 Mario Moreno-Pino1
Mario Moreno-Pino1 Juan A. Ugalde2
Juan A. Ugalde2 Jorge H. Valdés3
Jorge H. Valdés3 Susana Rodríguez-Marconi1
Susana Rodríguez-Marconi1 Génesis Parada-Pozo1
Génesis Parada-Pozo1 Nicole Trefault1*
Nicole Trefault1*- 1GEMA Center for Genomics, Ecology and Environment, Faculty of Sciences, Universidad Mayor, Santiago, Chile
- 2Millennium Initiative for Collaborative Research on Bacterial Resistance (MICROB-R), Santiago, Chile
- 3Center for Genomics and Bioinformatics, Faculty of Sciences, Universidad Mayor, Santiago, Chile
Antarctic sponges harbor a diverse range of microorganisms that perform unique metabolic functions for nutrient cycles. Understanding how microorganisms establish functional sponge–microbe interactions in the Antarctic marine ecosystem provides clues about the success of these ancient animals in this realm. Here, we use a culture-dependent approach and genome sequencing to investigate the molecular determinants that promote a dual lifestyle in three bacterial genera Sporosarcina, Cellulophaga, and Nesterenkonia. Phylogenomic analyses showed that four sponge-associated isolates represent putative novel bacterial species within the Sporosarcina and Nesterenkonia genera and that the fifth bacterial isolate corresponds to Cellulophaga algicola. We inferred that isolated sponge-associated bacteria inhabit similarly marine sponges and also seawater. Comparative genomics revealed that these sponge-associated bacteria are enriched in symbiotic lifestyle-related genes. Specific adaptations related to the cold Antarctic environment are features of the bacterial strains isolated here. Furthermore, we showed evidence that the vitamin B5 synthesis-related gene, panE from Nesterenkonia E16_7 and E16_10, was laterally transferred within Actinobacteria members. Together, these findings indicate that the genomes of sponge-associated strains differ from other related genomes based on mechanisms that may contribute to the life in association with sponges and the extreme conditions of the Antarctic environment.
Introduction
The sponge holobiont- or marine sponge-microorganism assemblage is considered one of the most basal and complex symbiotic relationships and proposed as one of the principal contributors to microbial diversity and functionality across global ocean ecosystems (Ruby, 2008; Pita et al., 2016; Thomas et al., 2016). The sponge holobiont is composed of a permanent, very dense, and diverse microbial community, dominated by the bacterial domain (Hentschel et al., 2006; Taylor et al., 2007). The sponge-associated bacteria participate actively in various fundamental metabolic processes including metabolizing sponge waste compounds, nutrient cycling (carbon, nitrogen, phosphorus, and sulfur), and production of secondary metabolites as chemical defensive molecules (Unson et al., 1994; Fan et al., 2012; Zhang et al., 2015).
Symbiotic functions are essential in the maintenance process of the host–microorganism relationship, which ultimately affects the health of the sponge holobiont. These functions, in turn, affect the health of the ecosystems (Pita et al., 2018). The establishment of the functional sponge–microbe interaction depends, in part, on a series of molecular determinants that mediate the symbiotic lifestyle adaptation (Gao et al., 2014; Tian et al., 2014, 2016Burgsdorf et al., 2015; Zhang et al., 2019). These symbiotic signatures include eukaryotic-like protein domains (ELPs), clustered regularly interspaced short palindromic repeats (CRISPR), restriction-modification (R-M), and toxin–antitoxin (T-A) systems, which are involved in avoiding the digestion by the sponge host and defending against incoming foreign DNA (Magnuson, 2007; Nguyen et al., 2014; Horn et al., 2016; Díez-Vives et al., 2017; Slaby et al., 2017; Jahn et al., 2019). Fundamental insights into the specific strategies that promote the symbiotic lifestyle have been provided by genome studies from sponge symbionts (Slaby et al., 2017; Tian et al., 2017). The genome from Candidatus Synechococcus spongiarum revealed singular differences from free-living Cyanobacteria including the following: genome streamlining, lack of amino acid biosynthesis genes and the partial loss of DNA repair mechanisms, antioxidant enzymes, and photosynthetic complex genes (Gao et al., 2014; Burgsdorf et al., 2015). In the case of the sponge symbiont Candidatus Poribacteria, evidence of symbiotic lifestyle adaptation was inferred based on the loss of the flagellar structure, the presence of exclusive adhesion-related proteins, and a broad set of carbohydrate-degrading enzymes (Siegl et al., 2011; Zhang et al., 2019).
The study of sponge–microorganism symbiotic relationship is particularly relevant in Antarctica, where sponges are pivotal members of the benthic system with a high abundance and species-level endemism (McClintock et al., 2005; Downey et al., 2012). Antarctic sponge microbiomes are diverse, host specific, temporally stable, and different from tropical and temperate sponges (Webster et al., 2004; Rodríguez-Marconi et al., 2015; Savoca et al., 2019; Steinert et al., 2019). Functional insights of the Antarctic sponge-associated bacteria revealed that these bacteria produce antibacterial compounds that could control microbial populations inside the sponges (Papaleo et al., 2012). These compounds include a broad range of antibacterial compounds and xenobiotics (Steinert et al., 2019). More recently, the metagenomic analysis of two Antarctic sponge microbiomes showed the potential for nutrient cycling (carbon, nitrogen, sulfur, and phosphorus) and a high versatility of autotrophic carbon fixation (Moreno-Pino et al., 2020). Furthermore, enrichment of genes related to transposons, phages, CRISPR, T-A, and R-M systems was detected in these metagenomes, supporting the notion of genomic adaptations to symbiotic lifestyle in Antarctic sponges (Moreno-Pino et al., 2020).
Here, we describe molecular determinants of the Antarctic sponge microbiome interactions of five bacterial strains isolated from the Antarctic sponge Iophon sp. using a culture-dependent approach and genome sequencing. This study characterizes the mechanisms that promote a dual lifestyle in bacterial members belonging to the Sporosarcina, Cellulophaga, and Nesterenkonia genera. We aim to understand the genomic adaptations of these bacteria to live in association with the Antarctic sponges and whether their genomes present differences to the free-living counterparts.
Materials and Methods
Bacterial Cultures and Physiological Characterization
Seventeen bacterial strains were originally isolated from the Antarctic sponge Iophon sp. (class Demospongiae). The sponge sample was obtained in January 2014 and collected from Fildes Bay, King George Island, Antarctica (62° 12′ 11′′ S, 58° 55′ 15′′ W) at a depth of 5 m. For bacterial cultures and isolation, triplicate pieces (1 cm3) from the inner structure of the sponge tissue were cut with a sterile scalpel blade and were rinsed three times with sterilized seawater. The sponge tissues were homogenized, and the obtained extracts were plated in Marine Agar 2216 (BD-Difco) and incubated in the dark at 4°C for 30 days. Bacterial growth was examined weekly, and the bacterial colonies were isolated and transferred to Marine Broth 2216 (BD-Difco). Pure isolates were cryopreserved in Marine Broth supplemented with 10% glycerol at −80°C. The bacterial strains selected for genome sequencing included in this study were chosen based on their preliminary taxonomic assignation and the availability of reference genomes for comparative genomic analyses.
Strains E16_2, E16_3, E16_7, E16_8, and E16_10 were initially characterized by Gram staining, according to Dussault (1955). Enzymatic activities from these strains were tested with the API-ZYM Test System (bioMérieux) at 21°C for 3 days.
Whole-Genome Sequencing and Bioinformatic Analysis
Genomic DNA was extracted using the GenElute bacterial genomic DNA kit (Sigma-Aldrich). DNA was used to prepare a sequencing library with NEBNext dsDNA Fragmentase kit (New England Biolabs, Ipswich, MA, United States) and quantified using a standard qPCR assay with a Library Quant Kit Illumina (Kapa). Libraries were sequenced using the Illumina Miseq platform with 2 × 150 cycles using the Miseq kit v.2. In order to improve sequence coverage of the strain E16_10, it was resequenced with a TruSeq Nano DNA LT Kit and using the Illumina HiSeq 4000 with 2 × 100 cycles. All raw whole-genome sequencing reads were filtered using PrinSeq (Schmieder and Edwards, 2011) (version 0.20.4). Adapters were removed with Cutadapt (version 1.10; -q 30) (Martin, 2011; Schmieder and Edwards, 2011). High-quality reads were assembled using SPAdes (version 3.10.1) with default parameters (Bankevich et al., 2012). Genome assembly of strain E16_10 was improved by performing genome scaffolding, combining the previously generated Miseq and Hiseq contigs with MeDuSa, using default parameters (Bosi et al., 2015). ORFs were predicted for all contigs >500 bp using Prodigal (version 2.6.3) (Hyatt et al., 2010). Functional annotation was done using eggNOG mapper v.1 against the eggNOG v.4.5 (Huerta-Cepas et al., 2017; see Table 1 for details).
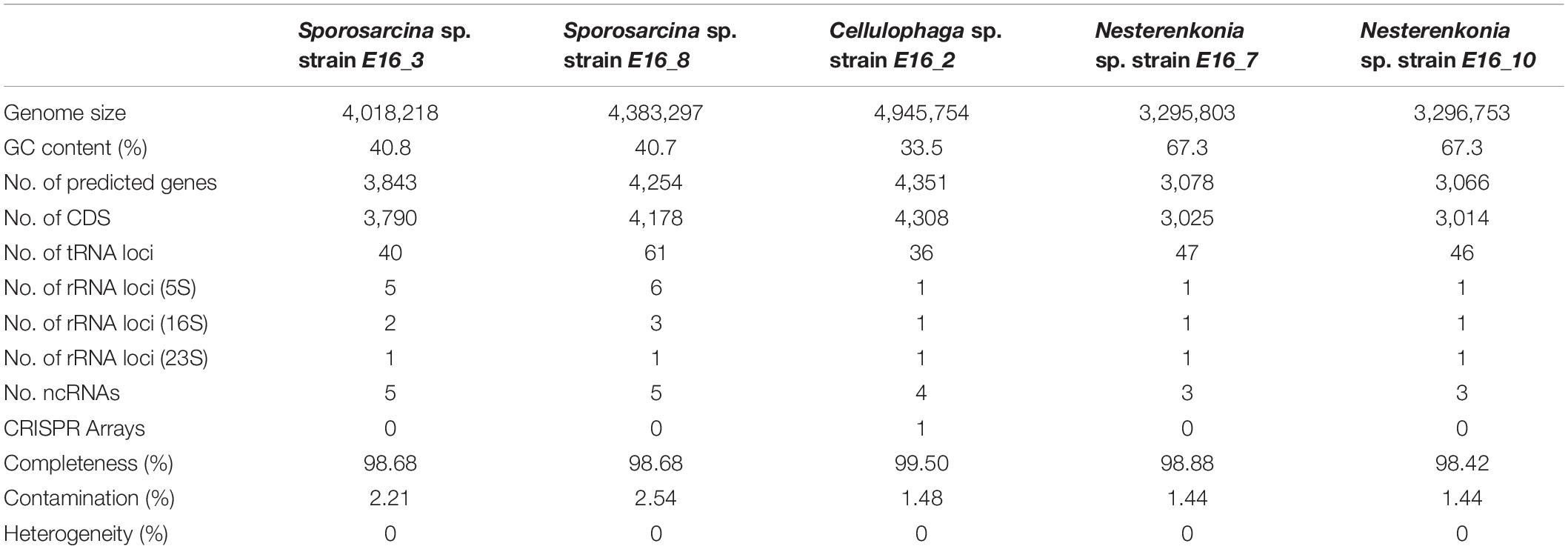
Table 1. Summary of the genomic features of the Antarctic sponge-associated isolates.
Taxonomic classification of strains was performed using the full-length 16S rRNA gene sequences detected with barrnap version 0.71, and RNAmmer 1.2 Server (Lagesen et al., 2007). Closely 16S rRNA gene sequences were found using BLASTn against NCBI-nr database (August 2019), with a cutoff of E-value < 10–5, in Geneious software version R10.2. The retrieved 10 best hits and sequences of neighboring representative lineages were aligned using MUSCLE (version 3.5) (Edgar, 2004), with 999 iterations, in Geneious version R10.2. The alignment was manually edited and used to construct Maximum Likelihood (ML) phylogenies. ML trees were generated using the best-fit models (K2 + GAMMA for Cellulophaga and Sporosarcina, and TN93+ GAMMA for Nesterenkonia) and were computed with 500 bootstrap replicates in MEGA7 (Kumar et al., 2016).
Comparative Genomic Analyses
Fragment Recruitment
To estimate the abundance of genomes related to the bacterial strains E16_2, E16_3, E16_7, E16_8, and E16_10 in marine sponge microbiomes and planktonic communities, we performed a fragment recruitment analysis using 23 publicly available metagenomic datasets. These metagenomes included microbiomes associated with Antarctic sponges (n = 3), microbiomes from tropical and temperate sponges (n = 11), and marine microbial communities (n = 9), including one sample from surrounding seawater from the Antarctic sponges (see Supplementary Table 1 for details).
Metagenomic datasets were filtered based on a Q > 30 quality threshold using Prinseq (Schmieder and Edwards, 2011). Non-ribosomal reads were obtained using SortMeRNA (Kopylova et al., 2012), fragmented and filtered applying a previously published script (Plominsky et al., 2018) to obtain high-quality sequences within 50–100 bp. These were randomly subsampled according to the sample with the lowest number of reads across metagenomes (i.e., Iophon sp.). The recruitment of metagenomic sequences was performed using the genome of the bacterial strains from the Antarctic sponges as reference with FR-HIT (Niu et al., 2011) with a cutoff of 90% identity and 75% coverage. To further evaluate the relative abundance of genera associated with the five isolates obtained from Antarctic sponges, we performed a protein sequence classification of the metagenomes mentioned above. Non-ribosomal sequences were randomly subsampled according to the lowest reads across samples (Iophon sp.). The subsampled sequences were classified with Kaiju with default settings, aligning them against the NCBI BLAST nr +euk database (version 2019-06-25) (Menzel et al., 2016).
Pangenome Analysis
Pangenome analysis was performed by comparing 73 genomes from the Sporosarcina, Cellulophaga, and Nesterenkonia genera using the workflow implemented in the Anvi’o package2. Twenty-one percent of these genomes were complete, 16% were in the scaffold level, and 46% in the contig level (NCBI Sequence Read Archive version March 2019). We included 44 genomes from Cellulophaga spp., 17 genomes from Sporosarcina spp., and 12 genomes from Nesterenkonia spp. (see Supplementary Table 2 for details). Genomes were transformed to the Anvi’o format and filtered by contig length >500 bp (anvi-script-reformat-fasta; -l 500). Protein clustering was performed using BLAST and Markov Clustering (MCL) (van Dongen and Abreu-Goodger, 2012) algorithm with an inflation value of 0.1 (anvi-pan-genome; -minbit 0.5 –mcl-inflation 0.1 –use-ncbi-blast). Average nucleotide identity (ANI) was computed using BLAST (anvi-compute-ani; –method ANIb). Pangenomes were analyzed by clustering the genomes depending on whether the strains were host associated or free living.
Core and accessory genes obtained from the pangenome analysis were classified according to the following parameters. For core genes: maximum number of genes from each genome, 1; maximum functional homogeneity index, 0.9; minimum geometric homogeneity index, 1, minimum functional homogeneity index, 0.9; and maximum functional homogeneity index, 1. For accessory genes, only genes that were present in the host-associated strains were selected.
To test for significant differences in COG abundances between sponge-associated and free-living and between host-associated and free-living genomes, we used a two-tailed Wilcoxon rank-sum test in R environment using rstatix and STAMP 2.0.9 (Parks et al., 2014). COGs with a p-value < 0.05 were considered enriched. Analysis of similitude (ANOSIM) was used to test for significant differences between host-associated and free-living genomes using Primer 6 (PRIMER-E Ltd.).
Phylogenomics
The phylogenomic analysis was performed using the core genes. For this, 756, 355, and 190 core proteins for Cellulophaga spp., Sporosarcina spp., and Nesterenkonia spp., respectively, were concatenated and aligned using MUSCLE (version 3.5) (Edgar, 2004) with 999 iterations, and ambiguous positions were removed using trimAl v1.3 (-automated 1) (Capella-Gutiérrez et al., 2009). ML trees were performed using RAxML version 8 (Stamatakis, 2014), with 1,000 bootstrap replicates under the best-fit model for the three genera (LG + GAMMA + F). A multilocus sequence analysis (MLSA) based on 23 single-copy genes (SCG) shared between all genomes analyzed was performed to support the phylogenomic inferences (Supplementary Table 3; Wu et al., 2013). SCG were concatenated, aligned using MUSCLE (version 3.5) (Edgar, 2004) with 999 iterations in Geneious software version R10.2, and trimmed using trimAl v1.3 (Capella-Gutiérrez et al., 2009). ML trees were performed using RAxML (Stamatakis, 2014), with 1,000 bootstrap replicates, under the best-fit model (for Nesterenkonia, LG + GAMMA + F and for Sporosarcina and Cellulophaga, DAYHOFF + F).
Horizontal Gene Transfer and Synteny Analyses
Genomic islands (GIs) were identified in the genomes of Sporosarcina sp. E16_3 and E16_8, Cellulophaga E16_2, and Nesterenkonia E16_7 and E16_10 using IslandViewer 4 with default settings (Bertelli et al., 2017). IslandViewer 4 integrates a comprehensive approach to predict GIs using four different methods: IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander.
The genomic GC content (%) from the GIs identified in Nesterenkonia E16_7 and E16_10 was computed with Geneious software version R10.2. Finally, the GI containing the ketopantoate reductase coding gene (panE) was analyzed to confirm horizontal gene transfer (HGT) events using phylogenetic and comparative genomic analyses. The closest relatives of panE gene were determined using bidirectional BLASTp against the NCBI-nr database (August 2019), retrieving the 50 non-redundant best hits. The panE genes were aligned using MUSCLE (version 3.5) with 999 iterations in Geneious software version R10.2, and ambiguous positions were removed using trimAl v1.3 (-automated 1) (Edgar, 2004; Capella-Gutiérrez et al., 2009). ML tree was performed using RAxML (Stamatakis, 2014), with 1,000 bootstrap replicates, under the LG + GAMMA + F model. Phylogenetic analysis of five neighboring genes to panE was also performed using 13--20 non-redundant best hits as described before. Additionally, to explore the synteny of panE-related sequences in E16_7 and E16_10 strains, and Nesterenkonia sp. AN1 (NZ_JEMO00000000), and Nesterenkonia aurantiaca DSM27373 (NZ_SOAN00000000), contigs containing ORFs related to panE were extracted using Geneious version R10.2 and were compared with BLASTn using Trebol3.
Results
Phylogenetic analyses using the full-length 16S rRNA gene sequences of the five bacterial strains isolated from the Antarctic sponge Iophon sp. showed that strains E16_3 and E16_8 form a strongly supported monophyletic group with Sporosarcina globispora and Sporosarcina psychrophila (Supplementary Figure 1A). A whole-genome comparison showed that these sponge-associated strains shared 92% of ANI between them and below 89% of ANI to all Sporosarcina genomes (Supplementary Figure 2A). Furthermore, core and SCG showed a separation between the genomes of Sporosarcina sp. E16_3 and E16_8, and S. psychrophila, suggesting a species divergence between them (Figure 1A and Supplementary Figure 3A).
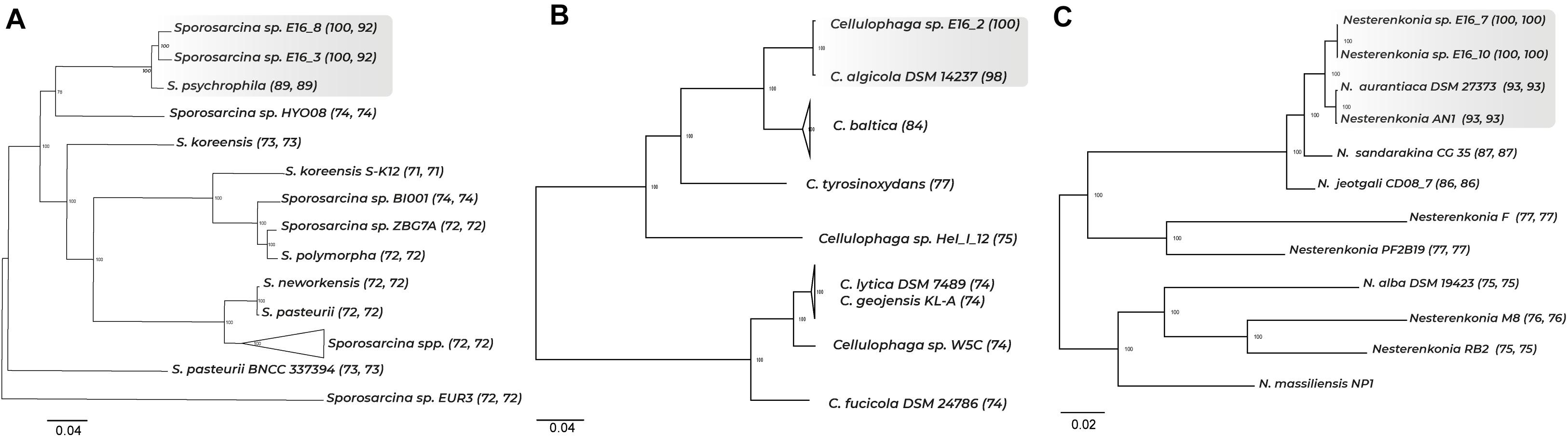
Figure 1. Phylogenomic tree of core genes and ANI among representative Sporosarcina, Cellulophaga, and Nesterenkonia genomes. A total of 756, 355, and 190 core genes were used to perform the phylogenomic tree for Sporosarcina, Cellulophaga, and Nesterenkonia. ANI values are shown in parenthesis. In (A), first and second ANI values correspond to comparison with Sporosarcina E16_8 and E16_3, respectively; in (B), ANI value corresponds to comparison with Cellulophaga E16_2, and in (C), first and second ANI values correspond to comparison with Nesterenkonia E16_7 and E16_10, respectively.
For the strain E16_2, the 16S rRNA gene displayed a high similarity with Cellulophaga algicola, Cellulophaga pacifica, and Cellulophaga baltica, which formed a well-supported monophyletic clade (Supplementary Table 4 and Supplementary Figure 1B). Cellulophaga sp. E16_2 displayed ANI values from 74 to 98% among Cellulophaga genomes (Supplementary Figure 2B). The close relationship between Cellulophaga sp. E16_2 and C. algicola DSM 14237 was confirmed with SCG (Figure 1B and Supplementary Figure 3B). These results indicate a close phylogenetic relationship between Cellulophaga E16_2 and C. algicola DSM 14237.
The 16S rRNA gene of strains E16_7 and E16_10 showed a high similarity with Nesterenkonia sp. AN1, Nesterenkonia aurantiaca, and Nesterenkonia lutea (Supplementary Table 4). ML tree showed that the strains E16_7 and E16_10 cluster together with Nesterenkonia sp. AN1 and N. aurantiaca (Supplementary Figure 1C). Nesterenkonia E16_7 and E16_10 showed 100% of ANI between them, and below 93% when compared with all Nesterenkonia genomes (Supplementary Figure 2C). Phylogenomic trees showed a clear separation between the sponge-associated strains and Antarctic strains Nesterenkonia sp. AN1 and N. aurantiaca (Figure 1C and Supplementary Figure 3C).
Altogether, these results indicate that some strains isolated from the Antarctic sponge Iophon sp. correspond to new species within two different phyla, i.e., Firmicutes and Actinobacteria. Sporosarcina sp. E16_3 and E16_8 represent two putative novel species. Nesterenkonia sp. E16_7 and E16_10 correspond to a single putative novel species, while the fifth strain (E16_2) corresponds to Cellulophaga algicola, a member of the phylum Bacteroidetes.
A general enzymatic characterization of these five strains reflected their taxonomic separation. The strains share the presence of proteases (valine arylamidase, leucine arylamidase) and a phosphohydrolase. Phosphatases were unevenly distributed between the sponge-associated strains (Supplementary Figure 4 and Supplementary Table 5).
Distribution of the Sponge-Associated Bacterial Genomes in Host-Associated Microbiomes and Seawater
Fragment recruitment against 23 metagenomes showed that three of the five strains isolated have a higher abundance in seawater than in sponge microbiomes (Figure 2A). The relative abundance of Sporosarcina E16_3 and E16_8 was higher in the Antarctic sponge microbiomes Myxilla sp. and Leucetta antarctica and in seawater from Antarctica (Fildes Bay, King George Island, and Cooperation Sea), the Arctic, and the British Columbia coast. The genome of Cellulophaga E16_2 was most abundant in the Antarctic seawater and the Northern Pacific Ocean. However, we found the highest abundance of Nesterenkonia E16_10 and E16_7 in the non-Antarctic sponges Sarcotragus foetidus, Aplysina aerophoba, and Petrosia ficiformis (Figure 2A).
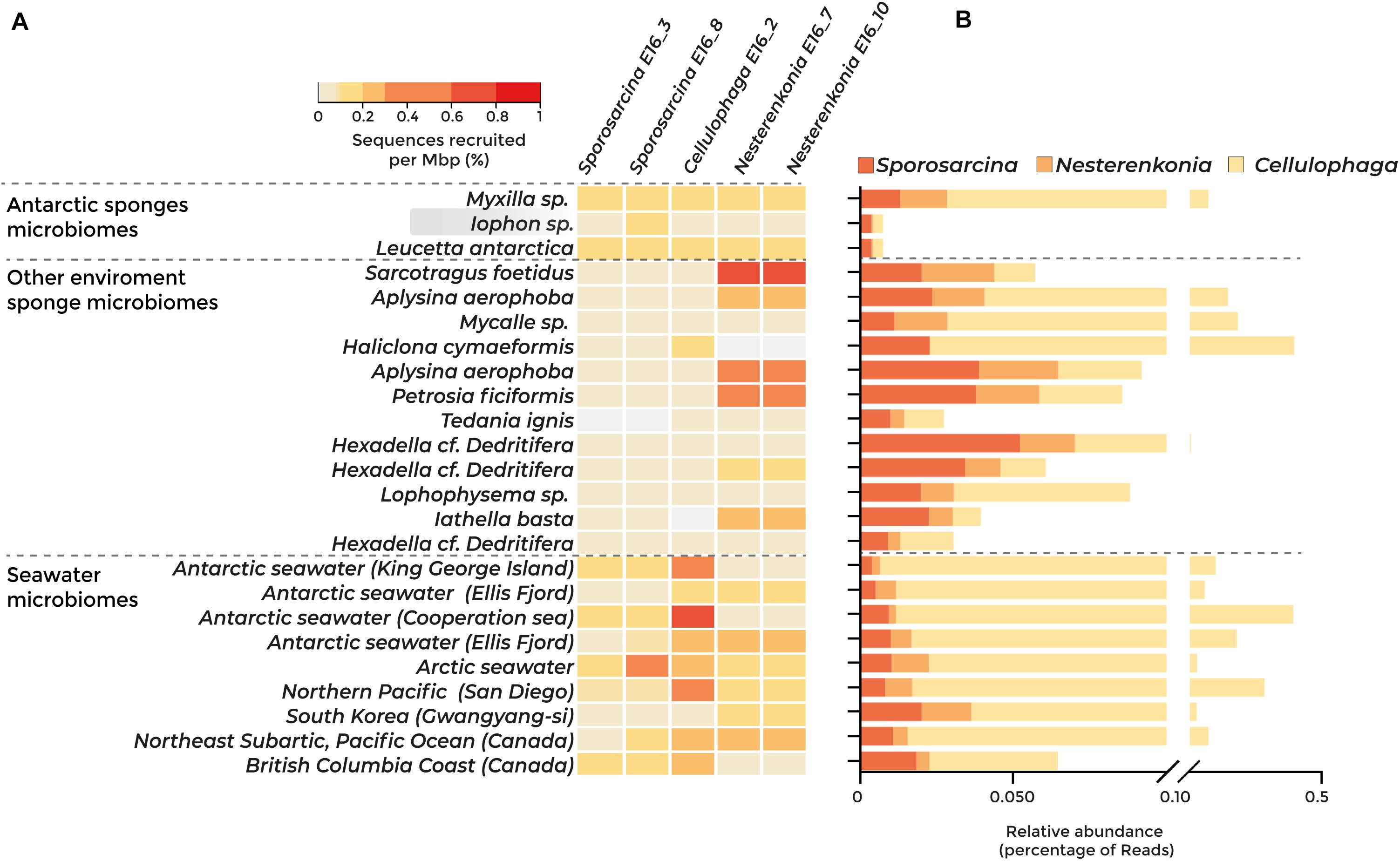
Figure 2. Abundance of the sponge-associated bacterial genomes among sponge and seawater microbiomes, determined by metagenomic fragment recruitment and taxonomic classification of protein sequences. Dashed lines separate Antarctic sponge microbiomes, sponge microbiomes from other environments, and seawater microbiomes. Highlighted in gray is the metagenome from the Antarctic sponge host of the isolated strains. (A) Heatmap of fragment recruitment analysis. The scale bar indicates the percentage of recruited sequences per megabase pair. (B) Abundance of sequences classified at the protein level belonging to Sporosarcina, Cellulophaga, and Nesterenkonia genera.
The taxonomic classification of the 23 metagenomic sequences revealed similar results, showing that Sporosarcina, Nesterenkonia, and Cellulophaga were in equal abundance in sponge microbiome and seawater samples (Figure 2B). Altogether, our results indicate that these sponge-associated bacteria inhabit marine sponges and their surrounding water, suggesting metabolic adaptations to thrive in both environments.
Genes Related to the Symbiotic Lifestyle in the Antarctic Sponge Bacterial Strains
To explore the genomic adaptations that allow these bacteria to live in association with marine invertebrates in an extremely cold environment, we performed comparative genomic analyses focused on the genomes from host-associated bacteria. Summary of genomic features of sponge-associated strains is shown in Table 1. Among all genomes of Sporosarcina spp., Cellulophaga spp., and Nesterenkonia spp., we generated a total of 9,632, 3,312, and 4,483 protein clusters, and we predicted 2,595, 1,481, and 1,701 COGs, respectively.
Symbiosis-related genes were widespread among the analyzed genomes (Supplementary Table 6). ANOSIM according to lifestyle indicates that host-associated and free-living groups were similar although with some differences (R: 0.2). Enriched functions in host-associated genomes were T-A and R-M systems, transposases, and ELPs (p-value < 0.05) (Figure 3 and Supplementary Table 7), as well as transporter-encoding genes (osmotic response, efflux of threonine/homoserine lactone, and arsenic detoxification system) (Supplementary Figure 5). Genes involved in cold and osmotic stress response were widely distributed in the genomes analyzed (Supplementary Table 8). However, COGs related to Na+/H+ antiporter, universal stress protein family, and binding-protein-dependent transport system, were significantly enriched in the bacteria isolated from Antarctica in comparison with the strains isolated from other environments (Supplementary Figure 6 and Supplementary Table 9).
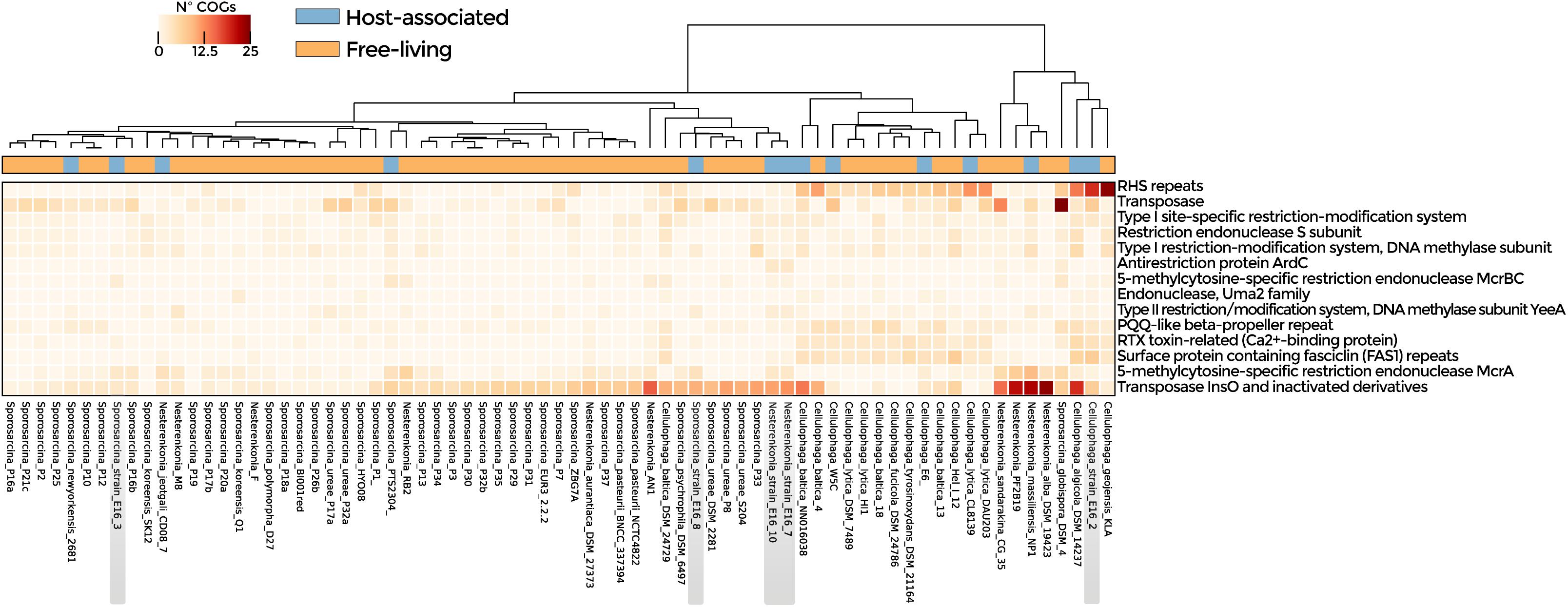
Figure 3. Symbiotic lifestyle functions detected in the genomes of Nesterenkonia, Sporosarcina, and Cellulophaga. COGs related to CRISPR, R-M, and T-A systems, transposases, and ELPs are shown. Heatmap represents the abundance of statistically significant COGs in host-associated and free-living genomes (p-value < 0.05).
The genomes of Sporosarcina sp. PTS2304, Sporosarcina sp. P1, Sporosarcina sp. E16_8, Nesterenkonia E16_7, Nesterenkonia E16_10, N. massiliensis, and N. jeotgali CD08_7 were enriched in COGs related to transposases. The genomes of Cellulophaga sp. E16_2, C. algicola DSM14237, and C. lytica CL8139 in proteins with rearrangement hotspot RHS repeats, members of the ELPs. Sporosarcina E16_3 and E16_8 shared three COGs related to carbohydrate degradation, virulence, and tRNA modifications (Figure 4A and Supplementary Table 10).
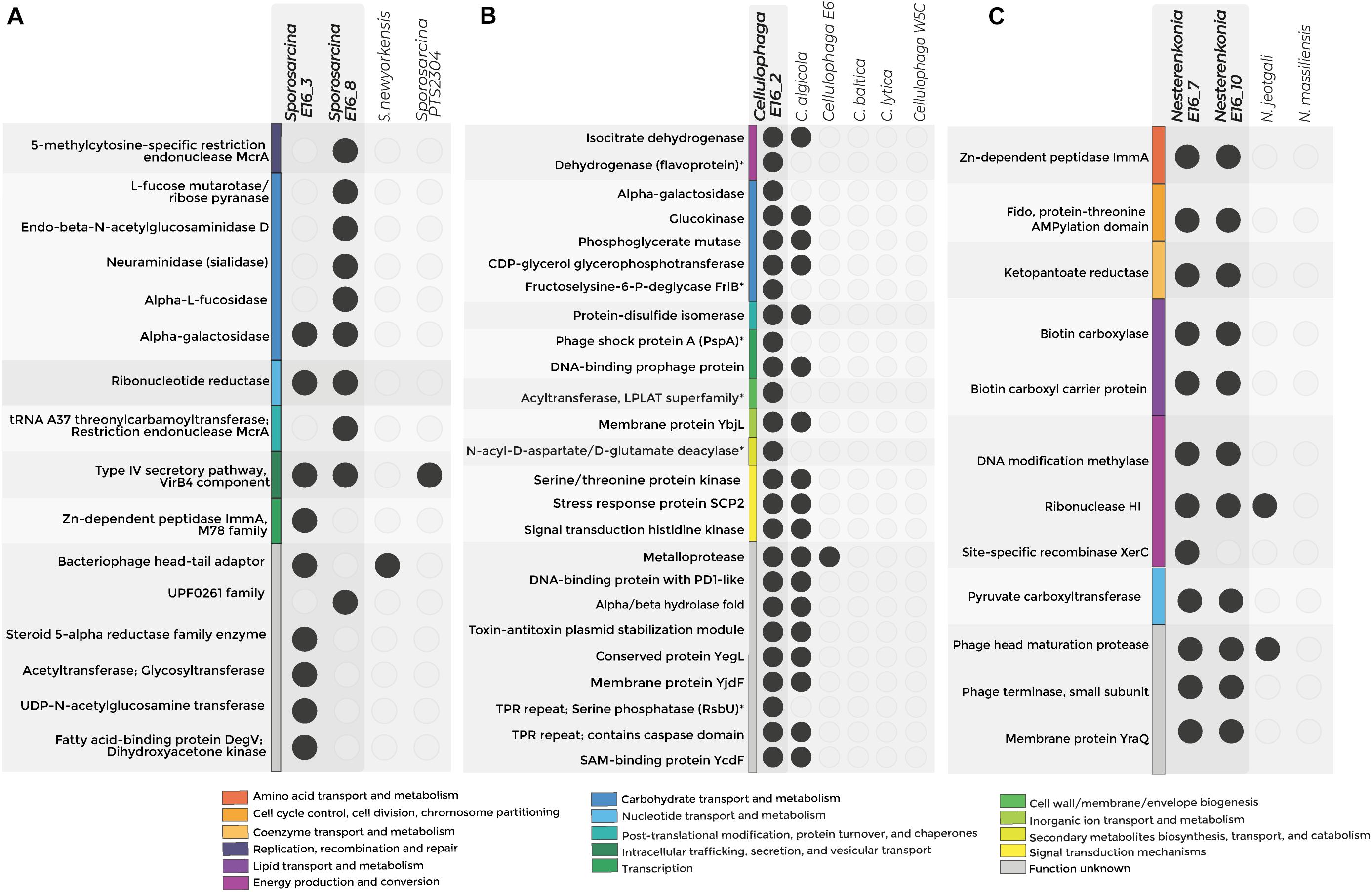
Figure 4. Exclusive COGs detected in the genome of host-associated bacteria. The figure shows the exclusive COG for Sporosarcina (A), Cellulophaga (B), and Nesterenkonia (C). Colors represent categories of exclusive COGs, and empty circles denote the absence of the COG in the genome. *p-value < 0.05—functions without statistical support.
Accessory COGs for Sporosarcina E16_3 were related to DNA recombination, degradation of sialic acids, and chitin. Specifically, accessory COGs related to neuraminidase and endo-β-N-acetylglucosaminidase suggest the potential for the digestion of structural polymers of sponge tissue (Figure 4A and Supplementary Tables 10,11).
For Sporosarcina E16_8, accessory COGs were related to cell wall biosynthesis and extracellular fatty acid incorporation into phospholipids. Specifically, UDP-N-acetylglucosamine enolpyruvyl transferase and the fatty acid kinase’s structural components, the fatty acid-binding protein, and dihydroxyacetone kinase were detected exclusively in Sporosarcina E16_8 (Figure 4A).
The Antarctic strains Cellulophaga E16_2 and C. algicola DSM 14237 shared COGs potentially involved in environmental adaptations to extremely cold conditions, e.g., correct formation of disulfide bonds for protein folding and tellurite resistance, as well as COGs linked to the symbiotic lifestyle, e.g., T-A system and protein-containing tetratricopeptide (TPR) repeats. From the seven accessory COGs that the genome from Cellulophaga sp. E16_2 contains, three were related to d-galactose degradation and environmental stressor response by the sigma factor (serine phosphatase RsbU) and phage shock protein (Psp) (Supplementary Table 10 and Figure 4B).
The pangenome analysis of the sponge-associated Nesterenkonia E16_7 and E16_10 revealed that their genomes shared COGs related to the T-A system (Zn-dependent peptidase ImmA), Mobile Genetic Elements (Phage head maturation protease and phage terminase), and pantothenate (vitamin B5) synthesis (Figure 4C and Supplementary Tables 10,11).
In all Sporosarcina genomes analyzed, the panBCDE cluster is complete (Supplementary Table 12). This cluster codes for the vitamin B5 synthesis pathway, which is absent in sponges. panBCDE cluster was complete in 26% of the available genomes of Cellulophaga. However, this cluster was exclusively found in the Antarctic sponge-associated Nesterenkonia strains (Supplementary Figure 7 and Supplementary Table 13).
Genes coding for the enzymes biotin carboxyl carrier protein of acetyl-CoA carboxylase (pycA), biotin carboxylase subunit of acetyl coenzyme A carboxylase (pycB), and pyruvate carboxylase (pycC) involved in the central anaplerotic step in the TCA cycle were also detected exclusively in both sponge-associated Nesterenkonia strain (Figure 4C). Altogether, all the functions exclusively found in the Antarctic sponge-associated bacteria’s genomes could be related to genomic adaptations to cold environments and the host-associated lifestyle in the form of virulence factor as well as degradation of skeletal components of the sponges.
Horizontal Gene Transfer Related to a Symbiotic Lifestyle
We found 20 and nine GIs in the genomes of Cellulophaga E16_3 and E16_2, respectively, 20 GIs in Sporosarcina E16_8 and 34 in the sponge-associated Nesterenkonia strains (Supplementary Table 14). The presence of GIs indicated putative horizontal transfer events of the panE gene in the genome of the sponge-associated Nesterenkonia strains. The genomic region containing the panE gene represents a GI of 8,497 bp, characterized by a codon usage bias variation and a lower GC percentage than the GC-content for the whole genomic region (57.7% GC). This region is also characterized by a loose in the synteny in the genome region where the GI is present (Figure 5A).
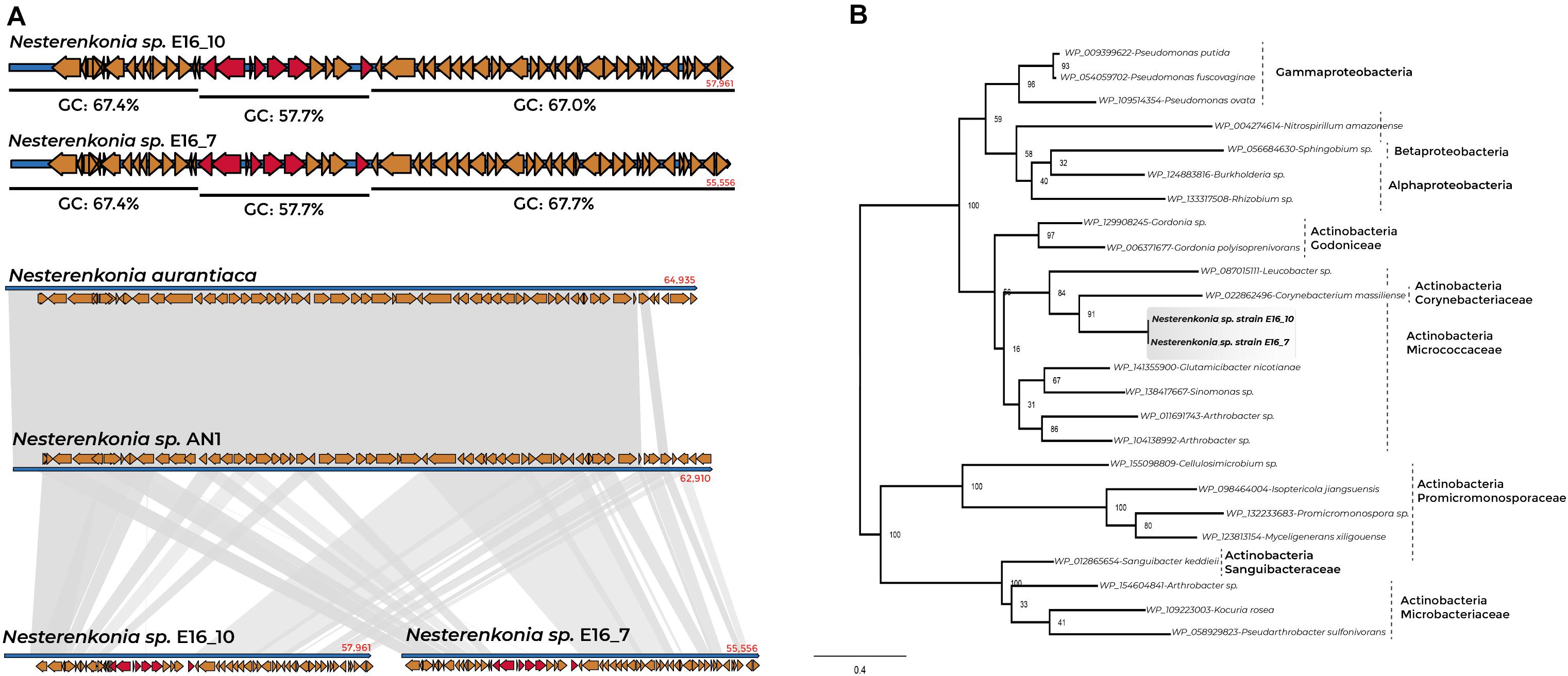
Figure 5. Synteny conservation and phylogenetic tree of panE gene for sponge-associated Nesterenkonia strains and Actinobacteria members. (A) %GC content of the contig containing the panE gene of Nesterenkonia E16_7 and E16_10 and genomic organization between sponge-associated Nesterenkonia, N. aurantiaca, and Nesterenkonia AN1. (B) Phylogenetic tree of panE genes of Nesterenkonia E16_7 and E16_10 and Actinobacteria members.
Phylogenetic analysis of panE-deduced protein from Nesterenkonia E16_7 and E16_10 revealed a relationship with panE of the Actinobacteria Corynebacterium massiliense (member of the family Corynebacteriaceae), Arthrobacter sp., and Glutamicibacter nicotianae (members of the family Micrococcaceae) (Figure 5B and Supplementary Table 15). The adjacent genes to the panE gene in the genomes of Nesterenkonia E16_7 and E16_10 show a close relationship with members belonging to the order Corynebacteriales (Rhodococcus sp., and Corynebacterium sp.), and Micrococcales (Arthrobacter sp., Kocuria sp., Mycobacterium sp., and Micrococcus sp.) (Supplementary Figure 8), indicating that most likely this complete gene cluster has undergone horizontal gene transfer within Actinobacteria members.
Discussion
The sponge holobiont corresponds to one of the most complex symbiotic units, characterized by a huge microbial diversity that is considered higher than in planktonic communities globally (Thomas et al., 2016). This intimate host–microorganisms relationship is supported by a series of genomic adaptations to the symbiotic lifestyle that are still not fully understood. The whole-genome analysis presented here allows us to interrogate the genomic adaptations to the symbiotic lifestyle acquired by five Antarctic sponge-associated bacteria belonging to the genera Cellulophaga, Sporosarcina, and Nesterenkonia.
We conclude that sponge-associated Sporosarcina E16_3 and E16_8 and Nesterenkonia E16_7 and E16_10 may correspond to previously undescribed species. We follow multiple approaches to strengthen the analyzed isolates’ taxonomic classification, including ANI analysis and phylogenomics using core genes and SCG. Based on the conventional threshold of ≥95 ANI used for species delineation, the sponge-associated Sporosarcina and Nesterenkonia strains showed identity values below the species-level cutoff threshold (Konstantinidis et al., 2017). ANI is a widely accepted criterion for bacterial species classification purposes (Konstantinidis and Tiedje, 2005; Yarza et al., 2014; Konstantinidis et al., 2017; Jain et al., 2018) and has been helpful for the classification of sponge symbionts as Ca. Poribacteria (Kamke et al., 2013), Ca. Synechococcus spongiarum (Gao et al., 2014), Pseudovibrio sp. POLY-S9 (Alex and Antunes, 2015), and Shewanella sp. OPT22 (Alex and Antunes, 2019). Phylogenomic analysis further supports that sponge-associated Sporosarcina and Nesterenkonia are putative novel species. However, it is important to note that all genome-based taxonomy approaches are strongly affected by genome availability in public databases, which also applies to our study. Thus, efforts to increase whole-genome sequencing of sponge-associated bacteria would be relevant to expand our taxonomic observations described here.
Notably, we observe that the five bacterial strains isolated from the Antarctic sponge Iophon sp. grouped in a separated phylogenetic cluster consisting exclusively of Antarctic strains. The phylogenetic separation between Antarctic and non-Antarctic clades could be associated with environmental adaptation, as observed in several bacterial members, including Arthrobacter, Pseudoalteromonas, Nesterenkonia, and Paenibacillus darwinianus (Dsouza et al., 2014, 2015; Aliyu et al., 2016). Although the underlying molecular mechanisms involved in cold adaptation in bacteria are still not fully understood, these bacteria display specialized metabolism to survive under extremely cold conditions, like heat-shock proteins, cold-active enzymes, extracellular polymeric substances, and specialized metabolites production (Pearce, 2017; Caruso et al., 2018a,b; Millán-Aguiñaga et al., 2019).
Fragment recruitment indicated that the relative abundance of the Antarctic sponge-associated strains is relatively low in Iophon compared with other sponges and seawater. This could be due to a low intrinsic microbial abundance in their tissues or due to the technical impairments of cultivability of marine bacteria and sponge-associated bacteria (Hardoim et al., 2014; Esteves et al., 2016). A common categorization for sponge microbiomes is between high microbial abundance (HMA) and low microbial abundance (LMA) and refers to the abundance and diversity of bacterial cells inside the sponges (Hentschel et al., 2003). This dichotomy has profound implications for sponge physiology, as HMA sponges tend to have higher pumping rates and higher nutrient exchanges (Weisz et al., 2008; Ribes et al., 2012). The HMA and LMA dichotomy has not been thoroughly studied in the sponge species Iophon sp.; however, Iophon methanophila from asphalt seeps can be considered both LMA and HMA, due to its low diversity but high abundance of associated microorganisms (Rubin-Blum et al., 2019).
Here, we show that the bacterial strains characterized contain genes relevant to the host–microorganisms relationship under cold conditions. Between them, we detect the gene coding for fatty acid-binding protein and dihydroxyacetone kinase, in Sporosarcina E16_8. These enzymes participate in fatty acid modification of bacterial cell membranes which mediate the adaptation to growth in cold temperatures, as well described in S. globispora (Yoon et al., 2001; Diomandé et al., 2015). However, fatty acid synthesis is an energy-demanding process, their exogenous uptake and assimilation would be particularly relevant for optimizing cellular metabolic processes. The same was described in the gram-positive pathogen Staphylococcus aureus, which incorporates host-origin unsaturated fatty acids using the fatty acid kinase (Parsons et al., 2014). This process allows S. aureus to lead energetic metabolism to virulence factor production (Parsons et al., 2014).
Another example is the general stress response sigma factor B, which modulates the expression of antibiotic resistance and cold stress genes (Price et al., 2001). The factor sigma factor B requires the serine phosphatase, RsbU, to release the anti-sigma factor RsbW and respond to stressors (Pané-Farré et al., 2009). Our results show that RsbU is present exclusively in the genome of Cellulophaga E16_2, which makes us speculate that sigma factor B-regulator would be key to respond to cold-conditions as has been proposed previously in the genome of other Antarctic bacterial strains (Aliyu et al., 2016). However, we do not rule out that RsbU could also promote the establishment of the sponge–microorganism relationship based on their role in regulating antibiotic resistance genes (Price et al., 2001). Antibiotic production in the sponge holobiont is an important process in the control of microbial pathogens and their resistance has been described as an adaptive mechanism to survive in the sponge microbiome (Phelan et al., 2011; Mangano et al., 2013; Versluis et al., 2016; Almeida et al., 2019). Thus, we can suppose that sigma B-regulator can mediate antibiotic resistance in Cellulophaga, as has been proposed for the pathogenic bacteria S. aureus and Listeria monocytogenes (Price et al., 2001; Kumar et al., 2014). Future studies, including transcriptomic analysis, would help clarify the role these genes play in establishing the host–microorganism relationship under cold conditions.
Furthermore, we demonstrate that the genomes from the sponge-associated strains described here have molecular determinants related to their host-associated lifestyle. These determinants include ELPs, CRISPR, and the R-M and T-A systems, which are typically enriched and highly active in sponge symbiont genomes (Slaby et al., 2017). Another accessory function detected in the establishment of microorganisms in the sponge is the degradation of structural biopolymers of sponge tissue. It has been detected in diverse pathogens of invertebrates and also in the sponge-symbiont Poribacteria (Kamke et al., 2013; Podell et al., 2019). The demosponge skeleton is composed of polysaccharides, such as chitin, and mucopolysaccharides in their cell surface (Brunner et al., 2009; Ehrlich et al., 2018). Accordingly, we found that the genomes of the sponge-associated Sporosarcina species are enriched in chitinases and neuraminidases, supporting their sponge-associated lifestyle.
Finally, we show that sponge-associated Nesterenkonia possesses all the genes responsible for vitamin B5 synthesis. Pantothenate (vitamin B5) forms the core of coenzyme A and acyl carrier protein, being crucial in energy and fatty acid metabolism (Leonardi and Jackowski, 2007). In contrast to animals, some sponge–symbionts can perform de novo synthesis of B vitamins such as thiamine (vitamin B1), riboflavin (vitamin B2), pantothenate (vitamin B5), pyridoxine (vitamin B6), biotin (vitamin B7), and cobalamin (vitamin B12) to likely provide to the sponge their vitamin requirements (Engelberts et al., 2020). Moreover, our results strongly suggest that sponge-associated Nesterenkonia E16_7 and E16_10 acquired the gene encoding the ketopantoate reductase within Actinobacteria members by HGT. It has been demonstrated that HGT within sponge microbiomes plays a relevant role in the metabolic versatility of sponge-associated microorganisms (Gauthier et al., 2016). Thus, microorganisms within the Antarctic sponge likely perform de novo pantothenate synthesis as a genomic adaptation to inhabit the sponge holobiont.
Conclusion
Our genomic analysis of five bacterial strains belonging to Cellulophaga, Sporosarcina, and Nesterenkonia reveals that their genomes contain unique genomic adaptations to life associated with the Antarctic sponges. The sponge-associated bacteria isolated differ with respect to their free-living counterparts by the presence of exclusive genes related to cold environment adaptation and host-associated lifestyle. These genomic features include fatty acids incorporation from host origin, antibiotic resistance, and degradation of sponge structural biopolymers. Furthermore, HGT in sponge-associated Nesterenkonia strains suggests a putative role of panE gene in the symbiotic lifestyle in the cold Antarctic waters. Overall, genomic adaptations described in the five strains suggest a high metabolic versatility that may be involved in the capability to inhabit the sponge tissue and live in extremely cold seawater environments.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
MM-P and NT conceived the experiments and drafted the manuscript. MM-P, GP-P, and SR-M conducted the experiments. MM-P analyzed the data. MM-P, NT, JU, and JV interpreted the data. All authors reviewed and approved the final manuscript.
Funding
This work has been funded by INACH grants RG_31-15, DG_12-17, RT_34-17, and ANID-FONDECYT grant no. 1190879. This work has also been funded by grants Puente “Linking microbial community structure with their functional traits in Antarctic sponge symbionts: insights into the mechanisms of host–microbe interactions,” FDP “Funcionalidad en Comunidades Microbianas Asociadas a esponjas Antárticas,” and the Ph.D. fellowship (to MM-P) from the Direction of Research and Art Creation of Universidad Mayor. JU was funded by the ANID Millennium Science Initiative/Millennium Initiative for Collaborative Research on Bacterial Resistance NCN17_081.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are infinitely grateful to Ignacio Garrido, María José Díaz, and Jorge Holtheuer for sponge sampling and Dr. Eduardo Hajdu (UFRJ) for sponge taxonomic identification. We also thank the staff at “Prof. Julio Escudero” INACH Research Station for the logistic support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.660779/full#supplementary-material
Footnotes
- ^ http://www.vicbioinformatics.com/software.barrnap.shtml
- ^ http://merenlab.org/2016/11/08/pangenomics-v2
- ^ https://inf.imo-chile.cl/software/trebol.html
References
Alex, A., and Antunes, A. (2015). Whole genome sequencing of the symbiont Pseudovibrio sp. from the intertidal marine sponge Polymastia penicillus revealed a gene repertoire for host-switching permissive lifestyle. Genome Biol. Evol. 7, 3022–3032. doi: 10.1093/gbe/evv199
Alex, A., and Antunes, A. (2019). Whole-genome comparisons among the genus shewanella reveal the enrichment of genes encoding ankyrin-repeats containing proteins in sponge-associated bacteria. Front. Microbiol. 10:5. doi: 10.3389/fmicb.2019.00005
Aliyu, H., De Maayer, P., and Cowan, D. (2016). The genome of the Antarctic polyextremophile Nesterenkonia sp. AN1 reveals adaptive strategies for survival under multiple stress conditions. FEMS Microbiol. Ecol. 92:fiw032. doi: 10.1093/femsec/fiw032
Almeida, E. L., Rincón, A. F. C., Jackson, S. A., and Dobson, A. D. W. (2019). Comparative genomics of marine sponge-derived Streptomyces spp. isolates sm17 and sm18 with their closest terrestrial relatives provides novel insights into environmental niche adaptations and secondary metabolite biosynthesis potential. Front. Microbiol. 10:1713. doi: 10.3389/fmicb.2019.01713
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35.
Bosi, E., Donati, B., Galardini, M., Brunetti, S., Sagot, M.-F., Lió, P., et al. (2015). MeDuSa: a multi-draft based scaffolder. Bioinformatics 31, 2443–2451. doi: 10.1093/bioinformatics/btv171
Brunner, E., Ehrlich, H., Schupp, P., Hedrich, R., Hunoldt, S., Kammer, M., et al. (2009). Chitin-based scaffolds are an integral part of the skeleton of the marine demosponge Ianthella basta. J. Struct. Biol. 168, 539–547. doi: 10.1016/j.jsb.2009.06.018
Burgsdorf, I., Slaby, B. M., Handley, K. M., Haber, M., Blom, J., Marshall, C. W., et al. (2015). Lifestyle evolution in cyanobacterial symbionts of sponges. MBio 6:e00391–15.
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Caruso, C., Rizzo, C., Mangano, S., Poli, A., Di Donato, P., Finore, I., et al. (2018a). Production and biotechnological potential of extracellular polymeric substances from sponge-associated antarctic bacteria. Appl. Environ. Microbiol. 84:e01624–17. doi: 10.1128/AEM.01624-17
Caruso, C., Rizzo, C., Mangano, S., Poli, A., Di Donato, P., Nicolaus, B., et al. (2018b). Extracellular polymeric substances with metal adsorption capacity produced by Pseudoalteromonas sp. MER144 from Antarctic seawater. Environ. Sci. Pollut. Res. Int. 25, 4667–4677. doi: 10.1007/s11356-017-0851-z
Díez-Vives, C., Moitinho-Silva, L., Nielsen, S., Reynolds, D., and Thomas, T. (2017). Expression of eukaryotic-like protein in the microbiome of sponges. Mol. Ecol. 26, 1432–1451. doi: 10.1111/mec.14003
Diomandé, S. E., Nguyen-The, C., Guinebretière, M.-H., Broussolle, V., and Brillard, J. (2015). Role of fatty acids in Bacillus environmental adaptation. Front. Microbiol. 6:813. doi: 10.3389/fmicb.2015.00813
Downey, R. V., Griffiths, H. J., Linse, K., and Janussen, D. (2012). Diversity and distribution patterns in high southern latitude sponges. PLoS One 7:e41672. doi: 10.1371/journal.pone.0041672
Dsouza, M., Taylor, M. W., Turner, S. J., and Aislabie, J. (2014). Genome-based comparative analyses of antarctic and temperate species of Paenibacillus. PLoS One 9:e108009. doi: 10.1371/journal.pone.0108009
Dsouza, M., Taylor, M. W., Turner, S. J., and Aislabie, J. (2015). Genomic and phenotypic insights into the ecology of Arthrobacter from Antarctic soils. BMC Genomics 16:36. doi: 10.1186/s12864-015-1220-2
Dussault, H. P. (1955). An improved technique for staining red halophilic bacteria. J. Bacteriol. 70, 484–485. doi: 10.1128/jb.70.4.484-485.1955
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ehrlich, H., Shaala, L. A., Youssef, D. T. A., Aksamitowska, S. Ż, Tsurkan, M., Galli, R., et al. (2018). Discovery of chitin in skeletons of non-verongiid Red Sea demosponges. PLoS One 13:e0195803. doi: 10.1371/journal.pone.0195803
Engelberts, J. P., Robbins, S. J., de Goeij, J. M., Aranda, M., Bell, S. C., and Webster, N. S. (2020). Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J. 14, 1100–1110. doi: 10.1038/s41396-020-0591-9
Esteves, A. I. S., Amer, N., Nguyen, M., and Thomas, T. (2016). Sample processing impacts the viability and cultivability of the sponge microbiome. Front. Microbiol. 7:499. doi: 10.3389/fmicb.2016.00499
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887.
Gao, Z.-M., Wang, Y., Tian, R.-M., Wong, Y. H., Batang, Z. B., Al-Suwailem, A. M., et al. (2014). Symbiotic adaptation drives genome streamlining of the cyanobacterial sponge symbiont “Candidatus Synechococcus spongiarum. mbio 5:e00079–14.
Gauthier, M.-E. A., Watson, J. R., and Degnan, S. M. (2016). Draft genomes shed light on the dual bacterial symbiosis that dominates the microbiome of the coral reef sponge Amphimedon queenslandica. Front. Mar. Sci. 3:196. doi: 10.3389/fmars.2016.00196
Hardoim, C. C. P., Cardinale, M., Cúcio, A. C. B., Esteves, A. I. S., Berg, G., Xavier, J. R., et al. (2014). Effects of sample handling and cultivation bias on the specificity of bacterial communities in keratose marine sponges. Front. Microbiol. 5:611. doi: 10.3389/fmicb.2014.00611
Hentschel, U., Fieseler, L., Wehrl, M., Gernert, C., Steinert, M., Hacker, J., et al. (2003). Microbial diversity of marine sponges. Prog. Mol. Subcell. Biol. 37, 59–88. doi: 10.1007/978-3-642-55519-0_3
Hentschel, U., Usher, K. M., and Taylor, M. W. (2006). Marine sponges as microbial fermenters. FEMS Microbiol. Ecol. 55, 167–177. doi: 10.1111/j.1574-6941.2005.00046.x
Horn, H., Slaby, B. M., Jahn, M. T., Bayer, K., Moitinho-Silva, L., Förster, F., et al. (2016). An enrichment of CRISPR and other defense-related features in marine sponge-associated microbial metagenomes. Front. Microbiol. 7:1751. doi: 10.3389/fmicb.2016.01751
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jahn, M. T., Arkhipova, K., Markert, S. M., Stigloher, C., Lachnit, T., Pita, L., et al. (2019). A phage protein aids bacterial symbionts in eukaryote immune evasion. Cell Host Microbe 26, 542–550.e5.
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Kamke, J., Sczyrba, A., Ivanova, N., Schwientek, P., Rinke, C., Mavromatis, K., et al. (2013). Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 7, 2287–2300. doi: 10.1038/ismej.2013.111
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kumar, P. S., Kumar, Y. N., Prasad, U. V., Yeswanth, S., Swarupa, V., Vasu, D., et al. (2014). Comparative structural and functional analysis of staphylococcus aureus glucokinase with other bacterial glucokinases. Indian J. Pharm. Sci. 76, 430–436.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Leonardi, R., and Jackowski, S. (2007). Biosynthesis of pantothenic acid and Coenzyme A. EcoSal Plus 2:10. doi: 10.1128/ecosalplus.3.6.3.4
Magnuson, R. D. (2007). Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189, 6089–6092. doi: 10.1128/jb.00958-07
Mangano, S., Michaud, L., Caruso, C., and Lo Giudice, A. (2013). Metal and antibiotic resistance in psychrotrophic bacteria associated with the Antarctic sponge Hemigellius pilosus (Kirkpatrick, 1907). Polar Biol. 37, 227–235. doi: 10.1007/s00300-013-1426-1
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
McClintock, J. B., Amsler, C. D., Baker, B. J., and van Soest, R. W. M. (2005). Ecology of Antarctic marine sponges: an overview. Integr. Comp. Biol. 45, 359–368. doi: 10.1093/icb/45.2.359
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257.
Millán-Aguiñaga, N., Soldatou, S., Brozio, S., Munnoch, J. T., Howe, J., Hoskisson, P. A., et al. (2019). Awakening ancient polar Actinobacteria: diversity, evolution and specialized metabolite potential. Microbiology 165, 1169–1180. doi: 10.1099/mic.0.000845
Moreno-Pino, M., Cristi, A., Gillooly, J. F., and Trefault, N. (2020). Characterizing the microbiomes of Antarctic sponges: a functional metagenomic approach. Sci. Rep. 10:645.
Nguyen, M. T. H. D., Liu, M., and Thomas, T. (2014). Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 23, 1635–1645. doi: 10.1111/mec.12384
Niu, B., Zhu, Z., Fu, L., Wu, S., and Li, W. (2011). FR-HIT, a very fast program to recruit metagenomic reads to homologous reference genomes. Bioinformatics 27, 1704–1705. doi: 10.1093/bioinformatics/btr252
Pané-Farré, J., Jonas, B., Hardwick, S. W., Gronau, K., Lewis, R. J., Hecker, M., et al. (2009). Role of RsbU in controlling SigB activity in Staphylococcus aureus following alkaline stress. J. Bacteriol. 191, 2561–2573. doi: 10.1128/jb.01514-08
Papaleo, M. C., Fondi, M., Maida, I., Perrin, E., Lo Giudice, A., Michaud, L., et al. (2012). Sponge-associated microbial Antarctic communities exhibiting antimicrobial activity against Burkholderia cepacia complex bacteria. Biotechnol. Adv. 30, 272–293. doi: 10.1016/j.biotechadv.2011.06.011
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Parsons, J. B., Broussard, T. C., Bose, J. L., Rosch, J. W., Jackson, P., Subramanian, C., et al. (2014). Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 111, 10532–10537. doi: 10.1073/pnas.1408797111
Pearce, D. A. (2017). “Extremophiles in antarctica: life at low temperatures,” in Adaption of Microbial Life to Environmental Extremes, eds H. Stan-Lotter and S. Fendrihan (Cham: Springer), 99–131. doi: 10.1007/978-3-319-48327-6_5
Phelan, R. W., Clarke, C., Morrissey, J. P., Dobson, A. D. W., O’Gara, F., and Barbosa, T. M. (2011). Tetracycline resistance-encoding plasmid from Bacillus sp. strain #24, isolated from the marine sponge Haliclona simulans. Appl. Environ. Microbiol. 77, 327–329. doi: 10.1128/aem.01239-10
Pita, L., Fraune, S., and Hentschel, U. (2016). Emerging sponge models of animal-microbe symbioses. Front. Microbiol. 7:2102. doi: 10.3389/fmicb.2016.02102
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 1–18.
Plominsky, A. M., Trefault, N., Podell, S., Blanton, J. M., De la Iglesia, R., Allen, E. E., et al. (2018). Metabolic potential and in situ transcriptomic profiles of previously uncharacterized key microbial groups involved in coupled carbon, nitrogen and sulfur cycling in anoxic marine zones. Environ. Microbiol. 20, 2727–2742. doi: 10.1111/1462-2920.14109
Podell, S., Blanton, J. M., Neu, A., Agarwal, V., Biggs, J. S., Moore, B. S., et al. (2019). Pangenomic comparison of globally distributed poribacteria associated with sponge hosts and marine particles. ISME J. 13, 468–481. doi: 10.1038/s41396-018-0292-9
Price, C. W., Fawcett, P., Cérémonie, H., Su, N., Murphy, C. K., and Youngman, P. (2001). Genome-wide analysis of the general stress response in Bacillus subtilis. Mol. Microbiol. 41, 757–774. doi: 10.1046/j.1365-2958.2001.02534.x
Ribes, M., Jiménez, E., Yahel, G., López-Sendino, P., Diez, B., Massana, R., et al. (2012). Functional convergence of microbes associated with temperate marine sponges. Environ. Microbiol. 14, 1224–1239. doi: 10.1111/j.1462-2920.2012.02701.x
Rodríguez-Marconi, S., De la Iglesia, R., Díez, B., Fonseca, C. A., Hajdu, E., and Trefault, N. (2015). Characterization of bacterial, archaeal and eukaryote symbionts from antarctic sponges reveals a high diversity at a three-domain level and a particular signature for this ecosystem. PLoS One 10:e0138837. doi: 10.1371/journal.pone.0138837
Rubin-Blum, M., Antony, C. P., Sayavedra, L., Martínez-Pérez, C., Birgel, D., Peckmann, J., et al. (2019). Fueled by methane: deep-sea sponges from asphalt seeps gain their nutrition from methane-oxidizing symbionts. ISME J. 13, 1209–1225. doi: 10.1038/s41396-019-0346-7
Ruby, E. G. (2008). Symbiotic conversations are revealed under genetic interrogation. Nat. Rev. Microbiol. 6, 752–762. doi: 10.1038/nrmicro1958
Savoca, S., Lo Giudice, A., Papale, M., Mangano, S., Caruso, C., Spanò, N., et al. (2019). Antarctic sponges from the Terra Nova Bay (Ross Sea) host a diversified bacterial community. Sci. Rep. 9:16135. doi: 10.1038/s41598-019-52491-0
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Slaby, B. M., Hackl, T., Horn, H., Bayer, K., and Hentschel, U. (2017). Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 11, 2465–2478. doi: 10.1038/ismej.2017.101
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steinert, G., Wemheuer, B., Janussen, D., Erpenbeck, D., Daniel, R., Simon, M., et al. (2019). Prokaryotic diversity and community patterns in Antarctic continental shelf sponges. Front. Mar. Sci. 6:297. doi: 10.3389/fmars.2019.00297
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870.
Tian, R.-M., Sun, J., Cai, L., Zhang, W.-P., Zhou, G.-W., Qiu, J.-W., et al. (2016). The deep-sea glass sponge Lophophysema eversa harbours potential symbionts responsible for the nutrient conversions of carbon, nitrogen and sulfur. Environ. Microbiol. 18, 2481–2494. doi: 10.1111/1462-2920.13161
Tian, R.-M., Wang, Y., Bougouffa, S., Gao, Z.-M., Cai, L., Zhang, W.-P., et al. (2014). Effect of copper treatment on the composition and function of the bacterial community in the sponge Haliclona cymaeformis. MBio 5:e01980.
Tian, R.-M., Zhang, W., Cai, L., Wong, Y.-H., Ding, W., and Qian, P.-Y. (2017). Genome reduction and microbe-host interactions drive adaptation of a sulfur-oxidizing bacterium associated with a cold seep sponge. mSystems 2:e00184–16. doi: 10.1128/mSystems.00184-16
Unson, M. D., Holland, N. D., and Faulkner, D. J. (1994). A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of the crystalline metabolite in the sponge tissue. Mar. Biol. 119, 1–11. doi: 10.1007/bf00350100
van Dongen, S., and Abreu-Goodger, C. (2012). Using MCL to extract clusters from networks. Methods Mol. Biol. 804, 281–295. doi: 10.1007/978-1-61779-361-5_15
Versluis, D., Rodriguez de Evgrafov, M., Sommer, M. O. A., Sipkema, D., Smidt, H., and van Passel, M. W. J. (2016). Sponge microbiota are a reservoir of functional antibiotic resistance genes. Front. Microbiol 7:1848. doi: 10.3389/fmicb.2016.01848
Webster, N. S., Negri, A. P., Munro, M. M. H. G., and Battershill, C. N. (2004). Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 6, 288–300. doi: 10.1111/j.1462-2920.2004.00570.x
Weisz, J. B., Lindquist, N., and Martens, C. S. (2008). Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia 155, 367–376. doi: 10.1007/s00442-007-0910-0
Wu, D., Jospin, G., and Eisen, J. A. (2013). Systematic identification of gene families for use as “markers” for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PLoS One 8:e77033. doi: 10.1371/journal.pone.0077033
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K.-H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Yoon, J. H., Lee, K. C., Weiss, N., Kho, Y. H., Kang, K. H., and Park, Y. H. (2001). Sporosarcina aquimarina sp. nov., a bacterium isolated from seawater in Korea, and transfer of Bacillus globisporus (Larkin and Stokes 1967), Bacillus psychrophilus (Nakamura 1984) and Bacillus pasteurii (Chester 1898) to the genus Sporosarcina as Sporosarcina globispora comb. nov., Sporosarcina psychrophila comb. nov. and Sporosarcina pasteurii comb. nov., and emended description of th. Int. J. Syst. Evol. Microbiol. 51, 1079–1086. doi: 10.1099/00207713-51-3-1079
Zhang, F., Blasiak, L. C., Karolin, J. O., Powell, R. J., Geddes, C. D., and Hill, R. T. (2015). Phosphorus sequestration in the form of polyphosphate by microbial symbionts in marine sponges. Proc. Natl. Acad. Sci. U.S.A. 112, 4381–4386. doi: 10.1073/pnas.1423768112
Keywords: Antarctic sponges, symbiotic lifestyles, sponge microbiome, Antarctic ecosystem, sponge-associated bacteria
Citation: Moreno-Pino M, Ugalde JA, Valdés JH, Rodríguez-Marconi S, Parada-Pozo G and Trefault N (2021) Bacteria Isolated From the Antarctic Sponge Iophon sp. Reveals Mechanisms of Symbiosis in Sporosarcina, Cellulophaga, and Nesterenkonia. Front. Microbiol. 12:660779. doi: 10.3389/fmicb.2021.660779
Received: 03 February 2021; Accepted: 21 April 2021;
Published: 10 June 2021.
Edited by:
Russell T. Hill, University of Maryland, Baltimore County, United StatesReviewed by:
Alan D. W. Dobson, University College Cork, IrelandFan Zhang, Baylor College of Medicine, United States
Copyright © 2021 Moreno-Pino, Ugalde, Valdés, Rodríguez-Marconi, Parada-Pozo and Trefault. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Trefault, bmljb2xlLnRyZWZhdWx0QHVtYXlvci5jbA==