 Xu Cheng
Xu Cheng Yitong Lu
Yitong Lu Haixia Liu
Haixia Liu- College of Animal Science and Technology, Northwest A&F University, Xianyang, China
The excessive use of antibiotics speeds up the dissemination and aggregation of antibiotic resistance genes (ARGs) in the environment. The ARGs have been regarded as a contaminant of serious environmental threats on a global scale. The constant increase in aquaculture production has led to extensive use of antibiotics as a means to prevent and treat bacterial infections; there is a universal concern about the environmental risk of ARGs in the aquaculture environment. In this study, a survey was conducted to evaluate the abundance and distributions of 10 ARGs, bacterial community, and environmental factors in sediment samples from aquatic farms distributed in Anhui (AP1, AP2, and AP3), Fujian (FP1, FP2, and FP3), Guangxi (GP1, GP2, and GP3), Hainan (HP1, HP2, and HP3), and Shaanxi (SP1, SP2, and SP3) Province in China. The results showed that the relative abundance of total ARGs was higher in AP1, AP2, AP3, FP3, GP3, HP1, HP2, and HP3 than that in FP1, FP2, GP1, GP2, SP1, SP2, and SP3. The sul1 and tetW genes of all sediment samples had the highest abundance. The class 1 integron (intl1) was detected in all samples, and the result of Pearson correlation analysis showed that the intl1 has a positive correlation with the sul1, sul2, sul3, blaOXA, qnrS, tetM, tetQ, and tetW genes. Correlation analysis of the bacterial community diversity and environmental factors showed that the Ca2+ concentration has a negative correlation with richness and diversity of the bacterial community in these samples. Of the identified bacterial community, Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidota were the predominant phyla in these samples. Redundancy analysis showed that environmental factors (TN, TP, Cl–, and Ca2+) have a positive correlation with the bacterial community (AP1, GP1, GP2, GP3, SP1, SP2, and SP3), and the abundance of ARGs (sul1, tetW, qnrS, and intl1) has a positive correlation with the bacterial community (AP2, AP3, HP1, HP2, and HP3). Based on the network analysis, the ARGs (sul1, sul2, blaCMY, blaOXA, qnrS, tetW, tetQ, tetM, and intl1) were found to co-occur with bacterial taxa from the phyla Chloroflexi, Euryarchaeota, Firmicutes, Halobacterota, and Proteobacteria. In conclusion, this study provides an important reference for understanding the environmental risk associated with aquaculture activities in China.
Introduction
Antibiotics are extensively used to prevent and control bacterial infections in medical care, livestock husbandry, and aquaculture (Kümmerer, 2009; Luo et al., 2010). Some of them are also used as growth promoters in aquaculture activities (Chen H. et al., 2018). However, excessive antibiotics and their metabolites would enter the environment, and they might be further absorbed into soil particles and eventually accumulated in sediments because aquatic animals cannot take full advantage of these antibiotics (Kümmerer, 2009). It is worth noting that the abundance of antibiotic resistance genes (ARGs) in soil was associated with the amount of antibiotic residues in the environment (Fahrenfeld et al., 2014), and the ARGs combining with minerals and humus from the environment might exist for a long time (Dang et al., 2017; Hurst et al., 2019; Ma et al., 2019). It is well known that the ARGs have unique biological characteristics, and they could spread by horizontal gene transfer among bacteria of different species and self-amplify among the same species (Guo et al., 2017; Kumar et al., 2017).
The sediments were regarded as an important plot for accumulation and transmission of ARGs (Marti et al., 2014). Shen et al. (2020) reported that several ARGs (sul1, tetG, tetW, tetX, and intl1 gene) were detected in water and sediment of aquaculture farms in Jiangsu Province, China. Chen B. et al. (2018) explored the ARGs in the sediments from bullfrog farms and confirmed that these identified ARGs were able to encode resistance to over 10 categories of antibiotics, such as aminoglycosides, beta-lactams, chloramphenicols, fluoroquinolones, macrolides, polypeptides, sulfonamides, and tetracyclines.
There is a universal concern that the presence of ARGs in sediments is a potential environmental threat (Wang et al., 2015). The antibiotic-resistant bacteria have constituted a huge repository of ARGs in sediments (Martínez, 2008). Once these ARGs have been transferred into the human symbiotic microbes, they would cause great risks of the ecological environment and human health (Smillie et al., 2011; Forsberg et al., 2012). Currently, a study about ARGs in the environment suggested that the existing forms of ARGs largely determine the ways in which these genes are acquired and disseminated among bacterial hosts (Mao et al., 2014). The conception of integron was first proposed by Stokes in 1989 (Stokes and Hall, 1989). It is a key pathway for bacteria to acquire ARGs, which influenced the removal and transfer of ARGs in the bacterial community (Gaze et al., 2011). As one of the most important mobile genetic materials, the integron could capture, rearrange, and express mobile gene cassettes responsible for the spread of ARGs (Zhang X. et al., 2020) and further accelerate the prevalence and transmission of ARGs in the environment (Martinez-Freijo et al., 1998; Cambray et al., 2010).
Previous studies have suggested that the nutrients also promote directly or indirectly the ARGs propagation (Zhao et al., 2017; Zhang J. et al., 2020). Furthermore, the long-time input of nitrogen and phosphorus not only changed the composition of the bacterial community but also drove the propagation of ARGs (Pan et al., 2020). Total organic carbon (TOC) and total dissolved nitrogen (TDN) were potentially important environmental factors, which affected the abundance and diversity of ARGs in urban river systems (Zhou et al., 2017). Moreover, some researches have indicated that bacterial communities shaped the distribution and abundance of ARGs (Huerta et al., 2013; Xiong et al., 2015). These findings suggested that the distribution and prevalence of ARGs are not only related to the use of antibiotics but also affected by many environmental factors. In this study, we aimed to (1) evaluate the relative abundance of 10 ARGs in sediment samples from different aquaculture farms; (2) elucidate the correlation between environmental factors, ARGs abundance, and bacterial community in different aquaculture farms; (3) identify the co-occurrence patterns between ARGs and bacterial taxa.
Materials and Methods
Sample Collection
A total of 15 sediment samples were collected from aquaculture ponds distributed in five Chinese provinces, including Anhui (Wuwei, Freshwater aquaculture farm), Fujian (Zhangzhou, Mariculture farm), Guangxi (Qinzhou, Mariculture farm), Hainan (Haikou, Freshwater aquaculture farm), and Shaanxi (Heyang, Freshwater aquaculture farm) between September and October 2019 (Supplementary Figure 1). Three ponds were selected in every aquaculture farm. These aquaculture ponds could produce aquatic products with average 4,000 kg or more per year. Due to the high stocking density, different antibiotics, including sulfonamides, tetracyclines, beta-lactams, and quinolones were used for prophylactic purposes on these farms. No bacterial infections occurred in the sampled ponds in the past year according to our investigation.
Each pond has an area of approximately 900–1,200 m2 with a depth of approximately 150–200 cm. The sediments of all sampled ponds have not been cleaned for at least 1 year to ensure that the samples meet the requirements. The samples were collected from water inlets, water outlets, and center areas of each pond (collected the top 10 cm of the sediment) using the CN-100 bottom sampler (Ruibin, China), and the samples of each pond were completely mixed to avoid heterogeneous differences caused by single sampling. After mixing, the sample was sealed in a sterile plastic bag and transported at 4°C to the laboratory. All the samples were divided into two parts and stored at −80°C for further analysis.
DNA Extraction and Qualitative PCR of Antibiotic Resistance Genes
The genomic DNA was extracted from 0.25 g lyophilized sediment samples using the TIANamp Soil DNA kit (Tiangen, China). All the operations were performed with the product instructions. The quality of DNA was detected using Ultramicro nucleic acid analyzer (Allsheng, China). The PCR amplification was performed to test 10 ARGs (sul1, sul2, sul3, tetM, tetQ, tetW, qnrB, qnrS, blaOXA, and blaCMY) and class 1 integron integrase gene (intl1) based on the investigation of antibiotic use in the aquaculture farms of this study. The primers of target genes were synthesized by Sangon Biotech (Shanghai, China), and the primer sequences are shown in Supplementary Tables 1, 2. The PCR conditions were as follows: pre-denaturation at 95°C for 3 min, then 35 cycles of denaturation at 95°C for 30 s, annealing at the specified temperature (Supplementary Table 1) for 30 s, extension at 72°C for 45 s, and a final extension at 72°C for 5 min. PCR products were subsequently detected by agarose gel electrophoresis analysis, and the results are shown in Supplementary Figure 2.
High-Throughput Sequencing
In order to further analyze the bacterial community composition in the sediment samples, the high-throughput sequencing of bacterial community was analyzed with an Illumina Mi Seq platform at Novogene (Beijing, China). The V3–V4 regions of bacterial 16S rRNA genes were amplified using the primer pair 341F and 806R. The sequencing analysis was processed using QIIME software for 16S rRNA datasets described in a previous literature (Caporaso et al., 2010).
Quantification of Antibiotic Resistance Genes
PCR products of the target gene were purified with the Universal DNA Purification Kit (Tiangen, China) and ligated into pGM-T vector (Tiangen, China). Subsequently, the pGEM-T vector carrying target gene was transformed into Escherichia coli DH5α (Tiangen, China), and the positive clones were acquired after PCR amplification and sequence analysis. Moreover, the recombinant plasmids with target gene were extracted with TIANprep Mini Plasmid Kit (Tiangen, China) and searched for homolog identity with NCBI Blast program. The concentration of recombinant plasmids was checked by Ultramicro nucleic acid analyzer (Allsheng, China), and the standard curves of recombinant plasmids were built with 10-fold serial diluted. The amplification efficiency of all primers ranged from 91.07 to 106.64% with R2 > 0.99 (Supplementary Table 3). The target gene copy numbers of the sediment samples were calculated with the CT values according to a previous study (Yuan et al., 2019).
The real-time quantitative PCR (Q-PCR) was performed on a LightCycler® 96 instrument (Roche Ltd., Italy) by utilizing SYBR® Green Pro Taq HS Premix (AG, China) according to the manufacturer’s protocol for further analysis. The 10-μl qPCR reaction system contained 2 × SYBR® Green Pro Taq HS Premix (5 μl), 10 μM primer (0.2 μl for each primer, Sangon Biotech, China), RNase-free water (4.1 μl), and DNA samples or standard plasmid (0.5 μl). The amplification condition of qPCR was as follows: initial enzyme activation at 95°C for 30 s, then 40 cycles of at 95°C for 5 s and at 60°C for 30 s.
Analysis of Environmental Factors
The contents of Ca2+, Mg2+, and Cl– were determined by ethylene diamine tetraacetic acid (EDTA) volumetric method (Wang et al., 2020). The concentrations of total nitrogen (TN) and total phosphorus (TP) were determined by spectrophotometric method (SEPA, 2002; Trolle et al., 2009), and the standard curve of TN and TP is shown in Supplementary Table 4.
Statistical Analysis
Pearson correlation analysis was used to analyze the correlation between environmental factors (TN, TP, Cl–, Ca2+, and Mg2+) with the relative abundance of bacterial community and ARGs. Non-metric multidimensional scaling (NMDS) analysis was used to evaluate the difference of bacteria between different sampling sites. Redundancy analysis (RDA) was employed to assess the effects of environmental factors and ARGs on the bacterial community. The co-occurrence between abundance of ARGs and bacterial taxa was analyzed using network analysis based on the Pearson correlation (Li et al., 2015).
Statistical analysis was performed using SPSS 19.0 (IBM, Chicago, IL, United States). The RDA, Mantel test, Pearson correlation, and heatmap were performed in RStudio (v1.2.5019) with several packages, including vegan and pheatmap packages. Co-occurrence networks were constructed by Gephi software (0.9.2; Gephi, WebAtlas, France).
Results
Environmental Factors in the 15 Sediment Samples
To study the effects of environmental factors on bacterial community in the sediments, the concentrations of five factors (TN, TP, Cl–, Ca2+, and Mg2+) at different sample sites were detected (Supplementary Table 5). Among the sampling sites, TN concentrations ranged from 48.841 ± 0.158 to 193.679 ± 0.45 mg/kg (Figure 1A). The lowest concentration of TN appeared in the sample of HP3 and the highest in the sample of GP2. The TP concentrations were relatively lower in other sampling sites, except GP1 (118.757 ± 0.956 mg/kg), GP3 (48.064 ± 1.373 mg/kg), and SP1 (48.938 ± 0.323 mg/kg) (Figure 1B). The concentration of Cl– has a higher correlation with the aquaculture environment of sampling sites. The Cl– concentration in the sample sites from mariculture farms was generally higher than that of other sampling sites (Figure 1C). The Ca2+ concentration of GP3 and FP2 was the highest (1.632 ± 0.889 g/kg) and lowest (0.0800 ± 0.0346 g/kg), respectively (Figure 1D). The concentrations of Mg2+ of all sampling sites were generally lower, with the exception of GP1 (0.720 ± 0.128 g/kg), GP2 (1.377 ± 0.159 g/kg), and GP3 (3.334 ± 0.703 g/kg) (Figure 1E).
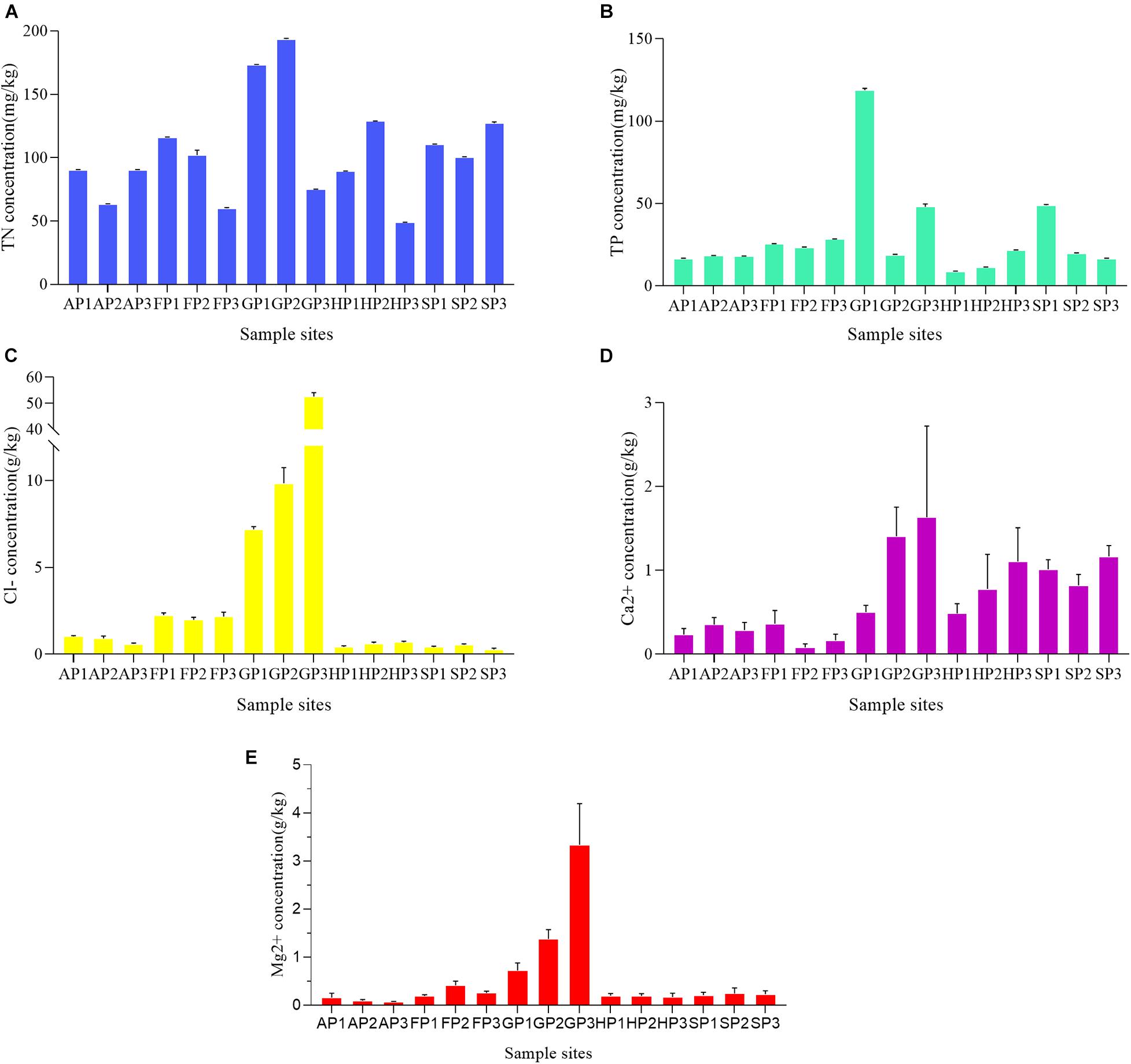
Figure 1. The TN, TP, Cl–, Ca2+, and Mg2+ concentrations (A–E) in the 15 sediment samples.
Bacterial Community in the 15 Sediment Samples
Diversity and Composition of Bacterial Community
With the sequencing of 16S rRNA gene, a total of 61,871 operational taxonomic units (OTUs) were identified from all the 15 sediment samples (Supplementary Table 6). The Rank Abundance curves of OTUs were saturated with all samples (Supplementary Figure 3), which indicated that the abundance and evenness of bacterial community were similar. The indices of ACE, Chao1, Shannon, and Simpson revealed the richness and diversity of the bacterial communities (Supplementary Table 6). The NMDS analysis based on the OTU abundance also indicated that there was no obvious geographic cluster of bacterial communities in different sediment samples (Supplementary Figure 4).
At the phylum level, four predominant phyla (Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidota) were detected in all sediment samples (Figure 2A). The Cyanobacteria has minor abundance, accounting for 0.30–9.39% of total bacterial 16S rRNA sequence libraries. At the genus level, the 16S rRNA sequence libraries detected 30 predominant bacterial genes from the 15 sediment samples. The bacterial community mainly includes Methanosaeta, Sphingomonas, Sulfurovum, and Thiobacillus (Figure 2B). In the phylum Proteobacteria, Dechloromonas, Pseudomonas, Sphingomonas, and Thiobacillus were the predominant genera of the bacterial community. In the sediment of FP1, FP2, and FP3, Sulfurovum and Sulfurimonas have a higher abundance (Supplementary Figure 5).
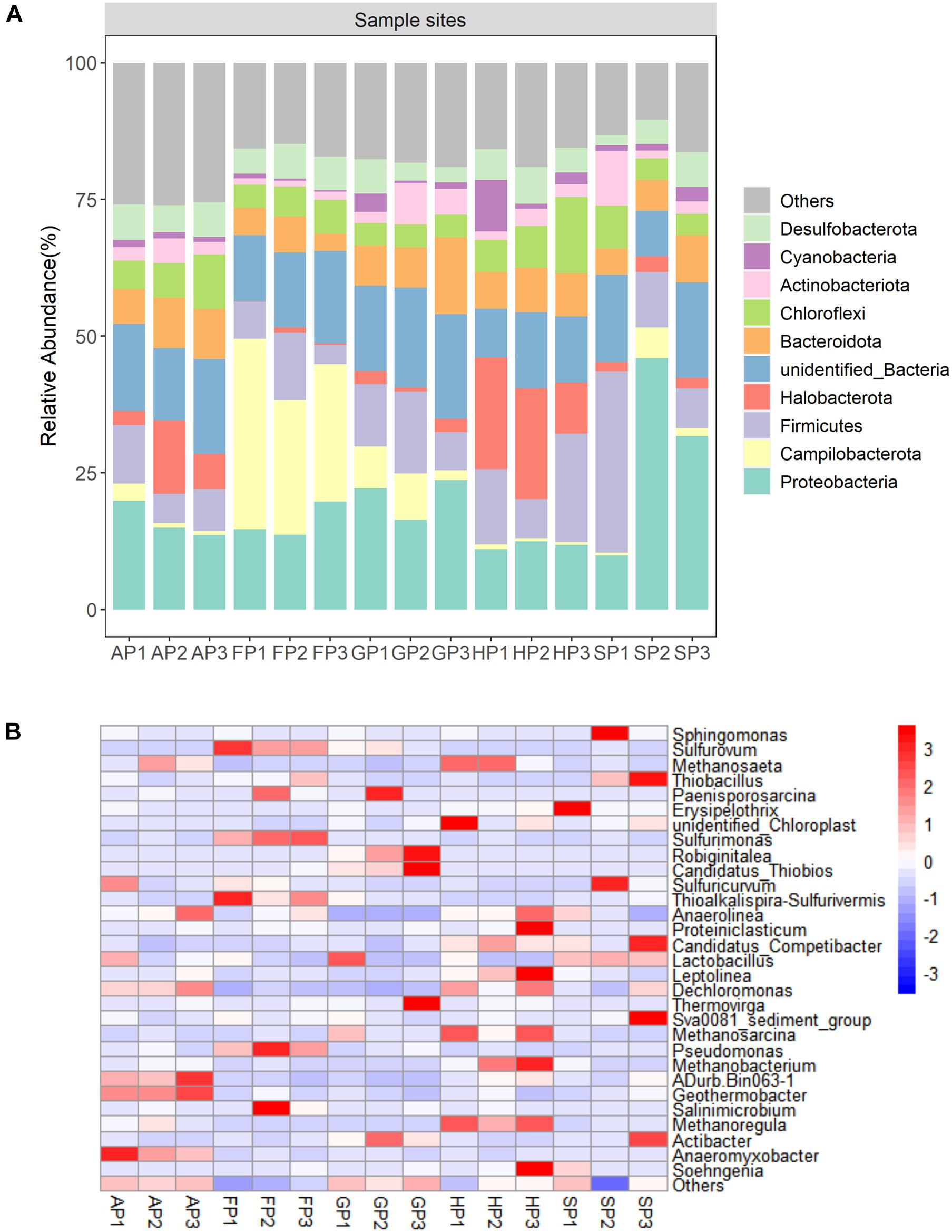
Figure 2. (A) Relative abundance of top 10 phyla in the 15 sediment samples. (B) The heatmap at the genus level of the 15 sediment samples.
Effect of Environmental Factors on the Diversity of the Bacterial Community
The Pearson correlation analysis was used to evaluate the effects of environmental factors on the bacterial community structure. The results indicated that there was no significant correlation between the concentration of TN, TP, Cl–, and Mg2+ with the richness and the diversity of bacterial community. However, the Ca2+ concentration had a significantly negative correlation with the richness and diversity of the bacterial community [OTU (r = −0.61, p < 0.05), ACE (r = −0.65, p < 0.05), and Chao1 index (r = −0.62, p < 0.05)].
Abundance and Distribution of Antibiotic Resistance Genes
To analyze the ARGs distribution in different aquaculture farms, 10 tested ARGs and 16S rRNA were investigated. The ARGs abundance was normalized to 16S rRNA genes to compare the difference of ARGs in different samples (Chen et al., 2019). As shown in Figure 3A, the ARGs were classified into four categories (sulfonamide, tetracycline, beta-lactam, and fluoroquinolone resistance genes) and integron. The higher abundance of sulfonamide resistance genes was detected in all these samples, but higher abundance of tetracycline resistance genes was detected in the samples of FP3, HP2, and HP3. The highest abundance of total ARGs was detected in the samples of HP (HP1, HP2, and HP3). The beta-lactam resistance genes have the highest abundance in the samples of FP (FP1, FP2, and FP3).
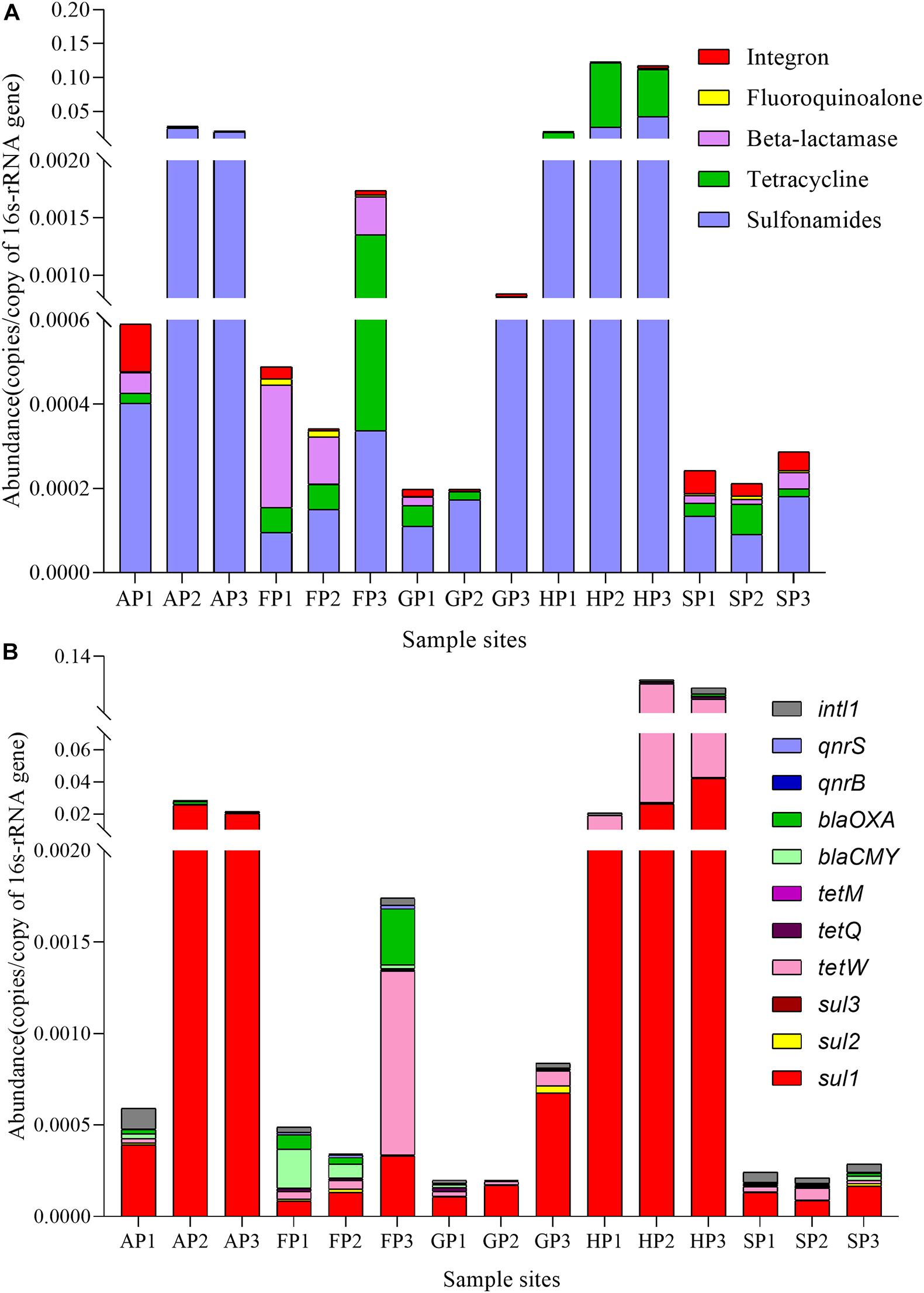
Figure 3. The abundance of antibiotic resistance genes (ARGs) in the 15 sediment samples. (A) Five categories. (B) 11 kinds.
The distribution of ARGs is shown in Figure 3B. Sul1, sul2, and tetM genes were the highest abundance in all sediment samples, while sul3 and qnrS genes were only detected in the sediments from HP (HP1, HP2, and HP3) and SP (SP1, SP2, and SP3), respectively. Overall, the ARGs levels in different sample sites were obviously different, which could be associated with the different types of antibiotics used in different aquaculture farms.
Correlations Among Bacterial Community, Antibiotic Resistance Genes, and Environmental Factors
The RDA analysis was performed to further explore the correlation between the bacterial communities of the 15 sediment samples, ARGs abundance, and environmental factors (Figure 4). The weights for variables making up canonical axes of RDA are summarized in Supplementary Table 7. We found that the TN, TP, Cl–, Ca2+, sul1, blaCMY, intl1, qnrS, and tetW have a significant correlation with the bacterial community in the 15 sediment samples (permutations = 999, p < 0.05), which explained 61.82% of overall variation in the bacterial community. The RDA1 and RDA2 explained 44.36 and 17.46% of the total variance, respectively. Positive correlation was found between environmental factors detected in this study and the bacterial community (AP1, GP1, GP2, GP3, SP1, SP2, and SP3), and the bacterial community of the sampling sites showed a negative correlation with ARGs. Moreover, the abundance of ARGs (sul1, tetW, qnrS, and intl1) has a much higher correlation with the bacterial community (AP2, AP3, HP1, HP2, and HP3), while blaCMY has a stronger correlation with the bacteria of FP (FP1, FP2, and FP3).
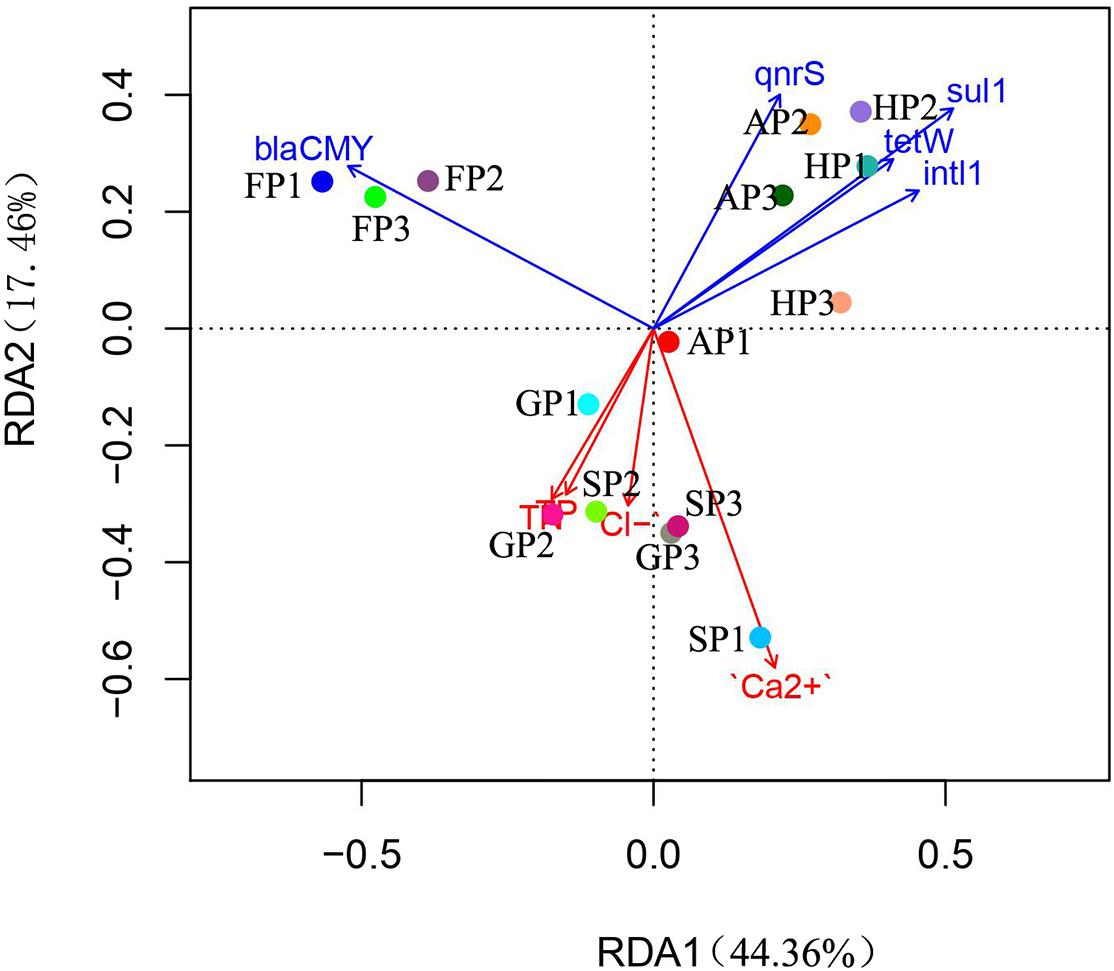
Figure 4. The redundancy analysis (RDA) of environmental factors, antibiotic resistance genes (ARGs), and bacterial communities.
Furthermore, the correlation analysis of 10 ARGs showed that there were significant correlations among multiple ARGs (Supplementary Figure 6). Similarly, the intl1 gene was correlated with the sul1, sul2, sul3, blaOXA, qnrS, and tetM, tetQ, and tetW genes. A Mantel test was also performed to demonstrate whether there were high correlations between the total ARGs and intl1. Results showed that a significant correlation (permutations = 999, r = 0.8013, p < 0.01) was found among the total ARGs and intl1.
Co-occurrence of Bacterial Community and Antibiotic Resistance Genes in Sediment Samples
The co-occurrence patterns between ARGs and bacterial community were further analyzed with the network analysis (Figures 5A,B). As shown in Figure 5B, the intl1 gene was found to co-occur with eight genera of bacteria taxa, followed by sul1 (7), sul2 (6), tetQ (6), tetM (6), qnrS (4), tetW (3), blaCMY (2), and blaOXA (2).
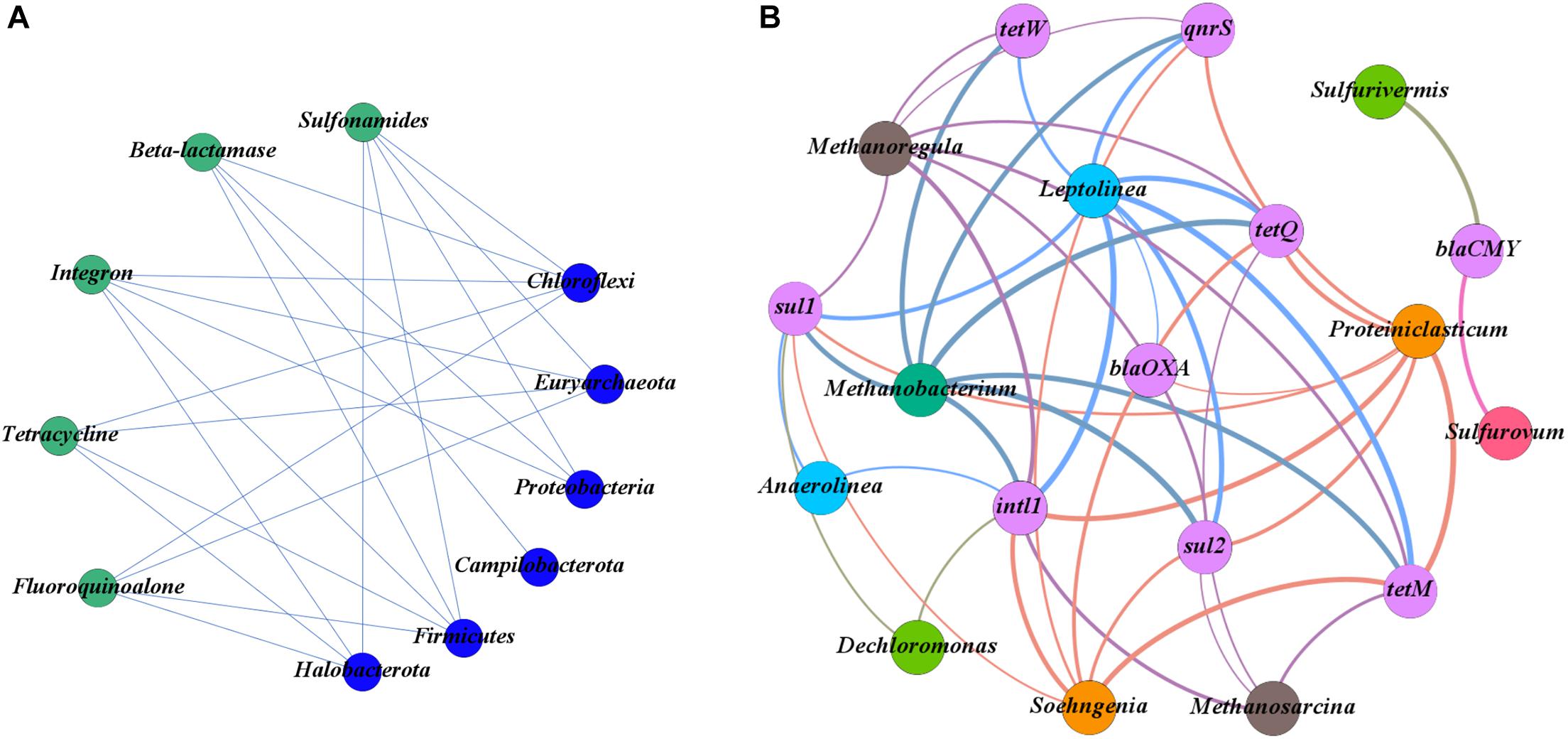
Figure 5. (A) Network plots between antibiotic resistance genes (ARGs) and the phyla of the bacterial community. (B) Network plots between ARGs and the genera of the bacterial community.
At the phylum level of the bacterial community, the main phyla potentially carrying the target ARGs were Chloroflexi, Euryarchaeota, Firmicutes, Halobacterota, and Proteobacteria (Figure 5A). There was a significant co-occurrence pattern in the bacterial taxa of Chloroflexi and eight subtype ARGs (sul1, sul2, blaOXA, qnrS, tetW, tetQ, tetM, and intl1) and a significant co-occurrence pattern in the bacterial taxa of Firmicutes and seven subtype ARGs (sul1, sul2, blaOXA, qnrS, tetQ, tetM, and intl1) (p < 0.05) (Figure 5B). At the genus level of the bacterial community, Proteiniclasticum, Leptolinea, Methanobacterium, and Methanoregula were the main potential hosts of ARGs (Figure 5B). Among them, the Leptolinea was found to have the most diverse connections with ARGs, including sul1, sul2, blaOXA, qnrS, tetW, tetQ, tetM, and intl1 (p < 0.05). Proteiniclasticum was also detected to carry seven ARGs (sul1, sul2, blaOXA, qnrS, tetQ, tetM, and intl1) (p < 0.05). In addition, Sulfurovum and Sulfurivermis have simply co-occurred with the blaCMY gene encoding resistance.
Discussion
Aquaculture ponds are regarded as a major reservoir for antibiotic resistant bacteria and ARGs due to the overreliant use of antibiotics (Boran et al., 2013). However, excessive antibiotics and their metabolites have been released into the environment due to the abuse and misuse of antibiotics (Bu et al., 2013; Liu and Wong, 2013). Relevant studies have indicated that antibiotic contamination could lead to the emergence of ARGs in the environment (Qiao et al., 2018; Yang et al., 2018). In this study, we explored the correlation between environmental factors, ARGs, and the bacterial community in different aquatic environments.
Sulfonamides and tetracyclines were used widely in aquatic farms (Luo et al., 2010), the abundance of sul and tet genes was significantly correlated with the use of the corresponding antibiotics (Gao et al., 2012). Generally, the establishment of ARGs requires selective pressure on antibiotics over a long period. However, once the selective pressure was established, the ARGs would persist and difficult to be eliminated even if the pressure is removed (Pei et al., 2006; Xiao et al., 2016). In this study, a higher abundance of sul and tet genes in sediment samples was detected, and it was consistent with previous studies that sul and tet genes were the dominant ARGs in aquaculture water environments (Hoa et al., 2008; Tamminen et al., 2011). Remarkably, the abundance of sul gene was higher than that of tet gene, except for the sample from FP3. A previous study also indicated that the sul gene persists longer than the tet gene (McKinney et al., 2010). The intl1 was one of the mobile genetic element genes and widely existed in Gram-positive and Gram-negative bacteria (Zeng et al., 2019), and it was regarded as an important pollution genetic marker caused by human activity (Gillings et al., 2015). The abundance of Intl1 gene is a proxy for anthropogenic pollution among many other factors are that they are linked to genes conferring resistance to antibiotics, and intl1 gene was also closely related to multidrug resistance (MDR). Our study revealed that the abundance of ARGs (sul1, sul2, sul3, tetW, tetQ, tetM, blaOXA, and qnrS) was significantly correlated with the abundance of Intl1. It was indicated that intl1 may play a key role in ARGs proliferation and diffusion from the sediment of aquaculture farms. Moreover, the previous study indicated that there were the co-occurrence patterns among many ARGs in pig farm wastewater (Yang et al., 2020). Correlation analysis in the study also showed that there was a significant positive correlation among the different types of ARGs. The bacteria carrying multiple ARGs could easily obtain the resistance to antibiotics (Trudel et al., 2016); therefore, the potential environmental risk of ARGs should be given attention.
In this study, a significant difference was observed in bacterial communities among different aquatic farms. The dominant phyla were Proteobacteria and Firmicutes in all sediment samples. Similar results were found in pig farms and the sediment of a shrimp farm (Yang et al., 2020; Zeng et al., 2020). Within the Proteobacteria phylum, the Sphingomonas was the main compositions of the genus. It was reported that the genome of Sphingomonas contains multiple efflux pumps (Jia et al., 2019), suggesting that Sphingomonas might better exist in the sediments of aquatic environments. A previous study implied that the physiochemical properties of the environment may influence the bacterial community by affecting the nutrient availability or physiological activity (Li et al., 2019). The present study found that the bacterial communities from AP1, GP1, GP2, GP3, SP1, SP2, and SP3 were significantly correlated with environmental factors. In addition, the concentrations of Mg2+, Ca2+, and Cl– in the environment influence the composition of the bacterial community (Xu et al., 2018). It is worth noting that the Ca2+ has a significant negative correlation with the richness and diversity of bacterial communities. Interestingly, the calcium carbonate was widely used in aquaculture farms, which led to the high accumulation of Ca2+ in the sediments. Therefore, the excessive use of calcium carbonate might lead to a decrease in the diversity and richness of bacterial communities in the environment.
The aquatic environment gradually becomes a reservoir of antibiotic-resistant bacteria because of the use and abuse of antibiotics in aquatic farms (Huang et al., 2017). Previous studies certified that some bacterial taxa from Firmicutes were the dominant ARGs-carrying bacteria (Zhang et al., 2021). We also found that the Proteiniclasticum and Soehngenia from Firmicutes might be the main potential hosts of ARGs, which have strong co-occurrence with sul1, sul2, blaOXA, qnrS, tetQ, tetM, and intl1 genes. Similarly, the Anaerolinea and Leptolinea from Chloroflexi have strong co-occurrence with ARGs (sul1, sul2, blaOXA, qnrS, tetW, tetQ, tetM, and intl1). However, the Sulfurovum from Campilobacterota only has a co-occurrence with blaCMY, and the Campilobacterota was the dominant phylum in the samples of FP (FP1, FP2, and FP3). Furthermore, there was a stronger co-occurrence between tetM gene and six bacterial taxa in these samples. A previous study revealed the similar results in the soil of swine feedlots (Li et al., 2019). It is worth noting that the tetM gene was regarded as a detection tool to track and monitor ARGs transport in agricultural systems (Cadena et al., 2018). Our research found that the ARGs have a complex co-occurrence correlation with the bacterial taxa in sediment, which indicated that some bacterial taxa could be resistant to multiple antibiotics in the sediments. Overall, this study indicated that the ARGs in the sediments of aquaculture farms have an impact on the environment and bacterial communities, and we must pay more attention to and take preventive measures.
Conclusion
The present study indicated that the sulfonamides and tetracycline resistance genes were the predominant ARGs in the sediments of the investigated aquatic farms. Some bacterial taxa from the phyla Chloroflexi, Euryarchaeota, Firmicutes, Halobacterota, and Proteobacteria might be the main potential hosts of ARGs in these aquatic farms. Moreover, the excessive Ca2+ might inhibit the diversity and richness of bacterial communities.
Data Availability Statement
The sequence raw datasets in this study can be found in the NCBI repository (http://www.ncbi.nlm.nih.gov/bioproject/708165).
Author Contributions
XC and HL played an important role in the conception of the study. CL and YS finished the part of the experiment. RZ and HX organized the original data. YL and XS performed the data analysis. XC wrote the first manuscript. HL edited the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Key Research and Development Project of Shaanxi Province (No. 2018NY-109).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.679805/full#supplementary-material
References
Boran, H., Terzi, E., Altinok, I., Capkin, E., and Bascinar, N. (2013). Bacterial diseases of cultured mediterranean horse mackerel (trachurus mediterraneus) in sea cages. Aquaculture 396-399, 8–13. doi: 10.1016/j.aquaculture.2013.02.025
Bu, Q., Wang, B., Huang, J., Deng, S., and Yu, G. (2013). Pharmaceuticals and personal care products in the aquatic environment in China: a review. J. Hazard. Mater. 262, 189–211. doi: 10.1016/j.jhazmat.2013.08.040
Cadena, M., Durso, L. M., Miller, D. N., Waldrip, H. M., Castleberry, B. L., Drijber, R. A., et al. (2018). Tetracycline and Sulfonamide Antibiotic Resistance Genes in Soils from Nebraska Organic Farming Operations. Front. Microbiol. 9:1283. doi: 10.3389/fmicb.2018.01283
Cambray, G., Guerout, A. M., and Mazel, D. (2010). Integrons. Annu. Rev. Genet. 44, 141–166. doi: 10.1146/annurev-genet-102209-163504
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmethf.303
Chen, B., Lin, L., Fang, L., Yang, Y., Chen, E., Yuan, K., et al. (2018). Complex pollution of antibiotic resistance genes due to beta-lactam and aminoglycoside use in aquaculture farming. Water Res. 134, 200–208. doi: 10.1016/j.watres.2018.02.003
Chen, H., Bai, X., Li, Y., Jing, L., Chen, R., and Teng, Y. (2019). Characterization and source-tracking of antibiotic resistomes in the sediments of a peri-urban river. Sci. Tot. Environ. 679, 88–96. doi: 10.1016/j.scitotenv.2019.05.063
Chen, H., Liu, S., Xu, X. R., Diao, Z. H., Sun, K. F., Hao, Q. W., et al. (2018). Tissue distribution, bioaccumulation characteristics and health risk of antibiotics in cultured fish from a typical aquaculture area. J. Hazard. Mater. 343, 140–148. doi: 10.1016/j.jhazmat.2017.09.017
Dang, B., Mao, D., Xu, Y., and Luo, Y. (2017). Conjugative multi-resistant plasmids in Haihe River and their impacts on the abundance and spatial distribution of antibiotic resistance genes. Water Res. 111, 81–91. doi: 10.1016/j.watres.2016.12.046
Fahrenfeld, N., Knowlton, K., Krometis, L. A., Hession, W. C., Xia, K., Lipscomb, E., et al. (2014). Effect of manure application on abundance of antibiotic resistance genes and their attenuation rates in soil: field-scale mass balance approach. Environ. Sci. Technol. 48, 2643–2650. doi: 10.1021/es404988k
Forsberg, K. J., Reyes, A., Wang, B., Selleck, E. M., Sommer, M. O., and Dantas, G. (2012). The shared antibiotic resist one of soil bacteria and human pathogens. Science 337, 1107–1111. doi: 10.1126/science.1220761
Gao, P., Mao, D., Luo, Y., Wang, L., Xu, B., and Xu, L. (2012). Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res. 46, 2355–2364. doi: 10.1016/j.watres.2012.02.004
Gaze, W. H., Zhang, L., Abdouslam, N. A., Hawkey, P. M., Calvo-Bado, L., Royle, J., et al. (2011). Impacts of anthropogenic activity on the ecology of class 1 integrons and integron-associated genes in the environment. ISME J. 5, 1253–1261. doi: 10.1038/ismej.2011.15
Gillings, M. R., Gaze, W. H., Pruden, A., Smalla, K., Tiedje, J. M., and Zhu, Y. G. (2015). Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 9, 1269–1279. doi: 10.1038/ismej.2014.226
Guo, J., Li, J., Chen, H., Bond, P. L., and Yuan, Z. (2017). Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 123, 468–478. doi: 10.1016/j.watres.2017.07.002
Hoa, P. T. P., Nonaka, L., Viet, P. H., and Suzuki, S. (2008). Detection of the sul1, sul2, and sul3 genes in sulfonamide-resistant bacteria from wastewater and shrimp ponds of north Vietnam. Sci. Tot. Environ. 405, 377–384. doi: 10.1016/j.scitotenv.2008.06.023
Huang, L., Xu, Y. B., Xu, J. X., Ling, J. Y., Chen, J. L., Zhou, J. L., et al. (2017). Antibiotic resistance genes (ARGs) in duck and fish production ponds with integrated or non-integrated mode. Chemosphere 168, 1107–1114. doi: 10.1016/j.chemosphere.2016.10.096
Huerta, B., Marti, E., Gros, M., López, P., Pompêo, M., Armengol, J., et al. (2013). Exploring the links between antibiotic occurrence, antibiotic resistance, and bacterial communities in water supply reservoirs. Sci. Tot. Environ. 45, 161–170. doi: 10.1016/j.scitotenv.2013.03.071
Hurst, J. J., Oliver, J. P., Schueler, J., Gooch, C., Lansing, S., Crossette, E., et al. (2019). Trends in Antimicrobial Resistance Genes in Manure Blend Pits and Long-Term Storage Across Dairy Farms with Comparisons to Antimicrobial Usage and Residual Concentrations. Environ. Sci. Technol. 53, 2405–2415. doi: 10.1021/acs.est.8b05702
Jia, S., Wu, J., Ye, L., Zhao, F., Li, T., and Zhang, X. X. (2019). Metagenomic assembly provides a deep insight into the antibiotic resistome alteration induced by drinking water chlorination and its correlations with bacterial host changes. J. Hazard. Mater. 379:120841. doi: 10.1016/j.jhazmat.2019.120841
Kumar, M., Prasad, R., Sharma, S., Varma, A., and Kumar, V. (2017). Dissemination Mechanism of AntibioticResistance Genes in Environment. Soil Biol. 51, 191–205. doi: 10.1007/978-3-319-66260-2_10
Kümmerer, K. (2009). Antibiotics in the aquatic environment–a review–part I. Chemosphere 75, 417–434. doi: 10.1016/j.chemosphere.2008.11.086
Li, B., Yang, Y., Ma, L., Ju, F., Guo, F., Tiedje, J. M., et al. (2015). Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 9, 2490–2502. doi: 10.1038/ismej.2015.59
Li, N., Zhu, C., Liu, C., Zhang, X., Ding, J., Zandi, P., et al. (2019). The persistence of antimicrobial resistance and related environmental factors in abandoned and working swine feedlots. Environ. Pollut. 255(Pt 1):113116. doi: 10.1016/j.envpol.2019.113116
Liu, J. L., and Wong, M. H. (2013). Pharmaceuticals and personal care products (PPCPs): a review on environmental contamination in China. Environ. Int. 59, 208–224. doi: 10.1016/j.envint.2013.06.012
Luo, Y., Mao, D., Rysz, M., Zhou, Q., Zhang, H., Xu, L., et al. (2010). Trends in antibiotic resistance genes occurrence in the Haihe River, China. Environ. Sci. Technol. 44, 7220–7225. doi: 10.1021/es100233w
Ma, L., Li, B., and Zhang, T. (2019). New insights into antibiotic resistome in drinking water and management perspectives: A metagenomic based study of small-sized microbes. Water Res. 152, 191–201. doi: 10.1016/j.watres.2018.12.069
Mao, D., Luo, Y., Mathieu, J., Wang, Q., Feng, L., Mu, Q., et al. (2014). Persistence of extracellular DNA in river sediment facilitates antibiotic resistance gene propagation. Environ. Sci. Technol. 48, 71–78. doi: 10.1021/es404280v
Marti, E., Variatza, E., and Balcazar, J. L. (2014). The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 22, 36–41. doi: 10.1016/j.tim.2013.11.001
Martínez, J. L. (2008). Antibiotics and antibiotic resistance genes in natural environments. Science 321, 365–367. doi: 10.1126/science.1159483
Martinez-Freijo, P., Fluit, A. C., Schmitz, F. J., Grek, V. S., Verhoef, J., and Jones, M. E. (1998). Class I integrons in Gram-negative isolates from different European hospitals and association with decreased susceptibility to multiple antibiotic compounds. J. Antimicrob. Chemother. 42, 689–696. doi: 10.1093/jac/42.6.689
McKinney, C. W., Loftin, K. A., Meyer, M. T., Davis, J. G., and Pruden, A. (2010). tet and sul antibiotic resistance genes in livestock lagoons of various operation type, configuration, and antibiotic occurrence. Environ. Sci. Technol. 44, 6102–6109. doi: 10.1021/es9038165
Pan, X., Lin, L., Zhang, W., Dong, L., and Yang, Y. (2020). Metagenome sequencing to unveil the resistome in a deep subtropical lake on the Yunnan-Guizhou Plateau, China. Environ. Pollut. 263(Pt B):114470. doi: 10.1016/j.envpol.2020.114470
Pei, R., Kim, S. C., Carlson, K. H., and Pruden, A. (2006). Effect of river landscape on the sediment concentrations of antibiotics and corresponding antibiotic resistance genes (ARG). Water Res. 40, 2427–2435. doi: 10.1016/j.watres.2006.04.017
Qiao, M., Ying, G. G., Singer, A. C., and Zhu, Y. G. (2018). Review of antibiotic resistance in China and its environment. Environ. Int. 110, 160–172. doi: 10.1016/j.envint.2017.10.016
SEPA (2002). Water and wastewater monitoring and analysis method, 4th Edn. Beijing: China Environmental Science Press.
Shen, X., Jin, G., Zhao, Y., and Shao, X. (2020). Prevalence and distribution analysis of antibiotic resistance genes in a large-scale aquaculture environment. Sci. Tot. Environ. 711:134626. doi: 10.1016/j.scitotenv.2019.134626
Smillie, C. S., Smith, M. B., Friedman, J., Cordero, O. X., David, L. A., and Alm, E. J. (2011). Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480, 241–244. doi: 10.1038/nature10571
Stokes, H. W., and Hall, R. M. (1989). A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol. Microbiol. 3, 1669–1683. doi: 10.1111/j.1365-2958.1989.tb00153.x
Tamminen, M., Karkman, A., Lõhmus, A., Muziasari, W. I., Takasu, H., Wada, S., et al. (2011). Tetracycline resistance genes persist at aquaculture farms in the absence of selection pressure. Environ. Sci. Technol. 45, 386–391. doi: 10.1021/es102725n
Trolle, D., Zhu, G., Hamilton, D., Luo, L., Mcbride, C., and Zhang, L. (2009). The influence of water quality and sediment geochemistry on the horizontal and vertical distribution of phosphorus and nitrogen in sediments of a large, shallow lake. Hydrobiologia 627, 31–44. doi: 10.1007/s10750-009-9713-0
Trudel, M. V., Vincent, A. T., Attéré, S. A., Labbé, M., Derome, N., Culley, A. I., et al. (2016). Diversity of antibiotic-resistance genes in Canadian isolates of Aeromonas salmonicida subsp. salmonicida: dominance of pSN254b and discovery of pAsa8. Sci. Rep. 6:35617. doi: 10.1038/srep35617
Wang, A., Kong, X., Song, X., Ju, B., and Li, D. (2020). Study on exchangeable cation determining base saturation percentage of soil in south china. Agricult. Sci. 11, 17–26. doi: 10.4236/as.2020.111002
Wang, R. X., Wang, J. Y., Sun, Y. C., Yang, B. L., and Wang, A. L. (2015). Antibiotic resistance monitoring in Vibrio spp. isolated from rearing environment and intestines of abalone Haliotis diversicolor. Mar. Pollut. Bul. 101, 701–706. doi: 10.1016/j.marpolbul.2015.10.027
Xiao, K. Q., Li, B., Ma, L., Bao, P., Zhou, X., Zhang, T., et al. (2016). Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMS Microbiol. Ecol. 92:fiw023. doi: 10.1093/femsec/fiw023
Xiong, W., Sun, Y., Ding, X., Wang, M., and Zeng, Z. (2015). Selective pressure of antibiotics on ARGs and bacterial communities in manure-polluted freshwater-sediment microcosms. Front. Microbiol. 6:194. doi: 10.3389/fmicb.2015.00194
Xu, Z., Woodhouse, J. N., Te, S. H., Yew-Hoong Gin, K., He, Y., Xu, C., et al. (2018). Seasonal variation in the bacterial community composition of a large estuarine reservoir and response to cyanobacterial proliferation. Chemosphere 202, 576–585. doi: 10.1016/j.chemosphere.2018.03.037
Yang, Y., Chen, Y., Cai, Y., Xing, S., Mi, J., and Liao, X. (2020). The relationship between culturable doxycycline-resistant bacterial communities and antibiotic resistance gene hosts in pig farm wastewater treatment plants. Ecotoxicol. Environ. Safety 206:111164. doi: 10.1016/j.ecoenv.2020.111164
Yang, Y., Song, W., Lin, H., Wang, W., Du, L., and Xing, W. (2018). Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 116, 60–73. doi: 10.1016/j.envint.2018.04.011
Yuan, K., Wang, X., Chen, X., Zhao, Z., Fang, L., Chen, B., et al. (2019). Occurrence of antibiotic resistance genes in extracellular and intracellular DNA from sediments collected from two types of aquaculture farms. Chemosphere. 234, 520–527. doi: 10.1016/j.chemosphere.2019.06.085
Zeng, Q., Sun, J., and Zhu, L. (2019). Occurrence and distribution of antibiotics and resistance genes in greenhouse and open-field agricultural soils in China. Chemosphere 224, 900–909. doi: 10.1016/j.chemosphere.2019.02.167
Zeng, S., Khoruamkid, S., Kongpakdee, W., Wei, D., Yu, L., Wang, H., et al. (2020). Dissimilarity of microbial diversity of pond water, shrimp intestine and sediment in Aquamimicry system. AMB Express 10:180. doi: 10.1186/s13568-020-01119-y
Zhang, J., Buhe, C., Yu, D., Zhong, H., and Wei, Y. (2020). Ammonia stress reduces antibiotic efflux but enriches horizontal gene transfer of antibiotic resistance genes in anaerobic digestion. Bioresour. Technol. 295:122191. doi: 10.1016/j.biortech.2019.122191
Zhang, R. M., Liu, X., Wang, S. L., Fang, L. X., Sun, J., Liu, Y. H., et al. (2021). Distribution patterns of antibiotic resistance genes and their bacterial hosts in pig farm wastewater treatment systems and soil fertilized with pig manure. Sci. Tot. Environ. 758:143654. doi: 10.1016/j.scitotenv.2020.143654
Zhang, X., Li, X., Wang, W., Qi, J., Wang, D., Xu, L., et al. (2020). Diverse Gene Cassette Arrays Prevail in Commensal Escherichia coli From Intensive Farming Swine in Four Provinces of China. Front. Microbiol. 11:565349. doi: 10.3389/fmicb.2020.565349
Zhao, Z., Wang, J., Han, Y., Chen, J., Liu, G., Lu, H., et al. (2017). Nutrients, heavy metals and microbial communities co-driven distribution of antibiotic resistance genes in adjacent environment of mariculture. Environ. Pollut. 220(Pt B), 909–918. doi: 10.1016/j.envpol.2016.10.075
Keywords: aquaculture, sediment, antibiotic resistance genes, environmental factors, bacterial community
Citation: Cheng X, Lu Y, Song Y, Zhang R, ShangGuan X, Xu H, Liu C and Liu H (2021) Analysis of Antibiotic Resistance Genes, Environmental Factors, and Microbial Community From Aquaculture Farms in Five Provinces, China. Front. Microbiol. 12:679805. doi: 10.3389/fmicb.2021.679805
Received: 12 March 2021; Accepted: 19 May 2021;
Published: 24 June 2021.
Edited by:
Jorge Andres Olivares Pacheco, Pontificia Universidad Católica de Valparaíso, ChileReviewed by:
Claudio D. Miranda, Catholic University of the North, ChileMichael P. Ryan, University of Limerick, Ireland
Copyright © 2021 Cheng, Lu, Song, Zhang, ShangGuan, Xu, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haixia Liu, bGl1aGFpeGlhMjA5QG53c3VhZi5lZHUuY24=