
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 02 June 2021
Sec. Microbial Symbioses
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.670383
This article is part of the Research TopicInsect Microbiome: From Diversity To ApplicationsView all 35 articles
 Hui Xue1,2
Hui Xue1,2 Xiangzhen Zhu1,2Li Wang1,2Kaixin Zhang1,2Dongyang Li1,2Jichao Ji1,2Lin Niu1,2Changcai Wu1,2Xueke Gao1,2*Junyu Luo1,2*
Xiangzhen Zhu1,2Li Wang1,2Kaixin Zhang1,2Dongyang Li1,2Jichao Ji1,2Lin Niu1,2Changcai Wu1,2Xueke Gao1,2*Junyu Luo1,2* Jinjie Cui1,2*
Jinjie Cui1,2*Bacteria and insects have a mutually beneficial symbiotic relationship. Bacteria participate in several physiological processes such as reproduction, metabolism, and detoxification of the host. Adelphocoris suturalis is considered a pest by the agricultural industry and is now a major pest in cotton, posing a serious threat to agricultural production. As with many insects, various microbes live inside A. suturalis. However, the microbial composition and diversity of its life cycle have not been well-studied. To identify the species and community structure of symbiotic bacteria in A. suturalis, we used the HiSeq platform to perform high-throughput sequencing of the V3–V4 region in the 16S rRNA of symbiotic bacteria found in A. suturalis throughout its life stages. Our results demonstrated that younger nymphs (1st and 2nd instar nymphs) have higher species richness. Proteobacteria (87.06%) and Firmicutes (9.43%) were the dominant phyla of A. suturalis. At the genus level, Erwinia (28.98%), Staphylococcus (5.69%), and Acinetobacter (4.54%) were the dominant bacteria. We found that the relative abundance of Erwinia was very stable during the whole developmental stage. On the contrary, the relative abundance of Staphylococcus, Acinetobacter, Pseudomonas, and Corynebacterium showed significant dynamic changes at different developmental stages. Functional prediction of symbiotic bacteria mainly focuses on metabolic pathways. Our findings document symbiotic bacteria across the life cycle of A. suturalis, as well as differences in both the composition and richness in nymph and adult symbiotic bacteria. Our analysis of the bacteria in A. suturalis provides important information for the development of novel biological control strategies.
Insects are found almost everywhere in the world. Most insects carry symbiotic microorganisms that are involved in the life cycle processes of the host (Yong et al., 2017; Zhao et al., 2019). There is an interactive relationship between insects and symbiotic bacteria, which play an important role in the health, survival, and behavior of the host (Dillon and Dillon, 2004; Kikuchi et al., 2007; Moran et al., 2008; Santos-Garcia et al., 2017). However, the vast majority of symbiotic microorganisms are concentrated in the intestines of insects, where they act as key regulators of the insect host’s multiple lifestyles (including diet and ecological niche; Gupta and Nair, 2020). Studies have shown that symbiotic bacteria in the intestinal tract of insects can promote host food consumption and digestion; provide immunity and protection against various predators, pathogens, and parasites; control the success rate of host mating and reproduction; provide essential amino acids, metabolic compounds, and nutrients (Russell et al., 2014; Douglas, 2015; Arbuthnott et al., 2016; Wielkopolan and Obrępalska-Stęplowska, 2016; Engl and Kaltenpoth, 2018; Grenier and Leulier, 2020; Horak et al., 2020). In a recent study, the main roles of insect gut bacteria were demonstrated to be providing essential nutrients, followed by digestion and detoxification (Jing et al., 2020). Therefore, insects are highly dependent on intestinal symbiotic bacteria to complete their own growth and development. In short, a very complex and interesting relationship is formed between intestinal symbiotic bacteria and their hosts.
Hemiptera insects have piercing and sucking mouthparts, which not only can directly suck plant juice and kill crops, but also cause serious economic losses by spreading plant viruses. They are notorious agricultural pests (Wang et al., 2015; Liu et al., 2018; Qin et al., 2018). Adelphocoris suturalis was originally a minor pest of cotton, but the widespread application of Bacillus thuringiensis (Bt) cotton and the reduction of broad-spectrum insecticides has made it a major problem in cotton-growing areas in China (Wu et al., 2008; Lu and Wu, 2011; Luo et al., 2017a). Bt plants can effectively control Lepidopteran pests and can significantly reduce the use of chemical pesticides that are often required in conventional planting systems (Wu et al., 2008; Wang et al., 2018). A major challenge in planting Bt crops to control pests is that insects may evolve resistance to Bt (Wu et al., 2008). Adelphocoris suturalis is a typical omnivorous insect, which regularly feeds on crop plants (e.g., cotton and garden pea), other insects like aphids, and occasionally its weaker siblings (Lu and Wu, 2011; Luo et al., 2021). Additionally, A. suturalis also has the characteristic of high liquidity (Wu et al., 2010), resulting in poor chemical controls. At present, chemical pesticides such as organophosphates and pyrethroids are widely used in China to control these insects (Zhen et al., 2016). The question of whether these insects are resistant or not needs to be resolved urgently. There is increasing evidence that there is a link between symbiotic bacteria in the insect gut and the evolution of drug resistance (Broderick et al., 2006; Kikuchi et al., 2012; Engel and Moran, 2013; Xia et al., 2013). In addition, the omnivorous nature of A.suturalis increases the difficulty of pest control. Previous studies have reported that some symbiotic bacteria help insect hosts form new feeding habits, thus expanding food sources and enhancing the adaptability of insects to the environment (Lu et al., 2001; Douglas, 2009), which may also increase the difficulty of A.suturalis control. There are currently few studies examining the symbiotic bacteria of A. suturalis, making the number and species of the symbiotic bacteria in A. suturalis uncertain (Luo et al., 2021; Ma et al., 2021). Therefore, it is necessary to investigate the distribution of symbiotic bacteria at different stages of A. suturalis, which will provide a framework for exploring the function of symbiotic bacteria and pest control in A. suturalis.
In this study, the bacterial community composition and relative abundance of 1st instar to 5th instar A. suturalis nymphs and 1, 6, and 9 days male and female adults of A. suturalis were investigated via high-throughput Illumina sequencing of the 16S rRNA gene. To identify new pest control strategies, we explored the cooperative coevolution of A. suturalis and its symbiotic bacteria to better understand how A. suturalis relates to the symbiotic bacteria community structure, to examine the symbiotic bacteria related to A. suturalis, and to provide a theoretical basis for revealing a series of principles such as its resistance regulation mechanism and feeding characteristics.
The A. suturalis used in this study were collected from the field in Wuhan (Hubei Province, China). Adelphocoris suturalis strains were maintained in climate chambers (75 ± 5% relative humidity, 26 ± 2°C temperature and a 16:8 h, light:dark cycle) and fed green beans and a 5% sugar solution (Lu et al., 2008). Adelphocoris suturalis started feeding cotton aphid from the third instar nymph. Cotton aphids are reared on non-transgenic cotton seedlings, living in the same environment as A. suturalis.
DNA was extracted from whole insects (1st instar nymph, 2nd instar nymph, 3rd instar nymph, 4th instar nymph, 5th instar nymph, and 1, 6, and 9 days male and female adults) using MagPure Stool DNA KF kit B (Magen, China) according to the manufacturer’s instructions. Six biological replicates were set for samples at each developmental stage (females and males were counted as two treatments). Insects were rinsed three times in distilled sterile water prior to DNA extraction (without soaking in ethanol). DNA was quantified using a Qubit Fluorometer with a Qubit dsDNA BR Assay kit (Invitrogen, United States) and the quality was checked by performing an aliquot on 1% agarose gel.
The variable regions V3–V4 of the bacterial 16S rRNA gene were amplified with the degenerate PCR primers 341F (5'-ACTCCTACGGGAGGCAGCAG-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3'). Both forward and reverse primers were tagged with Illumina adapter, pad, and linker sequences. PCR enrichment was performed in a 50 μl reaction containing a 30 ng template, a fusion PCR primer, and a PCR master mix. PCR cycling conditions were as follows: 94°C for 3 min, 30 cycles of 94°C for 30 s, 56°C for 45 s, 72°C for 45 s, and a final extension at 72°C for 10 min. The PCR products were purified with AmpureXP beads and eluted in an Elution buffer. Libraries were qualified by the Agilent 2100 bioanalyzer (Agilent, United States). The validated libraries were used for sequencing on an Illumina HiSeq platform (BGI, Shenzhen, China) according to standard Illumina procedures, and generated 2 × 300 bp paired-end reads. The sequences obtained in this study were deposited in the GenBank short-read archive (SRA), accession number PRJNA662509.
The data obtained from independent sequencing were analyzed separately. Samples were marked as follows: ZL1: 1st instar nymph; ZL2: 2nd instar nymph; ZL3: 3rd instar nymph; ZL4: 4th instar nymph; ZL5: 5th instar nymph; ZM1D: adult male eclosion at 1 day; ZF1D: adult female eclosion at 1 day; ZM6D: adult male eclosion at 6 days; ZF6D: adult female eclosion at 6 days; ZM9D: adult male eclosion at 9 days; and ZF9D: adult female eclosion at 9 days.
Raw reads were filtered to remove adaptors and low-quality and ambiguous bases, and paired-end reads were added to tags using the Fast Length Adjustment of Short reads program (FLASH, v1.2.11; Magoc and Salzberg, 2011) to obtain the tags. The tags were clustered into operational taxonomic units (OTUs) with a cutoff value of 97% using UPARSE software (v7.0.1090; Edgar, 2013) and we used UCHIME (v4.2.40) and Gold database for chimera sequence alignment and detection (Edgar et al., 2011). OTU representative sequences were then taxonomically classified using the Ribosomal Database Project (RDP) Classifier v.2.2 with a minimum confidence threshold of 0.6, and trained on the Greengenes database v201305 by QIIME v1.8.0 (Caporaso et al., 2010). The USEARCH_global (Edgar, 2010) was used to compare all tags to obtain an OTU statistical abundance table for each sample. Alpha and beta diversity were estimated by MOTHUR (v1.31.2; Schloss et al., 2009) and QIIME (v1.8.0; Caporaso et al., 2010), respectively, at the OTU level. Principal component analysis (PCA) in OTUs was plotted with the R package “ade4.” The difference in alpha diversity among groups was compared using Kruskal-Test, with values of p ≤ 0.05 considered statistically significant (*, 0.01 6< p ≤ 0.05; **, 0.001 < p ≤ 0.01; ***, p ≤ 0.001). We used PICRUSt to obtain the KO corresponding to the OTU through the greengene ID corresponding to each OTU, and calculated the abundance of the KO from the sum of the abundances of the OTU corresponding to the KO. We calculated the abundance of each functional category based on the information in the KEGG database and the OTU abundance information. In addition, PICRUSt was used to obtain the levels of metabolic pathway information, and the abundance of each level was obtained.
In order to explore the phylogenetic relationship of the two most abundant bacterial genera, we compared the Erwinia and Acinetobacter DNA sequences obtained by high-throughput sequencing in the NCBI nucleotide (nr) database. Six 16S rRNA fragments belonging to Erwinia and 16 16S rRNA fragments belonging to Acinetobacter were downloaded from GenBank to construct a phylogenetic tree. The phylogenetic tree analysis of 249 base pairs was carried out. Using MEGA7.0, the phylogenetic tree was constructed by Neighbor-joining method (1,000 bootstraps).
The bacteria of A. suturalis were analyzed by Illumina HiSeq via the sequencing of the 16S rRNA gene. We obtained a total of 800,836 raw reads and 707,474 clean reads, with an average length of 296 bp. Based on 97% species similarity, we clustered the spliced tags into OTU. The number of OTUs at each developmental stage is detailed in Table 1. We constructed dilution curves for Ace, Chao1, Shannon, Simpson, Good’s Coverage and Observed species, which demonstrated the quality and credibility of sequencing quantity (Supplementary Figure 1). Good’s coverage of all samples was above 99%, indicating that our sequencing results were sufficient to fully estimate the diversity of A. suturalis bacterial community (Table 1).
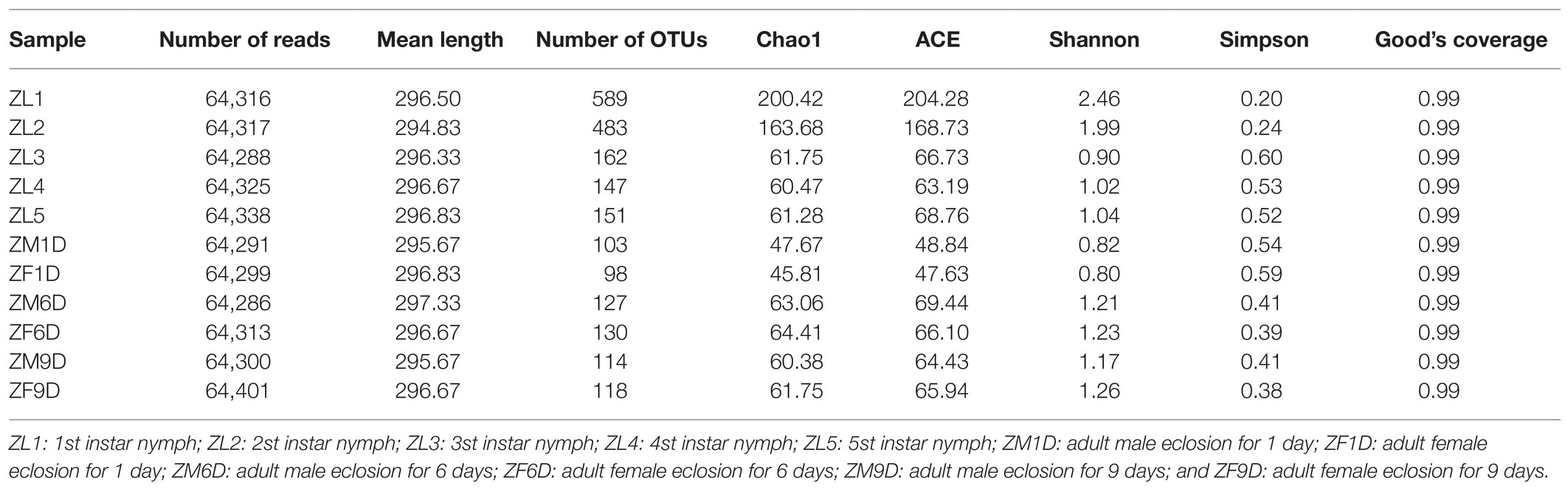
Table 1. 16S rRNA gene sequencing data.
Nymphs had higher species richness in ZL1 and ZL2 periods, and were significantly higher than other periods (Table 1; Figure 1A). Proteobacteria and Firmicutes were the dominant phyla during the nymphal stage, and their relative abundances were 85.57% (average value across all of the samples at nymphal stage) and 11.34%, respectively (Figure 1B). We compared the changes of A. suturalis nymphs in different developmental stages at the genus level. As the nymphs grew, the microbial community changed significantly. There were 63 common OTUs classifications in the five developmental stages of nymphs, and each developmental stage had characteristic OTUs (Figure 1C). Erwinia (17.65%) (average value across all of the samples at the nymphal stage), Staphylococcus (9.83%), and Acinetobacter (9.83%) were the top three genera in relative abundance during the nymphal stages. Erwinia was the dominant genus of bacteria throughout the nymph period, and the microbial community was relatively stable without significant dynamic changes. However, the relative abundance of Staphylococcus, Acinetobacter, Pseudomonas, and Corynebacterium changed significantly (Figure 1D). The relative abundance of Staphylococcus increased significantly, the lowest in the ZL1 (4.98%) stage, and the highest in the ZL5 (13.98%) stage. The trends for Acinetobacter, Pseudomonas, and Corynebacterium were opposite to those of Staphylococcus. Acinetobacter had the highest abundance in the ZL1 (24.69%) stage, and then gradually decreased (Figure 1D).
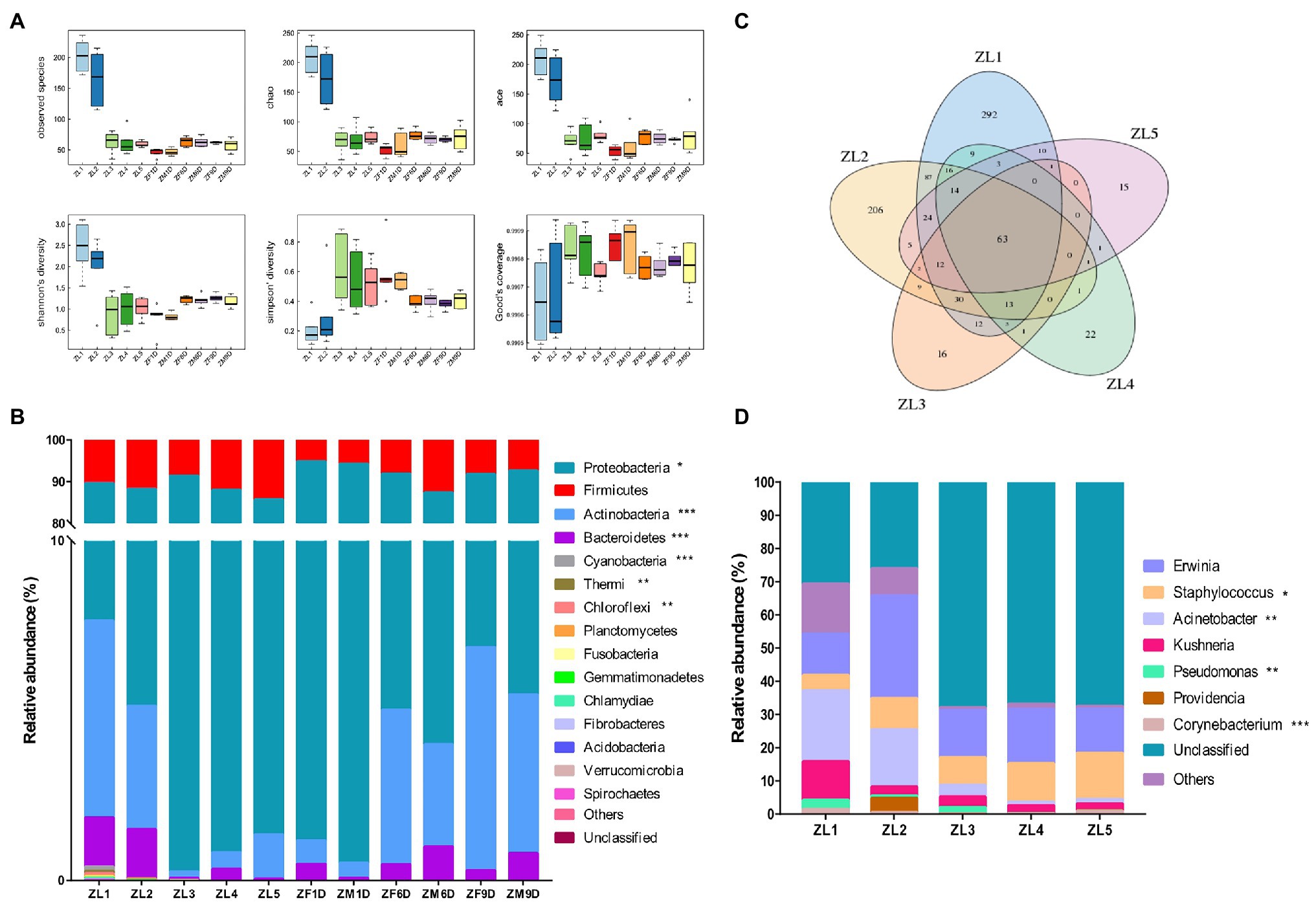
Figure 1. Bacterial community dynamics among different developmental stages in Adelphocoris suturalis. (A) Boxplot of α-diversity measured by the six indexs. (B) Relative abundance of bacteria communities at the phylum level in different groups. (C) Venn diagram showing operational taxonomic unit (OTU) classification in nymphal period. (D) Relative abundance of bacteria communities at the genus level in nymph stages. (Bacteria with relative abundance lower than 0.5% in all samples were all merged into others. Kruskal-Wallis test, *0.01 < p ≤ 0.05, **0.001 < p ≤ 0.01, and ***p ≤ 0.001).
As in the nymph stage, the Proteobacteria (88.31%; average value across all of the samples at adult stage) and Firmicutes (7.84%) were the dominant phyla in the adult stage, and the species richness of each developmental stage was similar (Figure 1B). Similarly, we analyzed the bacterial communities of A. suturalis adults at different developmental stages at the genus level. Erwinia (38.42%; average value across all of the samples at adult stage) and Lactococcus (5.53%) were the dominant genus in the entire adult stage and their relative abundance was relatively stable, without significant dynamic changes (Figure 2A). Interestingly, although the relative abundance of Corynebacterium was low, it was significantly increased, reaching the highest in ZF9D (5.41%) during the adult stage. Compared with females and males, only Staphylococcus had a significant difference on the 6th day of adult development. The relative abundance of Staphylococcus ZM6D (6.90%) was significantly higher than that of ZF6D (1.55%), and there was no significant difference in other periods (Figure 2A).
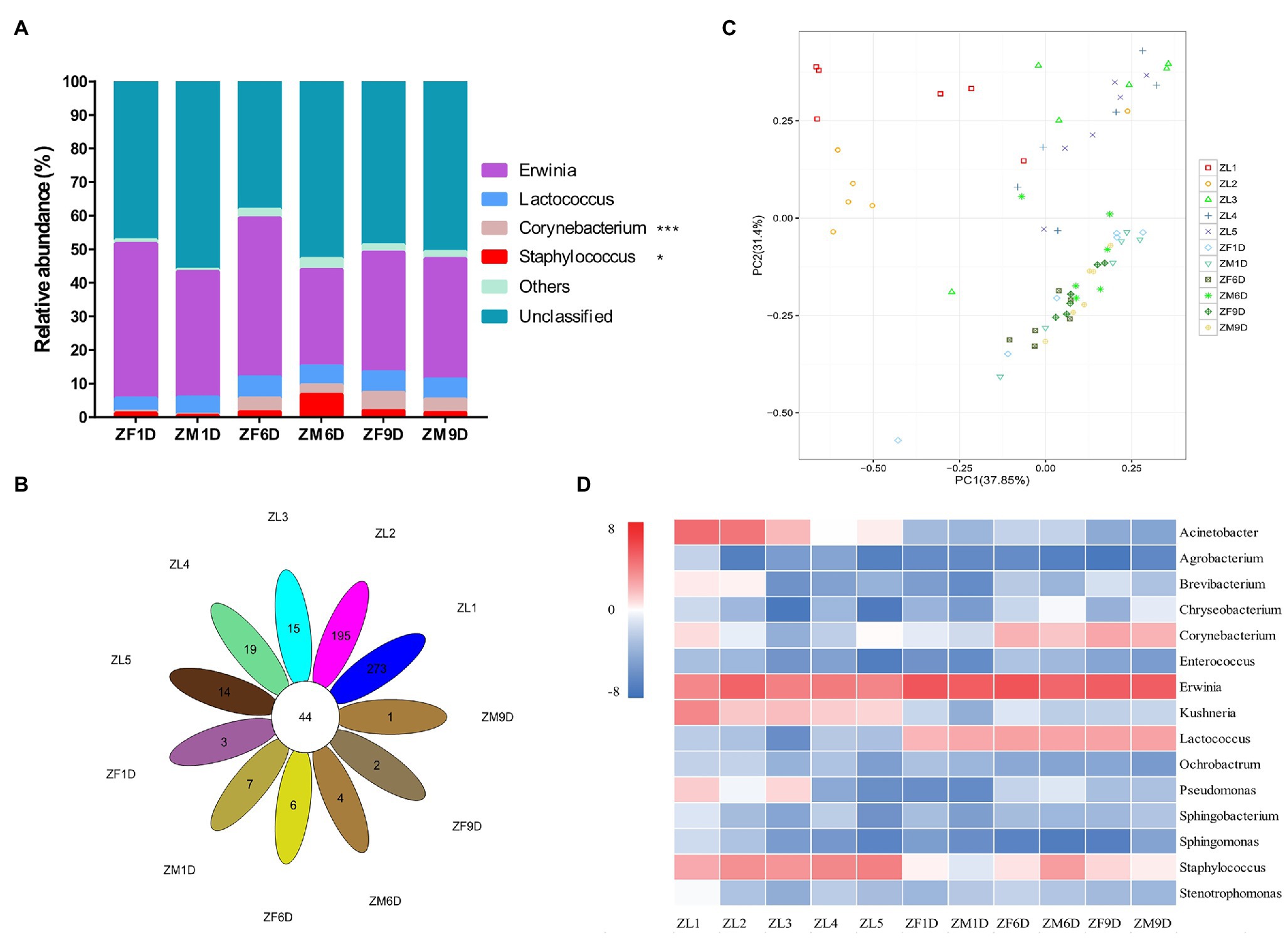
Figure 2. Bacterial community dynamics among different developmental stages in A. suturalis. (A) Relative abundance of bacteria communities at the genus level in adult stages. (B) Core-Pan OTU presents the common and unique OTU of all samples in petal diagram. (C) Difference of OTU types in different developmental stages based on Principal Components Analysis (PCA). (D) Heat map analysis of the top 15 microbial populations with relative abundance at different developmental stages. The data represented by color in the figure is represented by log2(relative abundance). Bacteria with relative abundance lower than 0.5% in all samples were all merged into others (Kruskal-Wallis test, *0.01 < p ≤ 0.05 and ***p ≤ 0.001).
Species richness of ZL1 and ZL2 was the highest (Figure 1A), with 44 common OTU species in all samples, and the unique OTU species of ZL1 and ZL2 was also the highest (Figure 2B). PCA analysis demonstrated that the OTUs of A. suturalis in different development stages showed dispersion and aggregation. The degree of dispersioncan show whether the sample composition under the same conditions is similar (Figure 2C). Each point on the PCA graph represented a stage of development; the closer the distance, the more similar the composition.
At the phylum level, taxonomic analysis of all samples showed that Proteobacteria was the most prevalent phylum. Among all samples, the top three phyla with the highest relative abundance were Proteobacteria, Firmicutes, and Actinobacteria, accounting for 87.06, 9.43, and 2.8% (average value across all of the samples), respectively. Proteobacteria had the highest abundance in adult stage compared with nymph stage, with the highest relative abundance in ZM1D (93.77%) stage of adult stage (Figure 1B). Throughout the different development stages of A. suturalis, the relative abundance of Proteobacteria increased significantly in the nymph stage, and in ZL3 (91.15%) the relative abundance of the period was the highest. After the first day of adult emergence, the relative abundance of the Proteobacteria showed a decreasing trend (Figure 1B). Compared with Proteobacteria, the change trend of Actinobacteria was the opposite. The relative abundance of Actinobacteria decreased significantly in the nymph stage, and then increased significantly from the first day of adult emergence. The relative abundance of Proteobacteria and Actinobacteria at 6 days (Proteobacteria, ZF6D: 86.86%, ZM6D: 83.39%; Actinobacteria, ZF6D: 4.55%, and ZM6D: 3.01%) and 9 days (Proteobacteria, ZF9D: 84.92%, ZM9D: 87.17%; Actinobacteria, ZF9D: 6.57%, and ZM9D: 4.67%) of adult stage was similar to that at nymph ZL1 (Proteobacteria: 81.97%; Actinobacteria: 5.80%) stage. Firmicutes were also ubiquitous in each developmental stage, and their relative abundance was relatively stable without significant change.
We selected the bacterial genera with the top 15 abundance ratios and drew heat maps based on their relative abundance at different developmental stages (Figure 2D; Supplementary Table 1). From the overall distribution of the microbial community at different developmental stages of A. suturalis, Erwinia (28.98%; average value across all of the samples) was still the dominant genus (Figure 2D; Supplementary Table 1). Acinetobacter, Kushneria, and Staphylococcus were relatively abundant in the nymph stage, and the number was very small in the adult stage. The abundance ratio of Lactococcus during the adult stage was significantly higher than that of the nymph, and the relative abundance during the nymph stage was all below 0.3% (Figure 2D).
Based on the predicted results of KEGG function, we showed the pathway abundance at two levels (level 1 and level 2). In level 1, metabolism accounted for about 41.61–47.01% at each developmental stage, followed by environmental information processing (16.46–21.61%) and genetic information processing (15.02–16.02%; Figure 3A). In level 2, we showed the richness of the top 20 pathways, and the other pathways are classified as Others (Figure 3B). The relative abundance of membrane transport (13.89–18.64%) was the highest at different developmental stages. In addition, pathways related to metabolism account for the vast majority, and carbohydrate metabolism, amino acid metabolism, energy metabolism, metabolism of cofactors and vitamins, nucleotide metabolism, lipid metabolism were abundantly enriched. The phylogenetic tree indicated the developmental relationship between Erwinia and Acinetobacter, the two important genera in our study, and the more closely related genera (Figure 4).
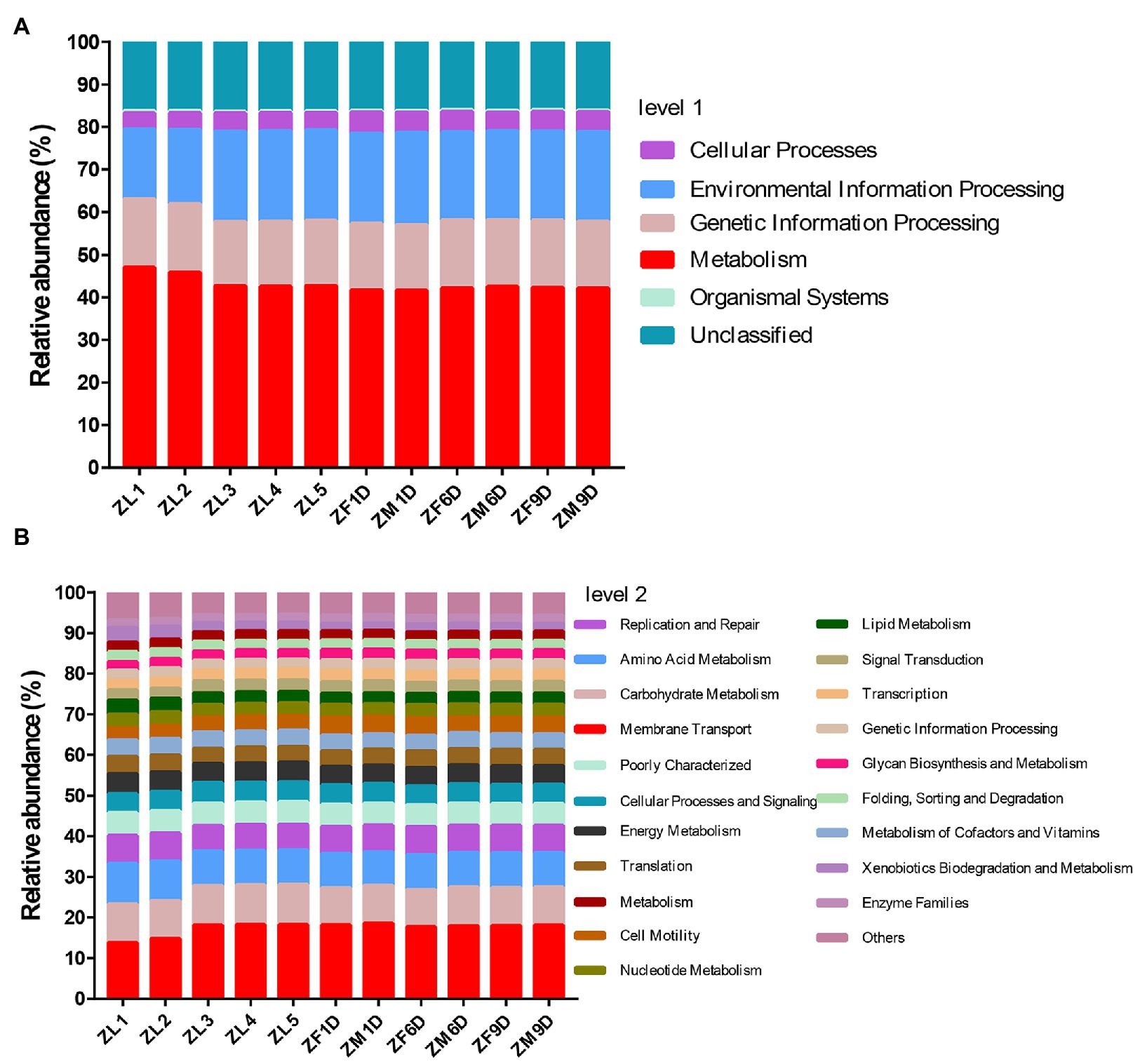
Figure 3. Prediction of functional pathway abundance at different developmental stages. (A) Function prediction based on Level 1. (B) Function prediction based on Level 2. In level 2, pathways ranked below 20 in total relative abundance were classified as “Others.”
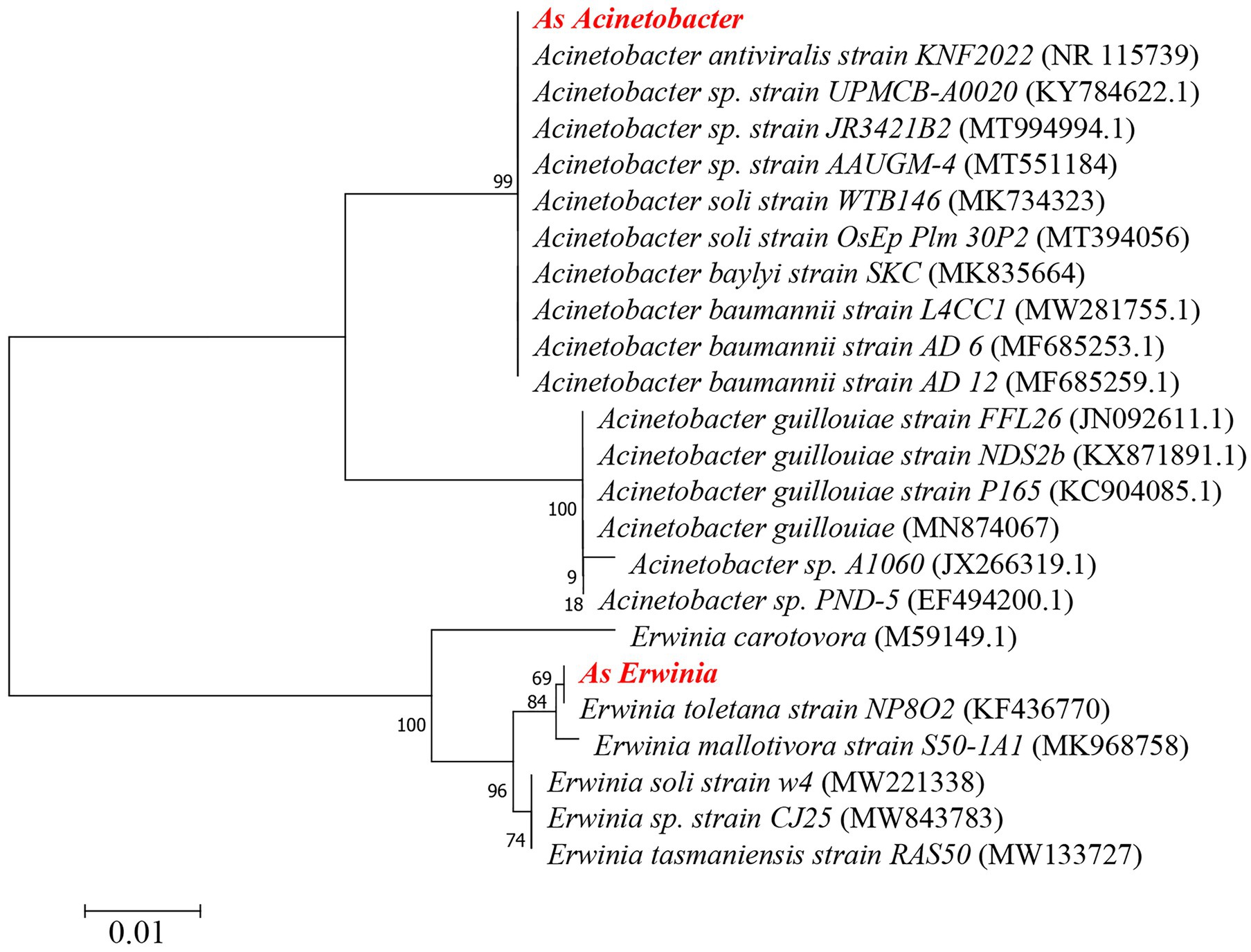
Figure 4. Phylogenetic analysis of Erwinia and Acinetobacter. As Erwinia: A. suturalis Erwinia, As Acinetobacter: A. suturalis Acinetobacter. The phylogenetic tree was made by MEGA7.0 software and constructed by the Neighbor-joining method. The number in parentheses indicates the GeneBank accession number of the 16S rRNA gene sequence.
Most insects contain a large number of symbiotic bacteria (Bosch and McFall-Ngai, 2011; Herzner et al., 2013; Ruffner et al., 2013; Marchesi et al., 2016). Although the microbial diversity of insects has been extensively studied, most of the research has focused on insect gut microbes (Roh et al., 2008; Hulcr et al., 2012; Salem et al., 2013), and there are few reports on the dynamic changes of microbial diversity and species richness in different developmental stages of insects. However, the vast majority of insect symbiotic bacteria are concentrated in the intestinal tract, which is rich in symbiotic bacterial communities (Gupta and Nair, 2020). Similarly, although our study detected the microbial community of the entire insect body, intestinal microbes accounted for almost all of them (Campbell et al., 2018; Karimi et al., 2019; Rossitto De Marchi and Smith, 2020). These bacteria and host insects have formed an interdependent relationship during the co-evolution process (Gao et al., 2019). However, we have not found any endosymbionts in A. suturalis, although endosymbionts have been widely reported in other Hemipterans (Campbell et al., 2018; Karimi et al., 2019; Rossitto De Marchi and Smith, 2020).
Interestingly, it can be observed from the aspect of microbial diversity that the first and second instar microbial community diversity of newly hatched nymphs is the highest, and its unique OTU species are also abundant. From the third instar nymph, the diversity of the microbial community began to decline. It is well known that bacterial diversities vary from field collected to lab-reared (Rani et al., 2009) as well as across different geographical regions (Zouache et al., 2011). In addition, the composition and diversity of bacteria are affected by the in vitro environment (Zouache et al., 2011) and artificial feed (Priya et al., 2012). It has been confirmed in Helicoverpa armigera and Lymantria dispar that food sources have a great influence on the microbial diversity of insects (Broderick et al., 2004; Priya et al., 2012). Adelphocoris suturalis began to prey on cotton aphid from the third instar nymph stage, which may be one of the reasons for the changes in microbial diversity, or it may be that the nymphal microbial community becomes highly simplified through the development of the host insect. Starting from the third instar nymph stage, the alpha diversity decreased significantly (Figure 1A), and diet is the best explanation for this effect. This is consistent with the results of recent studies on the influence of A. suturalis diet on microbial diversity (Luo et al., 2021). An increase in nutrient richness (such as protein quality) may lead to a decrease in alpha diversity, although refined diets are also associated with increased species richness (Erkosar et al., 2018; Kešnerová et al., 2020). PCA analysis also confirmed the difference of species diversity mentioned above. During the transition period from fifth instar nymph to adult, there was no significant change in alpha diversity, because A.suturalis is an incomplete metamorphosis insect. Nymphs and adults have small changes in their living environment, feeding habits, and food sources, which lead to changes in the intestinal bacterial community (Engel and Moran, 2013; Hammer et al., 2017; Luo et al., 2021). Complete metamorphosis involves complex structural changes, which leads to significant changes in microbial diversity from egg to adult (Chen et al., 2016; Hammer et al., 2017; Zhao et al., 2019).
We also found that Proteobacteria and Firmicutes dominated across the entire life cycle, which was similar to findings in other Hemipteran insects (Husseneder et al., 2017; Lim and Ab Majid, 2020), each of which contained a different proportion of microbes depending on the species or sample (Figure 1B). Similar results have been found in other insects (Chandler et al., 2011; Colman et al., 2012; Engel et al., 2012). These phyla are often listed as the most abundant bacterial communities associated with insect taxa (Colman et al., 2012; Thomas et al., 2013; Yun et al., 2014; Kim et al., 2017). Firmicutes and Proteobacteria are key to maintaining the growth and development of insects during the metabolism of secondary metabolites in host plants (Dillon and Charnley, 2002).
Erwinia in the phylum Proteobacteria is a genus of dominant bacteria in the nymphal and adult stages, a member of the Gram-negative Enterobacteriaceae family (Basset et al., 2000), and is a type of intestinal bacteria. Throughout the development cycle of A. suturalis, the Erwinia population was stable and continuous, with high abundance, indicating that Erwinia plays a lasting symbiotic role in the growth, development, and survival of A. suturalis. Erwinia can metabolize most sources of nitrogen, sulfur, and phosphorus (Friedl et al., 2008), and is thus an important microbial species in the intestinal tract of insects. Adelphocoris suturalis is a highly omnivorous insect (Luo et al., 2017b). Erwinia can enhance the adaptability of insect hosts to their plant hosts by regulating the diet of insect hosts (De Vries et al., 2004), which is critical for omnivorous insects. Erwinia have strong metabolic ability, which plays an important role in the digestion of food and body development of A. suturalis. Interestingly, Erwinia can secrete a variety of cell wall degrading enzymes, causing potato black leg disease, soft rot, fusarium wilt, and other plant diseases (Whitehead et al., 2002; Grenier et al., 2006). Drosophila melanogaster has been reported to be an important media for Erwinia carotovora (Vieira et al., 2020). The Hemiptera insect Creontiades signatus is a vector for the transmission of bacterial pathogenic bacteria Serratia marcescens (Bizio; Enterobacteriales: Enterobacteriaceae) that rots cotton seeds and bolls (Glover et al., 2020). Further research is needed to determine whether A.suturalis is a vector for the bacteria. Acinetobacter has a very high abundance in the nymph stage, but extremely low abundance in the adult stage. The vast majority of Acinetobacter bacteria have strong drug resistance (Li et al., 2021). In China, due to the large-scale planting of Bt crops, A. suturalis has risen from secondary pests in cotton fields to primary pests. Among them, Cry1, Cry2, and Cry9 toxins have been reported to show high insecticidal activity against lepidopteran pests (Palma et al., 2014; Silva et al., 2015). Once ingested by the susceptible insect larvae, these cry proteins (present in the form of protoxin) are proteolytically processed by midgut proteases to the active toxin that subsequently binds to specific protein receptors of the midgut epithelium leading to cell disruption and eventual death of the insect larvae (Pardo-López et al., 2013). The introduction of the intestinal isolate Acinetobacter guillouiae into Plutella xylostella significantly enhances its sensitivity to Bt Cry1Ac protoxin, and Acinetobacter plays an important role in the immune response of insects (Li et al., 2021). During the nymph period, its ability to resist the stimulation of external agents is relatively weak. It requires the support of symbiotic bacteria in the body to defend against unfavorable pesticide environments and Bt crops. We analyzed the developmental relationship of Erwinia and Acinetobacter with the aforementioned E. carotovora and A. guillouiae through phylogenetic tree development. As Erwinia and Erwinia toletana strain NP8O2 are the most closely related in evolution. At the same time, As Erwinia and E. carotovora have 94.47% identity. Although As Acinetobacter and A. guillouiae are not on the same branch, they have 94.86% identity. Staphylococcus and Lactococcus are the two genera with the highest relative abundance in Firmicutes. The content of Staphylococcus increased significantly in the nymph stage, and the relative abundance was the highest in ZL5 (13.98%) stage, but it was scarce in adult stage. Lactococcus has a relatively high abundance in the adult stage and exists in large numbers in the intestines (De Jonge et al., 2020). This type of bacteria is known for fermenting complex molecular carbohydrates to produce lactic acid. Lactococcus is a lactobacillus of firmicutes with high abundance in both male and female adults, which can decompose sugar to produce organic acids, reduce the pH value of its environment, and defend against some acid-sensitive pathogenic bacteria (Evans and Armstrong, 2006). This ensures that A. suturali can obtain the nutrients it needs in the complex environment. Our functional prediction also confirmed that in different stages of development, metabolic function is the main, whether amino acid metabolism or carbohydrate metabolism, is very important for the survival of insects.
Our results showed that the dominant bacteria genera (Erwinia, Acinetobacter, Staphylococcus, and Lactococcus) of Proteobacteria and Firmicutes were mostly concentrated in the intestinal tract of insects. These bacteria played an important role in nutrient uptake and adaptability to the environment, and were directly related to the growth, development, and reproduction of the insects. We sequenced the A. suturalis 16S rRNA gene through Illumina HiSeq, which directly revealed the structure of the bacterial community in the life cycle of A. suturali, predicted the biological functions of different bacterial communities, and provided a basis for further research on the role of bacteria in this and other insects. We provide a crucial theoretical basis for future research on A. suturalis symbiotic bacteria. These foundations can help formulate environmentally friendly management strategies for pest control and provide ideas for new pest control strategies.
The datasets presented in this study cans be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA662509.
JC and XG: conceptualization and writing – review and editing. XG: methodology. HX, LN, and CW: software. JC: validation and funding acquisition. HX: formal analysis and writing – original draft preparation. HX and XZ: investigation. JL: resources. HX, JJ, and XG: data curation. LW and DL: visualization. KZ: supervision and project administration. All authors contributed to the article and approved the submitted version.
This research was supported by National Special Transgenic Project of China (No. 2016ZX08012-004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.670383/full#supplementary-material
Supplementary Table 1 | Relative abundance of bacteria communities at the genus level in different group (Top 15).
Supplementary Figure 1 | Alpha diversity dilution curve.
Arbuthnott, D., Levin, T. C., and Promislow, D. E. (2016). The impacts of Wolbachia and the microbiome on mate choice in Drosophila melanogaster. J. Evol. Biol. 29, 461–468. doi: 10.1111/jeb.12788
Basset, A., Khush, R. S., Braun, A., Gardan, L., Boccard, F., Hoffmann, J. A., et al. (2000). The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc. Natl. Acad. Sci. U. S. A. 97, 3376–3381. doi: 10.1073/pnas.97.7.3376
Bosch, T. C., and Mcfall-Ngai, M. J. (2011). Metaorganisms as the new frontier. Zoology 114, 185–190. doi: 10.1016/j.zool.2011.04.001
Broderick, N. A., Raffa, K. F., Goodman, R. M., and Handelsman, J. (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 70, 293–300. doi: 10.1128/AEM.70.1.293-300.2004
Broderick, N. A., Raffa, K. F., and Handelsman, J. (2006). Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl. Acad. Sci. U. S. A. 103, 15196–15199. doi: 10.1073/pnas.0604865103
Campbell, M. A., Łukasik, P., Meyer, M. C., Buckner, M., Simon, C., Veloso, C., et al. (2018). Changes in endosymbiont complexity drive host-level compensatory adaptations in Cicadas. MBio 9, e02104–e02118. doi: 10.1128/mBio.02104-18
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
Chen, B., Teh, B. S., Sun, C., Hu, S., Lu, X., Boland, W., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Colman, D. R., Toolson, E. C., and Takacs-Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacteria communities? Mol. Ecol. 21, 5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x
De Jonge, N., Michaelsen, T. Y., Ejbye-Ernst, R., Jensen, A., Nielsen, M. E., Bahrndorff, S., et al. (2020). Housefly (Musca domestica L.) associated microbiota across different life stages. Sci. Rep. 10:7842. doi: 10.1038/s41598-020-64704-y
De Vries, E. J., Jacobs, G., Sabelis, M. W., Menken, S. B., and Breeuwer, J. A. (2004). Diet-dependent effects of gut bacteria on their insect host: the symbiosis of Erwinia sp. and western flower thrips. Proc. Biol. Sci. 271, 2171–2178. doi: 10.1098/rspb.2004.2817
Dillon, R., and Charnley, K. (2002). Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res. Microbiol. 153, 503–509. doi: 10.1016/S0923-2508(02)01361-X
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. doi: 10.1111/j.1365-2435.2008.01442.x
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engel, P., Martinson, V. G., and Moran, N. A. (2012). Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U. S. A. 109, 11002–11007. doi: 10.1073/pnas.1202970109
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects - diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Engl, T., and Kaltenpoth, M. (2018). Influence of microbial symbionts on insect pheromones. Nat. Prod. Rep. 35, 386–397. doi: 10.1039/C7NP00068E
Erkosar, B., Yashiro, E., Zajitschek, F., Friberg, U., Maklakov, A. A., Van Der Meer, J. R., et al. (2018). Host diet mediates a negative relationship between abundance and diversity of Drosophila gut microbiota. Ecol. Evol. 8, 9491–9502. doi: 10.1002/ece3.4444
Evans, J. D., and Armstrong, T. N. (2006). Antagonistic interactions between honey bee bacterial symbionts and implications for disease. BMC Ecol. 6:4. doi: 10.1186/1472-6785-6-4
Friedl, M. A., Kubicek, C. P., and Druzhinina, I. S. (2008). Carbon source dependence and photostimulation of conidiation in Hypocrea atroviridis. Appl. Environ. Microbiol. 74, 245–250. doi: 10.1128/AEM.02068-07
Gao, X., Li, W., Luo, J., Zhang, L., Ji, J., Zhu, X., et al. (2019). Biodiversity of the microbiota in Spodoptera exigua (Lepidoptera: Noctuidae). J. Appl. Microbiol. 126, 1199–1208. doi: 10.1111/jam.14190
Glover, J. P., Medrano, E. G., Isakeit, T., and Brewer, M. J. (2020). Transmission of cotton seed and boll rotting bacteria by the verde plant bug (Hemiptera: Miridae). J. Econ. Entomol. 113, 793–799. doi: 10.1093/jee/toz334
Grenier, A. M., Duport, G., Pagès, S., Condemine, G., and Rahbé, Y. (2006). The phytopathogen Dickeya dadantii (Erwinia chrysanthemi 3937) is a pathogen of the pea aphid. Appl. Environ. Microbiol. 72, 1956–1965. doi: 10.1128/AEM.72.3.1956-1965.2006
Grenier, T., and Leulier, F. (2020). How commensal microbes shape the physiology of Drosophila melanogaster. Curr. Opin. Insect Sci. 41, 92–99. doi: 10.1016/j.cois.2020.08.002
Gupta, A., and Nair, S. (2020). Dynamics of insect-microbiome interaction influence host and microbial symbiont. Front. Microbiol. 11:1357. doi: 10.3389/fmicb.2020.01357
Hammer, T. J., Janzen, D. H., Hallwachs, W., Jaffe, S. P., and Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proc. Natl. Acad. Sci. U. S. A. 114, 9641–9646. doi: 10.1073/pnas.1707186114
Herzner, G., Schlecht, A., Dollhofer, V., Parzefall, C., Harrar, K., Kreuzer, A., et al. (2013). Larvae of the parasitoid wasp Ampulex compressa sanitize their host, the American cockroach, with a blend of antimicrobials. Proc. Natl. Acad. Sci. U. S. A. 110, 1369–1374. doi: 10.1073/pnas.1213384110
Horak, R. D., Leonard, S. P., and Moran, N. A. (2020). Symbionts shape host innate immunity in honeybees. Proc. Biol. Sci. 287:20201184. doi: 10.1098/rspb.2020.1184
Hulcr, J., Rountree, N. R., Diamond, S. E., Stelinski, L. L., Fierer, N., and Dunn, R. R. (2012). Mycangia of ambrosia beetles host communities of bacteria. Microb. Ecol. 64, 784–793. doi: 10.1007/s00248-012-0055-5
Husseneder, C., Park, J. S., Howells, A., Tikhe, C. V., and Davis, J. A. (2017). Bacteria associated with Piezodorus guildinii (Hemiptera: Pentatomidae), with special reference to those transmitted by feeding. Environ. Entomol. 46, 159–166. doi: 10.1093/ee/nvw112
Jing, T. Z., Qi, F. H., and Wang, Z. Y. (2020). Most dominant roles of insect gut bacteria: digestion, detoxification, or essential nutrient provision? Microbiome 8:38. doi: 10.1186/s40168-020-00823-y
Karimi, S., Askari Seyahooei, M., Izadi, H., Bagheri, A., and Khodaygan, P. (2019). Effect of Arsenophonus endosymbiont elimination on fitness of the date palm hopper, Ommatissus lybicus (Hemiptera: Tropiduchidae). Environ. Entomol. 48, 614–622. doi: 10.1093/ee/nvz047
Kešnerová, L., Emery, O., Troilo, M., Liberti, J., Erkosar, B., and Engel, P. (2020). Gut microbiota structure differs between honeybees in winter and summer. ISME J. 14, 801–814. doi: 10.1038/s41396-019-0568-8
Kikuchi, Y., Hayatsu, M., Hosokawa, T., Nagayama, A., Tago, K., and Fukatsu, T. (2012). Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. U. S. A. 109, 8618–8622. doi: 10.1073/pnas.1200231109
Kikuchi, Y., Hosokawa, T., and Fukatsu, T. (2007). Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl. Environ. Microbiol. 73, 4308–4316. doi: 10.1128/AEM.00067-07
Kim, J. M., Choi, M.-Y., Kim, J.-W., Lee, S. A., Ahn, J.-H., Song, J., et al. (2017). Effects of diet type, developmental stage, and gut compartment in the gut bacterial communities of twoCerambycidae species(Coleoptera). J. Microbiol. 55, 21–30. doi: 10.1007/s12275-017-6561-x
Li, S., Xu, X., De Mandal, S., Shakeel, M., Hua, Y., Shoukat, R. F., et al. (2021). Gut microbiota mediate Plutella xylostella susceptibility to Bt Cry1Ac protoxin is associated with host immune response. Environ. Pollut. 271:116271. doi: 10.1016/j.envpol.2020.116271
Lim, L., and Ab Majid, A. H. (2020). Metagenomic 16S rDNA amplicon data of microbial diversity of guts of fully fed tropical bed bugs, Cimex hemipterus (F.) (Hemiptera: Cimicidae). Data Brief 30:105575. doi: 10.1016/j.dib.2020.105575
Liu, W., Hajano, J. U., and Wang, X. (2018). New insights on the transmission mechanism of tenuiviruses by their vector insects. Curr. Opin. Virol. 33, 13–17. doi: 10.1016/j.coviro.2018.07.004
Lu, Y. H., Qiu, F., Feng, H. Q., Li, H. B., Yang, Z. C., Wyckhuys,, et al. (2008). Species composition and seasonal abundance of pestiferous plant bugs (Hemiptera: Miridae) on Bt cotton in China. Crop Prot. 27, 465–472. doi: 10.1016/j.cropro.2007.07.017
Lu, Y., and Wu, K. (2011). Mirid bugs in China: Pest status and management strategies. Outlooks Pest Manage. 22, 248–252. doi: 10.1564/22dec02
Lu, Z. X., Yu, X. P., Chen, J. M., Zheng, X. S., Xu, H. X., Tao, L. Y., et al. (2001). Role of endosymbiote in virulence change of the brown planthopper to rice resistant varieties. Acta Entomol. Sin. 44, 197–204.
Luo, J., Cheng, Y., Guo, L., Wang, A., Lu, M., and Xu, L. (2021). Variation of gut microbiota caused by an imbalance diet is detrimental to bugs’ survival. Sci. Total Environ. 771:144880. doi: 10.1016/j.scitotenv.2020.144880
Luo, J., Li, Z., Ma, C., Zhang, Z., Hull, J. J., Lei, C., et al. (2017a). Knockdown of a metathoracic scent gland desaturase enhances the production of (E)-4-oxo-2-hexenal and suppresses female sexual attractiveness in the plant bug Adelphocoris suturalis. Insect Mol. Biol. 26, 642–653. doi: 10.1111/imb.12325
Luo, J., Liang, S., Li, J., Xu, Z., Li, L., Zhu, B., et al. (2017b). A transgenic strategy for controlling plant bugs (Adelphocoris suturalis) through expression of double-stranded RNA homologous to fatty acyl-coenzyme A reductase in cotton. New Phytol. 215, 1173–1185. doi: 10.1111/nph.14636
Ma, M., Guo, L., Tu, C., Wang, A., Xu, L., and Luo, J. (2021). Comparative analysis of Adelphocoris suturalis Jakovlev (Hemiptera: Miridae) immune responses to fungal and bacterial pathogens. Front. Physiol. 12:646721. doi: 10.3389/fphys.2021.646721
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Marchesi, J. R., Adams, D. H., Fava, F., Hermes, G. D., Hirschfield, G. M., Hold, G., et al. (2016). The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339. doi: 10.1136/gutjnl-2015-309990
Moran, N. A., Mccutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Palma, L., Muñoz, D., Berry, C., Murillo, J., and Caballero, P. (2014). Bacillus thuringiensis toxins: an overview of their biocidal activity. Toxins 6, 3296–3325. doi: 10.3390/toxins6123296
Pardo-López, L., Soberón, M., and Bravo, A. (2013). Bacillus thuringiensis insecticidal three-domain cry toxins: mode of action, insect resistance and consequences for crop protection. FEMS Microbiol. Rev. 37, 3–22. doi: 10.1111/j.1574-6976.2012.00341.x
Priya, N. G., Ojha, A., Kajla, M. K., Raj, A., and Rajagopal, R. (2012). Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS One 7:e30768. doi: 10.1371/journal.pone.0030768
Qin, F., Liu, W., Wu, N., Zhang, L., Zhang, Z., Zhou, X., et al. (2018). Invasion of midgut epithelial cells by a persistently transmitted virus is mediated by sugar transporter 6 in its insect vector. PLoS Pathog. 14:e1007201. doi: 10.1371/journal.ppat.1007201
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
Roh, S. W., Nam, Y. D., Chang, H. W., Kim, K. H., Kim, M. S., Ryu, J. H., et al. (2008). Phylogenetic characterization of two novel commensal bacteria involved with innate immune homeostasis in Drosophila melanogaster. Appl. Environ. Microbiol. 74, 6171–6177. doi: 10.1128/AEM.00301-08
Rossitto De Marchi, B., and Smith, H. A. (2020). Bacterial endosymbiont diversity among Bemisia tabaci (Hemiptera: Aleyrodidae) populations in Florida. Insects 11:179. doi: 10.3390/insects11030179
Ruffner, B., Péchy-Tarr, M., Ryffel, F., Hoegger, P., Obrist, C., Rindlisbacher, A., et al. (2013). Oral insecticidal activity of plant-associated pseudomonads. Environ. Microbiol. 15, 751–763. doi: 10.1111/j.1462-2920.2012.02884.x
Russell, C. W., Poliakov, A., Haribal, M., Jander, G., Van Wijk, K. J., and Douglas, A. E. (2014). Matching the supply of bacterial nutrients to the nutritional demand of the animal host. Proc. Biol. Sci. 281:20141163. doi: 10.1098/rspb.2014.1163
Salem, H., Kreutzer, E., Sudakaran, S., and Kaltenpoth, M. (2013). Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae). Environ. Microbiol. 15, 1956–1968. doi: 10.1111/1462-2920.12001
Santos-Garcia, D., Silva, F. J., Morin, S., Dettner, K., and Kuechler, S. M. (2017). The all-rounder sodalis: A new bacteriome-associated endosymbiont of the Lygaeoid bug Henestaris halophilus (Heteroptera: Henestarinae) and a critical examination of its evolution. Genome Biol. Evol. 9, 2893–2910. doi: 10.1093/gbe/evx202
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Silva, M. C., Siqueira, H. A., Silva, L. M., Marques, E. J., and Barros, R. (2015). Cry proteins from Bacillus thuringiensis active against diamondback moth and fall armyworm. Neotrop. Entomol. 44, 392–401. doi: 10.1007/s13744-015-0302-9
Thomas, J. R., Gonzales, S. L., Noah, F., and Anthony, G. J. (2013). A cross-taxon analysis of insect-associated bacterial diversity. PLoS One 8:e61218. doi: 10.1371/journal.pone.0061218
Vieira, F. J. D., Nadal-Jimenez, P., Teixeira, L., and Xavier, K. B. (2020). Erwinia carotovora quorum sensing system regulates host-specific virulence factors and development delay in Drosophila melanogaster. MBio 11, e01292–e01220. doi: 10.1128/mBio.01292-20
Wang, X., Liu, Q., Meissle, M., Peng, Y., Wu, K., Romeis, J., et al. (2018). Bt rice could provide ecological resistance against nontarget planthoppers. Plant Biotechnol. J. 16, 1748–1755. doi: 10.1111/pbi.12911
Wang, H., Wu, K., Liu, Y., Wu, Y., and Wang, X. (2015). Integrative proteomics to understand the transmission mechanism of barley yellow dwarf virus-GPV by its insect vector Rhopalosiphum padi. Sci. Rep. 5:10971. doi: 10.1038/srep10971
Whitehead, N. A., Byers, J. T., Commander, P., Corbett, M. J., Coulthurst, S. J., Everson, L., et al. (2002). The regulation of virulence in phytopathogenic Erwinia species: quorum sensing, antibiotics and ecological considerations. Antonie Van Leeuwenhoek 81, 223–231. doi: 10.1023/A:1020570802717
Wielkopolan, B., and Obrępalska-Stęplowska, A. (2016). Three-way interaction among plants, bacteria, and coleopteran insects. Planta 244, 313–332. doi: 10.1007/s00425-016-2543-1
Wu, D., Lin, F. M., Lu, Y. H., Liu, Y., Zhang, Y. J., Wu, K. M., et al. (2010). Selective preferences of Apolygus lucorum and Adelphocoris suturalis (Hemiptera: Miridae) to cotton plants with different resistance levels and damaging treatments. Acta Entomol. Sin. 53, 696–701. doi: 10.1016/S1002-0721(10)60377-8
Wu, K. M., Lu, Y. H., Feng, H. Q., Jiang, Y. Y., and Zhao, J. Z. (2008). Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin-containing cotton. Science 321, 1676–1678. doi: 10.1126/science.1160550
Xia, X., Zheng, D., Zhong, H., Qin, B., Gurr, G. M., Vasseur, L., et al. (2013). DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS One 8:e68852. doi: 10.1371/journal.pone.0068852
Yong, H. S., Song, S. L., Chua, K. O., and Lim, P. E. (2017). High diversity of bacterial communities in developmental stages of Bactrocera carambolae (Insecta: Tephritidae) revealed by Illumina MiSeq sequencing of 16S rRNA gene. Curr. Microbiol. 74, 1076–1082. doi: 10.1007/s00284-017-1287-x
Yun, J. H., Roh, S. W., Whon, T. W., Jung, M. J., and Bae, J.-W. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/AEM.01226-14
Zhao, C., Zhao, H., Zhang, S., Luo, J., Zhu, X., Wang, L., et al. (2019). The developmental stage symbionts of the pea aphid-feeding Chrysoperla sinica (Tjeder). Front. Microbiol. 10:2454. doi: 10.3389/fmicb.2019.02454
Zhen, C., Miao, L., Liang, P., and Gao, X. (2016). Survey of organophosphate resistance and an Ala216Ser substitution of acetylcholinesterase-1 gene associated with chlorpyrifos resistance in Apolygus lucorum (Meyer-Dür) collected from the transgenic Bt cotton fields in China. Pestic. Biochem. Physiol. 132, 29–37. doi: 10.1016/j.pestbp.2016.04.008
Zouache, K., Raharimalala, F. N., Raquin, V., Tran-Van, V., Raveloson, L. H., Ravelonandro, P., et al. (2011). Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 75, 377–389. doi: 10.1111/j.1574-6941.2010.01012.x
Keywords: 16S rRNA, microbial composition, symbiotic bacteria, pest control, life stages
Citation: Xue H, Zhu X, Wang L, Zhang K, Li D, Ji J, Niu L, Wu C, Gao X, Luo J and Cui J (2021) Gut Bacterial Diversity in Different Life Cycle Stages of Adelphocoris suturalis (Hemiptera: Miridae). Front. Microbiol. 12:670383. doi: 10.3389/fmicb.2021.670383
Edited by:
Adly M. M. Abdalla, International Atomic Energy Agency, Vienna, AustriaReviewed by:
Mohammad Mehrabadi, Tarbiat Modares University, IranCopyright © 2021 Xue, Zhu, Wang, Zhang, Li, Ji, Niu, Wu, Gao, Luo and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueke Gao, MTUwMzYxMzgzODlAMTYzLmNvbQ==; Junyu Luo, bHVvanVueXUxODE4QDEyNi5jb20=; Jinjie Cui, YXljdWlqaW5qaWVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.