 Xiaoying Han†
Xiaoying Han† Yangchun Cao
Yangchun Cao Junhu Yao
Junhu Yao- Country College of Animal Science and Technology, Northwest A&F University, Yangling, China
High starch diets have been proven to increase the risk of hindgut acidosis in high-yielding dairy animals. As an effective measurement of dietary carbohydrate for ruminants, studies on rumen degradable starch (RDS) and the effects on the gut microbiota diversity of carbohydrate-active enzymes (CAZymes), and Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology functional categories are helpful to understand the mechanisms between gut microbiota and carbohydrate metabolism in dairy goats. A total of 18 lactating goats (45.8 ± 1.54 kg) were randomly divided equally into three dietary treatments with low dietary RDS concentrations of 20.52% (LRDS), medium RDS of 22.15% (MRDS), and high RDS of 24.88% (HRDS) on a DM basis for 5 weeks. Compared with the LRDS and MRDS groups, HRDS increased acetate molar proportion in the cecum. For the HRDS group, the abundance of family Ruminococcaceae and genus Ruminococcaceae UCG-010 were significantly increased in the cecum. For the LRDS group, the butyrate molar proportion and the abundance of butyrate producer family Bacteroidale_S24-7, family Lachnospiraceae, and genus Bacteroidale_S24-7_group were significantly increased in the cecum. Based on the BugBase phenotypic prediction, the microbial oxidative stress tolerant and decreased potentially pathogenic in the LRDS group were increased in the cecum compared with the HRDS group. A metagenomic study on cecal bacteria revealed that dietary RDS level could affect carbohydrate metabolism by increasing the glycoside hydrolase 95 (GH95) family and cellulase enzyme (EC 3.2.1.4) in the HRDS group; increasing the GH13_20 family and isoamylase enzyme (EC 3.2.1.68) in the LRDS group. PROBIO probiotics database showed the relative gene abundance of cecal probiotics significantly decreased in the HRDS group. Furthermore, goats fed the HRDS diet had a lower protein expression of Muc2, and greater expression RNA of interleukin-1β and secretory immunoglobulin A in cecal mucosa than did goats fed the LRDS diet. Combined with the information from previous results from rumen, dietary RDS level altered the degradation position of carbohydrates in the gastrointestinal (GI) tract and increased the relative abundance of gene encoded enzymes degrading cellulose in the HRDS group in the cecum of dairy goats. This study revealed that the HRDS diet could bring disturbances to the microbial communities network containing taxa of the Lachnospiraceae and Ruminococcaceae and damage the mucus layer and inflammation in the cecum of dairy goats.
Introduction
Due to the complex and abundant microorganisms colonized in the rumen and cecum, feed fermentation organs in ruminants are more diverse than monogastric animals (Howie and Baker, 1952; Shen et al., 2020b). Rumen has attracted a tremendous amount of attention in ruminant nutrition studies, whereas limited attention has been placed on the microorganism of the hindgut (Seshadri et al., 2018; Xue et al., 2020). Dietary carbohydrate could affect the intestinal microbiome and metabolites by affecting the digesta composition in the hindgut of human and murine (Riva et al., 2019; Deehan et al., 2020). For ruminants, high starch intake tends to decrease ruminal fiber digestibility and increase the risk of dysbiosis and the depression of fiber degradation in the hindgut (Petri et al., 2019). Previously, we found rumen degradable starch (RDS) could be an effective evaluation for ruminant dietary carbohydrate and digestive health. Increased RDS could affect cellulolytic bacteria and amylolytic bacteria in rumen and increase the potential risk for sub-acute ruminal acidosis of goats (Li et al.,2014a,b). High RDS diet decreased the cellulose and starch branching enzymes in dairy goats. Meanwhile, large amount of RDS gathered in rumen not only enhance the risk of subacute ruminal acidosis (SARA), but also cause hindgut acidosis in dairy cows (Petri et al., 2019). Increased dietary RDS represents a rich amount of undigested starch passing into the intestine. Hindgut acidosis could be characterized by accumulation of short-chain fatty acids (SCFAs), decreased pH, and damage to the intestinal-barrier such as mucin (Plaizier et al., 2018). For ruminants suffering hindgut acidosis, due to feeding on a higher proportion of grains or concentrate and lesser amounts of fiber and forage, which will increase the flow of fermentable substrate to the hindgut, undigested carbohydrate and microbial products like LPS accumulate in the hindgut (Gressley et al., 2011; Shen et al., 2020a). Those compounds could cause intestinal mucosa injury and increases expression of cytokines, such as tumor necrosis factor-α and interleukin-1β (Tao L. et al., 2017). Over 99% of the microorganisms colonizing in the intestinal tract are bacteria, which could use the undigested nutrients and non-structural CHO to supply various material for host ruminants, like energy sources, neurotransmitters, and even toxins (Steele et al., 2016; Tao S. Y. et al., 2017). Although a lot of studies used metagenomic techniques to investigate the metagenome-assembled genomes in rumen (Stewart et al., 2018, 2019), the function of microbes colonized in the intestine still did not get enough attention. At present, most research about dietary treatments in ruminants characterizing the intestinal bacterial community based on 16s rRNA sequencing analysis also have shortcomings, in that they do not include sufficient information of microbial functions (Jiao et al., 2019; Li et al., 2019). Our study is the first one combining 16S rRNA sequencing and metagenomic technologies together to evaluate the effects of dietary RDS level on intestinal bacteria, immune barrier, and carbohydrate degradation by carbohydrate-active enzymes (CAZymes) and Kyoto Encyclopedia of Gene and Genomes (KEGG) functional analysis in dairy goats. In general, this study enhanced our understanding of hindgut microorganisms in response to the changes in dietary RDS level and cellulose degradation in dairy goats.
Materials and Methods
Ethics Statement
In this study, all animal procedures were approved by the Institutional Animal Care and Use Committee of Northwest A&F University. The protocols of this study were specifically approved with the protocol number NWAFAC1008.
Animals, Diets, and Experimental Design
A total of 18 lactating dairy goats with an average body weight of 45.8 ± 1.54 kg were used in this study. Two weeks before the experiment, all goats were fed ad libitum with a diet containing a forage-to-concentrate ratio (F: C) of 45:55 to ensure adaptation to the diet. After dietary adaptation, all goats were randomly divided equally into three dietary treatments with different RDS levels for 5 weeks. The three diets with different RDS levels were designed based on our previous study (Li et al., 2014c). All goats were fed twice per day at 0,830 and 1,630 h, and drinking water was offered ad libitum during the experimental period. The dietary treatments were isoenergetic, isonitrogenous, and isostarch diets with three levels of RDS: low RDS (LRDS = 20.52%), medium RDS (MRDS = 22.15%), and high RDS (HRDS = 24.88%). The calculation of RDS used the following formula: RDS = × ERDi, where Pi represents the proportion of dietary starch of the feed i in the diet, ERDi represents the effective starch degradability of the feed i, and n is the number of ingredients containing starch in the feed formula. A detailed method was reported in Li et al. (2018). Corn and wheat were used to obtain the required RDS level. The dietary composition and nutrient contents of the experimental diets are presented in Supplementary Table 1.
Sample Collection
After a 5-week feeding period, all goats were slaughtered 3 h after morning feeding under combined anesthesia {intramuscular injection of xylazole and dihydroetorphine hydrochloride [1.5 mL (100 kg body weight); Lumianning; HUAMU, Jilin, China]}. After death, the intestine (jejunum, cecum, colon, and rectum) were separated and the digesta of jejunum, cecum, and rectum were aseptically collected and frozen (frozen by liquid nitrogen in the first step then stored at −80°C) for further analysis. The intestinal mucous of the cecum was separated from the muscular layers and carefully harvested. The intestine tissues of the cecum were collected in 2 × 2 cm2 pieces and fixed in 4% paraformaldehyde to make paraffin-embedded tissues. The pH of the digesta in the jejunum and cecum were measured by mobile pH meter (Ohaus Instruments Co., Ltd., China).
Determination of Intestinal Digesta Composition
The cellulose content in the cecum was determined by Cellulose Content Kit (Nanjing Jiancheng Bioengineering Institute CO., Ltd., China). The amylose content in the cecum was determined by Megazyme Total Starch Kit (K-TSTA 04/2009, Biostest Co., Ltd., China). The acetate, propionate, butyrate, valerate, isobutyrate, and isovalerate concentrations of hindgut digesta were determined by Agilent 7820A GC system (Agilent, CA, United States) (Li et al., 2014b). About 0.5 g digesta was weighed and diluted with 1 mL ultrapure water. Then, a high-speed homogenizer Scientz-48 (Ningbo Scientz Bioscience Co., Inc., China) was used to make sure the combination was mixed completely. After being centrifuged by centrifugal 5810R (Eppendorf, United States) at 13,500 rpm about 10 min, 1.5 mL supernatant of diluted digesta was moved into 300 μL 25%w/v metaphosphoric solution for further purification. After repeating the previous centrifugation step, 1 mL supernatant was moved into 200 μL 25% crotonic acid, and the mixture was collected into an EP tube passed through a 0.5 μm filter to be measured.
Bacterial DNA Extraction and PCR Amplification
The total microbial DNA was extracted from intestinal digesta using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, United States.). The concentrations of all extracted nucleic acid samples were detected by spectrophotometry Nanodrop 2000 (Thermo Fisher Scientific, Wilmington, DE, United States), and purity was monitored with 1% agarose gels. All extracted DNA samples were stored at −80°C for further analysis. The amplicon library preparation was performed by polymerase chain reaction amplification of the V3-V4 region of the 16S rRNA gene using the primer pairs: 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCT-AAT-3′) by thermocycler polymerase chain reaction system ABI GeneAmp 9700 (Thermo Fisher Scientific, Wilmington, DE, United States) The polymerase chain reactions amplification refer to the previous study (Zhang et al., 2020).
16S rRNA Sequencing Analysis
After library construction, the samples were sequenced on an Illumina MiSeq platform using HiSeq 3000/4000 PE Cluster Kit and HiSeq 3000/4000 SBS Kit (Altschul et al., 1997). The raw reads were demultiplexed, quality-filtered, and analyzed by Quantitative Insight Into Microbial Ecology software [refer to previous study by Navas-Molina et al. (2013)]. The sequences were clustered into operational taxonomic units (OTUs) at a 97% similarity threshold using UPARSE (version 7.11) (Edgar et al., 2011). Silva v132 Database based on the mothur version 1.30.1 algorithm was used to select, annotate, and normalize OTUs (Schloss et al., 2009; Glöckner et al., 2017). Sequences were submitted to the NCBI short read archive (SRA) under accession numbers SRP295458.
The principle coordinate analysis (PCoA) graphs were enabled the between-samples comparison (beta diversity) of the microbial communities. The PCoA of weighted UniFrac matrices with statistical significance was determined by Permutational Multivariate Analysis of Variance (PERMANOVA) (Lozupone et al., 2011; Dong et al., 2019). The resulting OTUs were submitted to BugBase2 which could calculate differences between groups in terms of microbial phenotypes in the cecum: microbial oxidative stress tolerant and potentially pathogenic (Thomas et al., 2016). Differential abundance of family, genus, and predicted microbial phenotypes was tested by SPSS software (version 20.0) using the one-way ANOVA model across the three treatments.
Metagenome Library Preparation and Sequencing
Digesta DNA from the jejunum, caecum, and rectum were fragmented to obtain fragments of an average size of 400 bp, using Covaris M220 (Gene Company Limited, China) for paired-end library construction. Paired-end library preparations were performed according to the NEXTFLEX Rapid DNA-Seq (Bioo Scientific, Austin, TX, United States). Adapters containing the full complement of sequencing primer hybridization sites were ligated to the blunt end of fragments. After library construction, the paired-end sequencing was performed on Illumina HiSeq4000 platform (Illumina Inc., San Diego, CA, United States) located at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) using HiSeq 3000/4000 PE Cluster Kit and HiSeq 3000/4000 SBS Kit according to the manufacturer’s instructions3.
Sequence Quality Control and Genome Assembly
The data were analyzed on the free online platform of Majorbio Cloud Platform4. SeqPrep (Verision 1.1) was used to strip the adapter sequence from the 3′ and 5′ end of paired-end Illumina reads. The paired-end Illumina reads were trimmed of adaptors, and low-quality reads (shorter than 50 bp or with an average quality score < 20 or having N bases) were removed by Sickle (Verision 1.33). Then, host reads were filtered by aligning reads against Capra hircus genome with BWA (Version 0.7.9a) and removing reads with high scoring alignments host. The Megahit5 (version 1.1.2), which makes use of SdBG was used to assemble different depth sequences and get the statistical table of gene prediction results. Contigs with lengths of 300 bp or over were selected as the final assembling result, and then the contigs were used for further gene prediction and annotation. Sequences were submitted to the NCBI short read archive (SRA) under accession numbers PRJNA706869.
Gene Prediction, Taxonomy, and Functional Annotation
We used Metagene to predict the Open reading frames (ORFs) from each assembled contig (Noguchi et al., 2006). Then, we used the NCBI translation table to translate predicted ORFs over a length of ≥ 100 bp into amino acid sequences. Then we clustered the database with CD-HIT software6 (default parameters: 95% identity, 90% coverage) and took the longest gene of each class as a representative sequence (Fu et al., 2012). A non-redundant gene set was constructed and compared with the optimized reads (default parameter: 95% identity). The abundance information of genes was counted by Soapaligner software, and evaluated via reads per kilobase per million mapped (RPKM) in each sample. The taxonomic information was annotated by a BLASTp (Version 2.2.28 +) search in the NCBI NR database [refer to previous study by Altschul et al. (1997)]; the KEGG annotation was conducted by KOBAS 2.0 against the KEGG database (Version: 2018-07-30); the carbohydrate active enzymes annotation was conducted by hmmscan7 against CAZymes database Version 6.0. All these databases had an E-value cutoff of 1E-5 while annotating ORFs.
Quantitative Real-Time Quantitative PCR
The expression levels of six genes (IL1B, IL 12, IkB, NF-kB, and IFN-γ) associated with mucosal immunity were analyzed by RT-qPCR. The total RNA was extracted by the Trizol (Takara, Dalian, China) method from intestinal mucosa samples and reverse transcribed by the Prime Script® RT reagent kit (Takara, Dalian, China) (Chang et al., 2015). The housekeeping gene GAPDH was used as an internal normalization control (Vandesompele et al., 2002). All samples were examined in triplicate and analyzed using the 2–ΔΔ Ct method (Livak and Schmittgen, 2001).
Immunohistochemistry
Immunohistochemistry was performed [refer to the previous study by Dong et al. (2015)]. The cecum was removed and fixed in 4% paraformaldehyde/PBS overnight at 4°C. The tissue blocks were dehydrated and embedded in paraffin. The 5 μm thick serial sections were cut in a coronal plane under a Leica microtome (Leica 2016, Germany) and mounted on 0.1% polylysine reagent (Sigma, Germany) coated slides. Then, rehydrated and ethanol cleared sections were incubated with 3% H2O2-methanol (ZLI-9064, ZSGB-BIO, Beijing, China) for 10 min at room temperature, and washed in PBS for 3 min × 5 min. Slides were treated with microwaves (700 W) in 0.01 mol/L citrate-buffered saline (pH 6.0) for 2 min × 5 min for antigen retrieval and washed in PBS for 2 min × 5 min. Then, the sections were incubated in 10% normal goat serum (ZLI-9021, ZSGB-BIO, Beijing, China) in PBS for 20 min. Sections were next incubated with rabbit anti-Muc2 (1:50, GB11344,Servicebio, Wuhan, China) overnight at 4°C. Thereafter, they were exposed to biotinylated second antibody biotinylated goat anti-rabbit IgG (SP-9001, ZSGB-BIO, China) for 30 min at 37°C. Color development was performed using a diaminobenzidine (DAB) kit (K135925C, ZSGB-BIO, Beijing, China) according to the manufacturer’s instructions. Images were digitized using a microscope (BA400Digital, Motic, Xiamen, China) and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, Maryland, United States).
Enzyme-Linked Immunosorbent Assay in Cecum
Enzyme-Linked Immunosorbent Assay (ELISA) kits were employed to measure the concentrations of CD4+ T Cells, CD8+ T Cells (Shanghai Mlbio Institute Co., Ltd, China), and SIgA (Nanjing Jiancheng Bioengineering Institute Co., Ltd, China), following the manufacturer’s instructions. The absorbance of each solution was determined at a wavelength of 450 nm (Colón-Díaz et al., 2020).
Statistical Analysis
Statistical comparisons based on statistical software package (SPSS version20.0; United States) with a false discovery rate (FDR) value < 0.05 to correct the P values. Differential abundance of bacteria on family and genus level (jejunum, cecum, and rectum), CAZymes and KEGG enzymes (cecum) were tested by one-way ANOVA among three groups, Student’s t test was used between two groups, omics data was tested using the SPSS software (version20.0; Chicago, IL, United States), and the significance was defined by Duncan’s test among more than two groups. Taxonomic data in jejunum, cecum and rectum were analyzed on the online platform of Majorbio Cloud Platform8, and so did the functional data in cecum. The Differences were statistically significant at P < 0.05 or a tendency of difference at 0.05 ≤ P < 0.10. Spearman’s rank correlation coefficients were used to examine the correlations between bacterial abundance and SCFAs concentrations.
Results
Carbohydrates and SCFAs Composition in Hindgut
The concentration of total SCFAs and molar proportions of individual SCFAs were shown in Table 1. There was an increased tendency in the concentrations of total SCFAs in the LRDS group than the HRDS group at the cecum and colon (0.05 ≤ P < 0.10), and the molar proportion of various SCFAs were compared. The results showed that butyrate was decreased in the HRDS group at the colon and rectum (P < 0.05) compared with the MRDS group; acetate was increased in the HRDS group (P < 0.05) at each position of the hindgut (cecum, colon, and rectum) compared with the LRDS group. To quantify the contribution between the molar proportion of butyrate and acetate, we used butyrate: acetate (B:A) ratio in this study. Compared with the LRDS group, the B:A ratio was decreased in the HRDS group at the colon (P < 0.05), rectum (P < 0.05), and had a downward trend at the cecum (0.05 ≤ P < 0.10). We also found that the molar proportion of propionate was higher in the MRDS group (P < 0.05) compared with the HRDS group. There was no difference in pH between different RDS groups (Table 2; P > 0.05). The starch and cellulose are also analyzed in intestinal digesta. As Supplementary Figure 1 shows, the content of cellulose in the HRDS group was higher (P < 0.05) than the MRDS group. The determination of amylose content showed that cecum digesta in the HRDS group had a lower amylose content (P < 0.05) compared with the LRDS group.

Table 1. Effect of dietary RDS on hindgut SCFAs concentration.
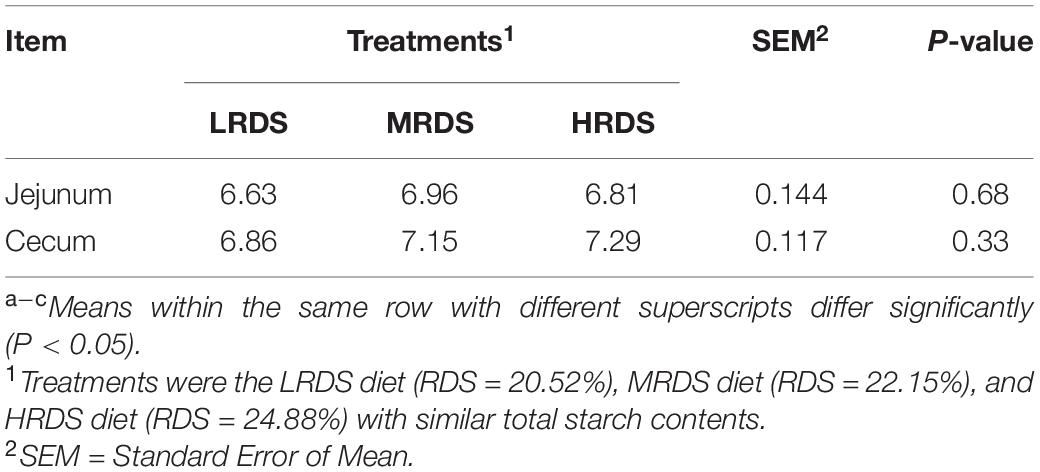
Table 2. Effect of dietary RDS on intestinal pH level.
High RDS Diet Induced Shift in SCFAs-Producing Community and Cellulolytic
The comparisons of microbial beta-diversity based on PCoA analysis and Adonis test were given in Figure 1. The LRDS, HRDS, and MRDS groups in the jejunum cannot be distinguished. Moreover, the HRDS group can be distinguished from LRDS (P < 0.05) and MRDS (P < 0.05) in the cecum. In the rectum, the HRDS group can be distinguished from LRDS (P < 0.05) and had a trend of separation with MRDS (0.05 ≤ P < 0.1).
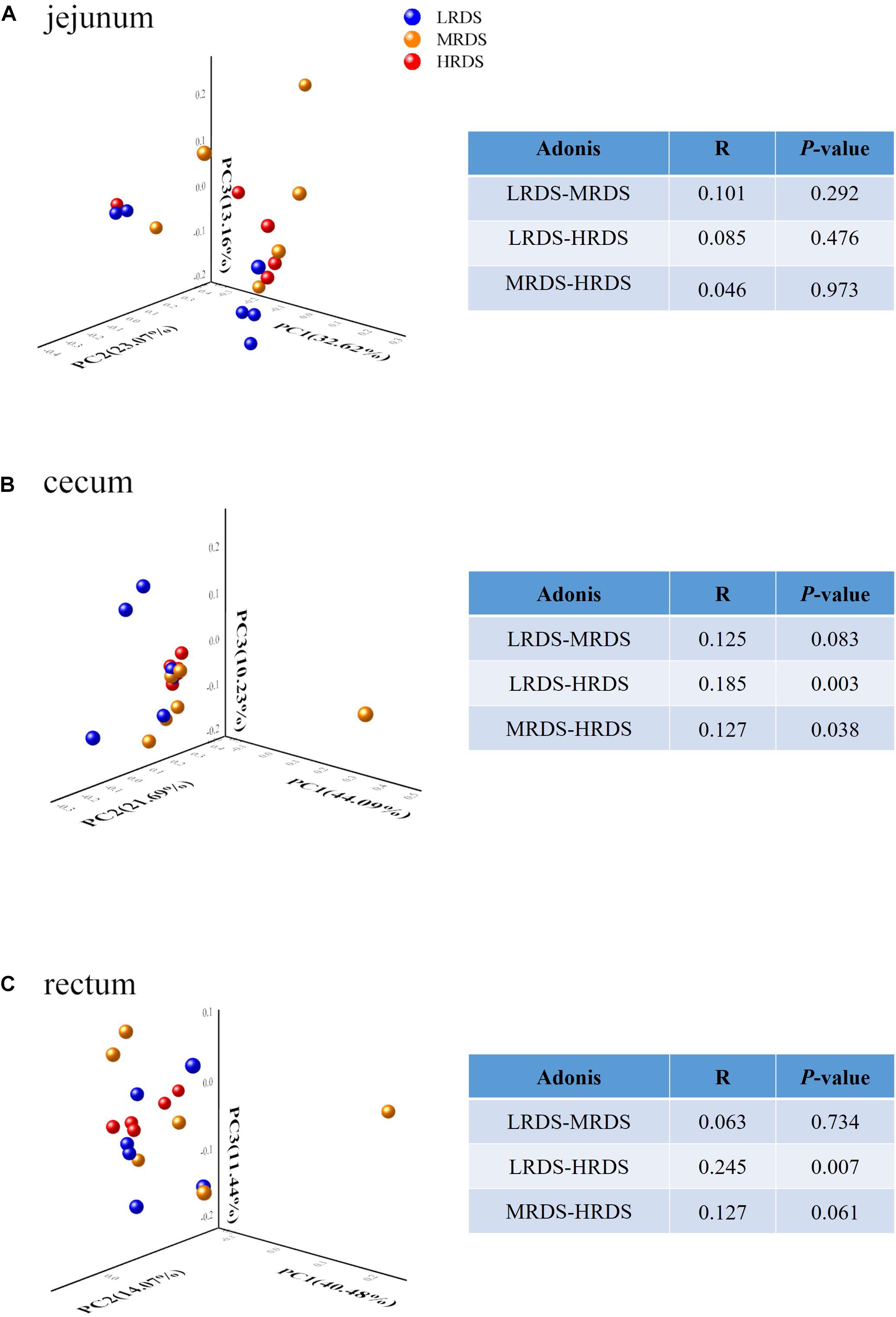
Figure 1. Microbial community difference among different groups in the jejunum microbiome (A), but showing obvious dysbiosis over positions in the cecum (B) and rectum microbiome (C). The data represent the comparison of the gut bacterial profiles among the LRDS (blue), MRDS (yellow), and HRDS (red) groups. Principal coordinate analysis and Adonis scores computed for OTU taxon illustrating the grouping patterns based on the unweighted UniFrac distances. Distances between each pair of samples represent their dissimilarities. The R value represents the effect size indicating that the percent variation can be explained by RDS level. Only taxa meeting an LDA significant threshold > 2 are shown.
To illustrate whether the changes of SCFAs were associated with the acid-produced bacteria, we compared the number of significantly changed bacteria based on 16s rRNA sequencing (Figure 2). A total of 22 phyla, 130 families, and 214 bacterial genera were identified in all samples, including the five most abundant phyla: Firmicutes, Bacteroidetes, Spirochetes, Tenericutes, and Proteobacteria (Figure 2A). To identify the specific bacterial taxa affected by RDS, we compared the families and genera based on one-way ANOVA analysis (Figures 2B,C). The results revealed the most abundant family was classified as Ruminococcaceae and the most abundant genus was classified as F_Ruminococcaceae. On a family level, compared to the LRDS group, HRDS increased the relative abundance of Ruminococcaceae (P < 0.05), decreased the relative abundance of Lachnospiraceae and Bacteroidales_S24-7 (P < 0.05) in samples collected from the cecum, and also increased the relative abundance of Bacteroidales_S24-7 (P < 0.05) in the rectum. MRDS increased the relative abundance of Succinivibrionaceae (P < 0.05) compared to the LRDS group in the cecum, and increased the relative abundance of Succinivibrionaceae (P < 0.05) compared to the HRDS group in rectum (Figure 2B).
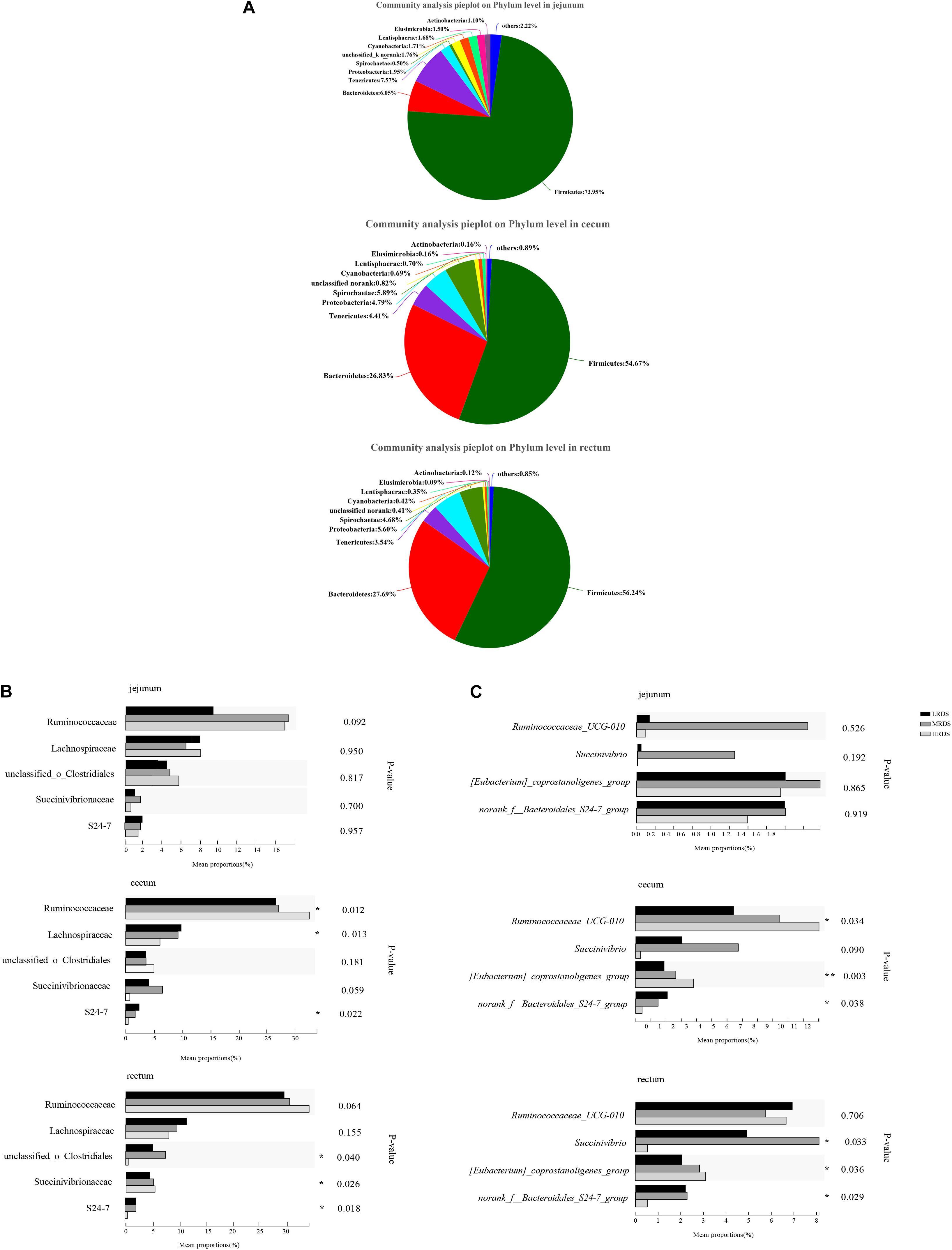
Figure 2. Bacterial community composition in each intestinal tract region (A) on phyla level, (B) family level and, (C) genus level. (B) Comparison of relative abundances of the five main bacterial families (Ruminococcaceae, Lachnospiraceae, O. Clostridiales, S24-7, and Succinivibrion- aceae) among groups throughout the GIT region. (C) Comparison of relative abundances of the four main bacterial genera (Ruminococcaceae UCG-010, Eubacterium_coprostanoligenes, Bacteroidale_S24-7_group, and Succinivibrio) among groups throughout the intestinal tract region. Asterisk (*) denotes significant correlations between hind-gut microbiome abundance with parameters *P < 0.05, **P < 0.01.
On a genus level, Ruminococcaceae UCG-010 was the most abundant with an increase in the HRDS group at the cecum (P < 0.05) compared with the LRDS group (Figure 2C). The HRDS group increased the relative abundance of Eubacterium_coprostanoligenes_group (P < 0.05) and decreased the relative abundance of Bacteroidales_S24-7_group_unidentified (P < 0.05) in the rectum and cecum. The MRDS group increased the relative abundance of Succinivibrio in the rectum (P < 0.05) and had an uptrend in the cecum (0.05 ≤ P < 0.10) compared with the HRDS group (Figure 2C). In the jejunum, there was no significant change (P > 0.05) in previous genera or 15% top abundance, reflecting that dietary RDS played an even more important role in the composition of bacterial in hindgut than the small intestine.
The BugBase phenotypes prediction was presented in Figure 3. The results show that the LRDS group increased the microbial oxidative stress tolerant phenotypes in the cecum (P < 0.05) compared with the MRDS and HRDS groups, as well as in the rectum (P < 0.05) compared to the MRDS group. Compared to the MRDS and HRDS groups, the potentially pathogenic phenotypes of the LRDS group were reduced (P < 0.05) in jejunum as well as in the cecum (0.05 ≤ P < 0.10).
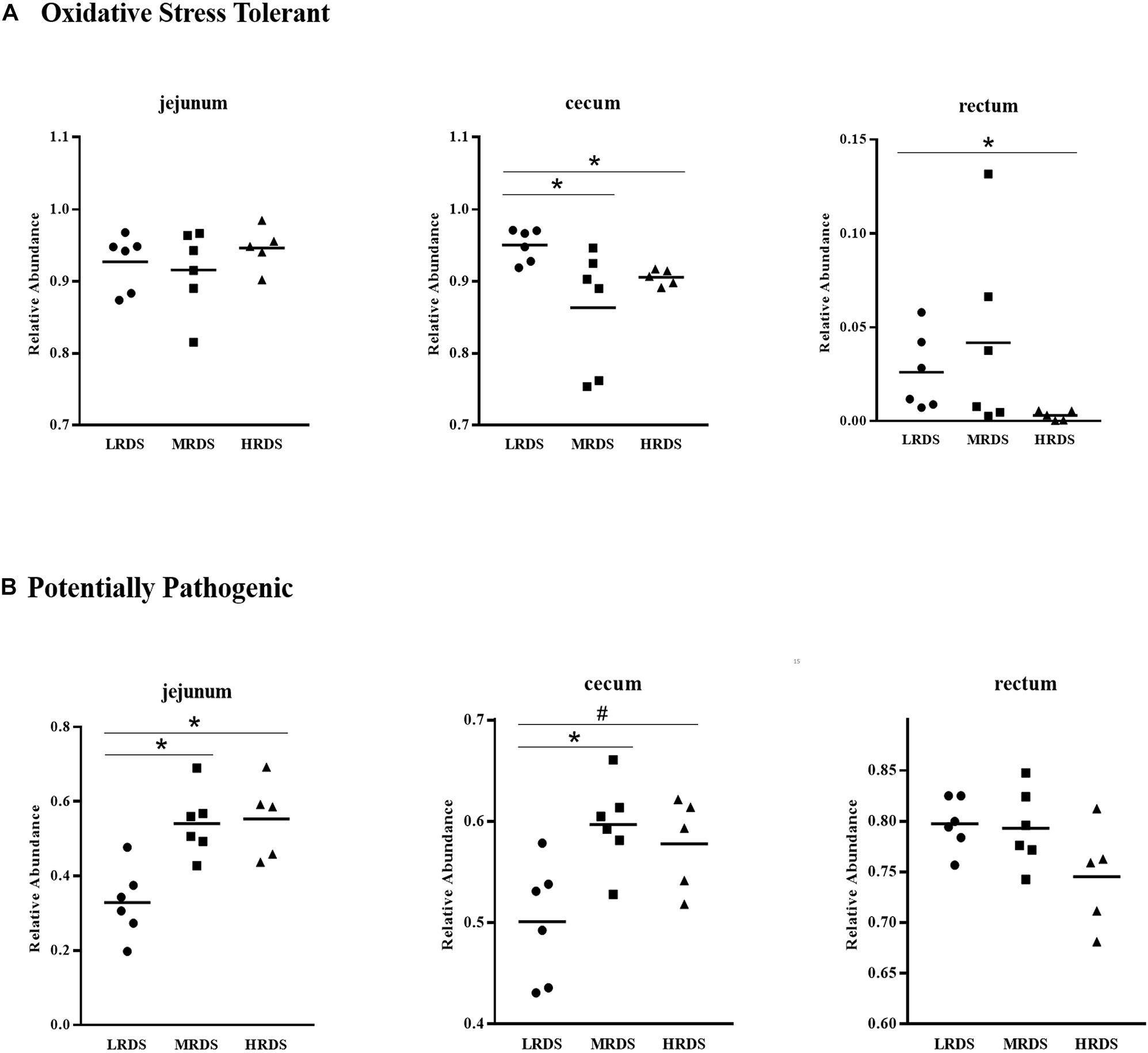
Figure 3. BugBase phenotypes prediction. (A) The oxidative stress tolerant. (B) The potentially pathogenic *P < 0.05, #0.05 < P < 0.10.
Correlation analysis of cecal microbiome abundance with microbial metabolites were shown in Figure 4. There were nine families related with SCFAs molar proportion (P < 0.05) (Figure 4A). The strongest positive correlation with the molar proportion of acetate was found in Bacteroidale_incertae_sedis, Rikenellaceae, and Ruminococcaceae; the strongest negative correlation with the molar proportion of acetate was found in Bacteroidale_S24-7, Erysipelotrichaceae, and Succinivibrionaceae. In addition, Lachnospiraceae and Succinivibrionaceae were positively related with butyrate molar proportion and B: A ratio. There were six genera significantly related with SCFAs molar proportion (P < 0.05; Figure 4B). Besides the genera Ruminococcaceae_UCG-010 and Rikenellaceae_RC9_gut_group we mentioned previously, the Eubacterium_coprostanoligenes group and Phocaeicola were also positively correlated (P < 0.05) with the molar proportion of acetate. Genus Succinivibrio was negatively correlated with the acetate molar proportion, while positively correlated with the molar proportion of butyrate, propionate, and B: A ratio. Genus Eubacterium_coprostano-ligenes_group and Ruminococcaceae_UCG-010 were negatively correlated with butyrate molar proportion and B: A ratio.
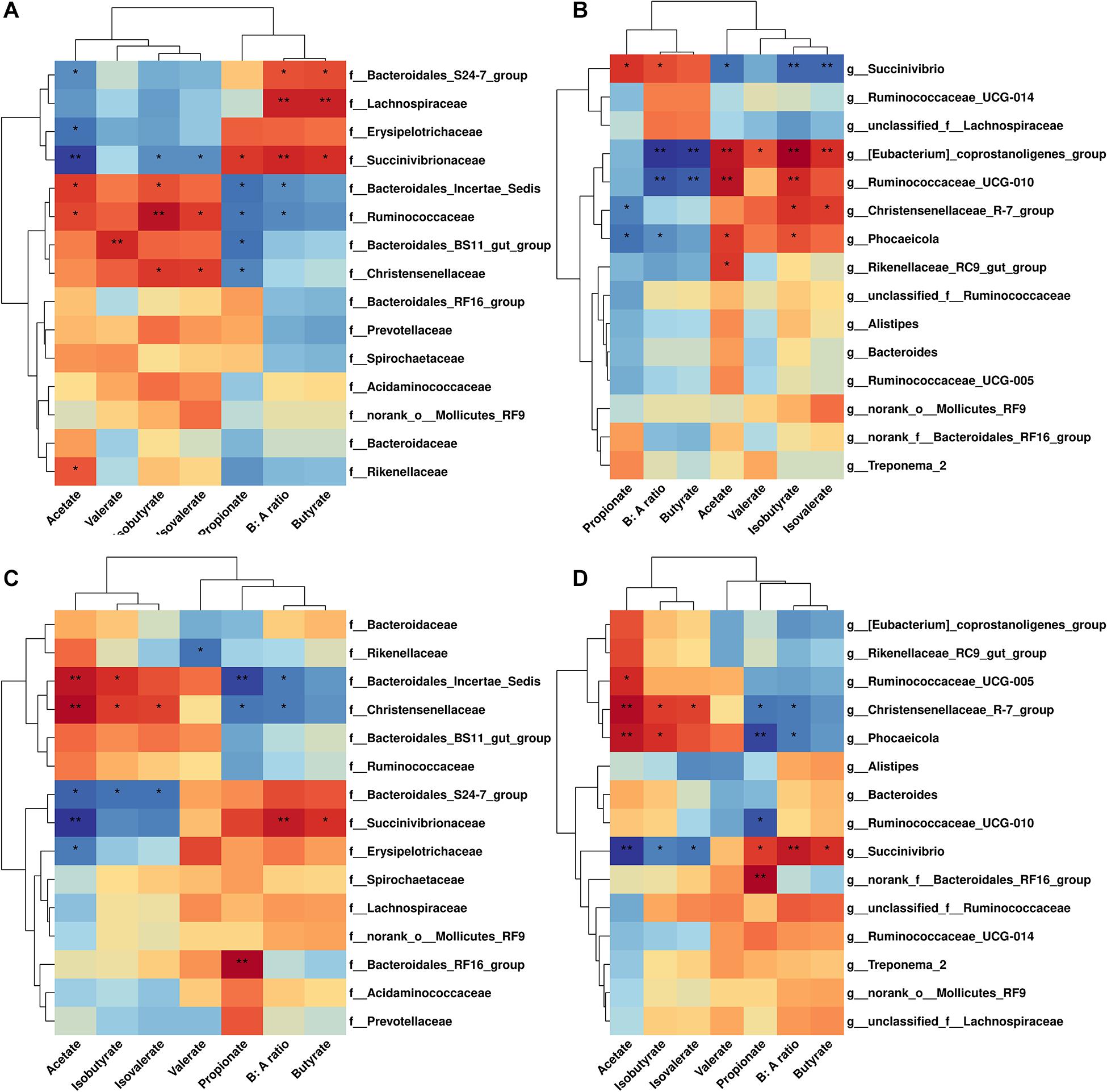
Figure 4. Correlation analysis between microbiome abundance and SCFAs concentration in cecum and rectum.(A) Family level in cecum. (B) Genus level in cecum. (C) Family level in rectum. (D) Genus level in rectum. Cells are colored based on spearman correlation coefficient. Only relative abundance of the 15 most-abundant bacterial families or genera are shown.
Metagenomic Analysis Found That High RDS Diet Disturbed Microbial Function by Affecting Genes Encoding Carbohydrate Enzymes
To gain insight into the molecular functions of gut microbiota, metagenomics analysis was used to analyze the contribution of microbial function in the cecum. Using BLASTp to compare non-redundant gene sets based on the NR database, we compared the relative abundance of microbial genes among groups. These results were shown in Figure 5. Of the 78 identified phyla, Firmicutes was predominant in cecum samples. On species level, Succinatimonas_sp._CAG:777 was the second most abundant in the cecum, and was increased in the MRDS group compared with the LRDS group (P < 0.05).
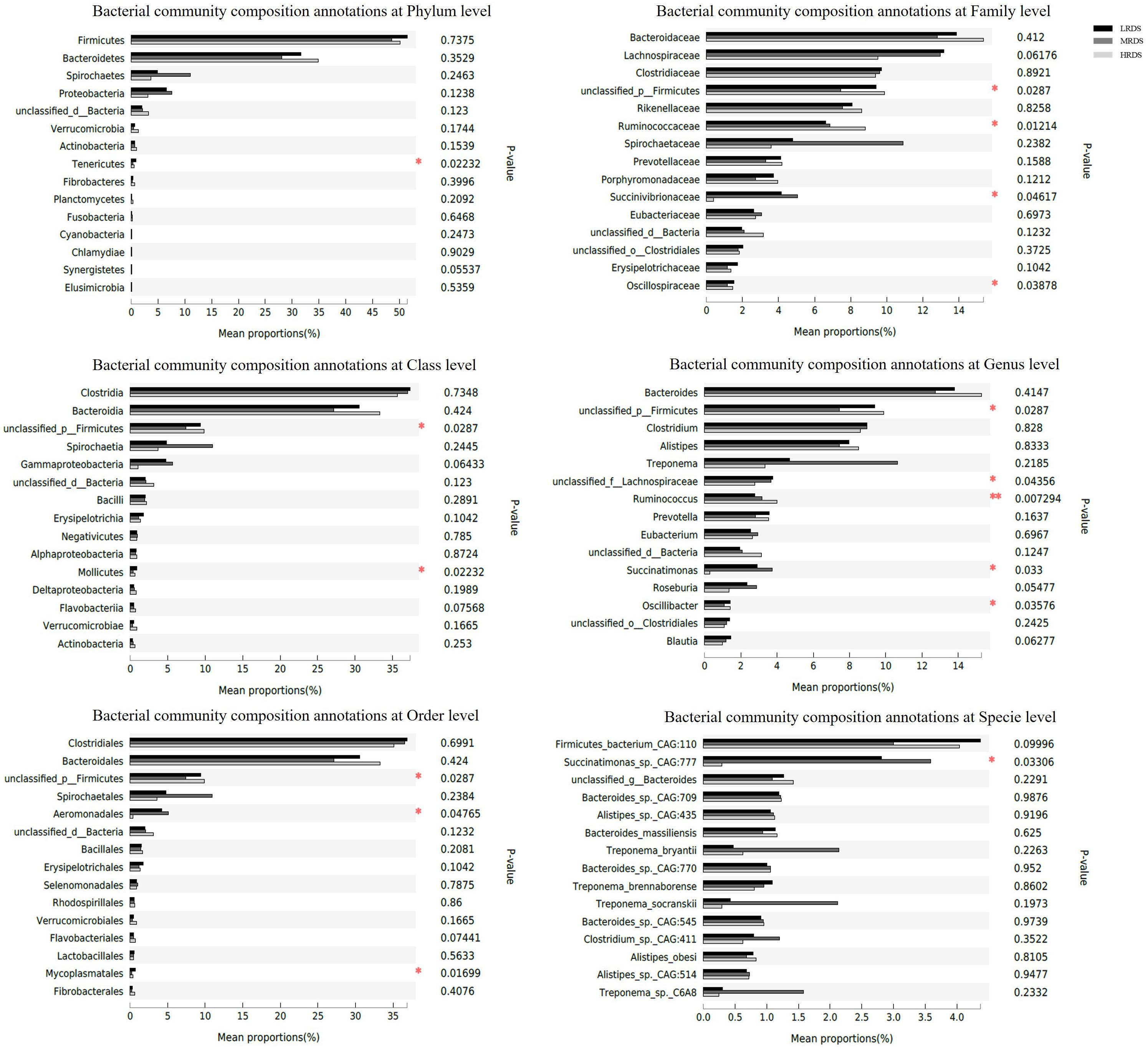
Figure 5. Relative gene abundance of microbial metagenomics compared with NR database in cecum. On phylum, class, order, family, genus, and species levels.
Compared to the LRDS and MRDS groups, the HRDS group decreased (P < 0.05) the relative abundance of class Mollicutes, order Mycoplasmatales, and phylum Tenericutes. Compared with the HRDS group, the MRDS group decreased (P < 0.05) the relative abundance of unclassified_p__Firmicutes. In addition, the MRDS group increased the relative abundance of the family Succinivibrionaceae (P < 0.05) and genus Succinatimonas compared to the HRDS group.
Comparison of the functional capacity of the microbiota could help us investigating the metabolic differences among groups. In this study, we used Gene set enrichment analysis (GSEA) to identify differentially abundant KEGG pathways. Finally, we identified 15 KEGG pathways in carbohydrate metabolism at level 3, five of them changed in mean proportion among groups (Figure 6), including starch and sucrose metabolism, Glycolysis/Gluconeogenesis, Pyruvate metabolism, Citrate cycle (TCA cycle), and C5-Branched dibasic acid metabolism (P < 0.05). The KEGG database showed that there were various enzymes encoded by corresponding functional genes which took part in carbohydrate metabolism in the cecum, including cellulase (endoglucanases, glucosidase, and glycosylase) and amylase (α-amylase, glycogen phosphorylase, and glucosidase) (Figures 7A,B). Within the process of cellulose degradation, the genes encoding cellulase enzyme (EC 3.2.1.4) were increased (0.05 ≤ P < 0.10) in the HRDS group compared with the LRDS group. The abundance of amylopectin-degrading enzyme isoamylase (EC 3.2.1.68) was lower (P < 0.05) in the HRDS group compared with the LRDS group.

Figure 6. KEGG pathways corresponded to carbohydrate metabolism with significant changes.
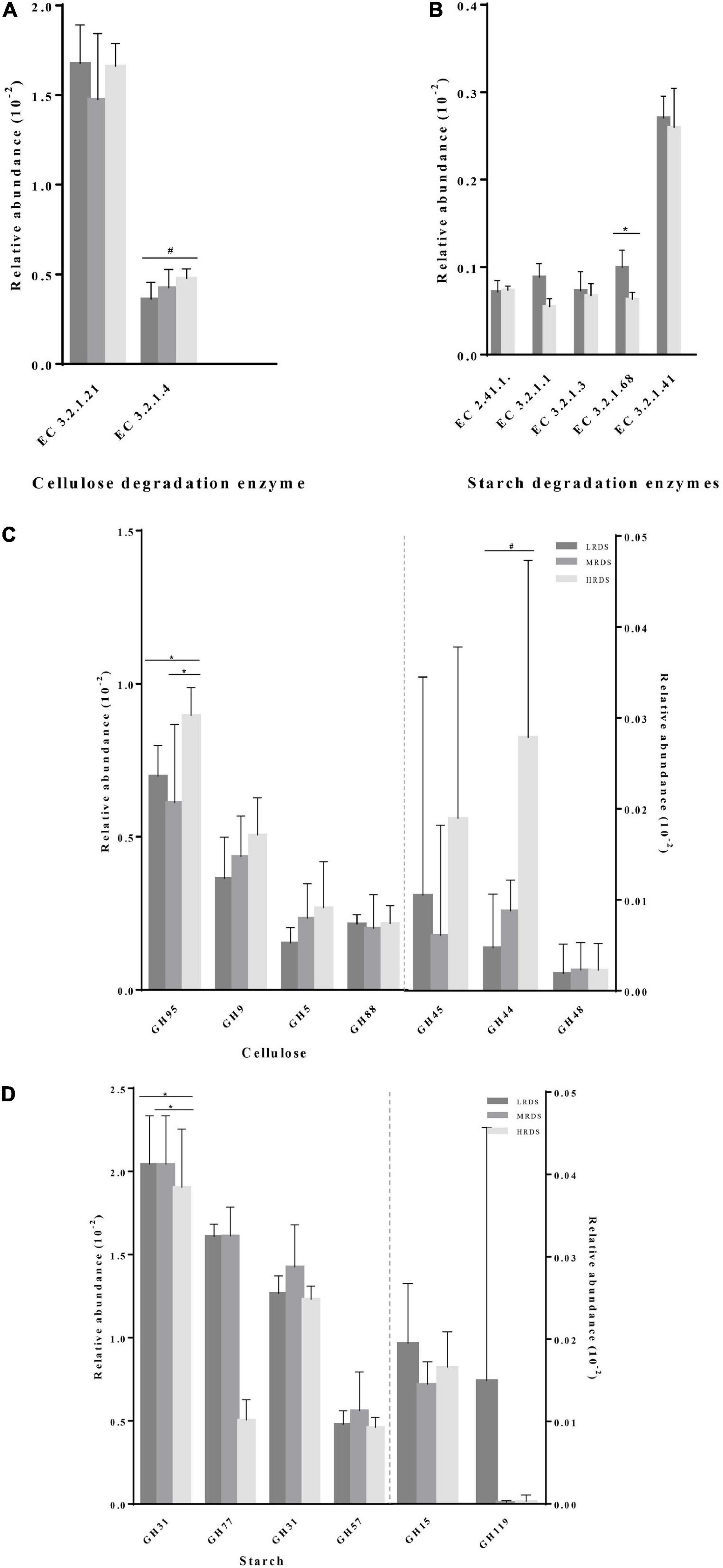
Figure 7. Comparisons of the gene abundance of KEGG enzymes and GH family for (A,C) cellulase and (B,D) amylase.
We also investigated the functional information of genes annotated by the CAZymes database, and the results corresponded with KEGG annotation. When comparing the gene abundance of the carbohydrate-active enzymes family, we found that there were seven GH families (GH95, GH9, GH5, GH88, GH45, GH44, and GH48) mainly found to be associated with cellulolytic functions. The abundance of GH95 was increased in the HRDS group (P < 0.05) compared with the LRDS group. Six GH families (GH13_20, GH77, GH31, GH57, GH15, and GH19) were found to be mainly associated with starch degradation, and the abundance of GH13_20 was decreased in the MRDS and LRDS groups (P < 0.05) compared with the HRDS group (Figures 7C,D).
We further analyzed the genes differentially expressed among groups by the Probio database, a database of probiotics functions and lineages (Figure 8). We found that compared to the MRDS and HRDS groups, the LRDS group had an increased (P < 0.05) gene abundance of Bacillus coagulans, Bacillus subtilis, Lactobacillus equigenerosi, Lactobacillus paracasei, and Lactococcus lactis ssp. Cremoris. Compared with the MRDS and HRDS groups, Clostridium butyricum and Pseudobutyrivibrio ruminis were enriched (P < 0.05) in the MRDS group and decreased (P < 0.05) in the HRDS group.
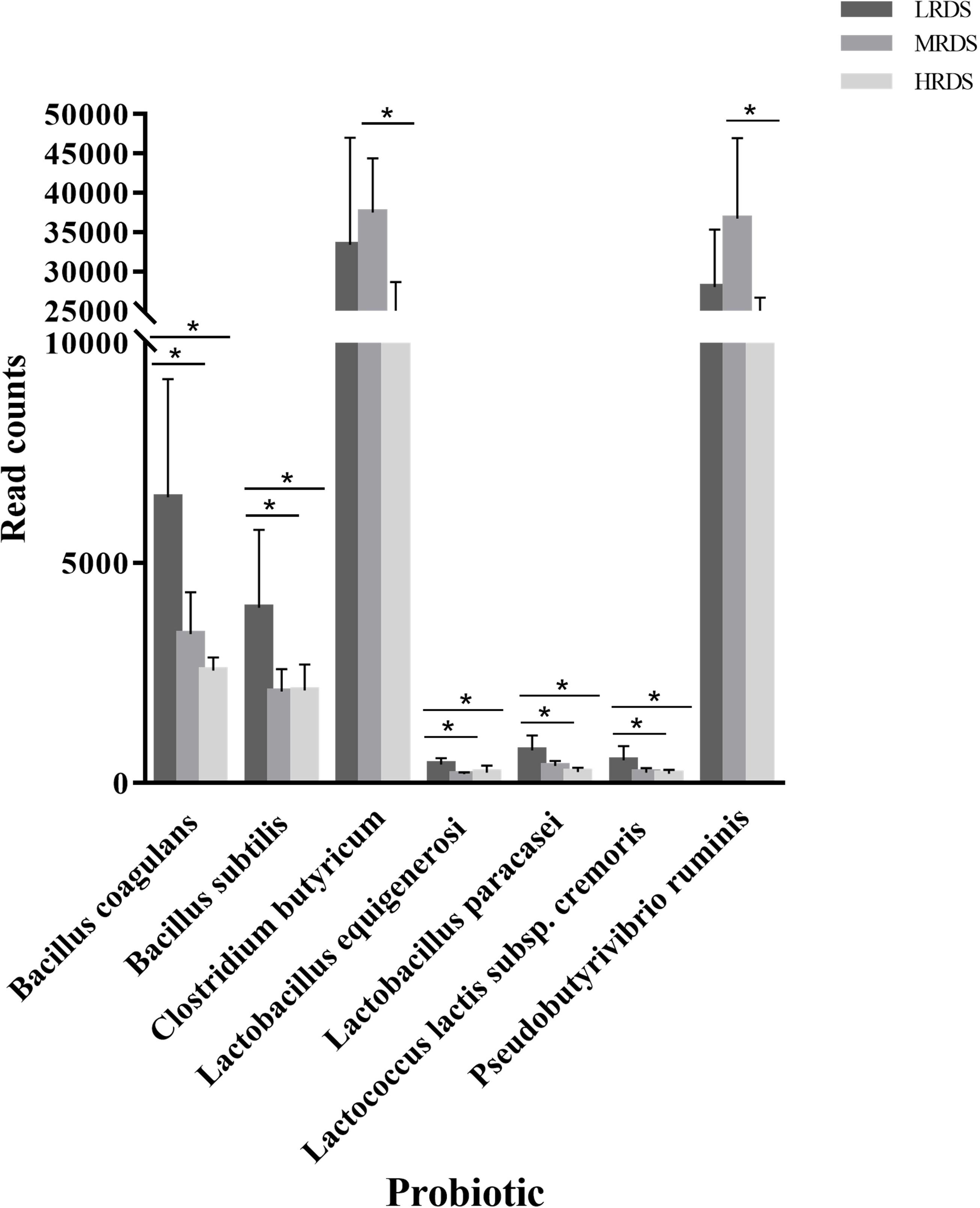
Figure 8. Comparisons of the abundance of the Probio annotated probiotics.
High RDS Diet Upregulated Healthy Risk by Increasing Intestinal Inflammation
We found that the relative expression of genes IL-1β (immunoregulatory cytokines interleukin 1 beta), IL-12 (interleukin 12), TNF-α (tumor necrosis factor alpha), IκB (inhibitor of nuclear factor kappa B kinase), NFκ-B (nuclear factor kappa B subunit), and IFN-γ (interferon gamma) were detectable in the cecum mucosa. The mRNA expression of IL-1β (P < 0.05) was decreased in the LRDS and MRDS groups (Figure 9A). We found that the MRDS and HRDS groups had a significant difference of relative concentration of secretory immunoglobulin A (SIgA) in cecum mucosa tissues compared with the MRDS and HRDS groups (Figure 9B). Dietary RDS did not influence (P > 0.05) the relative protein concentration of CD4+ T Cells, CD8+ T Cells, and CD4+: CD8+ ratio. As Figure 9C shows, Muc2 average optical density in crypt goblet cells of the HRDS group was decreased compared with the MRDS group (P < 0.05).
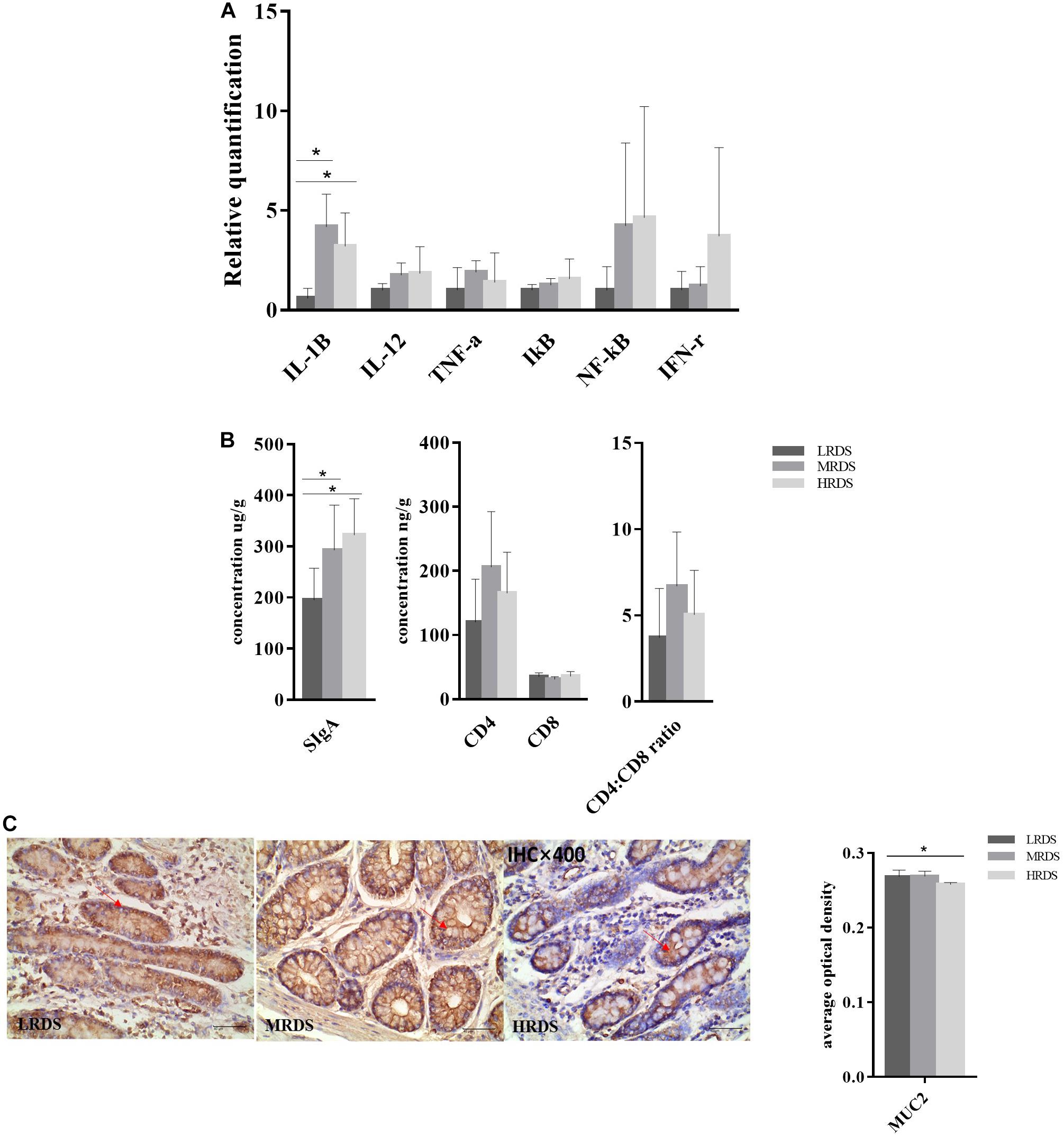
Figure 9. Intestinal mucosal immune in cecum. (A) Gene expression in the cecal mucosa. Glyeraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the reference gene for gene expression including IL-1β, IL-12, IκB, NF-κB, TNF-α, and IFN-γ. (B) Antigen specific proteins expression of SIgA, CD4+ T cell, CD8+ T cell, and CD4+: CD8+ ratio in the cecal mucosa. (C) Antigen specific proteins expression of Muc2 and tissue localization of Muc2 in the cecum. The goblet cells contain Muc2 (brown), and mucin stains are shown in serial sections on the same tissue area.
Discussion
We have found that RDS can be used as an effective measurement of dietary carbohydrate and digestive health in ruminants (Shen et al., 2020b). As the RDS level increased, more starch would be degraded in rumen and less starch would reach the intestine when the three treatment diets were formulated to be an isostarch (Li et al., 2014c).
It is generally known that large amounts of starch are rapidly degraded in the rumen when SARA happens (Plaizier et al., 2017). Some investigations indicated that damage to the hindgut could also occur in association with ruminal acidosis (Petri et al., 2019). In a previous study, we observed that the HRDS diet induced SARA in dairy goats (Zheng et al., 2020). However, no difference in pH level was found in any segments of intestine we detected, and no animals had diarrhea or other digestive problems, suggesting that rumen acid accumulation and inflammation induced by systemic entry of endotoxins did not cause observable hindgut acidosis in the HRDS group. Our previous study shown that HRDS altered the compositions of volatile fatty acid (VFAs) in rumen (Shen et al., 2020b). For ruminants, the fermentation site (rumen and hindgut) producing VFAs is more diverse than monogastric animals (Jin et al., 2018). The VFAs produced in the cecum account for 12% of the total VFA production due to approximately 17% cellulose degradation occurring in the cecum (Faichney, 1968). We hypothesized that a high cecal cellulose concentration in the HRDS group was one of the effects of SARA, which may explain the reduced cellulose digestive rate in rumen. However, we cannot prove conclusively why the MRDS group seems to have had a higher concentration of cellulose compared with the LRDS group, and the content may cause the high abundance of Succinivibrionaceae and Succinivibrio which could promote the digestion of cellulose and high propionate concentration in the hindgut.
Amylose in corn is recognized to be slowly digested in rumen because the linear structure and proteins wrapper allow it to be extensively passed into the hindgut (Larson and Hoffman, 2008). In our experiment, the increased cecal amylose concentration in LRDS-fed goats is speculated to be related to the more uniform distribution at each site of the gastrointestinal tract (GIT). Previous studies show that high amylose increased cecal SCFAs concentrations in rats and piglets (Nagata et al., 2019; Gao et al., 2020). Our study further verified that feeding diets with different RDS levels could alter the hindgut digesta composition of dairy goats thereby influencing the synthesis of SCFAs. Combined with the previous studies, we assumed that the SCFAs profile can be altered by the concentration of cellulose and amylose among groups (Matt et al., 2018; Kumar et al., 2020).
Gut microbiota plays an intermediary role between carbohydrate digestion and SCFAs production (Deehan et al., 2020). In the hindgut, microbial fermentation take over most assignments of digesting remaining fiber (Chen et al., 2019). With the increasing participation of fermentation, we found that the bacterial community in the hindgut was more easily affected than in the small intestine.
While our attention has been focused on the hindgut, some changes in microflora were observed. Family Ruminococcaceae was the most abundant family, genus Ruminococcacea_unidentified was the most abundant genus. Family Ruminococcaceae is a cellulolytic bacteria, which converts cellulose and sugars to acetate, propionate, and succinate (Liu et al., 2018). A recent study showed that inflammatory bowel diseases (IBD) could cause disturbances in an association network of the families Lachnospiraceae and Ruminococcaceae (Yilmaz et al., 2019). In our study, an increase of the relative abundance of the family Ruminococcaceae occurred with a decrease of the family Lachnospiraceae in HRDS in the cecum. Combined with butyrogenic capability of the family Lachnospiraceae (Zhang et al., 2019), we presumed that intestinal inflammation caused by a high RDS level diet might induce an imbalance between Lachnospiraceae and Ruminococcaceae in the hindgut. This intestinal flora disturbance resulted in a decrease of butyrate: acetate (B: A) ratio in HRDS. We also found an increase of relative abundance of the families Succinivibrionaceae and Succinivibrio accompanied by elevated concentration of propionate molar proportion in the MRDS group at the rectum. Succinivibrionaceae and Succinivibrio have been proven to be positively correlated with the propionate concentration in rumen, the one reason is that the genus Succinivibrio could promote the digestion of cellulose and hemicellulose, another reason is succinate can be further decarboxylated to form propionate (Pope et al., 2011; Sun et al., 2016; Ren et al., 2019). Previous study found that the concentration of propionate in the hindgut was higher in pigs fed with a high amylose starch diet (Gao et al., 2020). Considering Succinivibrionaceae and Succinivibrio were both positively correlated with the molar proportion of propionate, we speculated that the MRDS group with appropriate amounts of RDS in the hindgut could promote the synthesis of propionate. In addition, as butyrate-producing bacteria, Bacteroidales_S24-7 and Bacteroidales_S24-7_group_unidentified also alerted in the hindgut with an increased relative abundance in LRDS. Genome analysis revealed the Bacteroidale_S24-7_spp. is versatile with complex carbohydrate degradation and butyrate production (Lagkouvardos et al., 2019). Our findings supported this positive correlation between the Bacterfamilyoidale_S24-7 group (family Bacteroidale_S24-7) and butyrate in the hindgut. We hypothesized that the butyrate molar proportion was increased due to the high abundance of the Bacterfamilyoidale_S24-7 group (family Bacteroidale_S24-7) and high amylose in the LRDS group.
The microbial communities in the cecum were predicted with BugBase phenotypic prediction. We observed that the LRDS group had low cecal potentially pathogenic microflora abundance and strong oxidative stress tolerability compared with the HRDS group at the cecum. Against colonization of potentially harmful microorganisms in the intestine is a principal function for bacteria to protect the host (Rivera-Chavez et al., 2016). Some anaerobes frequently suffered from oxidative stress because anaerobic metabolic pathways were inhibited (Fu et al., 2015). Based on microbial community phenotypes, we concluded that LRDS is helpful for preventing the colonization of potentially pathogenic phenotypes and creating an environment for anaerobes in the hindgut.
Metagenomic approaches based on high-throughput sequencing have rapidly facilitated the study of intestinal microbiota in livestock right behind mice (Liu et al., 2019; Xue et al., 2020). Based on metagenomics, sequencing results showed a high abundance of specie Succinatimonas_sp._CAG:777 in MRDS group. In our study it was the second most abundant cecal specie belonging to the lineage of Proteobacteria (phylum), Gammaproteobacteria (class), Aeromonadales (order), Succinivibrionaceae (family), and Succinatimonas (genus), but the investigations on these bacteria in ruminants are very poor (Qi et al., 2019). Our study first found Succinatimonas_sp._CAG:777 present in ruminants’ hindguts. It was the predominantly affected species and proved the positive correlation between the molar proportion of propionate and genus Succinatimonas in the cecum. Considering succinate can be further decarboxylated to the form of propionate by Wood–Werkman cycle in propionate-producing bacteria (Wood and Werkman, 1938), we suggested that this study may provide a new direction to study the relationship between dietary carbohydrate and intestinal SCFAs-producing strain in ruminants. Similar to previous studies in goats, we found Firmicutes was the dominant phylum in the cecum (Jin et al., 2018). We observed an increase of genus Mycoplasma in the HRDS groups. The study showed that as fermentation patterns of cellulolytic fungi, Mycoplasma could produce acetate as major metabolites (Rimoldi et al., 2019). However, as no members of Mycoplasma could produce digestive enzymes without symbiotic bacteria (Conche and Greten, 2018), a further study of intestinal symbiosis bacteria is undoubtedly required.
Functional capacities were performed by metagenomic analysis. CAZymes-encoding gene abundance and KEGG enzyme gene abundance were helpful to characterize carbohydrate degradation for dairy goats (Stewart et al., 2018). CAZymes metagenomic analysis has shown that glycoside hydrolase (GH) families were the most abundant in the cecum, which is consistent with our published result in the rumen (Shen et al., 2020b). Non-starch polysaccharides including cellulose are the main available energy source for microbes in the cecum (Gray, 1947). During degradation, the cellulose fibrils attacked by exoglucanase (EC3.2.1.91) are generally from GH families 6, 7, and 48. The endoglucanases (EC 3.2.1.4), which cleaved the cellulose chain internally, are generally from GH families 5, 6, 7, 9, 10, and 45. The β-1,4-glucosidase(EC 3.2.1.21), which hydrolyzed the non-reducing β–D-glucose bond, are generally from GH families 1, 3, 5, and 9 (Cairo et al., 2013; Morais and Mizrahi, 2019). In our study, gene-encoded cellulases mainly belonged to GH9, GH10, GH5, and GH88 families. The second most abundant family GH10 has β-1,4-glucanase activity (Zhao et al., 2019). Adding recombinant endoglucanase of GH10 can increase the digestibility of DM, NDF, and ADF in vitro ruminal culture (Ribeiro et al., 2018). We found that the abundance of the GH10 family was higher under HRDS treatment. Consistent with early research in rumen, we concluded that the HRDS diet could decrease cellulase activity by down-regulating endoglucanases in rumen, and the un-degraded cellulose which flowed into the hindgut could lead to an increasement of cecal cellulose content.
In addition, we found that LRDS and MRDS diets significantly increased gene abundance of GH13_11 and GH13_20. A previous study has shown that GH13_11 (isoamylase and glycogen debranching enzymes) and GH13_20 (pullulanase and pullulan hydrolase) belong to GH13 subfamilies as amylopectin degradation enzymes (Møller et al., 2016). Isoamylase could specifically attack α-1,6-glucosidic linkages at the branch to leave amylose (Li et al., 2017). We found a high concentration of amylose which was consistent with the high isoamylase gene abundance in the LRDS group. Combined with our previous study in rumen, we believed the decreased starch branching enzymes could cause some of the starch which escapes from the rumen to be passed into the hindgut in the LRDS group (Shen et al., 2020b). We speculated that the extensive amylose made efficient microorganisms in the hindgut for amylopectin degradation.
Using PROBIO database to help characterize the contribution of host probiotics is a new trend to improve gut health (Tao L. et al., 2017). In the present study, a total of 128 probiotics were detected and seven changed noticeably with the same trend, which was decreased in the HRDS group compared with the LRDS and MRDS groups. We found five probiotics related to intestinal anti-inflammation which were decreased in the HRDS group. A previous study has shown that Bacillus subtilis and Bacillus Coagulans could improve abdominal pain and diarrhea in irritable bowel syndrome (IBS) patients (Rogha et al., 2014). Lactobacillus equigenerosi and Lactobacillus paracasei belonging to the genus Lactobacillus could activate macrophages to suppress inflammation in IBS patients (Koryszewska-Baginska et al., 2019). Lactococcus lactis ssp. Cremoris could limit tissue injury in the intestine (Naudin et al., 2020). In our study, we found that goats fed the HRDS diet had higher free LPS concentrations in plasma than under LRDS or MRDS diets. We also found a significant correlation between the dietary RDS content and plasma LPS concentration (Shen et al., 2020a). Based on the above studies, we speculated that a lack of anti-inflammatory probiotics might cause intestinal tissue damage and inflammation in HRDS, and the increased free LPS in the circulatory system may be responsible.
In addition to microbiota dysbiosis, inflammatory response was another important consequence of a high-concentrate diet for goats (Tao S. Y. et al., 2017). In our previous study, we observed lipopolysaccharide (LPS) concentration increased in the jugular vein of the HRDS group (Shen et al., 2020a). The increase of circulating LPS is mainly absorbed from the large intestine rather than the rumen (Chang et al., 2019). We measured mRNA level of inflammatory markers and observed the expression of the cytokine IL-1β and SIgA were upregulated in the cecal mucosa of the HRDS group. A previous studies has shown inflammation could be promoted by IL-1β and stimulated the expression SIgA against the adhesion of pathogens in colonic inflammation (Liu et al., 2014; Tao S. Y. et al., 2017). We found that the expression of Muc2 in the cecal epithelium was decreased by immunohistochemistry, which could protect the intestinal epithelial cells from spontaneous colitis (Johansson et al., 2014). In this study, the phenotypes such as decreased probiotics, increased butyrate-producing bacteria, and expression of IL-1β were identically observed in mice with deleted Muc2 (Johansson et al., 2008). Overall, we speculated that feeding a HRDS diet to dairy goats could cause hindgut mucosal injuries via epithelial damage and inflammation.
Conclusion
In conclusion, this study investigated changes in intestinal fermentation, composition of bacterial in the hindgut, and CAZymes-encoding gene and KEGG enzyme gene related to fiber and starch degradation in the cecum in dairy goats with different dietary RDS. We revealed that dietary RDS altered intestinal bacterial community and carbohydrate digestibility in dairy goats. The HRDS group increased the gene abundance of cellulase enzyme, and then acetate proportion in the cecum. The LRDS group increased the butyrate molar proportion by increasing the abundance of butyrate producer bacterial families and gene abundance of probiotics, and then increased the expression of Muc2 SIgA in cecal mucosa at the cecum. The MRDS group increased the propionate proportion by increased succinate-producing bacteria abundance. The HRDS group decreased amylose content and increased cellulose content in the cecum. Therefore, this study can enhance our understanding of hindgut starch and cellulose degradation, and cecal epithelial inflammation in dairy goats with the change of dietary RDS level.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm. nih.gov/, PRJNA706869.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Northwest A&F University.
Author Contributions
XH, JS, LZ, and JY designed the research. XH, XY, JS, LZ, and CJ performed the experiment. XH, XL, and XY analyzed the data and wrote the manuscript. XL undertook revision work. JY, YC, and XL finalized the manuscript. All authors read and approved the final version of the manuscript.
Funding
This research was financially supported by the National Key Research and Development Program of China (2017YFD0500500) and the Science & Technological Project of Shaanxi Province, China (award number: 2017 TSCXL-NY-04-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the members of the Innovative Research Team of Animal Nutrition & Healthy Feeding of Northwest A&F University for providing valuable assistance and care of the goats.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.651631/full#supplementary-material
Supplementary Figure 1 | Effect of dietary RDS on the content of cellulose and amylose in the cecum digesta.
Supplementary Figure 2 | A significant number of bacterial based on 16s rRNA sequence dataset between LRDS, MRDS, and HRDS groups.
Footnotes
- ^ http://drive5.com/uparse/
- ^ https://bugbase.cs.umn.edu/index.html
- ^ www.illumina.com
- ^ https://github.com/voutcn/megahit
- ^ http://www.bioinformatics.org/cd-hit/
- ^ https://github.com/weizhongli/cdhit
- ^ http://www.hmmer.org/
- ^ www.majorbio.com
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Cairo, J. P. L. F., Oliveira, L. C., Uchima, C. A., Alvarez, T. M., Citadini, A. P. D., Cota, J., et al. (2013). Deciphering the synergism of endogenous glycoside hydrolase families 1 and 9 from coptotermes gestroi. Insect Biochem. Mol. Biol. 43, 970–981. doi: 10.1016/j.ibmb.2013.07.007
Chang, G., Petzl, W., Vanselow, J., Günther, J., Shen, X., and Seyfert, H. M. (2015). Epigenetic mechanisms contribute to enhanced expression of immune response genes in the liver of cows after experimentally induced Escherichia coli mastitis. Vet. J. 203, 339–341. doi: 10.1016/j.tvjl.2014.12.023
Chang, G. J., Zhang, H. M., Wang, Y., Ma, N. N., Chandra, R. A., Ye, G. P., et al. (2019). Microbial community shifts elicit inflammation in the caecal mucosa via the gpr41/43 signalling pathway during subacute ruminal acidosis. BMC Vet. Res. 15:298. doi: 10.1186/s12917-019-2031-5
Chen, T. T., Chen, D. W., Tian, G., Zheng, P., Mao, X. B., Yu, J., et al. (2019). Soluble fiber and insoluble fiber regulate colonic microbiota and barrier function in a piglet model. Biomed Res. Int. 2019:7809171. doi: 10.1155/2019/7809171
Colón-Díaz, M., Ortiz-Santana, J., and Craig, Z. R. (2020). Data on the activity of DNA methyltransferase in the uteri of cd-1 mice exposed to dibutyl phthalate. Data Brief. 28:105061. doi: 10.1016/j.dib.2019.105061
Conche, C., and Greten, F. R. (2018). Fungi enter the stage of colon carcinogenesis. Immunity 49, 384–386. doi: 10.1016/j.immuni.2018.09.002
Deehan, E. C., Yang, C., Perez-Munoz, M. E., Nguyen, N. K., Cheng, C. C., Triador, L., et al. (2020). Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe 27, 389–404.e6. doi: 10.1016/j.chom.2020.01.006
Dong, W., Guo, W., Zheng, X., Wang, F., Chen, Y., Zhang, W., et al. (2015). Electroacupuncture improves cognitive deficits associated with ampk activation in samp8 mice. Metab. Brain Dis. 30, 777–784. doi: 10.1007/s11011-014-9641-1
Dong, W., Yang, W., Li, F., Guo, W., Qian, C., Wang, F., et al. (2019). Electroacupuncture improves synaptic function in samp8 mice probably via inhibition of the ampk/eef2k/eef2 signaling pathway. Evid. Based Complement. 2019:8260815. doi: 10.1155/2019/8260815
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). Uchime improves sensitivity and speed of chimera detection. Bioinformatics. 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Faichney, G. J. (1968). Volatile fatty acids in the caecum of the sheep. Aust. J. Biol. Sci. 21, 177–180. doi: 10.1071/bi9680177
Fu, H. H., Yuan, J., and Gao, H. C. (2015). Microbial oxidative stress response: novel insights from environmental facultative anaerobic bacteria. Arch. Biochem. Biophys. 584, 28–35. doi: 10.1016/j.abb.2015.08.012
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, X. Q., Yu, B., Yu, J., Mao, X. B., Huang, Z. Q., Luo, Y. H., et al. (2020). Effects of dietary starch structure on growth performance, serum glucose-insulin response, and intestinal health in weaned piglets. Animals 10:543. doi: 10.3390/ani10030543
Glöckner, F. O., Yilmaz, P., Quast, C., Gerken, J., Beccati, A., Ciuprina, A., et al. (2017). 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176. doi: 10.1016/j.jbiotec.2017.06.1198
Gray, F. V. (1947). The digestion of cellulose by sheep - the extent of cellulose digestion at successive levels of the alimentary tract. J. Exp. Biol. 24, 15–19. doi: 10.1242/jeb.24.1-2.15
Gressley, T. F., Hall, M. B., and Armentano, L. E. (2011). Ruminant nutrition symposium: productivity, digestion, and health responses to hindgut acidosis in ruminants. J. Anim. Sci. 89, 1120–1130. doi: 10.2527/jas.2010-3460
Howie, J. W., and Baker, F. (1952). Rumen and caecal micro-organisms as symbionts. Proc. R. Soc Lond. B Biol. Sci. 139, 193–196. doi: 10.1098/rspb.1952.0005
Jiao, J. Z., Zhang, X. L., Wang, M., Zhou, C. S., Yan, Q. X., and Tan, Z. L. (2019). Linkages between epithelial microbiota and host transcriptome in the ileum during high-grain challenges: implications for gut homeostasis in goats. J. Agric. Food Chem. 67, 551–561. doi: 10.1021/acs.jafc.8b05591
Jin, W., Li, Y., Cheng, Y. F., Mao, S. Y., and Zhu, W. Y. (2018). The bacterial and archaeal community structures and methanogenic potential of the cecal microbiota of goats fed with hay and high-grain diets. Antonie Van Leeuwenhoek 111, 2037–2049. doi: 10.1007/s10482-018-1096-7
Johansson, M. E. V., Gustafsson, J. K., Holmen-Larsson, J., Jabbar, K. S., Xia, L. J., Xu, H., et al. (2014). Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63, 281–291. doi: 10.1136/gutjnl-2012-303207
Johansson, M. E. V., Phillipson, M., Petersson, J., Velcich, A., Holm, L., and Hansson, G. C. (2008). The inner of the two muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 15064–15069. doi: 10.1073/pnas.0803124105
Koryszewska-Baginska, A., Gawor, J., Nowak, A., Grynberg, M., and Aleksandrzak-Piekarczyk, T. (2019). Comparative genomics and functional analysis of a highly adhesive dairy lactobacillus Paracasei subsp. Paracasei ibb3423 strain. Appl. Microbiol. Biotechnol. 103, 7617–7634. doi: 10.1007/s00253-019-10010-1
Kumar, J., Rani, K., and Datt, C. (2020). Molecular link between dietary fibre, gut microbiota and health. Mol. Biol. Rep. 47, 6229–6237. doi: 10.1007/s11033-020-05611-3
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Galvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and cultivation study of muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7:28. doi: 10.1186/s40168-019-0637-2
Larson, J., and Hoffman, P. C. (2008). Technical note: a method to quantify prolamin proteins in corn that are negatively related to starch digestibility in ruminants. J. Dairy Sci. 91, 4834–4839. doi: 10.3168/jds.2008-1378
Li, B. B., Zhang, K., Li, C., Wang, X. L., Chen, Y. L., and Yang, Y. X. (2019). Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (capra hircus) during preweaning development. Front. Microbiol. 10:2125. doi: 10.3389/fmicb.2019.02125
Li, F., Cao, Y., Liu, N., Yang, X., Yao, J., and Yan, D. (2014a). Subacute ruminal acidosis challenge changed in situ degradability of feedstuffs in dairy goats. J. Dairy Sci. 97, 5101–5109. doi: 10.3168/jds.2013-7676
Li, F., Li, Z. J., Li, S. X., Ferguson, J. D., Cao, Y. C., Yao, J. H., et al. (2014b). Effect of dietary physically effective fiber on ruminal fermentation and the fatty acid profile of milk in dairy goats. J. Dairy Sci. 97, 2281–2290. doi: 10.3168/jds.2013-6895
Li, F., Yang, X. J., Cao, Y. C., Li, S. X., Yao, J. H., Li, Z. J., et al. (2014c). Effects of dietary effective fiber to rumen degradable starch ratios on the risk of sub-acute ruminal acidosis and rumen content fatty acids composition in dairy goat. Anim. Feed Sci. Technol. 189, 54–62. doi: 10.1016/j.anifeedsci.2013.12.011
Li, Y. R., Xu, J. J., Zhang, L., Ding, Z. Y., Gu, Z. H., and Shi, G. Y. (2017). Investigation of debranching pattern of a thermostable isoamylase and its application for the production of resistant starch. Carbohydr. Res. 446, 93–100. doi: 10.1016/j.carres.2017.05.016
Li, Z., Liu, N., Cao, Y., Jin, C., Li, F., Cai, C., et al. (2018). Effects of fumaric acid supplementation on methane production and rumen fermentation in goats fed diets varying in forage and concentrate particle size. J. Anim. Sci. Biotechnol. 9:21. doi: 10.1186/s40104-018-0235-3
Liu, J. H., Xu, T. T., Zhu, W. Y., and Mao, S. Y. (2014). High-grain feeding alters caecal bacterial microbiota composition and fermentation and results in caecal mucosal injury in goats. Br. J. Nutr. 112, 416–427. doi: 10.1017/S0007114514000993
Liu, J. X., Taft, D. H., Maldonado-Gomez, M. X., Johnson, D., Treiber, M. L., LemayQ, D. G., et al. (2019). The fecal resistome of dairy cattle is associated with diet during nursing. Nat. Commun. 10:4406. doi: 10.1038/s41467-019-12111-x
Liu, X., Fan, P., Che, R., Li, H., Yi, L., Zhao, N., et al. (2018). Fecal bacterial diversity of wild sichuan snub-nosed monkeys (Rhinopithecus roxellana). Am. J. Primatol. 80:e22753. doi: 10.1002/ajp.22753
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). Unifrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. doi: 10.1038/ismej.2010.133
Matt, S. M., Allen, J. M., Lawson, M. A., Mailing, L. J., Woods, J. A., and Johnson, R. W. (2018). Butyrate and dietary soluble fiber improve neuroinflammation associated with aging in mice. Front. Immunol. 9:1832. doi: 10.3389/fimmu.2018.01832
Møller, M. S., Henriksen, A., and Svensson, B. (2016). Structure and function of α-glucan debranching enzymes. Cell. Mol. Life Sci. 73, 2619–2641. doi: 10.1007/s00018-016-2241-y
Morais, S., and Mizrahi, I. (2019). Islands in the stream: from individual to communal fiber degradation in the rumen ecosystem. FEMS Microbiol. Rev. 43, 362–379. doi: 10.1093/femsre/fuz007
Nagata, R., Innami, N., Pelpolage, S., Shimada, K., Koaze, H., Tani, M., et al. (2019). Effects of raw potato starch with high resistant starch levels on cecal fermentation properties in rats. J. Nutr. Sci. Vitaminol. 65, S192–S195. doi: 10.3177/jnsv.65.S192
Naudin, C. R., Maner-Smith, K., Owens, J. A., Wynn, G. M., Robinson, B. S., Matthews, J. D., et al. (2020). Lactococcus lactis subspecies cremoris elicits protection against metabolic changes induced by a western-style diet. Gastroenterology 159, 639–651. doi: 10.1053/j.gastro.2020.03.010
Navas-Molina, J. A., Peralta-Sánchez, J. M., González, A., McMurdie, P. J., Vázquez-Baeza, Y., Xu, Z., et al. (2013). Advancing our understanding of the human microbiome using qiime. Methods Enzymol. 531, 371–444. doi: 10.1016/b978-0-12-407863-5.00019-8
Noguchi, H., Park, J., and Takagi, T. (2006). Metagene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630. doi: 10.1093/nar/gkl723
Petri, R. M., Munnich, M., Zebeli, Q., and Klevenhusen, F. (2019). Graded replacement of corn grain with molassed sugar beet pulp modulates the fecal microbial community and hindgut fermentation profile in lactating dairy cows. J. Dairy Sci. 102, 5019–5030. doi: 10.3168/jds.2018-15704
Plaizier, J. C., Danesh Mesgaran, M., Derakhshani, H., Golder, H., Khafipour, E., Kleen, J. L., et al. (2018). Review: enhancing gastrointestinal health in dairy cows. Animal 12, S399–s418. doi: 10.1017/s1751731118001921
Plaizier, J. C., Li, S. C., Danscher, A. M., Derakshani, H., Andersen, P. H., and Khafipour, E. (2017). Changes in microbiota in rumen digesta and feces due to a grain-based subacute ruminal acidosis (sara) challenge. Microb. Ecol. 74, 485–495. doi: 10.1007/s00248-017-0940-z
Pope, P. B., Smith, W., Denman, S. E., Tringe, S. G., Barry, K., Hugenholtz, P., et al. (2011). Isolation of Succinivibrionaceae implicated in low methane emissions from tammar wallabies. Science 333, 646–648. doi: 10.1126/science.1205760
Qi, K., Men, X., Wu, J., and Xu, Z. (2019). Rearing pattern alters porcine myofiber type, fat deposition, associated microbial communities and functional capacity. BMC Microbiol. 19:181. doi: 10.1186/s12866-019-1556-x
Ren, H., Su, X. D., Bai, H. X., Yang, Y. T., Wang, H. R., Dan, Z., et al. (2019). Specific enrichment of microbes and increased ruminal propionate production: the potential mechanism underlying the high energy efficiency of holstein heifers fed steam-flaked corn. AMB Expr. 9:209. doi: 10.1186/s13568-019-0937-8
Ribeiro, G. O., Badhan, A., Huang, J., Beauchemin, K. A., Yang, W., Wang, Y., et al. (2018). New recombinant fibrolytic enzymes for improved in vitro ruminal fiber degradability of barley straw1. J. Anim. Sci. 96, 3928–3942. doi: 10.1093/jas/sky251
Rimoldi, S., Gini, E., Iannini, F., Gasco, L., and Terova, G. (2019). The effects of dietary insect meal from Hermetia illucens prepupae on autochthonous gut microbiota of rainbow trout (Oncorhynchus mykiss). Animals 9:143. doi: 10.3390/ani9040143
Riva, A., Kuzyk, O., Forsberg, E., Siuzdak, G., Pfann, C., Herbold, C., et al. (2019). A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome. Nat. Commun. 10:4366. doi: 10.1038/s41467-019-12413-0
Rivera-Chavez, F., Zhang, L. F., Faber, F., Lopez, C. A., Byndloss, M. X., Olsan, E. E., et al. (2016). Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of salmonella. Cell Host Microbe 19, 443–454. doi: 10.1016/j.chom.2016.03.004
Rogha, M., Esfahani, M. Z., and Zargarzadeh, A. H. (2014). The efficacy of a synbiotic containing Bacillus Coagulans in treatment of irritable bowel syndrome: a randomized placebo-controlled trial. Gastroenterol. Hepatol. Bed Bench 7, 156–163.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Seshadri, R., Leahy, S. C., Attwood, G. T., Teh, K. H., Lambie, S. C., Cookson, A. L., et al. (2018). Cultivation and sequencing of rumen microbiome members from the hungate1000 collection. Nat. Biotechnol. 36, 359–367. doi: 10.1038/nbt.4110
Shen, J., Han, X., Zheng, L., Liu, S., Jin, C., Liu, T., et al. (2020a). High rumen-degradable starch diet promotes hepatic lipolysis and disrupts enterohepatic circulation of bile acids in dairy goats. J. Nutr. 150, 2755–2763. doi: 10.1093/jn/nxaa238
Shen, J., Zheng, L. X., Chen, X. D., Han, X. Y., Cao, Y. C., and Yao, J. H. (2020b). Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of dairy goats fed different rumen degradable starch. Front. Microbiol. 11:1003. doi: 10.3389/fmicb.2020.01003
Steele, M. A., Penner, G. B., Chaucheyras-Durand, F., and Guan, L. L. (2016). Development and physiology of the rumen and the lower gut: targets for improving gut health. J. Dairy Sci. 99, 4955–4966. doi: 10.3168/jds.2015-10351
Stewart, R. D., Auffret, M. D., Warr, A., Walker, A. W., Roehe, R., and Watson, M. (2019). Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961. doi: 10.1038/s41587-019-0202-3
Stewart, R. D., Auffret, M. D., Warr, A., Wiser, A. H., Press, M. O., Langford, K. W., et al. (2018). Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9:870. doi: 10.1038/s41467-018-03317-6
Sun, B., Wang, X., Bernstein, S., Huffman, M. A., Xia, D. P., Gu, Z., et al. (2016). Marked variation between winter and spring gut microbiota in free-ranging tibetan macaques (Macaca thibetana). Sci. Rep. 6:26035. doi: 10.1038/srep26035
Tao, L., Wang, B. H., Zhong, Y. F., Pow, S. H., Zeng, X., Qin, C., et al. (2017). Database and bioinformatics studies of probiotics. J. Agric. Food Chem. 65, 7599–7606. doi: 10.1021/acs.jafc.7b01815
Tao, S. Y., Tian, P., Luo, Y. W., Tian, J., Hua, C. F., Geng, Y. L., et al. (2017). Microbiome-metabolome responses to a high-grain diet associated with the hind-gut health of goats. Front. Microbiol. 8:1764. doi: 10.3389/fmicb.2017.01764
Thomas, A. M., Jesus, E. C., Lopes, A., Aguiar, S. Jr., Begnami, M. D., Rocha, R. M., et al. (2016). Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16s rrna community profiling. Front. Cell. Infect. Microbiol. 6:179. doi: 10.3389/fcimb.2016.00179
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., et al. (2002). Accurate normalization of real-time quantitative rt-pcr data by geometric averaging of multiple internal control genes. Genome. Biol. 3:Research0034. doi: 10.1186/gb-2002-3-7-research0034
Wood, H. G., and Werkman, C. H. (1938). The utilization of co2 by the propionic acid bacteria. Biochem. J. 32, 1262–1271. doi: 10.1042/bj0321262
Xue, M. Y., Sun, H. Z., Wu, X. H., Liu, J. X., and Guan, L. L. (2020). Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 8:64. doi: 10.1186/s40168-020-00819-8
Yilmaz, B., Juillerat, P., Øyås, O., Ramon, C., Bravo, F. D., Franc, Y., et al. (2019). Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 25, 323–336. doi: 10.1038/s41591-018-0308-z
Zhang, C., Zhang, T., Lu, W., Duan, X., Luo, X., Liu, S., et al. (2020). Altered airway microbiota composition in patients with pulmonary hypertension. Hypertension 76, 1589–1599. doi: 10.1161/hypertensionaha.120.15025
Zhang, J. D., Song, L. J., Wang, Y. J., Liu, C., Zhang, L., Zhu, S. W., et al. (2019). Beneficial effect of butyrate-producing lachnospiraceae on stress-induced visceral hypersensitivity in rats. J. Gastroenterol. Hepatol. 34, 1368–1376. doi: 10.1111/jgh.14536
Zhao, F., Cao, H.-Y., Zhao, L.-S., Zhang, Y., Li, C.-Y., Zhang, Y.-Z., et al. (2019). A novel subfamily of endo-β-1,4-glucanases in glycoside hydrolase family 10. Appl. Environ. Microbiol. 85:e01029-19. doi: 10.1128/aem.01029-19
Zheng, L. X., Wu, S. R., Shen, J., Han, X. Y., Jin, C. J., Chen, X. D., et al. (2020). High rumen degradable starch decreased goat milk fat via trans-10, cis-12 conjugated linoleic acid-mediated downregulation of lipogenesis genes, particularly, INSIG1. J. Anim. Sci. Biotechnol. 11:30. doi: 10.1186/s40104-020-00436-3
Keywords: dairy goat, GH family, Muc2, rumen degradable starch, short-chain fatty acids
Citation: Han X, Lei X, Yang X, Shen J, Zheng L, Jin C, Cao Y and Yao J (2021) A Metagenomic Insight Into the Hindgut Microbiota and Their Metabolites for Dairy Goats Fed Different Rumen Degradable Starch. Front. Microbiol. 12:651631. doi: 10.3389/fmicb.2021.651631
Received: 10 January 2021; Accepted: 20 April 2021;
Published: 07 June 2021.
Edited by:
Antonio Faciola, University of Florida, United StatesReviewed by:
Xiaoxia Dai, Royal Veterinary College (RVC), United KingdomZhigang Zhou, Feed Research Institute (CAAS), China
Copyright © 2021 Han, Lei, Yang, Shen, Zheng, Jin, Cao and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junhu Yao, eWFvanVuaHUyMDA0QHNvaHUuY29t
†These authors have contributed equally to this work and share first authorship