 Jonathan Asante1,2*
Jonathan Asante1,2* Bakoena A. Hetsa1,2
Bakoena A. Hetsa1,2 Daniel G. Amoako1
Daniel G. Amoako1 Akebe L. K. Abia1
Akebe L. K. Abia1 Linda A. Bester3
Linda A. Bester3 Sabiha Y. Essack1
Sabiha Y. Essack1- 1Antimicrobial Research Unit, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
- 2School of Laboratory Medicine and Medical Sciences, University of KwaZulu-Natal, Durban, South Africa
- 3Biomedical Research Unit, University of KwaZulu-Natal, Durban, South Africa
Staphylococcus epidermidis has become an important nosocomial pathogen. Multidrug resistance makes S. epidermidis infections difficult to treat. The study aims to describe the genomic characteristics of methicillin-resistant S. epidermidis (MRSE) isolated from clinical sources, to comprehend the genetic basis of antibiotic resistance, virulence, and potential pathogenicity. Sixteen MRSE underwent whole-genome sequencing, and bioinformatics analyses were carried out to ascertain their resistome, virulome, mobilome, clonality, and phylogenomic relationships. In all, 75% of isolates displayed multidrug resistance and were associated with the carriage of multiple resistance genes including mecA, blaZ, tet(K), erm(A), erm(B), erm(C), dfrG, aac(6′)-aph(2′′), and cat(pC221) conferring resistance to β-lactams, tetracyclines, macrolide–lincosamide–streptogramin B, aminoglycosides, and phenicols, which were located on both plasmids and chromosomes. Their virulence profiles were evidenced by the presence of genes involved in adherence/biofilm formation (icaA, icaB, icaC, atl, ebh, and ebp), immune evasion (adsA, capC, and manA), and antiphagocytosis (rmlC, cdsA, and A). The community-acquired SCCmec type IV was the most common SCCmec type. The CoNS belonged to seven multilocus sequence types (MLSTs) and carried a diversity of mobile genetic elements such as phages, insertion sequences, and plasmids. The bacterial anti-phage defense systems clustered regularly interspaced short palindromic repeats/CRISPR-associated (CRISPR-Cas) immunity phage system and restriction-modification system (R-M system) and the arginine catabolic mobile element (ACME) involved in immune evasion and transport of virulence genes were also found. The insertion sequence, IS256, linked with virulence, was found in 56.3% of isolates. Generally, the isolates clustered according to STs, with some similarity but also considerable variability within isolates. Whole-genome sequencing and bioinformatics analysis provide insights into the likely pathogenicity and antibiotic resistance of S. epidermidis, necessitating surveillance of this emerging pathogen.
Introduction
Staphylococcus epidermidis are coagulase-negative staphylococci (CoNS) that are commensals of the skin microbiome. Among the CoNS, S. epidermidis and Staphylococcus haemolyticus, together referred to as the S. epidermidis group, are the most prevalent in clinical settings (Azih and Enabulele, 2013) and can behave as pathogens by colonizing medical devices, infecting surgical wounds, and causing bacteremia (Cabrera-Contreras et al., 2019). CoNS infection is commonly associated with device-associated healthcare infection. S. epidermidis is considered clinically relevant, moderately pathogenic, and known to display multidrug and methicillin resistance that complicates treatment (Becker et al., 2014; Xu et al., 2015). mecA gene, which mediates methicillin resistance, is highly conserved in S. epidermidis, easily transferred to other staphylococcal species by horizontal transmission (Méric et al., 2015), and borne on the staphylococcal cassette chromosome, SCCmec, which is shared between S. epidermidis and Staphylococcus aureus (Méric et al., 2015). It is estimated that globally, S. epidermidis together with other CoNS and S. aureus causes 30% of hospital-associated infections (Xu et al., 2018).
Staphylococcus epidermidis is an important carrier of antibiotic resistance genes (ARGs), which can be transferred between staphylococcal species (Xu et al., 2018). The pathogenicity of S. epidermidis is further enhanced by virulence genes associated with adherence/biofilm formation, phenol-soluble modulins, and various mobile genetic elements (MGEs) such as plasmids, insertion sequences (ISs), transposons, pathogenicity islands, and phages that are involved in the acquisition and transmission of resistance and virulence characteristics (Bouchami et al., 2016; Rolo et al., 2017). The arginine catabolic mobile element (ACME) system, a pathogenicity island thought to facilitate host colonization and immune evasion, has generated interest in recent years (O’Connor et al., 2018). According to recent phylogenetic studies, the ACME most likely originated from S. epidermidis and transmitted to S. aureus through horizontal transfer (Onishi et al., 2013; Planet et al., 2013). Other factors that have been associated with pathogenicity in S. epidermidis include the metabolic state of the bacterial cell, genomic rearrangements in pathogenic isolates facilitated by IS256, and the conjugative transfer of antibiotic resistance (Cabrera-Contreras et al., 2019).
Analyses of S. epidermidis from diverse geographic locations and clinical origins have shown that the most common STs were ST2, ST59, and ST23, with 74% of screened isolates belonging to the clonal complex 2 (Miragaia et al., 2007). The sequence type ST2, a single S. epidermidis lineage, is dominant in hospital environments (Lee et al., 2018). The widespread sequence types ST5, ST12, and ST23 have been reported to exhibit high resistance against most antibiotic drug classes (Martínez-Meléndez et al., 2016). Additionally, there is increasing rifampicin resistance in S. epidermidis isolates belonging to ST2 and ST23 in Europe, the United States, and Australia (Lee et al., 2018). This observed resistance conferred by mutations in rpoB gene has independently emerged, supporting the assertion that few, well-adapted clonal lineages of S. epidermidis are abound in clinical environments (Lee et al., 2018).
In South Africa, a study on catheter-related bloodstream infections (CRBSIs) in a hospital in Pretoria, identified S. epidermidis as the causative organism in 31% of CRBSI cases (Ehlers et al., 2018). All the S. epidermidis isolates identified were resistant to β-lactams, all carried mecA gene (100%), and 83% carried the IS element IS256. There is however a paucity of studies characterizing the virulence and resistance mechanisms of S. epidermidis in South Africa and Africa more generally.
In this study, we describe the genomic characteristics of methicillin-resistant S. epidermidis (MRSE) isolated from hospitals within the KwaZulu-Natal province in South Africa, specifically their resistome, virulome, mobilome, clonality, and phylogenies together with associations between them and other parameters to gain insights into the genetic basis of antibiotic resistance, virulence, and potential pathogenicity.
Materials and Methods
Description of Strains and Antimicrobial Susceptibility Testing
Sixteen MRSE isolates, selected as a subsample from a previous study (Asante et al., 2021) based on their resistance profiles, were subjected to whole-genome sequencing (WGS). Briefly, an initial 89 suspected CoNS isolates were collected from blood cultures sourced from hospitals within the uMgungundlovu District in the KwaZulu-Natal Province. Isolates were selected based on their resistance to cefoxitin and to multiple antibiotics. MRSE isolates were selected based on their well-documented role as a frequent cause of nosocomial infection and ability to form biofilms (Becker et al., 2014). The 16 clinical MRSE isolates collected from patients in three hospitals in the uMgungundlovu District in the KwaZulu-Natal Province, South Africa, had been subjected to initial identification by Gram staining, colony characteristics, and the StaphaurexTM Latex Agglutination Test (Thermo Scientific, Kent, United Kingdom). Speciation of isolates was done using the automated VITEK 2 system (BioMérieux, Marcy-L’Etoile, France).
The antibiotypes for 20 antibiotics were determined by the Kirby–Bauer disk-diffusion method or by the broth microdilution method. The Clinical and Laboratory Standards Institute (CLSI) guidelines were used to interpret their antimicrobial susceptibility (Clinical and Laboratory Standards Institute, 2016). Antibiotics tested (disk diffusion) included penicillin G (10 μg), cefoxitin (30 μg), ceftaroline (30 μg), ciprofloxacin (5 μg), moxifloxacin (5 μg), azithromycin (15 μg), erythromycin (15 μg), gentamicin (120 μg), amikacin (30 μg), chloramphenicol (30 μg), tetracycline (30 μg), doxycycline (30 μg), tigecycline (15 μg), teicoplanin (30 μg), linezolid (30 μg), clindamycin (10 μg), rifampicin (5 μg), sulfamethoxazole/trimethoprim (1.25/23.75 μg), and nitrofurantoin (300 μg). The minimum inhibitory concentration (MIC) of vancomycin was determined by the broth microdilution method according to CLSI guidelines (Clinical and Laboratory Standards Institute, 2016). The cefoxitin disk diffusion was used to detect methicillin resistance and molecularly confirmed by PCR detection of mecA gene.
Phenotypic Detection of Biofilm Formation
The biofilm-forming abilities of isolates were assessed by the tissue culture plate method described by Mitchell et al. (2010). Briefly, isolates were grown in trypticase soy broth (TSB) supplemented with glucose for 24 h at 37°C. Sterile 96-well microtiter plates were inoculated with bacterial suspension adjusted to 0.5 MacFarland standard and incubated at 37°C for 24 h. The plates were washed after incubation and dried at room temperature. The wells then stained with 0.1% crystal violet solution, incubated at room temperature for 10 min, and washed thrice with distilled water. A 30% acetic acid solution was added to solubilize the crystal violet retained by the biofilm. The optical densities of samples in each well were read at 570 nm using a microtiter plate reader (BMG LABTECH, Offenburg, Germany). S. epidermidis ATCC 35984 was used as positive control. Isolates were categorized as strong, moderate, weak, and non-biofilm formers, using the formula OD < ODC = no biofilm producer, ODC < OD ≤ (2 × ODC) = weak biofilm producer, (2ODC) < OD ≤ (4 × ODC) = moderate biofilm producer, and (4 × ODC) < OD = strong biofilm producer, where ODC is the average OD of the negative control.
Whole-Genome Sequencing
Genomic DNA (gDNA) from pure colonies of MRSE isolates grown from overnight cultures was extracted and purified using the GenElute Bacterial Genomic DNA kit (Sigma Aldrich, St. Louis, MO, United States), according to the manufacturer’s instructions. DNA was checked by agarose gel electrophoresis, while the concentration and purity were determined using NanodropTM 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, United States). For library preparation, the Nextera XT DNA Sample Preparation Kit was used to generate paired-end libraries, followed by WGS on the Illumina MiSeq platform (Illumina, San Diego, CA, United States) at the Sequencing Core Facility, National Institute for Communicable Diseases, Johannesburg, South Africa.
Quality trimming of the sequence reads was done by the use of Sickle version 1.331, while assembler, SPAdes version 3.11 (Bankevich et al., 2012), and the CLC Genomics Workbench version 10.1 (CLC, Bio-QIAGEN, Aarhus, Denmark) were used for de novo assembly of the reads. The assembled contiguous sequences were submitted via the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline to GenBank for gene annotation. The generated contigs were analyzed further to investigate genetic elements of interest.
Bioinformatics Analyses
Pathogenicity, Single-Nucleotide Polymorphism Calling, Resistome, and Virulome Analyses
The identities of isolates and observed phenotypic resistance were confirmed by the genomic data using the Pathogenwatch platform2. The prediction of isolates’ pathogenicity toward human hosts was determined by PathogenFinder available at https://cge.cbs.dtu.dk/services/PathogenFinder/. The assembled genomes from the WGS data were annotated to predict and identify the resistome using ResFinder 4.1 (with a minimum length and threshold of 60% and 90%, respectively), and the Comprehensive Antibiotic Resistance Database (CARD)3 (Alcock et al., 2020), using the default selection criteria “perfect and strict hits only.” The platforms were used side by side to compensate for the inherent deficits in individual platforms.
We determined the genetic basis (chromosomal single-nucleotide polymorphism [SNP]) for observed fluoroquinolone and rifampicin resistance from the assembled genomes by investigating mutations conferring resistance to fluoroquinolones and rifampicin using BLASTn (Altschul et al., 1990). Briefly, gyrA, gyrB, parC, parE, and rpoB genes in a reference susceptible S. epidermidis (S. epidermidis strain ATCC 12228) were aligned with the corresponding genes from resistant isolates in this study with BLASTn to call for SNPs in those genes using the Clustal Omega tool (European Molecular Biology Laboratory). Thus, the mutations in the genomes of the study isolates were manually curated.
VirulenceFinder 2.0 (using a minimum length of 60% and a threshold of 90%)4 (Joensen et al., 2014), virulence factor database (VFDB)5, and BacWGSTdb6 were used to screen for the presence of virulence genes. Various virulence determinants consistent with different major virulence factors (including adherence, enzymes, immune evasion, secretion system, toxins, anti-phagocytosis, biofilm formation/adherence, and intracellular survival) associated with S. epidermidis were investigated.
In silico Multilocus Sequence Typing
Multilocus sequence typing (MLST) was performed in silico using MLST 2.0 program software7 available on the website of the Center for Genomic Epidemiology (Larsen et al., 2012) and the public molecular typing database, PubMLST8. Sequence types were assigned by matching the internal fragments of the seven housekeeping genes (arcC, aroE, gtr, mutS, pyrR, tpiA, and yqiL) from S. epidermidis to identify alleles (Thomas et al., 2007). We performed eBURST analyses (Feil et al., 2004) in the MLST database to identify clones similar to obtained STs.
Identification of Mobile Genetic Elements and Genetic Support Environment
Mobile genetic elements associated with ARGs and their genetic context were investigated using NCBI annotations. RAST 2.0 (Aziz et al., 2008) was used to ascertain MGEs and the genetic support environment. The web-based typing tool SCCmecFinder9 was used for the in silico determination of the SCCmec types and their structural position in the MRSE isolates. In silico detection of plasmid replicon types was done out using PlasmidFinder 2.1, available at https://cge.cbs.dtu.dk/services/PlasmidFinder/ (Carattoli et al., 2014).
The PHASTER tool10 was used to identify and annotate prophage sequences within the genomes (Arndt et al., 2016). Only the prophage regions identified as “intact” by PHASTER were considered. The region positions of the prophages were BLASTED on CARD to determine if the prophages harbored resistance genes. ISs and transposons flanking the resistance genes were identified using the MobileElementFinder v1.0.3 (2020-10-09)11 (Johansson et al., 2020), available on the website of the Center for Genomic Epidemiology12. We used NCBI annotations to determine the support environment of the resistance genes.
Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated (CRISPR-Cas System), Arginine Catabolic Mobile Element, and Restriction-Modification System
We searched for clustered regularly interspaced short palindromic repeats (CRISPR) and cas genes in the sequence data using the CRISPRCasFinder, available at https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index, using the default advanced settings for CRISPR and the clustering model “SubTyping” for Cas. Restriction-ModificationFinder 1.1, available at https://cge.cbs.dtu.dk/services/Restriction-ModificationFinder/, was used to investigate the presence of the restriction-modification system (R-M system), using a minimum length of 60% and a threshold for%ID of 95% (Roer et al., 2016). The ACME genes within the genomes were detected and aligned. Alignment of the ACME components made up of the arc operon, the opp3 operon, and the kdp operon, which was used to classify the ACME components as follows: arc and opp3 operons (type I), the arc operon only (type II), the opp3 operon only (type III), the arc and kdp operons (type IV), and all three arc, opp, and kdp operons (type V), using Pathosystems Resource Integration Center (PATRIC)13 annotations.
Phylogenetic Analyses Using Whole-Genome Sequencing Single-Nucleotide Polymorphisms and Whole-Genome Sequencing Multilocus Sequence Type Trees
Phylogenetic trees were constructed based on the maximum likelihood method using the CSIPhylogeny14 (Kaas et al., 2014), which performs SNP calling, filtering of the SNPs, and inferring phylogeny based on the concatenated alignment of the high-quality SNPs, using the assembled contigs. The analysis was performed on the platform using default parameters, as follows: minimum depth at SNP positions of 10×; minimum relative depth at SNP positions of 10%; minimum distance between SNPs (prune) at 10 bp; minimum SNP quality of 30; minimum read mapping quality of 25; and minimum Z-score of 1.96. S. aureus ATCC 25923 was used to root the tree, facilitating the configuration of the phylogenetic distance between the isolates on the branches. Assembled genomes for comparison were uploaded. To see how our isolates compare with S. epidermidis genomes from Africa, we searched and downloaded S. epidermidis genomes reported in Africa and curated on the PATRIC website and included them in the analysis. To edit and visualize the phylogenetic tree, we used the Figtree program15. We used Phandango (Hadfield et al., 2018) to visualize the phylogeny in association with the isolate demographics, resistance mechanisms, and in silico WGS typing metadata.
Nucleotide Sequence Accession Numbers
The nucleotide sequences of the 16 MRSE strains (C35, C36, C38, C40, C68, C81, C119, C122, C127, C133, C135, C137, C138, C145, C146, and C148) used in this work were uploaded in GenBank database in the Bioproject number PRJNA667485.
Results
Phenotypic Characteristics of Methicillin-Resistant Staphylococcus epidermidis Isolates
Sixteen blood culture MRSE isolates were used in the WGS analysis. Methicillin resistance was confirmed by PCR detection of mecA gene. Twelve (75%) were multidrug-resistant, defined as resistance to at least one antibiotic in three or more distinct antibiotic classes (Magiorakos et al., 2012). Two isolates were resistant to the anti-MRSE cephalosporin ceftaroline, while one and two isolates were resistant to tigecycline and teicoplanin, respectively. None of the isolates were resistant to vancomycin; however, one isolate showed intermediate susceptibility to vancomycin, while one isolate each was resistant to linezolid and nitrofurantoin. Antibiotic susceptibility profiles of MRSE isolates used in this study are shown in Table 1.
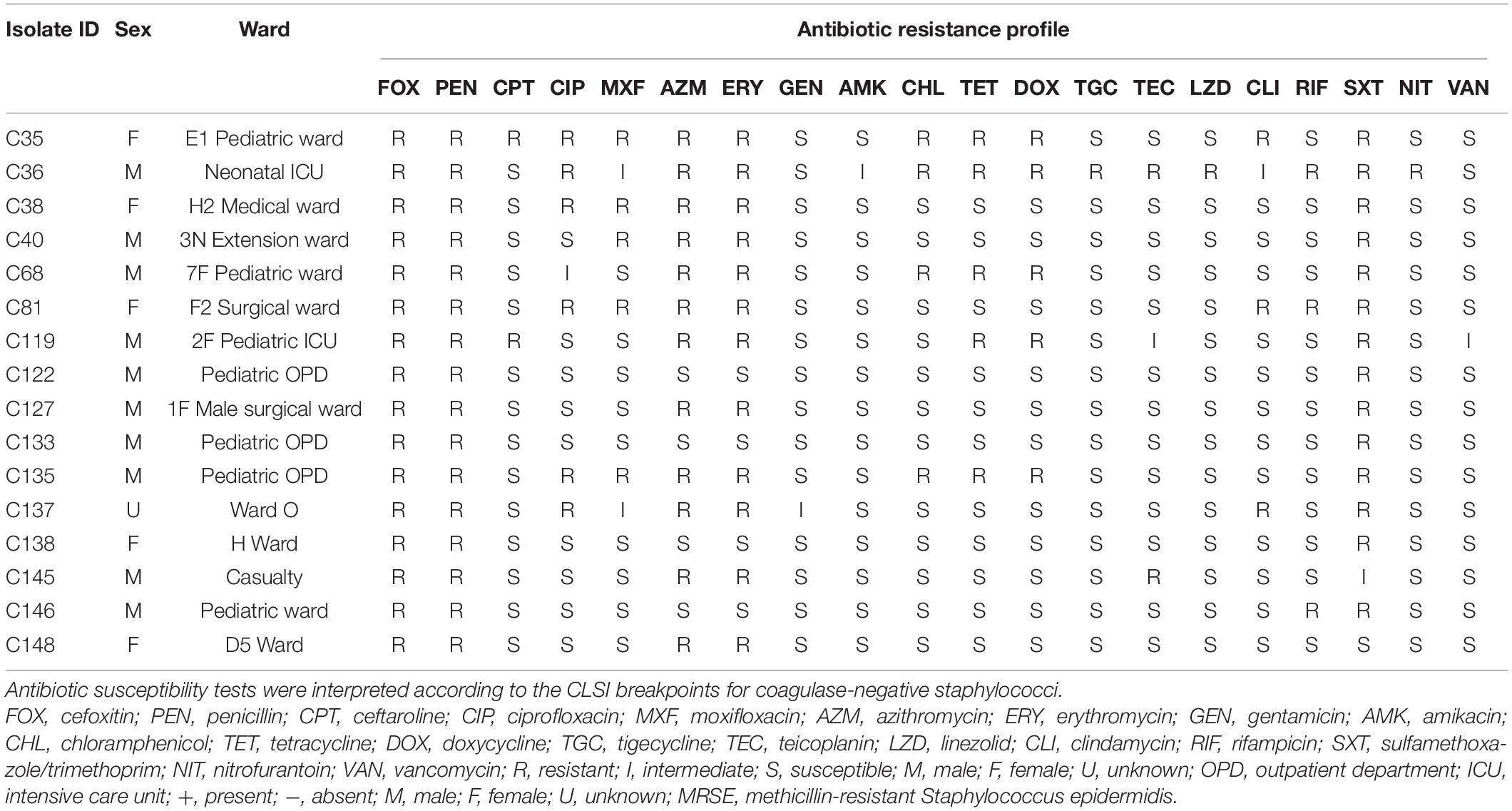
Table 1. Antibiotic susceptibility and demographic characteristics of the MRSE collected from blood cultures.
Genome and Assembly Features, and Characterization of Resistome
The genome and assembly characteristics of the sequences, including size, number of contigs, number of RNAs, guanine–cytosine (GC) content (%), number of coding sequences, N50, and L50 are shown in Table 2. The isolates’ draft genome size ranged from 1.9 Mb to 2.9 Mb, with a GC content of 31.7% to 32.5%. ARGs conferring resistance to β-lactams (mecA and blaZ), tetracyclines [tet(K) and tet(M)], macrolide–lincosamide–streptogramin B antibiotic (MLSB) [erm(A), erm(B), erm(C), msr(A), and mph(C)], trimethoprim-sulfamethoxazole (dfrG), aminoglycosides [aac(6′)-aph(2″), aph(3′)-III, and aadD], chloramphenicol [cat(pC221) and cat(pC233)], and fosfomycin [fosB] were detected in isolates (Table 3). All MRSE isolates possessed either mecA or blaZ gene. Genetic resistance determinants for tigecycline-, teicoplanin-, linezolid-, and nitrofurantoin-resistant phenotypes are currently under investigation (He et al., 2019; Wardenburg et al., 2019; Yushchuk et al., 2020). There was discordance between resistance phenotype and ARGs, relating to trimethoprim- sulfamethoxazole-, tetracycline-, doxycycline-, rifampicin-, and erythromycin-resistant phenotypes. Even though some isolates were phenotypically resistant to these antibiotics, no corresponding ARGs were detected.
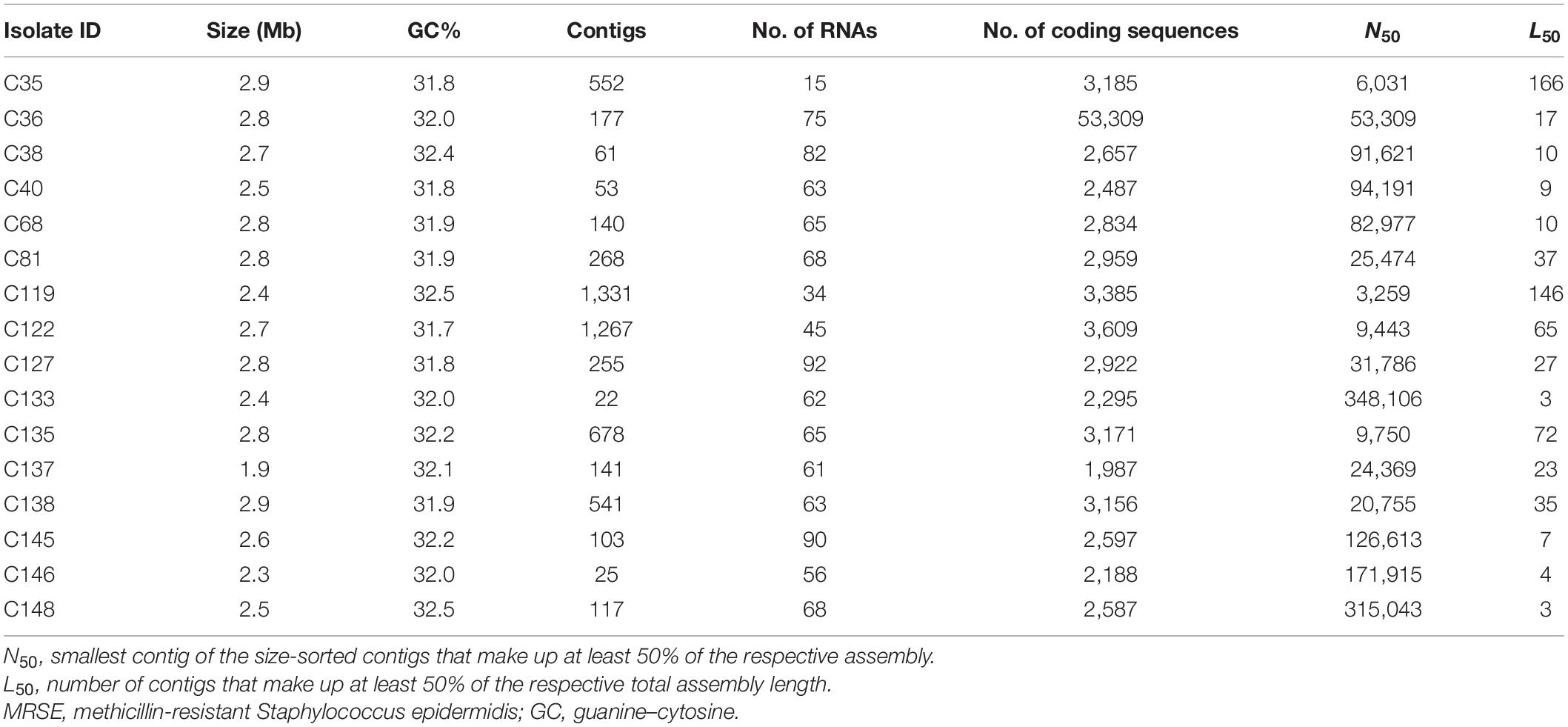
Table 2. Genome and assembly characteristics of sequenced MRSE isolates from clinical sources.
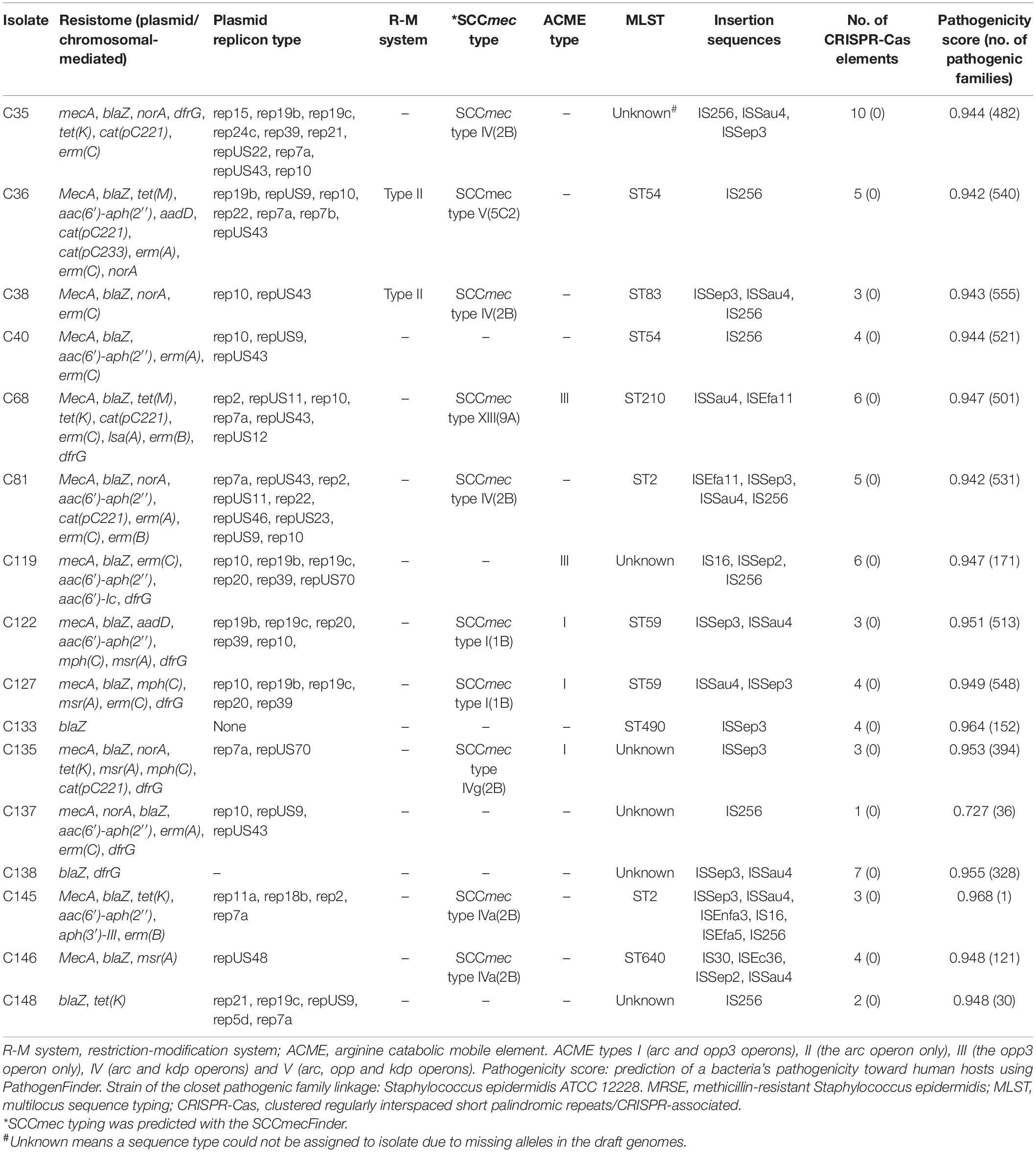
Table 3. Genotypic characteristics of the MRSE isolates.
Thirteen out of the 16 isolates showed an agreement between the cefoxitin-resistant phenotype and mecA gene. The tetracycline-resistant genes tet(K) and tet(M) were found in 4/5 (80%) of isolates phenotypically resistant to tetracycline. The aminoglycoside resistance mechanisms aac(6′)-aph(2″), aad, and aph(3′)-III were found in 6/16 (37.5%) of isolates; however, none of those isolates were phenotypically resistant to gentamicin or amikacin. Furthermore, the MLSB resistance mechanisms msr(A), mph(C), erm(A), erm(B), and erm(C) were also detected in 11/12 (91.6%) of phenotypically resistant isolates. dfrG gene was found in 9/14 (62.3%) of isolates resistant to sulfamethoxazole/trimethoprim. We identified known and putatively novel mutations in gryA, parC, and parE quinolone resistance-determining region (QRDR) genes in some fluoroquinolone-resistant isolates (Table 4). We detected no mutation in the drug target in one of the three isolates found to be resistant to rifampicin. Resistance, in this case, may be mediated by a mechanism yet to be described. We identified the major facilitator superfamily (MFS) antibiotic efflux pump (norA), which can also confer resistance to fluoroquinolones. Seven mutations were found in gyrA and four mutations in parC, but no mutations were detected in gyrB. We further found two mutations (S486Y and Y737S) in rpoB gene.
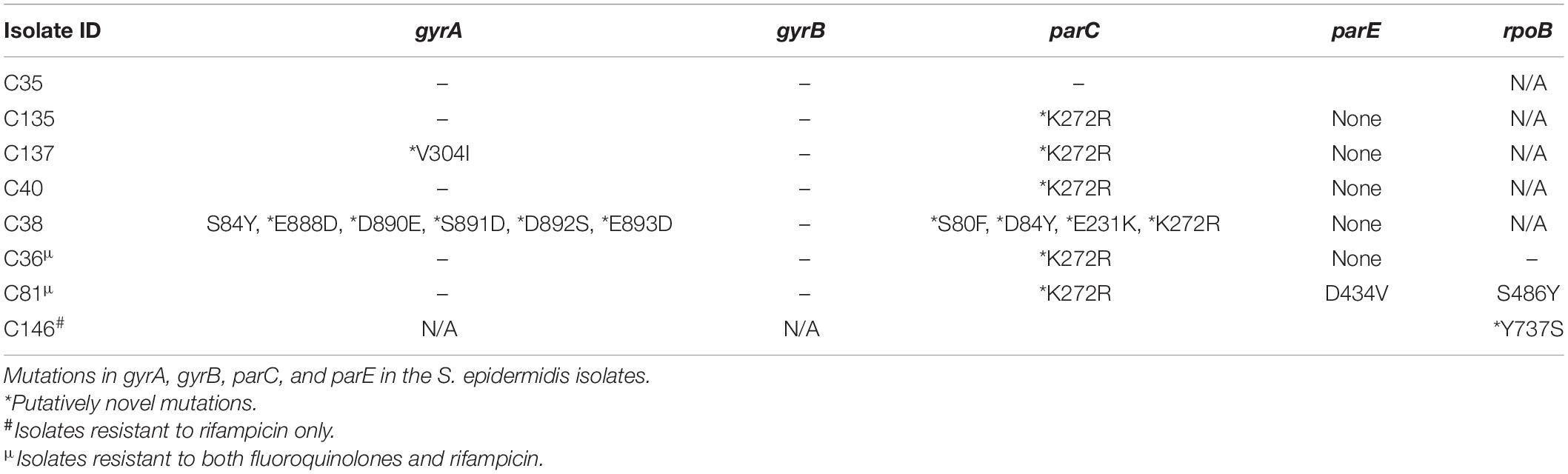
Table 4. Mutations in gyrA, gyrB, parC, and parE in the Staphylococcus epidermidis isolates.
Pathogenicity and Virulome
The mean probability of isolates being pathogenic to humans ranged from 0.727 to 0.968 as predicted by PathogenFinder and matched several pathogenic families. The virulome analysis revealed putative virulence genes encoding proteins belonging to multiple virulence categories of S. epidermidis, i.e., adherence/biofilm formation, enzymes, immune evasion, secretion, toxin, anti-phagocytosis, intracellular survival, and stress adaptation (Table 5).
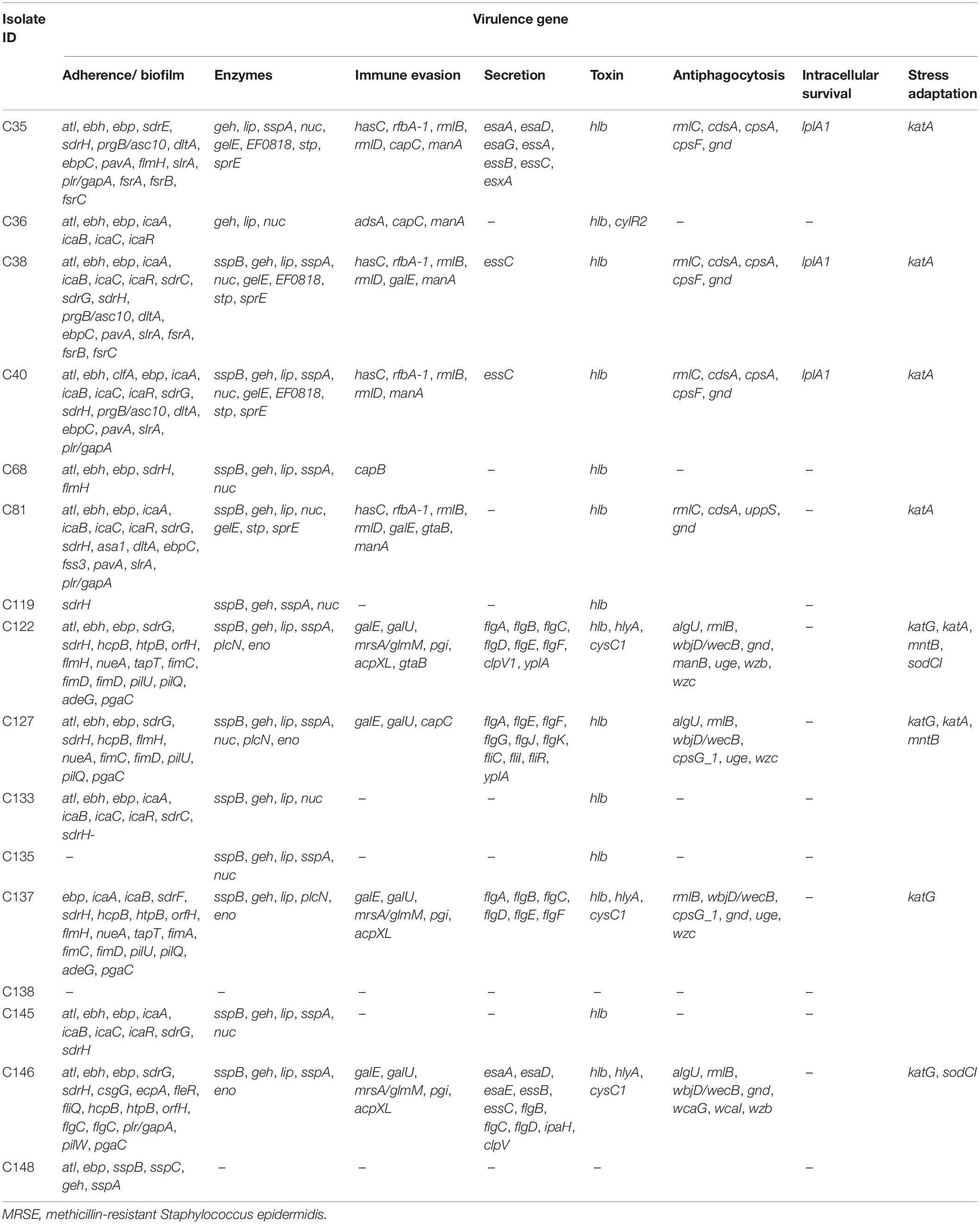
Table 5. Virulence genes identified in MRSE isolates in this study.
Whole-Genome Sequencing-Based Multilocus Sequence Typing
In silico MLST analyses identified seven different MLST types, namely, sequence types (ST) ST54 (two), ST83 (one), ST2 (two), ST490 (one), ST640 (one), ST210 (one), and ST59 (two). The most resistant isolate belonged to ST54 and harbored nine ARGs encoding resistance to five antibiotic drug classes (Table 3). The eBURST analyses matched the various STs to the closest global ancestry STs. ST54 matched STs originating from human and animal sources from Denmark, Italy, Japan, India, and Russia. The eBURST analyses also matched ST2 to the highest number of similar clones originating from several countries including Argentina, Cape Verde, Denmark, Germany, Spain, Italy, and Japan (Supplementary Table 1).
Mobilome and the Genetic Support Environment
In silico SCCmec typing/subtyping revealed six SCCmec types/subtypes: SCCmec type IV(2B), SCCmec type IVg(2B), SCCmec type V(5C2), SCCmec type IVa(2B), SCCmec type XIII(9A), and SCCmec type I(1B). Isolates with SCCmec type I(1B) (n = 2) belonged to sequence type (ST59), while SCCmec type V(5C2) isolates (n = 2) belonged to sequence type ST54. The community-acquired SCCmec type IV (in various subtypes) was the most common type found. mecA gene (but not mecC) was detected by SCCmecFinder as the sole mechanism of resistance in MRSE isolates. All β-lactam-resistant isolates possessed blaZ gene (encoding β-lactamase) and their regulator genes blaR and blaI.
Plasmid analysis by PlasmidFinder and BacWGSTdb16 revealed 24 different plasmid replicon types. Rep10 (10), rep7a (7), and repUS43 (6) were the most predominant plasmid replicon types. Replicon types rep39, repUS9, and rep19b were found in four (25%), four (25%), and five (31.2%) of isolates, respectively.
We found ISs in all 16 isolates. In total, 10 different IS types belonging to six different IS families were detected (Supplementary Figure 1). The most predominant IS families were IS256, 1S200/IS605, and IS3. IS256, closely linked to biofilm formation and virulence in pathogenic MRSE isolates, was found in nine isolates, seven of which had the ica operon involved in biofilm formation. The resistance gene aac(6′)-aph(2″) was found in association with IS256, while the virulence gene gelE (predicted to be linked with Enterococcus) was found in association with ISEfa11 in one isolate (C81). Furthermore, the resistance gene erm(A) was also found in association with the transposon Tn554 in some isolates. Using NCBI annotation, we found blaZ gene surrounded by regulator genes blaR and blaI. Similarly, mecA gene was frequently found with regulatory genes mecI (a repressor) and mecR1 (a sensor inducer) and ISs (IS257 and IS1182). Table 6 describes the genetic support environment of some resistance genes found in this study, with the focus on the association MGEs with the ARGs and virulence genes.
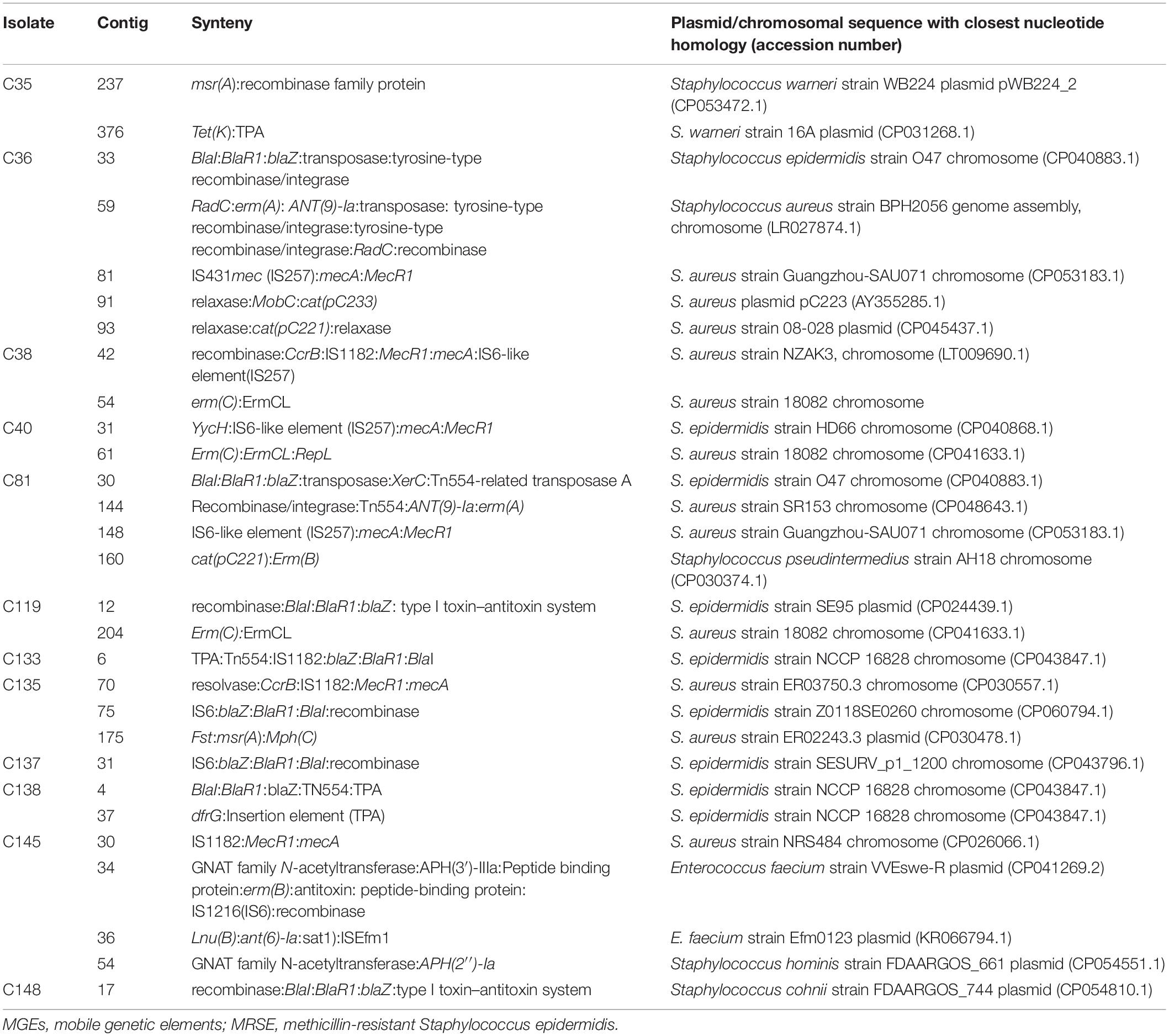
Table 6. MGEs associated with antibiotic resistance genes in the MRSE strains.
The PHASTER tool identified intact prophages integrated into the genomes of 10 isolates. PHAGE_Staphy_StB20 (n = 9) and PHAGE_Staphy_187 (n = 5) were the most predominant prophages. Prophages did not harbor resistance genes. Prophage characteristics, including GC content and number of coding sequences, are shown in Supplementary Table 2.
Identification and Classification of Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated Elements, Arginine Catabolic Mobile Element, and Restriction-Modification System
The CRISPRCasFinder identified sequences with CRISPR. All isolates possessed at least one sequence with CRISPR. However, CRISPR-associated (Cas) genes were not detected. Two isolates possessed the R-M system, and both were classified as type II. ACME was identified in five isolates and were classified as type I (three) and type III (two).
Phylogenetic Relationship Among Staphylococcus epidermidis Isolates in This Study and With Other African Staphylococcus epidermidis Isolates
The phylogenetic relationship between the 16 study isolates (Figure 1) and five collected isolates from African countries, together with a reference strain, was determined (Figure 2). Generally, the isolates clustered according to STs, with the whole-genome phylogenetics showing higher resolution than the MLST typing scheme. For instance, C145 (ST2) was phylogenetically closer to both C36 and C40 (ST54) than the other ST2 isolate (C81), which was found on different mini branch (Figures 1, 2). However, not much inference could be drawn from the tree analysis due to the small number of deposited S. epidermidis genomes from Africa.
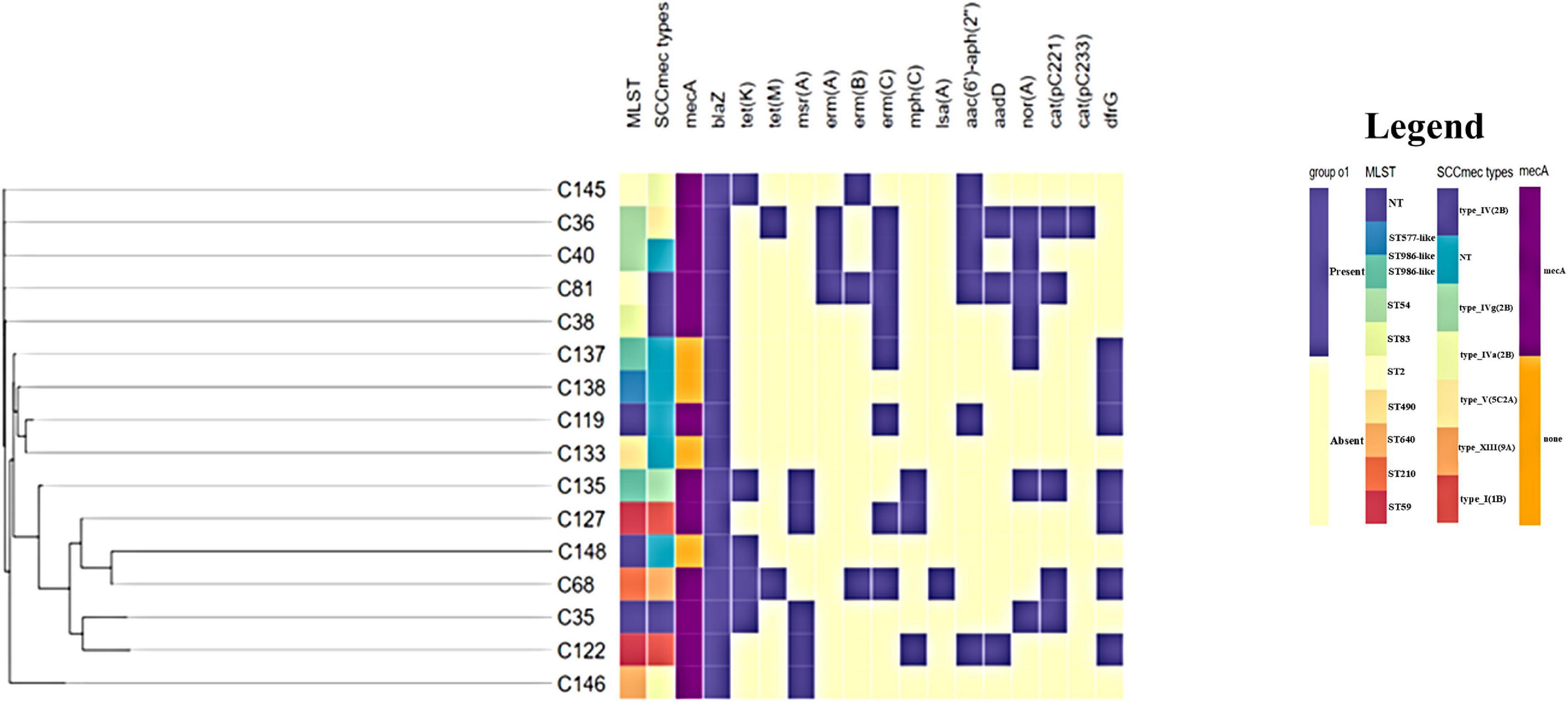
Figure 1. Whole-genome phylogenetic tree annotated with sequence type (ST) assignment, SCCmec types, and antibiotic resistance carriage. Gene names above the annotation according to antibiotic class, as follows: β-lactams: mecA and blaZ; tetracyclines: tet(K) and tet(M); macrolide, lincosamide streptogramin B: msr(A), erm(A), erm(B), erm(C), lsa(A), and mph(C); aminoglycosides: aac(6′)-aph(2″) and aadD; efflux pump: norA; trimethoprim: dfrG; chloramphenicol: cat(pC221) and cat(pC233). The profile was generated using Phandango (https://jameshadfield.github.io/phandango/#/). Heat map at the middle indicates antibiotic resistance gene presence (purple) and absence (yellow). NT, non-typeable.
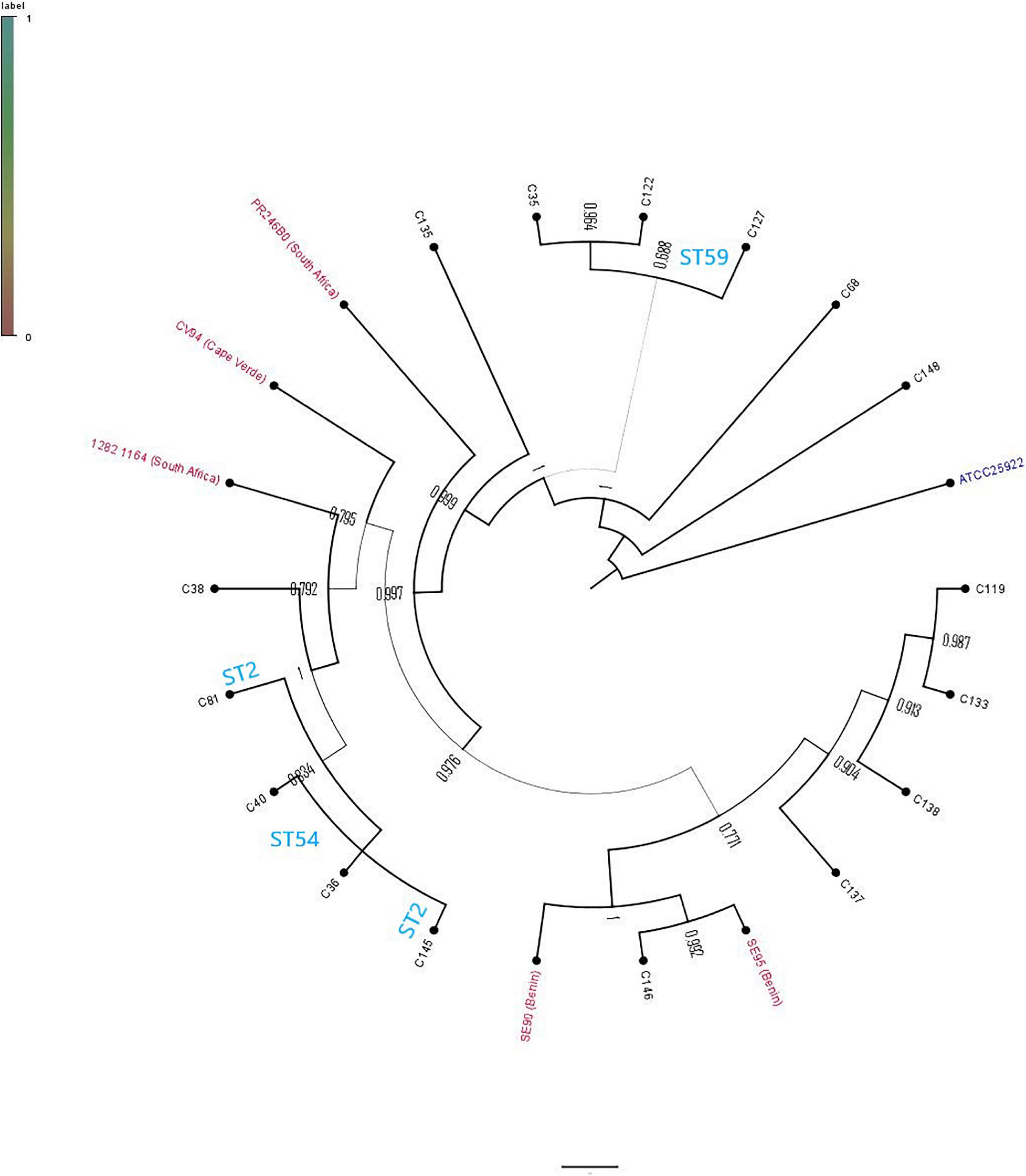
Figure 2. Phylogenetic relationship of Staphylococcus epidermidis isolates in this study and African S. epidermidis (with their countries indicated) obtained from PATRIC.
Discussion
In this study, we sequenced MRSE isolates from clinical sources from hospitals in the uMgungundlovu District in the KwaZulu-Natal Province in South Africa. Using WGS, we studied the genomic characteristics, including resistance and virulence determinants, MGEs, and the genetic environments of the resistance genes observed.
Concerning pathogenicity, it has been suggested that there is no apparent genetic difference between commensal non-pathogenic and pathogenic S. epidermidis strains, albeit nosocomial S. epidermidis strains are boosted with resistance and virulence genes (Méric et al., 2015). The 16 genomes herein analyzed were mostly from pediatric/neonatal patients. S. epidermidis is commonly recovered from bacterial bloodstream infections from neonatal units as a probable causative agent (Asante et al., 2020). Children are particularly susceptible to acquiring S. epidermidis in perinatal hospitals (Cabrera-Contreras et al., 2019).
We detected various resistance genes encoding resistance to several antibiotic drug classes that explained the observed phenotypic resistance in isolates. Resistance genes found in this study encode enzyme inactivation (β-lactamases), enzyme modification of antibiotic target such as erm genes that mediate macrolide–lincosamide–streptogramin B (MLSB) resistance, the aminoglycoside-modifying enzymes, replacement of antibiotic target site as in mecA-mediated resistance to β-lactams in staphylococci, and MFS antibiotic efflux pump (norA), which can also confer resistance to fluoroquinolones (Foster, 2017). There was no association between ARG type and hospital/department. However, mecA, blaZ, and norA genes were found in nearly all isolates from all three hospitals. As well, erm(C), msr(A), dfrG, and mph(C) genes were distributed across the regional hospital but were not ward specific, while dfrG, tet(K), and erm(B) were frequently found in the district hospital. The detection of blaZ in all isolates and the regulator genes blaR and blaI may explain the resistance against penicillin and cephalosporins observed in antibiotic susceptibility tests, which is in agreement with a previous study (Cabrera-Contreras et al., 2019). S. epidermidis isolates carrying similar resistance gene profiles have been previously reported (Xu et al., 2018; Cabrera-Contreras et al., 2019; Raue et al., 2020).
Generally, there was considerable agreement between resistance phenotypes and genotypes observed with ARGs affirming the phenotype, except in a few instances. The discordance between cefoxitin-resistant phenotype and the mecA genotype could be due to alternative mechanisms of resistance, hetero-resistance, or drawback of the present phenotypic testing methods (Band et al., 2019; Harrison et al., 2019).
The lack of phenotypic resistance to gentamicin and amikacin even though aminoglycoside resistance genes were detected could be due the lack of expression of these genes. This could also be due to the fact that amikacin is not affected by most aminoglycoside-modifying enzymes (Doyle et al., 2020). Other factors that can cause discordance between phenotype and genotype include sequence quality of the sample and the read depth of the sequencing platform; insufficient read depth leads to lower coverage (Doyle et al., 2020). The molecular mechanism of resistance in the tigecycline-resistant isolate in this study is unknown, and there was no tet(X) gene detected. Similarly, even though resistance phenotypes were observed for teicoplanin, linezolid, and nitrofurantoin, molecular resistance mechanisms were not detected in their genomes. Limitations of current phenotypic detection methods could be responsible for such discrepancies (Cabrera-Contreras et al., 2019). Novel resistance mechanisms may explain these observed phenotypes and are subject to further studies (Osei Sekyere and Asante, 2018).
We found mutations in gyrA, gyrB, parC, parE, and rpoB genes. In gyrA gene, the substitution S84Y, a mutation known to confer fluoroquinolone resistance (Yamada et al., 2008), was detected, while putatively novel mutations V304I, E888D, D890E, S891D, D892S, and E893D were also detected. The effects of the individual mutations on fluoroquinolone resistance were not investigated in this study.
Several virulence genes are shared by both pathogenic and commensal S. epidermidis strains (Otto, 2009). Consistent with hospital and commensal S. epidermidis strains isolated worldwide (Cabrera-Contreras et al., 2019), our isolates were characterized by adherence/biofilm-forming genes and multidrug resistance. Twelve out of the 16 isolates were biofilm formers as determined by the phenotypic tissue culture plate method, most of which were corroborated by the detection of genes involved in adherence. The ica operon and IS256 used as measures of pathogenicity in S. epidermidis (Murugesan et al., 2018) were not detected in nine and seven isolates, respectively. This is similar to what was observed in a study in Mexico where 50% of analyzed S. epidermidis isolates lacked the ica operon (Cabrera-Contreras et al., 2019). The isolates, however, cannot be dismissed as non-pathogenic, as they can deploy several other virulence factors such as immune evasion (encoded by hasC, rfbA-1, adsA, and capC), toxins (encoded by hlb, hlyA, and cysC1) anti-phagocytosis (rmlC, cdsA, cpsA, and cpsF), stress adaptation (katA, katA, mntB, and sodCI), and intracellular survival (lplA1). Previous studies in S. epidermidis have reported similar biofilm/adherence-associated genes to those reported in this study, including elastin binding protein gene ebp, serine protease gene sspA, autolysin gene atlE, lipase gene geh, the cell wall-associated fibronectin-binding protein gene ebh, nuclease gene nuc, and the ica genes (Salgueiro et al., 2017; Xu et al., 2018; Raue et al., 2020).
These antibiotic resistance and virulence genes have been shown to form part of the accessory genome organized within and between species. Prediction of isolates’ pathogenicity toward human hosts yielded a high average probability score (Pscore ≈ 0.937), close to 1.00. This pathogenicity score juxtaposed with the several virulence genes possessed by isolates, supporting their pathogenic potential to humans (Adzitey et al., 2020).
Methicillin-resistant staphylococci have been associated with MGEs, such as the SCCmec and ACME. MGEs may be repositories of resistance and virulence genes (Sheppard et al., 2016; Foster, 2017). Their importance is related to their mobile nature, which allows them to transfer from cell to cell, within and between bacterial species through horizontal gene transfer, resulting in frequent exchange of genetic material within the population. The variability observed in the genome of S. epidermidis points to the active gene exchange. We thus looked for prophages, CRISPR-Cas system, transposons, and ISs to investigate this phenomenon. The study isolates possessed various ISs that belong to different families. IS256 has been found to be present in pathogenic S. epidermidis strains and closely linked with virulence and biofilm formation among MRSE (Murugesan et al., 2018). This observation was affirmed by the fact that all but two of our isolates with IS256 also contained the ica operon involved in biofilm formation. Indeed, only one isolate (C133) harbored the ica gene that did not have IS256. IS256 has been shown to facilitate genomic rearrangements in pathogenic S. epidermidis isolates (Cabrera-Contreras et al., 2019). The detection of IS families IS110, IS200/IS605, and IS3 agrees with a study of Raue et al. (2020), which also found the IS family IS6 was not detected in the current study. Again, the detection of IS256 and the ica operon in isolates belonging to ST2 agrees with a study conducted in China, which analyzed S. epidermidis from community and hospital environments (Du et al., 2013).
In this study, plasmids rep10, repUS43, rep7a, rep7b, and repUS70 frequently carried resistance genes erm(C), tet(M), tet(K), catpC233, and blaZ, respectively, whereas other genes were chromosomally mediated. These plasmid-borne genes can easily be transferred by conjugation between cells, spreading resistance (Cabrera-Contreras et al., 2019). Furthermore, although prophages can transfer DNA between cells by transduction, no resistance genes were carried by prophages detected in this study. Transposons detected in the genome of two isolates, like plasmids, may carry genes beneficial to bacteria, such as those involved in antibiotic resistance (Mbelle et al., 2019). They can transpose from the chromosome and can move to different sites of the DNA within a cell. In this study, the transposon Tn554 was found to flank the resistance gene erm(A) in some isolates, which may allow it to jump between the chromosome and the plasmid. The small sample size of isolates in this study limits associations between ARGs, virulence genes, MGEs, and isolate demographics. However, the carriage of ARGs on diverse MGEs enhances the mobilization and dissemination of these genes.
CRISPR-Cas system is a defense mechanism deployed by bacteria against phage infection. After surviving a viral infection, certain bacteria imprint a piece of the viral genetic code as a memory of the infection. Bacteria may use this to neutralize future infections caused by similar viruses by cleaving the viral genetic sequence before they can take control of the bacterial host (Makarova et al., 2020). All isolates bore at least one sequence with CRISPR (Table 3), 10 of which contained at least one intact prophage. In a previous study (Raue et al., 2020), four candidates for CRISPR elements were found while investigating the genome of S. epidermidis O47 strain, which is biofilm-positive and methicillin-susceptible. The S. epidermidis O47 strain from that study lacked the CRISPR-associated genes just, as was observed in this study. The two isolates found with the R-M system were classified as type II. Like the CRISPR-Cas system, the R-M system is a defense system developed by bacteria against invasion by bacteriophages (Vasu and Nagaraja, 2013).
The ACME system, a pathogenicity island, has generated interest and is thought to facilitate the host colonization and immune evasion and to transport virulence or survival genes (O’Connor et al., 2018). ACME elements were detected in 31.3% of study isolates. This is lower than the prevalence of 40% to 65.4% reported in MRSE in a study that investigated the diversity of the ACME in S. epidermidis from the oral cavity and periodontal pockets (O’Connor et al., 2018). In comparison, an ACME carriage of 16% was detected in a study that compared the resistance and virulence profile of S. epidermidis isolates from bloodstream infections and nares of neonates (Salgueiro et al., 2017). ACME shows a higher prevalence and greater diversity in S. epidermidis compared with S. aureus. In S. aureus, studies have proved that ACME is usually incorporated in bacterial chromosome adjoining the SCCmecIV element (Diep et al., 2006; Ellington et al., 2008). In this study, however, no association between ACME and SCCmec type IV was found, which is consistent with results obtained by Du et al. (2013). Most of the resistance genes were bracketed by either transposases or ISs or a combination of both, and these can transfer resistance genes within and between plasmids and chromosomes (Mbelle et al., 2019) potentially within and between bacterial species.
cat(pC233) gene was bracketed by plasmid mobilization relaxosome protein MobC in isolate C36, while erm(C) gene was bracketed by the 23S rRNA methylase leader peptide ErmCL and replication/maintenance protein RepL. In two isolates (C148 and C119), blaZ gene was surrounded by the type I toxin–antitoxin system, which may play a role in biofilm and persister cell formation (Wang and Wood, 2011; Table 6 and Figure 1).
Multilocus sequence typing has shown the population structure of S. epidermidis to be clonal (Thomas et al., 2007). The clonal lineages ST2, ST5, and ST23, which are the most commonly reported in hospital environments as well as other sequence types of S. epidermidis are globally distributed (Miragaia, 2018). ST2, in particular, is predominant in the hospital environment. In this study, 2/16 isolates belonged to ST2. Both ST2 isolates in this study possessed icaA gene and IS256, both of which are linked to enhanced pathogenicity (Du et al., 2013). ST35, ST81, and ST89 were not represented in this study, consistent with global data (Cabrera-Contreras et al., 2019). Despite the relatedness of isolates, there is still considerable variation within individual isolates pointing to their mobilization on diverse MGEs.
Conclusion
Staphylococcus epidermidis isolates from public hospitals in uMgungundlovu exhibited several permutations and combinations of ARGs, virulence genes, and MGEs pointing to a complex milieu of mobilized antibiotic resistance and pathogenic characteristics in clonal and multiclonal strains. The study thus reiterates the need for the genomic surveillance of CoNS as emerging pathogens to gain insights into their potential pathogenicity.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
Ethical approval for the study was obtained from the Biomedical Research Ethics Committee of the University of KwaZulu-Natal under reference number BREC/00001302/2020. This study was a sub-study of the overarching research program on Antibiotic Resistance and One Health (Reference: 473 BCA444/16).
Author Contributions
JA, DA, AA, and SE conceptualized the work. JA carried out the experiments, analyzed the data, and drafted the manuscript. DA and AA analyzed the data and undertook critical revision of the manuscript. BH, LB, and SE undertook critical revision of the manuscript. All authors have contributed to the final version.
Funding
The research reported in this publication was funded by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (Grant No. 98342), the SAMRC under a Self-Initiated Research Grant and the College of Health Sciences, University of KwaZulu-Natal, South Africa.
Conflict of Interest
SE is the chairperson of the Global Respiratory Infection Partnership and member of the Global Hygiene Council, both sponsored by unrestricted educational grants from Reckitt and Benckiser (Pty.) UK.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to Sumayya Haffejee of the National Health Laboratory Service for her assistance during sample collection and in obtaining demographic data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.656306/full#supplementary-material
Footnotes
- ^ https://github.com/najoshi/sickle
- ^ https://pathogen.watch
- ^ https://card.mcmaster.ca/analyze/rgi
- ^ https://cge.cbs.dtu.dk/services/VirulenceFinder/
- ^ http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi?func=VFanalyzer
- ^ http://bacdb.cn/BacWGSTdb
- ^ https://cge.cbs.dtu.dk/services/MLST/
- ^ https://pubmlst.org/
- ^ https://cge.cbs.dtu.dk/services/SCCmecFinder/
- ^ https://phaster.ca/
- ^ https://cge.cbs.dtu.dk/services/MobileElementFinder/
- ^ https://cge.cbs.dtu.dk/services/
- ^ https://www.patricbrc.org/
- ^ https://cge.cbs.dtu.dk/services/CSIPhylogeny/
- ^ http://tree.bio.ed.ac.uk/software/figtree/
- ^ http://bacdb.org/BacWGSTdb/
References
Adzitey, F., Asante, J., Kumalo, H. M., Khan, R. B., Somboro, A. M., and Amoako, D. G. (2020). Genomic investigation into the virulome, pathogenicity, stress response factors, clonal lineages, and phylogenetic relationship of Escherichia coli strains isolated from meat sources in Ghana. Genes 11:1504. doi: 10.3390/genes11121504
Alcock, B. P., Raphenya, A. R., Lau, T. T., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21.
Asante, J., Amoako, D. G., Abia, A. L., Somboro, A. M., Govinden, U., Bester, L. A., et al. (2020). Review of clinically and epidemiologically relevant coagulase-negative Staphylococci in Africa. Microb. Drug Resist. 26, 951–970. doi: 10.1089/mdr.2019.0381
Asante, J., Hetsa, B. A., Amoako, D. G., Abia, A. L. K., Bester, L. A., and Essack, S. Y. (2021). Multidrug-Resistant coagulase-negative staphylococci isolated from bloodstream in the umgungundlovu district of kwazulu-natal province in south africa: emerging pathogens. Antibiotics 10:198. doi: 10.3390/antibiotics10020198
Azih, A., and Enabulele, I. (2013). Species distribution and virulence factors of coagulase negative Staphylococci isolated from clinical samples from the University of Benin Teaching hospital, Edo State, Nigeria. J. Nat. Sci. Res. 3, 38–43.
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Band, V. I., Hufnagel, D. A., Jaggavarapu, S., Sherman, E. X., Wozniak, J. E., Satola, S. W., et al. (2019). Antibiotic combinations that exploit heteroresistance to multiple drugs effectively control infection. Nat. Microbiol. 4, 1627–1635. doi: 10.1038/s41564-019-0480-z
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Becker, K., Heilmann, C., and Peters, G. (2014). Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27, 870–926.
Bouchami, O., De Lencastre, H., and Miragaia, M. (2016). Impact of insertion sequences and recombination on the population structure of Staphylococcus haemolyticus. PLoS One 11:e0156653. doi: 10.1371/journal.pone.0156653
Cabrera-Contreras, R., Santamaría, R. I., Bustos, P., Martínez-Flores, I., Meléndez-Herrada, E., Morelos-Ramírez, R., et al. (2019). Genomic diversity of prevalent Staphylococcus epidermidis multidrug-resistant strains isolated from a Children’s Hospital in México City in an eight-years survey. PeerJ 7, 1–27.
Carattoli, A., Zankari, E., Garcìa-Fernandez, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). PlasmidFinder and pMLST: in silico detection and typing of plasmids. Antimicrob. Agents Chemother. AAC 58, 02412–02414.
Clinical and Laboratory Standards Institute (2016). Performance Standards for Antimicrobial Susceptibility Testing. CLSI Documents Information Supplement M100-S127, 26th Edn. Wayne, PA: Clinical and Laboratory Standards Institute, 74–80.
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/s0140-6736(06)68231-7
Doyle, R. M., O’sullivan, D. M., Aller, S. D., Bruchmann, S., Clark, T., Pelegrin, A. C., et al. (2020). Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study. Microb. Genomics 6:e000335.
Du, X., Zhu, Y., Song, Y., Li, T., Luo, T., Sun, G., et al. (2013). Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One 8:e62742. doi: 10.1371/journal.pone.0062742
Ehlers, M. M., Strasheim, W., Lowe, M., Ueckermann, V., and Kock, M. M. (2018). Molecular epidemiology of Staphylococcus epidermidis implicated in catheter-related bloodstream infections at an Academic Hospital in Pretoria, South Africa. Front. Microbiol. 9:417. doi: 10.3389/fmicb.2018.00417
Ellington, M. J., Yearwood, L., Ganner, M., East, C., and Kearns, A. M. (2008). Distribution of the ACME-arcA gene among methicillin-resistant Staphylococcus aureus from England and Wales. J. Antimicrob. Chemother. 61, 73–77. doi: 10.1093/jac/dkm422
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/jb.186.5.1518-1530.2004
Foster, T. J. (2017). Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 41, 430–449. doi: 10.1093/femsre/fux007
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Harrison, E. M., Ba, X., Coll, F., Blane, B., Restif, O., Carvell, H., et al. (2019). Genomic identification of cryptic susceptibility to penicillins and β-lactamase inhibitors in methicillin-resistant Staphylococcus aureus. Nat. Microbiol. 4, 1680–1691.
He, T., Wang, R., Liu, D., Walsh, T. R., Zhang, R., Lv, Y., et al. (2019). Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol. 4, 1450–1456. doi: 10.1038/s41564-019-0445-2
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/jcm.03617-13
Johansson, M. H., Bortolaia, V., Tansirichaiya, S., Aarestrup, F. M., Roberts, A., and Petersen, T. N. (2020). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/jac/dkaa390
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. doi: 10.1371/journal.pone.0104984
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/jcm.06094-11
Lee, J. Y., Monk, I. R., Da Silva, A. G., Seemann, T., Chua, K. Y., Kearns, A., et al. (2018). Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat. Microbiol. 3, 1175–1185. doi: 10.1038/s41564-018-0230-7
Magiorakos, A.-P., Srinivasan, A., Carey, R., Carmeli, Y., Falagas, M., Giske, C., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J., et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Martínez-Meléndez, A., Morfín-Otero, R., Villarreal-Treviño, L., Camacho-Ortíz, A., González-González, G., Llaca-Díaz, J., et al. (2016). Molecular epidemiology of coagulase-negative bloodstream isolates: detection of Staphylococcus epidermidis ST2, ST7 and linezolid-resistant ST23. Braz. J. Infect. Dis. 20, 419–428. doi: 10.1016/j.bjid.2016.05.007
Mbelle, N. M., Feldman, C., Sekyere, J. O., Maningi, N. E., Modipane, L., and Essack, S. Y. (2019). The resistome, mobilome, virulome and phylogenomics of multidrug-resistant Escherichia coli clinical isolates from Pretoria, South Africa. Sci. Rep. 9:16457.
Méric, G., Miragaia, M., De Been, M., Yahara, K., Pascoe, B., Mageiros, L., et al. (2015). Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 7, 1313–1328.
Miragaia, M. (2018). Factors contributing to the evolution of Meca-mediated β-lactam resistance in staphylococci: update and new insights from whole genome sequencing (WGS). Front. Microbiol. 9:2723. doi: 10.3389/fmicb.2018.02723
Miragaia, M., Thomas, J., Couto, I., Enright, M., and De Lencastre, H. (2007). Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J. Bacteriol. 189, 2540–2552. doi: 10.1128/jb.01484-06
Mitchell, G., Séguin, D. L., Asselin, A.-E., Déziel, E., Cantin, A. M., Frost, E. H., et al. (2010). Staphylococcus aureus sigma B-dependent emergence of small-colony variants and biofilm production following exposure to Pseudomonas aeruginosa 4-hydroxy-2-heptylquinoline-N-oxide. BMC Microbiol. 10:33. doi: 10.1186/1471-2180-10-33
Murugesan, S., Mani, S., Kuppusamy, I., and Krishnan, P. (2018). Role of insertion sequence element IS256 as a virulence marker and its association with biofilm formation among methicillin-resistant Staphylococcus epidermidis from hospital and community settings in Chennai, South India. Indian J. Med. Microbiol. 36, 124–126. doi: 10.4103/ijmm.ijmm_17_276
O’Connor, A. M., Mcmanus, B. A., Kinnevey, P. M., Brennan, G. I., Fleming, T. E., Cashin, P. J., et al. (2018). Significant enrichment and diversity of the staphylococcal arginine catabolic mobile element ACME in Staphylococcus epidermidis isolates from subgingival peri-implantitis sites and periodontal pockets. Front. Microbiol. 9:1558. doi: 10.3389/fmicb.2018.01558
Onishi, M., Urushibara, N., Kawaguchiya, M., Ghosh, S., Shinagawa, M., Watanabe, N., et al. (2013). Prevalence and genetic diversity of arginine catabolic mobile element (ACME) in clinical isolates of coagulase-negative staphylococci: identification of ACME type I variants in Staphylococcus epidermidis. Infect. Genet. Evol. 20, 381–388. doi: 10.1016/j.meegid.2013.09.018
Osei Sekyere, J., and Asante, J. (2018). Emerging mechanisms of antimicrobial resistance in bacteria and fungi: advances in the era of genomics. Future Microbiol. 13, 241–262. doi: 10.2217/fmb-2017-0172
Otto, M. (2009). Staphylococcus epidermidis—the’accidental’pathogen. Nat. Rev. Microbiol. 7, 555–567. doi: 10.1038/nrmicro2182
Planet, P. J., Larussa, S. J., Dana, A., Smith, H., Xu, A., Ryan, C., et al. (2013). Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. mBio 4:e00889-13.
Raue, S., Fan, S.-H., Rosenstein, R., Zabel, S., Luqman, A., Nieselt, K., et al. (2020). The Genome of Staphylococcus epidermidis O47. Front. Microbiol. 11:2061. doi: 10.3389/fmicb.2020.02061
Roer, L., Hendriksen, R., Leekitcharoenphon, P., Lukjancenko, O., Kaas, R., Hasman, H., et al. (2016). Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? mSystems 1, 1–15. doi: 10.1128/mSystems.00009-16
Rolo, J., Worning, P., Nielsen, J. B., Bowden, R., Bouchami, O., Damborg, P., et al. (2017). Evolutionary origin of the staphylococcal cassette chromosome mec (SCCmec). Antimicrob. Agents Chemother. 61, 1–16.
Salgueiro, V. C., Iorio, N. L. P., Ferreira, M. C., Chamon, R. C., and Dos Santos, K. R. N. (2017). Methicillin resistance and virulence genes in invasive and nasal Staphylococcus epidermidis isolates from neonates. BMC Microbiol. 17:15. doi: 10.1186/s12866-017-0930-9
Sheppard, A. E., Stoesser, N., Wilson, D. J., Sebra, R., Kasarskis, A., Anson, L. W., et al. (2016). Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob. Agents Chemother. 60, 3767–3778. doi: 10.1128/aac.00464-16
Thomas, J. C., Vargas, M. R., Miragaia, M., Peacock, S. J., Archer, G. L., and Enright, M. C. (2007). Improved multilocus sequence typing scheme for Staphylococcus epidermidis. J. Clin. Microbiol. 45, 616–619. doi: 10.1128/jcm.01934-06
Vasu, K., and Nagaraja, V. (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77, 53–72. doi: 10.1128/mmbr.00044-12
Wang, X., and Wood, T. K. (2011). Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583. doi: 10.1128/aem.05068-11
Wardenburg, K. E., Potter, R. F., D’souza, A. W., Hussain, T., Wallace, M. A., Andleeb, S., et al. (2019). Phenotypic and genotypic characterization of linezolid-resistant Enterococcus faecium from the USA and Pakistan. J. Antimicrob. Chemother. 74, 3445–3452. doi: 10.1093/jac/dkz367
Xu, Z., Misra, R., Jamrozy, D., Paterson, G. K., Cutler, R. R., Holmes, M. A., et al. (2018). Whole genome sequence and comparative genomics analysis of multi-drug resistant environmental Staphylococcus epidermidis ST59. G3 Genes Genomes Genet. 8, 2225–2230. doi: 10.1534/g3.118.200314
Xu, Z., Mkrtchyan, H. V., and Cutler, R. R. (2015). Antibiotic resistance and mecA characterization of coagulase-negative staphylococci isolated from three hotels in London, UK. Front. Microbiol. 6:947. doi: 10.3389/fmicb.2015.00947
Yamada, M., Yoshida, J., Hatou, S., Yoshida, T., and Minagawa, Y. (2008). Mutations in the quinolone resistance determining region in Staphylococcus epidermidis recovered from conjunctiva and their association with susceptibility to various fluoroquinolones. Br. J. Ophthalmol. 92, 848–851. doi: 10.1136/bjo.2007.129858
Keywords: Staphylococcus epidermidis, antibiotic resistance, whole-genome sequencing, genomics, coagulase-negative staphylococci
Citation: Asante J, Hetsa BA, Amoako DG, Abia ALK, Bester LA and Essack SY (2021) Genomic Analysis of Antibiotic-Resistant Staphylococcus epidermidis Isolates From Clinical Sources in the Kwazulu-Natal Province, South Africa. Front. Microbiol. 12:656306. doi: 10.3389/fmicb.2021.656306
Received: 20 January 2021; Accepted: 07 July 2021;
Published: 05 August 2021.
Edited by:
Karsten Becker, Friedrich Loeffler Institute of Medical Microbiology, University Medicine Greifswald, GermanyReviewed by:
Eike Joachim Steinig, James Cook University, AustraliaFranziska Layer, Robert Koch Institute (RKI), Germany
Copyright © 2021 Asante, Hetsa, Amoako, Abia, Bester and Essack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Asante, am9zYW50ZTMzQHlhaG9vLmNvbQ==