
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 July 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.679894
 Neža Čadež1*
Neža Čadež1* Nicolas Bellora2
Nicolas Bellora2 Ricardo Ulloa3
Ricardo Ulloa3 Miha Tome1
Miha Tome1 Hrvoje Petković1Marizeth Groenewald4
Hrvoje Petković1Marizeth Groenewald4 Chris Todd Hittinger5
Chris Todd Hittinger5 Diego Libkind2*
Diego Libkind2*During a survey of Nothofagus trees and their parasitic fungi in Andean Patagonia (Argentina), genetically distinct strains of Hanseniaspora were obtained from the sugar-containing stromata of parasitic Cyttaria spp. Phylogenetic analyses based on the single-gene sequences (encoding rRNA and actin) or on conserved, single-copy, orthologous genes from genome sequence assemblies revealed that these strains represent a new species closely related to Hanseniaspora valbyensis. Additionally, delimitation of this novel species was supported by genetic distance calculations using overall genome relatedness indices (OGRI) between the novel taxon and its closest relatives. To better understand the mode of speciation in Hanseniaspora, we examined genes that were retained or lost in the novel species in comparison to its closest relatives. These analyses show that, during diversification, this novel species and its closest relatives, H. valbyensis and Hanseniaspora jakobsenii, lost mitochondrial and other genes involved in the generation of precursor metabolites and energy, which could explain their slower growth and higher ethanol yields under aerobic conditions. Similarly, Hanseniaspora mollemarum lost the ability to sporulate, along with genes that are involved in meiosis and mating. Based on these findings, a formal description of the novel yeast species Hanseniaspora smithiae sp. nov. is proposed, with CRUB 1602H as the holotype.
The yeasts of the genus Hanseniaspora (Saccharomycodaceae, Ascomycota) are characterized by their apiculate form, bipolar budding, and basal phylogenetic placement relative to the family Saccharomycetaceae. Currently there are 20 recognized taxa of Hanseniaspora, including three recently assigned anamorphic species formerly known as Kloeckera spp. (Čadež et al., 2019). The latest incorporations to the genus were Hanseniaspora mollemarum from The Netherlands (Groenewald et al., 2018), Hanseniaspora gamundiae from Argentina (Čadež et al., 2019), and Hanseniaspora terricola from Tibet (Liu et al., 2021). Species of Hanseniaspora are widespread and particularly abundant on various fruits, as well as on flowers and bark, which are their primary habitats (Saubin et al., 2020). The vectors for their dispersal are mostly insects, such as Drosophila spp., which are attracted by the aromatic volatile compounds produced by these yeasts (Hamby et al., 2012; Becher et al., 2018). Due to their ability to ferment simple sugars, they contribute to the beginning phases of various spontaneous food fermentation processes, and thus, many Hanseniaspora spp. have been intensively studied to determine their potential to improve sensorial complexity of fermented products (Steensels and Verstrepen, 2014), primarily for wine (Martin et al., 2018), but more recently for beer production (Burini et al., 2021). Strains of the species Hanseniaspora uvarum, Hanseniaspora guilliermondii, Hanseniaspora viniae, and Hanseniaspora valbyensis are probably the most relevant for bioflavoring in the beverage industry.
Recently, it was shown that species of the genus Hanseniaspora have evolved unusually rapidly, likely due to the loss of many genes associated with DNA repair and maintenance (Steenwyk et al., 2019). Increased mutation rates can accelerate adaptation to fluctuating and stressful conditions (Giraud et al., 2001; Healey et al., 2016) and represent one of the drivers of intrinsic reproductive isolation among diverging populations (Seehausen et al., 2014). Furthermore, the observed pervasive gene loss in Hanseniaspora was shown to have a biased pattern in the functional categories of metabolism and the cell cycle. This bias might be associated with the lifestyle of these yeasts, as they are dominant on ripe fruit where simple sugars are available as a food source for only one period each year. Hanseniaspora species may have adapted to novel environments of flowering plants that produce sugar-based fruit once a year by the conditional dispensability of genes that are not required under specific conditions. At the same time, the loss of cell-cycle checkpoint genes may have led to rapid growth as an advantage over competitors.
To gain insight into the ecological causes of gene loss during adaptation of Hanseniaspora species to novel ecological niches, we continue our exploration of the yeast communities associated with Nothofagus trees and its parasitic fungi in Andean Patagonia. We discovered a group of apiculate yeast strains that are genetically distinct from their sister species H. valbyensis based on rRNA gene sequence and genomic data. In the present paper, we formally describe a novel species of apiculate yeast where their delineation is additionally based on data of complete genomic sequences of 20 type strains of Hanseniaspora species, two of which were sequenced during the present study. The genomic contents of the new species and its closest relatives, H. valbyensis, H. mollemarum, Hanseniaspora lindneri, Hanseniaspora singularis, and Hanseniaspora jakobsenii, correlate with their physiological and ecological backgrounds.
The strains examined in this study are listed in Supplementary Table 1. These included five isolates from the stromata of Cyttaria species and the bark of Nothofagus betuloides and Nothofagus antarctica, which were collected from sites very distant from each other (ca. 2,000 km) in the Andean forests in Patagonia, Argentina. Isolations were obtained according to the enrichment protocol described by Sampaio and Gonçalves (2008) and Libkind et al. (2011a). The Argentinean National Park Administration issued permission to Diego Libkind for this sampling. The strains were phenotypically characterized by methods described by Kurtzman et al. (2011). Assimilation tests were performed in liquid media following standard procedures. Sporulation was investigated on 2% malt extract (BD Difco) and 5% malt extract (Sigma) agars at 25°C, and cells were examined weekly for up to 4 weeks.
MasterPure Yeast DNA Purification kits (Lucigen) were used to extract DNA from cultures grown on yeast extract–peptone–glucose (YPD, Conda) agar plates for 2 days. Amplification of DNA sequences that encode the internal transcribed spacer (ITS; 668 bp), the large subunit rRNA (LSU) D1/D2 domain (573 bp), and actin (encoded by ACT1; 949 bp) was performed as described by Cadez et al. (2006). These DNA sequences were determined using a commercial sequencing facility (Macrogen Inc., Netherlands). The sequences were assembled in BioNumerics 7.6 and aligned using the MEGA X software (Kumar et al., 2018) with the built-in MUSCLE algorithm (Edgar, 2004).
Phylogenetic relationships among the five investigated yeast strains and their closest relatives were inferred using the maximum likelihood (ML) method with the substitution model of the Kimura two-parameter model calculated for actin, ITS, and the LSU D1/D2 concatenated datasets. The basic maximum parsimony network (Huson and Bryant, 2006), which finds the most parsimonious network using simulated annealing, was chosen using BioNumerics 7.6. Bootstrap support was determined from 1,000 pseudoreplicates (Felsenstein, 1985). For formal delineation of the phylogenetic species, a parsimony network using aligned ITS-D1/D2 sequences, excluding gaps at a 95% connection limit, was constructed using the TCS 1.21 program (Clement et al., 2000).
The genomes of almost all type strains of Hanseniaspora species are publicly available, including the type strain of Hanseniaspora smithiae (Shen et al., 2018; Steenwyk et al., 2019). In the course of the present study, the genomes of H. mollemarum CBS 15034T and H. lindneri CBS 285T were sequenced. The genomic DNA was isolated according to a protocol published by Schwartz and Sherlock (2016) that is based on protein removal using phenol:chloroform. A sequencing library was constructed using TruSeq DNA PCR Free (350) kits (Illumina), and run on an Illumina NovaSeq instrument at the Macrogen Inc. sequencing facility.
To generate the whole-genome assembly, the Illumina reads were used as input to the meta-assembler pipeline of iWGS v1.1 (Zhou et al., 2016). The pipeline first trimmed the adapters and low-quality bases using Trimmomatic v.038 (Bolger et al., 2014), and calculated the optimal k-mer length for a range of assemblers. The quality of each assembly was evaluated by N50 statistics and genome size, using Quast v5.0.2 (Gurevich et al., 2013). The assembly produced by SPAdes v.3.11.1 (Bankevich et al., 2012) gave the best results, and was used further. The completeness of the genome was evaluated using the BUSCO v3.0.2 software (Simão et al., 2015).
For annotation of the H. mollemarum genome, the MAKER genome annotation pipeline v3.01.02 was used (Holt and Yandell, 2011). For the homology evidence for genome annotation, the proteomes and transcripts of Saccharomyces cerevisiae, H. valbyensis, and H. uvarum were used. To soft-mask repeats, RepeatMasker was used with the fungal RepBase repeat library. Furthermore, three ab initio gene predictions were used with the MAKER pipeline: SNAP (Korf, 2004), GeneMark-ES v4.57 (Ter-Hovhannisyan et al., 2008) trained for the H. mollemarum or H. lindneri genomes, and AUGUSTUS v3.3.2 (Stanke et al., 2008) with “Saccharomyces” as an Augustus pre-existing species model. All of the resulting gene models, together with the homology evidence and masked repeats, were used to perform the final set of annotations of the genome.
For the phylogenomic reconstruction of the genus Hanseniaspora, protein sequences of conserved single-copy orthologs were identified by BUSCO v3.0.2 using the “Saccharomycetales” reference dataset. A total of 251 conserved genes present in all Hanseniaspora species were extracted, aligned and trimmed using MAFFT v7.310 (Katoh and Standley, 2013) and trimAl v1.4 (Capella-Gutiérrez et al., 2009), which are part of the BUSCO USECASE genomic utilities pipeline (Waterhouse et al., 2018). The alignments were concatenated into a single data matrix that was used as input for ML phylogenetic inference with RAxML v8.2.11 (Stamatakis, 2014). To determine the best-fit phylogenetic model, the ModelFinder option of IQ-TREE v. 1.6.1 was used (Kalyaanamoorthy et al., 2017). Branch support was evaluated with 100 rapid bootstrap inferences, while internode certainty (IC) was used to evaluate the degree of conflicting bipartitions among the 251 individual gene trees (Salichos and Rokas, 2013; Salichos et al., 2014); both were carried out in RAxML. For calculations, the ML gene-based trees were constructed in RAxML with the best-fit model of amino-acid substitutions that were separately estimated for each gene dataset in IQ-TREE. The gene-based partitions were used to calculate the IC values on the concatenated gene tree in RAxML (option –f i).
Similarities between the genomes of Hanseniaspora species were calculated using average nucleotide identity (ANI) metrics with the web-based calculator available at https://www.ezbiocloud.net/tools/ani (Yoon et al., 2017) and with command-line tool FastANI (Jain et al., 2018). To calculate distances between genomes, digital DNA-DNA homology (dDDH) values were calculated using the Genome-to-Genome Distance Calculator 2.1, provided by the German Collection of Microorganisms and Cell Cultures website1. The dDDH values presented here were calculated using Formula 2, which estimates them based on the identities of high-scoring segment pairs (Meier-Kolthoff et al., 2013). Heatmap visualization and clustering based on dDDH and ANI values were created using Orange v3.23.1 (Demšar et al., 2013).
To determine the presence or absence and the copy number of genes related to carbon source assimilation, their profiles in the genomes of H. smithiae and its closest relatives were defined using the National Center for Biotechnology Information BLAST + v2.8.1 BLASTP and TBLASTX tools with query protein sequences from S. cerevisiae S288C (all proteins except Lac4 and Lac12) and Kluyveromyces marxianus DMKU3-1042 (Lac4 and Lac12) against subject databases built from the Hanseniaspora proteomes and transcriptomes. The E-value threshold was set to 10–10 to assign copy numbers of each gene as described by Haase et al. (2017).
Steenwyk et al. (2019) reported the presence and absence of S. cerevisiae homologs in Hanseniaspora genomes. The results are available from figshare: https://doi.org/10.6084/m9.figshare.7670756.v2. However, since the genomes of H. mollemarum and H. smithiae were sequenced later, we searched for the genes using a hidden Markov model, as described in detail by Steenwyk et al. (2019).
The matrix of present/absent orthologs of H. smithiae and five of its closest relatives was imported into Orange v3.23.1 (Demšar et al., 2013) for data mining, in which a Venn diagram was used to select the unique genes present in H. smithiae and its closest relatives. To determine in which functional categories these genes clustered, we conducted a functional grouping of genes based on gene ontology (GO) annotations (Ashburner et al., 2000; The Gene Ontology Consortium, 2019), using the “Yeast” subset on the Saccharomyces Genome Database (Cherry et al., 2012). To further visualize the species-specific GO annotations, we built a GO network using REViGO (Supek et al., 2011) with the following settings: medium list size (0.7) with SimRel semantic similarity measures to calculate the p-values.
Growth was followed manually under semi-aerobic conditions in shake flasks on a rotary shaker (Multitron, Infors) in triplicates. First, overnight cultures suspended in saline solution were inoculated to OD 0.1 in 50 ml YPD (Conda) in 250 ml shake flasks and cultivated on an orbital shaker at high intensity (220 rpm) at 28°C for 24 h. During the exponential growth phase, optical density measurements were taken manually at time intervals of 1–2 h at 600 nm on the Spark microplate reader (Tecan).
At the end of the exponential phase, 1 ml cell suspension was filtered through 0.2 μm filters for HPLC analysis to determine ethanol production and sugar consumption. Filtered samples were analyzed using an UltiMate 3000 HPLC system (Thermo Fisher) with 5 mM H2SO4 in water as the mobile phase at a flow rate of 0.6 ml/min. Relevant standards for quantification were prepared with ethanol (Merck, for analysis, ACS, ISO, Reag. Ph Eur, 100%) and glucose (anhydrous for biochemistry Reag. Ph Eur).
During an exploration of fermentative yeast diversity in the natural Andean forests in Patagonia, Argentina, from 2006 to 2014, we collected ca. 800 samples of bark, soil, and stromata of the parasitic Cyttaria species of fungi that are associated with Nothofagus species of trees (Libkind et al., 2007, 2011a,b; Almeida et al., 2014; Eizaguirre et al., 2018). Although Saccharomyces eubayanus and Saccharomyces uvarum prevailed among the fermentative yeasts, non-Saccharomyces species of Lachancea, Kregervanrija, Torulaspora, Zygosaccharomyces, Phaffia, and Hanseniaspora were occasionally isolated as well. For Hanseniaspora, two strains that were collected from stromata of Cyttaria were recently formally described as Hanseniaspora gamundiae (Čadež et al., 2019). Furthermore, identification based on sequence comparisons of the LSU D1/D2 gene and the ITS sequences revealed that an additional five apiculate strains from Cyttaria stromata and Nothofagus tree bark were genetically divergent, as they differed by 5 (0.9% sequence divergence) and 11 (1.7% sequence divergence) nucleotide substitutions, respectively, from their closest relative of H. valbyensis. As the guidelines of Kurtzman and Robnett, 1998, which suggest that 1% substitutions in the LSU D1/D2 domain represent separate species, have been found to differ for closely related species of this rapidly evolving genus (Cadez et al., 2003, 2006), we searched for genetic discontinuities that would indicate interruption of gene flow between the species (Lachance et al., 2010). Operationally, based on the concatenated sequences of the actin gene (ACT1) and the ITS and LSU D1/D2 regions of rRNA, we constructed a maximum parsimony network (Figure 1) of five strains isolated from Cyttaria, the type strain of H. lindneri, and 10 strains from different collections (CBS, NCAIM) and from recent surveys in South Africa that were previously identified as H. valbyensis. The network analysis revealed reticulation that originated from incongruence between single gene trees (Supplementary Figure 1) between nine of the 10 H. valbyensis strains, which likely indicates gene flow within H. valbyensis (Lee et al., 2020). However, the five strains from Cyttaria and strain CBS 2590 (isolated from English draft beer and identified as H. valbyensis based on DNA-DNA reassociation measurements; Meyer et al., 1978) showed clear genetic discontinuities from their closest relatives. Based on these data, to accommodate these strains, we propose the novel species Hanseniaspora smithiae sp. nov.
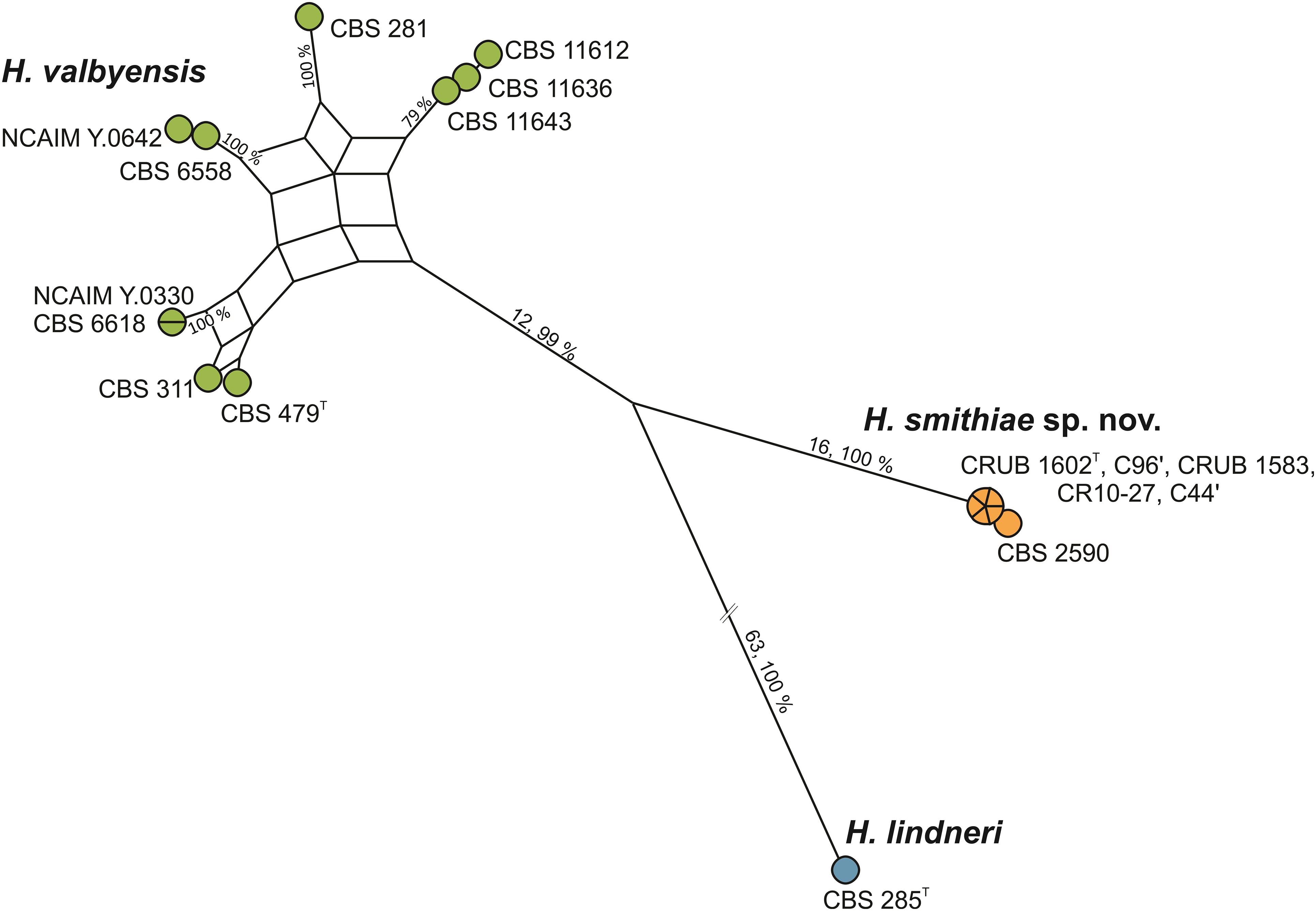
Figure 1. Maximum parsimony network for H. smithiae (6 strains), H. valbyensis (9 strains), and H. lindneri (one strain) based on the concatenated sequences of the actin gene and of the ITS and D1/D2 regions of rRNA (2,190 bp). Boxes (nodes subtended by multiple edges) indicate incongruence in the data. The numbers on the branches show the number of nucleotide substitutions and the bootstrap values (%).
To phylogenetically place H. smithiae within the genus, we selected the type or representative strains from each species of Hanseniaspora that had publicly available genome sequences (Riley et al., 2016; Steenwyk et al., 2019). The genomes of the type strains of the recently described species H. mollemarum (Groenewald et al., 2018) and of H. lindneri were sequenced in this study. Phylogenetic reconstruction was based on concatenated protein sequences of 251 conserved single-copy BUSCO orthologs, which yielded a tree fully consistent with the dichotomous tree of slower- and faster-evolving lineages (SEL and FEL, respectively) of Steenwyk et al. (2019) and placed H. smithiae within the faster-evolving lineage and sister to H. valbyensis (Figure 2).
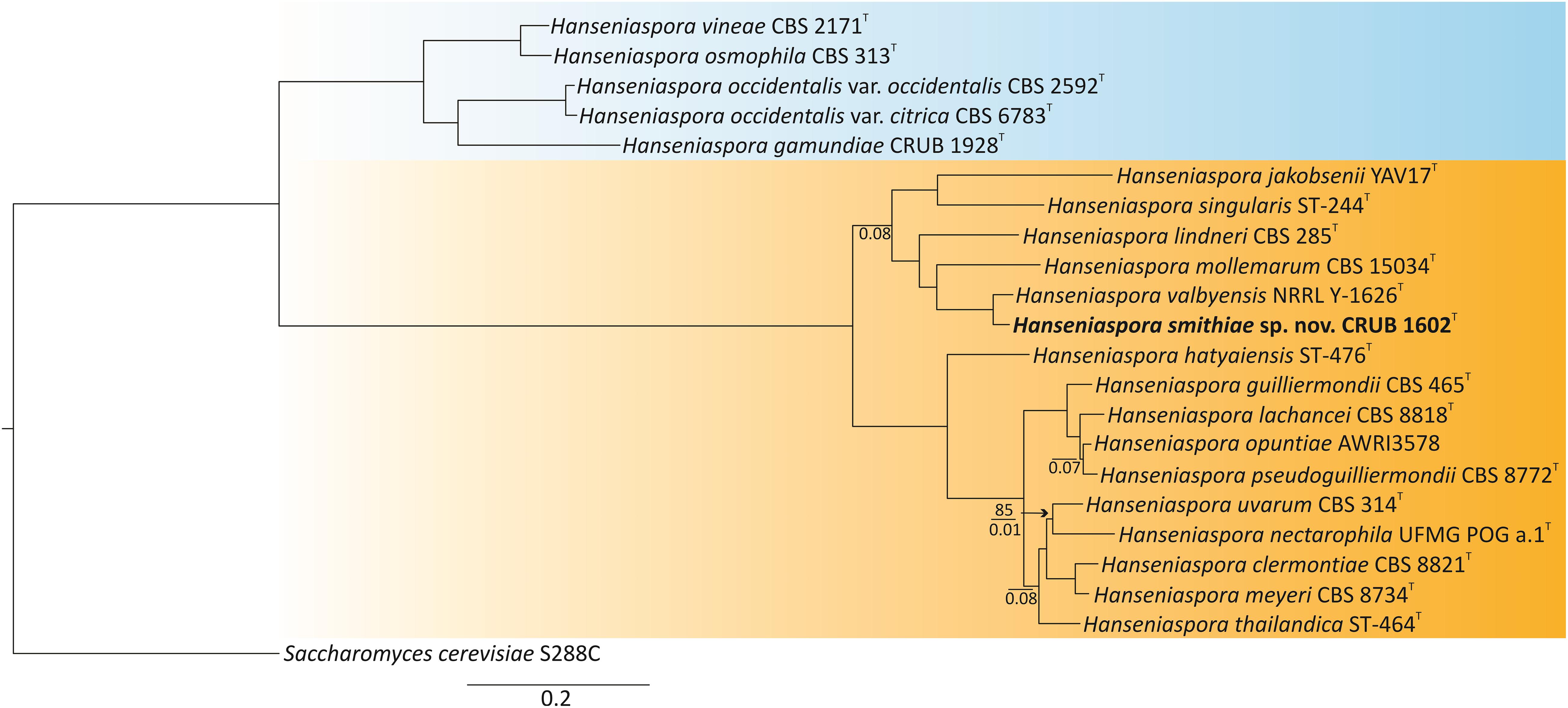
Figure 2. Phylogenomic tree showing the placement of the novel species Hanseniaspora smithiae sp. nov. CRUB 1602T. The ML tree was constructed based on a concatenated alignment of 251 orthologous, single-copy BUSCO proteins. Bootstrap values are shown only on branches where it is lower than 100% (above the line), while internode certainty (IC) values below 0.1 are shown below the line. Slower-evolving (SEL) and faster-evolving (FEL) lineages, according to Steenwyk et al. (2019), are colored in blue and orange, respectively. Saccharomyces cerevisiae was used as the outgroup.
Before genomic sequences of the type strains were available, the phylogeny of the closely related Hanseniaspora species of the FEL was poorly resolved, which was evident from the low bootstrap values (Čadež et al., 2019). However, because concatenation of a large number of genes nearly always produces a phylogeny with high bootstrap support, as shown in Figure 2, Salichos and Rokas (2013) suggested the use of internode certainty (IC) as an alternative measure to quantify congruence between the genes from which the phylogeny is reconstructed. All of the internodes had IC values > 0.1, which indicates that the majority of the individual gene tree topologies support the concatenated phylogeny. However, four out of 18 internodes had IC values < 0.1, which suggested considerable conflict among the gene trees. These conflicts were mostly concerned with the placement of Hanseniaspora nectarophila as sister to H. uvarum and within the species complex of Hanseniaspora pseudoguilliermondii, Hanseniaspora opunitae, and Hanseniaspora lachancei. The challenges in resolving this species complex may be because a lineage split into three descendant lineages at approximately the same time. Thus, the phylogenetic reconstruction of the species tree appears to be very difficult in this genus, which has highly variable rates of nucleotide substitution (Cadez et al., 2006; Steenwyk et al., 2019).
The power of genomic data also lies in their use as tools that can calculate genome-wide genetic distances between a novel taxon and its closest relatives (Libkind et al., 2020b). For estimation of genetic distances between H. smithiae and its closest relatives (Figure 3), we used two tools that are available as web interfaces: the ANI calculator and the Genome-to-Genome Distance Calculator. The overall genome relatedness indices (OGRI) of the ANI and dDDH values of 87 and 31%, respectively, both confirmed the close relationship between H. smithiae and H. valbyensis, which is higher than between any of the other species of the H. valbyensis clade (ranges: 78–79% and 23–27%, respectively). To calibrate the utilities of the ANI and dDDH indices for delimiting species of a yeast genus for which an operational species concept was based on DNA reassociation values (Meyer et al., 1978), we arranged both types of values into distance matrices to reconstruct trees representing genomic relatedness within the genus (Supplementary Figures 2A,B). In contrast to dDDH, the ANI tree represents phylogenetic relationships between species very well, as also observed by Lachance et al. (2020) for the genus Metschnikowia. In general, we can divide the ANI -values into three categories: (1) between FEL and SEL (Figure 2), the values were below 77; (2) interspecific values within both lineages were between 77.3 and 81.1; and (3) between closely related species and varieties, values were between 83.2 and 91.8. In view of these results, we support the proposal of Libkind et al. (2020b) and Lachance et al. (2020) that OGRI values, preferably the ANI, should accompany any formal yeast species description.
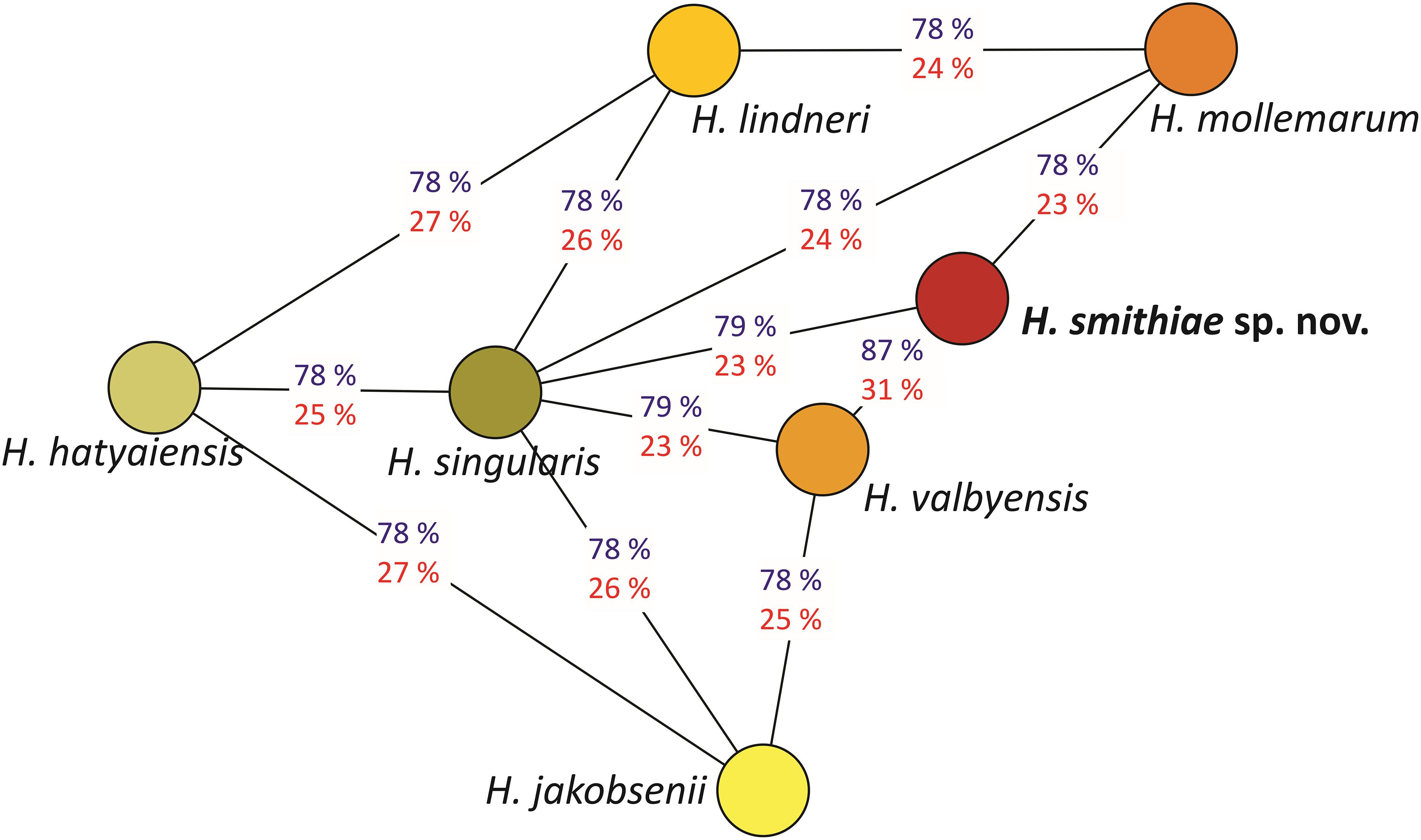
Figure 3. Genomic relationships between species closely related to Hanseniaspora smithiae sp. nov., based on the average nucleotide identity (ANI; blue) and the digital DNA-DNA homology values (red). The network was inferred using the minimum spanning tree algorithm on the ANI values.
One of the main forces in the evolution of Hanseniaspora species has been their widespread loss of genes (Steenwyk et al., 2019). Therefore, we examined which genes have been retained or lost during speciation for H. smithiae in comparison to its closest relatives, H. valbyensis, H. mollemarum, H. jakobsenii, and H. singularis. From the list of genes present in the H. smithiae, H. valbyensis, H. jakobsenii, and H. singularis genomes obtained from Steenwyk et al. (2019) and the list of genes in the H. mollemarum genome prepared during this study, we performed comparative analyses of gene gains and losses using Venn diagrams following previously published procedures (Supplementary Figures 3A,B). In general, during speciation, H. smithiae retained more genes than its closest relatives. For example, H. smithiae lost 24 genes, but it retained 120 genes lost by its closest relative, H. valbyensis. To determine whether this phenomenon was biased toward specific groups of genes (Albalat and Cañestro, 2016), we performed gene ontology (GO) enrichment analysis (Figure 4 and Supplementary Table 2). The genes lost by H. valbyensis compared to H. smithiae were those involved in processes, such as the electron transport chain and ATP synthesis, as well as the generation of precursor metabolites and energy. It appears that, after branching apart from of H. smithiae, H. valbyensis lost mitochondrial and other genes involved in respiration, which could explain differences in growth rate and ethanol production during aerobic growth on fermentable sugar-rich media.
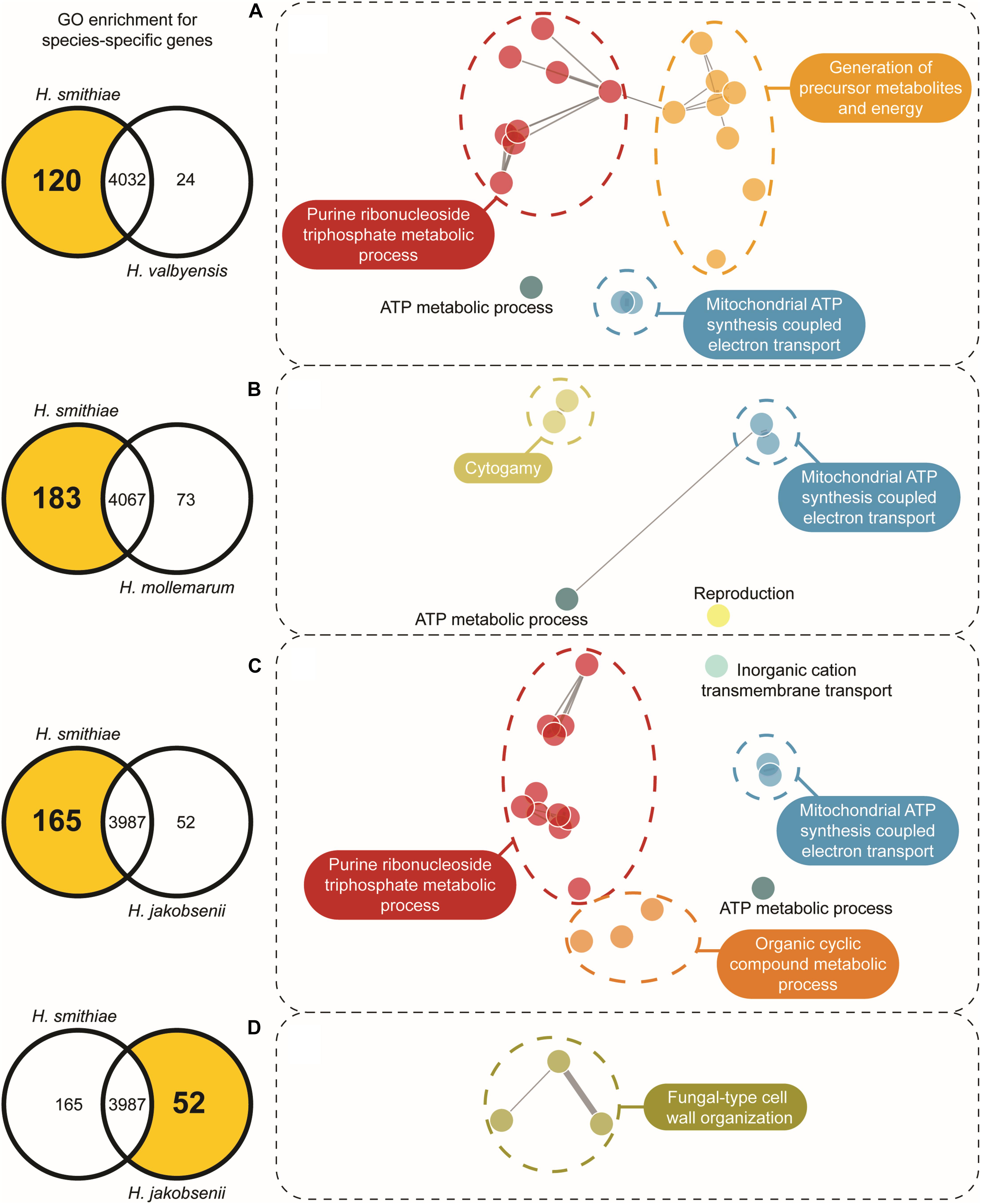
Figure 4. Gene ontology (GO) network based on enriched species-specific GO terms for biologic processes. The selected number of genes is indicated on the left side in the Venn diagram for each species comparison. For each highlighted zone of the Venn diagram in (A–D), the GO networks were constructed using REViGO with clustering based on shared ontology groups, indicated by their unique color.
Therefore, we compared the specific growth rates, doubling times, and ethanol yields per cell of the two type strains of the species in exponential growth phase in rich medium (YPD) under semi-aerobic conditions (Table 1). Notably, H. smithiae grew significantly faster and produced less ethanol per cell than H. valbyensis, which is consistent with an impairment of respiratory metabolism by the latter. A similar observation was reported by Merico et al. (2007), who noted the absence of re-oxidation of NADH at the mitochondrial level in H. valbyensis. The present data suggest that H. valbyensis adapted to fermentative environments, resulting in the loss of many genes associated with respiration.
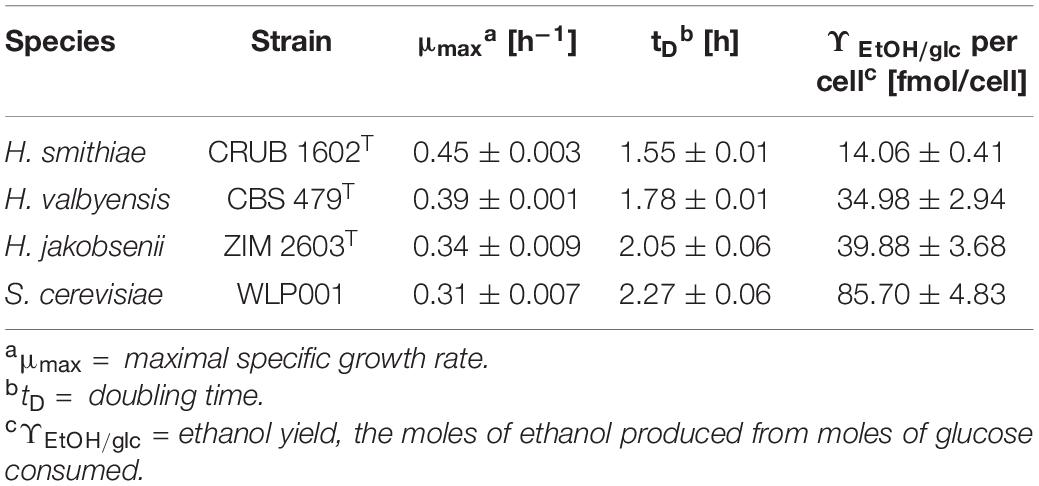
Table 1. Growth characteristics of H. smithiae, H. valbyensis, H. jakobsenii, and S. cerevisiae.
A similar but more profound loss was revealed by comparing the genes of H. smithiae and H. jakobsenii, a species isolated so far only from African palm wine in Burkina Faso (Ouoba et al., 2015). In comparison to H. smithiae, H. jakobsenii lost 95 genes associated with mitochondrial ATP synthesis and nuclear genes involved in the electron transport chain (Figure 4C). Similar to H. valbyensis, H. jakobsenii grew more slowly and produced higher amounts of ethanol when grown under aerobic conditions, suggesting that fermentative metabolism is a major source of energy for growth (Table 1). Furthermore, compared to H. jakobsenii (Figure 4D), the Patagonian species H. smithiae lost 10 genes involved in cell wall organization, most of which (7 of 10) belong to the PAU family, which are genes that are induced by anaerobiosis (Rachidi et al., 2000). These gene losses indicate a functional bias in the speciation of H. smithiae, which appears to be less adapted to man-made environments.
We also examined the gene loss in H. mollemarum compared to H. smithiae (i.e., 85 lost genes). H. mollemarum is a recently described new species from Europe that was isolated from garden soil in The Netherlands, home-made elderberry syrup in Hungary, and home-made cider in England (Groenewald et al., 2018). In H. mollemarum, genes involved in the reproductive process are significantly overrepresented in the GO categories of lost genes (Figure 4B and Supplementary Table 2). Specifically, after mapping these to KEGG pathways, we found that many are involved in the MAP kinase intracellular signal transduction pathway, which is involved in responses to pheromones (e.g., via FUS1) and/or is associated in processes that coordinate cell cycle (e.g., via BUB1, MAD3 and MCD1) and meiosis (e.g., via SPO22 and GIP1). In addition, the mating type locus gene MATα1 and a silenced mating cassette gene HMLα1 were lost in H. mollemarum. The absence of these genes is most likely correlated with the inability of H. mollemarum to sporulate (Groenewald et al., 2018). Since H. smithiae is a homothallic species that producing two to four hat-shaped ascospores per ascus on malt extract agar (Figure 5), the loss of genes involved in meiosis could be a possible mechanism of genetic isolation of H. mollemarum from its ancestors.
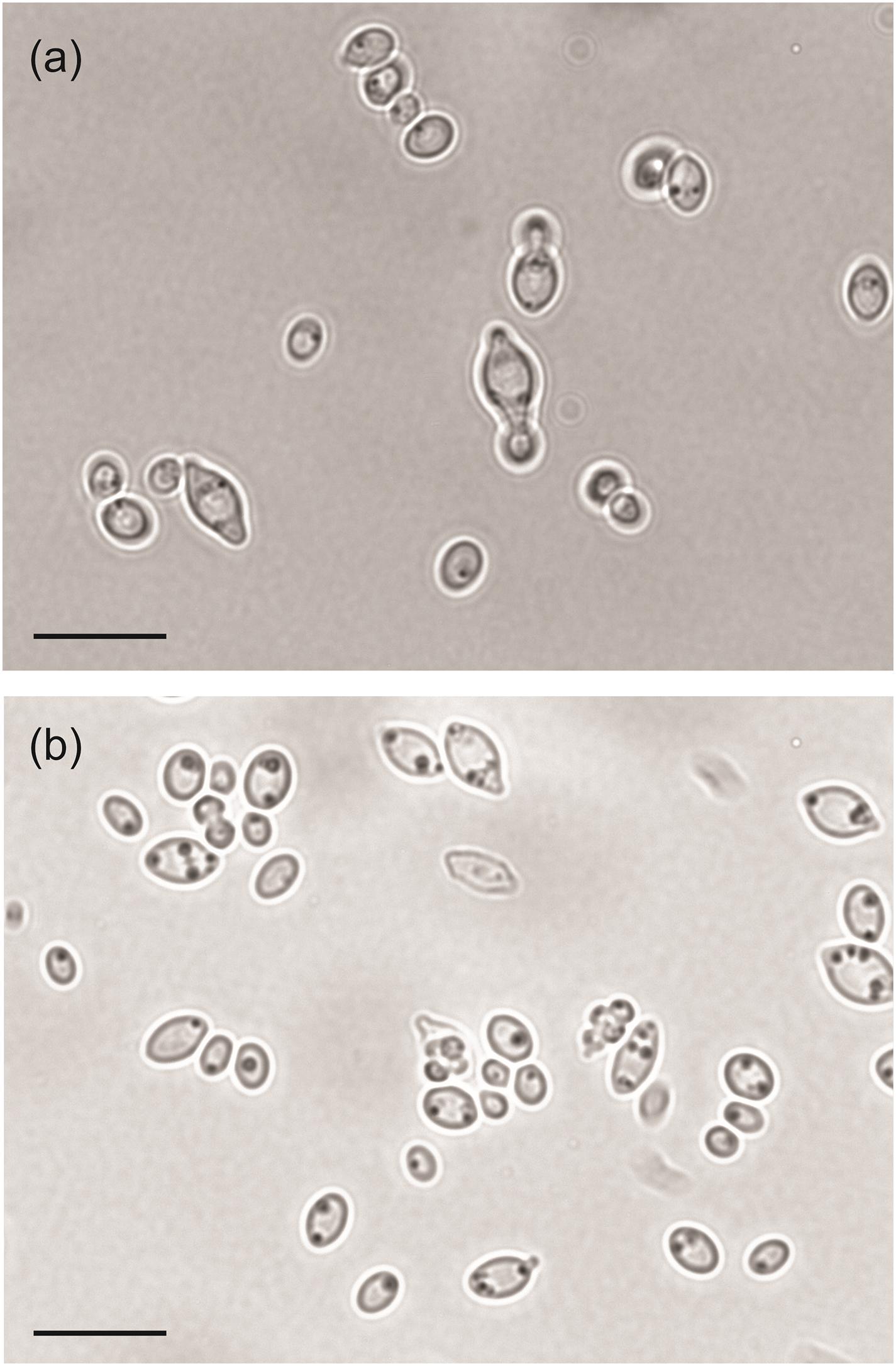
Figure 5. Microphotographs of Hanseniapora smithiae sp. nov. CRUB 1928T. (a) Budding cells; yeast–malt broth, 25°C, 220 rpm, 2 days. (b) Hat-to-helmet shaped ascospores released from the ascus; 5% malt extract agar, 14 days at 25°C. Scale bars, 10 μm.
Although it is particularly appealing to explain the process of speciation only by comparing the lists of genes that are present and absent between two species, the loss of genes is most likely not the only driver of their evolution. Nevertheless, most of the Hanseniaspora species are sexual diploids that can outcross, as shown recently by the discovery of interspecies hybrids isolated from various fruit in distinct regions (Saubin et al., 2020).
Five of the six strains of H. smithiae were associated with parasitic fungi of Cyttaria on the Nothofagus beech tree, where they share their ecological niche with other fermentative yeasts, such as S. uvarum, S. eubayanus (Libkind et al., 2011a; Almeida et al., 2014; Eizaguirre et al., 2018), and Phaffia rhodozyma (Libkind et al., 2007, 2011b). Phenotypically, H. smithiae differs from its closest relative, H. valbyensis, in its ability to assimilate trehalose, albeit slowly and weakly. Trehalose is one of the sugars in the stromata of Cyttaria (Waksman, 1977), and this sugar is also assimilated by other sympatric species, such as S. eubayanus, although this ability varies among strains (Libkind et al., 2011a). Thus, the ability to assimilate trehalose might be one of the phenotypic advantages that allowed H. smithiae to thrive in stromata as its primary habitat.
To better understand the genetic causes of yeast metabolic diversity, we searched the H. smithiae genome for candidate genes that underlie the physiological traits used for traditional taxonomic identification. We analyzed the genome content of H. smithiae and its closest relatives for genes required for the use of disaccharides, as well as for the use of raffinose and glucono-δ-lactone (Figure 6). As noted previously, since Hanseniaspora species assimilate only a limited number of carbon sources, they also lack the genes required for the use of many carbon sources (Riley et al., 2016; Čadež et al., 2019; Steenwyk et al., 2019). Two notable exceptions are enzymes accurately predicting the utilization of cellobiose (BGL2) and glucono-δ-lactone. Another exception is for the genes predicted to encode acid trehalase (ATH1) and neutral trehalase (NTH1), two enzymes that are required for the utilization of extracellular trehalose (Jules et al., 2004). However, these genes are also present in Hanseniaspora species that cannot assimilate trehalose as their sole carbon source. This discordance is an additional example that illustrates that our knowledge of the genotype–phenotype relationships of non-conventional yeasts remains limited (Libkind et al., 2020a).
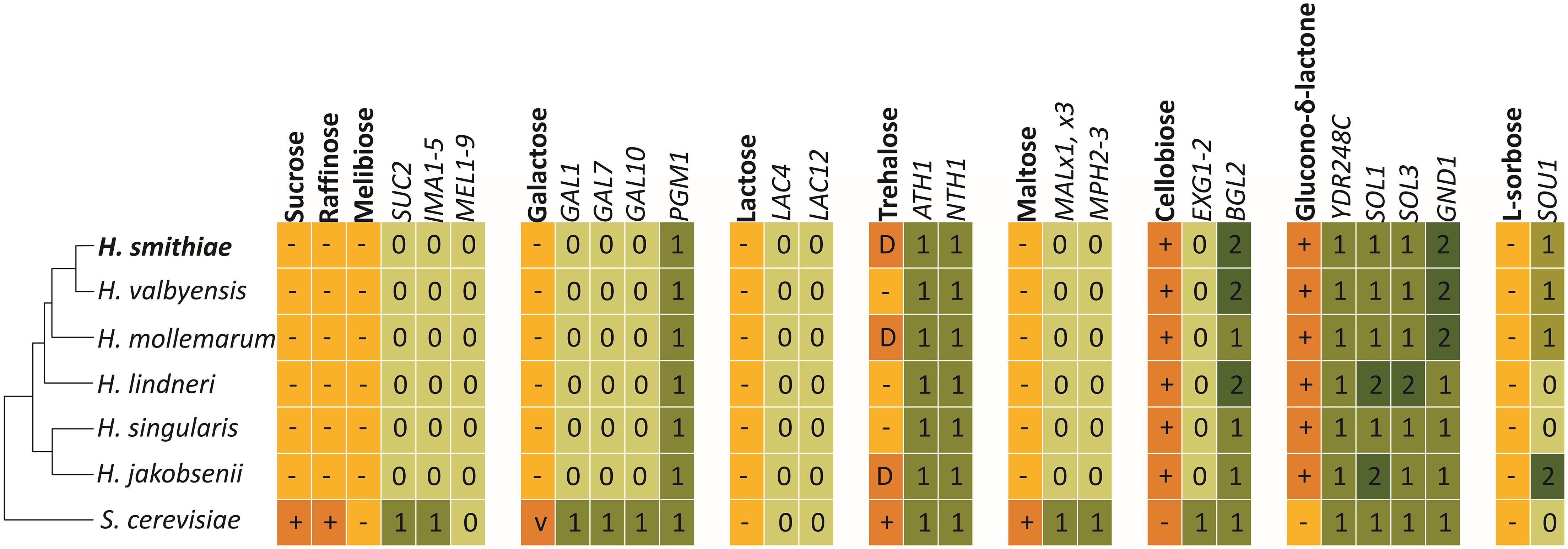
Figure 6. Metabolic traits and genes involved in the assimilation of carbon sources included in standard phenotypic characterization by Hanseniaspora smithiae, its closest relatives, and Saccharomyce cerevisiae (S288C). Numbered boxes indicate presence (1) or absence (0) of putative homologous genes, as determined using the BLASTP and TBLASTX tools.
In recognition of Dr. Maudy Th. Smith (Netherlands), for her valuable contributions to Hanseniaspora taxonomy.
In yeast extract–malt extract liquid medium after 48 h at 25°C, the cells are apiculate, ovoid, or elongate (2.0–7.5) μm × (1.2–3.2) μm, and they occur singly or in pairs. Budding is bipolar. Sediment is present, and a very thin ring is formed after 1 month. On yeast extract–malt extract agar after 1 month at 25°C, the steak culture is cream-colored, butyrous, smooth, glossy and flat to slightly raised at the center, with an entire margin. Asci contain two to four hat-shaped to helmet-shaped ascospores (1.4–4.3 μm), which are usually released at maturity (Figure 5). Released ascospores usually aggregate. They were observed after 2 weeks or more on 5% malt extract agar (Difco) at 25°C. Glucose is fermented; D-galactose, maltose, lactose, and cellobiose are not fermented. The carbon compounds that are assimilated are glucose, α,α-trehalose, cellobiose, salicin, arbutin, glucono-δ-lactone, and D-gluconate; no growth occurs on galactose, L-sorbose, D-glucosamine, N-acetyl-D-glucosamine, D-ribose, D-xylose, L-arabinose, D-arabinose, L-rhamnose, sucrose, maltose, methyl α-D-glucoside, melibiose, lactose, raffinose, melezitose, inulin, starch, glycerol, erythritol, ribitol, xylitol, L-arabinitol, D-glucitol, D-mannitol, galactitol, myo-inositol, 2-keto-D-gluconate, D-glucuronate, D-galacturonate, DL-lactate, succinate, citrate, methanol, ethanol, propane-1,2-diol, butane-2,3-diol, and hexadecane. Assimilation of nitrogen compounds is positive for ethylamine hydrochloride, lysine, and cadaverine, but it is negative for potassium nitrate, sodium nitrite, creatine, creatinine, glucosamine, and imidazole. Growth in vitamin-free medium is absent. Growth occurs at 30°C, but it is absent at 35°C. Growth with 10% NaCl is positive, but it is absent with 16% NaCl, on 50% (w/w) glucose–yeast extract agar, with 1% acetic acid, and with 0.01% cycloheximide. The diazonium blue B reaction is negative.
The holotype (CRUB 1602H) is permanently maintained in a metabolically inactive state at the Centro Regional Universitario Bariloche Yeast Culture Collection, Argentina, and the type strain was deposited in the same collection (CRUB 1602T), in the ARS Culture Collection, National Center for Agricultural Utilization Research, IL, United States (NRRL Y-63759T), in the Portuguese Yeast Culture Collection, Caparica, Portugal (PYCC 7263T), and in the Collection of Industrial Microorganisms, Slovenia (ZIM 2538T). The strain CRUB 1602T was isolated from Cyttaria hariotii stromata infecting Nothofagus antarctica in December 2006. The Mycobank number is MB839330. The BioProject number for raw genome sequencing reads is PRJNA529215 (BioSample SAMN11259523), and the GenBank accession number for the assembled genome is GCA_004919795.1 (Steenwyk et al., 2019).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.mycobank.org/, MB839330. The GenBank accession numbers are listed in Supplementary Table 1.
NČ, MT, DL, and CH conceived the study and wrote the manuscript. NČ, MT, and NB performed the experiments and analyzed the data. RU and MG isolated the cultures. NČ and MT prepared the figures. All authors have reviewed and edited the manuscript.
The work was supported by the Slovenian Research Agency (P4–0116, and MRIC-UL ZIM, IP-0510 to NČ). MT was supported by grant C3330-19-952047 funded by the Republic of Slovenia Ministry of Education, Science and Sport, and European Union European Regional Development Fund. DL was funded by CONICET (PIP11220130100392CO) and Universidad Nacional del Comahue (B199). CH was supported by the National Science Foundation under Grant No. DEB-1442148; in part by the Great Lakes Bioenergy Research Center, US. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018409; and the USDA National Institute of Food and Agriculture (Hatch Project 1020204). CH was a Pew Scholar in the Biomedical Sciences and a H. I. Romnes Faculty Fellow, supported by the Pew Charitable Trusts and Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation, respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Argentinean National Park Administration for sample collection permits, as well as Martina Podgoršek for DNA isolations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.679894/full#supplementary-material
Albalat, R., and Cañestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Almeida, P., Gonçalves, C., Teixeira, S., Libkind, D., Bontrager, M., Masneuf-Pomarède, I., et al. (2014). A Gondwanan imprint on the global diversity and domestication of the wine and cider yeast Saccharomyces uvarum. Nat. Commun. 5, 1–12. doi: 10.1038/ncomms5044
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Gene Ontol. Consort. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Becher, P. G., Hagman, A., Verschut, V., Chakraborty, A., Rozpêdowska, E., Lebreton, S., et al. (2018). Chemical signaling and insect attraction is a conserved trait in yeasts. Ecol. Evol. 8, 2962–2974. doi: 10.1002/ece3.3905
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burini, J., Eizaguirre, J. I., Loviso, C., and Libkind, D. (2021). Non-conventional yeasts as tools for innovation and differentiation in brewing. Rev. Argent. Microbiol. 2021:3. doi: 10.1016/j.ram.2021.01.003
Čadež, N., Bellora, N., Ulloa, R., Hittinger, C. T., and Libkind, D. (2019). Genomic content of a novel yeast species Hanseniaspora gamundiae sp. nov. from fungal stromata (Cyttaria) associated with a unique fermented beverage in Andean Patagonia, Argentina. PLoS One 14:e0210792. doi: 10.1371/journal.pone.0210792
Cadez, N., Poot, G. A., Raspor, P., and Smith, M. T. (2003). Hanseniaspora meyeri sp. nov., Hanseniaspora clermontiae sp. nov., Hanseniaspora lachancei sp. nov. and Hanseniaspora opuntiae sp. nov., novel apiculate yeast species. Int. J. Syst. Evol. Microbiol. 53, 1671–1680. doi: 10.1099/ijs.0.02618-0
Cadez, N., Raspor, P., and Smith, M. T. (2006). Phylogenetic placement of Hanseniaspora-Kloeckera species using multigene sequence analysis with taxonomic implications: descriptions of Hanseniaspora pseudoguilliermondii sp. nov. and Hanseniaspora occidentalis var. citrica var. nov. Int. J. Syst. Evol. Microbiol. 56, 1157–1165. doi: 10.1099/ijs.0.64052-0
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp34
Cherry, J. M., Hong, E. L., Amundsen, C., Balakrishnan, R., Binkley, G., Chan, E. T., et al. (2012). Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–D705. doi: 10.1093/nar/gkr1029
Clement, M., Posada, D., and Crandall, K. A. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Demšar, J., Curk, T., Erjavec, A., Gorup, È, Hoèevar, T., Milutinović, M., et al. (2013). Orange : data mining toolbox in Python. J. Mach. Learn. Res. 14, 2349–2353.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eizaguirre, J. I., Peris, D., Rodriguez, M. E., Lopes, C. A., De Los Rios, P., Hittinger, C. T., et al. (2018). Phylogeography of the wild Lager-brewing ancestor (Saccharomyces eubayanus) in Patagonia. Environ. Microbiol. 20, 3732–3743. doi: 10.1111/1462-2920.14375
Felsenstein, J. (1985). Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Giraud, A., Matic, I., Tenaillon, O., Clara, A., Radman, M., Fons, M., et al. (2001). Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291, 2606–2608. doi: 10.1126/science.1056421
Groenewald, M., Lombard, L., de Vries, M., Lopez, A. G., Smith, M., and Crous, P. W. (2018). Diversity of yeast species from Dutch garden soil and the description of six novel Ascomycetes. FEMS Yeast Res. 18:foy076. doi: 10.1093/femsyr/foy076
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haase, M. A. B., Kominek, J., Langdon, Q. K., Kurtzman, C. P., and Hittinger, C. T. (2017). Genome sequence and physiological analysis of Yamadazyma laniorum f.a. sp. nov. and a re-evaluation of the apocryphal xylose fermentation of its sister species, Candida tenuis. FEMS Yeast Res. 7:fox019. doi: 10.1093/femsyr/fox019
Hamby, K. A., Hernández, A., Boundy-Mills, K., and Zalom, F. G. (2012). Associations of yeasts with spotted-wing Drosophila (Drosophila suzukii; Diptera: Drosophilidae) in cherries and raspberries. Appl. Envir. Microbiol. 78, 4869–4873. doi: 10.1128/AEM.00841-12
Healey, K. R., Zhao, Y., Perez, W. B., Lockhart, S. R., Sobel, J. D., Farmakiotis, D., et al. (2016). Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 7:11128. doi: 10.1038/ncomms11128
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome- database management tool for second-generation genome projects. BMC Bioinform. 12:491. doi: 10.1186/1471-2105-12-491
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jules, M., Guillou, V., François, J., and Parrou, J. L. (2004). Two distinct pathways for trehalose assimilation in the yeast Saccharomyces cerevisiae. Appl. Environ. Microbiol. 70, 2771–2778. doi: 10.1128/AEM.70.5.2771-2778.2004
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Haeseler von, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtzman, C. P., Fell, J. W., and Boekhout, T. (2011). “Methods for isolation, phenotypic characterization and maintenance of yeasts,” in The Yeasts, a Taxonomic Study, 5 Edn, eds C. P. Kurtzman, J. W. Fell, and T. Boekhout (Amsterdam: Elsevier), 87–110.
Kurtzman, C. P., and Robnett, C. J. (1998). Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie van Leeuwenhoek 73, 331–371. doi: 10.1023/a:1001761008817
Lachance, M.-A., Dobson, J., Wijayanayaka, D. N., and Smith, A. M. (2010). The use of parsimony network analysis for the formal delineation of phylogenetic species of yeasts: Candida apicola, Candida azyma, and Candida parazyma sp. nov., cosmopolitan yeasts associated with floricolous insects. Antonie van Leeuwenhoek 97, 155–170. doi: 10.1007/s10482-009-9399-3
Lachance, M.-A., Lee, D. K., and Hsiang, T. (2020). Delineating yeast species with genome average nucleotide identity: a calibration of ANI with haplontic, heterothallic Metschnikowia species. Antonie van Leeuwenhoek 113, 2097–2106. doi: 10.1007/s10482-020-01480-9
Lee, D. K., Santos, A. R. O., Hsiang, T., Rosa, C. A., and Lachance, M. A. (2020). Catching speciation in the act-act 2: Metschnikowia lacustris sp. nov., a sister species to Metschnikowia dekortorum. Antonie van Leeuwenhoek 113, 753–762. doi: 10.1007/s10482-020-01395-5
Libkind, D., Hittinger, C. T., Valério, E., Gonçalves, C., Dover, J., Johnston, M., et al. (2011a). Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc. Natl. Acad. Sci. USA 108, 14539–14544. doi: 10.1073/pnas.1105430108
Libkind, D., Peris, D., Cubillos, F. A., Steenwyk, J. L., Opulente, D. A., Langdon, Q. K., et al. (2020a). Into the wild: new yeast genomes from natural environments and new tools for their analysis. FEMS Yeast Res. 20:foaa008. doi: 10.1093/femsyr/foaa008
Libkind, D., Čadež, N., Opulente, D. A., Langdon, Q. K., Rosa, C. A., Sampaio, J. P., et al. (2020b). Towards yeast taxogenomics: lessons from description of novel species based on complete genome sequences. FEMS Yeast Res. 20:foaa042. doi: 10.1093/femsyr/foaa042
Libkind, D., Ruffini, A., van Broock, M., Alves, L., and Sampaio, J. P. (2007). Biogeography, host-specificity, and molecular phylogeny of Phaffia rhodozyma and its sexual form, Xanthophyllomyces dendrorhous. Appl. Environ. Microbiol. 73, 1120–1125. doi: 10.1128/AEM.01432-06
Libkind, D., Tognetti, C., Ruffini, A., Sampaio, J. P., and van Broock, M. R. (2011b). Xanthophyllomyces dendrorhous (Phaffia rhodozyma) on stromata of Cyttaria hariotii in Patagonian Nothofagus forests. Rev. Argent. Microbiol. 43, 226–232. doi: 10.1590/S0325-75412011000300011
Liu, Z., Wang, M. M., Wang, G. S., Li, A. H., Wangmu, Wang, Q. M., et al. (2021). Hanseniaspora terricola sp. nov., an ascomycetous yeast isolated from Tibet. Int. J. Syst. Evol. Microbiol. 71:4741. doi: 10.1099/ijsem.0.004741
Martin, V., Jose Valera, M., Medina, K., Boido, E., and Carrau, F. (2018). Oenological impact of the Hanseniaspora/Kloeckera yeast genus on wines – a review. Fermentation 4:76. doi: 10.3390/fermentation4030076
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Merico, A., Sulo, P., Piškur, J., and Compagno, C. (2007). Fermentative lifestyle in yeasts belonging to the Saccharomyces complex. FEBS J. 274, 976–989. doi: 10.1111/j.1742-4658.2007.05645.x
Meyer, S. A., Smith, M. T., and Simione, F. P. (1978). Systematics of Hanseniaspora Zikes and Kloeckera Janke. Antonie van Leeuwenhoek 44, 79–96. doi: 10.1007/Bf00400078
Ouoba, L. I., Nielsen, D. S., Anyogu, A., Kando, C., Diawara, B., Jespersen, L., et al. (2015). Hanseniaspora jakobsenii sp. nov., a yeast isolated from Bandji, a traditional palm wine of Borassus akeassii. Int. J. Syst. Evol. Microbiol. 65, 3576–3579. doi: 10.1099/ijsem.0.000461
Rachidi, N., Martinez, M. J., Barre, P., and Blondin, B. (2000). Saccharomyces cerevisiae PAU genes are induced by anaerobiosis. Mol. Microbiol. 35, 1421–1430. doi: 10.1046/j.1365-2958.2000.01807.x
Riley, R., Haridas, S., Wolfe, K. H., Lopes, M. R., Hittinger, C. T., Göker, M., et al. (2016). Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA 113, 9882–9887. doi: 10.1073/pnas.1603941113
Salichos, L., and Rokas, A. (2013). Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497, 327–331. doi: 10.1038/nature12130
Salichos, L., Stamatakis, A., and Rokas, A. (2014). Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 31, 1261–1271. doi: 10.1093/molbev/msu061
Sampaio, J. P., and Gonçalves, P. (2008). Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl. Environ. Microbiol. 74, 2144–2152. doi: 10.1128/AEM.02396-07
Saubin, M., Devillers, H., Proust, L., Brier, C., Grondin, C., Pradal, M., et al. (2020). Investigation of genetic relationships between Hanseniaspora species found in grape musts revealed interspecific hybrids with dynamic genome structures. Front. Microbiol. 10:2960. doi: 10.3389/fmicb.2019.02960
Schwartz, K., and Sherlock, G. (2016). Preparation of yeast DNA sequencing libraries. Cold Spring Harb Protoc. 2016:88930. doi: 10.1101/pdb.prot088930
Seehausen, O., Butlin, R. K., Keller, I., Wagner, C. E., Boughman, J. W., Hohenlohe, P. A., et al. (2014). Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192. doi: 10.1038/nrg3644
Shen, X.-X., Opulente, D. A., Kominek, J., Zhou, X., Steenwyk, J. L., Buh, K. V., et al. (2018). Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175, 1533–1545. doi: 10.1016/j.cell.2018.10.023
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Steensels, J., and Verstrepen, K. J. (2014). Taming wild yeast: potential of conventional and nonconventional yeasts in industrial fermentations. Annu. Rev. Microbiol. 68, 61–80. doi: 10.1146/annurev-micro-091213-113025
Steenwyk, J. L., Opulente, D. A., Kominek, J., Shen, X.-X., Zhou, X., Labella, A. L., et al. (2019). Extensive loss of cell cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. Plos Biol. 17:e3000255. doi: 10.1371/journal.pbio.3000255
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
The Gene Ontology Consortium. (2019). The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
Waksman, N. H. (1977). Hidratos de carbono en Cyttaria hariotii Fischer: Determinación de la estructura de un Pululano. Estudio de azúcares simples, aislamiento e identificación de ácido D-arabino hexulosónico y alfa, alfa-D-trehalosa. Buenos Aires: Facultad de Ciencias Exactas y Naturales, UBA.
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Yoon, S. H., Ha, S. M., Lim, J. M., Kwon, S. J., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhou, X. F., Peris, D., Kominek, J., Kurtzman, C. P., Hittinger, C. T., and Rokas, A. (2016). In silico Whole Genome Sequencer and Analyzer (iWGS): a computational pipeline to guide the design and analysis of de novo genome sequencing studies. G3-Genes Genom. Genet. 6, 3655–3662. doi: 10.1534/g3.116.034249
Keywords: apiculate yeast, new species, phylogenomics, OGRI, gene loss, speciation, domestication
Citation: Čadež N, Bellora N, Ulloa R, Tome M, Petković H, Groenewald M, Hittinger CT and Libkind D (2021) Hanseniaspora smithiae sp. nov., a Novel Apiculate Yeast Species From Patagonian Forests That Lacks the Typical Genomic Domestication Signatures for Fermentative Environments. Front. Microbiol. 12:679894. doi: 10.3389/fmicb.2021.679894
Received: 12 March 2021; Accepted: 28 June 2021;
Published: 21 July 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesCopyright © 2021 Čadež, Bellora, Ulloa, Tome, Petković, Groenewald, Hittinger and Libkind. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neža Čadež, bmV6YS5jYWRlekBiZi51bmktbGouc2k=; Diego Libkind, bGlia2luZGZkQGNvbWFodWUtY29uaWNldC5nb2IuYXI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.