
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 05 July 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.651520
This article is part of the Research TopicBacterial Pathogen Genomics: Recent Achievements, Current Applications and Future ChallengesView all 16 articles
 Xingxing Xu1,2†
Xingxing Xu1,2† Qiao Bian3†
Qiao Bian3† Yun Luo4Xiaojun Song5Shan Lin2Huan Chen6,7
Yun Luo4Xiaojun Song5Shan Lin2Huan Chen6,7 Qian Liang6,7Meixia Wang6,7Guangyong Ye1Bo Zhu1
Qian Liang6,7Meixia Wang6,7Guangyong Ye1Bo Zhu1 Liang Chen8,9Yi-Wei Tang10
Liang Chen8,9Yi-Wei Tang10 Xianjun Wang11*
Xianjun Wang11* Dazhi Jin2,5*
Dazhi Jin2,5*Clostridioides difficile sequence type 2 (ST2) has been increasingly recognized as one of the major genotypes in China, while the genomic characteristics and biological phenotypes of Chinese ST2 strains remain to be determined. We used whole-genome sequencing and phylogenetic analysis to investigate the genomic features of 182 ST2 strains, isolated between 2011 and 2017. PCR ribotyping (RT) was performed, and antibiotic resistance, toxin concentration, and sporulation capacity were measured. The core genome Maximum-likelihood phylogenetic analysis showed that ST2 strains were distinctly segregated into two genetically diverse lineages [L1 (67.0% from Northern America) and L2], while L2 further divided into two sub-lineages, SL2a and SL2b (73.5% from China). The 36 virulence-related genes were widely distributed in ST2 genomes, but in which only 11 antibiotic resistance-associated genes were dispersedly found. Among the 25 SL2b sequenced isolates, RT014 (40.0%, n = 10) and RT020 (28.0%, n = 7) were two main genotypes with no significant difference on antibiotic resistance (χ2 = 0.024–2.667, P > 0.05). A non-synonymous amino acid substitution was found in tcdB (Y1975D) which was specific to SL2b. Although there was no significant difference in sporulation capacity between the two lineages, the average toxin B concentration (5.11 ± 3.20 ng/μL) in SL2b was significantly lower in comparison to those in L1 (10.49 ± 15.82 ng/μL) and SL2a (13.92 ± 2.39 ng/μL) (χ2 = 12.30, P < 0.05). This study described the genomic characteristics of C. difficile ST2, with many virulence loci and few antibiotic resistance elements. The Chinese ST2 strains with the mutation in codon 1975 of the tcdB gene clustering in SL2b circulating in China express low toxin B, which may be associated with mild or moderate C. difficile infection.
Clostridioides difficile is an anaerobic, spore-forming Gram-positive bacillus that is able to colonize and proliferate in the human gut, especially following changes in the indigenous colonic microbiota after antibiotic use (Knight et al., 2015). The C. difficile genomes have been well documented with a high proportion of mobile genetic elements and an ultra-low level of genetic conservation (as low as 16%) (Knight et al., 2016), and its phylogenetics and evolutionary clades have been recognized as well (Knight et al., 2015). A whole-genome sequencing (WGS)-based phylogenetic tree showed that C. difficile has six main clades (Knight et al., 2015), in which clade 1 is the largest one with a variety of sequence types (STs), including ST2, which usually resulted in mild or moderate C. difficile infection (CDI) (Griffiths et al., 2010; Cheng et al., 2016). ST2 exhibits high genetic diversity with various PCR ribotypes (RT) (Griffiths et al., 2010; Jin et al., 2017), two toxin genes (tcdA and tcdB) are located on a 19.6-kb pathogenicity locus (PaLoc), and no binary toxins are found (Knight et al., 2015). ST2 also belongs to RT014, which is a successful lineage of C. difficile as significant reservoirs in both human and porcine populations in Australia, and there were obviously different genomic features and biological phenotypes in ST2 including antibiotic resistance elements and antibiotic resistance phenotype between different resources (Knight et al., 2016). However, the genomic characteristics and biological phenotypes remain to be determined in other regions.
Recent studies showed that the molecular epidemiology of C. difficile in China has its own characteristics with specific antibiotic resistance and genotype profiles as below (Tang et al., 2016; Jin et al., 2017; Luo et al., 2019). A meta-analysis displayed that the pooled incidence of toxigenic C. difficile in diarrhea patients in Mainland China was approximately 14%, and ST2 was one of the dominant genotypes in China (Tang et al., 2016). However, ST2 usually led to mild or moderate CDI with no other severe symptoms, making it be easily overlooked in clinical treatment (Jin et al., 2017; Luo et al., 2018). Even though antimicrobial resistance phenotypes of C. difficile ST2 in Australia have been described as reported previously, there is still a paucity of data on ST2 genomic characteristics and the biological characteristics, including toxin expression and sporulation in other regions.
WGS provides an ultra-fine scale resolution tool for analysis of genomic characteristics, evaluation of bacterial genetic diversity, identification of subtle genetic variability, identification of signatures of clonal transmission, and assessments of C. difficile epidemiology of strains implicated in infection recurrences and outbreaks (Didelot et al., 2012; Eyre and Walker, 2013; Martin et al., 2016). Here, a collection of 182 ST2 strains from different regions of human origin with isolation dates between 2011 and 2017 were studied. Genome characteristics associated with virulence and antibiotic resistance genotypes were studied in silico, and biological features, including antibiotic resistance phenotypes, toxin expression, and sporulation, were also measured in vitro.
A total of 30 ST2 strains were isolated from CDI patients as part of different CDI surveillance programs conducted in different sites including Zhejiang (n = 18) between February 2013 and January 2017, Hebei (n = 10) between January 2011 and December 2014, Hunan (n = 2) in 2014. The remaining 10 ST2 strains were sourced from CDI patients as part of an international collaboration on molecular characteristics of C. difficile. Isolates originated from six sites related to hospitals, including Hong Kong (n = 2) in China, Pusan in South Korea (n = 1), Fukuoka in Japan (n = 1), Singapore (n = 3), and Sydney (n = 2) and Perth (n = 1) in Australia. All the 40 available ST2 isolates with no epidemiological relationships were included in this study by the time the study started. Clinical data were collected after the study was approved by the Ethics Committee of the Hangzhou Medical College. All available genomic data for ST2 strains were included in this study up to January 2017. The genome data of a total of 139 ST2 strains published between 2001 and 2016 were downloaded from the National Center for Biotechnology (NCBI1). Raw reads data of 3 ST2 strains (SRR1519431, SRR1519374, and SRR1519422) isolated in 1995 and 1997, respectively, were downloaded from the NCBI database (up to January 2017). The information of the 182 strain was provided in Supplementary Material.
Six reference C. difficile strains (ATCC 43255, ATCC 43598, BAA-1870, BAA-1803, BAA-1801, and ATCC 700057) were used as controls. PCR ribotyping was performed by using PCR followed by capillary gel electrophoresis described previously (Indra et al., 2008). The 16S rRNA gene primers were labeled at the 5′ end with carboxyfluorescein. After PCR amplification, PCR fragments were analyzed using an ABI 3100 genetic analyzer (Applied Biosystems, Foster City, CA, United States) with a 36-cm capillary loaded with a POP4 gel (Applied Biosystems). The size of each peak was determined using GeneMapper ID-X 1.3 (Applied Biosystems). RT assignment was performed after the data were deposited in the WEBRIBO database2.
Genomic DNA was extracted as previously described (Stabler et al., 2009). WGS libraries were created using TruePrep DNA Library PrepTM Kit V2 (Illumina, Santiago, CA, United States). WGS was performed using the Illumina Hiseq X-ten with 150-base paired-end reads. The sequence data were processed and quality controlled according to a standard pipeline as previously described (Preston et al., 2014). Briefly, FASTQ-formatted sequencing reads were quality controlled with a minimum quality Phred score of 30 (as a rolling average over 4 bases) using trimmomatic v0.36. Trimmomatic v0.36 was also used to remove adapters and low-quality sequences, and 63.051 Gb clean bases were finally generated (1.616 Gb/per isolate, Q20 ≥ 95%) (Bolger et al., 2014). Genome data for the 40 isolates from this study, and the downloaded raw reads for three genomes, were de novo assembled using Velvet (version 1.2.10). Optimal k-mers fell between 47 and 93 bp, according to the mean value for median contig size of genome assembly (n50). The genomic sequences of the remaining 139 strains were downloaded from the NCBI.
All genome sequences were aligned to the C. difficile W0022a reference genome (GCF_002812625.1) and SNPs were identified using MUMmer (v3.23) with default parameters (Kurtz et al., 2004; Li et al., 2016). Following the removal of SNPs within 5 bp of the location interval (less than five bases existed between any two SNPs) by using the perl script (file name: filter_dist.pl.3), high-quality SNPs were annotated according to demographic information including clades and locations.
Gubbins was used to detect recombination in newly built whole-genome sequences (Croucher et al., 2015) as previously reported (Knight et al., 2015, 2016), and SNPs located in the recombination regions were identified and removed by using the perl script (file name: gubbinssns.vcf_to_genotype.pl., see text footnote 3). After that, an alignment of non-recombinant SNPs was obtained. A total of 182 C. difficile genomes were used to generate core genes by using BLAST with the thresholds of 80% nucleotide sequence identity and 80% of the query length. A total of 1,685 genes were defined as the core gene sets using BLASTp with an E-value of 1e-10, out of which the final SNPs were detected from all non-recombinant SNPs. The Maximum likelihood (ML) tree topology and branch length were inferred using IQ-TREE multicore v1.6.6. The recombination/mutation (r/m) ratio was calculated within the deep-branching phylogeny, which gives the relative probability that a nucleotide has changed as a result of recombination relative to a point mutation. Two phylogenetic trees before and after removal of SNPs located in the recombination regions by using Gubbins were compared, and the consistency indexes for two phylogenetic trees were analyzed by using the phangorn package in R (Schliep, 2011).
Antibiotic resistance-associated genes were determined using RGI (version 5.1.0) analysis and the CARD antibiotic resistance gene database4 (Alcock et al., 2020). Virulence loci were determined by BLAST (Ye et al., 2006; Johnson et al., 2008) analysis of genome sequences in the virulence factors database (VFDB5), which aims to provide up-to-date knowledge of virulence factors from various bacteria and serves as a comprehensive warehouse of bacterial pathogenesis knowledge for the scientific community (Liu et al., 2019). The cutoff values were set as 90% nucleotide identity and 90% of the query length for gene coverage to screen virulence loci (Dingle et al., 2019).
Twelve antibiotics including vancomycin, metronidazole, moxifloxacin, erythromycin, clindamycin, rifampicin, levofloxacin, gatifloxacin, ciprofloxacin, fusidic acid, tetracycline, and piperacillin-tazobactam (PIP-TAZ) were used to test the minimum inhibition concentration (MIC) of the 40 isolates by the agar dilution method according to standard clinical and laboratory guidelines (CLSI) (Clinical and Laboratory Standards Institute, 2011). The reference strain (ATCC 700057) was included in each test as a control. MIC breakpoints were chosen according to a previous report (Jin et al., 2017).
Clostridioides difficile toxin B concentration was quantitatively detected using the real-time cell analysis (RTCA) system according to a previously reported method (Ryder et al., 2010). The toxin B concentration was calculated by a formula which was derived based on the testing results of a panel of purified toxin B standards with known concentrations ranging from 0.02 to 200 ng/μL. The sporulation capacity was measured as reported previously (Qin et al., 2017). A count between 30 and 300 spores on each plate was considered significant.
Data analysis was performed using Statistical Package for Social Sciences (SPSS, Chicago, IL, United States) version 22.0. The difference in toxin concentration and sporulation capacity was analyzed using a t-test or nonparametric test. A P-value of <0.05 was considered statistically significant.
All 182 WGS data were analyzed after sequence quality control and mapping to the C. difficile strain W0022a (NCBI accession: GCF_002812625.1). A total of 14,795 high-quality SNPs were identified after the resulting alignments. Gubbins analysis revealed 10,324 spatially clustered SNPs within 1,352 homologous recombination events. After removing these SNPs, 4,471 high-quality biallelic SNPs were extracted. Of these, 1,249 non-rare (core) SNPs (27.9%, 1,249/4,471) were present in the core genome, which included 966 (77.3%, 966/1,249) non-synonymous SNPs. The r/m ratio was approximately 2.31, which was determined through 10,324 SNPs divided by 4,471 SNPs. The phylogenetic tree was explored by high-resolution core genome SNPs analysis as briefly shown in Figure 1A. Firstly, the tcdB gene was conserved across all ST2 strains, and 98.4% (179/182) of ST2 had an intact tcdA gene, and the remainder carried truncated tcdA genes with an in-frame deletion of 1,633, 1,732, and 2,371 bp, respectively. All strains harbored cdtA/B pseudogene located in a 4.2-kb pathogenicity locus (CdtLoc) as described (Gerding et al., 2014). The remaining 33 virulence-related genes were distributed in the 182 genomes. Of them, 25 virulence-related genes were found in all ST2 strains. The putative flagellar protein gene (CD0259), which was associated with C. difficile virulence (Aubry et al., 2012), exists in only three of the ST2 genomes. The remaining seven virulence loci including groEL, which plays a role in C. difficile cell adherence (Hennequin et al., 2001) and decreases C. difficile intestinal colonization (Péchiné et al., 2013), CD0255A, flgE, fliK, fliL, fliP, and motA were widely distributed in ST2 genomes (Figure 1B).
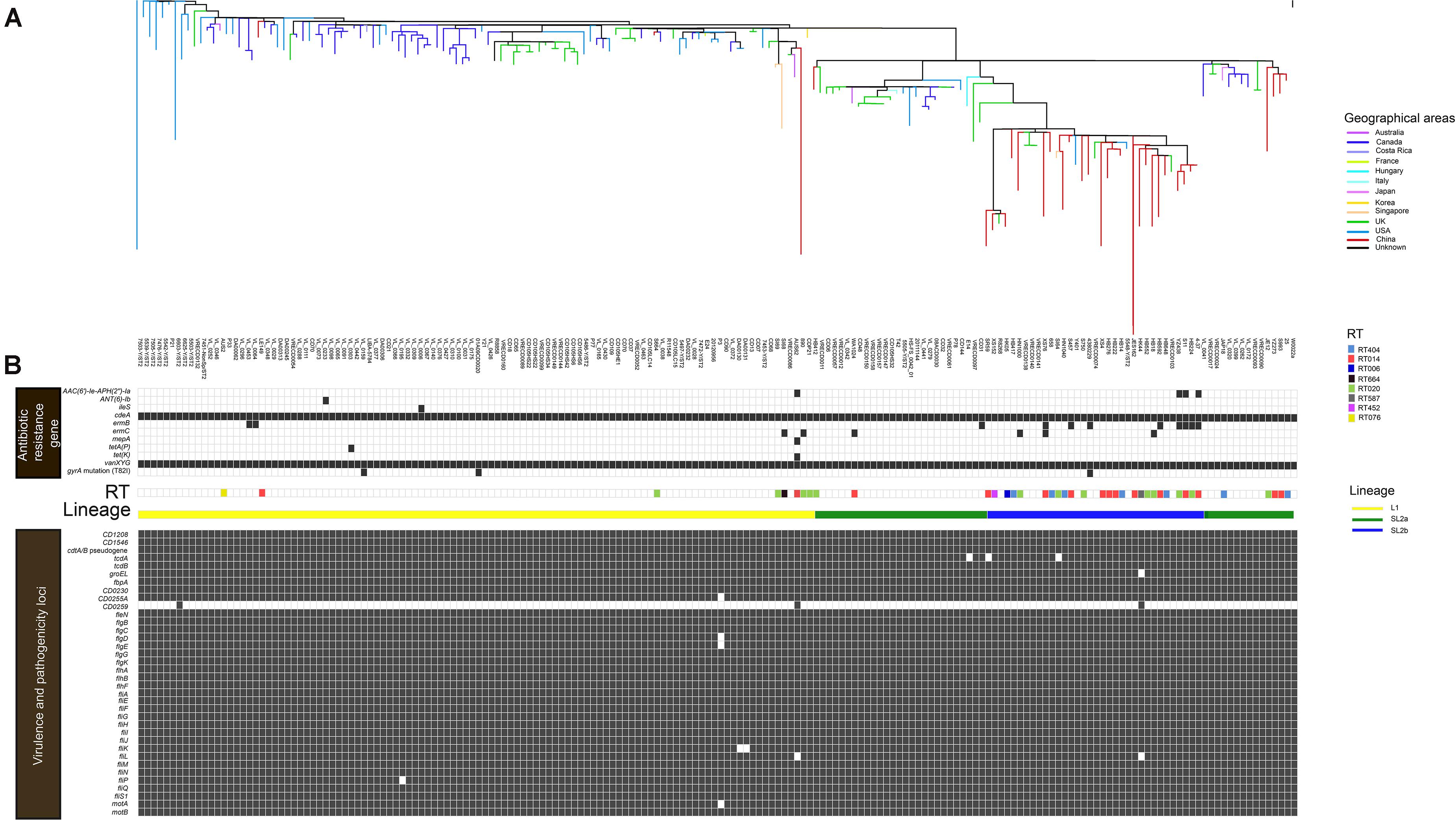
Figure 1. In silico predicted antibiotic resistance genes and virulence loci of 182 C. difficile ST2 strains. The heat map showed the distribution of antibiotic resistance genes and virulence loci in the genome-wide data. (A) The phylogenetic tree. The different colors in the figure represent the different origins. (B) Presence is indicated by black bars and absence by white bars. Colored by RT: light blue, RT404; orange, RT014; navy blue, RT006; black, RT664; green, RT020; gray, RT587; rose red, RT452; and yellow, RT076. Sorted by lineage: yellow line, L1; green line, SL2a; and blue line, SL2b.
A total of 10 putative antibiotic resistance genes and one gyrA gene variant with amino acid substitution (T82I) were identified within the 182 genomes (Figure 1B). The vanXY variant gene, vanXYG, mediated aminoglycoside resistance, and the cdeA gene, associated with fluoroquinolone resistance, were found in all 182 isolates. The remaining nine genes were found in several genomes as described below. The tetracycline resistance elements, tetA(P) and tet(K), were only found in two different C. difficile strains (one from Canada, another from Australia) (Figure 1B). For macrolide–lincosamide–streptogramin B (MLSB) resistance, the ermB and ermC genes were scattered in 6.0% (11/182) and 3.3% (6/182) of strains, respectively. Only three strains carried the gyrA mutation (T82I). The AAC(6′)-1e-APH(2″)-1a cassette associated with aminoglycoside resistance was found in 2.2% (4/182) of strains. The above results showed that C. difficile ST2 genomes dispersedly harbored a small number of antibiotic resistance genes and associated mutations.
A ML phylogeny indicated that the 182 ST2 genomes were obviously divided into two genetically diverse lineages, L1 and L2 (Figure 2), primarily differentiated by 18 unique SNPs (Supplementary Table 1). L2 could be further divided into two sub-lineages, SL2a and SL2b, based on eight specific SNPs (Supplementary Table 2). SL2b primarily comprised most of the C. difficile strains from China (25/34, 73.5%), while North America had a high proportion of strains in L1 (66.4%, 71/107) and there was a high diversity of strain sources in SL2a. Various RTs were dispersedly distributed in different sub-lineages as Figure 1B shown. The geographical distribution showed significant differences among L1, SL2a, and SL2b in this ML phylogeny. Interestingly, it has been found that SL2b has only one sublineage-specific non-synonymous mutation, which occurred in the tcdB gene (W0022a reference genome, base position: T671,314G; amino acid position: Tyr1975Asp). The r/m value of the Chinese SL2b was 2.60, which was higher than those of strains from SL2a (2.33) and SL1 (1.26). There were 28 homologous recombination regions found in all strains belonging to SL2, of which 14 were specific to SL2b; six homologous recombination regions existed in almost all of the strains (Supplementary Figure 1 and Supplementary Table 3).
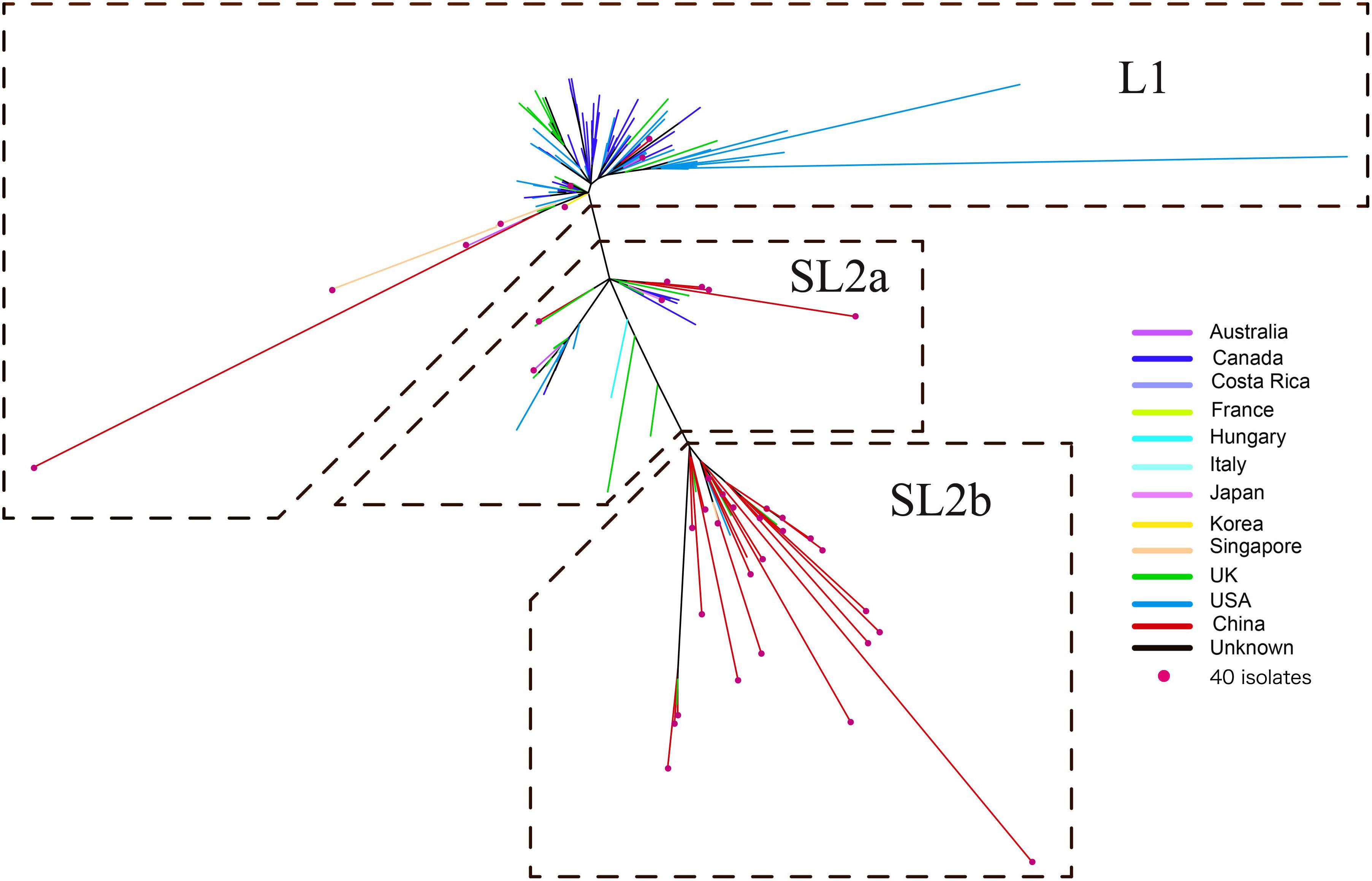
Figure 2. Maximum likelihood phylogeny tree of C. difficile ST2. The different colors represented the different origins; the red dot marker was used for 40 C. difficile isolates by whole genome sequencing in this study.
Antibiotic susceptibility testing was performed in the 40 isolates sequenced in this study (Table 1). Summary MIC distributions for 12 antibiotic agents, by RT lineage, were presented in Figure 3. The rate of resistance to MLSB was 90.0% (36/40). The rates of resistance to the fourth-generation fluoroquinolones, gatifloxacin (15.0%, 6/40) and moxifloxacin (10.0%, 4/40) were lower than those to the third-generation fluoroquinolones, levofloxacin (82.5%, 33/40) (χ2 = 36.47 and 42.29, P < 0.001) and ciprofloxacin (92.5%, 37/40) (χ2 = 48.32 and 54.48, P < 0.001). No isolates were resistant to vancomycin or metronidazole. The rates of resistance to MLSB, fluoroquinolone, and fusidic acid (62.5%, 25/40) were distinctly higher than those to rifampicin (5.0%, 2/40) (χ2 = 29.57, P < 0.001), tetracycline (7.5%, 3/40) (χ2 = 26.59, P < 0.001), and piperacillin (2.5%, 1/40) (χ2 = 32.82, P < 0.001). All the RT020 isolates were susceptible to PIP-TAZ, and resistant to MLSB and fluoroquinolone. Of them, 61.5% (8/13), 7.7% (1/13), and 7.7% (1/13) were resistant to fusidic acid, and rifampicin, tetracycline, respectively. All of the RT014 isolates were susceptible to rifampicin; 86.7% (13/15) and 93.3% (14/15) were resistant to fluoroquinolone and MLSB, respectively; 60.0% (9/15) were resistant to fusidic acid; 6.7% (1/15) were resistant to tetracycline and piperacillin. The multidrug-resistant (MDR: resistant to ≥3 of these agents) rate was 65.0% (26/40), noted to be high in this study. Even though no significant differences in the antibiotic patterns were found among the different RTs and SLbs, L1 and SL2a, with no strains resistant to rifampin, tetracycline, PIP-TAZ, and moxifloxacin, they obviously presented different resistance patterns from SL2b (Supplementary Table 4).
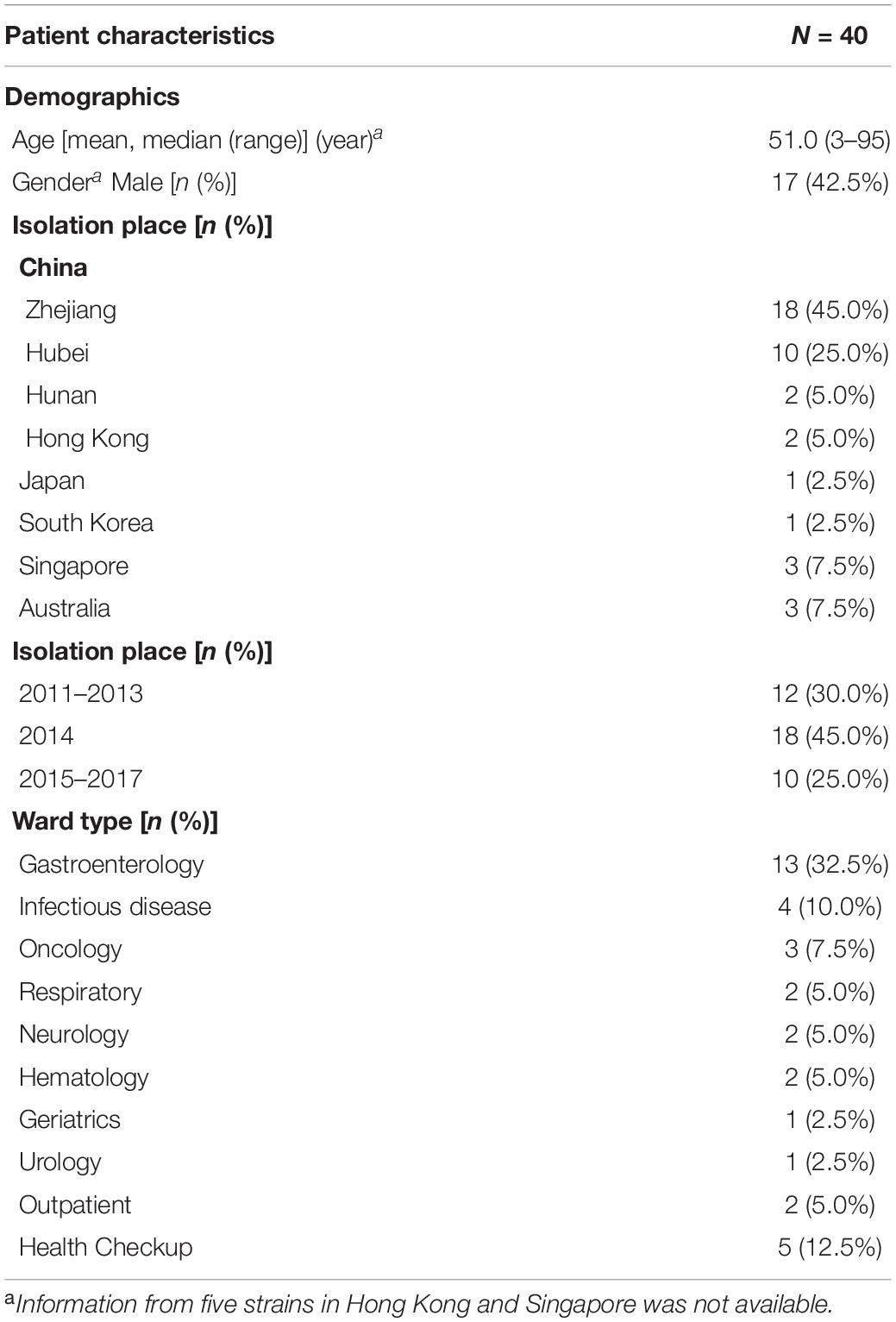
Table 1. Clinical information of 40 C. difficile ST2 isolate.
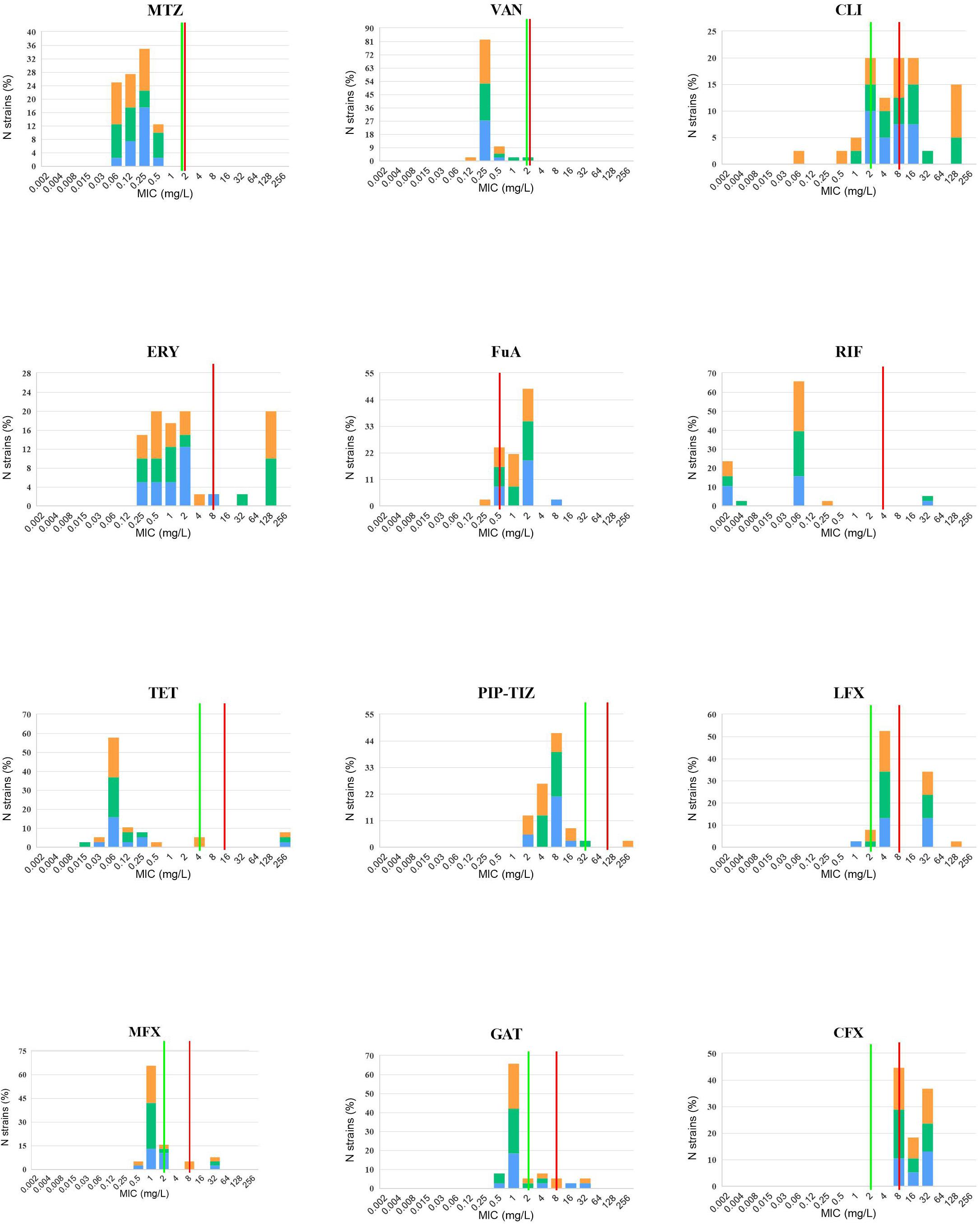
Figure 3. In vitro 12 antibiotic susceptibility with susceptible and resistant breakpoints. MIC distributions for 12 antibiotic agents against 40 C. difficile isolates, orange: RT014, green: RT020, and blue: RT664, RT006, RT076, RT404, RT452, and RT587. VAN, vancomycin; MTZ, metronidazole; ERY, erythromycin; CLI, Clindamycin; RIF, rifaximin; TET, tetracycline; PIP-TAZ, piperacillin-tazobactam; LFX, levofloxacin; MFX, moxifloxacin; GAT, gatifloxacin; CFX, ciprofloxacin. Where available, established susceptible and resistant breakpoints were indicated by vertical green and red lines, respectively.
The clinical information for the 40 patients is shown in Table 1. All the 40 ST2 isolates had no epidemiological relationships in spite that some of them belonged to the same RT. Some of the isolates were from the same province but not from the same location. Thus, no same clones existed in these ST2 genomes. The RTCA data showed that the average concentrations of toxin B in groups L1, SL2a, and SL2b were 10.49 ± 15.82, 13.92 ± 2.39, and 5.11 ± 3.20 ng/μL, respectively (Figure 4). C. difficile isolates in SL2a had significantly higher toxin concentration abilities than those in SL2b (t = 6.709, P < 0.001). Spore resuscitation abilities were represented in the mode of InterQuartile Range [M (P25, P75)]. The numbers for C. difficile sporulation in L1, SL2a and SL2b were 1.1 × 106 (5.2 × 105, 2.3 × 106)/mL, 1.2 × 106 (7.8 × 104, 1.3 × 106)/mL, and 6.6 × 105 (2.5 × 105, 1.2 × 106)/mL, respectively (Figure 4). There were no statistical differences in sporulation in the three groups (χ2 = 2.13, P = 0.346).
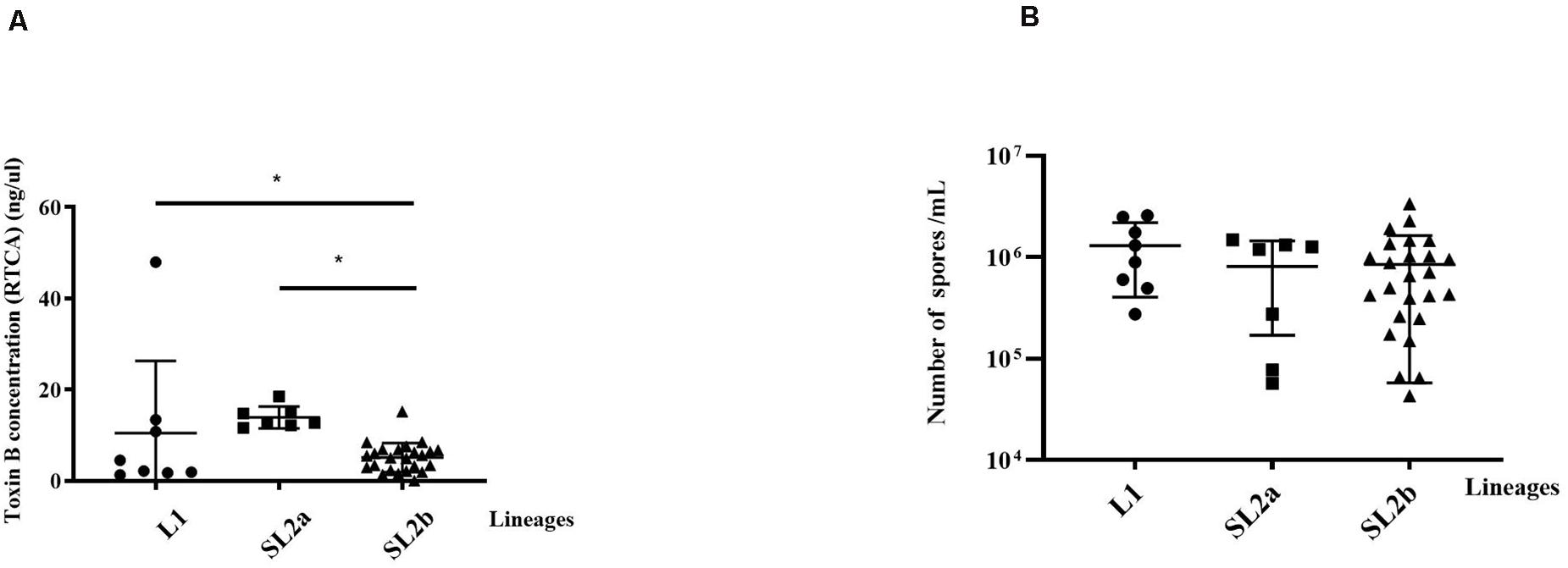
Figure 4. Comparison of toxin production and Sporulation determination in the 40 C. difficile ST2 strains with different sub-lineages. (A) Toxin B concentrations by the RTCA system; (B) sporulation capacity by the heat-induced experiment. The toxin concentrations and sporulation capacity were shown as the mean ± standard deviation. Significant differences were marked with *P < 0.05.
Clostridioides difficile ST2 is one of the most dominant genotypes associated with mild CDI or asymptomatic carriage in China (Tian et al., 2016; Zhang et al., 2016; Jin et al., 2017), whereas the biological phenotypes and genomic characteristics of Chinese ST2 strains remain unexplored thus far. The ST2 lineage might be overshadowed by increasing prevalence of ST37/RT017 worldwide and outbreaks of ST1/RT027 lineage. As the ML phylogenetic trees on ST1/RT027 and ST37/RT017, our data revealed two distinct lineages of C. difficile ST2 with multiple independent clonal expansions after removal of SNPs located in the recombination regions. We also compared two phylogeny trees before and after the removal of SNPs located in the recombination regions by using Gubbins. The ML phylogeny tree before removal of SNPs located in the recombination regions showed similarity with SL1, SL2a, and SL2b (data not shown). The results showed that the consistency indexes of the ML phylogenetic trees with and without removal of SNPs located in the recombination regions were 0.9796078 and 0.937613, which was analyzed by using the phangorn package in R, indicating that removal of recombinant SNPs made the remaining SNPs more consistent with the resulting phylogeny. Notably, further analysis indicated that there was a sub-lineage of SL2b divided under L2, most of which (73.5%, 25/34) originated from China, and the average toxin B concentration in SL2b was significantly lower in comparison to those in L1 and SL2a.
ST2 and ST37 are the predominant genotypes in China (Tang et al., 2016; Jin et al., 2017). The T82I gene mutation in gyrA found in our study has been reported in the global outbreak of RT027 and RT017 (He et al., 2013; Cairns et al., 2017). The T82I substitution in RT027 and RT017 was globally distributed in both of their two sublineages but was found in silico in three ST2 genomes. In addition, a substitution in the gyrB gene, also associated with fluoroquinolone resistance, and the substitutions in the rifampin resistance-determining region of the rpoB gene (He et al., 2013; Cairns et al., 2017) were not found in ST2 genomes as the RT014 lineage shown (Knight et al., 2016), indicating that ST2 genomes had their own specific molecular characteristics with low diversity of antibiotic resistance-related gene mutations.
The data showed the average toxin production in SL2b was significantly lower than those in L1 and SL2a, and then a sublineage specific non-synonymous mutation in the tcdB gene (Y1975D) was found to be located in the C-terminus receptor-binding domain (RBD) of tcdB, which is a critical region for interaction with host epithelial cell membranes (Carter et al., 2012; Orth et al., 2014). The demographic information of all 40 isolates was checked, and no epidemiological relationships were found, even in the same RT and the same location. Thus, the ST2 genomes with the mutation of Y1975D were not from the same clone. The tcdB gene showed high genetic diversity with classification into eight subtypes (tcdB1-8). The subtype tcdB1 has the maximum within-subtype variation and consists of three clusters (tcdB1a, tcdB1b, and tcdB1c) as shown in our previous study, and all ST2 strains carried tcdB1a (Shen et al., 2020). Most of the C. difficile strains from North America, East Asia, and European countries express tcdB1, which was closely related to human and animal diseases with more cytotoxicity than other subtypes (Shen et al., 2020). The sequences of RT027-tcdB-RBD are genetically divergent from other genotypes, and probably associated with rapid cell entry (Dingle et al., 2011). Although the biological function of the gene mutation (Y1975D) in tcdB was unknown as this mutation was not located in the 712bp amplicon previously analyzed (Dingle et al., 2011), the structure of the tcdB protein might be impacted, leading to the prevention of toxin B binding to cell surface receptors and decreasing C. difficile ST2 cytotoxicity. Thus, we speculated that it was reasonable that CDI led by C. difficile ST2 in SL2b had mild or moderate clinical symptoms, and the level of pathological lesion was low, especially for strains in China. However, the mutation in the tcdB gene-specific to SL2b from China was not found to be significantly associated with toxin B concentration, probably due to the sampling bias in this study. Thus, more studies should be performed to clarify the relationship between this SNP and low toxin B cytotoxicity and examine its contribution to the decreased ST2 strain virulence.
Clostridioides difficile sporulation capacity was associated with bacterial viability, successfully determining the transmission ability of epidemic clones (Qin et al., 2017; Zhu et al., 2018). In this study, no significant difference was found on sporulation capacity of C. difficile ST2 between SL2b and other lineages. A previous report provided the first epidemiological evidence on an outbreak of epidemic C. difficile genotype ST81 in a general teaching hospital in China (Qin et al., 2017). The data showed that ST81 had similar sporulation capacity in comparison to ST2, but significantly less toxin production than ST2 in China (Qin et al., 2017). Obviously, ST2 strains had a potentially high risk of transmission, leading to a nosocomial outbreak, and the mild clinical symptoms were easily overlooked by clinicians. Moreover, C. difficile ST2 with high sporulation capacity might contribute to cross-species transmission from animal to human. Therefore, C. difficile ST2 should be closely monitored in future surveillance.
Antibiotic resistance elements are the key factors driving genetic diversity and epidemiological changes in C. difficile (Knight et al., 2015). In our study, only two antibiotic resistance loci (cdeA and vanXYG) existed in all ST2 strains and other antibiotic resistance loci were dispersedly distributed in ST genomes. Similar data were also presented in the previous study on RT014 lineage, mainly including ST2, ST13, and ST49 (Knight et al., 2016). In comparison to other STs in RT014, the ST2 genome has fewer antibiotic resistance genes and associated SNPs. The previous study also showed that only one ST2 strain simultaneously presented phenotypic resistance to tetracycline and MLSB with positivity for tetM and ermB, and ST2, except two strains that were resistant to aminoglycoside-streptothricin, but without any resistance cassettes (Knight et al., 2016). Similarly, our data showed that only two strains carried tetA(P) and tet(K), and two aminoglycoside-streptothricin resistance cassettes [AAC(6′)-1e-APH(2″)-1a and ANT(6)-1b] existed in each of five strains. However, ermB was present in the 11 strains, of which 83.3% (10/12) were from SL2b. The rates of resistance to the third-generation fluoroquinolones were significantly higher than those to the fourth-generation fluoroquinolones. According to in silico antibiotic resistance analysis, the T82I SNP in gyrA was only found in three strains, but cdeA existed in all ST2 strains. Therefore, we predicted that the cdeA gene was the main reason for third-generation fluoroquinolones resistance in C. difficile ST2, and but did not mediate resistance to fourth-generation fluoroquinolones. Based on the MIC data reported here, only 30.6% (11/36) of MLSB resistant isolates contained the ermB or ermC gene, suggesting that these antibiotic resistance genes alone did not always lead to phenotypic resistance, or other alternative antibiotic resistance mechanisms might exist (Knight et al., 2019). We did not find vancomycin-resistant strains in this study despite finding the vanXYG gene in all 182 isolates, demonstrating that in silico vancomycin genotyping was a poor predictor of vancomycin phenotype in C. difficile.
There is still a paucity of antibiotic resistance data for C. difficile ST2 strains in China. In this study, we found all ST2 isolates susceptible to the first-line human CDI therapies vancomycin and metronidazole. The antibiotic resistance pattern of C. difficile ST2 observed in this study differed dramatically from that in our previous studies (Jin et al., 2017). The resistance rate for clindamycin (17/18, 94.4%) was markedly higher than that reported previously, but that for fusidic acid, erythromycin, rifampicin, and tetracycline were lower than in previous studies (Jin et al., 2017). The antibiotic resistance pattern in ST strains observed in this study also differed dramatically from that in a systematic review and meta-analysis in China in 2016 (Tang et al., 2016), in which data on erythromycin, clindamycin, and rifampicin showed higher resistance rates than those in ST2, but our data reported here shows high rates of resistance to fusidic acid and levofloxacin in comparison to the systematic review data (Tang et al., 2016). However, the above review data did not provide detailed information about RTs, thus more data on antibiotic resistance specific to C. difficile ST2 should be investigated in future studies.
Our study had some limitations as described below. Firstly, the number of isolates investigated (n = 40) was low relative to the phylogenetic analysis of ST2 genomes, and the number of strains from other regions or countries is limited. Thus, greater numbers of strains from various regions in China and other countries would enhance the understanding of the phylogenetic analysis of ST2 genomes. Secondly, clinical information and biological features from the other 142 strains were unavailable, including RT types and phenotypic antibiotic resistance. The antibiotic resistance phenotypic-genotypic concordance was still unclear in C. difficile ST2. We are going to collecting more ST2 strains from different provinces in China and other countries in order to confirm the biological features of ST2 strains in SL2b in the near future.
Our study revealed two distinct lineages in C. difficile ST2 genomes with many virulence loci and few antibiotic resistance elements. SL2b was exclusively identified with a sub-lineage-dependent genome mutation (Y1975D) in tcdB, mainly in C. difficile strains from China expressing low toxin B, which might be associated with mild or moderate CDI.
The original contributions presented in the study are publicly available. The genomic data of the 39 isolates sequenced in this study were deposited in the NCBI database under study accession number PRJNA591265. The accession number of C. difficile W0022a is GCF_002812625.1.
The studies involving human participants were reviewed and approved by the Hangzhou Medical College. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
DJ, Y-WT, and XW conceived the study, designed the experiments, and revised the manuscript. XX, QB, and XS collected the samples and performed the experiments. XX, YL, QB, QL, and MW analyzed the data. QB, SL, BZ, and GY performed the statistical analysis. XX, YL, and QB drafted the manuscript. DJ, LC, and Y-WT supervised the study. All authors edited and approved the final version of the manuscript. All corresponding authors had full access to all the data in this study and had final responsibility for the decision to submit for publication.
This work was supported in part by the Program for Major Science and Technology Medicine and Healthcare in Zhejiang (WKJ-ZJ-2107) and by a Key Research and Development Program of Shandong (2019JZZY011018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.651520/full#supplementary-material
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525.
Aubry, A., Hussack, G., Chen, W., KuoLee, R., Twine, S. M., Fulton, K. M., et al. (2012). Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect. Immun. 80, 3521–3532. doi: 10.1128/iai.00224-12
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cairns, M. D., Preston, M. D., Hall, C. L., Gerding, D. N., Hawkey, P. M., Kato, H., et al. (2017). Comparative genome analysis and global phylogeny of the toxin variant clostridium difficile PCR Ribotype 017 reveals the evolution of two independent sublineages. J. Clin. Microbiol. 55, 865–867. doi: 10.1128/jcm.01296-16
Carter, G. P., Rood, J. I., and Lyras, D. (2012). The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol. 20, 21–29. doi: 10.1016/j.tim.2011.11.003
Cheng, J. W., Xiao, M., Kudinha, T., Kong, F., Xu, Z. P., Sun, L. Y., et al. (2016). Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile Isolates from a university teaching hospital in China. Front. Microbiol. 7:1621. doi: 10.3389/fmicb.2016.01621
Clinical and Laboratory Standards Institute (2011). Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria. Approved Standard, Seventh Edn. Wayne, PA: Clinical and Laboratory Standards Institute.
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Didelot, X., Eyre, D. W., Cule, M., Ip, C. L., Ansari, M. A., Griffiths, D., et al. (2012). Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 13:R118.
Dingle, K. E., Didelot, X., Quan, T. P., Eyre, D. W., Stoesser, N., Marwick, C. A., et al. (2019). A role for tetracycline selection in recent evolution of agriculture-associated Clostridium difficile PCR Ribotype 078. mBio 10:e2790-18.
Dingle, K. E., Griffiths, D., Didelot, X., Evans, J., Vaughan, A., Kachrimanidou, M., et al. (2011). Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One 6:e19993. doi: 10.1371/journal.pone.0019993
Eyre, D. W., and Walker, A. S. (2013). Clostridium difficile surveillance: harnessing new technologies to control transmission. Expert Rev. Anti Infect. Ther. 11, 1193–1205. doi: 10.1586/14787210.2013.845987
Gerding, D. N., Johnson, S., Rupnik, M., and Aktories, K. (2014). Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microb. 5, 15–27. doi: 10.4161/gmic.26854
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778.
He, M., Miyajima, F., Roberts, P., Ellison, L., Pickard, D. J., Martin, M. J., et al. (2013). Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 45, 109–113.
Hennequin, C., Porcheray, F., Waligora-Dupriet, A., Collignon, A., Barc, M., Bourlioux, P., et al. (2001). GroEL (Hsp60) of Clostridium difficile is involved in cell adherence. Microbiology (Reading) 147, 87–96. doi: 10.1099/00221287-147-1-87
Indra, A., Huhulescu, S., Schneeweis, M., Hasenberger, P., Kernbichler, S., Fiedler, A., et al. (2008). Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 57, 1377–1382. doi: 10.1099/jmm.0.47714-0
Jin, D., Luo, Y., Huang, C., Cai, J., Ye, J., Zheng, Y., et al. (2017). Molecular epidemiology of clostridium difficile infection in hospitalized patients in Eastern China. J. Clin. Microbiol. 55, 801–810. doi: 10.1128/jcm.01898-16
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9.
Knight, D. R., Elliott, B., Chang, B. J., Perkins, T. T., and Riley, T. V. (2015). Diversity and evolution in the genome of Clostridium difficile. Clin. Microbiol. Rev. 28, 721–741. doi: 10.1128/cmr.00127-14
Knight, D. R., Kullin, B., Androga, G. O., Barbut, F., Eckert, C., Johnson, S., et al. (2019). Evolutionary and genomic insights into Clostridioides difficile Sequence Type 11: a diverse zoonotic and antimicrobial-resistant lineage of global one health importance. mBio 10:e00446-19.
Knight, D. R., Squire, M. M., Collins, D. A., and Riley, T. V. (2016). Genome analysis of Clostridium difficile PCR Ribotype 014 Lineage in Australian pigs and humans reveals a diverse genetic repertoire and signatures of long-range interspecies transmission. Front. Microbiol. 7:2138. doi: 10.3389/fmicb.2016.02138
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12.
Li, P., Li, X., Gu, Q., Lou, X. Y., Zhang, X. M., Song, D. F., et al. (2016). Comparative genomic analysis of Lactobacillus plantarum ZJ316 reveals its genetic adaptation and potential probiotic profiles. J. Zhejiang Univ. Sci. B 17, 569–579. doi: 10.1631/jzus.b1600176
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692.
Luo, Y., Cheong, E., Bian, Q., Collins, D. A., Ye, J., Shin, J. H., et al. (2019). Different molecular characteristics and antimicrobial resistance profiles of Clostridium difficile in the Asia-Pacific region. Emerg. Microbes Infect. 8, 1553–1562. doi: 10.1080/22221751.2019.1682472
Luo, Y., Zhang, W., Cheng, J. W., Xiao, M., Sun, G. R., Guo, C. J., et al. (2018). Molecular epidemiology of Clostridium difficile in two tertiary care hospitals in Shandong Province. China. Infect. Drug Resist. 11, 489–500. doi: 10.2147/idr.s152724
Martin, J. S., Monaghan, T. M., and Wilcox, M. H. (2016). Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 13, 206–216. doi: 10.1038/nrgastro.2016.25
Orth, P., Xiao, L., Hernandez, L. D., Reichert, P., Sheth, P. R., Beaumont, M., et al. (2014). Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 289, 18008–18021. doi: 10.1074/jbc.m114.560748
Péchiné, S., Hennequin, C., Boursier, C., Hoys, S., and Collignon, A. (2013). Immunization using GroEL decreases Clostridium difficile intestinal colonization. PLoS One 8:e81112. doi: 10.1371/journal.pone.0081112
Preston, M. D., Assefa, S. A., Ocholla, H., Sutherland, C. J., Borrmann, S., Nzila, A., et al. (2014). PlasmoView: a web-based resource to visualise global Plasmodium falciparum genomic variation. J. Infect. Dis. 209, 1808–1815. doi: 10.1093/infdis/jit812
Qin, J., Dai, Y., Ma, X., Wang, Y., Gao, Q., Lu, H., et al. (2017). Nosocomial transmission of Clostridium difficile Genotype ST81 in a General Teaching Hospital in China traced by whole genome sequencing. Sci. Rep. 7:9627.
Ryder, A. B., Huang, Y., Li, H., Zheng, M., Wang, X., Stratton, C. W., et al. (2010). Assessment of Clostridium difficile Infections by quantitative detection of tcdB Toxin by use of a real-time cell analysis system. J. Clin. Microbiol. 48, 4129–4134. doi: 10.1128/jcm.01104-10
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Shen, E., Zhu, K., Li, D., Pan, Z., Luo, Y., Bian, Q., et al. (2020). Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun. Biol. 3:347.
Stabler, R. A., He, M., Dawson, L., Martin, M., Valiente, E., Corton, C., et al. (2009). Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102.
Tang, C., Cui, L., Xu, Y., Xie, L., Sun, P., Liu, C., et al. (2016). The incidence and drug resistance of Clostridium difficile infection in Mainland China: a systematic review and meta-analysis. Sci. Rep. 6:37865.
Tian, T. T., Zhao, J. H., Yang, J., Qiang, C. X., Li, Z. R., Chen, J., et al. (2016). Molecular characterization of Clostridium difficile isolates from human subjects and the environment. PLoS One 11:e0151964. doi: 10.1371/journal.pone.0151964
Ye, J., McGinnis, S., and Madden, T. L. (2006). BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9.
Zhang, X., Wang, X., Yang, J., Liu, X., Cai, L., and Zong, Z. (2016). Colonization of toxigenic Clostridium difficile among ICU patients: a prospective study. BMC Infect. Dis. 16:397. doi: 10.1186/s12879-016-1729-2
Keywords: Clostridioides difficile, whole genome sequencing, ST2, tcdB, genomic characteristics
Citation: Xu X, Bian Q, Luo Y, Song X, Lin S, Chen H, Liang Q, Wang M, Ye G, Zhu B, Chen L, Tang Y-W, Wang X and Jin D (2021) Comparative Whole Genome Sequence Analysis and Biological Features of Clostridioides difficile Sequence Type 2‡. Front. Microbiol. 12:651520. doi: 10.3389/fmicb.2021.651520
Received: 20 January 2021; Accepted: 31 May 2021;
Published: 05 July 2021.
Edited by:
Guido Werner, Robert Koch Institute (RKI), GermanyReviewed by:
Jason Sahl, Northern Arizona University, United StatesCopyright © 2021 Xu, Bian, Luo, Song, Lin, Chen, Liang, Wang, Ye, Zhu, Chen, Tang, Wang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dazhi Jin, amluZEBobWMuZWR1LmNu; Xianjun Wang, d2FuZ3hqMDUyNUAxMjYuY29t
†These authors have contributed equally to this work
‡This study was presented in part at the ASM Microbe 2019 (2019).
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.