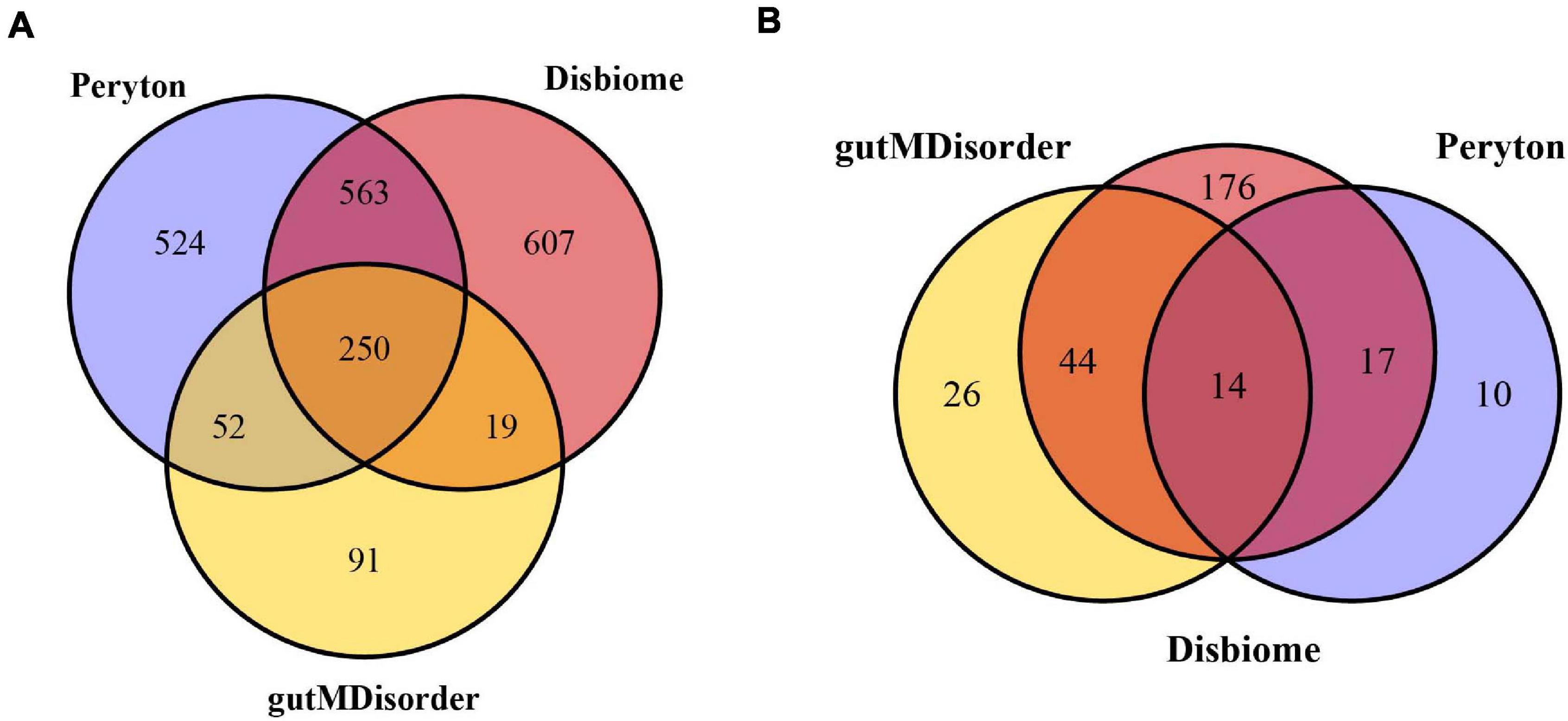
Figure 1. Venn diagram of overlapping microbes (A) and diseases (B) from the Peryton, Disbiome, and gutMDisorder databases.
Based on the assumption that microbes with similar functions tend to share similar interactions or non-interaction patterns with diseases (Chen X. et al., 2017), we obtained the microbe similarity via known human microbe–disease associations using the Gaussian interaction profile kernel. The interaction profile (IP) of a microbe represented the associations between this microbe and 287 human diseases. The IP of microbe mi was denoted as a vector, IP(mi), in which the jth element was set to be 1 when the disease dj was confirmed to be associated with mi; otherwise, it was set as 0. According to the interaction profiles, the Gaussian interaction profile kernel microbe similarity was defined as follows:
In the formula mentioned above, γm denotes the normalized kernel bandwidth, which can be calculated by a new bandwidth . In this study, we set = 1 according to previous relevant research (Chen X. et al., 2017). nm denotes the number of microbes in this study. KM(mi,mj) denotes the Gaussian interaction profile kernel similarity between two microbes, mi and mj. We constructed a microbe–microbe network, in which 2,106 microbes and the similarity between them were represented by nodes and edges, respectively.
Compared with microbe similarity, disease similarity has been widely investigated. A variety of disease similarity in Cheng’s study (Cheng et al., 2018) and the Gaussian interaction profile kernel disease similarity were used in this study to obtain the disease similarity. Firstly, we calculated the Gaussian interaction profile kernel similarity between disease di and dj as follows:
In the formula mentioned above, was also set to be 1 and nd denotes the number of diseases in this study. KD(di,dj) denotes the Gaussian interaction profile kernel similarity between two diseases, di and dj.
Cheng et al. (2018) provided DincRNA, a comprehensive bioinformatics resource for disease similarity calculation and non-coding RNA functional analysis. They utilized five methods, i.e., those of Wang et al. (2007), Resnik (1995), Lin (1998), PSB (Mathur and Dinakarpandian, 2012), and SemFunSim (Cheng et al., 2014) to calculate the similarity of pairwise diseases (SPWD). These methods took into consideration semantic associations, information content (IC), biological processes, and functional associations. The disease similarity score between di and dj in Cheng’s study was defined as SPWD(di,dj). Finally, the average value of Gaussian interaction profile kernel similarity as well as Cheng’s SPWD was taken as disease similarity, which is shown as follows:
Finally, we constructed a disease–disease network, comprising 287 human diseases, and the similarity between them was represented by edges.
We constructed a composite heterogeneous weighted network by integrating the microbe–disease, microbe–microbe, and disease–disease associations mentioned above. In the composite network, there were two types of nodes, 2,106 microbes and 287 human diseases. The edges between microbes and diseases represented 11,042 distinct microbe–disease associations, and the edge weight was set to be 1 when the microbe mi was confirmed to be associated with disease dj; otherwise, it was 0. The edges between different microbes were based on microbe similarity, and the edge weight between node mi and mj was denoted by KM(mi,mj). The edges between different diseases were based on disease similarity, and the edge weight between nodes di and dj was denoted by SD(di,dj).
Based on the composite heterogeneous weighted network, we used the RWR to prioritize candidate disease-related microbes by fully exploiting the heterogeneous biological associations. The RWR algorithm simulates a random walker that starts from the seed nodes and then moves to their immediate neighbors or stays at the current nodes according to the probability transition matrix. The iterative transition is repeated until all vertices achieve a steady state. In this study, the formula of RWR is defined as:
In the abovementioned formula, r∈(0,1) denotes the restart probability. Pt denotes a vector in which the ith element holds the probability of being at node i at step t. W denotes the transition matrix, which is a column-normalized adjacency matrix of the composite network. Here, we defined the adjacency matrix W as follows:
B is a probability transition matrix from microbe network to disease network. Accordingly, BT is the transpose of B. Let λ be the probability of the random walker jumping from microbe network to disease network or vice versa. We defined the transition probability from microbe network to disease network as follows:
AM is the microbe network transition matrix. The element of AM(i,j) represents the probability of the random walker transition from mi to mj, which is defined as follows:
Similarly, AD is the disease network transition matrix. The element of AD represents the probability of the random walker transition from di to dj, which is defined as follows:
P0 denotes the initial probability vector, which is a normalized unit vector. P0 = represents the initial probability vector for the heterogeneous network. m0 and d0 represent the initial probabilities of the microbe network and the disease network, respectively. After many iterations, when the difference between Pt and Pt+1 falls below 10–10, it achieves a steady state. Then, microbes and diseases are ranked based on the steady probability. The flowchart of this work is shown in Figure 2.
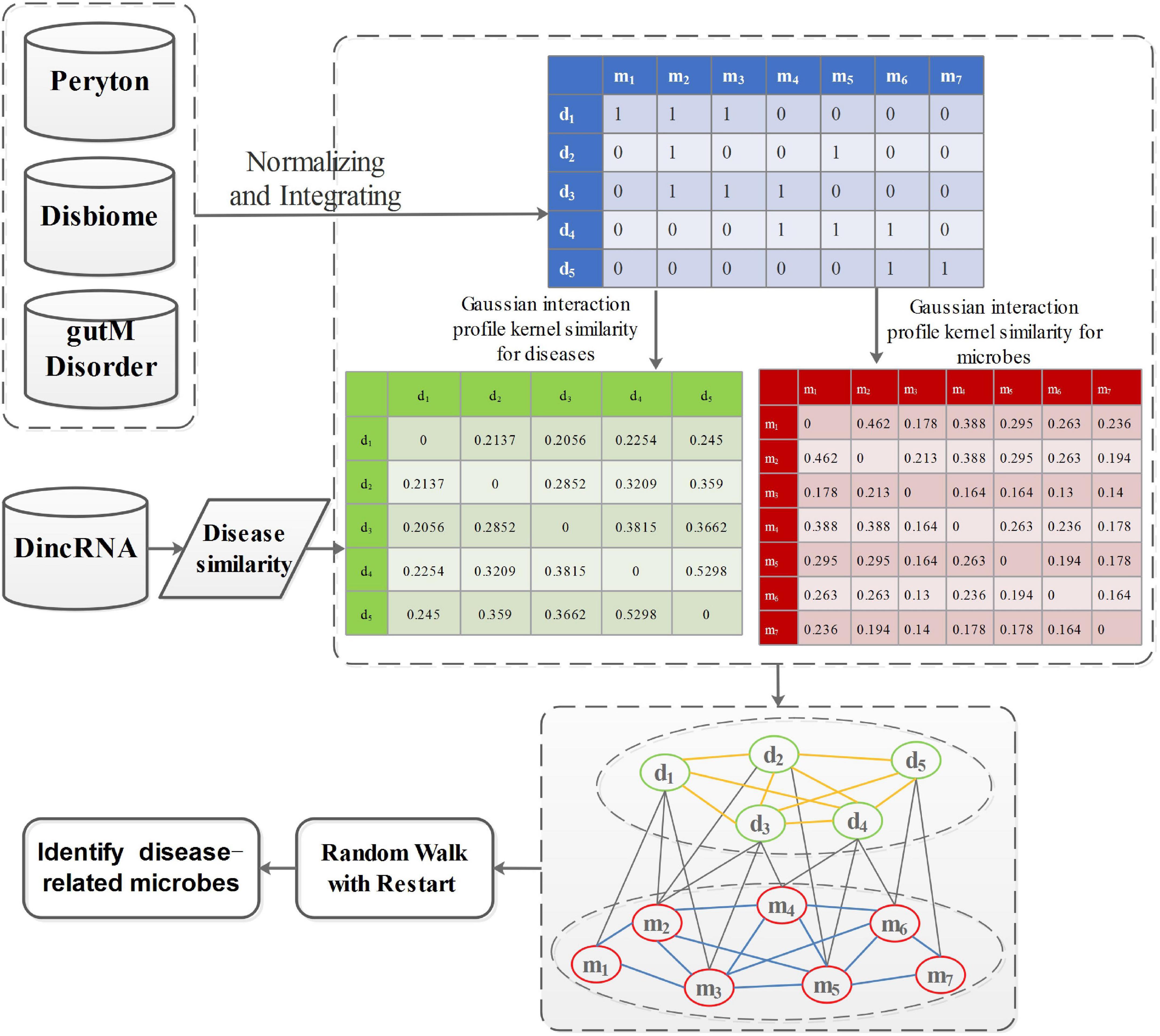
Figure 2. The flowchart of prioritization of candidate disease-related microbes.
To assess the performance of our method, we determined its ability to identify known disease-related microbes. The leave-one-out cross-validation (LOOCV) and fivefold cross-validation (fivefold CV) methods (Dao et al., 2020; Wang et al., 2021) were applied on known microbe–disease associations for 236 diseases, which included at least five known microbes. The receiver operating characteristic curve (ROC) plots the true-positive rate (sensitivity) versus false-positive rate (1 - specificity) at different cutoffs, and the area under the curve (AUC) was used to represent the results of cross-validation (Feng et al., 2019; Lv et al., 2020).
For LOOCV, for every disease, each known disease-related microbe was considered as one test sample, the remaining known disease-related microbes were considered as training samples, and all other unknown disease-related microbes in the composite network were considered as candidate samples. Then, we obtained a rank list of the test samples and all candidate samples according to prediction scores by performing our method. The model would achieve high prediction performance when the test samples ranked higher than the given threshold. The ROC and AUC values indicated the performance of the method. In our study, we found that all diseases achieved high predictive performance and the AUC values of LOOCV ranged from 0.9370 to 1 (see Supplementary Table 1).
For fivefold CV, for every disease, a set of known disease-related microbes was equally and randomly divided into five subparts. Each subpart was considered as the test sample in turn, and the other four subparts were considered as training samples; all of the other unknown disease-related microbes in the composite network were considered as candidate samples. Considering the potential bias caused by random sample division, we repeated this process 10 times to obtain an average AUC. Similar to LOOCV, we found that the AUC values of fivefold CV ranged from 0.9366 to 1 (Supplementary Table 2). The high predictive power indicated that the approach utilizing integrated interactions from the composite two-layer network was highly efficient in prioritizing candidate disease-related microbes.
There are two parameters in our method, one is the restart probability denoted as r, and the other is the probability of the random walker jumping between different networks denoted as λ. We set various values under the framework of LOOCV and fivefold CV to evaluate the impact of these parameters and found that the method achieved its best performance when r was set as 0.1 and λ was set as 0.5.
We integrated a composite network that included 2,393 nodes (2,106 microbes and 287 human diseases) and 11,037 edges. The RWR algorithm, which makes full use of the network topology, was applied to identify candidate microbes involved in diseases among the composite network of 236 diseases. To verify the ability of our method to discover unknown associations, we implemented case studies on IBD, asthma, and obesity. The resulting list of the top 30 candidate microbes associated with these diseases is shown in Supplementary Table 3.
Inflammatory bowel disease, mainly in the form of ulcerative colitis and Crohn’s disease, is a chronic relapsing inflammatory disease of the colon and small intestine that affects an increasing number of people (Jostins et al., 2012). When considering case studies of IBD, ROC curves were obtained (Figure 3A) and the AUC values of LOOCV and fivefold CV for IBD were both 0.9913. Although there have been many studies on IBD–microbe associations (with 106 known IBD-related microbes), 16 of the top 30 prioritized IBD–microbe associations were manually confirmed by newly published literature (Table 1). For example, Roseburia is a top-ranked microbe in the prioritized IBD–related microbe list. Kim E.S. et al. (2020) found higher fecal calprotectin (FC) levels in pregnant patients with IBD through pregnancy, and Roseburia was positively correlated with maternal FC levels at T3. Sokol et al. (2018) found that IBD patients with Clostridium difficile infection (CDI) had more pronounced dysbiosis of Dorea, which was also a top-ranked microbe in the prioritized IBD-related microbe list. Toyonaga et al. (2015) found that compared with IL-10 knockout mice, the level of Clostridium cluster XVIII was significantly higher in OPN/IL-10 double knockout mice, when the role of osteopontin in the pathophysiology of IBD was investigated.
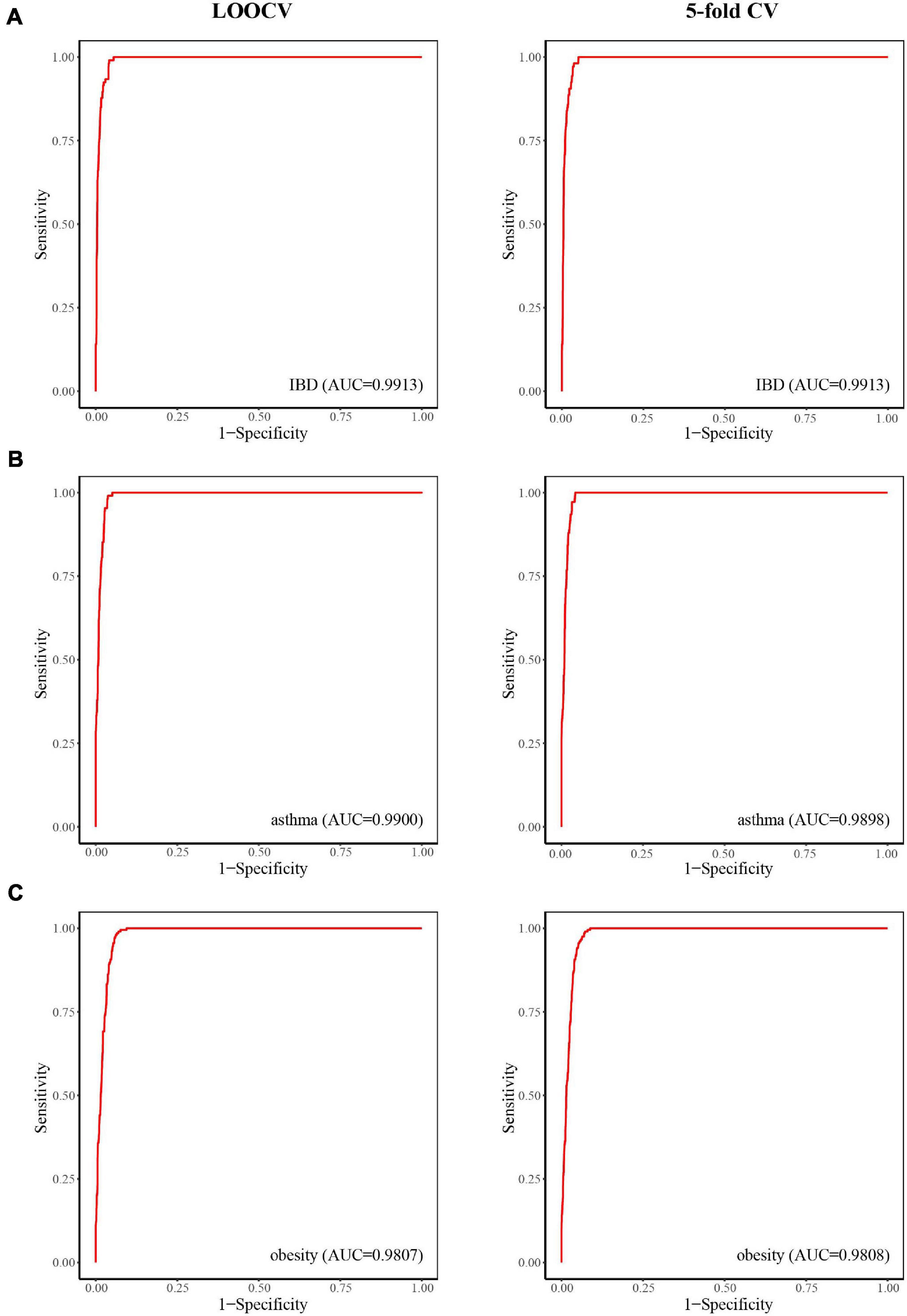
Figure 3. The predictive power of LOOCV (left) and fivefold CV (right) for IBD (A), asthma (B), and obesity (C).

Table 1. Literature verification of the predicted IBD-related microbes.
Asthma is a common chronic inflammatory disease caused by a variety of factors, including genetic and environment factors. Microorganisms may also play a role in the pathogenesis of asthma. Here, we considered asthma case studies, the ROC curves for which are displayed in Figure 3B, and the AUC values of LOOCV and fivefold CV for asthma were 0.9900 and 0.9898, respectively. Since asthma and its related microbes have been widely studied (with 108 known asthma-related microbes), seven of the top 30 prioritized asthma–microbe associations were manually confirmed by newly published literature (Table 2). Blautia, a top-ranked microbe in the prioritized IBD-related microbe list, was found to be present at high concentration in asthma patients (Fu et al., 2021). Dong et al. (2020) showed that treatment with Gu–Ben–Fang–Xiao Decoction (GBFXD) increased the abundance of Lachnospiraceae in asthmatic mice, which consequently led to elevated levels of short-chain fatty acids. Patricia et al. found that the abundance of Epicoccum was negatively associated with male asthma patients (Segura-Medina et al., 2019).

Table 2. Literature verification of the predicted asthma–related microbes.
Obesity is a disease associated with a body mass index of 30 kg/m2 or higher. It is prevalent in both adults and children worldwide and has been linked to health complications such as rheumatoid arthritis, nonallergic rhinitis, and cancer (Apovian, 2016). Here, we considered obesity case studies, the ROC curves for which are displayed in Figure 3C, and the AUC values of LOOCV and fivefold CV for obesity were 0.9807 and 0.9808, respectively. Although obesity and its related microbes have been widely studied (with 204 known obesity-related microbes), seven of the top 30 prioritized obesity–microbe associations were manually confirmed by newly published literature (Table 3). Raman et al. (2013) found that Robinsoniella, a top-ranked microbe in the obesity-related microbe list, was present at higher levels in nonalcoholic fatty liver disease patients and was implicated in the etiology of, and complications related to, obesity. Zeng et al. (2019) showed that Dorea was positively correlated with bodyweight and serum lipids, which were two significant clinical indicators of obesity.

Table 3. Literature verification of the predicted obesity-related microbes.
A wide variety of microbes have been found to be parasitic within the human body. Such microbes play important roles in various physiological processes, such as metabolism regulation and immune defense. Research has also revealed that imbalances in microbial communities are closely associated with human diseases. Thus, identifying novel disease-related microbes is vital when investigating disease pathogenesis, and computational methods have been effective in achieving this. To date, the computational methods that have been applied to identify novel microbe–disease associations have all been based on the HMDAD database, which only recorded 483 microbe–disease entries from 61 publications before July 2014. In this study, we constructed a comprehensive microbe–disease network by integrating known microbe–disease associations from three novel large-scale databases (Peryton, Disbiome, and gutMDisorder), and extended the RWR to the network for prioritizing candidate disease-related microbes. The AUC values of the LOOCV and fivefold CV for 236 human diseases exceeded 0.9370 and 0.9366, respectively, indicating the high performance of our method. Furthermore, we considered case studies of IBD, asthma, and obesity. Although these three diseases have been widely studied, some prioritized disease-related microbes were validated by new publications. This finding suggested that our method is an effective method for prioritizing novel disease-related microbes, thereby aiding our understanding of disease pathogenesis.
There were some limitations in our current study. Firstly, the number of diseases considered in our study was small. This reflects the fact that large-scale microbe studies across a wide range of diseases are lacking, although the development of high-throughput sequencing technologies, such as 16S rRNA, may address this. Secondly, the microbe similarity used in this study was only based on known human microbe–disease associations using a Gaussian interaction profile kernel, which may lead to a defective heterogeneous network. This limitation may be addressed by further research into microbial functions and by integrating the functional similarities of microbes.
The known microbe-disease associations used in this study were downloaded from Peryton database (https://dianalab.e-ce.uth.gr/peryton/#/associations), Disbiome database (https://disbiome.ugent.be/export), and gutMDisorder database (http://bio-annotation.cn/gutMDisorder/resource.dhtml). The raw data used in this study were downloaded from the databases mentioned above, which is open source without any accession number. Other dataset presented in the study are included in the article/Supplementary Material.
LC and JL conceived and designed the study. HY and CQ collected and processed the data. HY and PW performed the experiments. HY and FT wrote the manuscript. All authors read and approved the final manuscript.
This work was supported by the Tou-Yan Innovation Team Program of the Heilongjiang Province (2019–15), the National Natural Science Foundation of China (61902095 and 61871160), the Heilongjiang Province Postdoctoral Fund (LBH-Q20030), and the Young Innovative Talents in Colleges and Universities of Heilongjiang Province (2018–69).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.685549/full#supplementary-material
Althani, A. A., Marei, H. E., Hamdi, W. S., Nasrallah, G. K., El Zowalaty, M. E., Al Khodor, S., et al. (2016). Human microbiome and its association with health and diseases. J. Cell Physiol. 231, 1688–1694. doi: 10.1002/jcp.25284
Apovian, C. M. (2016). Obesity: definition, comorbidities, causes, and burden. Am. J. Manag. Care 22(Suppl. 7), s176–s185.
Chen, H., Peng, S., Dai, S., Zou, Q., Yi, B., Yang, X., et al. (2017a). Oral microbial community assembly under the influence of periodontitis. Plos One 12:e0182259. doi: 10.1371/journal.pone.0182259
Chen, X., Huang, Y. A., You, Z. H., Yan, G. Y., and Wang, X. S. (2017b). A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739.
Chen, Y., and Blaser, M. J. (2007). Inverse associations of helicobacter pylori with asthma and allergy. Arch. Intern. Med. 167, 821–827. doi: 10.1001/archinte.167.8.821
Cheng, L. (2019). Computational and biological methods for gene therapy. Curr. Gene Ther. 19:210. doi: 10.2174/156652321904191022113307
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 34, 1953–1956. doi: 10.1093/bioinformatics/bty002
Cheng, L., Li, J., Ju, P., Peng, J., and Wang, Y. (2014). SemFunSim: a new method for measuring disease similarity by integrating semantic and gene functional association. PLoS One 9:e99415. doi: 10.1371/journal.pone.0099415
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2020). gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 48, D554–D560.
Cheng, L., Zhao, H., Wang, P., Zhou, W., Luo, M., Li, T., et al. (2019). Computational methods for identifying similar diseases. Mol. Ther. Nucleic Acids 18, 590–604. doi: 10.1016/j.omtn.2019.09.019
Dao, F. Y., Lv, H., Zhang, D., Zhang, Z. M., Liu, L., Lin, H., et al. (2020). DeepYY1: a deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. bbaa356. [Epub ahead of print].
Das, B., and Nair, G. B. (2019). Homeostasis and dysbiosis of the gut microbiome in health and disease. J. Biosci. 44:117.
Deng, L., Wang, J., and Zhang, J. (2019). Predicting gene ontology function of human microRNAs by integrating multiple networks. Front. Genet. 10:3. doi: 10.3389/fgene.2019.00003
Dong, Y., Yan, H., Zhao, X., Lin, R., Lin, L., Ding, Y., et al. (2020). Gu-Ben-Fang-Xiao decoction ameliorated murine asthma in remission stage by modulating microbiota-acetate-tregs axis. Front. Pharmacol. 11:549. doi: 10.3389/fphar.2020.00549
Feng, C. Q., Zhang, Z. Y., Zhu, X. J., Lin, Y., Chen, W., Tang, H., et al. (2019). iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics 35, 1469–1477. doi: 10.1093/bioinformatics/bty827
Fu, X., Li, Y., Meng, Y., Yuan, Q., Zhang, Z., Wen, H., et al. (2021). Derived habitats of indoor microbes are associated with asthma symptoms in Chinese university dormitories. Environ. Res. 194:110501. doi: 10.1016/j.envres.2020.110501
Gevers, D., Knight, R., Petrosino, J. F., Huang, K., McGuire, A. L., Birren, B. W., et al. (2012). The human microbiome project: a community resource for the healthy human microbiome. PLoS Biol. 10:e1001377. doi: 10.1371/journal.pbio.1001377
Huang, S. Y., Xiang, X., Qiu, L., Wang, L., Zhu, B., Guo, R., et al. (2020). Transfection of TGF-beta shRNA by using ultrasound-targeted microbubble destruction to inhibit the early adhesion repair of rats wounded achilles tendon in vitro and in vivo. Curr. Gene Ther. 20, 71–81. doi: 10.2174/1566523220666200516165828
Huang, Y. A., You, Z. H., Chen, X., Huang, Z. A., Zhang, S., Yan, G. Y., et al. (2017). Prediction of microbe-disease association from the integration of neighbor and graph with collaborative recommendation model. J. Transl. Med. 15:209.
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Janssens, Y., Nielandt, J., Bronselaer, A., Debunne, N., Verbeke, F., Verbeke, F., et al. (2018). Disbiome database: linking the microbiome to disease. BMC Microbiol. 18:50. doi: 10.1186/s12866-018-1197-5
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., McGovern, D. P., Hui, K. Y., et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124.
Kim, D. J., Yang, J., Seo, H., Lee, W. H., Lee, D. H., Lee, S., et al. (2020b). Colorectal cancer diagnostic model utilizing metagenomic and metabolomic data of stool microbial extracellular vesicles. Sci. Rep. 10:2860.
Kim, E. S., Tarassishin, L., Eisele, C., Barre, A., Nair, N., Rendon, A., et al. (2020a). Longitudinal changes in fecal calprotectin levels among pregnant women with and without inflammatory bowel disease and their babies. Gastroenterology 160, 1118–1130.e3.
Ley, R. E., Bäckhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., Gordon, J. I., et al. (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U. S. A. 102, 11070–11075.
Lin, D. (1998). “An information-theoretic definition of similarity, in Proceedings of the 15th International Conference on Machine Learning, ed M. Kaufman (San Francisco, CA), 296–304.
Liu, H., Zhang, W., Zou, B., and Wang, J. (2020a). DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res. 48, D871–D881.
Liu, W., Haran, J. P., Allison, J. J., Ye, S., Tjia, J., Bucci, V., et al. (2020b). High-dimensional causal mediation analysis with a large number of mediators clumping at zero to assess the contribution of the microbiome to the risk of bacterial pathogen colonization in older adults. Curr. Bioinform. 15, 671–696. doi: 10.2174/1574893614666191115123219
Liu, Y., Wang, S., Zhang, J., and Zhang, W. (2020c). DMFMDA: prediction of microbe-disease associations based on deep matrix factorization using bayesian personalized ranking. IEEE/ACM Trans. Comput. Biol. Bioinform. [Epub ahead of print].
Luo, Y., Zhao, X., Zhou, J., Yang, J., Zhang, Y., Kuang, W., et al. (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8:573.
Lv, H., Dao, F. Y., Guan, Z. X., Li, Y. W., Lin, H., Yang, H., et al. (2020). Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief. Bioinform. bbaa255. [Epub ahead of print].
Ma, W., Zhang, L., Zeng, P., Huang, C., Li, J., Geng, B., et al. (2017). An analysis of human microbe-disease associations. Brief. Bioinform. 18, 85–97.
Mathur, S., and Dinakarpandian, D. (2012). Finding disease similarity based on implicit semantic similarity. J. Biomed. Inform. 45, 363–371. doi: 10.1016/j.jbi.2011.11.017
Munir, A., Malik, S. I., and Malik, K. A. (2019). Proteome mining for the identification of putative drug targets for human pathogen clostridium tetani. Curr. Bioinform. 14, 532–540. doi: 10.2174/1574893613666181114095736
Nadia, and Ramana, J. (2020). The human oncobiome database: a database of cancer microbiome datasets. Curr. Bioinform. 15, 472–477. doi: 10.2174/1574893614666190902152727
Qi, C., Wang, P., Fu, T., Lu, M., Cai, Y., Chen, X., et al. (2021). A comprehensive review for gut microbes: technologies, interventions, metabolites and diseases. Brief. Funct. Genomics 20, 42–60. doi: 10.1093/bfgp/elaa029
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65.
Qu, K., Guo, F., Liu, X., Lin, Y., and Zou, Q. (2019). Application of machine learning in microbiology. Front. Microbiol. 10:827. doi: 10.3389/fmicb.2019.00827
Raman, M., Ahmed, I., Gillevet, P. M., Probert, C. S., Ratcliffe, N. M., Smith, S., et al. (2013). Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 11868-75.e1-3.
Resnik, P. (1995). Using information content to evaluate semantic similarity in a taxonomy. arXiv [Preprint]. cmp-lg/9511007.
Segura-Medina, P., Vargas, M. H., Aguilar-Romero, J. M., Arreola-Ramírez, J. L., Miguel-Reyes, J. L., and Salas-Hernández, J. (2019). Mold burden in house dust and its relationship with asthma control. Respir. Med. 150, 74–80. doi: 10.1016/j.rmed.2019.02.014
Shen, X., Chen, Y., Jiang, X., Hu, X., He, T., Yang, J., et al. (2017). Prioritizing disease-causing microbes based on random walking on the heterogeneous network. Methods 124, 120–125. doi: 10.1016/j.ymeth.2017.06.014
Skoufos, G., Alexiou, A., Kavakiotis, L., Lambropoulou, A., Kotsira, V., Tastsoglou, S., et al. (2021). Peryton: a manual collection of experimentally supported microbe-disease associations. Nucleic Acids Res. 49. 1328–1333. doi: 10.1093/nar/gkaa902
Sokol, H., Jegou, S., McQuitty, C., Straub, M., Leducq, V., Landman, C., et al. (2018). Specificities of the intestinal microbiota in patients with inflammatory bowel disease and Clostridium difficile infection. Gut Microbes 9, 55–60. doi: 10.1080/19490976.2017.1361092
Sommer, F., and Backhed, F. (2013). The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Toyonaga, T., Nakase, H., Ueno, S., Matsuura, M., Yoshino, T., Honzawa, Y., et al. (2015). Osteopontin deficiency accelerates spontaneous colitis in mice with disrupted gut microbiota and macrophage phagocytic activity. PLoS One 10:e0135552. doi: 10.1371/journal.pone.0135552
Wang, D., Jiang, Y., Wang, D., Zhang, Z., Mao, Z., Lin, H., et al. (2021). DM3Loc: multi-label mRNA subcellular localization prediction and analysis based on multi-head self-attention mechanism. Nucleic Acids Res. 49:e46. doi: 10.1093/nar/gkab016
Wang, J., Wang, H., Wang, X., and Chang, H. (2020). Predicting drug-target interactions via FM-DNN learning. Curr. Bioinform. 15, 68–76. doi: 10.2174/1574893614666190227160538
Wang, J. Z., Du, Z., Payattakool, R., Yu, P. S., and Yu, P. S. (2007). A new method to measure the semantic similarity of GO terms. Bioinformatics 23, 1274–1281. doi: 10.1093/bioinformatics/btm087
Wang, L., Wang, Y., Li, H., Feng, X., Yuan, D., and Yang, J. (2019b). A bidirectional label propagation based computational model for potential microbe-disease association prediction. Front. Microbiol. 10:684. doi: 10.3389/fmicb.2019.00684
Wang, L., Xuan, Z., Zhou, S., Kuang, L., and Pei, T. (2019a). A novel model for predicting LncRNA-disease associations based on the LncRNA-MiRNA-disease interactive network. Curr. Bioinform. 14, 269–278. doi: 10.2174/1574893613666180703105258
Yang, F., and Zou, Q. (2020). mAML: an automated machine learning pipeline with a microbiome repository for human disease classification. Database 2020:baaa050.
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk prediction of diabetes: big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi: 10.1016/j.inffus.2021.02.015
Yang, H., Tang, H., Chen, X. X., Zhang, C. J., Zhu, P. P., Ding, H., et al. (2016). Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. Biomed. Res. Int. 2016:5413903.
Yang, H., Xu, Y., Shang, D., Shi, H., Zhang, C., Dong, Q., et al. (2020). ncDRMarker: a computational method for identifying non-coding RNA signatures of drug resistance based on heterogeneous network. Ann. Transl. Med. 8:1395. doi: 10.21037/atm-20-603
Yousef, M., Jung, S., Kossenkov, A. V., Showe, L. C., and Showe, M. K. (2007). Naive bayes for microRNA target predictions–machine learning for microRNA targets. Bioinformatics 23, 2987–2992. doi: 10.1093/bioinformatics/btm484
Zeng, Q., Li, D., He, Y., Li, Y., Yang, Z., Zhao, X., et al. (2019). Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 9:13424.
Zhang, J., Zhang, Z., Chen, Z., and Deng, L. (2019a). Integrating multiple heterogeneous networks for novel lncrna-disease association inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 396–406. doi: 10.1109/tcbb.2017.2701379
Zhang, L., Liu, Y., Zheng, H. J., and Zhang, C. P. (2019b). The oral microbiota may have influence on oral cancer. Front. Cell Infect. Microbiol. 9:476. doi: 10.3389/fcimb.2019.00476
Zhang, Z., Zhang, J., Fan, C., Tang, Y., and Deng, L. (2019c). KATZLGO: large-scale prediction of LncRNA functions by using the KATZ measure based on multiple networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 407–416. doi: 10.1109/tcbb.2017.2704587
Zhao, T., Hu, Y., Peng, J., and Cheng, L. (2020). DeepLGP: a novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 36, 4466–4472. doi: 10.1093/bioinformatics/btaa428
Zheng, Y., Fang, Z., Xue, Y., Zhang, J., Zhu, J., Gao, R., et al. (2020). Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microbes 11, 1030–1042. doi: 10.1080/19490976.2020.1737487
Keywords: microbe, disease, heterogeneous network, random walk with restart, microbe-disease associations
Citation: Yang H, Tong F, Qi C, Wang P, Li J and Cheng L (2021) Prioritizing Disease-Related Microbes Based on the Topological Properties of a Comprehensive Network. Front. Microbiol. 12:685549. doi: 10.3389/fmicb.2021.685549
Received: 25 March 2021; Accepted: 10 May 2021;
Published: 08 July 2021.
Edited by:
Jialiang Yang, Geneis (Beijing) Co., Ltd., ChinaReviewed by:
Hui Ding, University of Electronic Science and Technology of China, ChinaCopyright © 2021 Yang, Tong, Qi, Wang, Li and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangyu Li, bGlqaWFuZ3l1QGJtaS5hYy5jbg==; Liang Cheng, bGlhbmdjaGVuZ0BocmJtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.