 James S. Griffiths*
James S. Griffiths* Giorgio Camilli
Giorgio Camilli Natalia K. Kotowicz
Natalia K. Kotowicz Jemima Ho
Jemima Ho Jonathan P. Richardson
Jonathan P. Richardson Julian R. Naglik
Julian R. Naglik
- Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral and Craniofacial Sciences, King’s College London, London, United Kingdom
Fungal pathogens kill approximately 1.5 million individuals per year and represent a severe disease burden worldwide. It is estimated over 150 million people have serious fungal disease such as recurrent mucosal infections or life-threatening systemic infections. Disease can ensue from commensal fungi or new infection and involves different fungal morphologies and the expression of virulence factors. Therefore, anti-fungal immunity is complex and requires coordination between multiple facets of the immune system. IL-1 family cytokines are associated with acute and chronic inflammation and are essential for the innate response to infection. Recent research indicates IL-1 cytokines play a key role mediating immunity against different fungal infections. During mucosal disease, IL-1R and IL-36R are required for neutrophil recruitment and protective Th17 responses, but function through different mechanisms. During systemic disease, IL-18 drives protective Th1 responses, while IL-33 promotes Th2 and suppresses Th1 immunity. The IL-1 family represents an attractive anti-fungal immunotherapy target. There is a need for novel anti-fungal therapeutics, as current therapies are ineffective, toxic and encounter resistance, and no anti-fungal vaccine exists. Furthering our understanding of the IL-1 family cytokines and their complex role during fungal infection may aid the development of novel therapies. As such, this review will discuss the role for IL-1 family cytokines in fungal infections.
Introduction
Fungal pathogens represent an increasingly severe disease burden and are responsible for ∼1.5 million deaths per year. Patients who are immunocompromised, have undergone invasive clinical procedures or suffered trauma are particularly susceptible to fungal infection. Fungi can be frequently encountered, such as Aspergillus through inhalation or Candida, which colonizes mucosal barriers (Brown et al., 2012). Anti-fungal responses must strike a careful balance to provide protection and maintain homeostasis. Regularly encountered fungal pathogens must be cleared with minimal effect on the host, while commensal fungi must be maintained without reducing barrier integrity. The majority of serious fungal disease arises from poorly cleared infection or disrupted barrier integrity (Rautemaa-Richardson and Richardson, 2017). Here, the IL-1 family play a crucial role mediating both barrier and systemic anti-fungal immunity. As such, modulating IL-1 family cytokines to enhance anti-fungal immunity may provide valuable therapeutic strategies that overcome current therapeutic inadequacies.
The IL-1 family possess numerous potent biological activities and mediate a wide range of immunological responses (Garlanda et al., 2013). IL-1 was identified in the 1980’s but had been investigated for many years under various aliases. The discovery of IL-1 was initially met with skepticism that a molecule at such low concentration could have potent, systemic effect. Since then, our understanding of the IL-1 family has grown and now comprises four sub-families containing eleven signaling members, five primary receptors and six co-receptors (Figure 1). Of the eleven signaling members, seven are pro-inflammatory and four are anti-inflammatory (Table 1). The signaling members of the IL-1 family share a highly conserved gene sequence and structure and (except for IL-18 and IL-33) are clustered on human chromosome 2 (Sims and Smith, 2010). Due to these similarities, the genomic identification of IL-1 family members largely preceded the discovery of their function. Aside from IL-1 receptor antagonist (IL-1Ra), all other IL-1 family cytokines lack a secretion signal peptide and either require cleaving and activation or are active in their precursor form. Signaling typically occurs when cytokines bind their primary receptor and recruit a co-receptor, which induces signaling through the Toll/interleukin 1 receptor (TIR) domain resulting in mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) activation (Fields et al., 2019). In this review, we describe each IL-1 subfamily (IL-1, IL-18, and IL-36) and investigate the mechanism of induction and functional role of each subfamily member within the context of fungal disease.
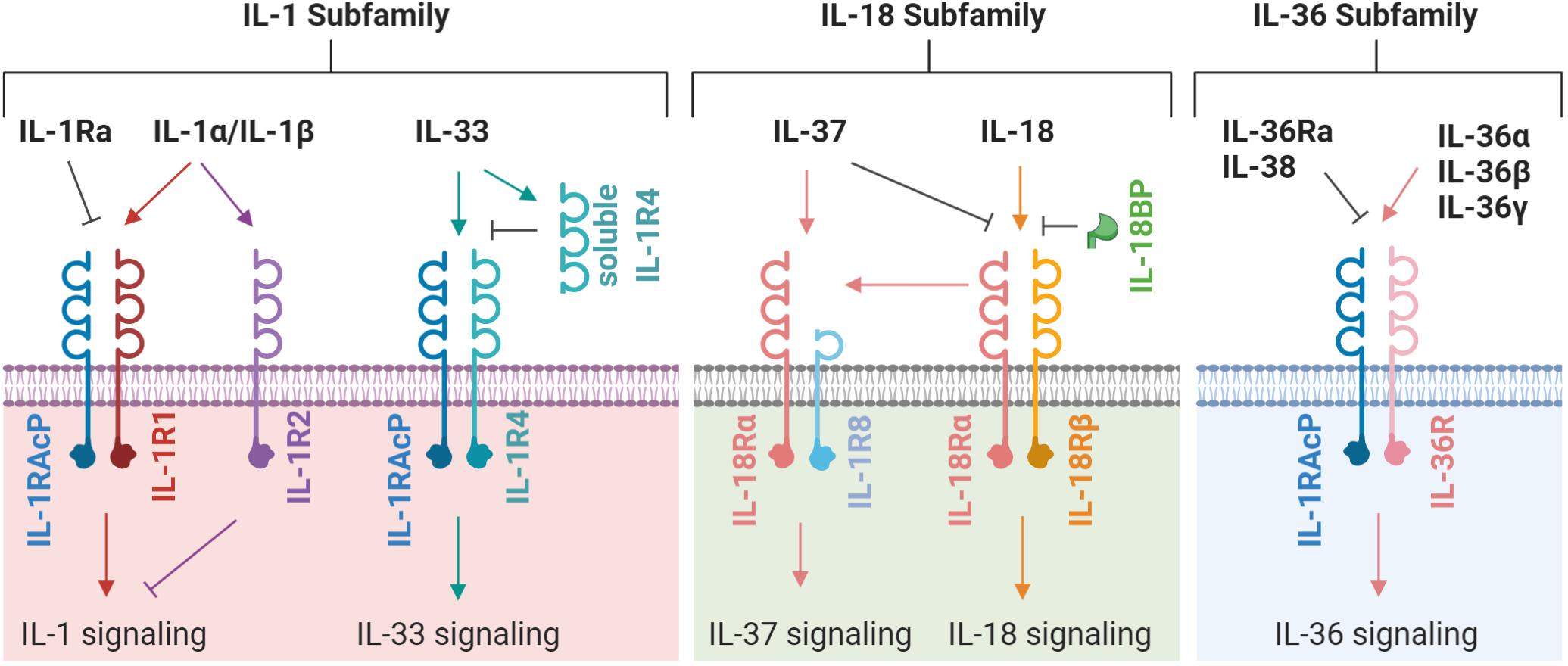
Figure 1. A schematic representation of IL-1 family receptors, co-receptors, and ligands. Each ligand, receptor and co-receptor are separated into their appropriate IL-1 subfamily. An arrow indicates the induction of signaling; a flat line indicates the blocking of signaling. The latest nomenclature is used, for previous/alternative nomenclature see Table 1.
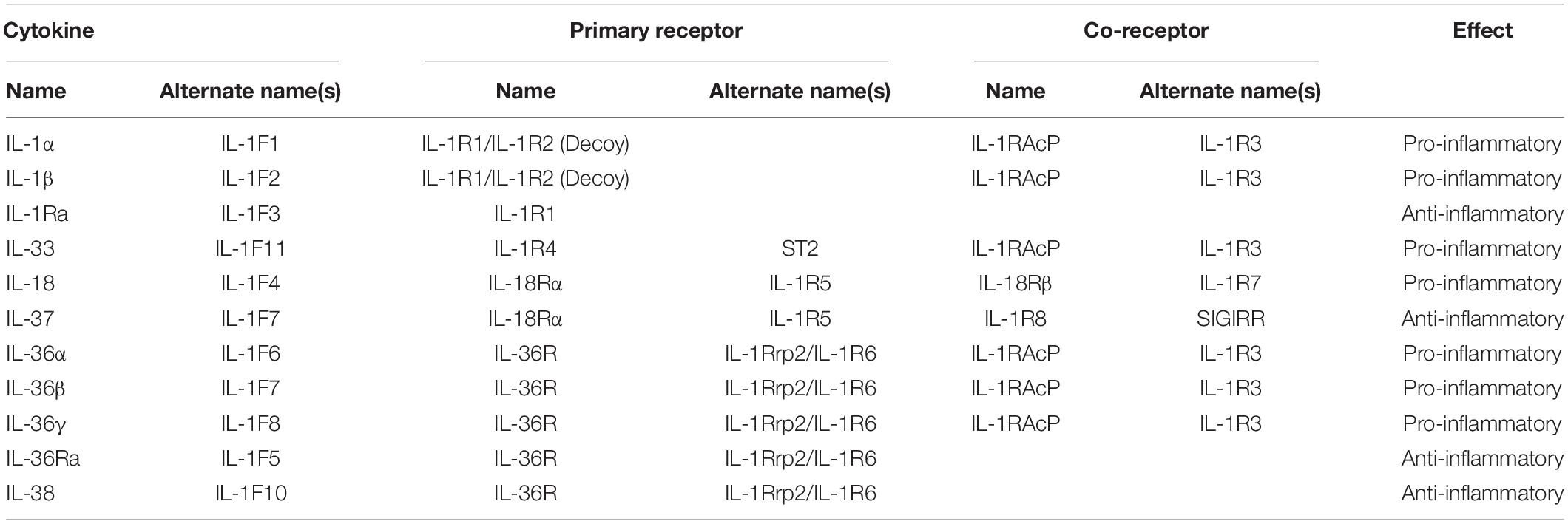
Table 1. Nomenclature and function of IL-1 family members.
The IL-1 Subfamily
Interleukin-1 was the first interleukin to be identified and represented a huge leap forward in immunology. The IL-1 gene cluster encodes the pro-inflammatory cytokines IL-1α and IL-1β, and anti-inflammatory receptor antagonist IL-1Ra (Garlanda et al., 2013). IL-1α/β bind their receptor IL-1R1, which recruits IL-1RAcP and permits signaling through both receptor’s TIR domain (Fields et al., 2019). This domain is well conserved throughout IL-1 and Toll-like receptor (TLR) signaling where it drives inflammation (Heguy et al., 1992). IL-1α/β are recognized as major inflammatory cytokines that mediate innate and adaptive immunity, and also general health. IL-33, a recent addition to the IL-1 subfamily, was discovered in 2005 and signals through its receptor IL-1R4 (formerly ST2) in complex with IL-1RAcP. IL-33 has many functional capabilities which influence barrier integrity and inflammation (Mehraj et al., 2016). IL-33 signaling has been implicated in numerous allergic-type diseases and, along with IL-1α/β, has recently been shown as a mediator of adaptive immunity (Kamijo et al., 2013).
IL-1 Subfamily Expression and Processing
Although IL-1α and IL-1β signal through the same receptor and have similar biological activities, these two pro-inflammatory cytokines differ in several aspects. IL-1α is constitutively expressed in epithelial and mesenchymal cell types and expression increases in response to growth factors, inflammation or stress-associated stimuli (Di Paolo and Shayakhmetov, 2016). Here, IL-1α is released from cells during damage or necrosis (Chen et al., 2007). IL-1α also possesses a nuclear localization signal and can interact with histone acetyltransferase complexes to mediate transcription of cytokines including IL-6 and IL-8 (Werman et al., 2004). However, nuclear translocation of IL-1α during apoptosis inactivates IL-1α signaling, likely sequestering it and blocking inflammatory effects (Cohen et al., 2010). IL-1α also functions as an active membrane bound precursor promoting inflammation through IL-1R1 binding. Membrane-associated IL-1α is present on the surface of numerous immune cells including macrophages (Kurt-Jones et al., 1985), monocytes, and B lymphocytes (Zola et al., 1993).
Interleukin-1β is mainly produced by mononuclear phagocytes as an inactive precursor and activated via a two-step process. During the initial priming step, pro-IL-1β is induced by the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRR) (Takeuchi and Akira, 2010). Activation requires pro-IL-1β cleavage by the intracellular cysteine protease caspase-1 and is regulated by inflammasomes (Franchi et al., 2009). Although caspase-1 is the main protease responsible for pro-IL-1β activation, Fas stimulation is also able to induce secretion of biologically active IL-1β from caspase-1-deficient murine macrophages (Bossaller et al., 2012). Once activated, IL-1β is secreted through one or more non-conventional secretory pathways (Lopez-Castejon and Brough, 2011). Here, another role for caspase-1 has been described in that it can cleave the pore forming toxin gasdermin D, which induces pore formation in the plasma membrane, and pyroptosis to enhance IL-1β release (Heilig et al., 2018). Importantly, the release of pro-IL-1β upon membrane disruption has functional consequences. Multiple proteases, mainly derived from neutrophils and mast cells, can cleave and activate pro-IL-1β in the extracellular environment which drives inflammation (Stehlik, 2009).
Interleukin-33 is constitutively expressed in multiple cell types but is mainly found in fibroblasts, epithelial and endothelial cells (Moussion et al., 2008), with expression further increasing during inflammation (Liew et al., 2016). In macrophages, IL-33 induction was dependent on glutaredoxin-1/TRAF6 and NF-κB signaling (Weinberg et al., 2019). Similar to IL-1α, IL-33 has dual functions acting as a transcriptional repressor of NF-κB following nuclear localization (Ali et al., 2011) and enhancing inflammation after being released from damaged or necrotizing cells (Moussion et al., 2008). Again like IL-1α, IL-33 is also sequestered during apoptosis (Bessa et al., 2014). Interestingly, caspase-1 IL-33 cleavage attenuated inflammation (Luthi et al., 2009), while neutrophil proteases enhanced biological activity (Lefrancais et al., 2012). Following the release of IL-1α and IL-33, and the release and activation of IL-1β, the cytokines drive potent immunological functions.
The Immunological Function of the IL-1 Subfamily
The first discovered IL-1 cytokine was named hemopoietin-1 after its signaling drove myeloid “emergency” responses (Pietras et al., 2016). Since then, the sub-family has expanded and IL-1α/β are now known to promote myelopoiesis and inflammation, lead to the release of antimicrobial compounds, and mediate immunity (Dinarello, 2018). Following the release of IL-1α and active IL-1β, the cytokines bind their receptor IL-1R1 and drive MAPK and NF-κB signaling through the myeloid differentiation primary response (MyD88) adaptor protein (Cohen, 2014). Two mechanisms antagonize the effects of IL-1α/β. Firstly, IL-1Ra binds IL-1R1 to prevent IL-1α/β-IL-1R1 interactions. The importance of IL-1Ra is highlighted in IL-1Ra deficient mice, which spontaneously develop Th17-associated rheumatoid arthritis (Koenders et al., 2008). Secondly, IL-1R2 acts as a decoy receptor and lacks a TIR domain, therefore blocking downstream signaling. Overexpressing IL-1R2 in mice reduced inflammation in numerous IL-1-induced inflammatory diseases (Peters et al., 2013).
While IL-1α/β have central roles in driving inflammation and mediating immune responses, each cytokine possesses distinct function. IL-1α acts as an alarmin and is the principal trigger of inflammation following cell membrane disruption or cellular necrosis. In this context, constitutively expressed IL-1α is released from cells and drives neutrophil recruitment and local inflammation (Chen et al., 2007; Eigenbrod et al., 2008). IL-1β signaling promotes the recruitment of monocytes, macrophages, and neutrophils; enhanced phagocytosis and killing; increased reactive oxygen species/nitrogen oxide synthase (ROS/NOS) production; and Th1 and Th17 immunity (Garlanda et al., 2013; Altmeier et al., 2016; Dinarello, 2018). Mice deficient in caspase-1 were protected from Th1 and Th17-associated experimental autoimmune encephalomyelitis (Gris et al., 2010) and mice deficient in IL-1Ra developed spontaneous Th17-associated autoimmunity (Horai et al., 2000). IL-1 is also associated with numerous autoinflammatory diseases, with blockade of IL-1 signaling rapidly improving symptoms (Dinarello et al., 2012). Interestingly, even when the autoimmune disease is primarily tumor necrosis factor (TNF)/IL-6 driven (as in rheumatoid arthritis), blocking IL-1β signaling reduced disease severity (Dinarello, 2011). This highlights the central role of IL-1α/β mediating immunity and health.
Interleukin-33 acts in a similar fashion to IL-1α, being released from barrier cells and functioning as a damage-associated molecular pattern (DAMP). Here, IL-33 activates T-cells directly through PRR/IL-1R4 binding or indirectly through local inflammatory responses. Immune cells such as mast cells, basophils, dendritic cells, macrophages, natural killer cells, and Th2 cells are receptive to IL-33 and express IL-1R4 (Martin and Martin, 2016). The soluble form of IL-1R4 sequesters “off-target” IL-33 and antagonizes IL-33 activity (Kakkar and Lee, 2008). IL-33 was originally identified as driving allergic and anti-helminthic immunity but is also a key mediator of adaptive immunity. During acute infection, IL-33 promotes tissue remodeling and drives Th1, Th2, and T regulatory (Treg) responses. In contrast, during chronic infection, IL-33 enhanced local inflammation, tissue damage and fibrosis (Li et al., 2014). IL-33 signaling has been implicated in numerous diseases associated with exacerbated inflammation including allergic-asthma (Salter et al., 2016), Crohn’s disease (Pastorelli et al., 2010), and rheumatoid arthritis (Tang et al., 2013). While the broad and potent consequences of IL-1α/β and IL-33 signaling are known, the induction of these cytokines during fungal infection is less clear.
Fungal Induction of the IL-1 Subfamily
Numerous fungal pathogens infect mucosal barrier sites and systemic disease often results from a loss in barrier integrity. It is therefore unsurprising that IL-1α/β and IL-33 have been implicated in anti-fungal immunity. All three cytokines are induced following Aspergillus fumigatus infection, a pathogen that typically infects mucosal surfaces. IL-1α expression in the lung is positively correlated with A. fumigatus strain virulence (Caffrey-Carr et al., 2017). This is likely a result of conidial germination and subsequent damage to mucosal barriers. IL-1β was rapidly induced and sustained for 2 days in an A. fumigatus keratitis model. Here, IL-1β induction was dependent on Dectin-1 signaling and c-Jun N-terminal kinase (JNK) phosphorylation (Yuan et al., 2017). Numerous PRRs that induce IL-1β signaling, including C-type lectin-like receptors (CLRs) and TLRs, are involved during A. fumigatus infection (Steele et al., 2005). The requirement of CLRs for IL-33 induction appears more complex. During acute A. fumigatus infection the induction of IL-33 occurred independently of Dectin-1 (Garth et al., 2017), while induction during chronic allergic-type infection required Dectin-1 (Lilly et al., 2012).
Candida albicans infection also results in IL-1α/β and IL-33 induction. IL-1α induction has been described in oral and vaginal epithelial cells (Steele and Fidel, 2002) and required membrane disruption and Ca2+ influx (Gross et al., 2012). Here, the peptide toxin candidalysin, secreted when C. albicans forms hyphae, disrupts host membranes resulting in IL-1α release (Moyes et al., 2016). IL-33 is also induced during C. albicans infection at barrier sites (Le et al., 2012) and likely requires candidalysin expression in a similar manner to IL-1α. The induction of IL-1β in monocytes was only achieved with live Candida and does not require CLR involvement (Castro et al., 1996). In contrast, dendritic cells (that require two signals for IL-1β induction and activation) were dependent on spleen tyrosine kinase (Syk) signaling for IL-1β induction during C. albicans infection (Gross et al., 2009). In macrophages, IL-1β is induced by C. albicans, C. tropicalis, and C. krusei, although the requirement for CLRs is disputed (Joly et al., 2009; Kasper et al., 2018). As such, there are several mechanisms by which Candida induce inflammasome activation and IL-1β release (Camilli et al., 2020). One particularly interesting mechanism involves the non-tyrosine kinase Tec which was found to activate non-canonical caspase-8 exclusively following fungal challenge (Zwolanek et al., 2014). IL-1α/β induction during mucosal C. albicans infection occurs through NF-κB and a biphasic MAPK response. Activation of NF-κB and the first MAPK c-Jun phase was dependent on fungal PAMP recognition, while the second MAPK MKP1/c-Fos phase was dependent on hyphae formation and fungal burden. This identifies an interesting mechanism by which the host may detect the switch from commensalism to pathogenicity (Moyes et al., 2010). A similar biphasic recognition mechanism has not yet been described for other fungal infections, although such a mechanism may also differentiate between A. fumigatus conidia and hyphae during infection.
Interleukin-1 subfamily members are also induced by Cryptococcus neoformans, Paracoccidioides brasiliensis, and Paracoccidioides lutzii [the causative agents of paracoccidioidomycosis (PCM)], and Sporothrix schenckii (the causative agent of sporotrichosis). IL-1α/β and IL-33 are induced in both mucosal and systemic compartments during C. neoformans infection (Flaczyk et al., 2013; Alvarez et al., 2019). During P. brasiliensis challenge, IL-1β was induced in human monocytes, macrophages and plasmacytoid dendritic cells (pDC). As was found with Candida, CLRs and Syk signaling were required for pDC IL-1β induction (Kurokawa et al., 2007; Preite et al., 2018). IL-1β release from macrophages was caspase-1 dependent; however, a non-canonical IL-1β processing pathway requiring Dectin-1 and caspase-8 activity has been described (Ketelut-Carneiro et al., 2018). P. brasiliensis challenge also induced IL-1α release through a macrophage-derived IFN-β/procaspase-11 pathway which enhanced pore-mediated cell lysis (Ketelut-Carneiro et al., 2019). Patients with severe PCM possessed high levels of IL-1β, IL-33, and IL-1R4 in serum which decreased following anti-fungal therapy (Silva et al., 1995; Alves et al., 2018). Recently, IL-1β has been implicated in S. schenckii infection. Here, IL-1β was induced in a caspase-1 dependent manner with increased IL-1β correlating with higher fungal burden (Goncalves et al., 2015). While it is clear the IL-1 subfamily is induced by fungal pathogens and contributes to fungal immunity, the sequence and mechanism of IL-1 subfamily induction during fungal challenge is still unclear.
The Role of the IL-1 Subfamily in Fungal Immunology
Interleukin-1α/β drive crucial immune mechanisms and are central mediators of immunity. Following their induction during fungal infection, both IL-1α/β play a key role orchestrating the immune response. While IL-33 is induced during fungal disease, its functional role is not yet fully delineated. Much of our understanding of the IL-1 subfamily during fungal disease has been revealed using mice deficient in IL-1 components. Mice lacking IL-1R1 exhibit reduced neutrophil recruitment and emergency granulopoieses following oropharyngeal candidiasis (OPC) challenge. Here, both IL-1β from hematopoietic cells and IL-1α from non-hematopoietic cells promoted neutrophil recruitment (Altmeier et al., 2016). However, neutrophil recruitment is not always protective, since in vulvovaginal candidiasis (VVC) IL-1α promotes damaging inflammation and neutrophil recruitment (Barousse et al., 2004). In agreement, mutations increasing Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activity are associated with VVC in patients (Jaeger et al., 2016; Roselletti et al., 2017).
Mice deficient in IL-1α or IL-1β were more susceptible to systemic C. albicans infection, with mice lacking both IL-1α/β possessing the highest fungal burdens and lowest survival. Interestingly, IL-1α/β deficient mice continued to possess higher fungal burdens in a neutropenic model of systemic C. albicans infection, suggesting the role of IL-1α/β is not just in neutrophil recruitment. Instead, IL-1α was found to activate macrophages and IL-1β enhanced neutrophil killing, and both IL-1α and IL-1β were involved in the development of IFN-γ Th1 and IL-10 Th2 responses (Vonk et al., 2006). A role for epithelial and hematopoietic IL-1α/β has also been described to promote essential Th17 immunity during mucosal Candida infection (Bishu et al., 2014; Verma et al., 2017, 2018). In patients, mutations in caspase recruitment domain-containing protein 9 (CARD9) or TLR1 (pathways that lead to IL-1β induction) resulted in defective Th17 responses and increased Candida susceptibility (Glocker et al., 2009; Plantinga et al., 2012). Th17 immunity is also vital during Coccidioides infection. Here, Th17 responses were dependent on CARD9 and IL-1R1, with IL-1R1 deficient mice lacking Th17 immunity and being highly susceptible to C. immitis infection (Hung et al., 2014; Viriyakosol et al., 2018). IL-1R1 deficient mice are highly susceptible to C. neoformans infection; however, susceptibility was associated with the induction of harmful Th2 immunity (Shourian et al., 2017).
Both IL-1α and IL-1β drive protective immunity during PCM. In vivo models of P. brasiliensis infection determined mice with impaired IL-1β responses had higher fungal burden and a dysregulated inflammatory response. IL-1α promoted local inflammatory responses, nitric oxide production and Th17 immunity (Ketelut-Carneiro et al., 2018, 2019). Whilst signaling through IL-1R1 is required for protective immunity against pulmonary A. fumigatus infection; unlike Candida and P. brasiliensis infection, only IL-1α was required. Here, IL-1α promoted neutrophil and macrophage recruitment through monocyte-induced CXCL1 signaling. Administration of CXCL1 partially restored neutrophil recruitment in IL-1R1 deficient mice (Caffrey et al., 2015), while mice lacking inflammasome components, and thus IL-1β activation, cleared A. fumigatus infection (Caffrey et al., 2015). However, IL-1β may play a key role during systemically disseminated Aspergillus infection, as was described with systemic Candida infection (Vonk et al., 2006). The Dectin-1 Y238X polymorphism, which results in diminished Dectin-1 activity and reduced IL-1β responses, significantly enhanced patient’s Aspergillus and Candida susceptibility (Plantinga et al., 2009; Cunha et al., 2010).
While the potent effects of IL-1α/β are clear during fungal disease, the effect of IL-1Ra and IL-1R2 are less well explored. Patients with severe fungal-sensitized asthma have higher levels of IL-1α/β and IL-1Ra, indicating the antagonist is involved. In an animal model of fungal-sensitized asthma, a lack of IL-1Ra enhanced Th1 and Th17 immunity, increased neutrophil recruitment and exacerbated disease. Treatment with recombinant IL-1Ra (Anakinra) reduced these responses and resolved disease. In the same model, IL-1R1 deficient mice displayed improved lung function, suggesting IL-1α/β enhance chronic disease (Godwin et al., 2019). IL-1Ra also has a protective role in VVC by restraining activation of damage-enhancing NLRP3 through an IL-22/NLR family CARD domain-containing protein 4 (NLRC4)/IL-1Ra pathway. Here, mice deficient in IL-1Ra displayed enhanced disease that could be rescued with the administration of Anakinra (Borghi et al., 2015). While the role of IL-1Ra and IL-1R2 in balancing IL-1 agonist activity during fungal disease requires further investigation, early indications suggest IL-1Ra and IL-1R2 play important roles in mediating and resolving potentially damaging inflammation during acute and chronic disease.
The role of IL-33 signaling is now being explored in the context of fungal infection. During C. neoformans infection, IL-1α/β drives protective Th17 immunity and reduces Treg responses, while IL-33 promotes the suppressive function of Treg cells (Alvarez et al., 2019) and enhances IL-5 and IL-13-derived Th2 immunity. This IL-33-Th2 response resulted in increased fungal burdens and reduced survival in murine models (Flaczyk et al., 2013). A deleterious role for IL-33 has also been described during acute and chronic A. fumigatus disease. Mice deficient in IL-33 produced more IL-17A and IL-22 and displayed enhanced fungal clearance when challenged with acute A. fumigatus infection. Furthermore, blocking IL-1R4 in a model of Aspergillus-sensitized asthma improved airway hyperresponsiveness and fibrosis (Ramaprakash et al., 2011). In contrast, pre-treatment of mice with IL-33 prior to peritoneal and systemic C. albicans infection resulted in fungal clearance and improved survival. Here, IL-33 enhanced the recruitment of neutrophils and increased their killing capability while also mediating T-cell tolerance (Le et al., 2012; Tran et al., 2015).
Therapeutic Potential of the IL-1 Subfamily
The activity of IL-1α, IL-1β, and IL-33 must be carefully regulated. Protection from pathogen-derived and autoinflammatory diseases likely involves a balance of IL-1 subfamily agonist and antagonist activity. Excessive IL-1/Th17 signaling results in numerous diseases including asthma, chronic obstructive pulmonary disease (Gurczynski and Moore, 2018), inflammatory bowel disease (IBD), and psoriasis (Beringer et al., 2016). As such, the IL-1/Th17 pathway has been therapeutically targeted. Anakinra blocks IL-1α/β activity and is used as a therapy in multiple inflammatory diseases (Dinarello et al., 2012). Similarly, targeting IL-1 signaling components is known to reduce inflammation and pathology in fungal-induced respiratory disease (Gresnigt et al., 2016; Griffiths et al., 2018; Godwin et al., 2019). Alongside Anakinra, numerous other IL-1 therapeutic strategies have been investigated. Examples include EBI-005, an IL-1R1 antagonist (Hou et al., 2013), Rilonacept (trade name Arcalyst), a decoy IL-1R1 (Economides et al., 2003), SL1067, a DNA aptamer that disrupts IL-1α (Ren et al., 2017), AF10847, a peptide inhibitor of IL-1R1 (Vigers et al., 2000), and AMG108, an antibody inhibitor of IL-1R1 (Cohen et al., 2011). IL-33 also has therapeutic potential and was effective in supporting immune defenses prior to C. albicans infection (Tran et al., 2015). The exogenous administration of IL-1 sub-family agonists or antagonists has potential to enhance protective anti-fungal immunity or resolve excessive, damaging immune responses. Another therapeutic route may involve the modulation of receptors or proteins that regulate IL-1 sub-family signaling. A clear link between CLRs and IL-1α/β signaling exists, as evidenced by CARD9-deficient patient’s reduced IL-1α/β expression and susceptibility to fungal infection (Drummond et al., 2015). Furthermore, Dectin-1-induced IL-1β protected against mucosal Candida infections becoming systemic and lethal (Hise et al., 2009). In summary, modulating mediators of IL-1 sub-family induction, translation and processing may provide novel therapeutic targets in the fight against fungal infections.
The IL-18 Subfamily
Interleukin-18 was first described in 1989 as IFN-γ inducing factor before it was re-named when its function was described as pro-inflammatory (Dinarello, 2018). IL-18 and IL-1β share similar structures and signaling pathways, and are produced as inactive precursors requiring caspase-1 activation; however, the two cytokines are functionally distinct (Dinarello, 2019). IL-18 forms a low affinity signaling complex by binding IL-18Rα, the ligand for mature IL-18. A high affinity signaling complex is formed in cells that also express the co-receptor IL-18Rβ (Kaplanski, 2018). Similar to IL-1α/β, signal transduction requires the TIR domain and drives NF-κB and MAPK signaling (Hoshino et al., 1999; Wyman et al., 2002). IL-18 is regulated by IL-18 binding protein (IL-18BP), a soluble protein with high affinity for IL-18 (Krumm et al., 2014), and IL-37 which binds IL-18Rα and inhibits recruitment of IL-18Rβ. Moreover, IL-37 binding of IL-18Rα induces recruitment of IL-1R8 (also called SIGIRR), which induces anti-inflammatory signaling (Nakanishi, 2018).
IL-18 Subfamily Expression and Processing
Interleukin-18 is a pleiotropic cytokine that plays a central role in immunity (Novick et al., 2013). Like IL-1β, IL-18 is produced as an inactive precursor and must be processed by the inflammasome/caspase-1 complex in order to be activated and secreted (Franchi et al., 2009). However, unlike IL-1β, IL-18 is constitutively expressed by several hemopoietic and non-hemopoietic cells, including macrophages, dendritic cells, Kupffer cells, keratinocytes, osteoblasts, adrenal cortex cells, intestinal epithelial cells, microglial cells, and synovial fibroblasts (Gracie et al., 2003). Fas stimulation was able to induce secretion of biologically active IL-18 in caspase-1-deficient murine macrophages (Bossaller et al., 2012). In addition, proteases secreted by the inflammatory cell infiltrate contribute to inflammation via activation of IL-18 (Omoto et al., 2006, 2010). IL-18 can be secreted from monocytes and macrophages in its active form (Fantuzzi et al., 1999), or released from dying endothelial and epithelial cells in its precursor form before being activated outside the cell (Sugawara et al., 2001). IL-37 acts as an IL-18 antagonist and is found in many human tissues (Dinarello et al., 2016), although more specific roles for IL-37 reducing inflammation in the lung, spleen, plasma and in dendritic cells have been described (Nold et al., 2010). IL-37 is similar to IL-1α and IL-33, functioning as a dual-action cytokine that does not require activation (Li et al., 2015), however, caspase-1 processing was required for IL-37 to translocate to the nucleus to modulate transcription. IL-37 can be released in a processed or precursor form, resulting from exogenous administration of ATP (Bulau et al., 2014). IL-18BP also acts as an IL-18 antagonist and contains a signal peptide that directs secretion into the extracellular environment (Novick et al., 2001).
The Immunological Function of the IL-18 Subfamily
Following induction and activation, IL-18 signaling drives inflammation and promotes innate and adaptive immunity. Accordingly, IL-18 signaling can direct robust anti-pathogen immunity but is also associated with numerous autoinflammatory diseases. A clear role for IL-18 promoting Th1 immunity through IFN-γ induction has been described during models of viral and bacterial infection. In addition, IL-18BP is targeted by viruses to reduce host-protective IL-18 mediated immunity (Born et al., 2000; Reading and Smith, 2003). Mice deficient in IL-18 components were highly susceptible to viral and bacterial challenge (Mastroeni et al., 1999; Kinjo et al., 2002; Freeman et al., 2015). However, IL-18 can only induce IFN-γ and Th1 immunity in combination with IL-12 or IL-15 signaling; IL-18 signaling alone enhances Th2 immunity (Nakanishi et al., 2001), which is typically detrimental for fungal clearance. An IL-18 synergy primarily with IL-12 but also IL-15 exists with each cytokine modulating the others transcription, expression, and receptor expression (Fantuzzi et al., 1999). Recently, IL-18 has also been shown to drive IL-17 production from γδ T-cells and promote Th17 responses (Sutton et al., 2012), modulate adhesion molecules in endothelium (Carrascal et al., 2003), promote nitric oxide (NO) synthesis (critical for viral and bacterial killing) and enhance production of numerous chemokines (Kaplanski, 2018). IL-18 contributes to autoimmune disease including Type-1 diabetes, psoriasis, IBD, asthma and numerous myocardial and kidney diseases (Nakanishi et al., 2001; Garlanda et al., 2013). Here, increased expression of IL-18 and IL-18BP has been observed suggesting that poor agonist/antagonist balance may result in disease. In agreement, the majority of IL-18 associated autoimmune diseases result from excess Th2 immune responses (Monteleone et al., 1999; Tanaka et al., 2001; Gerdes et al., 2002), potentially from IL-18 activity without IL-12 or IL-15.
Interleukin-37 has a broad anti-inflammatory effect limiting IL-18 responses. Reducing IL-37 in human cells increased the production of cytokines (IL-1β/TNF/IL-6) induced by IL-1 and TLRs (Nold et al., 2010). IL-37 is the only member of the IL-1 family not to have a mouse homolog, as such in vivo work is only possible with transgenic IL-37 models. Here, the anti-inflammatory effects of IL-37 have been demonstrated to reduce colitis, metabolic syndrome, acute lung injury, myocardial infarction, and asthma (Wu et al., 2014; Li et al., 2015). IL-18BP also acts to reduce IL-18 agonist activity and an imbalance of IL-18 and IL-18BP has been described in Wegener’s granulomatosis and systemic lupus erythematosus (Novick et al., 2009, 2010). Administration of IL-18BP reduced inflammation in a model of rheumatoid arthritis; however, at high concentrations IL-18BP also bound IL-37 and the anti-inflammatory effect was lost (Banda et al., 2003). The functional implications of IL-18 signaling driving inflammation, innate and adaptive immune responses in autoimmune and anti-pathogen disease are clear. However, the induction, function, and balance of the IL-18 subfamily during fungal disease is less well explored.
Fungal Induction of the IL-18 Subfamily
Interleukin-18 is a crucial cytokine that mediates innate and adaptive immunity and likely plays a key role during fungal infection. The cytokine is expressed in cells of mesenchymal origin and hematopoietic cells and therefore may share functions with both IL-1α and IL-1β. In agreement, the induction of IL-18 has been observed during systemic and mucosal fungal infections. IL-18 was induced following acute Aspergillus challenge and during a chronic model of fungal-sensitized asthma (Cenci et al., 1997, 1998). A recent study revealed IL-18 expression increased rapidly following Aspergillus challenge peaking at 24 h before resolving over the next 48 h (Cheng et al., 2020). However, the signaling events leading to IL-18 induction in these models has not been defined. Aspergillus conidia frequently interact with barrier surfaces and systemic disease arises from germination in this setting. Understanding the induction of IL-18 as Aspergillus conidia persist, germinate, and promote disease may lead to important findings.
Interleukin-18 is also induced during Candida infection. Oral epithelial cells constitutively expressed IL-18 mRNA and precursor IL-18. During C. albicans challenge expression of IL-18 mRNA and pre-IL-18 was reduced while active IL-18 was released in a caspase-1 dependent manner (Rouabhia et al., 2002). A similar effect was seen in a model of human oral mucosa where C. albicans challenge resulted in active IL-18 expression. In agreement, patients with oral candidiasis possess increased levels of active IL-18 in saliva samples (Tardif et al., 2004). There is limited evidence describing the signaling events required for IL-18 induction in fungal disease. Recent investigation determined that Dectin-1 signaling and activation of the non-canonical NF-κB subunit RelB resulted in IL-18 induction (Shen et al., 2020); however, little else is known. Investigating the mechanism of IL-18 induction during mucosal and systemic Candida disease may provide insight into protective anti-Candida immunity. Aside from the induction of IL-18 during Aspergillus and Candida disease, IL-18 has been functionally implicated in P. brasiliensis infection (Panagio et al., 2008; Alves et al., 2018), S. schenckii infection (Goncalves et al., 2015), and C. neoformans infection (Kawakami et al., 2000b; Wang et al., 2011), suggesting at the very least a general role in host immune response. Whether IL-37 and IL-18BP are induced during fungal disease currently unclear; however, increased serum IL-37 was identified in PCM patients with severe disease (Alves et al., 2018).
The Role of the IL-18 Subfamily in Fungal Immunology
While the induction of IL-18 requires further investigation, the functional consequences of IL-18 signaling during fungal disease highlight the importance of this IL-1 subfamily member. During acute Aspergillus lung infection, IL-18 promoted protective immunity and enhanced Th1 immunity and neutrophil recruitment in concert with IL-12 and IFN-γ (Blease et al., 2001). Skewing toward Th2 immunity during acute Aspergillus infection is non-protective and promotes chronic disease (Cenci et al., 1998). A study of acute Aspergillus infection determined 72 h after infection IL-18 mediates protection independently of IFN-γ, suggesting that the IL-18/IFN-γ axis occurs rapidly and IL-18 continues to mediate immunity independently of IFN-γ if the infection persists (Brieland et al., 2001). However, in immunocompromised models, IFN-γ was essential throughout infection and exogenous administration of IFN-γ was consistently protective (Nagai et al., 1995; Cenci et al., 1997, 1998). In an Aspergillus-sensitized asthma model, IL-18 was again found to be protective and acted without IL-12 or IFN-γ to enhance Aspergillus clearance. Depleting IL-18 in this model increased fungal burden and resulted in persistent airway hyperactivity and fibrosis (Blease et al., 2001). These results suggest IL-18 is protective during both acute and chronic Aspergillus disease but provides protection through distinct mechanisms.
Interleukin-18 is also vital for protection during Candida infection. Similar to Aspergillus infection, IL-18 induced protective Th1 immunity against Candida. Mice deficient in caspase-1 displayed reduced Th1 responses and were susceptible to Candida challenge. Here, the exogenous administration of IL-18, without IL-1β, restored Th1 responses and protection (Mencacci et al., 2000). In addition, the exogenous administration of IL-18BP reduced IFN-γ-derived Th1 immunity in human whole blood cultures (Netea et al., 2002) and in mice (Fantuzzi et al., 1999). In agreement, administration of anti-IL-18 antibodies prior to systemic Candida infection depleted IFN-γ responses and enhanced fungal disease (Stuyt et al., 2002), whereas increasing IL-18 enhanced IFN-γ and Th1 responses which ultimately promoted protection (Stuyt et al., 2004). Interestingly, the role for IL-18 mediating neutrophil recruitment is unclear with one study suggesting IL-18 is uncoupled from neutrophil recruitment (Netea et al., 2003). This aligns with the protective role of IL-18 during VVC, where neutrophil recruitment enhances disease. VVC disease results from unrestrained NLRP3 activation/continuous IL-1β stimulation and is regulated by the IL-22/NLR4C axis. In this setting, IL-18 acts in a cross-circuit with IL-22 with both cytokines regulating each other and reducing NLRP3 activity (Borghi et al., 2019). IL-18 appears to be broadly protective during Candida infection, although, as was found with Aspergillus, IL-18 acts through many distinct mechanisms to promote immunity.
Interleukin-18 signaling has also been described during C. neoformans and P. brasiliensis infection. IL-18 deficient mice exhibit increased C. neoformans fungal burdens and reduced IFN-γ/IL-12 responses (Kawakami et al., 2000a, b). In addition, IL-18R deficient mice were more susceptible to C neoformans than IL-1R deficient mice, suggesting IL-18 and not IL-1α/β signaling mediates C. neoformans immunity (Wang et al., 2011). While IFN-γ and Th1 immunity are vital during PCM, the role of IL-18 is controversial. IL-18 deficient mice on a BALB/c background were protected from PCM challenge and displayed increased survival and reduced fungal burden (Panagio et al., 2008). In contrast, IL-18 deficient mice on a C57BL/6 background were susceptible to PCM and displayed enhanced fungal burdens (Ketelut-Carneiro et al., 2015). In patients with PCM, increased IL-18 in serum correlated with more severe forms of disease (Corvino et al., 2007), suggesting the C57BL/6 mouse model may be more appropriate.
A role for IL-37 has been described in a murine model of pulmonary aspergillosis. Here, administration of IL-37 decreased NLRP3 activity and IL-1β expression through the SIGIRR signaling pathway and resulted in reduced inflammatory cell recruitment. This reduced tissue damage during acute Aspergillus infection, and dampened adaptive responses in chronic Aspergillus infections (Moretti et al., 2014). SIGIRR signaling has been described to prevent lethal dysregulated IL-1 dependent Th17 responses in fungal disease (Warris et al., 2005). The induction and function of IL-18BP and IL-37 during fungal disease requires investigation but these antagonists may provide important functions mediating IL-18 signaling, enhancing immune responses, and resolving inflammatory effect. Numerous viruses target IL-18BP as an immune evasion strategy; whether fungi can do the same would be interesting to determine.
Therapeutic Potential of the IL-18 Subfamily
The exogenous administration of IL-18 enhances immunity in systemic models of C. albicans infection. This strategy may also provide protection during Aspergillus and Cryptococcus infection where IL-18 responses also confer protection. IL-18 therapy may be targeted at barrier sites or systemically once any difference in IL-18 induction and function at these two sites is determined. Although the exogenous administration of IL-18BP and anti-IL-18 antibodies enhanced acute fungal susceptibility, both these IL-18 depleting strategies promote the resolution of autoinflammatory disease and improve chronic autoimmune disease (Kanai et al., 2001; Sivakumar et al., 2002). Although no IL-18 therapeutics are currently licensed, IL-18BP therapy was examined in rheumatoid arthritis and psoriasis patients with positive tolerance and safety profiles (Tak et al., 2006). IL-37 has clear therapeutic potential as the administration of IL-37 during Aspergillus infection promoted beneficial inflammatory resolution in both acute and chronic disease (Moretti et al., 2014).
The IL-36 Subfamily
Interleukin-36 is a recent addition to the IL-1 superfamily that was discovered and characterized 20 years ago. Initially, IL-36 was thought to be similar to IL-1 as the two members of the IL-1 family shared similar gene sequences, exon-intron arrangements and predicted protein structure (Smith et al., 2000). Intriguingly, however, these new IL-36 cytokines were unable to bind IL-1R or any known orphan receptors in the IL-1 superfamily (O’Neill and Dinarello, 2000). Shortly after, two studies determined that IL-36 cytokines signal through a complex of IL-36R and IL-1RAcP leading to NF-κB and MAPK activation, and IL-6 and IL-8 production (Debets et al., 2001; Towne et al., 2004). We now know the IL-36 subfamily is comprised of four IL-36 isoforms, three agonists IL-36α, IL-36β, and IL-36γ driving proinflammatory functions (Towne et al., 2004), and the IL-36Ra antagonist mediating inflammation (Debets et al., 2001). It is worth noting that although the IL-36 subfamily was renamed in 2010, the previous nomenclature is still frequently encountered. IL-36α, IL-36β, IL-36γ, and IL-36Ra were known as IL-1F6, IL-1F8, IL-1F9, and IL-1F5, respectively, and IL-36R was named IL-1Rrp2 (Dinarello et al., 2010). IL-38 also belongs to the IL-36 sub-family, signals through IL-36R and functions as a receptor antagonist similar to IL-36Ra (van de Veerdonk et al., 2012).
IL-36 Subfamily Expression and Processing
The IL-36 subfamily plays a key role driving immune responses at mucosal barriers in the skin, respiratory tract and intestine and are the only cytokines constitutively expressed in epithelium (D’Erme et al., 2015). This location-specific expression is not found with IL-1 or IL-18 (Gresnigt and van de Veerdonk, 2013). IL-36 agonist expression is predominantly restricted to epithelial cells (Towne et al., 2004; Blumberg et al., 2007), although expression has also been observed in macrophages, dendritic cells and monocytes (Smith et al., 2000; Vigne et al., 2011; Mutamba et al., 2012; Boutet et al., 2016). IL-36R transcripts are highly prevalent in keratinocytes and epithelial cell types (Kumar et al., 2000; Towne et al., 2004), and have been found in naïve CD4 + T-cell subsets (Vigne et al., 2012), monocytes and dendritic cells (Vigne et al., 2011). IL-36 agonists bind the same receptor complex and are expressed in similar cells; adding each IL-36 agonist to keratinocytes resulted in similar immunological outcomes (Swindell et al., 2018). However, certain isoforms have been described in specific disease settings such as IL-36α in arthritis (Frey et al., 2013) and IL-36γ in psoriasis (D’Erme et al., 2015), with the blocking of IL-36γ achieving a reduction in psoriasis-associated inflammation in a 3D skin model (Todorovic et al., 2019). Whether there is isoform redundancy or whether each isoform has its own role, potentially associated with location or stimulus, remains unclear.
Interleukin-36 cytokines, like all members of the IL-1 family, are expressed without a signal peptide and are not secreted via the classical secretory pathway (Rubartelli et al., 1990). Unlike IL-1 and IL-18, IL-36 cytokines do not possess a leading peptide sequence required for caspase-1 cleavage and are therefore regulated independently of the inflammasome (Barton et al., 2000). However, a role for IL-36 cytokines facilitating activation of the NLRP3 inflammasome has been described (Chi et al., 2017). IL-36 cytokines are produced as inactive, full-length proteins that must undergo N-terminal truncation 9 amino acid residues upstream of a conserved A-X-D motif for biological activity. This precise cleavage of IL-36 cytokines increased their receptor affinity over 10,000-fold (Towne et al., 2011). It is thought that IL-36 expression is regulated by epidermal growth factor receptor (EGFR) signaling (Satoh et al., 2020) and positive feedback loops associated with Th17 cytokines (Carrier et al., 2011). There has been a recent focus on proteases that can cleave and activate IL-36 cytokines. Interestingly, blocking these proteases in inflammatory disease may have therapeutic potential. The neutrophil-derived proteases elastase and proteinase 3 appear to cleave IL-36 agonist cytokines, although their non-specific protease activity rarely activated IL-36. While neutrophils are not abundantly resident at mucosal barrier surfaces, neutrophil elastase was able to specifically activate IL-36Ra (Macleod et al., 2016), suggesting neutrophils may mediate inflammation once recruited. A clear role for the cysteine protease cathepsin S activating IL-36 agonists has also been described. Crucially, cathepsin S is found in fibroblasts and keratinocytes and its activity was increased in psoriatic lesions (Ainscough et al., 2017). IL-38 lacks a signal peptide and caspase-1 cleavage site, suggesting that, unlike IL-36Ra, activation is not required. IL-38 is expressed mostly in the skin but has also been found in B-cells (Lin et al., 2001).
The Immunological Function of the IL-36 Subfamily
Following their induction and activation, IL-36 signaling has potent effects on barrier immunity and can lead to protective responses against pathogens or drive autoinflammatory disease. IL-36 agonist signaling leads to activation of MAPK and NF-κB pathways (He et al., 2013). Downstream this results in anti-microbial peptide release from keratinocytes (Nguyen et al., 2012), increased recruitment and maturation of myeloid cells (Foster et al., 2014), increased macrophage phagocytosis and microbial killing (Tao et al., 2017), and the robust production of IL-6, IL-8, TNF, CCL3, CCL4, CCL5, CCL20, CXCL1, CXCL2, and CXCL8 (Carrier et al., 2011; Ramadas et al., 2012; Foster et al., 2014; Dietrich et al., 2016). It is likely a finely tuned balance of IL-36 agonist and antagonist activity promotes protective immunity. This is evidenced in generalized pustular psoriasis (the most severe form of psoriasis) where patients lack IL-36Ra due to a missense mutation in the IL-36Ra gene. Furthermore, IL-36Ra deficiency is associated with systemic inflammation, suggesting that uncontrolled IL-36 agonist signaling has systemic effect (Marrakchi et al., 2011).
The IL-36 subfamily also bridges innate and adaptive immunity. IL-36α/γ secreted from immune and epithelial cells directly acts on CD4+ T-cells and results in the release of IL-36β, which through a feedback loop promoted IL-2 secretion, T-cell expansion, and Th1 differentiation (Vigne et al., 2012). More recently, IL-36 gene expression has been associated with Th17 immunity. Here, IL-36 agonist cytokines regulated their own expression and drove the expression and function of Th17 cytokines and immunity (Carrier et al., 2011). While our knowledge of IL-36 is increasing, little information exists about the function of IL-38. The induction of IL-38 occurs in apoptotic cells to limit inflammation (Mora et al., 2016). However, as yet no induction of IL-38 in disease settings has been described apart from the inhibition of Candida-induced Th17 immunity in a similar manner to IL-36Ra (van de Veerdonk et al., 2012).
Fungal Induction of the IL-36 Subfamily
Although there is good evidence describing the function of the IL-36 subfamily, there is limited evidence for the induction and function of IL-36 during fungal infection. Given the important barrier function of IL-36, these cytokines likely mediate interactions with both commensal and pathogenic fungi. C. albicans induced IL-36γ expression in human keratinocytes (Braegelmann et al., 2018), IL-36α/γ in TR146 cells (a human epithelial cell line), and all IL-36 agonists during an in vivo OPC model (Verma et al., 2018). IL-36α/γ expression was significantly increased within 24 h of OPC challenge, while IL-36β increased at 48 h. The induction of IL-36 following Candida challenge was dependent on candidalysin, with a candidalysin-null Candida strain inducing drastically reduced IL-36 expression (Verma et al., 2018). The induction of IL-36 during systemic Candida infection has yet to be demonstrated. However, systemic clinical infections typically arise from disrupted barrier integrity. Replicating this in vivo is challenging and systemic infections are achieved through intravenous injection.
The signaling events that lead to IL-36 induction following Candida infection have been investigated using the OPC model. Multiple signaling pathways are activated during OPC including MAPK, PI3K, and NF-κB (Moyes et al., 2010, 2014; Verma et al., 2018) and blocking p38-MAPK but not JNK-MAPK or ERK1/2 MAPK impaired IL-36α/γ expression (Verma et al., 2018). Further investigation revealed that blocking c-Fos impaired IL-36α/γ expression, reducing c-Jun increased IL-36α expression without effecting IL-36γ. In addition, when the MAPK phosphatase MKP1 (which negatively regulates p38-MAPK and JNK-MAPK) was knocked down, IL-36α/γ expression increased (Verma et al., 2018). While these results suggest p38 MAPK induces IL-36 expression and this is negatively regulated by MKP1, MAPK pathways did not fully account for IL-36 expression. Here, while NF-κB was able to mediate some IL-36α/γ expression, blocking PI3K reduced IL-36α/γ to resting levels. These data suggest significant roles for p38-MAPK, NF-KB and PI3K in inducing IL-36 expression, with PI3K playing the most prominent role (Verma et al., 2018).
Aspergillus fumigatus infection also resulted in IL-36 induction, interestingly in a morphology and-time dependent manner. IL-36γ was induced in human peripheral blood mononuclear cells (PBMCs) following incubation with live conidia and heat-killed hyphae, while IL-36Ra was induced following incubation with live conidia, heat-killed conidia and live hyphae. As expected, IL-36α was not induced in PBMCs (Gresnigt et al., 2013). The induction of IL-36γ during Aspergillus challenge was dependent on Dectin-1 Syk signaling and TLR4 (Gresnigt et al., 2013). Dectin-1 and TLR4 signaling results in NF-κB activation which, as with Candida, controlled IL-36α/γ expression. The CLR Dectin-2 signals through PI3K and has an important role driving Th17 immunity against Candida (Saijo et al., 2010; Lee et al., 2016). Aside from Candida and Aspergillus, only Trichophyton mentagrophytes has been shown to induce IL-36 expression (Braegelmann et al., 2018). Although not extensively investigated, the induction of IL-36 cytokines appears to be cell-type, fungal morphology and time-dependent.
The Role of the IL-36 Subfamily in Fungal Immunology
Following their induction, the role of IL-36 cytokines during fungal infection is poorly understood. IL-36 can be solely responsible for inflammatory disease and clearly has potent immunological effects. Therefore, following fungal induction of IL-36, the cytokines likely mediate important immunological responses. In agreement with this, IL-36 is protective during mucosal Candida disease. IL-36R deficient mice when challenged with OPC displayed increased fungal burden and reduced IL-23 expression (Verma et al., 2018). IL-36 also induces IL-23 in macrophages isolated from psoriasis patients, suggesting a consistent link between the two cytokines (Bridgewood et al., 2018). IL-23 drives the proliferation and survival of Th17 cells vital for anti-Candida immunity. Mice deficient in IL-23 (IL-23p19-/-) experienced severe OPC disease associated with a lack of neutrophil recruitment and anti-microbial peptides (Conti et al., 2009). It was thought IL-1 and IL-36 worked in tandem to enhance protective Th17 immunity (Verma et al., 2017); however, IL-36R deficient mice had normal IL-17 gene expression suggesting a distinct, unconnected role for each (Verma et al., 2018). As such, the IL-36/IL-23 axis may complement the IL-1/Th17 response through an uncoupled mechanism (Verma et al., 2018). This contrasts with Aspergillus infection which showed blockade of IL-36R with IL-36Ra reduced IL-17 and IFN-γ responses (Gresnigt et al., 2013).
The role of IL-36 during Candida infection appears to be tightly linked with candidalysin activity, which was required for IL-36 induction (Verma et al., 2018). Commensal (yeast) Candida does not produce candidalysin and subsequently does not initiate inflammation. Instead, candidalysin is expressed when Candida becomes invasive (through hypha formation) and results in inflammation and the loss of barrier integrity (Moyes et al., 2016). Here, IL-36 signaling may facilitate host discrimination between commensal and pathogenic Candida. Furthermore, excessive IL-36 signaling is damaging and leads to inflammatory disease. Thus, Candida may induce IL-36 to promote inflammation, disrupt barrier integrity and enhance disease. This may potentially explain why psoriasis patients are particularly susceptible to Candida infection (Pietrzak et al., 2018).
The function of IL-36 during Aspergillus infection has been less well explored. IL-36γ is induced following A. fumigatus infection in a Dectin-1 Syk dependent manner. In support of this, Dectin-1 deficient mice produced defective Th17 immune responses (LeibundGut-Landmann et al., 2007) and are highly susceptible to Aspergillus lung infection (Werner et al., 2009). Many systemic fungal infections are acquired across mucosal surfaces where IL-36 induction has potent effects. Understanding the function of IL-36 signaling and the balance of protective and excessive responses may provide valuable therapeutic targets to mediate barrier inflammation and integrity.
Therapeutic Potential of the IL-36 Subfamily
Modulating IL-36 signaling has been investigated with some early success. Recently, a small molecule inhibitor of IL-36γ successfully attenuated IL-36γ induced responses (Todorovic et al., 2019). A phase 1 study in generalized pustular psoriasis patients has been completed showing a monoclonal antibody against IL-36R rapidly reduced patient pustules and psoriasis severity score (Bachelez et al., 2019). Anti-IL-23 antibodies have also been trialed with success in psoriasis patients (Reich et al., 2011). While therapeutically inhibiting IL-36 signaling may produce rapid disease improvement in autoimmune settings, IL-36 has a protective role during infectious disease and a careful balance of IL-36 signaling must be achieved.
Discussion
The IL-1 family of cytokines are central to immunity and health. It is no surprise that this family plays a crucial role during fungal infection and in determining fungal disease outcomes. Lacking IL-1 agonist activity during acute fungal disease is often severely detrimental for the immunocompromised host, resulting in fungal growth and dissemination. However, excessive IL-1 family agonist activity, either through over expression/activation or through a lack of antagonist activity, can be equally destructive. Excessive IL-1 family signaling is associated with numerous inflammatory disorders and in the context of fungal infection, can exacerbate chronic disease and lead to barrier disruption and fungal dissemination. Therefore, potent IL-1 signaling must be carefully regulated through balancing levels of protease activation and antagonist activity, to successfully promote protection and immunity.
Understanding of the functional role, mechanism of induction and downstream regulators of each IL-1 family member would greatly improve our knowledge of anti-fungal immunity. Here, we have reviewed the effects of different IL-1 family members that provide action at various locations and in response to multiple stimuli. We graphically summarize our current understanding of this topic in Figure 2. It is also important that our investigations of IL-1 family members consider collaboration and redundancy that occurs throughout the IL-1 family. Although individual IL-1 family members appear to have clearly defined roles in specific locations and disease settings, the interaction between IL-1 family signaling likely contributes to the overall immune response and disease outcome. While enhancing individual IL-1 family agonists or antagonists to promote disease resolution is therapeutically effective, targeting multiple members at once may provide the best outcome. Furthermore, while the exogenous administration of IL-1 family members is being investigated with some success, targeting the receptors or enzymes that drive IL-1 family induction and activation is another strategy that may provide therapeutic benefit.
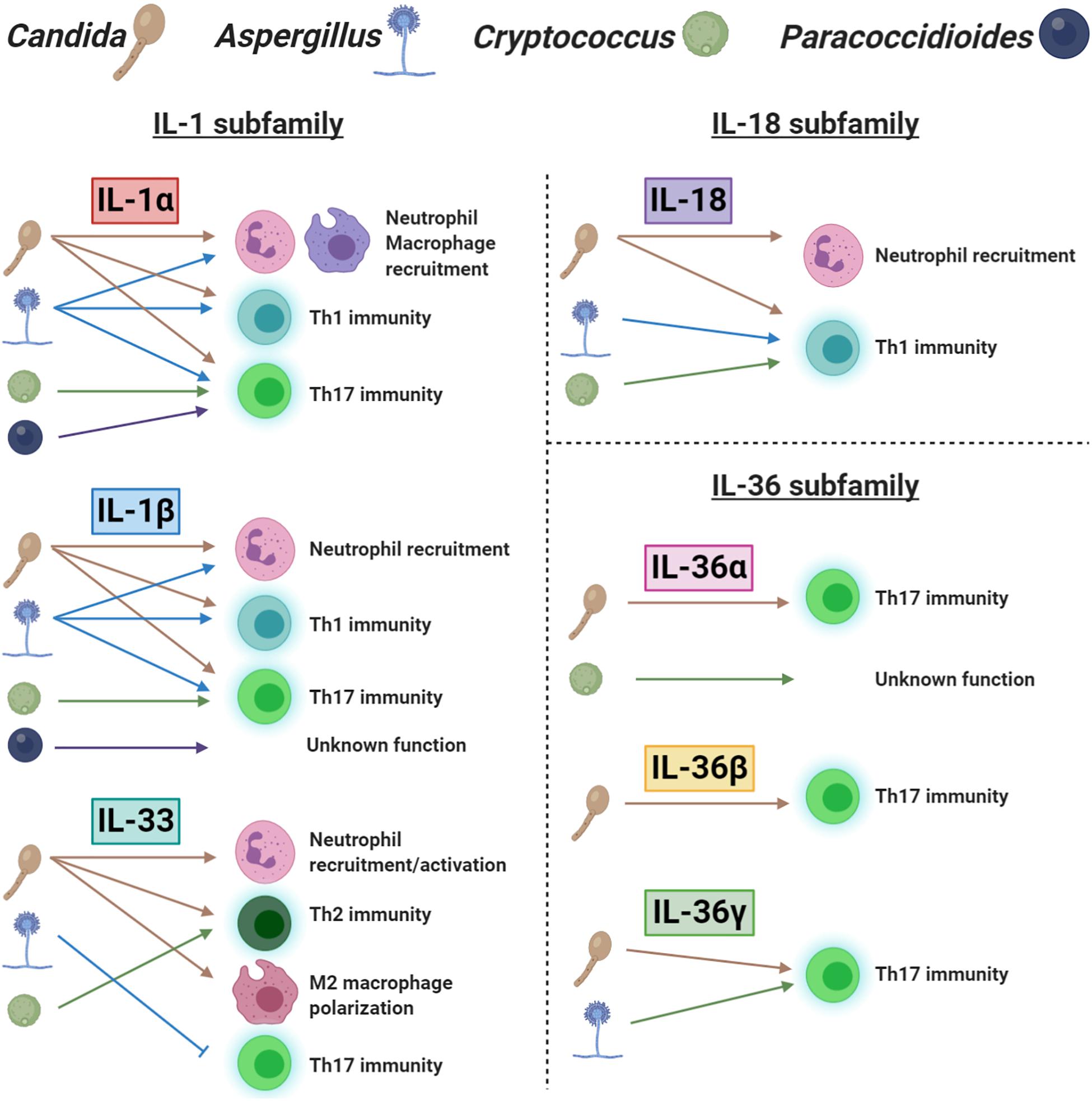
Figure 2. A schematic representation of the role of IL-1 family cytokines in fungal immunity. Each ligand is separated into their appropriate IL-1 subfamily. The icons for Candida, Aspergillus, Cryptococcus, and Paracoccidioides are labeled at the top of the figure. The presence of a fungal icon underneath a cytokine indicates induction. Arrows and blocked lines represent the functional outcome of this cytokine following induction.
As fungal diseases become an increasingly severe worldwide burden contributing to millions of deaths per year, the extensive use of immune-modulating therapies also continues to increase. Current therapies are inadequate, toxic, highly drug interactive, and frequently encounter resistance. In addition, there is no current fungal vaccine available for use. Therefore, immunotherapies that enhance anti-fungal immunity will be a vital component of future anti-fungal therapies.
Author Contributions
JG conceptualized and wrote the manuscript. GC contributed sections and produced figures. NK and JH contributed sections. JR and JN edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Wellcome Trust (214229_Z_18_Z), Biotechnology and Biological Sciences Research Council (BB/N014677/1), National Institutes of Health (R37-DE022550), and the NIH Research at Guys and St. Thomas’s NHS Foundation Trust and the King’s College London Biomedical Research Centre (IS-BRC-1215-20006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures were created using BioRender.
References
Ainscough, J. S., Macleod, T., McGonagle, D., Brakefield, R., Baron, J. M., Alase, A., et al. (2017). Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc. Natl. Acad. Sci. U S A. 114, E2748–E2757. doi: 10.1073/pnas.1620954114
Ali, S., Mohs, A., Thomas, M., Klare, J., Ross, R., Schmitz, M. L., et al. (2011). The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J. Immunol. 187, 1609–1616. doi: 10.4049/jimmunol.1003080
Altmeier, S., Toska, A., Sparber, F., Teijeira, A., Halin, C., and LeibundGut-Landmann, S. (2016). IL-1 Coordinates the neutrophil response to C. albicans in the Oral Mucosa. PLoS Pathog. 12:e1005882. doi: 10.1371/journal.ppat.1005882
Alvarez, F., Istomine, R., Shourian, M., Pavey, N., Al-Aubodah, T. A., Qureshi, S., et al. (2019). The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3(+) regulatory T cells during mucosal inflammation. Mucosal Immunol. 12, 746–760. doi: 10.1038/s41385-019-0153-155
Alves, A., David, M. A., de Castro, L. F., da Silva, R. M., Longhi, L. N. A., Blotta, M., et al. (2018). Differential production of interleukin-1 family cytokines (IL-1beta, IL-18, IL-33 and IL-37) in patients with paracoccidioidomycosis: correlation with clinical form and antifungal therapy. Med. Mycol. 56, 332–343. doi: 10.1093/mmy/myx050
Bachelez, H., Choon, S. E., Marrakchi, S., Burden, A. D., Tsai, T. F., Morita, A., et al. (2019). Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N. Engl. J. Med. 380, 981–983. doi: 10.1056/NEJMc1811317
Banda, N. K., Vondracek, A., Kraus, D., Dinarello, C. A., Kim, S. H., Bendele, A., et al. (2003). Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J. Immunol. 170, 2100–2105. doi: 10.4049/jimmunol.170.4.2100
Barousse, M. M., Van Der Pol, B. J., Fortenberry, D., Orr, D., et al. (2004). Vaginal yeast colonisation, prevalence of vaginitis, and associated local immunity in adolescents. Sex Transm. Infect. 80, 48–53. doi: 10.1136/sti.2002.003855
Barton, J. L., Herbst, R., Bosisio, D., Higgins, L., and Nicklin, M. J. (2000). A tissue specific IL-1 receptor antagonist homolog from the IL-1 cluster lacks IL-1, IL-1ra, IL-18 and IL-18 antagonist activities. Eur. J. Immunol. 30, 3299–3308. doi: 10.1002/1521-4141(200011)30:11<3299::aid-immu3299>3.0.co;2-s
Beringer, A., Noack, M., and Miossec, P. (2016). IL-17 in chronic inflammation: from discovery to targeting. Trends Mol. Med. 22, 230–241. doi: 10.1016/j.molmed.2016.01.001
Bessa, J., Meyer, C. A., de Vera Mudry, M. C., Schlicht, S., Smith, S. H., Iglesias, A., et al. (2014). Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J. Autoimmun. 55, 33–41. doi: 10.1016/j.jaut.2014.02.012
Bishu, S., Hernández-Santos, N., Simpson-Abelson, M. R., Huppler, A. R., Conti, H. R., Ghilardi, N., et al. (2014). The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect. Immun. 82, 1173–1180. doi: 10.1128/IAI.01335-1313
Blease, K., Kunkel, S. L., and Hogaboam, C. M. (2001). IL-18 modulates chronic fungal asthma in a murine model; putative involvement of Toll-like receptor-2. Inflamm. Res. 50, 552–560. doi: 10.1007/PL00000233
Blumberg, H., Dinh, H., Trueblood, E. S., Pretorius, J., Kugler, D., Weng, N., et al. (2007). Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 204, 2603–2614. doi: 10.1084/jem.20070157
Borghi, M., De Luca, A., Puccetti, M., Jaeger, M., Mencacci, A., Oikonomou, V., et al. (2015). Pathogenic NLRP3 inflammasome activity during candida infection is negatively regulated by IL-22 via activation of NLRC4 and IL-1Ra. Cell Host Microbe. 18, 198–209. doi: 10.1016/j.chom.2015.07.004
Borghi, M., Pariano, M., Solito, V., Puccetti, M., Bellet, M. M., Stincardini, C., et al. (2019). Targeting the Aryl hydrocarbon receptor with Indole-3-Aldehyde protects from vulvovaginal candidiasis via the IL-22-IL-18 cross-Talk. Front. Immunol. 10:2364. doi: 10.3389/fimmu.2019.02364
Born, T. L., Morrison, L. A., Esteban, D. J., VandenBos, T., Thebeau, L. G., Chen, N., et al. (2000). A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164, 3246–3254. doi: 10.4049/jimmunol.164.6.3246
Bossaller, L., Chiang, P. I., Schmidt-Lauber, C., Ganesan, S., Kaiser, W. J., Rathinam, V. A., et al. (2012). Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 189, 5508–5512. doi: 10.4049/jimmunol.1202121
Boutet, M. A., Bart, G., Penhoat, M., Amiaud, J., Brulin, B., Charrier, C., et al. (2016). Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 184, 159–173. doi: 10.1111/cei.12761
Braegelmann, J., Braegelmann, C., Bieber, T., and Wenzel, J. (2018). Candida induces the expression of IL-36gamma in human keratinocytes: implications for a pathogen-driven exacerbation of psoriasis? J. Eur. Acad. Dermatol. Venereol. 32, e403–e406. doi: 10.1111/jdv.14994
Bridgewood, C., Fearnley, G. W., Berekmeri, A., Laws, P., Macleod, T., Ponnambalam, S., et al. (2018). IL-36γ is a strong inducer of IL-23 in psoriatic cells and activates angiogenesis. Front. Immunol. 9:200. doi: 10.3389/fimmu.2018.00200
Brieland, J. K., Jackson, C., Menzel, F., Loebenberg, D., Cacciapuoti, A., Halpern, J., et al. (2001). Cytokine networking in lungs of immunocompetent mice in response to inhaled Aspergillus fumigatus. Infect. Immun. 69, 1554–1560. doi: 10.1128/IAI.69.3.1554-1560.2001
Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G., and White, T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4:165rv13. doi: 10.1126/scitranslmed.3004404
Bulau, A. M., Nold, M. F., Li, S., Nold-Petry, C. A., Fink, M., Mansell, A., et al. (2014). Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc. Natl. Acad. Sci. U S A. 111, 2650–2655. doi: 10.1073/pnas.1324140111
Caffrey, A. K., Lehmann, M. M., Zickovich, J. M., Espinosa, V., Shepardson, K. M., Watschke, C. P., et al. (2015). IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog. 11:e1004625. doi: 10.1371/journal.ppat.1004625
Caffrey-Carr, A. K., Kowalski, C. H., Beattie, S. R., Blaseg, N. A., Upshaw, C. R., Thammahong, A., et al. (2017). Interleukin 1alpha is critical for resistance against highly virulent aspergillus fumigatus isolates. Infect. Immun. 85:e00661-17. doi: 10.1128/IAI.00661-617
Camilli, G., Griffiths, J. S., Ho, J., Richardson, J. P., and Naglik, J. R. (2020). Some like it hot: Candida activation of inflammasomes. PLoS Pathog. 16:e1008975. doi: 10.1371/journal.ppat.1008975
Carrascal, M. T., Mendoza, L., Valcarcel, M., Salado, C., Egilegor, E., Telleria, N., et al. (2003). Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res. 63, 491–497.
Carrier, Y., Ma, H. L., Ramon, H. E., Napierata, L., Small, C., O’Toole, M., et al. (2011). Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J. Invest. Dermatol. 131, 2428–2437. doi: 10.1038/jid.2011.234
Castro, M., Bjoraker, J. A., Rohrbach, M. S., and Limper, A. H. (1996). Candida albicans induces the release of inflammatory mediators from human peripheral blood monocytes. Inflammation 20, 107–122. doi: 10.1007/BF01487749
Cenci, E., Mencacci, A., Fe, d’Ostiani, C., Del Sero, G., Mosci, P., et al. (1998). Cytokine- and T helper-dependent lung mucosal immunity in mice with invasive pulmonary aspergillosis. J. Infect. Dis. 178, 1750–1760. doi: 10.1086/314493
Cenci, E., Perito, S., Enssle, K. H., Mosci, P., Latge, J. P., Romani, L., et al. (1997). Th1 and Th2 cytokines in mice with invasive aspergillosis. Infect. Immun. 65, 564–570. doi: 10.1128/IAI.65.2.564-570.1997
Chen, C. J., Kono, H., Golenbock, D., Reed, G., Akira, S., and Rock, K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856. doi: 10.1038/nm1603
Cheng, Z., Li, M., Wang, Y., Chai, T., Cai, Y., and Li, N. (2020). Pathogenicity and immune responses of Aspergillus fumigatus infection in chickens. Front. Vet. Sci. 7:143. doi: 10.3389/fvets.2020.00143
Chi, H. H., Hua, K. F., Lin, Y. C., Chu, C. L., Hsieh, C. Y., Hsu, Y. J., et al. (2017). IL-36 signaling facilitates activation of the NLRP3 inflammasome and IL-23/IL-17 axis in renal inflammation and fibrosis. J. Am. Soc. Nephrol. 28, 2022–2037. doi: 10.1681/ASN.2016080840
Cohen, I., Rider, P., Carmi, Y., Braiman, A., Dotan, S., White, M. R., et al. (2010). Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. U S A. 107, 2574–2579. doi: 10.1073/pnas.0915018107
Cohen, P. (2014). The TLR and IL-1 signalling network at a glance. J. Cell Sci. 127:2383. doi: 10.1242/jcs.149831
Cohen, S. B., Proudman, S., Kivitz, A. J., Burch, F. X., Donohue, J. P., Burstein, D., et al. (2011). A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 13:R125. doi: 10.1186/ar3430
Conti, H. R., Shen, F., Nayyar, N., Stocum, E., Sun, J. N., Lindemann, M. J., et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311. doi: 10.1084/jem.20081463
Corvino, C. L., Mamoni, R. L., Fagundes, G. Z., and Blotta, M. H. (2007). Serum interleukin-18 and soluble tumour necrosis factor receptor 2 are associated with disease severity in patients with paracoccidioidomycosis. Clin. Exp. Immunol. 147, 483–490. doi: 10.1111/j.1365-2249.2006.03308.x
Cunha, C., Di Ianni, M., Bozza, S., Giovannini, G., Zagarella, S., Zelante, T., et al. (2010). Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116, 5394–5402. doi: 10.1182/blood-2010-04-279307
Debets, R., Timans, J. C., Homey, B., Zurawski, S., Sana, T. R., Lo, S., et al. (2001). Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J. Immunol. 167, 1440–1446. doi: 10.4049/jimmunol.167.3.1440
D’Erme, A. M., Wilsmann-Theis, D., Wagenpfeil, J., Holzel, M., Ferring-Schmitt, S., Sternberg, S., et al. (2015). IL-36gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J. Invest. Dermatol. 135, 1025–1032. doi: 10.1038/jid.2014.532
Di Paolo, N. C., and Shayakhmetov, D. M. (2016). Interleukin 1alpha and the inflammatory process. Nat. Immunol. 17, 906–913. doi: 10.1038/ni.3503
Dietrich, D., Martin, P., Flacher, V., Sun, Y., Jarrossay, D., Brembilla, N., et al. (2016). Interleukin-36 potently stimulates human M2 macrophages, langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 84, 88–98. doi: 10.1016/j.cyto.2016.05.012
Dinarello, C., Arend, W., Sims, J., Smith, D., Blumberg, H., O’Neill, L., et al. (2010). IL-1 family nomenclature. Nat. Immunol. 11:973. doi: 10.1038/ni1110-973
Dinarello, C. A. (2011). Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732. doi: 10.1182/blood-2010-07-273417
Dinarello, C. A. (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27. doi: 10.1111/imr.12621
Dinarello, C. A. (2019). The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 15, 612–632. doi: 10.1038/s41584-019-0277-278
Dinarello, C. A., Nold-Petry, C., Nold, M., Fujita, M., Li, S., Kim, S., et al. (2016). Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 46, 1067–1081. doi: 10.1002/eji.201545828
Dinarello, C. A., Simon, A., and van der Meer, J. W. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652. doi: 10.1038/nrd3800
Drummond, R. A., Collar, A. L., Swamydas, M., Rodriguez, C. A., Lim, J. K., Mendez, L. M., et al. (2015). CARD9-Dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 11:e1005293. doi: 10.1371/journal.ppat.1005293
Economides, A. N., Carpenter, L. R., Rudge, J. S., Wong, V., Koehler-Stec, E. M., Hartnett, C., et al. (2003). Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat. Med. 9, 47–52. doi: 10.1038/nm811
Eigenbrod, T., Park, J. H., Harder, J., Iwakura, Y., and Nunez, G. (2008). Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J. Immunol. 181, 8194–8198. doi: 10.4049/jimmunol.181.12.8194
Fantuzzi, G., Reed, D. A., and Dinarello, C. A. (1999). IL-12-induced IFN-gamma is dependent on caspase-1 processing of the IL-18 precursor. J. Clin. Invest. 104, 761–767. doi: 10.1172/JCI7501
Fields, J. K., Günther, S., and Sundberg, E. J. (2019). Structural basis of IL-1 family cytokine signaling. Front. Immunol. 10:1412. doi: 10.3389/fimmu.2019.01412
Flaczyk, A., Duerr, C. U., Shourian, M., Lafferty, E. I., Fritz, J. H., and Qureshi, S. T. (2013). IL-33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans. J. Immunol. 191, 2503–2513. doi: 10.4049/jimmunol.1300426
Foster, A. M., Baliwag, J., Chen, C. S., Guzman, A. M., Stoll, S. W., Gudjonsson, J. E., et al. (2014). IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 192, 6053–6061. doi: 10.4049/jimmunol.1301481
Franchi, L., Eigenbrod, T., Munoz-Planillo, R., and Nunez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. doi: 10.1038/ni.1703
Freeman, B. E., Raue, H. P., Hill, A. B., and Slifka, M. K. (2015). Cytokine-Mediated activation of NK cells during viral infection. J. Virol. 89, 7922–7931. doi: 10.1128/JVI.00199-115
Frey, S., Derer, A., Messbacher, M. E., Baeten, D. L., Bugatti, S., Montecucco, C., et al. (2013). The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis. 72, 1569–1574. doi: 10.1136/annrheumdis-2012-202264
Garlanda, C., Dinarello, C. A., and Mantovani, A. (2013). The interleukin-1 family: back to the future. Immunity 39, 1003–1018. doi: 10.1016/j.immuni.2013.11.010
Garth, J. M., Reeder, K. M., Godwin, M. S., Mackel, J. J., Dunaway, C. W., Blackburn, J. P., et al. (2017). IL-33 signaling regulates innate IL-17A and IL-22 production via suppression of prostaglandin E2 during lung fungal infection. J. Immunol. 199, 2140–2148. doi: 10.4049/jimmunol.1602186
Gerdes, N., Sukhova, G. K., Libby, P., Reynolds, R. S., Young, J. L., and Schonbeck, U. (2002). Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J. Exp. Med. 195, 245–257. doi: 10.1084/jem.20011022
Glocker, E. O., Hennigs, A., Nabavi, M., Schaffer, A. A., Woellner, C., Salzer, U., et al. (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 361, 1727–1735. doi: 10.1056/NEJMoa0810719
Godwin, M. S., Reeder, K. M., Garth, J. M., Blackburn, J. P., Jones, M., Yu, Z., et al. (2019). IL-1RA regulates immunopathogenesis during fungal-associated allergic airway inflammation. JCI Insight 4:e129055. doi: 10.1172/jci.insight.129055
Goncalves, A. C., Maia, D. C., Ferreira, L. S., Monnazzi, L. G., Alegranci, P., Placeres, M. C., et al. (2015). Involvement of major components from Sporothrix schenckii cell wall in the caspase-1 activation, nitric oxide and cytokines production during experimental sporotrichosis. Mycopathologia 179, 21–30. doi: 10.1007/s11046-014-9810-9810
Gracie, J. A., Robertson, S. E., and McInnes, I. B. (2003). Interleukin-18. J. Leukoc Biol. 73, 213–224. doi: 10.1189/jlb.0602313
Gresnigt, M. S., Rekiki, A., Rasid, O., Savers, A., Jouvion, G., Dannaoui, E., et al. (2016). Reducing hypoxia and inflammation during invasive pulmonary Aspergillosis by targeting the interleukin-1 receptor. Sci. Rep. 6:26490. doi: 10.1038/srep26490
Gresnigt, M. S., Rosler, B., Jacobs, C. W., Becker, K. L., Joosten, L. A., van der Meer, J. W., et al. (2013). The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur. J. Immunol. 43, 416–426. doi: 10.1002/eji.201242711
Gresnigt, M. S., and van de Veerdonk, F. L. (2013). Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 25, 458–465. doi: 10.1016/j.smim.2013.11.003
Griffiths, J. S., Thompson, A., Stott, M., Benny, A., Lewis, N. A., Taylor, P. R., et al. (2018). Differential susceptibility of Dectin-1 isoforms to functional inactivation by neutrophil and fungal proteases. FASEB J. 32, 3385–3397. doi: 10.1096/fj.201701145R
Gris, D., Ye, Z., Iocca, H. A., Wen, H., Craven, R. R., Gris, P., et al. (2010). NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 185, 974–981. doi: 10.4049/jimmunol.0904145
Gross, O., Poeck, H., Bscheider, M., Dostert, C., Hannesschlager, N., Endres, S., et al. (2009). Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436. doi: 10.1038/nature07965
Gross, O., Yazdi, A. S., Thomas, C. J., Masin, M., Heinz, L. X., Guarda, G., et al. (2012). Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400. doi: 10.1016/j.immuni.2012.01.018
Gurczynski, S. J., and Moore, B. B. (2018). IL-17 in the lung: the good, the bad, and the ugly. Am. J. Physiol. Lung. Cell Mol. Physiol. 314, L6–L16. doi: 10.1152/ajplung.00344.2017
He, Q., Chen, H. X., Li, W., Wu, Y., Chen, S. J., Yue, Q., et al. (2013). IL-36 cytokine expression and its relationship with p38 MAPK and NF-kappaB pathways in psoriasis vulgaris skin lesions. J. Huazhong Univ. Sci. Technol. Med. Sci. 33, 594–599. doi: 10.1007/s11596-013-1164-1161
Heguy, A., Baldari, C. T., Macchia, G., Telford, J. L., and Melli, M. (1992). Amino acids conserved in interleukin-1 receptors (IL-1Rs) and the Drosophila toll protein are essential for IL-1R signal transduction. J. Biol. Chem. 267, 2605–2609. doi: 10.1016/s0021-9258(18)45924-8
Heilig, R., Dick, M. S., Sborgi, L., Meunier, E., Hiller, S., and Broz, P. (2018). The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 48, 584–592. doi: 10.1002/eji.201747404
Hise, A. G., Tomalka, J., Ganesan, S., Patel, K., Hall, B. A., Brown, G. D., et al. (2009). An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497. doi: 10.1016/j.chom.2009.05.002
Horai, R., Saijo, S., Tanioka, H., Nakae, S., Sudo, K., Okahara, A., et al. (2000). Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J. Exp. Med. 191, 313–320. doi: 10.1084/jem.191.2.313
Hoshino, K., Tsutsui, H., Kawai, T., Takeda, K., Nakanishi, K., Takeda, Y., et al. (1999). Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 162, 5041–5044.
Hou, J., Townson, S. A., Kovalchin, J. T., Masci, A., Kiner, O., Shu, Y., et al. (2013). Design of a superior cytokine antagonist for topical ophthalmic use. Proc. Natl. Acad. Sci. U S A. 110, 3913–3918. doi: 10.1073/pnas.1217996110
Hung, C. Y., Jimenez-Alzate Mdel, P., Gonzalez, A., Wuthrich, M., Klein, B. S., and Cole, G. T. (2014). Interleukin-1 receptor but not Toll-like receptor 2 is essential for MyD88-dependent Th17 immunity to Coccidioides infection. Infect. Immun. 82, 2106–2114. doi: 10.1128/IAI.01579-1513
Jaeger, M., Carvalho, A., Cunha, C., Plantinga, T. S., van de Veerdonk, F., Puccetti, M., et al. (2016). Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur. J. Clin. Microbiol. Infect. Dis. 35, 797–801. doi: 10.1007/s10096-016-2600-2605
Joly, S., Ma, N., Sadler, J. J., Soll, D. R., Cassel, S. L., and Sutterwala, F. S. (2009). Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J. Immunol. 183, 3578–3581. doi: 10.4049/jimmunol.0901323
Kakkar, R., and Lee, R. T. (2008). The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 7, 827–840. doi: 10.1038/nrd2660
Kamijo, S., Takeda, H., Tokura, T., Suzuki, M., Inui, K., Hara, M., et al. (2013). IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J. Immunol. 190, 4489–4499. doi: 10.4049/jimmunol.1201212
Kanai, T., Watanabe, M., Okazawa, A., Sato, T., Yamazaki, M., Okamoto, S., et al. (2001). Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology 121, 875–888. doi: 10.1053/gast.2001.28021
Kaplanski, G. (2018). Interleukin-18: biological properties and role in disease pathogenesis. Immunol. Rev. 281, 138–153. doi: 10.1111/imr.12616
Kasper, L., Konig, A., Koenig, P. A., Gresnigt, M. S., Westman, J., Drummond, R. A., et al. (2018). The fungal peptide toxin candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 9, 4260. doi: 10.1038/s41467-018-06607-6601
Kawakami, K., Koguchi, Y., Qureshi, M. H., Kinjo, Y., Yara, S., Miyazato, A., et al. (2000a). Reduced host resistance and Th1 response to Cryptococcus neoformans in interleukin-18 deficient mice. FEMS Microbiol. Lett. 186, 121–126. doi: 10.1111/j.1574-6968.2000.tb09092.x
Kawakami, K., Koguchi, Y., Qureshi, M. H., Miyazato, A., Yara, S., Kinjo, Y., et al. (2000b). IL-18 contributes to host resistance against infection with Cryptococcus neoformans in mice with defective IL-12 synthesis through induction of IFN-gamma production by NK cells. J. Immunol. 165, 941–947. doi: 10.4049/jimmunol.165.2.941
Ketelut-Carneiro, N., Ghosh, S., Levitz, S. M., Fitzgerald, K. A., and da Silva, J. S. (2018). A Dectin-1-Caspase-8 pathway licenses canonical caspase-1 inflammasome activation and interleukin-1beta release in response to a pathogenic fungus. J. Infect. Dis. 217, 329–339. doi: 10.1093/infdis/jix568
Ketelut-Carneiro, N., Silva, G. K., Rocha, F. A., Milanezi, C. M., Cavalcanti-Neto, F. F., Zamboni, D. S., et al. (2015). IL-18 triggered by the Nlrp3 inflammasome induces host innate resistance in a pulmonary model of fungal infection. J. Immunol. 194, 4507–4517. doi: 10.4049/jimmunol.1402321
Ketelut-Carneiro, N., Souza, C. O. S., Benevides, L., Gardinassi, L. G., Silva, M. C., Tavares, L. A., et al. (2019). Caspase-11-dependent IL-1alpha release boosts Th17 immunity against Paracoccidioides brasiliensis. PLoS Pathog. 15:e1007990. doi: 10.1371/journal.ppat.1007990
Kinjo, Y., Kawakami, K., Uezu, K., Yara, S., Miyagi, K., Koguchi, Y., et al. (2002). Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: a comparative study with IL-12p40. J. Immunol. 169, 323–329. doi: 10.4049/jimmunol.169.1.323
Koenders, M. I., Devesa, I., Marijnissen, R. J., Abdollahi-Roodsaz, S., Boots, A. M., Walgreen, B., et al. (2008). Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum. 58, 3461–3470. doi: 10.1002/art.23957
Krumm, B., Xiang, Y., and Deng, J. (2014). Structural biology of the IL-1 superfamily: key cytokines in the regulation of immune and inflammatory responses. Protein Sci. 23, 526–538. doi: 10.1002/pro.2441
Kumar, S., McDonnell, P. C., Lehr, R., Tierney, L., Tzimas, M. N., Griswold, D. E., et al. (2000). Identification and initial characterization of four novel members of the interleukin-1 family. J. Biol. Chem. 275, 10308–10314. doi: 10.1074/jbc.275.14.10308
Kurokawa, C. S., Araujo, J. P. Jr., Soares, A. M., Sugizaki, M. F., and Peracoli, M. T. (2007). Pro- and anti-inflammatory cytokines produced by human monocytes challenged in vitro with Paracoccidioides brasiliensis. Microbiol. Immunol. 51, 421–428. doi: 10.1111/j.1348-0421.2007.tb03929.x
Kurt-Jones, E. A., Beller, D. I., Mizel, S. B., and Unanue, E. R. (1985). Identification of a membrane-associated interleukin 1 in macrophages. Proc. Natl. Acad. Sci. U S A. 82, 1204–1208. doi: 10.1073/pnas.82.4.1204
Le, H. T., Tran, V. G., Kim, W., Kim, J., Cho, H. R., and Kwon, B. (2012). IL-33 priming regulates multiple steps of the neutrophil-mediated anti-Candida albicans response by modulating TLR and dectin-1 signals. J. Immunol. 189, 287–295. doi: 10.4049/jimmunol.1103564
Lee, M. J., Yoshimoto, E., Saijo, S., Iwakura, Y., Lin, X., Katz, H. R., et al. (2016). Phosphoinositide 3-Kinase delta regulates Dectin-2 signaling and the generation of Th2 and Th17 Immunity. J. Immunol. 197, 278–287. doi: 10.4049/jimmunol.1502485
Lefrancais, E., Roga, S., Gautier, V., Gonzalez-de-Peredo, A., Monsarrat, B., Girard, J. P., et al. (2012). IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. U S A. 109, 1673–1678. doi: 10.1073/pnas.1115884109
LeibundGut-Landmann, S., Gross, O., Robinson, M. J., Osorio, F., Slack, E. C., Tsoni, S. V., et al. (2007). Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8, 630–638. doi: 10.1038/ni1460
Li, D., Guabiraba, R., Besnard, A. G., Komai-Koma, M., Jabir, M. S., Zhang, L., et al. (2014). IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin Immunol. 134, 1422-1432.e11. doi: 10.1016/j.jaci.2014.05.011
Li, S., Neff, C. P., Barber, K., Hong, J., Luo, Y., Azam, T., et al. (2015). Extracellular forms of IL-37 inhibit innate inflammation in vitro and in vivo but require the IL-1 family decoy receptor IL-1R8. Proc. Natl. Acad. Sci. U S A. 112, 2497–2502. doi: 10.1073/pnas.1424626112
Liew, F. Y., Girard, J. P., and Turnquist, H. R. (2016). Interleukin-33 in health and disease. Nat. Rev. Immunol. 16, 676–689. doi: 10.1038/nri.2016.95
Lilly, L. M., Gessner, M. A., Dunaway, C. W., Metz, A. E., Schwiebert, L., Weaver, C. T., et al. (2012). The beta-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J. Immunol. 189, 3653–3660. doi: 10.4049/jimmunol.1201797
Lin, H., Ho, A. S., Haley-Vicente, D., Zhang, J., Bernal-Fussell, J., Pace, A. M., et al. (2001). Cloning and characterization of IL-1HY2, a novel interleukin-1 family member. J. Biol. Chem. 276, 20597–20602. doi: 10.1074/jbc.M010095200
Lopez-Castejon, G., and Brough, D. (2011). Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 22, 189–195. doi: 10.1016/j.cytogfr.2011.10.001
Luthi, A. U., Cullen, S. P., McNeela, E. A., Duriez, P. J., Afonina, I. S., Sheridan, C., et al. (2009). Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98. doi: 10.1016/j.immuni.2009.05.007
Macleod, T., Doble, R., McGonagle, D., Wasson, C. W., Alase, A., Stacey, M., et al. (2016). Neutrophil elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 receptor antagonist. Sci. Rep. 6:24880. doi: 10.1038/srep24880
Marrakchi, S., Guigue, P., Renshaw, B. R., Puel, A., Pei, X. Y., Fraitag, S., et al. (2011). Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628. doi: 10.1056/NEJMoa1013068
Martin, N. T., and Martin, M. U. (2016). Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 17, 122–131. doi: 10.1038/ni.3370
Mastroeni, P., Clare, S., Khan, S., Harrison, J. A., Hormaeche, C. E., Okamura, H., et al. (1999). Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium. Infect. Immun. 67, 478–483. doi: 10.1128/IAI.67.2.478-483.1999
Mehraj, V., Ponte, R., and Routy, J. P. (2016). The dynamic role of the IL-33/ST2 axis in chronic viral-infections: alarming and adjuvanting the immune response. EBioMedicine 9, 37–44. doi: 10.1016/j.ebiom.2016.06.047
Mencacci, A., Bacci, A., Cenci, E., Montagnoli, C., Fiorucci, S., Casagrande, A., et al. (2000). Interleukin 18 restores defective Th1 immunity to Candida albicans in caspase 1-deficient mice. Infect. Immun. 68, 5126–5131. doi: 10.1128/iai.68.9.5126-5131.2000
Monteleone, G., Trapasso, F., Parrello, T., Biancone, L., Stella, A., Iuliano, R., et al. (1999). Bioactive IL-18 expression is up-regulated in Crohn’s disease. J. Immunol. 163, 143–147.
Mora, J., Schlemmer, A., Wittig, I., Richter, F., Putyrski, M., Frank, A. C., et al. (2016). Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell Biol. 8, 426–438. doi: 10.1093/jmcb/mjw006
Moretti, S., Bozza, S., Oikonomou, V., Renga, G., Casagrande, A., Iannitti, R. G., et al. (2014). IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog. 10:e1004462. doi: 10.1371/journal.ppat.1004462
Moussion, C., Ortega, N., and Girard, J. P. (2008). The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. 3:e3331. doi: 10.1371/journal.pone.0003331
Moyes, D. L., Runglall, M., Murciano, C., Shen, C., Nayar, D., Thavaraj, S., et al. (2010). A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8, 225–235. doi: 10.1016/j.chom.2010.08.002
Moyes, D. L., Shen, C., Murciano, C., Runglall, M., Richardson, J. P., Arno, M., et al. (2014). Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J. Infect. Dis. 209, 1816–1826. doi: 10.1093/infdis/jit824
Moyes, D. L., Wilson, D., Richardson, J. P., Mogavero, S., Tang, S. X., Wernecke, J., et al. (2016). Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68. doi: 10.1038/nature17625
Mutamba, S., Allison, A., Mahida, Y., Barrow, P., and Foster, N. (2012). Expression of IL-1Rrp2 by human myelomonocytic cells is unique to DCs and facilitates DC maturation by IL-1F8 and IL-1F9. Eur. J. Immunol. 42, 607–617. doi: 10.1002/eji.201142035
Nagai, H., Guo, J., Choi, H., and Kurup, V. (1995). Interferon-gamma and tumor necrosis factor-alpha protect mice from invasive aspergillosis. J. Infect. Dis. 172, 1554–1560. doi: 10.1093/infdis/172.6.1554
Nakanishi, K. (2018). Unique action of interleukin-18 on T Cells and other immune cells. Front. Immunol. 9:763. doi: 10.3389/fimmu.2018.00763
Nakanishi, K., Yoshimoto, T., Tsutsui, H., and Okamura, H. (2001). Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 12, 53–72. doi: 10.1016/s1359-6101(00)00015-10
Netea, M. G., Stuyt, R. J., Kim, S. H., Van der Meer, J. W., Kullberg, B. J., and Dinarello, C. A. (2002). The role of endogenous interleukin (IL)-18, IL-12, IL-1beta, and tumor necrosis factor-alpha in the production of interferon-gamma induced by Candida albicans in human whole-blood cultures. J. Infect. Dis. 185, 963–970. doi: 10.1086/339410
Netea, M. G., Vonk, A. G., van den Hoven, M., Verschueren, I., Joosten, L. A., van Krieken, J. H., et al. (2003). Differential role of IL-18 and IL-12 in the host defense against disseminated Candida albicans infection. Eur. J. Immunol. 33, 3409–3417. doi: 10.1002/eji.200323737
Nguyen, T. T., Niyonsaba, F., Ushio, H., Akiyama, T., Kiatsurayanon, C., Smithrithee, R., et al. (2012). Interleukin-36 cytokines enhance the production of host defense peptides psoriasin and LL-37 by human keratinocytes through activation of MAPKs and NF-kappaB. J. Dermatol. Sci. 68, 63–66. doi: 10.1016/j.jdermsci.2012.07.010
Nold, M. F., Nold-Petry, C. A., Zepp, J. A., Palmer, B. E., Bufler, P., and Dinarello, C. A. (2010). IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 11, 1014–1022. doi: 10.1038/ni.1944
Novick, D., Elbirt, D., Dinarello, C. A., Rubinstein, M., and Sthoeger, Z. M. (2009). Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis. J. Clin. Immunol. 29, 38–45. doi: 10.1007/s10875-008-9217-9210
Novick, D., Elbirt, D., Miller, G., Dinarello, C. A., Rubinstein, M., and Sthoeger, Z. M. (2010). High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J. Autoimmun 34, 121–126. doi: 10.1016/j.jaut.2009.08.002
Novick, D., Kim, S., Kaplanski, G., and Dinarello, C. A. (2013). Interleukin-18, more than a Th1 cytokine. Semin Immunol. 25, 439–448. doi: 10.1016/j.smim.2013.10.014
Novick, D., Schwartsburd, B., Pinkus, R., Suissa, D., Belzer, I., Sthoeger, Z., et al. (2001). A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine 14, 334–342. doi: 10.1006/cyto.2001.0914
Omoto, Y., Tokime, K., Yamanaka, K., Habe, K., Morioka, T., Kurokawa, I., et al. (2006). Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J. Immunol. 177, 8315–8319. doi: 10.4049/jimmunol.177.12.8315
Omoto, Y., Yamanaka, K., Tokime, K., Kitano, S., Kakeda, M., Akeda, T., et al. (2010). Granzyme B is a novel interleukin-18 converting enzyme. J. Dermatol. Sci. 59, 129–135. doi: 10.1016/j.jdermsci.2010.05.004
O’Neill, L. A., and Dinarello, C. A. (2000). The IL-1 receptor/toll-like receptor superfamily: crucial receptors for inflammation and host defense. Immunol. Today 21, 206–209. doi: 10.1016/s0167-5699(00)01611-x
Panagio, L. A., Tristao, F. S., Moreira, A. P., Pereira, M. S., Cavassani, K. A., Milanezi, C. M., et al. (2008). Role of interleukin (IL)-18 in experimental paracoccidioidomycosis. Med. Mycol. 46, 435–442. doi: 10.1080/13693780801895444
Pastorelli, L., Garg, R. R., Hoang, S. B., Spina, L., Mattioli, B., Scarpa, M., et al. (2010). Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. U S A. 107, 8017–8022. doi: 10.1073/pnas.0912678107
Peters, V. A., Joesting, J. J., and Freund, G. G. (2013). IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 32, 1–8. doi: 10.1016/j.bbi.2012.11.006
Pietras, E. M., Mirantes-Barbeito, C., Fong, S., Loeffler, D., Kovtonyuk, L. V., Zhang, S., et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 18, 607–618. doi: 10.1038/ncb3346
Pietrzak, A., Grywalska, E., Socha, M., Rolinski, J., Franciszkiewicz-Pietrzak, K., Rudnicka, L., et al. (2018). Prevalence and possible role of candida species in patients with psoriasis: a systematic review and meta-analysis. Med. Inflamm. 2018:9602362. doi: 10.1155/2018/9602362
Plantinga, T. S., Johnson, M. D., Scott, W. K., van de Vosse, E., Velez Edwards, D. R., Smith, P. B., et al. (2012). Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J. Infect. Dis. 205, 934–943. doi: 10.1093/infdis/jir867
Plantinga, T. S., van der Velden, W. J., Ferwerda, B., van Spriel, A. B., Adema, G., Feuth, T., et al. (2009). Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 49, 724–732. doi: 10.1086/604714
Preite, N. W., Feriotti, C., Souza, de Lima, D., da Silva, B. B., Condino-Neto, A., et al. (2018). The syk-coupled C-type lectin receptors Dectin-2 and Dectin-3 are involved in Paracoccidioides brasiliensis recognition by human plasmacytoid dendritic cells. Front. Immunol. 9:464. doi: 10.3389/fimmu.2018.00464
Ramadas, R. A., Ewart, S. L., Iwakura, Y., Medoff, B. D., and LeVine, A. M. (2012). IL-36alpha exerts pro-inflammatory effects in the lungs of mice. PLoS One 7:e45784. doi: 10.1371/journal.pone.0045784
Ramaprakash, H., Shibata, T., Duffy, K. E., Ismailoglu, U. B., Bredernitz, R. M., Moreira, A. P., et al. (2011). Targeting ST2L potentiates CpG-mediated therapeutic effects in a chronic fungal asthma model. Am. J. Pathol. 179, 104–115. doi: 10.1016/j.ajpath.2011.03.032
Rautemaa-Richardson, R., and Richardson, M. D. (2017). Systemic fungal infections. Medicine 45, 757–762. doi: 10.1016/j.mpmed.2017.09.007
Reading, P. C., and Smith, G. L. (2003). Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 77, 9960–9968. doi: 10.1128/jvi.77.18.9960-9968.2003
Reich, K., Langley, R. G., Papp, K. A., Ortonne, J. P., Unnebrink, K., Kaul, M., et al. (2011). A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N. Engl. J. Med. 365, 1586–1596. doi: 10.1056/NEJMoa1010858
Ren, X., Gelinas, A. D., von Carlowitz, I., Janjic, N., and Pyle, A. M. (2017). Structural basis for IL-1alpha recognition by a modified DNA aptamer that specifically inhibits IL-1alpha signaling. Nat. Commun. 8:810. doi: 10.1038/s41467-017-00864-862
Roselletti, E., Perito, S., Gabrielli, E., Mencacci, A., Pericolini, E., Sabbatini, S., et al. (2017). NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans. Sci. Rep. 7:17877. doi: 10.1038/s41598-017-17649-17648
Rouabhia, M., Ross, G., Page, N., and Chakir, J. (2002). Interleukin-18 and gamma interferon production by oral epithelial cells in response to exposure to Candida albicans or lipopolysaccharide stimulation. Infect. Immun. 70, 7073–7080. doi: 10.1128/iai.70.12.7073-7080.2002
Rubartelli, A., Cozzolino, F., Talio, M., and Sitia, R. (1990). A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. Embo J. 9, 1503–1510. doi: 10.1002/j.1460-2075.1990.tb08268.x
Saijo, S., Ikeda, S., Yamabe, K., Kakuta, S., Ishigame, H., Akitsu, A., et al. (2010). Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32, 681–691. doi: 10.1016/j.immuni.2010.05.001
Salter, B. M., Oliveria, J. P., Nusca, G., Smith, S. G., Tworek, D., Mitchell, P. D., et al. (2016). IL-25 and IL-33 induce Type 2 inflammation in basophils from subjects with allergic asthma. Respir Res. 17:5. doi: 10.1186/s12931-016-0321-z
Satoh, T. K., Mellett, M., Meier-Schiesser, B., Fenini, G., Otsuka, A., Beer, H. D., et al. (2020). IL-36γ drives skin toxicity induced by EGFR/MEK inhibition and commensal Cutibacterium acnes. J. Clin. Invest. 130, 1417–1430. doi: 10.1172/jci128678
Shen, H., Yu, Y., Chen, S. M., Sun, J. J., Fang, W., Guo, S. Y., et al. (2020). Dectin-1 Facilitates IL-18 production for the generation of protective antibodies against Candida albicans. Front. Microbiol. 11:1648. doi: 10.3389/fmicb.2020.01648
Shourian, M., Ralph, B., Angers, I., Sheppard, D. C., and Qureshi, S. T. (2017). Contribution of IL-1RI signaling to protection against cryptococcus neoformans 52D in a mouse model of infection. Front. Immunol. 8:1987. doi: 10.3389/fimmu.2017.01987
Silva, C. L., Silva, M. F., Faccioli, L. H., Pietro, R. C., Cortez, S. A., and Foss, N. T. (1995). Differential correlation between interleukin patterns in disseminated and chronic human paracoccidioidomycosis. Clin. Exp. Immunol. 101, 314–320. doi: 10.1111/j.1365-2249.1995.tb08357.x
Sims, J. E., and Smith, D. E. (2010). The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89–102. doi: 10.1038/nri2691
Sivakumar, P. V., Westrich, G. M., Kanaly, S., Garka, K., Born, T. L., Derry, J. M., et al. (2002). Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 50, 812–820. doi: 10.1136/gut.50.6.812
Smith, D. E., Renshaw, B. R., Ketchem, R. R., Kubin, M., Garka, K. E., and Sims, J. E. (2000). Four new members expand the interleukin-1 superfamily. J. Biol. Chem. 275, 1169–1175. doi: 10.1074/jbc.275.2.1169
Steele, C., Fidel, P. L., and Jr. (2002). Cytokine and chemokine production by human oral and vaginal epithelial cells in response to Candida albicans. Infect. Immun. 70, 577–583. doi: 10.1128/iai.70.2.577-583.2002
Steele, C., Rapaka, R. R., Metz, A., Pop, S. M., Williams, D. L., Gordon, S., et al. (2005). The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathogens 1:323–334. doi: 10.1371/journal.ppat.0010042
Stehlik, C. (2009). Multiple interleukin-1beta-converting enzymes contribute to inflammatory arthritis. Arthritis Rheum. 60, 3524–3530. doi: 10.1002/art.24961
Stuyt, R. J., Netea, M. G., van Krieken, J. H., van der Meer, J. W., and Kullberg, B. J. (2004). Recombinant interleukin-18 protects against disseminated Candida albicans infection in mice. J. Infect. Dis. 189, 1524–1527. doi: 10.1086/382955
Stuyt, R. J., Netea, M. G., Verschueren, I., Fantuzzi, G., Dinarello, C. A., Van Der Meer, J. W., et al. (2002). Role of interleukin-18 in host defense against disseminated Candida albicans infection. Infect. Immun. 70, 3284–3286. doi: 10.1128/iai.70.6.3284-3286.2002
Sugawara, S., Uehara, A., Nochi, T., Yamaguchi, T., Ueda, H., Sugiyama, A., et al. (2001). Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 167, 6568–6575. doi: 10.4049/jimmunol.167.11.6568
Sutton, C. E., Mielke, L. A., and Mills, K. H. (2012). IL-17-producing gammadelta T cells and innate lymphoid cells. Eur. J. Immunol. 42, 2221–2231. doi: 10.1002/eji.201242569
Swindell, W. R., Beamer, M. A., Sarkar, M. K., Loftus, S., Fullmer, J., Xing, X., et al. (2018). RNA-Seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-Dependent gene signature. Front. Immunol. 9:80. doi: 10.3389/fimmu.2018.00080
Tak, P. P., Bacchi, M., and Bertolino, M. (2006). Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur. J. Drug Metab. Pharmacokinet. 31, 109–116. doi: 10.1007/BF03191127
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tanaka, H., Miyazaki, N., Oashi, K., Teramoto, S., Shiratori, M., Hashimoto, M., et al. (2001). IL-18 might reflect disease activity in mild and moderate asthma exacerbation. J. Allergy Clin. Immunol. 107, 331–336. doi: 10.1067/mai.2001.112275
Tang, S., Huang, H., Hu, F., Zhou, W., Guo, J., Jiang, H., et al. (2013). Increased IL-33 in synovial fluid and paired serum is associated with disease activity and autoantibodies in rheumatoid arthritis. Clin. Dev. Immunol. 2013:985301. doi: 10.1155/2013/985301
Tao, X., Song, Z., Wang, C., Luo, H., Luo, Q., Lin, X., et al. (2017). Interleukin 36alpha attenuates sepsis by enhancing antibacterial functions of macrophages. J. Infect. Dis. 215, 321–332. doi: 10.1093/infdis/jiw535
Tardif, F., Goulet, J. P., Zakrazewski, A., Chauvin, P., and Rouabhia, M. (2004). Involvement of interleukin-18 in the inflammatory response against oropharyngeal candidiasis. Med. Sci. Monit. 10, BR239–BR249.
Todorovic, V., Su, Z., Putman, C. B., Kakavas, S. J., Salte, K. M., McDonald, H. A., et al. (2019). Small molecule IL-36gamma antagonist as a novel therapeutic approach for plaque psoriasis. Sci. Rep. 9:9089. doi: 10.1038/s41598-019-45626-w
Towne, J. E., Garka, K. E., Renshaw, B. R., Virca, G. D., and Sims, J. E. (2004). Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 279, 13677–13688. doi: 10.1074/jbc.M400117200
Towne, J. E., Renshaw, B. R., Douangpanya, J., Lipsky, B. P., Shen, M., Gabel, C. A., et al. (2011). Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602. doi: 10.1074/jbc.M111.267922
Tran, V. G., Kim, H. J., Kim, J., Kang, S. W., Moon, U. J., Cho, H. R., et al. (2015). IL-33 enhances host tolerance to Candida albicans kidney infections through induction of IL-13 production by CD4+ T Cells. J. Immunol. 194, 4871–4879. doi: 10.4049/jimmunol.1402986
van de Veerdonk, F. L., Stoeckman, A. K., Wu, G., Boeckermann, A. N., Azam, T., Netea, M. G., et al. (2012). IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. U S A. 109, 3001–3005. doi: 10.1073/pnas.1121534109
Verma, A. H., Richardson, J. P., Zhou, C., Coleman, B. M., Moyes, D. L., Ho, J., et al. (2017). Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2:eaam8834. doi: 10.1126/sciimmunol.aam8834
Verma, A. H., Zafar, H., Ponde, N. O., Hepworth, O. W., Sihra, D., Aggor, F. E. Y., et al. (2018). IL-36 and IL-1/IL-17 drive immunity to oral candidiasis via parallel mechanisms. J. Immunol. 201, 627–634. doi: 10.4049/jimmunol.1800515
Vigers, G. P., Dripps, D. J., Edwards, C. K. III, and Brandhuber, B. J. (2000). X-ray crystal structure of a small antagonist peptide bound to interleukin-1 receptor type 1. J. Biol. Chem. 275, 36927–36933. doi: 10.1074/jbc.M006071200
Vigne, S., Palmer, G., Lamacchia, C., Martin, P., Talabot-Ayer, D., Rodriguez, E., et al. (2011). IL-36R ligands are potent regulators of dendritic and T cells. Blood 118, 5813–5823. doi: 10.1182/blood-2011-05-356873
Vigne, S., Palmer, G., Martin, P., Lamacchia, C., Strebel, D., Rodriguez, E., et al. (2012). IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 120, 3478–3487. doi: 10.1182/blood-2012-06-439026
Viriyakosol, S., Walls, L., Okamoto, S., Raz, E., Williams, D. L., and Fierer, J. (2018). Myeloid differentiation factor 88 and interleukin-1R1 signaling contribute to resistance to Coccidioides immitis. Infect. Immun. 86:e00028-18. doi: 10.1128/IAI.00028-18
Vonk, A. G., Netea, M. G., van Krieken, J. H., Iwakura, Y., van der Meer, J. W., and Kullberg, B. J. (2006). Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J. Infect. Dis. 193, 1419–1426. doi: 10.1086/503363
Wang, J. P., Lee, C. K., Akalin, A., Finberg, R. W., and Levitz, S. M. (2011). Contributions of the MyD88-dependent receptors IL-18R, IL-1R, and TLR9 to host defenses following pulmonary challenge with Cryptococcus neoformans. PLoS One 6:e26232. doi: 10.1371/journal.pone.0026232
Warris, A., Netea, M. G., Verweij, P. E., Gaustad, P., Kullberg, B. J., Weemaes, C. M., et al. (2005). Cytokine responses and regulation of interferon-gamma release by human mononuclear cells to Aspergillus fumigatus and other filamentous fungi. Med. Mycol. 43, 613–621. doi: 10.1080/13693780500088333
Weinberg, E. O., Ferran, B., Tsukahara, Y., Hatch, M. M. S., Han, J., Murdoch, C. E., et al. (2019). IL-33 induction and signaling are controlled by glutaredoxin-1 in mouse macrophages. PLoS One 14:e0210827. doi: 10.1371/journal.pone.0210827
Werman, A., Werman-Venkert, R., White, R., Lee, J. K., Werman, B., Krelin, Y., et al. (2004). The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. U S A. 101, 2434–2439. doi: 10.1073/pnas.0308705101
Werner, J. L., Metz, A. E., Horn, D., Schoeb, T. R., Hewitt, M. M., Schwiebert, L. M., et al. (2009). Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 182, 4938–4946. doi: 10.4049/jimmunol.0804250
Wu, B., Meng, K., Ji, Q., Cheng, M., Yu, K., Zhao, X., et al. (2014). Interleukin-37 ameliorates myocardial ischaemia/reperfusion injury in mice. Clin. Exp. Immunol. 176, 438–451. doi: 10.1111/cei.12284
Wyman, T. H., Dinarello, C. A., Banerjee, A., Gamboni-Robertson, F., Hiester, A. A., England, K. M., et al. (2002). Physiological levels of interleukin-18 stimulate multiple neutrophil functions through p38 MAP kinase activation. J. Leukoc Biol. 72, 401–409.
Yuan, K., Zhao, G., Che, C., Li, C., Lin, J., Zhu, G., et al. (2017). Dectin-1 is essential for IL-1beta production through JNK activation and apoptosis in Aspergillus fumigatus keratitis. Int. Immunopharmacol. 52, 168–175. doi: 10.1016/j.intimp.2017.09.008
Zola, H., Flego, L., Wong, Y. T., Macardle, P. J., and Kenney, J. S. (1993). Direct demonstration of membrane IL-1 alpha on the surface of circulating B lymphocytes and monocytes. J. Immunol. 150, 1755–1762.
Keywords: fungi, fungal immunology, IL-1, Candida, Aspergillus
Citation: Griffiths JS, Camilli G, Kotowicz NK, Ho J, Richardson JP and Naglik JR (2021) Role for IL-1 Family Cytokines in Fungal Infections. Front. Microbiol. 12:633047. doi: 10.3389/fmicb.2021.633047
Received: 24 November 2020; Accepted: 15 January 2021;
Published: 10 February 2021.
Edited by:
Allan J. Guimaraes, Fluminense Federal University, BrazilReviewed by:
Flavio Vieira Loures, Federal University of São Paulo, BrazilTeresa Zelante, University of Perugia, Italy
Agostinho Carvalho, University of Minho, Portugal
Copyright © 2021 Griffiths, Camilli, Kotowicz, Ho, Richardson and Naglik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James S. Griffiths, amFtZXMuZ3JpZmZpdGhzQGtjbC5hYy51aw==