
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 26 February 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.631140
 T. Barani Devi1Krishnadas Devadas2
T. Barani Devi1Krishnadas Devadas2 Meekha George1A. Gandhimathi3
Meekha George1A. Gandhimathi3 Deepak Chouhan1,4
Deepak Chouhan1,4 R. J. Retnakumar1,4Sneha Mary Alexander1
R. J. Retnakumar1,4Sneha Mary Alexander1 Jijo Varghese2
Jijo Varghese2 Sanjai Dharmaseelan1
Sanjai Dharmaseelan1 Sivakumar Krishnankutty Chandrika1V. T. Jissa5
Sivakumar Krishnankutty Chandrika1V. T. Jissa5 Bhabatosh Das6G. Balakrish Nair1
Bhabatosh Das6G. Balakrish Nair1 Santanu Chattopadhyay1*
Santanu Chattopadhyay1*Helicobacter pylori infection in stomach leads to gastric cancer, gastric ulcer, and duodenal ulcer. More than 1 million people die each year due to these diseases, but why most H. pylori-infected individuals remain asymptomatic while a certain proportion develops such severe gastric diseases remained an enigma. Several studies indicated that gastric and intestinal microbiota may play a critical role in the development of the H. pylori-associated diseases. However, no specific microbe in the gastric or intestinal microbiota has been clearly linked to H. pylori infection and related gastric diseases. Here, we studied H. pylori infection, its virulence genes, the intestinal microbiota, and the clinical status of Trivandrum residents (N = 375) in southwestern India by standard H. pylori culture, PCR genotype, Sanger sequencing, and microbiome analyses using Illumina Miseq and Nanopore GridION. Our analyses revealed that gastric colonization by virulent H. pylori strains (vacAs1i1m1cagA+) is necessary but not sufficient for developing these diseases. Conversely, distinct microbial pools exist in the lower gut of the H. pylori-infected vs. H. pylori-non-infected individuals. Bifidobacterium (belonging to the phylum Actinobacteria) and Bacteroides (belonging to the phylum Bacteroidetes) were present in lower relative abundance for the H. pylori+ group than the H. pylori- group (p < 0.05). On the contrary, for the H. pylori+ group, genus Dialister (bacteria belonging to the phylum Firmicutes) and genus Prevotella (bacteria belonging to the phylum Bacteroidetes) were present in higher abundance compared to the H. pylori- group (p < 0.05). Notably, those who carried H. pylori in the stomach and had developed aggressive gastric diseases also had extremely low relative abundance (p < 0.05) of several Bifidobacterium species (e.g., B. adolescentis, B. longum) in the lower gut suggesting a protective role of Bifidobacterium. Our results show the link between lower gastrointestinal microbes and upper gastrointestinal diseases. Moreover, the results are important for developing effective probiotic and early prognosis of severe gastric diseases.
One of the most intriguing fundamental challenges in infectious disease research is to understand the combination of “microbial factors” and “other factors” that collectively determine clinical outcomes. For almost all bacterial infections, the pathogenic bacteria themselves and their virulence genes are extensively investigated, but the related “other factors” are often less studied or even ignored.
The gastric pathogen Helicobacter pylori is the main causative agent for gastric cancer (782,685 deaths/year) and gastric and duodenal ulcers (246,700 deaths/year), which together takes more than 1 million lives per year (GBD 2016 Causes of Death Collaborators, 2017; Bray et al., 2018). It is well known that the vacuolating cytotoxin A (vacA) and the cytotoxin-associated gene A (cagA) are the most critical bacterial genes that contribute to clinical outcomes (Isomoto et al., 2010; Hatakeyama, 2014). Both genes have polymorphic allelic structures and encode multitasking toxins. The VacA is a secreted toxin that penetrates gastric epithelial cells, produces large acidic vacuoles, and promotes cell death by stimulating intrinsic and extrinsic pathways of apoptosis, necrosis, and autophagy (Isomoto et al., 2010). In contrast, the CagA protein is injected by the bacterium into cells, where it interferes with actin cytoskeleton and tight junctions, and subverts pathways that regulate cell cycles (Hatakeyama, 2014). The role of these two toxins in gastric cancer and peptic ulcer were evaluated and confirmed by in vitro, in vivo, and clinical studies (Malfertheiner et al., 2014). For vacA, the s1, i1, and m1 alleles encode the signal sequence, intermediate and mid regions of the protein, respectively, and these alleles are considered more toxigenic than the alternative s2, i2, and m2 alleles (Atherton et al., 1995; Rhead et al., 2007; Basso et al., 2008; Chung et al., 2010). For cagA, the alleles that encode East-Asian CagA with “D” type-segments flanking its tyrosine phosphorylation motif EPIYA, rather than the alleles that encode Western type or “C” type-segments, are associated with more aggressive clinical outcomes (Isomoto et al., 2010; Hatakeyama, 2014). That said, however, ∼80–90% of the H. pylori-infected people, including those with the most virulent vacA and cagA alleles, do not develop any symptoms, while ∼10–20% suffer from different gastric diseases (Covacci et al., 1999). This implies that additional factors must contribute to the risks of overt diseases (Dorer et al., 2009).
The “other factors” that possibly play critical roles in determining the clinical outcomes are geography, host genetics, lifestyle, and gastrointestinal microbiota (Noto and Peek, 2017). Appreciating that the stomach harbors many bacteria other than H. pylori, and some of them may have a role in gastric diseases, the gastric microbiota was under intense study in the past decade (Bik et al., 2006; Li et al., 2009; Jo et al., 2016; Noto and Peek, 2017). Irrespective of the geographical locations, it was found that H. pylori colonization is associated with alteration of gastric microbiota, which is demonstrated by decreased microbial diversity and increased relative abundance of the bacteria under the phylum Proteobacteria, but the significance of these alterations are unknown (Andersson et al., 2008; Maldonado-Contreras et al., 2011; Das et al., 2017; Noto and Peek, 2017). Surprisingly, although the influences of intestinal microbiota in many non-communicable diseases including several cancers (e.g., colorectal cancer) are well-appreciated, not much attention was paid until recently to understand its significance in relation to H. pylori infection and associated gastric cancer (Saus et al., 2019). In murine model, it was shown that H. pylori colonization has distal effects including modulation in intestinal microbiota (Kienesberger et al., 2016). In human, H. pylori colonization in stomach was found to be associated with decreased abundance of the bacteria under the phylum Bacteroidetes as well as increased abundance of the bacteria under the phylum Proteobacteria and Firmicutes in intestine (Gao et al., 2018). However, no specific microbial species in the intestine has been shown to have a link with H. pylori infection and associated gastric diseases. This lack of information was the impetus for us to study the interrelations among the gastric H. pylori and its genotypes, the intestinal microbiota and the clinical status of hosts.
Patients included in this study were having various upper gastrointestinal symptoms and seeking care at the Department of Gastroenterology, Government Medical College, Trivandrum (TMC). Trivandrum is the capital of Indian state Kerala, which is located in extreme South-West part of the country (Menon, 2017). The entire state including Trivandrum has Arabian Sea to the West and Western Ghats Mountain to the East. It is suggested that humans lived in this geographical region during Neolithic Age (Menon, 2000, 2017). Later, peopling of Kerala happened during 2–3 AD through land and sea. The modern Keralite community has diverse (Negroid, Proto-Australoid, Dravidian, and Aryan) lineages (Menon, 2017).
Two gastric biopsies were collected during upper GI endoscopy. One of them was taken in 600 μl of autoclaved Brucella broth containing glycerol and the other in 200 μl of phosphate-buffered saline (PBS; 0.22 μm membrane filtered; autoclaved). A stool sample was also collected. The biopsy and stool samples were transported to the Microbiome Laboratory of Rajiv Gandhi Centre for Biotechnology (RGCB) at 4°C and were stored immediately in a −80°C freezer until further processing. Written informed consents from patients were taken. The study was approved by the Institutional Human Ethical Committee of TMC (approval number: 05/07/2016/MCT) and RGCB (approval number: IHEC/01/2017/18).
DNA was extracted from gastric biopsy as described elsewhere (Das et al., 2017). The DNA was diluted to 4 ng/μl, and 1 μl of DNA was used in 20 μl of reaction volume containing 10 μl of EmeraldAmp GT PCR Master Mix (TaKaRa) and 2 μl of forward and reverse primers specific for H. pylori ureB (Supplementary Figure S1). A patient is considered to have H. pylori infection if the collected gastric biopsy showed the presence of H. pylori either by ureB PCR or by culture (described below) or by both techniques (Table 1 and Supplementary Tables S1, S2).
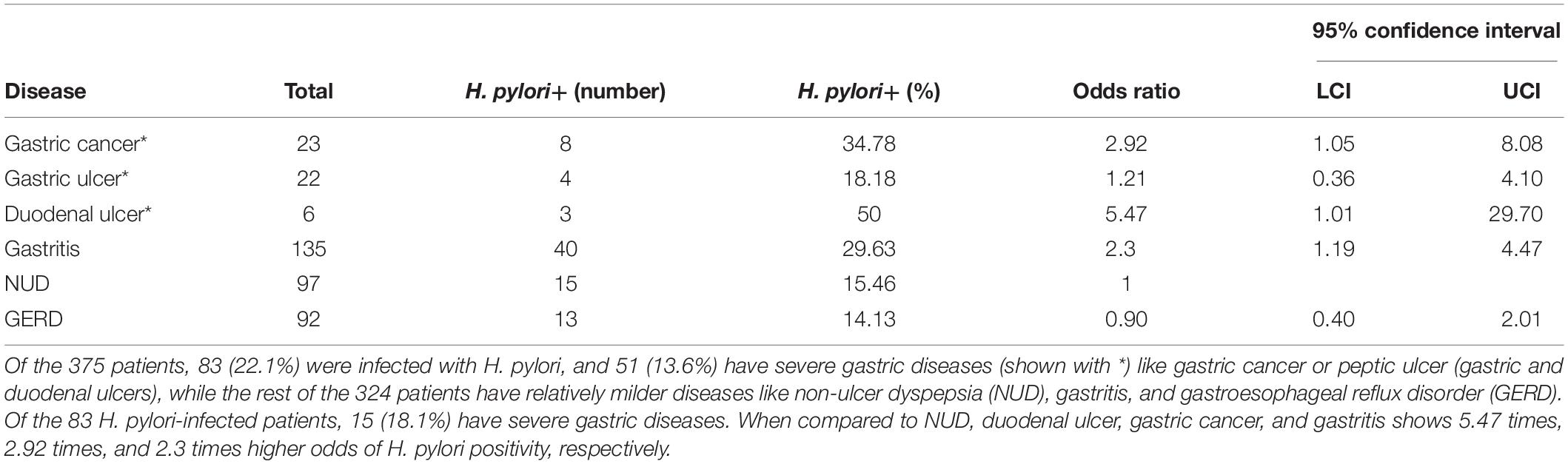
Table 1. The incidence of different upper GI diseases and the prevalence of H. pylori infection within the study group (N = 375).
The biopsies collected in Brucella broth were used for isolating H. pylori strains on Brain Heart Infusion (BHI) agar (2%) containing Dent (Oxoid), 0.4% IsoVitaleX (BBL), and 7% sheep blood. The inoculated plates were incubated in microaerobic condition (10% CO2, 5% O2, and 85% N2) at 37°C. H. pylori colonies (one colony for each patient) were further propagated as pure culture. H. pylori was identified by typical translucent colony morphology, Gram staining, as well as biochemical tests like urease, catalase, and oxidase (Supplementary Figure S2).
Genomic DNA was extracted from H. pylori strains as described elsewhere (Berg et al., 1997). RAPD-PCR was carried out in 25 μl of reaction volume containing 2.5 μl of primer 1,281 (10 pmole), 4 mM MgCl2, and 1.5 U of rTaq polymerase (TaKaRa) using previously described conditions (Berg et al., 1997; Mukhopadhyay et al., 2000). The multiplex PCR for vacA and cagA was performed using a modified protocol with 10 pmol of VA1-F/VA1-R, 10 pmol of VAG-F/VAG-R and 25 pmol of cag5c-F/cag5c-R, and 10 μl EmeraldAmp GT PCR Master Mix in 20 μl of reaction mix (Chattopadhyay et al., 2004). Characterization of other alleles of vacA and cagA by PCR and sequencing was done by previously described methods (Mukhopadhyay et al., 2000; Rhead et al., 2007; Chattopadhyay et al., 2012). The nucleotide sequences of the primers are given in Supplementary Table S3. For phylogenic analyses, PhyML 3.0 maximum likelihood trees were generated using bootstrapped (100 iterations) following estimation of an evolutionary model using full_modeltest_bootstrap genetic workflow in ETE3 python package (Guindon et al., 2010; Huerta-Cepas et al., 2016). The generated Newick tree files were used with the phylogram package for R to plot the phylogenetic trees (Wilkinson and Davy, 2018).
DNA was extracted from 200 mg of stool following a previously described protocol (Bag et al., 2016). For the preparation of metagenome library, 30 ng DNA was used to amplify the V3–V4 region of bacterial 16S rRNA genes for 26 cycles using KAPA HiFi HotStart PCR kit (KAPA Biosystems Inc., Boston, MA, United States) (Supplementary Table S3). The products were further amplified for 10 cycles by index PCR to add the Illumina sequencing barcoded adapters (Nextera XT v2 Index Kit, Illumina, United States). The products were sequenced using Illumina MiSeq following manufacturer’s protocol. The paired end V3–V4 reads (275 × 2) were demultiplexed using bcl2fastq, quality checked using FastQC, stitched using Fastq-join and analyzed using QIIME. The query sequences were clustered using UCLUST method against a curated chimera-free 16s rRNA database (Greengenes v.13.8). The taxonomies were assigned using RDP classifier to these clusters at ≥97% sequence similarity against the reference database. The generated BIOM file was used for further analysis and visualization. The box plot analysis was done by R.
For whole genome metagenome sequencing, 300 ng of genomic DNA was used after end-repairing (NEBnext ultra II end repair kit, New England Biolabs, MA, United States) and cleaning up with 1× AmPure beads (BeckmannCoulter, United States). The DNA samples were barcoded (LongAmp Taq 2× New England Biolabs, MA, United States) and cleaned up with 1.6× AmPure beads (Beckmann-Coulter, United States). The end-repairing was performed using NEBnext (New England Biolabs, MA, United States) and adapter ligation was performed for 10 min using NEB blunt/TA ligase (New England Biolabs, MA, United States). Library mix was cleaned up using 0.6× Ampure beads and finally eluted in 15 μl of elution buffer. The processed DNA samples were sequenced on GridION X5 (Oxford Nanopore Technologies, Oxford, United Kingdom) using SpotON flow cell (R9.4) in a 48 h sequencing protocol on MinKNOW 2.1 v18.05.5. Nanopore raw reads (“fast5” format) were base called (“fastq5” format) and demultiplexed using Albacore v2.3.1. The reads were compared against NCBI nr database using the diamond tool. The diamond BLASTX alignments were further converted to MEGAN readable format by using the NCBI taxonomy to summarize and order the results. MEGAN GUI is then used to estimate and interactively explore the taxonomical content by checking the read assignment from phylum to species level classification.
PCR with primers specific for Bifidobacterium species (Supplementary Table S3) was done using stool metagenomic DNA in a 20 μl PCR reaction. Similarly, PCR with DNA extracted metagenomically from gastric biopsies was also performed. We also performed quantitative PCR (qPCR) in triplicate for the stool DNA as well as the gastric biopsy DNA using Bifdobacterium species-specific primers. The qPCR was performed with Thermo Power SYBR Green Master Mix using 200 nM primers and 50 ng DNA. Standard program with annealing temperature of 55°C in Applied Biosystems QuantStudio 7 instrument was used.
For analyzing H. pylori infection status, clinical status and sex of the individual Chi-squared test was performed using Intercooled Stata 14.1 software to test the significance of the association between variables. Binary logistic regression was used to estimate the odds ratios with 95% confidence intervals. For metagenomics analysis, the statistical significances among the patient groups were calculated using the Kruskal–Wallis test (Kruskal–Wallis, p < 0.05).
The study includes a total of 375 adult (male: 181; female: 194; average age: 48.5 years) residents of Trivandrum city and suburbs. As shown in Table 1, the prevalence of H. pylori infection is remarkably low (83 of 375; 22.1%) in Trivandrum. Within the H. pylori-infected group, the total prevalence of severe gastric diseases (15 of 83; 18.1%) like gastric cancer and peptic ulcer (duodenal and gastric ulcers) are similar to other geographic regions. However, the distributions of different diseases were noticeably different from the rest of the country. For example, it is known that for most Indian states that duodenal ulcer is the major clinical outcome and gastric cancers are relatively less prevalent (Supplementary Table S4; Dhakal and Dhakal, 2018). In contrast, for Trivandrum, although the prevalence of total gastric cancer (23/375 or 6.1%) and gastric ulcer (22 or 5.9%) are high, the prevalence of duodenal ulcer (6/375 or 1.6%) is low (Table 1 and Supplementary Table S4).
Of the 83 H. pylori-infected patients, 35 were male (42.2%) and 48 were female (57.8%) (Table 2). The prevalence of severe gastric diseases like gastric cancer and peptic ulcer are significantly more in males (Table 2). Among the H. pylori-infected males, 34.3% had severe disease types, while in the corresponding female population, it was only 6.25%. The observed association between sex and disease status is statistically significant (p = 0.001) (Table 2).
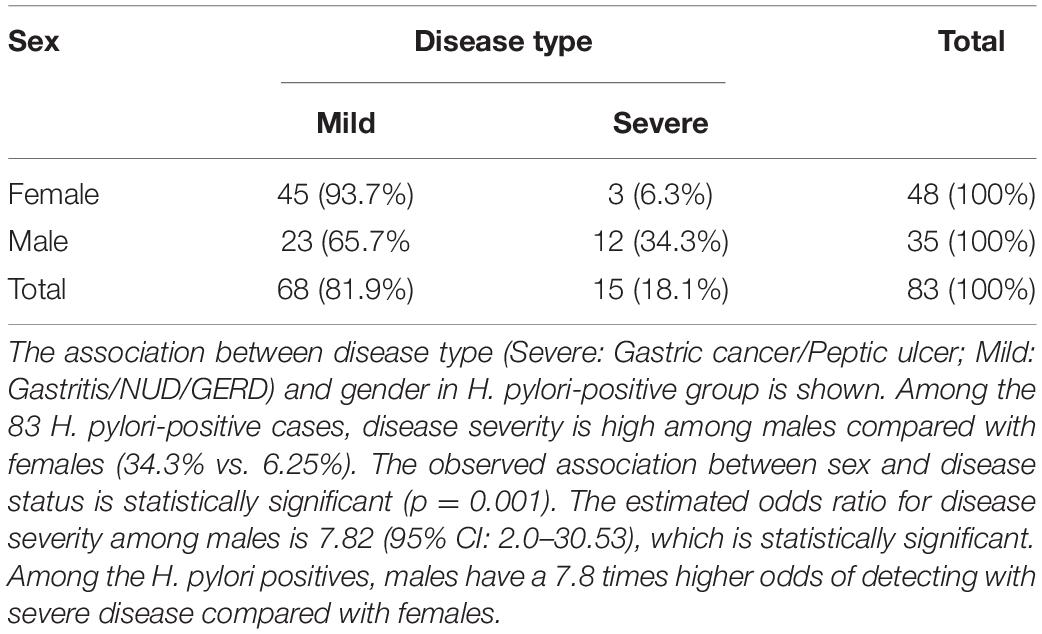
Table 2. The disease types of the H. pylori positive (N = 83), males (N = 35) and females (N = 48) in the study.
Of the 83 H. pylori positive cases, 42 were positive by culture. DNA extracted from all 42 isolated H. pylori strains were used for genotyping the vacA signal sequence (s), mid (m), and intermediate (i) region alleles as well as the cagA 5′end conserved region and 3′end variable region. Of the 42 strains, 39 (92.9%) carried the toxigenic vacAs1 allele, while three strains (7.1%) carried the non-toxigenic vacAs2 allele (Figures 1A,B and Table 3). The prevalence of vacAm1 (32 of 42; 76.2%) was higher than the prevalence of vacAm2 (10 of 42; 23.8%). The cagA gene is present in 38 (90.5%) of 42 strains (Figures 1A,B and Table 3). Four strains (9.5%), which were negative for cagA gene, gave the 550 bp amplicon in cag-empty site PCR confirming that these four strains lacked the entire cag-PAI (Figure 1C and Table 3). The prevalence of vacAi1 (37 of 42; 88.1%) allele was higher than the prevalence of vacAi2 (5 of 42; 11.9%) allele (Figures 1D,E and Table 3). When the genotype data were combined, it was found that in Trivandrum, the H. pylori strains predominantly carry the most toxigenic vacAs1i1m1cagA+ genotype (73.8%), followed by the vacAs1i1m2cagA+ genotype (11.9%) (Table 3 and Supplementary Table S5). The strains that carry the less toxigenic vacAs1i2m2cagA+ (4.8%) and non-toxigenic vacAs2i2m2cagA− (7.1%) genotypes are relatively uncommon. One strain (TMC280) carried a rare vacAs1i1m1cagA− genotype (Figure 2, Table 3, and Supplementary Table S5). DNA fingerprinting analysis using randomly amplified polymorphic DNA (RAPD)-PCR for 11 representative strains of different genotypes showed unique pattern for each strain, suggesting that the Trivandrum H. pylori strains, like the H. pylori strains from other geographic regions, are highly diverse (Supplementary Figure S3). Phylogenetic analyses revealed that the vacA of Trivandrum H. pylori strains are related to the vacA of H. pylori strains isolated from South Asia (India, Bangladesh, etc.), while the cagA of the Trivandrum H. pylori strains formed cluster with the Western cagA (Figure 3).
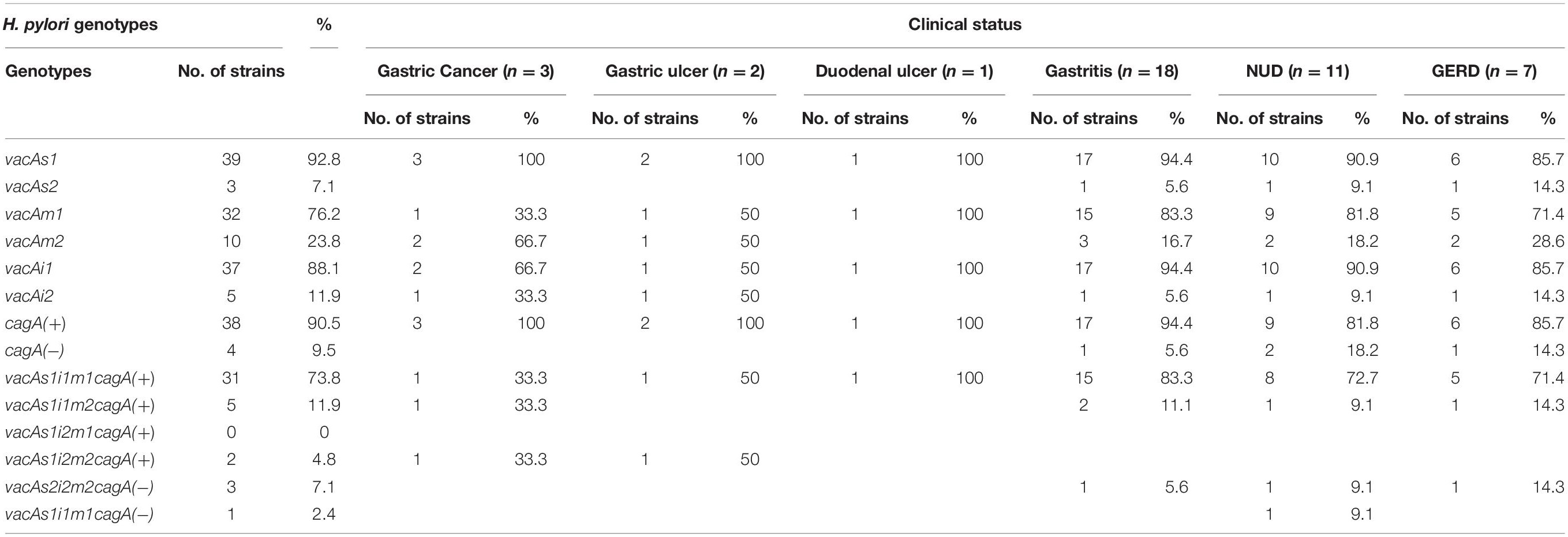
Table 3. H. pylori genotypes and the clinical status of the host.
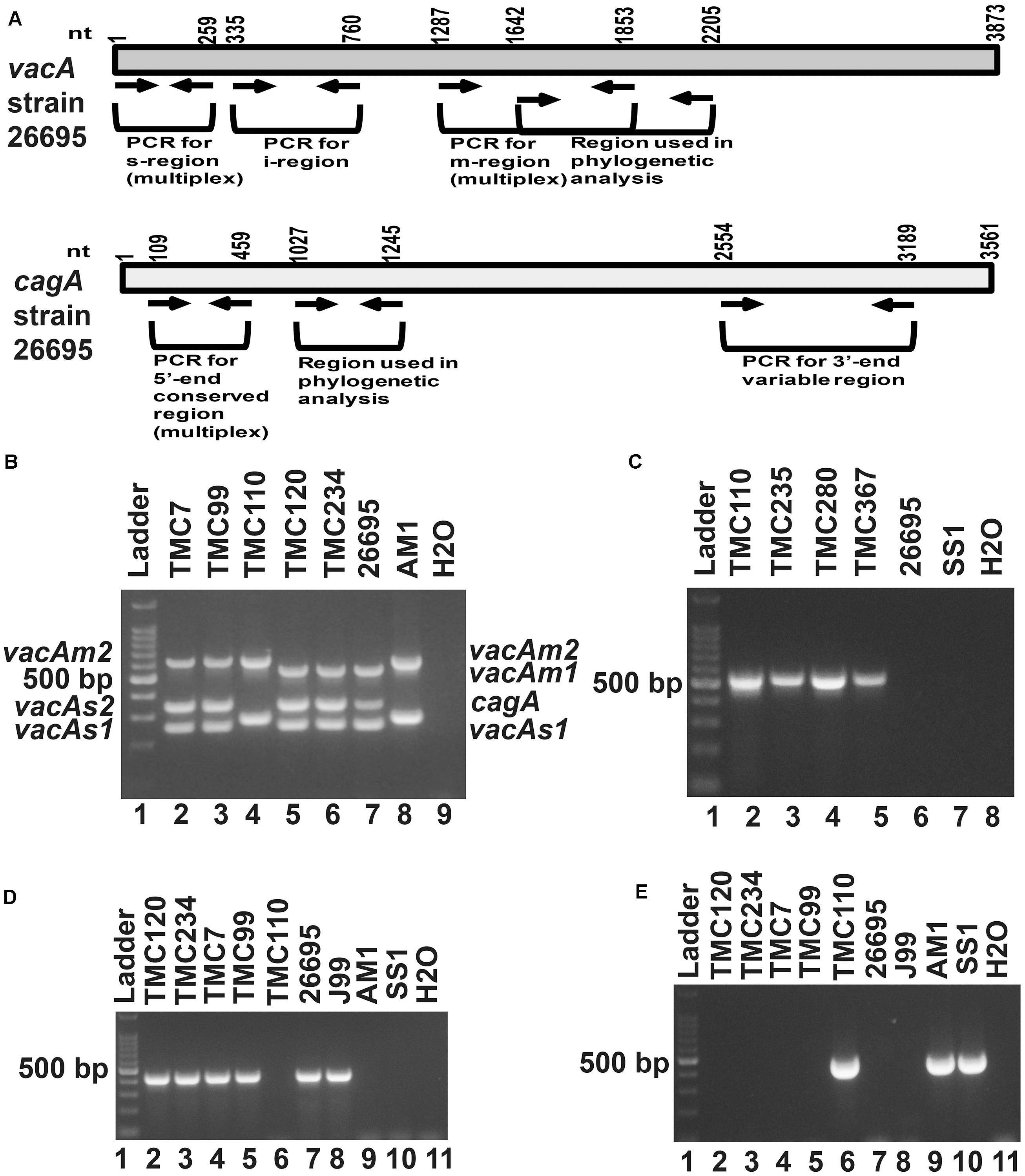
Figure 1. (A) Schematic diagram of the vacuolating cytotoxin A (vacA) and cytotoxin associated gene A (cagA) of the genome sequenced strain, 26,695 showing the nucleotide positions. The regions used in the analyses are indicated. (B) Multiplex PCR for genotyping the vacAs1, vacAs2, vacAm1, vacAm2, and cagA of H. pylori. The strains TMC7 and TMC99 (lane 2 and 3) have vacAs1m2cagA+; the strain TMC110 (lane 4) has vacAs2m2cagA–; the strains TMC120 and TMC234 (Lanes 5 and 6) have vacAs1m1cagA+ genotypes. The strains 26,695 (lane 7) and AM1 (lane 8) were used as positive controls for vacAs1m1cagA+ and vacAs2m2cagA- genotypes, respectively. Water (lane 9) was used as negative control. (C) The strains, TMC110, TMC235, TMC280, TMC367, which did not give amplicon for cagA in multiplex PCR, gave amplicon for the cag-empty site PCR. (D) PCR for the detection of vacAi1 allele. The strains TMC120, TMC 234, TMC7, and TMC99 were found to carry the vacAi1 allele, while the strain TMC110 did not give the amplicon. (E) PCR for the detection of vacAi2 allele. The strain TMC110 was found to carry the vacAi2 allele.
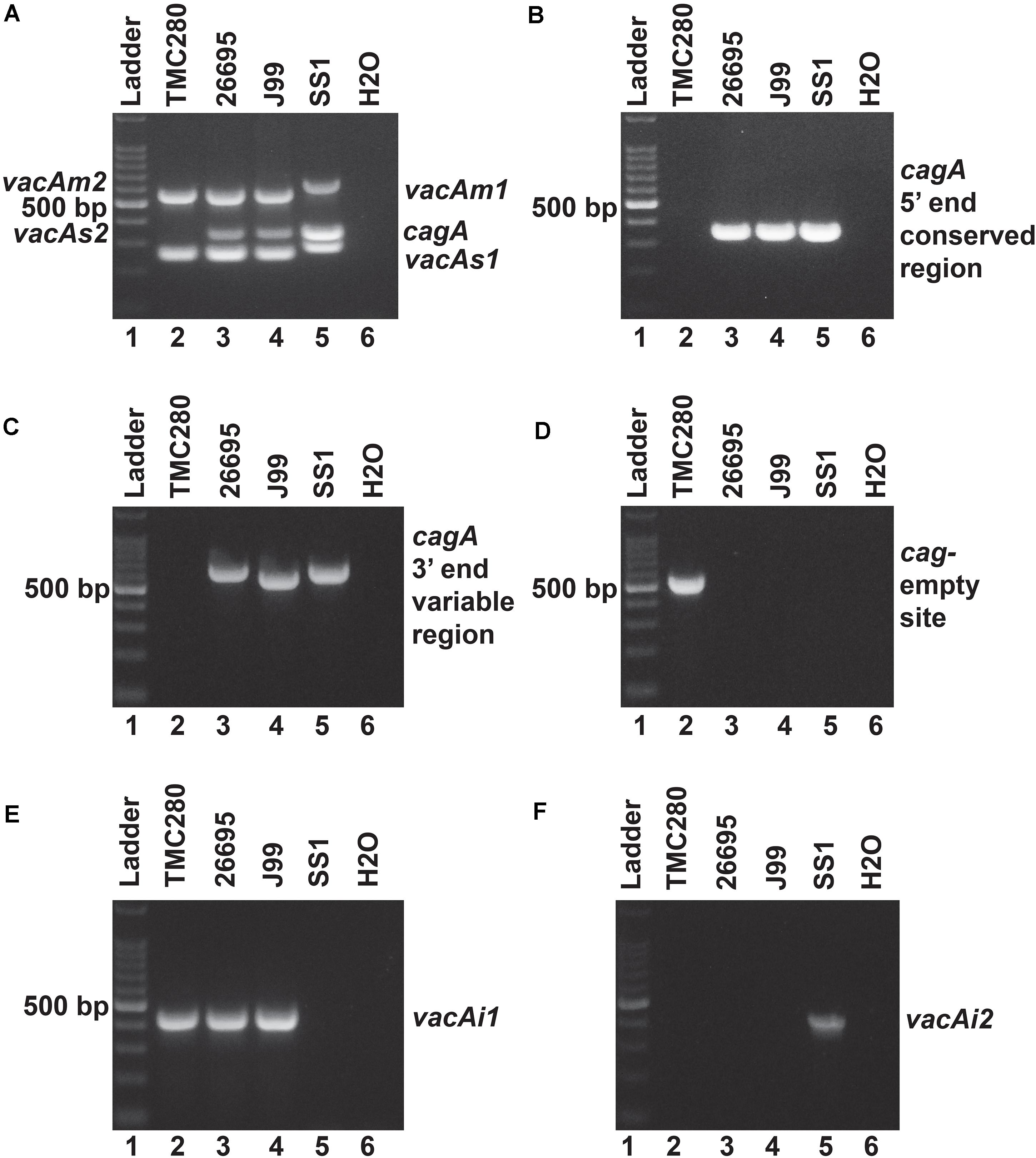
Figure 2. (A) Multiplex PCR for the characterization of vacA alleles and cagA gene of strain TMC280. The strain TMC280 carry vacAs1m1cagA- genotype, while the strains used as positive controls like 26,695 (vacAs1m1cagA+), J99 (vacAs1m1cagA+), and SS1 (vacAs2m2cagA–) gave amplicons at expected sizes. (B) No cagA specific amplicon were obtained for the strain TMC280 when the primers are targeted at the 5′ end conserved regions of the cagA gene. The positive controls, 26,695, J99, and SS, as expected, gave specific amplicons. (C) The PCR targeting the 3′ end variable region of the cagA gene. The strain TMC280 did not give any amplification with these primers, while the positive controls, 26,695, J99, and SS1, did. (D) The cag-empty site PCR. Only the strain TMC280, but not the strains 26,695, J99, and SS, gave amplicon for this PCR. (E) The PCR for the vacAi1 allele. The strain TMC280, along with 26,695 and J99, gave amplicon for the vacAi1 allele. The strain SS1, as expected did not give the vacAi1 specific amplicon. (F) The PCR for the vacAi2 allele. The strain TMC280 as well as the strains 26,695 and J99 were negative for this PCR, but the strain SS1 was positive. Therefore, it is confirmed that the strain TMC280 has vacAs1i1m1cagA- genotype.
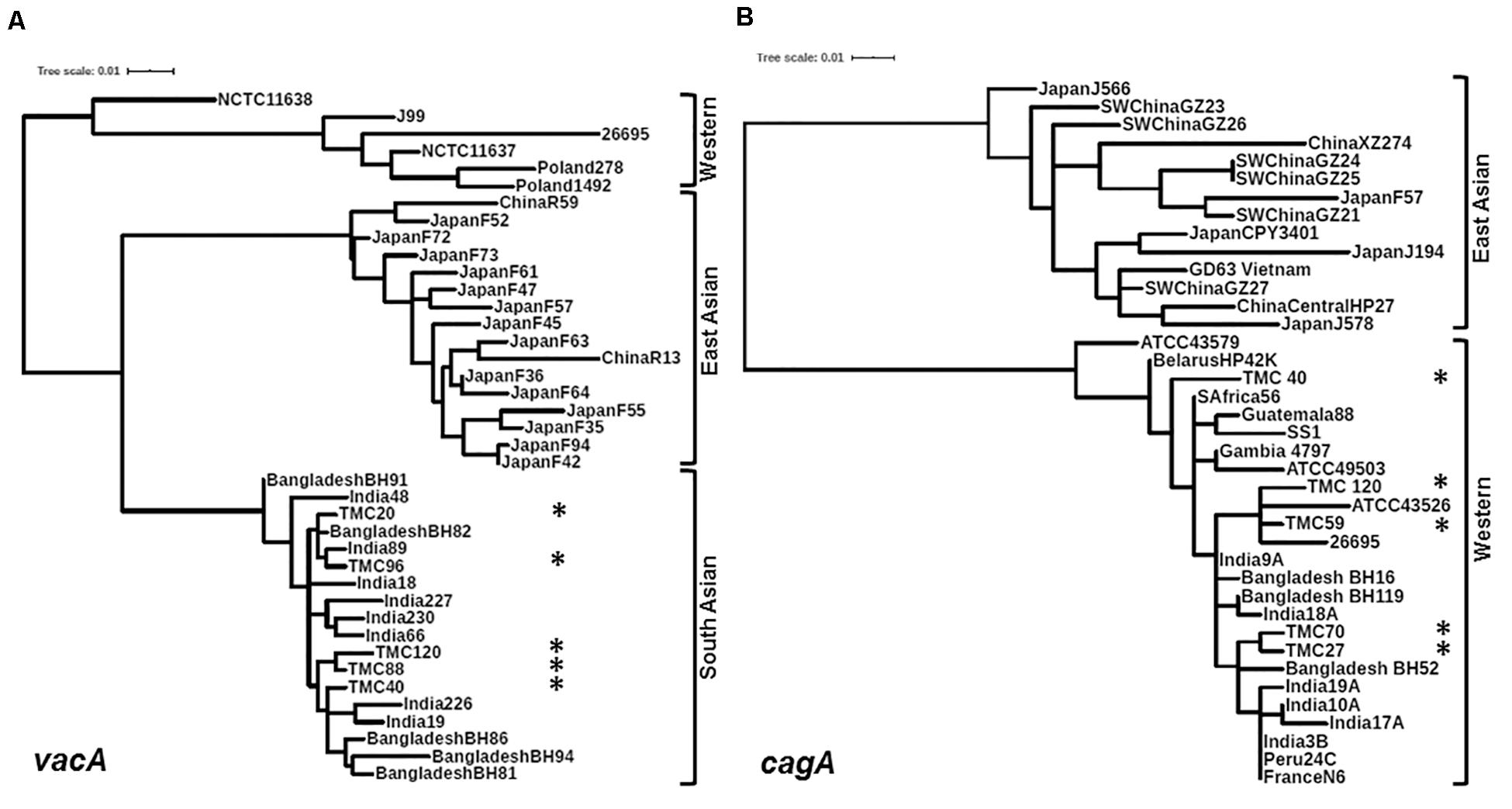
Figure 3. (A) Phylogenetic analysis of the vacAm1. The vacAm1 of Trivandrum H. pylori strains (named as TMC and shown as asterisks) are closely related to the vacAm1 of the H. pylori strains from South Asia, but differ from the vacAm1 of H. pylori strains from Western world and East Asia. The H. pylori vacAm1 sequences and their GenBank accession numbers are: TMC20 (MN968508); TMC40 (MN968509); TMC88 (MN968510); TMC96 (MN968511); TMC120 (MN968512); NCTC 11638 (HPU07145); Poland 1492 (AF097570); Poland 278 (AF097571); NCTC 11637 (AF049653); J99 (AE001511); 26695 (AE000598); Japan F73 (AF049652); Japan F72 (AF049651); Japan F52 (AF049631); China R59 (AF035611); Japan F61 (AF049645); Japan F57 (AF049634); Japan F35 (AF049625); Japan F55 (AF049632); Japan F47 (AF049629); Japan F45 (AF049628); Japan F94 (AF049640); Japan F42 (AF049626); Japan F36 (AF049642); Japan F64 (AF049647); Japan F63 (AF049635); China R13 (AF035610); Bangladesh BH91 (LC187447); India 48 (AF220112); India 18 (AF220110); India 227 (AF220116); India 230 (AF220117); India 66 (AF220113); India 89 (AF220114); Bangladesh BH82 (LC187444); Bangladesh BH94 (LC187448); India 226 (AF220115); India 19 (AF220111); Bangladesh BH81 (LC187443); Bangladesh BH86 (LC187446). (B) Phylogenetic analysis of the cagA 5′ end conserved region. The cagA of H. pylori strains isolated from Trivandrum (named as TMC and shown as asterisks) formed cluster within the cagA of Western H. pylori strains and not with the East Asian H. pylori strains. The H. pylori cagA sequences and their accession numbers are: TMC27 (MN968503); TMC40 (MN968504); TMC120 (MN968505); TMC59 (MN968506); TMC70 (MN968507); SW China GZ26 (KR154755); SW China GZ23 (KR154752); XZ274 China (NC_017926); Japan J566 (AB017922); SW China GZ27 (KR154756); GD63 Vietnam (CP031558); Japan J578 (AB017923); China Central HP27 (DQ306710); Japan CPY3401 (AY121840); Japan J194 (AB017921); Japan F57 (AB190935); SW China GZ21 (KR154750); SW China GZ24 (KR154753); SW China GZ25 (KR154754); ATCC 43579 (AB015414); HP42K Belarus (NZ_CP034314); India 17A (AF202223); S Africa 56 (AF198471); Guatemala 88 (AF198472); ATCC 49503 (AB015415); Gambia 4797 (AF198469); SS1 (KR154757); Bangladesh BH16 (LC187624); India 9A (AF202221); Bangladesh BH119 (LC187626); India 18A (AF202224); ATCC 43526 (AB015413); 26695 (AE000511); Bangladesh BH52 (LC187641); India 3B (AF202219); India 10A (AF202222); India 19A (AF202225); Peru 24C (AF198473); N6 France (CAHX01000001 to CAHX01000054).
The 3′end variable region of the cagA gene that encodes variable numbers of EPIYA motifs and spacer sequences was studied by PCR and sequencing (Figure 4A). All cagA + H. pylori strains carried Western type-specific sequence (WSS) with C-segment (Figure 4B). Of the 38 cagA + strains, 28 (73.7%) were AB-C type with three EPIYA motifs, and seven (18.4%) were AB-C-C type with four EPIYA motifs. Two strains (5.2%) did not carry EPIYA motifs at the C segment, and they were AB- type with only two EPIYA motifs. One strain (2.7%) was found to have one EPIYA motif at the A site and two EPIYA motifs at the C sites, but no EPIYA motif at the B site, and therefore, this was the A-C-C type CagA. However, no association with the number of EPIYA motifs at the C-segment was found to have any clinical correlation (Supplementary Table S6).
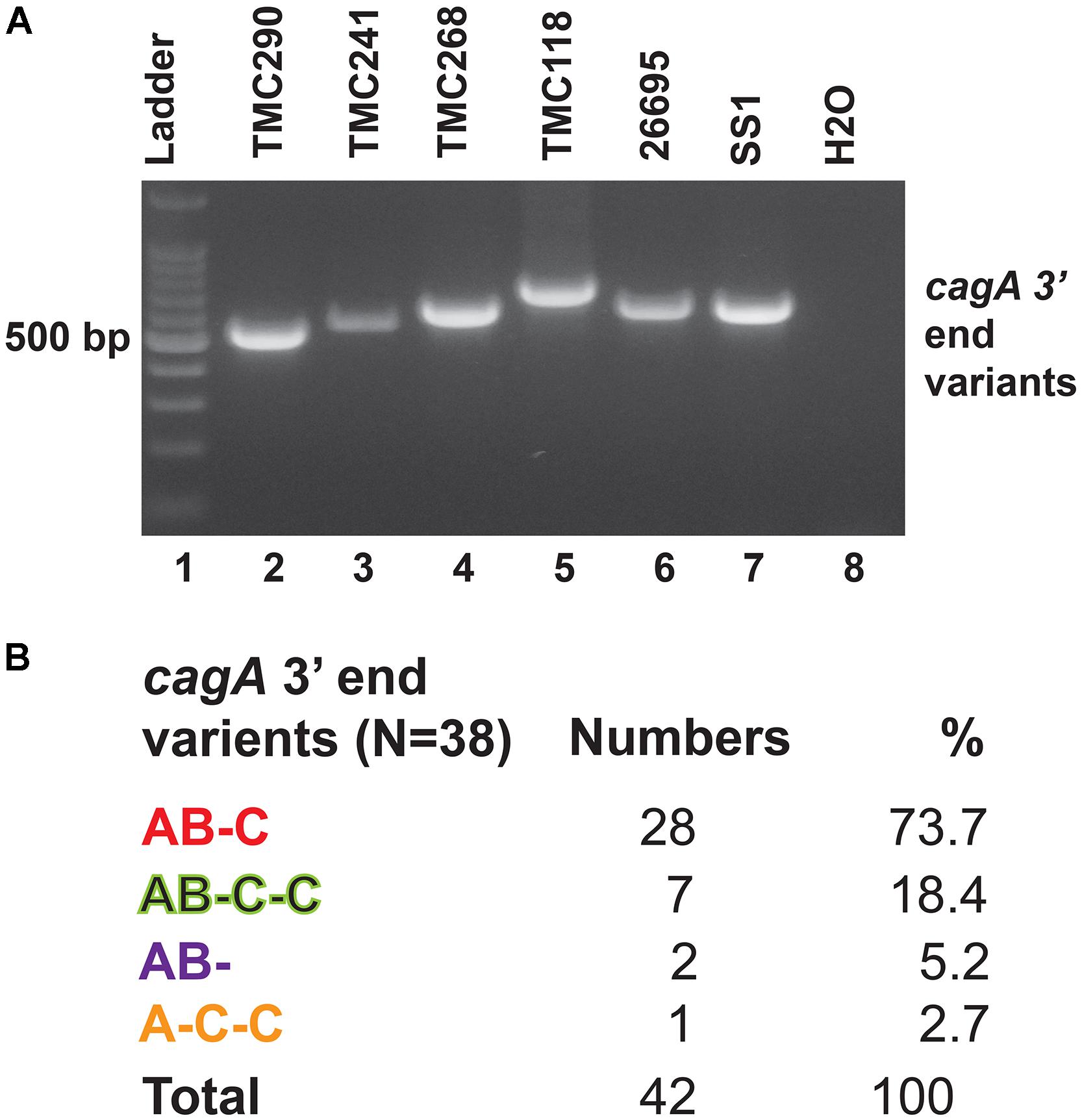
Figure 4. (A) PCR targeting the 3′ end variable regions of the cagA gene that encodes the EPIYA motifs. The amplicon sizes varied due to the variable number of EPIYA motifs and the spacer regions. (B) Different types of CagA types and their prevalence in Trivandrum. The nucleotide sequences (GenBank accession numbers MN968513 to MN968550) of the cagA genes were converted to the amino acid sequences and analyzed.
Likewise, no association with clinical status of the host and genotypes of the H. pylori strains could be made (Table 3 and Supplementary Tables S6, S7). Therefore, the infection with putatively pathogenic types of H. pylori strains is dominant in all patient populations, but only certain people develop the severe gastric diseases like gastric cancer and peptic ulcer. These data suggest that, in addition to H. pylori infection and associated virulence genes, other factors contribute in determining clinical outcomes of the infected individuals.
As subsets of the study population, a total of 60 patients (30 H. pylori+ with average age 50.5 years; H. pylori- with average age of 50.4 years; same male to female ratio) were included in the gut microbiota analysis using Illumina MiSeq (275 × 2) platform (Supplementary Table S8). The rarefaction curve used as a measure of depth of sequencing is shown in Supplementary Figure S4. It is evident that for most patients, the abundant gut bacterial phyla is Firmicutes, followed by Bacteroidetes, Proteobacteria, Tenericutes, and Actinobacteria (Supplementary Figures S5, S6). However, for few patients like TMC27 (a patient with GERD; infected with H. pylori), TMC50 (a patient with gastritis; not infected with H. pylori), TMC58 (a patient with non-ulcer dyspepsia; not infected with H. pylori), TMC131 and TMC156 (two patients with gastritis; infected with H. pylori). Proteobacteria is the dominant phylum in the gut (Figure 5A and Supplementary Figure S6). No patient with severe gastric diseases like gastric cancer or peptic ulcer was found to have Proteobacteria as dominant phylum in the fecal microbiome. Overall, as shown in the heat map, four phyla, Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria showed wide variation in abundance among individuals (Figure 5A). Principal Coordinates Analysis (PCoA) showed wide variations among subjects, but many H. pylori+ cases are found to be closely related to each other (Supplementary Figure S7). Likewise, many H. pylori- cases are also closely related to each other. This strongly suggests that the H. pylori+ group and the H. pylori- group may have respective unique components, which are distinct from the other group.
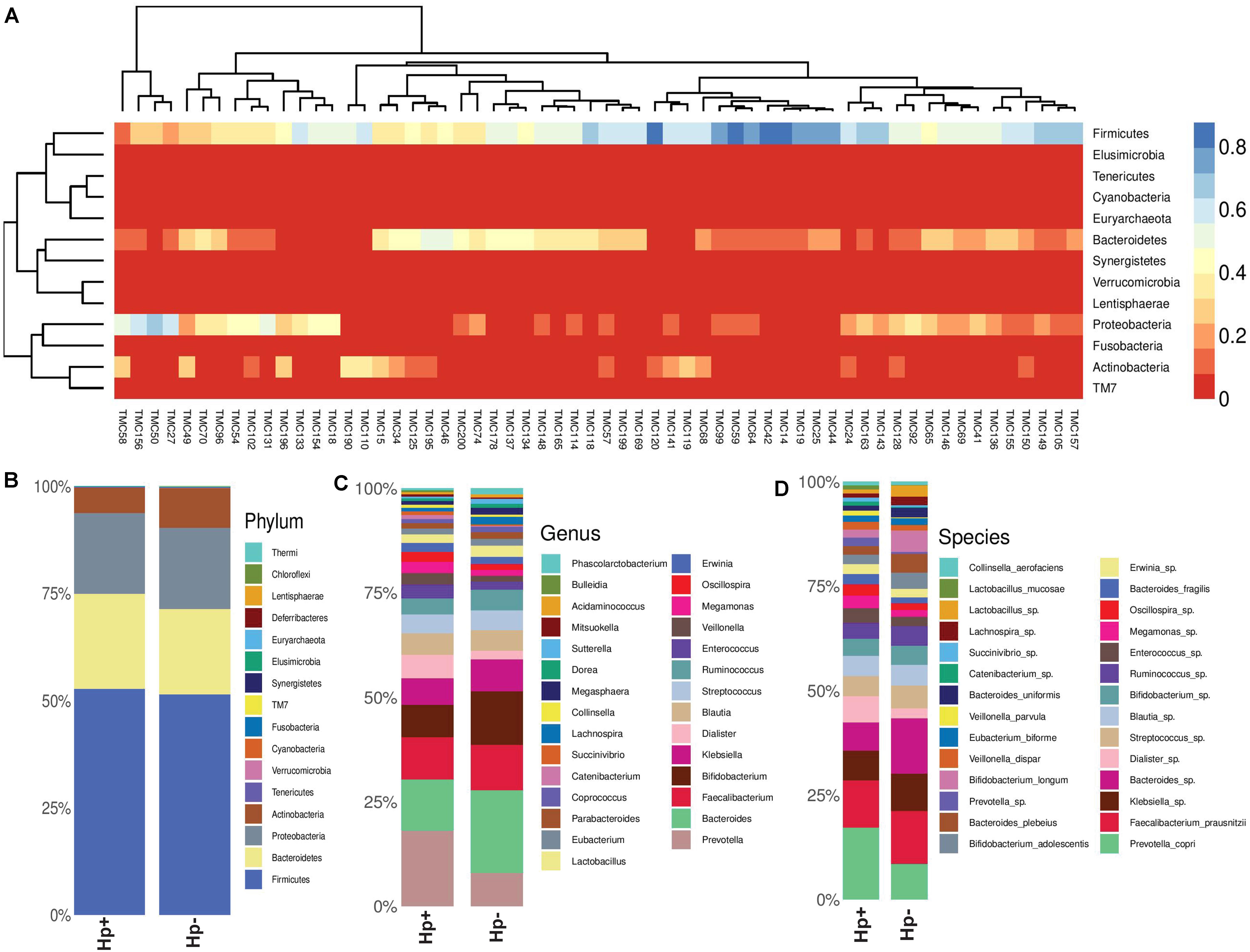
Figure 5. (A) Heat map showing the relative abundances of different phyla for each sample (N = 60). (B) Relative abundances of different phyla in gut shown in stacked column for the H. pylori+ (Hp+) and the H. pylori− (Hp−) groups. The Hp+ group has less relative abundance of the Phylum Actinobacteria. (C) Relative abundances of different genera in gut shown in stacked column for the H. pylori+ (Hp+; N = 30) and the H. pylori− (Hp−; N = 30) groups. The Hp+ group has less relative abundance of the genus Bifidobacterium. (D) Relative abundances of different bacterial species in gut shown in stacked column for the H. pylori+ (Hp+; N = 30) and the H. pylori− (Hp−; N = 30) groups. The Hp+ group has less relative abundance of the Bifidobacterium longum and B. adolescentis. The bacterial species that vary between the H. pylori+ and the H. pylori− groups are B. longum (Kruskal-Wallis, p = 0.009), B. adolescentis (p = 0.03), B. bividum (p = 0.004), B. plebeius (p = 0.05), B. uniformis (p = 0.04), and P. copri (p = 0.003).
To this end, we looked into comparative analysis of the gut microbiota composition for the H. pylori+ (N = 30) and H. pylori− (N = 30) patients irrespective of clinical status of the host. Most of the H. pylori+ patients have more diverse gut microbiota than their respective age- and sex-matched H. pylori- counterparts (Supplementary Table S9). Collectively, the H. pylori+ group has more alpha diversity than the H. pylori- group as was discerned by the Shannon index, Simpson index, and Chao1 index analyses (Supplementary Figure S8). When the bacterial relative abundance was taken into account between two groups, it was noticed that the abundance of phylum Actinobacteria was lower and phylum TM7 (Saccharibacteria) was higher in the H. pylori+ group than in the H. pylori- group (Figure 5B). Similar analysis at the genus level showed that Bifidobacterium (belonging to the phylum Actinobacteria) and Bacteroides (belonging to the phylum Bacteroidetes) were present in lower relative abundance for the H. pylori+ group than the H. pylori- group (Figure 5C). Conversely, for the H. pylori+ group, genus Dialister (bacteria belonging to the phylum Firmicutes) and genus Prevotella (bacteria belonging to the phylum Bacteroidetes) were present in higher abundance as compared to the H. pylori- group (Figure 5C). The species of the Bifidobacterium that vary between the H. pylori+ and the H. pylori- groups are found to be B. longum (Kruskal–Wallis, p = 0.009), B. adolescentis (p = 0.03), and B. bividum (p = 0.004), although some of the species remained unidentified at this point of the analyses (Figure 5D). Similarly, for the Bacteroides and Prevotella, the species that could be identified by the 16S rRNA gene analyses are B. plebeius (p = 0.05), B. uniformis (p = 0.04), and P. copri (p = 0.003), respectively, while it was noticed that other species were also present but were not identifiable by V3–V4 regions of the 16S rRNA gene sequence analyses (Figure 5D). The species of the genus Dialister (p = 0.05) also could not be identified by this analysis (Figure 5D).
Since our analyses pointed out differences in the composition of the gut microbiota between the H. pylori+ and the H. pylori- patients, our next aim was to find the distinctiveness in the gut microbiota of the H. pylori+ patients with severe gastric disorders. Therefore, we compared H. pylori+ patients with gastric cancer or gastric ulcer (CA/GU-Hp+) with patients having milder clinical outcomes like non-ulcer dyspepsia or gastritis with H. pylori infection (NUD/GAS-Hp+), non-ulcer dyspepsia, or gastritis without H. pylori infection (NUD/GAS-Hp−) and gastroesophageal reflux disease without H. pylori infection (GERD-Hp−). The metadata related to these four patient groups are given in Supplementary Table S10. It was noticed that the CA/GU-Hp+ group has remarkably low relative abundance of bacteria belonging to the phylum Actinobacteria (Figure 6A). Further analyses revealed that the most significant uniqueness of the CA/GU-Hp+ patients is the low relative abundance of the genus Bifidobacterium (under the phylum Actinobacteria) (Figure 6B). The abundance of Bifidobacterium in the CA/GU-Hp+ group was found to be significantly lower than the GERD-Hp− (p = 0.0181), NUD/GAS-Hp− (p = 0.0117), and NUD/GAS-Hp+ (p = 0.0229) groups (Figure 6C). This finding was further confirmed by heat map (Supplementary Figure S9). Further species level analysis revealed that several Bifidobacterium species like B. adolescentis (p = 0.005 with respect to NUD/GAS-Hp−; p = 0.003 with respect to NUD/GAS-Hp+; p = 0.009 with respect to GERD-Hp−), B. longum (p = 0.002 with respect to NUD/GAS-Hp−; p = 0.008 with respect to NUD/GAS-Hp+; p = 0.02 with respect to GERD-Hp−), and B. bifidum (p = 0.01 with respect to NUD/GAS-Hp−; p = 0.01 with respect to GERD-Hp−) were present at significantly lower relative abundance specifically for the CA/GU-Hp+ group compared with the other groups (Figure 6D). The CA/GU-Hp+ group also have high Oscillospira (p = 0.007 with respect to NUD/GAS-Hp−; p = 0.002 with respect to NUD/GAS-Hp+; p = 0.02 with respect to GERD-Hp−). However, we noticed that 16S rRNA gene analyses were not able to identify all Bifidobacterium species that are present at high abundance in the gut of NUD/GAS-Hp+, NUD/GAS- Hp−, and GERD-Hp− patients but were present at a significantly lower abundance in the gut of CA/GU-Hp+ patients (Figure 6D).
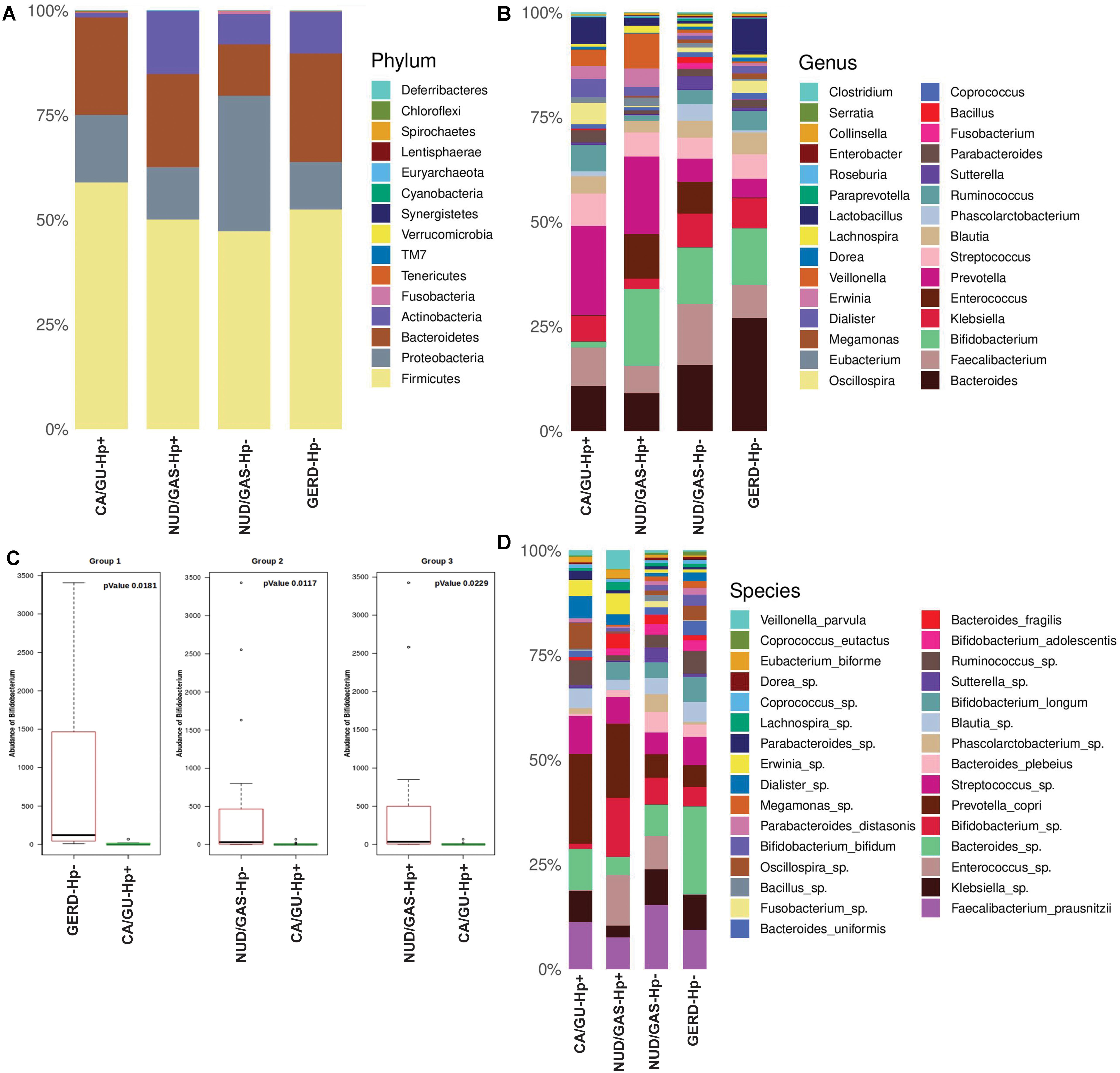
Figure 6. (A) Relative abundances of different phyla in gut shown in stacked column for the CA/GU-Hp+, NUD/GAS-Hp+, NUD/GAS− Hp−, and GERD-Hp− groups. The CA/GU-Hp+ group have low relative abundance of Actinobacteria as compared to the other groups. (B) Relative abundances of different genera in gut shown in stacked column plot for the CA/GU-Hp+, NUD/GAS-Hp+, NUD/GAS- Hp−, and GERD-Hp− groups. Relative abundance of Bifidobacterium for the CA/GU-Hp+ group is significantly low. (C) Box plot analysis showing that the abundance of Bifidobacterium is significantly lower in the CA/GU-Hp+ group than the NUD/GAS-Hp+, NUD/GAS- Hp−, and GERD-Hp− groups. (D) Relative abundances of different bacterial species in gut shown in stacked column for the CA/GU-Hp+, NUD/GAS-Hp+, NUD/GAS- Hp−, and GERD-Hp− groups. Significantly low relative abundances of Bifidobacterium bifidum, Bifidobacterium adolescentis, and Bifidobacterium longum are identified for the CA/GU-Hp+ group.
The 16S rRNA gene analyses could not resolve all Bifidobacterium species. Therefore, we decided to identify them by whole genome metagenome sequencing. For this experiment, we have chosen the Oxford Nanopore technology in GridION X5 platform for its longer read length. A total of six patients were chosen. Three of them are CA/GU-Hp+ and three are NUD/GAS-Hp+. The samples are age and sex matched and all individuals are H. pylori+ to avoid any bias. Details of the metadata are given in Supplementary Table S11. The Nanopore read statistics for each sample is given in Supplementary Table S12. The abundance of each domain in each sample is shown in Supplementary Table S13. Our analysis has identified a total of eight Bifidobacterium species, which were remarkably different between the two groups and for each age- and sex-matched pairs (Table 4). Seven (B. adolescentis, B. bifidum, B. breve, B. longum, B. moukalabense, B. pseudocatenulatum, and B. reuteri) of the eight Bifidobacterium species were present at a very low abundance in the intestine of the CA/GU-Hp + individuals than the corresponding age- and sex-matched NUD/GAS-Hp+ individuals (Table 4). The lower abundance of the Bifidobacterium in CA/GU-Hp+ individuals compared with the NUD/GAS-Hp+ individuals is also confirmed with regular PCR (Supplementary Figure 10) and quantitative PCR (Supplementary Figure 11 and Supplementary Table S14).
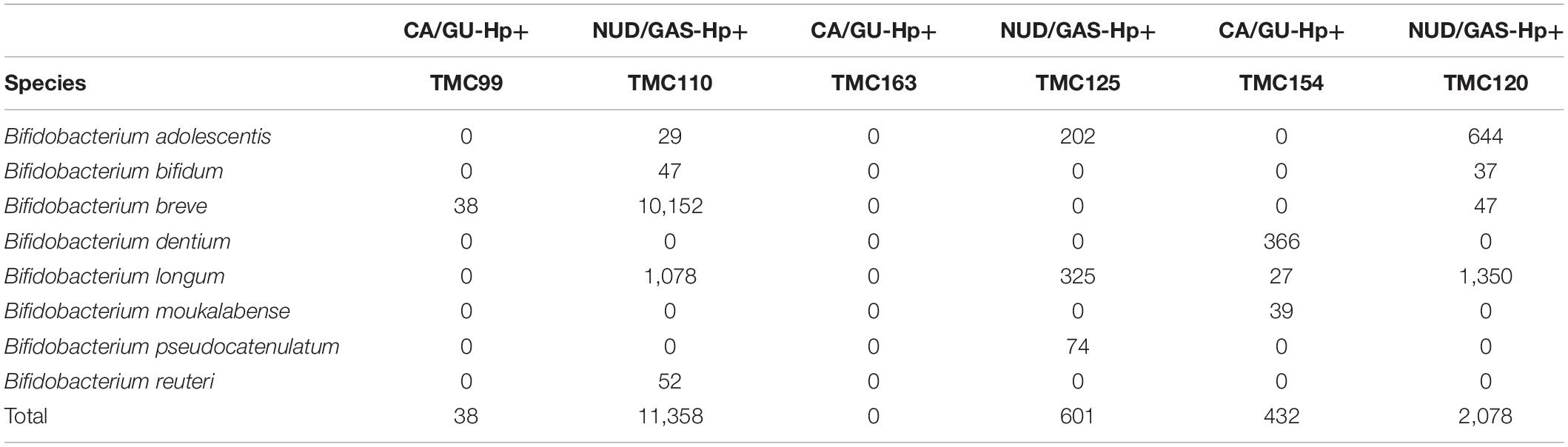
Table 4. The read assignment to Bifidobacterium species in whole genome metagenome sequencing and analysis for three paired (age and sex matched) CA/GU-Hp+ vs. NUD/GAS-Hp+ subjects.
Why most H. pylori-infected individuals remain asymptomatic while a certain proportion develops gastric ulcer, duodenal ulcer, and gastric cancer had remained enigmatic. Substantial literature has accumulated on the H. pylori virulence genes like vacA and cagA to understand their roles in pathogenesis, but relatively fewer attempts were made to understand the “other factors” that influence and determine clinical outcomes (Isomoto et al., 2010; Palframan et al., 2012; Hatakeyama, 2014). The other factors that are likely to be involved in determining the clinical status are polymorphism of several cytokine genes and lifestyle of the individuals, which includes diet, alcohol consumption, and smoking (Ladeiras-Lopes et al., 2008; Moy et al., 2010; Ren et al., 2012; Datta De and Roychoudhury, 2015).
It is hypothesized that the gastrointestinal microbiota also contributes to the clinical outcome. Dysbiosis of the gastrointestinal microbiota is associated with many communicable and non-communicable as well as chronic and acute diseases, and therefore, it is likely that it could also be involved in gastric cancer and peptic ulcer (Hsiao et al., 2014; Forslund et al., 2015; Rogers, 2015; Zhang X. et al., 2015; Bratburd et al., 2018; Saus et al., 2019). It is also not unlikely that a beneficial microbe in the gastrointestinal microbiota is involved in protecting certain individuals from H. pylori-mediated pathogenesis. A similar protective role of Ruminococcus obeum (later classified as Blautia obeum) in the human gut was found against the infection of intestinal pathogen Vibrio cholerae (Hsiao et al., 2014). It has been shown that H. pylori infection tends to decrease the overall microbial diversity in stomach with a preferential increase in the relative abundance of bacteria belonging to the phylum Proteobacteria, but no specific association between a particular microbial species in stomach and H. pylori-related gastric diseases was found (Andersson et al., 2008; Das et al., 2017). Fewer studies on intestinal microbiota in the context of H. pylori infection and gastric diseases also could not identify the same (Gao et al., 2018). The present study, which involved analyses of clinical data, H. pylori genotype data, and metagenomics data, revealed that gastric colonization of virulent H. pylori strain is necessary but not sufficient for developing severe gastric diseases. Rather, reduced abundance of several species of Bifidobacterium in intestinal microbiota (but not in the stomach microbiota) is linked to H. pylori infection and related severe gastric diseases (Figure 6 and Supplementary Table S14).
The bacteria belonging to the genus Bifidobacterium are one of the first colonizers in human gut after birth, and the overall health benefits (including anti-tumor immunity) of these lactose fermenting bacteria are well appreciated (Sivan et al., 2015; O’Callaghan and van Sinderen, 2016; Oki et al., 2018). Therefore, several clinical trials were conducted using Bifidobacterium, along with Lactobacillus or Streptococcus as probiotic supplements with antibiotics and proton pump inhibitors to eradicate H. pylori (Zhang M.M. et al., 2015; Wang et al., 2017). These studies showed moderate levels of improvements with H. pylori eradication and reductions of the side effects of therapy (Zhang M.M. et al., 2015; Wang et al., 2017). The abundance of Bifidobacterium in the gut was increased after a successful eradication of H. pylori (Guo et al., 2019). Likewise, gastric ulcers induced by acetic acid or ethanol in mice healed faster when the mice were colonized with Bifidobacterium (Nagaoka et al., 1994). Although the entire mechanism is not revealed, it is known that multiple mechanisms like modulation of NFkB signaling and synthesis of antimicrobial peptides are involved in the Bifidobacterium-mediated inhibition of H. pylori (Collado et al., 2005; Shirasawa et al., 2010). However, it was not known whether or not a real difference in the abundance of Bifidobacterium exists between the H. pylori-infected human with severe gastric diseases and the H. pylori-infected human without these diseases. Our data filled that gap by clearly showing that no virulence gene of H. pylori is associated with severe gastric diseases unless the relative abundance of Bifidobacterium in the lower gut is significantly low. To the best of our knowledge, this is the first study, which has identified a specific lower gut microbe is linked to the gastric disorders that take over a million lives every year. The lower abundance of the beneficial microbe Bifidobacterium in the lower gut may serve as non-invasive assessment of gastric cancer and gastric ulcer risks. Furthermore, some of the Bifidobacterium strains may also serve as effective probiotics against gastric cancer and gastric ulcer.
Numerous in vitro and in vivo studies have convincingly proved the pathogenic potential of H. pylori vacA and cagA. However, molecular epidemiology data showed that only 10–20% of the H. pylori-infected individuals develop gastroduodenal diseases. Why 80–90% H. pylori infections remain benign is unknown. The present study involving H. pylori vacA and cagA genotypes and fecal microbiota analyses revealed that apart from the virulence genes of the H. pylori strains, the intestinal microbiota is also involved in the context of H. pylori infection and the related gastric diseases. We have identified several species of Bifidobacterium (phylum Actinobacteria) that are present at very low abundance specifically in the gut of H. pylori-infected patients with severe gastric diseases suggesting a protective role of this beneficial microbe against severe gastric diseases. This finding may lead to developing early prognosis of severe gastric diseases or developing probiotics conferring protection against these diseases.
The GenBank accession numbers of nucleotide sequences of H. pylori genes are as follows: the 3′ end variable regions of cagA genes are MN968513 to MN968550; the 5′ end regions of cagA genes are MN968503 to MN968507; the m1 alleles of vacA genes are MN968508 to MN968512. The NGS (Illumina MiSeq) dataset related to microbial 16S rRNA gene is available at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA605091. The NGS (GridION X5) data related to the whole genome metagenome sequencing is available at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA648297.
The studies involving human participants were reviewed and approved by the Institutional Human Ethical Committee of RGCB. The patients/participants provided their written informed consent to participate in this study.
SC conceived the idea. TD, KD, MG, DC, RR, SA, JV, SD, and SC performed the experiments. TD, AG, SKC, VTJ, and SC analyzed the data. TD, KD, AG, RR, BD, GN, and SC wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant (ECR/2016/00017) from the Science and Engineering Research Board (SERB) of Department of Science and Technology (DST), Government of India, to SC as well as by the institutional support from Rajiv Gandhi Centre for Biotechnology, an autonomous institute under the Department of Biotechnology, Government of India.
AG was employed by the company Genotypic Technology Pvt. Ltd, Bengaluru, Karnataka, India. The NGS was outsourced to this company. Some of the analysis was done by AG upon request in the absence of any commercial or financial relationships.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Asish Mukhopadhyay (Scientist F at National Institute of Cholera and Enteric Diseases, Kolkata, West Bengal, India) for sending the reference H. pylori strains (26695, J99, SS1, and AM1) that were used as controls in the study. This study was benefitted from the critical discussions with Professor M. Radhakrishna Pillai (Former Director of Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala, India). We also thank Dr. Douglas E. Berg (Adjunct Professor at University of California, San Diego, La Jolla, CA, United States, and Professor Emeritus at the Department of Molecular Microbiology, Washington University School of Medicine in St. Louis, MO, United States) and Dr. Dhruba Chattoraj (Senior Investigator at NCI/CCR, National Institute of Health, Bethesda, MD, United States) for several scientific suggestions and for critically editing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.631140/full#supplementary-material
Andersson, A. F., Lindberg, M., Jakobsson, H., Backhed, F., Nyren, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. doi: 10.1371/journal.pone.0002836
Atherton, J. C., Cao, P., Peek, R. M. Jr., Tummuru, M. K., Blaser, M. J., and Cover, T. L. (1995). Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 270, 17771–17777. doi: 10.1074/jbc.270.30.17771
Bag, S., Saha, B., Mehta, O., Anbumani, D., Kumar, N., Dayal, M., et al. (2016). An improved method for high quality metagenomics DNA extraction from human and environmental samples. Sci. Rep. 6:26775. doi: 10.1038/srep26775srep26775
Basso, D., Zambon, C. F., Letley, D. P., Stranges, A., Marchet, A., Rhead, J. L., et al. (2008). Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 135, 91–99. doi: 10.1053/j.gastro.2008.03.041S0016-5085(08)00479-4
Berg, D. E., Lelwala-Guruge, J., Incecik, E. T., Srivastava, K., and Akopyants, N. S. (1997). H. pylori DNA fingerprinting using the arbitrarily primed PCR (AP-PCR) or random amplified polymorphic DNA (RAPD) method. Methods Mol. Med. 8, 117–132. doi: 10.1385/0-89603-381-3:117
Bik, E. M., Eckburg, P. B., Gill, S. R., Nelson, K. E., Purdom, E. A., Francois, F., et al. (2006). Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U.S.A. 103, 732–737. doi: 10.1073/pnas.0506655103
Bratburd, J. R., Keller, C., Vivas, E., Gemperline, E., Li, L., Rey, F. E., et al. (2018). Gut microbial and metabolic responses to Salmonella enterica serovar typhimurium and Candida albicans. MBio 9:e02032-18. doi: 10.1128/mBio.02032-18mBio.02032-18
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Chattopadhyay, S., Patra, R., Chatterjee, R., De, R., Alam, J., Ramamurthy, T., et al. (2012). Distinct repeat motifs at the C-terminal region of CagA of Helicobacter pylori strains isolated from diseased patients and asymptomatic individuals in West Bengal, India. Gut Pathog. 4:4. doi: 10.1186/1757-4749-4-41757-4749-4-4
Chattopadhyay, S., Patra, R., Ramamurthy, T., Chowdhury, A., Santra, A., Dhali, G. K., et al. (2004). Multiplex PCR assay for rapid detection and genotyping of Helicobacter pylori directly from biopsy specimens. J. Clin. Microbiol. 42, 2821–2824. doi: 10.1128/JCM.42.6.2821-2824.200442/6/2821
Chung, C., Olivares, A., Torres, E., Yilmaz, O., Cohen, H., and Perez-Perez, G. (2010). Diversity of VacA intermediate region among Helicobacter pylori strains from several regions of the world. J. Clin. Microbiol. 48, 690–696. doi: 10.1128/JCM.01815-09JCM.01815-09
Collado, M. C., Gonzalez, A., Gonzalez, R., Hernandez, M., Ferrus, M. A., and Sanz, Y. (2005). Antimicrobial peptides are among the antagonistic metabolites produced by Bifidobacterium against Helicobacter pylori. Int. J. Antimicrob. Agents 25, 385–391. doi: 10.1016/j.ijantimicag.2005.01.017
Covacci, A., Telford, J. L., Del Giudice, G., Parsonnet, J., and Rappuoli, R. (1999). Helicobacter pylori virulence and genetic geography. Science 284, 1328–1333.
Das, A., Pereira, V., Saxena, S., Ghosh, T. S., Anbumani, D., Bag, S., et al. (2017). Gastric microbiome of Indian patients with Helicobacter pylori infection, and their interaction networks. Sci. Rep. 7:15438. doi: 10.1038/s41598-017-15510-6
Datta De, D., and Roychoudhury, S. (2015). To be or not to be: the host genetic factor and beyond in Helicobacter pylori mediated gastro-duodenal diseases. World J. Gastroenterol. 21, 2883–2895. doi: 10.3748/wjg.v21.i10.2883
Dhakal, O. P., and Dhakal, M. (2018). Prevalence of Helicobacter pylori infection & pattern of gastrointestinal involvement in patients undergoing upper gastrointestinal endoscopy in Sikkim. Indian J. Med. Res. 147, 517–520. doi: 10.4103/ijmr.IJMR_1482_16IndianJMedRes_2018_147_5_517_238233
Dorer, M. S., Talarico, S., and Salama, N. R. (2009). Helicobacter pylori’s unconventional role in health and disease. PLoS Pathog. 5:e1000544. doi: 10.1371/journal.ppat.1000544
Forslund, K., Hildebrand, F., Nielsen, T., Falony, G., Le Chatelier, E., Sunagawa, S., et al. (2015). Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266. doi: 10.1038/nature15766
Gao, J. J., Zhang, Y., Gerhard, M., Mejias-Luque, R., Zhang, L., Vieth, M., et al. (2018). Association between gut microbiota and Helicobacter pylori-related gastric lesions in a high-risk population of gastric cancer. Front. Cell. Infect. Microbiol. 8:202. doi: 10.3389/fcimb.2018.00202
GBD 2016 Causes of Death Collaborators (2017). Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1151–1210. doi: 10.1016/S0140-6736(17)32152-9
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010syq010
Guo, Y., Zhang, Y., Gerhard, M., Gao, J. J., Mejias-Luque, R., Zhang, L., et al. (2019). Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut 69, 1598–1607. doi: 10.1136/gutjnl-2019-319696
Hatakeyama, M. (2014). Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15, 306–316. doi: 10.1016/j.chom.2014.02.008S1931-3128(14)00066-3
Hsiao, A., Ahmed, A. M., Subramanian, S., Griffin, N. W., Drewry, L. L., Petri, W. A. Jr., et al. (2014). Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature 515, 423–426. doi: 10.1038/nature13738nature13738
Huerta-Cepas, J., Serra, F., and Bork, P. (2016). ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638. doi: 10.1093/molbev/msw046msw046
Isomoto, H., Moss, J., and Hirayama, T. (2010). Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J. Exp. Med. 220, 3–14.
Jo, H. J., Kim, J., Kim, N., Park, J. H., Nam, R. H., Seok, Y. J., et al. (2016). Analysis of gastric microbiota by pyrosequencing: minor role of bacteria other than Helicobacter pylori in the gastric carcinogenesis. Helicobacter 21, 364–374. doi: 10.1111/hel.12293
Kienesberger, S., Cox, L. M., Livanos, A., Zhang, X. S., Chung, J., Perez-Perez, G. I., et al. (2016). Gastric Helicobacter pylori infection affects local and distant microbial populations and host responses. Cell. Rep. 14, 1395–1407. doi: 10.1016/j.celrep.2016.01.017
Ladeiras-Lopes, R., Pereira, A. K., Nogueira, A., Pinheiro-Torres, T., Pinto, I., Santos-Pereira, R., et al. (2008). Smoking and gastric cancer: systematic review and meta-analysis of cohort studies. Cancer Causes Control 19, 689–701. doi: 10.1007/s10552-008-9132-y
Li, X. X., Wong, G. L., To, K. F., Wong, V. W., Lai, L. H., Chow, D. K., et al. (2009). Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One 4:e7985. doi: 10.1371/journal.pone.0007985
Maldonado-Contreras, A., Goldfarb, K. C., Godoy-Vitorino, F., Karaoz, U., Contreras, M., Blaser, M. J., et al. (2011). Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 5, 574–579. doi: 10.1038/ismej.2010.149ismej2010149
Malfertheiner, P., Link, A., and Selgrad, M. (2014). Helicobacter pylori: perspectives and time trends. Nat. Rev. Gastroenterol. Hepatol. 11, 628–638. doi: 10.1038/nrgastro.2014.99nrgastro.2014.99
Menon, T. M. (2000). A Handbook of Kerala. Thiruvananthapuram: The International School of Dravidian Linguistics.
Moy, K. A., Fan, Y., Wang, R., Gao, Y. T., Yu, M. C., and Yuan, J. M. (2010). Alcohol and tobacco use in relation to gastric cancer: a prospective study of men in Shanghai, China. Cancer Epidemiol. Biomarkers Prev. 19, 2287–2297. doi: 10.1158/1055-9965.EPI-10-03621055-9965.EPI-10-0362
Mukhopadhyay, A. K., Kersulyte, D., Jeong, J. Y., Datta, S., Ito, Y., Chowdhury, A., et al. (2000). Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol. 182, 3219–3227. doi: 10.1128/jb.182.11.3219-3227.2000
Nagaoka, M., Hashimoto, S., Watanabe, T., Yokokura, T., and Mori, Y. (1994). Anti-ulcer effects of lactic acid bacteria and their cell wall polysaccharides. Biol. Pharm. Bull. 17, 1012–1017. doi: 10.1248/bpb.17.1012
Noto, J. M., and Peek, R. M. Jr. (2017). The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog. 13:e1006573. doi: 10.1371/journal.ppat.1006573PPATHOGENS-D-17-00804
O’Callaghan, A., and van Sinderen, D. (2016). Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 7:925. doi: 10.3389/fmicb.2016.00925
Oki, K., Akiyama, T., Matsuda, K., Gawad, A., Makino, H., Ishikawa, E., et al. (2018). Long-term colonization exceeding six years from early infancy of Bifidobacterium longum subsp. longum in human gut. BMC Microbiol. 18:209. doi: 10.1186/s12866-018-1358-6
Palframan, S. L., Kwok, T., and Gabriel, K. (2012). Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2:92. doi: 10.3389/fcimb.2012.00092
Ren, J. S., Kamangar, F., Forman, D., and Islami, F. (2012). Pickled food and risk of gastric cancer–a systematic review and meta-analysis of English and Chinese literature. Cancer Epidemiol. Biomarkers Prev. 21, 905–915. doi: 10.1158/1055-9965.EPI-12-0202
Rhead, J. L., Letley, D. P., Mohammadi, M., Hussein, N., Mohagheghi, M. A., Eshagh Hosseini, M., et al. (2007). A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 133, 926–936. doi: 10.1053/j.gastro.2007.06.056
Rogers, G. B. (2015). Germs and joints: the contribution of the human microbiome to rheumatoid arthritis. Nat. Med. 21, 839–841. doi: 10.1038/nm.3916
Saus, E., Iraola-Guzman, S., Willis, J. R., Brunet-Vega, A., and Gabaldon, T. (2019). Microbiome and colorectal cancer: roles in carcinogenesis and clinical potential. Mol. Aspects Med. 69, 93–106. doi: 10.1016/j.mam.2019.05.001
Shirasawa, Y., Shibahara-Sone, H., Iino, T., and Ishikawa, F. (2010). Bifidobacterium bifidum BF-1 suppresses Helicobacter pylori-induced genes in human epithelial cells. J. Dairy Sci. 93, 4526–4534. doi: 10.3168/jds.2010-3274S0022-0302(10)00483-2
Sivan, A., Corrales, L., Hubert, N., Williams, J. B., Aquino-Michaels, K., Earley, Z. M., et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. doi: 10.1126/science.aac4255
Wang, F., Feng, J., Chen, P., Liu, X., Ma, M., Zhou, R., et al. (2017). Probiotics in Helicobacter pylori eradication therapy: systematic review and network meta-analysis. Clin. Res. Hepatol. Gastroenterol. 41, 466–475. doi: 10.1016/j.clinre.2017.04.004
Wilkinson, S., and Davy, S. (2018). Phylogram: an R package for phylogenetic analysis with nested lists. J. Open Source Softw. 3:790.
Zhang, M. M., Qian, W., Qin, Y. Y., He, J., and Zhou, Y. H. (2015). Probiotics in Helicobacter pylori eradication therapy: a systematic review and meta-analysis. World J. Gastroenterol. 21, 4345–4357. doi: 10.3748/wjg.v21.i14.4345
Keywords: H. pylori, gastric ulcer and cancer, gut microbiome, Bifidobacterium, probiotic
Citation: Devi TB, Devadas K, George M, Gandhimathi A, Chouhan D, Retnakumar RJ, Alexander SM, Varghese J, Dharmaseelan S, Chandrika SK, Jissa VT, Das B, Nair GB and Chattopadhyay S (2021) Low Bifidobacterium Abundance in the Lower Gut Microbiota Is Associated With Helicobacter pylori-Related Gastric Ulcer and Gastric Cancer. Front. Microbiol. 12:631140. doi: 10.3389/fmicb.2021.631140
Received: 19 November 2020; Accepted: 19 January 2021;
Published: 26 February 2021.
Edited by:
Alain Pierre Gobert, Vanderbilt University Medical Center, United StatesReviewed by:
Zhongming Ge, Massachusetts Institute of Technology, United StatesCopyright © 2021 Devi, Devadas, George, Gandhimathi, Chouhan, Retnakumar, Alexander, Varghese, Dharmaseelan, Chandrika, Jissa, Das, Nair and Chattopadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Santanu Chattopadhyay, c2FudGFudUByZ2NiLnJlcy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.