 Gilda Varliero
Gilda Varliero Alexandre M. Anesio
Alexandre M. Anesio Gary L. A. Barker
Gary L. A. Barker
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 16 September 2021
Sec. Extreme Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.627437
The Arctic environment is particularly affected by global warming, and a clear trend of the ice retreat is observed worldwide. In proglacial systems, the newly exposed terrain represents different environmental and nutrient conditions compared to later soil stages. Therefore, proglacial systems show several environmental gradients along the soil succession where microorganisms are active protagonists of the soil and carbon pool formation through nitrogen fixation and rock weathering. We studied the microbial succession of three Arctic proglacial systems located in Svalbard (Midtre Lovénbreen), Sweden (Storglaciären), and Greenland (foreland close to Kangerlussuaq). We analyzed 65 whole shotgun metagenomic soil samples for a total of more than 400 Gb of sequencing data. Microbial succession showed common trends typical of proglacial systems with increasing diversity observed along the forefield chronosequence. Microbial trends were explained by the distance from the ice edge in the Midtre Lovénbreen and Storglaciären forefields and by total nitrogen (TN) and total organic carbon (TOC) in the Greenland proglacial system. Furthermore, we focused specifically on genes associated with nitrogen fixation and biotic rock weathering processes, such as nitrogenase genes, obcA genes, and genes involved in cyanide and siderophore synthesis and transport. Whereas we confirmed the presence of these genes in known nitrogen-fixing and/or rock weathering organisms (e.g., Nostoc, Burkholderia), in this study, we also detected organisms that, even if often found in soil and proglacial systems, have never been related to nitrogen-fixing or rock weathering processes before (e.g., Fimbriiglobus, Streptomyces). The different genera showed different gene trends within and among the studied systems, indicating a community constituted by a plurality of organisms involved in nitrogen fixation and biotic rock weathering, and where the latter were driven by different organisms at different soil succession stages.
Due to global warming, a clear trend of glacial ice retreat has been observed in recent decades worldwide (Moon et al., 2018; Maurer et al., 2019). This rapid loss of the cryosphere is leading to an expansion of proglacial systems, exposing bedrocks that have been covered by ice for thousands of years (Fountain et al., 2012; Heckmann et al., 2016). Chemical, physical, and biological processes lead to the formation of soil from the mineralization of the bedrocks during weathering (Uroz et al., 2009, 2015; Xi et al., 2018). Biological rock weathering is a key process in environments where soil microbiota and vegetation roots help the release of nutrients and major ions into the soil with their metabolism and mechanical actions (Kelly et al., 1998; Borin et al., 2010; Porder, 2019). The release of nutrients and major ions represents a source of enzyme cofactors and energy for the soil microbiota, especially in nutrient-poor soils, such as the early stage glacial forefield soils (Uroz et al., 2015), giving microbes a pivotal role in soil formation (Rousk and Bengtson, 2014; Koshila Ravi et al., 2019). Furthermore, other key environmental and ecological processes, such as nitrogen fixation, take place in proglacial systems. These environments are habitats for diverse diazotrophic communities that, using the enzyme nitrogenase, progressively enrich the soil with ammonia and bioavailable nitrogen sources to non-diazotrophic organisms (Bradley et al., 2014; Nash et al., 2018).
Rock weathering and nitrogen fixation create gradient conditions in the proglacial environments. Whereas the ground in the ice margins is dominated by rocks, the soil content increases and deepens going farther away from it, with an associated increase in vegetation. The bioavailability of nutrients, such as organic carbon and nitrogen, also increases with the formation of the soil. The presence of such gradients and the progressive ground exposure to the atmosphere make proglacial systems very suitable for the study of microbial succession (Edwards and Cook, 2015). Microbial communities show trends along chronosequences (Schmalenberger and Noll, 2010; Zumsteg et al., 2012; Bajerski and Wagner, 2013; Jiang et al., 2019b). Previous work has shown that the microbial communities close to the ice edge are usually dominated by autotrophs and chemolithotrophs that are able to use soil minerals and sunlight as energy source and enrich the soil with biological available organic carbon and nutrients (Schmidt et al., 2008; Liu et al., 2012; Fernández-Martínez et al., 2017). These first stages of the succession are also the most influenced by the glacier inputs and discharges in the environment (Hotaling et al., 2017). Going farther from the ice, different studies have observed a decrease of the autotroph component and an increase in the heterotroph microbial component, where the latter takes advantage of the progressive organic-enriched soil (Bradley et al., 2016). These trends are also accompanied by an increase of the vegetation complexity with the distance from the ice edge, establishing also symbiotic and mutualistic relationships with the soil microbiome (Knelman et al., 2012; Rime et al., 2016).
Despite the pivotal role of microbial communities in rock weathering, the protagonists and the mechanisms of these processes are not very clear. Different mechanisms and rock weathering-enhancing organisms have been reported in studies of soil isolates of both bacteria (Frey et al., 2010; Lepleux et al., 2012; Liu et al., 2012; Olsson-Francis et al., 2015; Xi et al., 2018; Wang et al., 2019) and fungi (Brunner et al., 2011), observing the production of organic acids (e.g., oxalate) and hydrogen cyanide (HCN) to mobilize the nutrients such as iron sulfur and phosphorus (El-Tarabily et al., 2008), and an increase in siderophore production to import iron into the cell (Frey et al., 2010; Wongfun et al., 2014; Olsson-Francis et al., 2015). Compared to rock weathering processes, diazotrophic organisms are better understood and characterized. Organisms spanning more than 13 phyla have been identified as nitrogen fixers (Addo and Dos Santos, 2020). Diazotrophic assemblages vary in relation to the soil and rhizosphere characteristics (Duc et al., 2009) and are more abundant in the first stages of the forefield succession where they enrich the soil with nitrogen, a key nutrient for cellular growth (Brankatschk et al., 2011). Despite the importance of this process in forefield dynamics, there is a lack of understanding of the diazotrophic organism variation along the proglacial microbial succession (Brankatschk et al., 2011; Nash et al., 2018).
Whereas previous studies have analyzed forefield microbial communities with several approaches, such as 16S rRNA gene sequencing (e.g., Bajerski and Wagner, 2013; Fernández-Martínez et al., 2017), clone library sequencing (e.g., Zumsteg et al., 2012), GeoChip microarray (Fernández-Martínez et al., 2016), and ecoenzymatic stoichiometry (Jiang et al., 2019a; Li et al., 2020). Only a few studies use a whole shotgun metagenomic sequencing approach (e.g., Nash et al., 2018); none of these, however, report taxon-wise functional profiles along forefield successions. We reanalyzed the whole shotgun sequencing dataset reported in Nash et al. (2018) to obtain a comprehensive picture of microbial diversity and gene function over glacial chronosequences. In particular, we analyzed 65 different metagenomes from three different proglacial systems: two forefields from two small glacier valleys, the Midtre Lovénbreen in Svalbard and the Storglaciären in Sweden, and a proglacial field of the Greenland ice sheet (GrIS) in proximity of point 601. In this work, we report (i) how taxonomical groups varied along the different proglacial successional gradients and (ii) which organisms were involved in different proglacial processes. We focused on two of the processes that shape forefield dynamics and nutrient bioavailability the most: nitrogen fixation, exploring nitrogenase genes, and rock weathering processes, looking at the obcA genes that are involved in the first step of oxalate biosynthesis (Nakata, 2011), genes involved in cyanide synthesis, and genes involved in siderophore synthesis and transport.
This dataset was previously analyzed in Nash et al. (2018) to show diazotrophic community variations among the three proglacial systems and in relation to the measured total nitrogen (TN) and total organic carbon (TOC) concentrations for each of the systems. However, no microbial succession along the different proglacial systems was analyzed in Nash et al. (2018). Therefore, we reanalyzed the dataset (i) to obtain information on relevant microbial successional patterns observed in the studied glacial forefields and (ii) to study the functional patterns related to proglacial soil formation focusing on not only nitrogen fixation but also rock weathering phenomena (Table 1).

Table 1. Comparative table on the main aspects of Nash et al. (2018) and this study.
Sample collection, soil geochemical characterization, sample preparation, and sequencing methods are reported in Nash et al. (2018). We briefly report details about sampling and forefield characterization in Site Characterization.
Samples were collected from the Midtre Lovénbreen forefield during summer 2013 and from the Storglaciären forefield and the proglacial field of the GrIS in proximity of point 601 during summer 2014. The sampling was conducted with the same approach in all the systems: only the top 10 cm of the soil was collected in a sterile Whirl-pack bag (Whirl-Pak, Nasco, United States) and then frozen at −20°C until processing. For Midtre Lovénbreen and Storglaciären, samples were transferred to −20°C storage on the day of collection, whereas the remote location of the Greenland field sites meant that samples were stored on ice in a cool box for up to 2 days before returning to the freezer at the Kangerlussuaq International Science Station. The soil samples were collected along transects starting in the proximity of the ice edge, going farther away. Where possible, multiple samples were taken at the same distance from the ice edge. Samples were not collected close to vegetation patches, rivers, or discontinuous soil patches to minimize site-specific effects. Samples were collected from three different proglacial systems sited in the Arctic circle (Nash et al., 2018; Figure 1A). These systems present different morphologies and have a different deglaciation rate, the ice sheet being much slower than the glaciers in the small valleys and the collected soil having different ages since deglaciation. The sampling size area and the geographical characteristics in the three different systems were also considerably different. The samples in Greenland were taken up to 10 km away from the closest ice point (Figure 1B). The samples in Svalbard were taken up to 1,600 m from the glacier toe and, as the forefield faces a fjord, this last point of the succession is sited close to the seawater (Figure 1C). Samples in the forefield from Sweden were taken up to a river that delimits the end of the small forefield area, at 350 m from the ice margin (Figure 1D). This diversity and the geographical dispersion of these sites allowed to compare functional and taxonomical trends between different systems. Even if the three forefield successions span different soil distances from the ice edge and different soil developmental stages, the generic comparison between young and older soils in different chronosequences (even if they represent different age increments) still provides insights into overall trends in soil development. The Greenland proglacial system is referred to here as G, while Svalbard is referred to as SV, and the forefield in Sweden as SW.
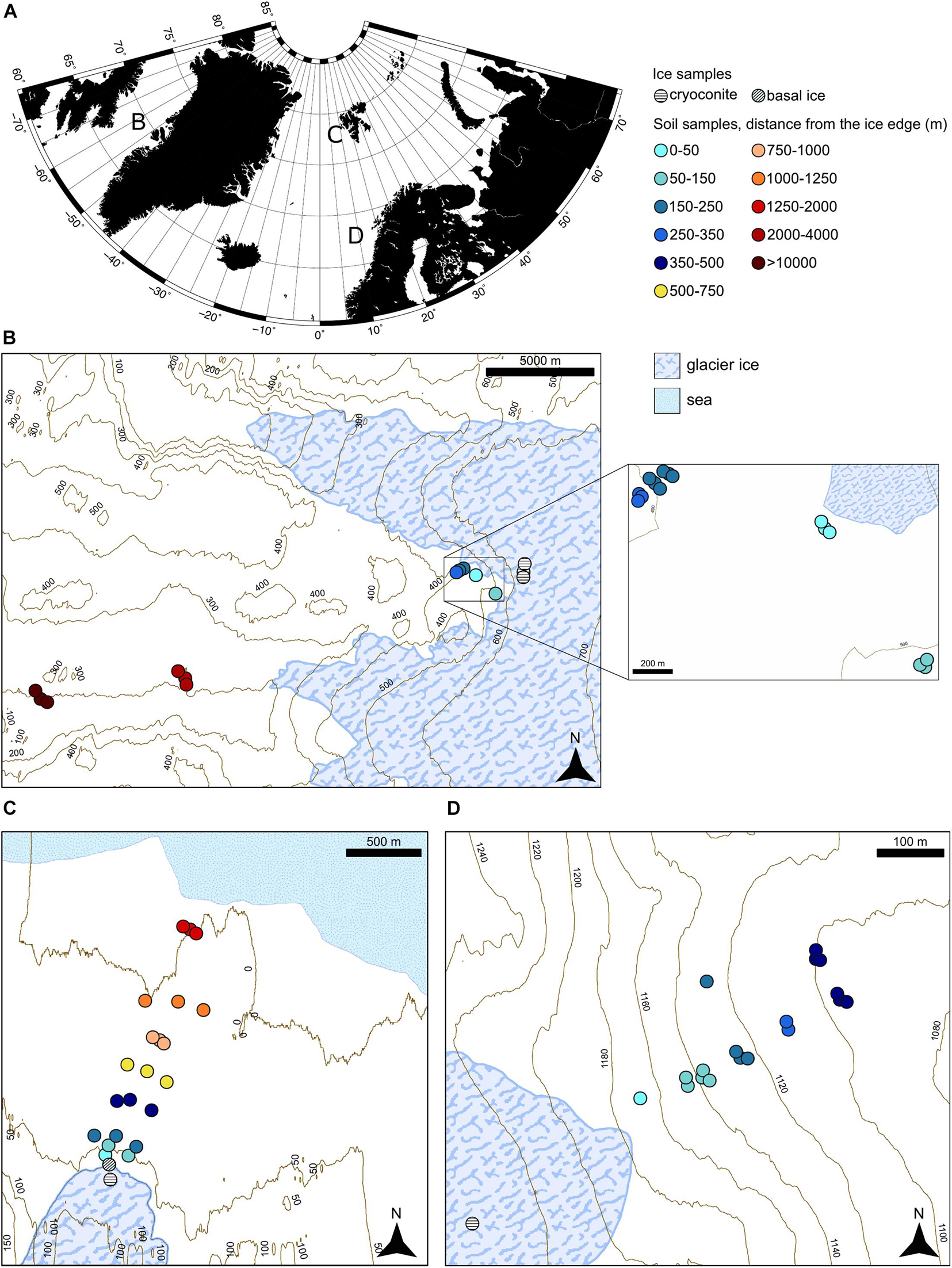
Figure 1. Sampling site locations. (A) Overview of the sampling site. (B) Greenland forefield sampling site. (C) Svalbard forefield sampling site. (D) Sweden forefield sampling site.
The samples were classified by distance from the glacier toe, and the distance was calculated as the distance between the sampling site and the closest ice edge point. Each sample was also characterized by TN and TOC values. The latter were measured by Nash et al. (2018) using mass spectrometry and carbon elemental analyzer.
A total of 65 samples were collected and Illumina whole shogun sequenced (Nash et al., 2018). The Illumina reads were checked, and quality was trimmed with FastQC v 0.11.71 and Trimmomatic v 0.36 (Bolger et al., 2014). The latter was run with the command options ILLUMINACLIP:TruSeq2-PE.fa:2:30:10 MINLEN:26, and the additional options TRAILING:10 for SV, SLIDINGWINDOW:5:20 CROP:149 for SW, and SLIDINGWINDOW:5:24 MINLEN:26 CROP:149 for G. The quality-checked sequences were then co-assembled from the three different datasets with the software MEGAHIT v 1.1.3 (Li et al., 2015) and parameters –k-min 25 –k-max 145 –k-step 8 -t 64.
Only assembly contigs longer than 300 bp were retrieved and aligned against the NCBI non-redundant (nr) protein database v 5 (Sayers et al., 2020) with Diamond 0.9.22 (Buchfink et al., 2015) using the command line options -e 0.000001 -F 15 –range-culling –range-cover 20 –id 50 –top 10 -f 6 -c1 -b4.0.
LongMeta, a custom pipeline whose scripts are publicly available on GitHub2, was used to detect and split chimeric contigs and to assign taxonomy and gene-coding regions to the assembly using the known protein alignment information. Functionality information was then associated with both nr (Sayers et al., 2020) and Gene Ontology (GO) nomenclature (Ashburner et al., 2000; Carbon et al., 2019). Reads were mapped back to the assembly with Bowtie2 v 2.3.4.3 (Langmead and Salzberg, 2012) and parameters –phred33 –local -I 100 -X 800 –no-hd –no-unal -D 30 -R 3 -N 0 -L 20 -i S,1,0.25 –non-deterministic -p 20. LongMeta was then used to assign taxonomic and function abundance information to each sample. LongMeta outputs taxonomy and functionality data as base coverage. In this article, whereas gene information is reported as base coverage, taxonomy information is reported as relative abundance (calculated from base coverage).
The geochemical dataset and LongMeta abundance and coverage datasets were imported to the R environment (R Core Team 2019, 2019) where all the statistical analyses were performed. In all the graphical visualizations and the principal component analysis (PCA) (Abdi and Williams, 2010), the samples were categorized in relation to their distances from the ice edge and divided into different groups: 0–50, 50–150, 150–250, 250–350, 350–500, 500–750, 750–1,000, 1,000–1,250, 1,250–2,000, 2,000–4,000, and >10,000 m (Supplementary Table 1).
Diversity indices were calculated on genus-level base coverage values with Shannon’s (H) and inverse Simpson’s (1/D) diversity indices. The diversity values were fitted with locally weighted scatterplot smoothing (LOWESS) curves in order to detect diversity trends in relation to the site distance from the ice edge.
Functional data were reported and analyzed as weighted gene coverage. The coverage of a specific gene in a specific sample was divided by the base coverage of the sample. The coverage scaling was necessary because different samples assembled with different efficiencies depending on their sequencing depth and community complexity. This led to a differential base coverage in different samples, and therefore, the gene coverages, if not scaled, could reflect the sample coverage and not the real gene coverage in the microbial community. Here, in addition to the gene coverage, functional profiles as GO biological categories are also presented.
Permutational multivariate analysis of variance (PERMANOVA) (Anderson, 2017) was performed with 9,999 permutations on Bray–Curtis dissimilarity matrices for the taxonomy, diversity, and functional datasets and Euclidean distance matrices for the geochemical dataset. The statistical tests were performed against the fixed factor Forefield that has three levels: Greenland, Svalbard, and Sweden. In order to show how the four different datasets varied with the sample distance from the ice edge and in relation to the geochemical dataset (i.e., TN and TOC), Mantel test statistic r using 9,999 permutations and the Spearman’s rank correlation coefficient were calculated (Smouse et al., 1986). Mantel tests were conducted on either Bray–Curtis or Euclidean matrices as for the PERMANOVA. Whereas the Mantel test was used to compare two different datasets (i.e., two different sets of variables), the Spearman’s rank correlation coefficient rs (with 9,999 permutations) was directly calculated when compared with only two different variables (e.g., a specific phylum vs. distance from the ice edge). A linear model calculation was used only when comparing two variables with the same units (i.e., Proteobacteria vs. Actinobacteria relative coverage). All the statistical analyses reported in the main text were performed without the inclusion of ice samples, unless otherwise specified. Statistical tests were interpreted as significant if p-value < 0.05.
Several R packages were used to perform statistical analyses and plot the graphs: vegan v 2.5.6 (Oksanen, 2017), ggpubr v 0.3.0 (Kassambara, 2018), ggplot2 v 3.3.0 (Wickham, 2016), gplots v 3.0.3 (Warnes, 2012), reshape v 0.8.8 (Wickham, 2007), and gridExtra v 2.3 (Auguie, 2017).
Total nitrogen and TOC showed different trends among the three proglacial systems (Figure 2). The Greenland (G) dataset showed low values in the first stages of the succession, maximum values at 150–250 m, and a gradual decrease going farther away from the glacier. The Svalbard (SV) and Swedish (SW) datasets showed an increase from the ice edge to more distant samples.
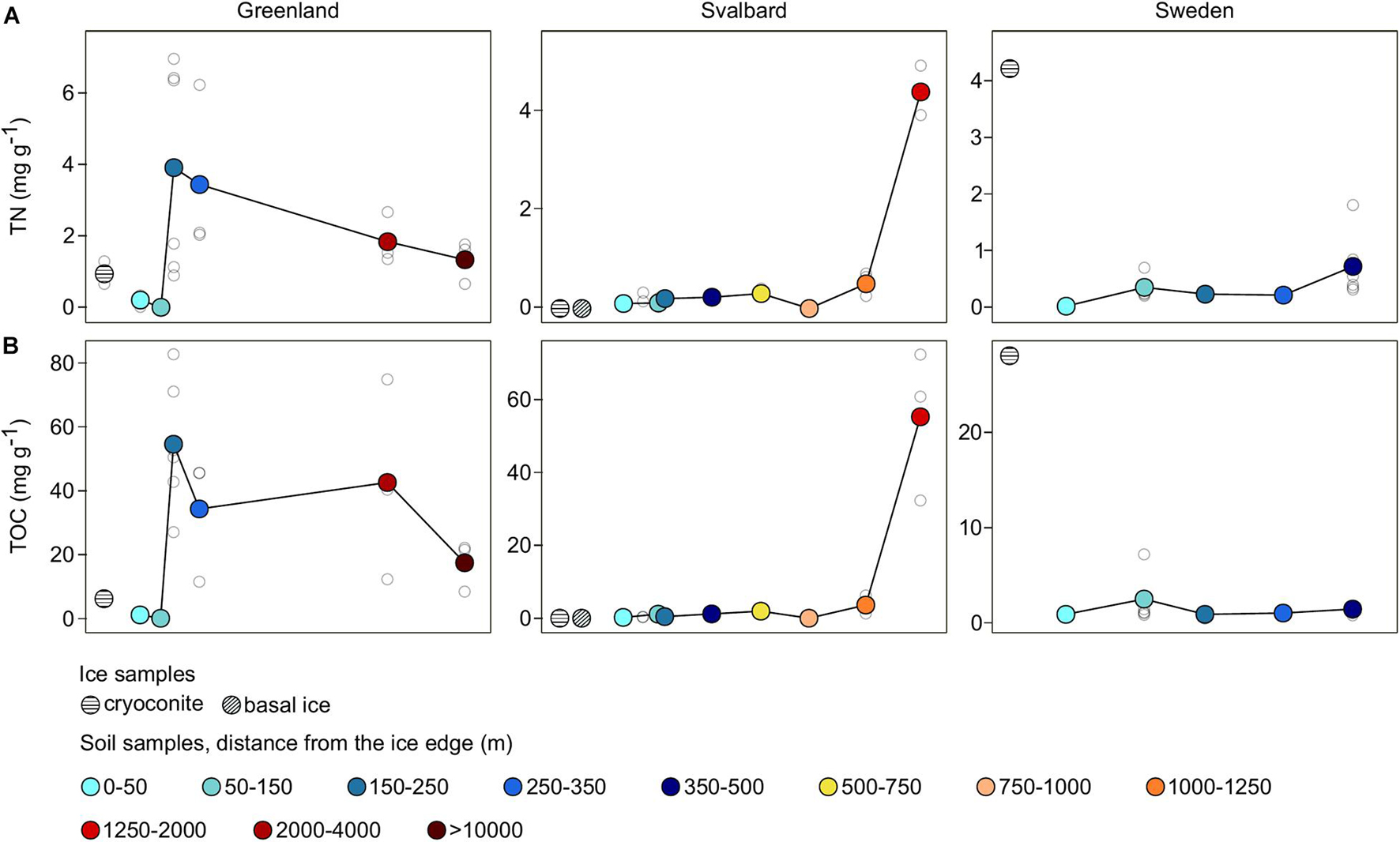
Figure 2. Total nitrogen (TN) (A) and total organic carbon (TOC) (B) trends across the three different proglacial systems. Samples were grouped into different categories and colored in relation to sample distance from the ice edge. Colored dots indicate the average values for each distance, whereas white dots are the values of the individual samples.
The SW dataset showed the lowest TN and TOC concentrations with values 0–2 and 0–7 mg g–1 in all the soil samples. The SV had a similar value range with 0–1 and 0–6 mg g–1, respectively, in all the soil samples except from the SV samples collected at 1,650 m from the ice edge (the closest samples to the sea; Figure 1C) where the TN and TOC mean values were 4 and 55 mg g–1. Compared to the SV and SW systems, G showed the highest TN and TOC values, ranging 0–7 and 0–82 mg g–1.
The two variables, TN and TOC, showed a positive Spearman’s correlation across all the systems (rs = 0.89; p-value < 0.05). Sample separation among the different forefield systems (factor Forefield) explained 26% of the observed variance in this dataset (p-value < 0.05; Table 2A). The correlation between these two variables and the distance from the ice edge was then explored with a Mantel test statistic r that was equal to 0.22 (p-value < 0.05). Different r values were obtained when Mantel tests were performed on the different forefield datasets: correlation with the sample distance from the ice edge was significant only in the SV and SW forefields with an r of 0.39 and 0.33, respectively (p-value < 0.05; Table 2B).
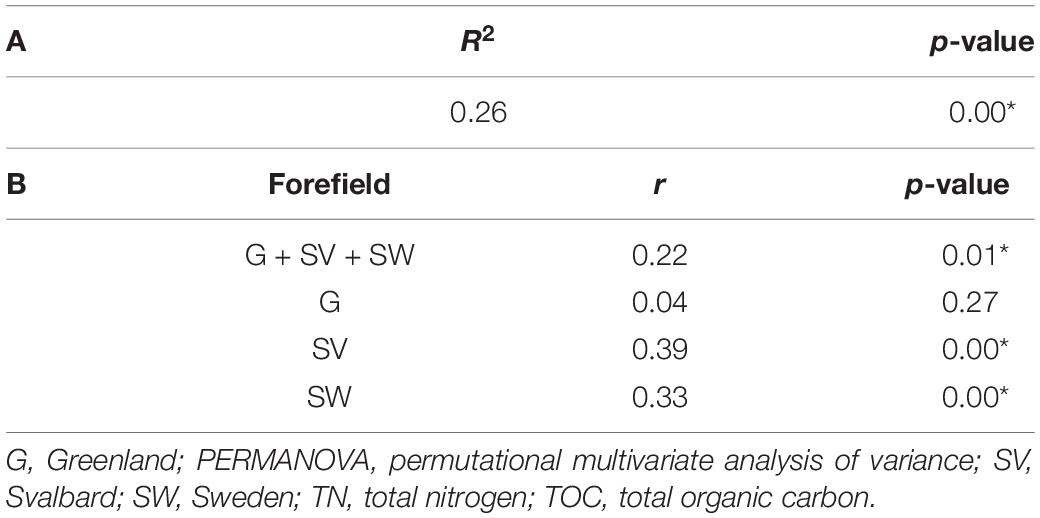
Table 2. PERMANOVA performed between the distance from the ice edge and the geochemical dataset (A). Mantel test performed to calculate the correlation between the distance from the ice edge and the geochemical dataset (i.e., TN + TOC) for the samples from the three different proglacial systems (G + SV + SW), only the G system, only the SV system, and only the SW system (B). The symbol ∗ is reported for significant R2 and r values where the statistic p-value < 0.05.
The quality-checked reads that were used to perform assembly were between 4 and 119 million (Supplementary Table 1) with 433 billion bases (433 Gb) used in total. The assembly was 30 Gb with an N50 length of 841 bases, minimum contig length of 300 bases, and maximum contig length of 561,967 bases. Reads mapped back to the assembly with a higher efficiency for ice samples (93–94%, 75–93%, and 92% for the G, SV, and SW datasets, respectively) compared to soil samples (50–78%, 34–64%, and 65–79%) (Supplementary Table 1). The average base coverage of the assembly ranged between 0.3× and 1.5× in the samples from the SV dataset, 0.7× and 3.7× in the SW dataset, and 0.7× and 3.4× in the G dataset. In each dataset, the highest coverages were observed in the ice samples. In the forefield assembly, 1% of the contigs were defined as chimeric and therefore split into shorter contigs.
Here, 1/D ranged between 25 and 128 across all the samples, whereas H ranged between 4 and 5. In all the proglacial systems, both indices were lower in the ice samples where 1/D reached a maximum of 54 and H a maximum of 4.4. Both the diversity indices had overall lower values in SV compared to SW and G datasets. However, PERMANOVA performed on the diversity dataset (i.e., 1/D and H) showed that only 20% of the variance was explained by the factor Forefield (p-value < 0.05) (Table 3A).
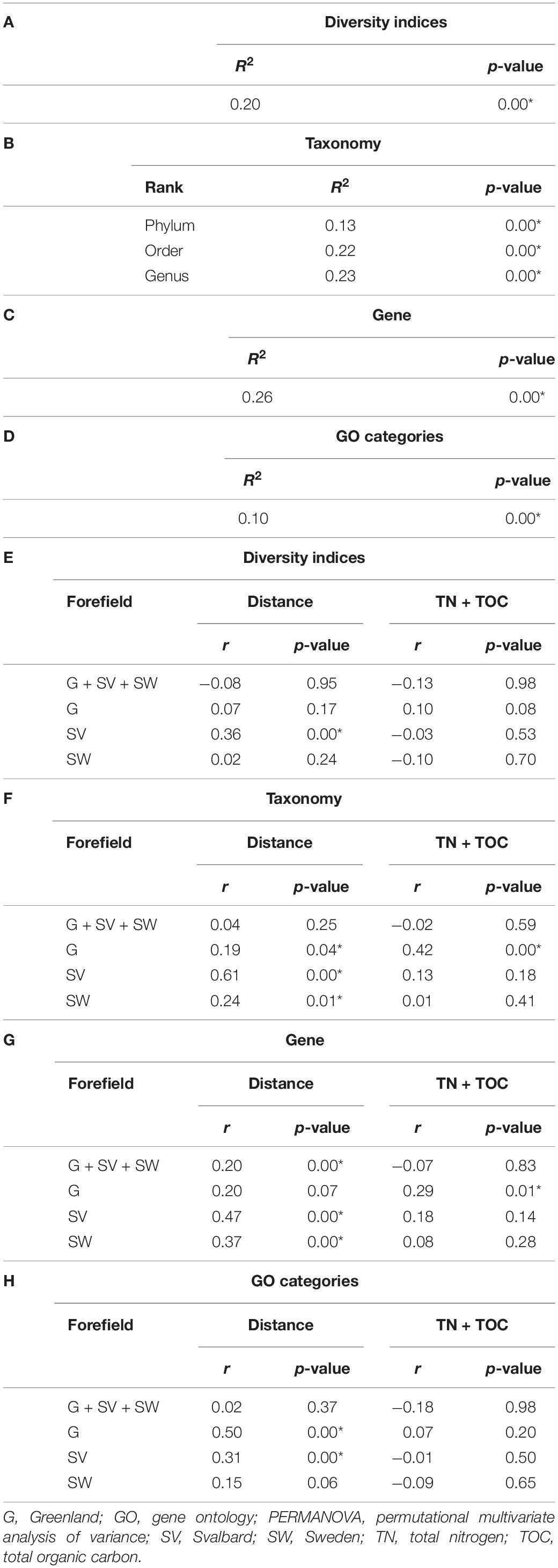
Table 3. PERMANOVA performed between the distance from the ice edge and the diversity index dataset (A); taxonomy dataset at the phylum, order, and genus level (B); gene dataset (C); and the GO dataset (D). Mantel test performed to calculate the correlation between the distance from the ice edge (distance) and the geochemical dataset (i.e., TN + TOC) with the diversity index dataset (E), taxonomy dataset at the genus level (F), gene dataset (G), and the GO dataset (H). Each of these four datasets were tested with all the samples from the three different proglacial systems (G + SV + SW), only the G system, only the SV system, and only the SW system. The symbol * is reported for significant R2 and r values where the statistic p-value < 0.05.
Fitted LOWESS lines showed increasing diversity trends along all the forefields (Figure 3). Diversity indices did not show a correlation with the distance from the ice edge (nor with TOC or TN concentration values). However, when performed on the different forefield datasets, the Mantel test statistic r was significant (p-value < 0.05) for the SV dataset (r = 0.36) (Table 3E). When ice samples were included together with the forefield samples, it was significant for all the datasets, where the r for G was 0.20, for SV was 0.45, and for SW was 0.18 (Supplementary Table 2E).
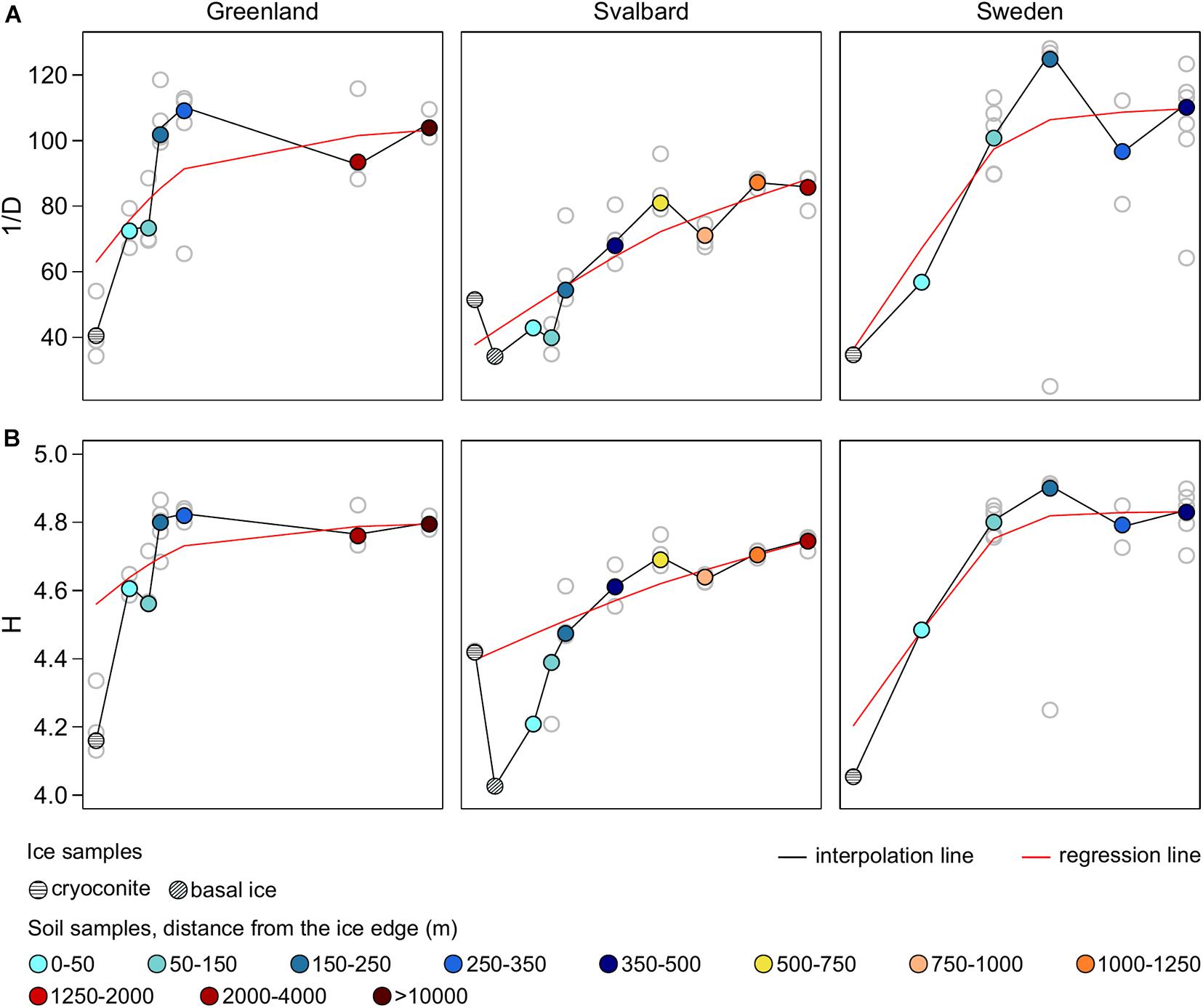
Figure 3. Inverse Simpson’s (A) and Shannon’s (B) diversity indices (1/D and H) calculated on the genus-level taxonomic dataset.
Microbial communities showed a minor sample clustering distribution across different forefields (Figure 4A). Ice and ice edge samples (<50 m from the glacier) clearly separated from the soil samples in the PCA representation, whereas soil samples collected at different distances from the ice edge did not show clear distinct clusters between each other (Figure 4B).
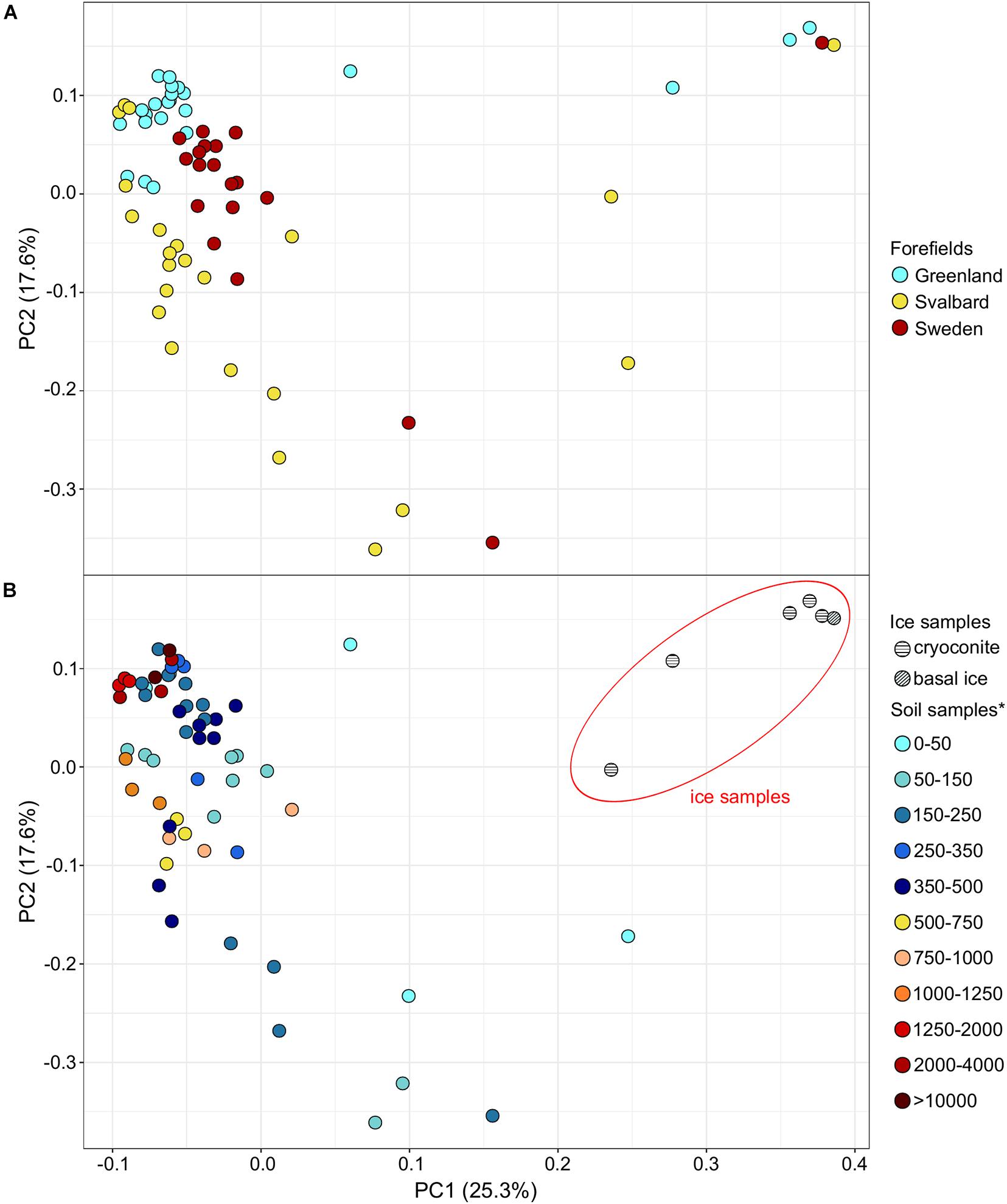
Figure 4. Principal component analysis (PCA) showing the sample distribution based on the genus taxonomic dataset for the forefield color-based samples (A) and ice and soil samples (B). *Soil samples are categorized in relation to their distance from the ice edge (m).
Permutational multivariate analysis of variance performed on the taxonomy dataset was significant (p-value < 0.05) for the factor Forefield that explained the 13, 22, and 23% of the variance at phylum, order, and genus levels (Table 3B). Less variance was explained when the PERMANOVA was performed including the ice samples (Supplementary Table 2B). The Mantel test performed between distance and taxonomical dataset (genus level) was not significant. When the Mantel test statistic r was calculated on separate forefield datasets (i.e., G, SV, and SW), the variable distance showed a significant (p-value < 0.05) r of 0.19, 0.61, and 0.24 for G, SV, and SW, respectively (Table 3F). Only the G dataset showed a significant correlation to the geochemical dataset (i.e., TN and TOC) with r equal to 0.42 (Table 3F). Supplementary Figure 1 reports how the taxonomic dataset (genus level) is influenced by the variables Distance, TN, and TOC. Ice communities and early stage soil communities were mainly influenced by the genera Thiobacillus, Purpureocillium, Methylotenera, and Cryobacterium, whereas samples sited more distant in the microbial succession were conditioned by Solirubribacter, Hyphomicrobium, Chthoniobacter, and Mycolicibacterium. TOC and TN shaped the distribution of several genera, such as Pseudolabrys, Bradyrhizobium, and Rhodoplanes.
At the phylum level, the two most abundant phyla, representing between 58 and 84% in the soil and 47 and 62% in the ice samples, were Proteobacteria and Actinobacteria, respectively. In all the proglacial system communities, these two phyla showed opposite trends in their abundance, especially in the latest stages of the succession (Figure 5A). The linear model showed a significant negative correlation (R2 = 0.62) between Proteobacteria and Actinobacteria (Figure 5B). The highest correlation was between Alphaproteobacteria and Actinobacteria at class level (Supplementary Table 3).
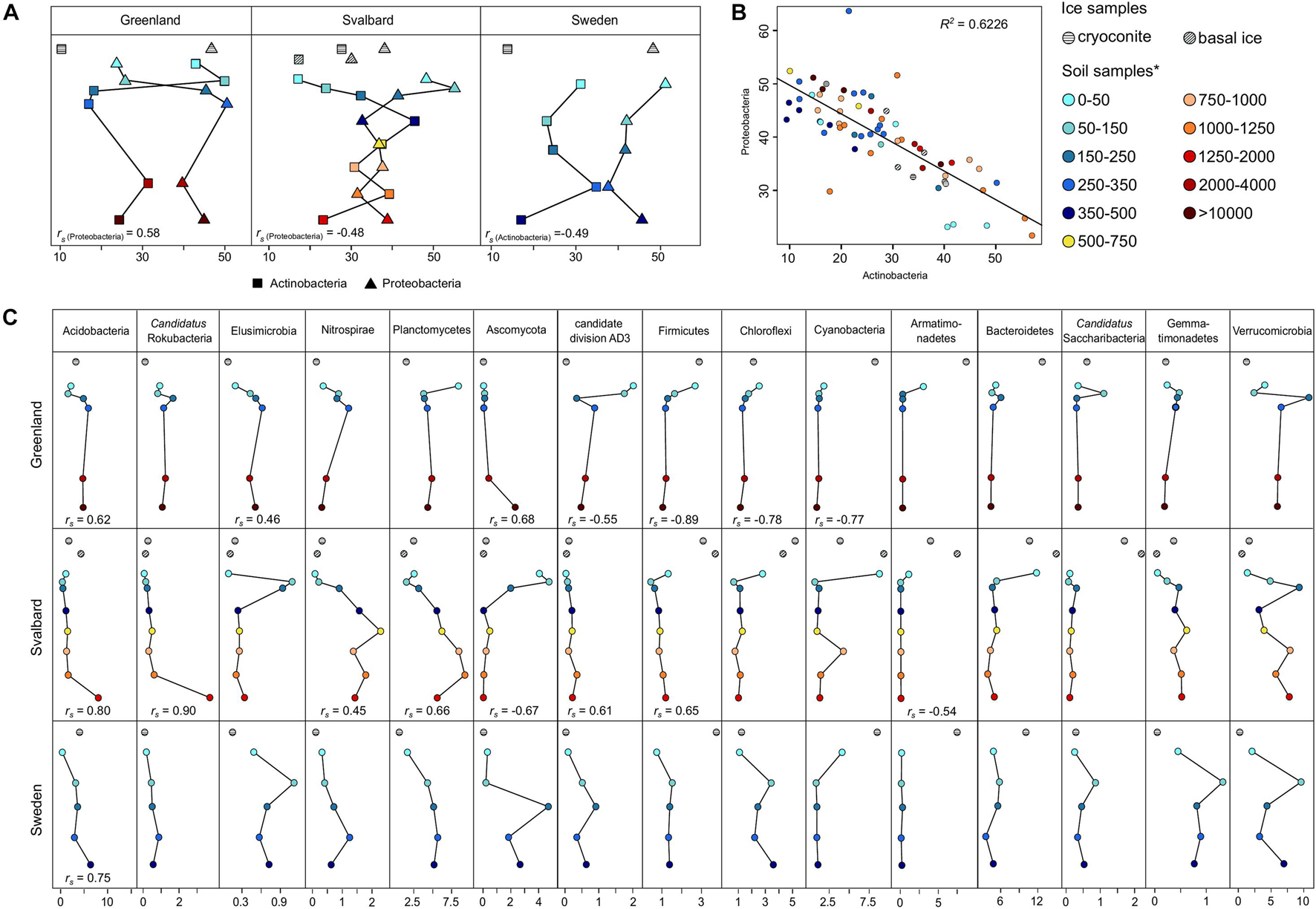
Figure 5. Taxonomy classification at the phylum level. (A) Actinobacteria and Proteobacteria trends in the dataset. (B) Linear model relation between Actinobacteria and Proteobacteria relative abundances (p-value = 3 × 10– 15). (C) Remaining phylum trends in the datasets. Spearman’s rank correlation coefficient (rs) was calculated between the sample distance and the relative abundance data for each phylum. rs is reported below the appropriate line plot only when significant (p-value < 0.05). *Soil samples are categorized in relation to their distance from the ice edge (m).
The phyla Acidobacteria, Candidatus Rokubacteria, Elusimicrobia, Nitrospirae, and Planctomycetes showed a significant positive Spearman’s rank correlation coefficient (rs) in at least one of the three forefield systems, showing an increase in the phyla relative abundance going farther away from the glacier toe, whereas Ascomycota, candidate division AD3, and Firmicutes showed both positive and negative rs in different proglacial systems. The phyla Armatimonadetes, Bacteroidetes, Cyanobacteria, and Firmicutes showed a high abundance in the ice samples and the sites proximal to the ice edge and then decreased in more distant soil (Figure 5C).
The PERMANOVA performed on the gene dataset (Table 3C) explained 26% of the observed variance of the samples across different forefields (p-value < 0.05) but only 10% of the variance in the GO dataset (Table 3D). Both values decreased when we looked at the datasets without ice (Supplementary Tables 2C,D). The Mantel test statistic performed between the distance from the ice edge and the entire dataset showed a correlation of 0.20. The gene dataset was then significant (p-value < 0.05) for SV and SW when correlated to the distance from the ice edge, with r equal to 0.47 and 0.37, respectively. In G, the geochemical datasets (TN and TOC) correlated with an r of 0.29 (Table 3G).
The 2,422 GO categories found in the proglacial dataset were then checked for statistical correlations with the distances from the ice edge, TN, and TOC. Out of a total of more than 2,400 GO categories, 431 and 110 were positively and negatively correlated to distance, 233 and 374 to TN, and 202 and 328 to TOC. All the GO classes that showed a significant correlation (p-value < 0.05) higher than 0.4 or lower than −0.4 were reported in Supplementary Tables 4–6. For example, the distance from the ice edge had a significant positive correlation with oxalate metabolic processes (r = 0.61). Also, genes involved in starvation responses (e.g., cellular response to amino acid starvation) or RNA and DNA repair decreased with the increase of the soil distance from the ice front. Furthermore, more genes indicating photosynthetic metabolism were present in early stages of the succession. Finally, distance, TN and TOC positively correlated with the distribution of genes involved in the response to drug and antimicrobial compounds.
No common and clear trends of the nitrogenase gene coverages were observed across the three forefields, where the coverage values peaked at different forefield stages. In the G forefield, the highest coverage of nitrogenase genes was observed at 150–250 m distance, SV showed higher coverage values in the medium soil stages (i.e., 250–750 m), and SW showed a gradual coverage increase with the site distance from the ice edge. The number of genera associated with nitrogenase genes followed the coverage trends (Figure 6A). Whereas Spearman’s correlation coefficient for gene coverage vs. distance was not significant for any of the datasets, it was significant (p-value < 0.05) for the G, SV, and SW datasets when performed against the TN data (r equals 0.50, 0.30, and 0.54, respectively).
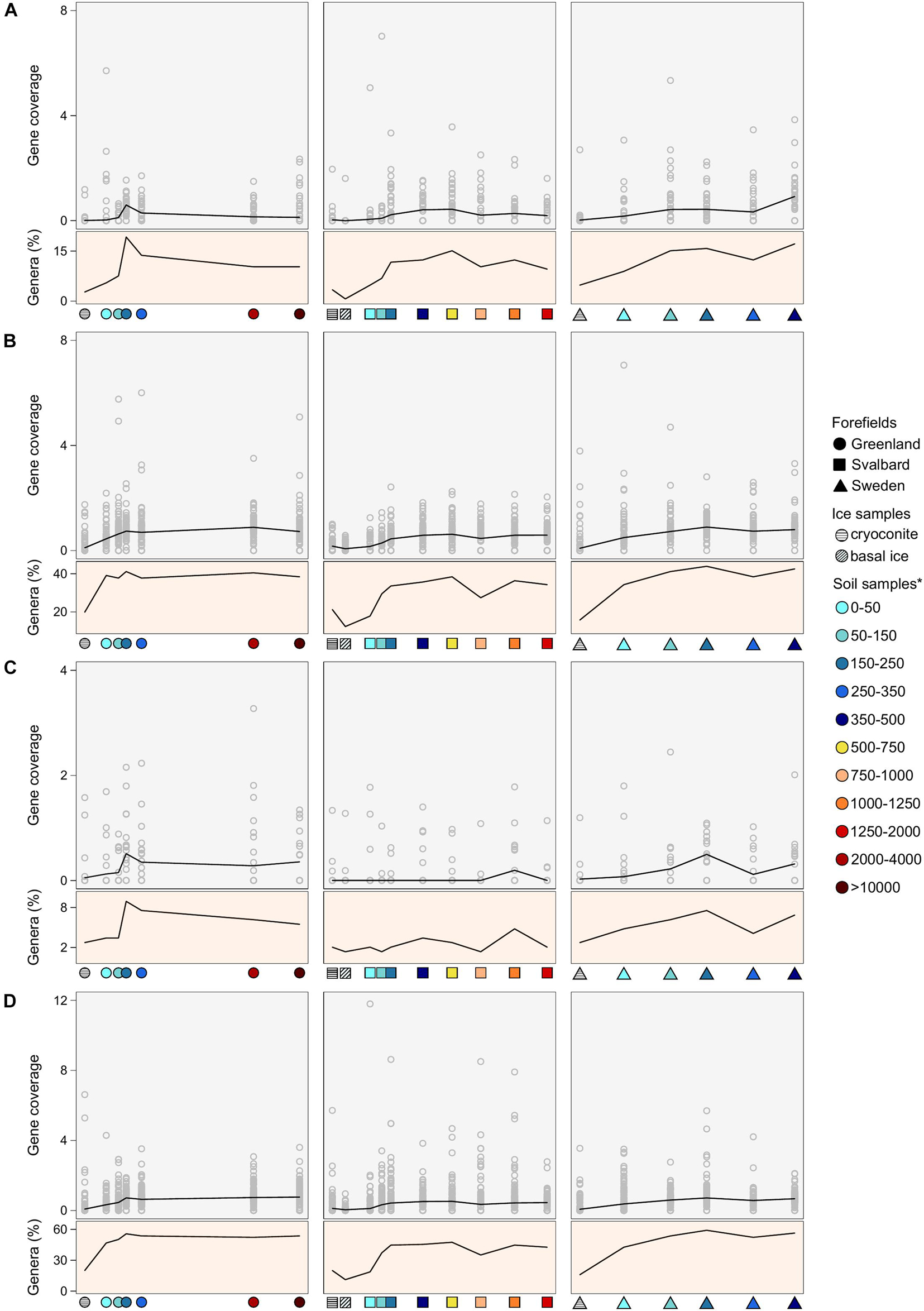
Figure 6. Gene coverage and number of genera trends along the microbial succession for (A) nitrogenase genes, (B) obcA genes that are involved in oxalate biosynthesis, (C) cyanide synthase genes, and (D) siderophore-related genes. The percentage of genera is calculated as the number of genera that possess the studied gene divided by the total number of genera at a certain distance. *Soil samples are categorized in relation to their distance from the ice edge (m).
Thirty genera were found to have at least one assigned region of the assembly with a nitrogenase coding region. The distance where the majority of taxa had a peak in the nitrogenase coverage was 150–250 m in the G succession, 500–750 m in the SV, and 50–150 m in the SW datasets. Geobacter, Bradyrhizobium, Nostoc, and Paraburkholderia had the highest number of genes associated with nitrogenase activity, 156, 101, 53, and 49, respectively (Figure 7). Nostoc was the most abundant genus in the ice samples and in the samples closer to the glacier edge, whereas the other three genera showed similar coverages across the soil succession. Frankia had 17 contig regions associated with nitrogenase and showed the highest coverage in the early and medium soil stages of the SV and SW soil successions, but it was not present in the ice samples. The other nitrogen-fixing organisms identified from our pipeline were Polaromonas, Rhizobium, Rhodopseudomonas, Fimbriiglobus, Streptomyces, and Mesorhizobium that showed different trends along the soil succession.
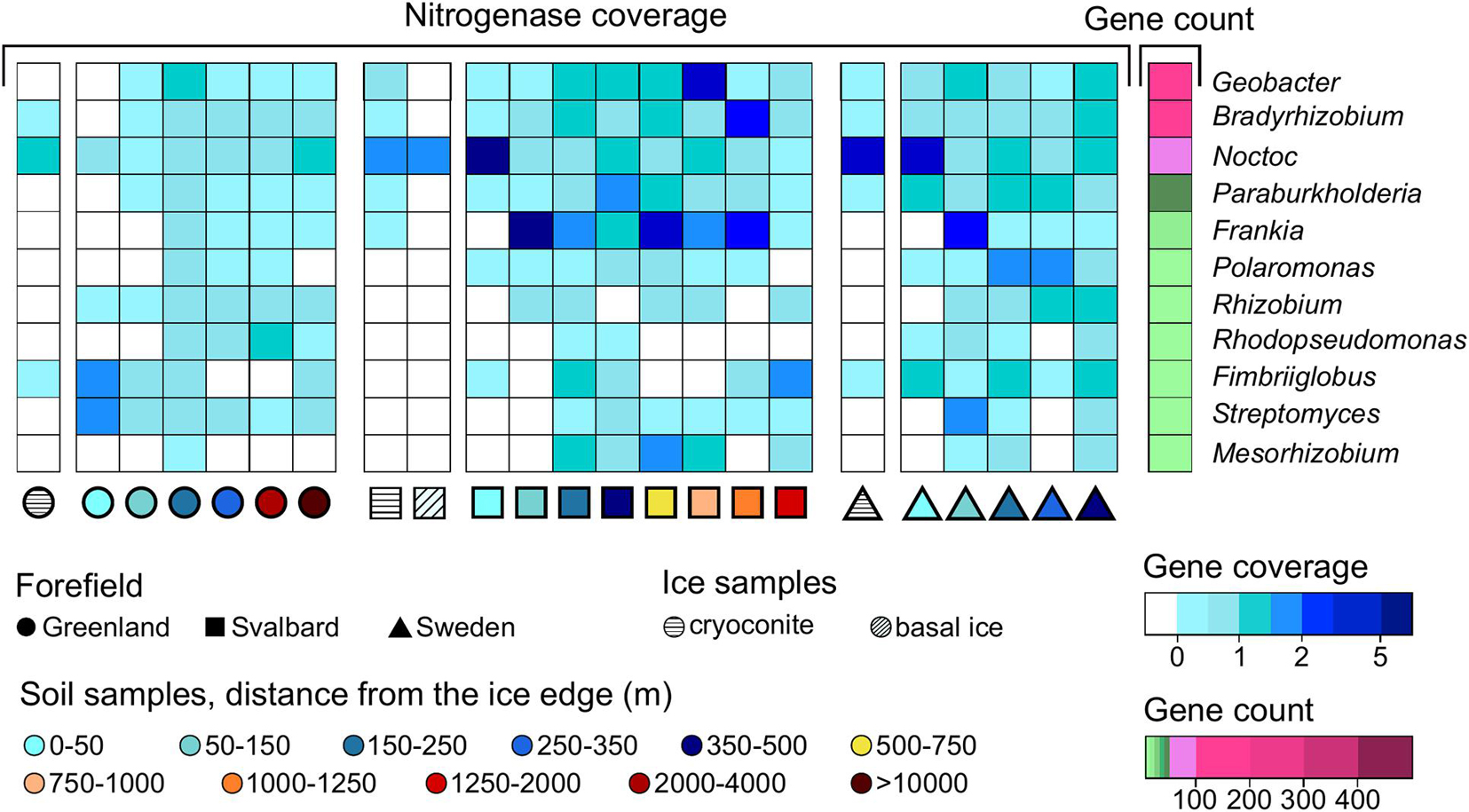
Figure 7. Nitrogenase gene trends at the genus level along the microbial succession. Only genera with more than one coding region associated with a nitrogenase are reported.
Two hundred two assembled coding regions associated with nitrogenases were also associated with unclassified contigs at the genus level. Whereas 50% of the genes were assigned to unknown organisms at the phylum level, 40% of these nitrogenase coding regions were assigned to Proteobacteria and another 5% to Verrucomicrobia (Supplementary Figure 2A).
Genes related to rock weathering processes (i.e., obcA, cyanide synthase, and genes involved in siderophore synthesis and transport) showed a general lower coverage in the ice and early soil samples and an increase with microbial succession. The number of genera containing these genes followed the coverage trends, increasing with the distance from the ice edge (Figures 6B–D). When looking at the gene distribution at the genus level (Figures 8, 9 and Supplementary Figure 3), we did not observe a common trend of gene distribution. For all the three rock weathering genes, different taxa showed different trends in the same forefield and the same taxon showed different trends in different forefields.
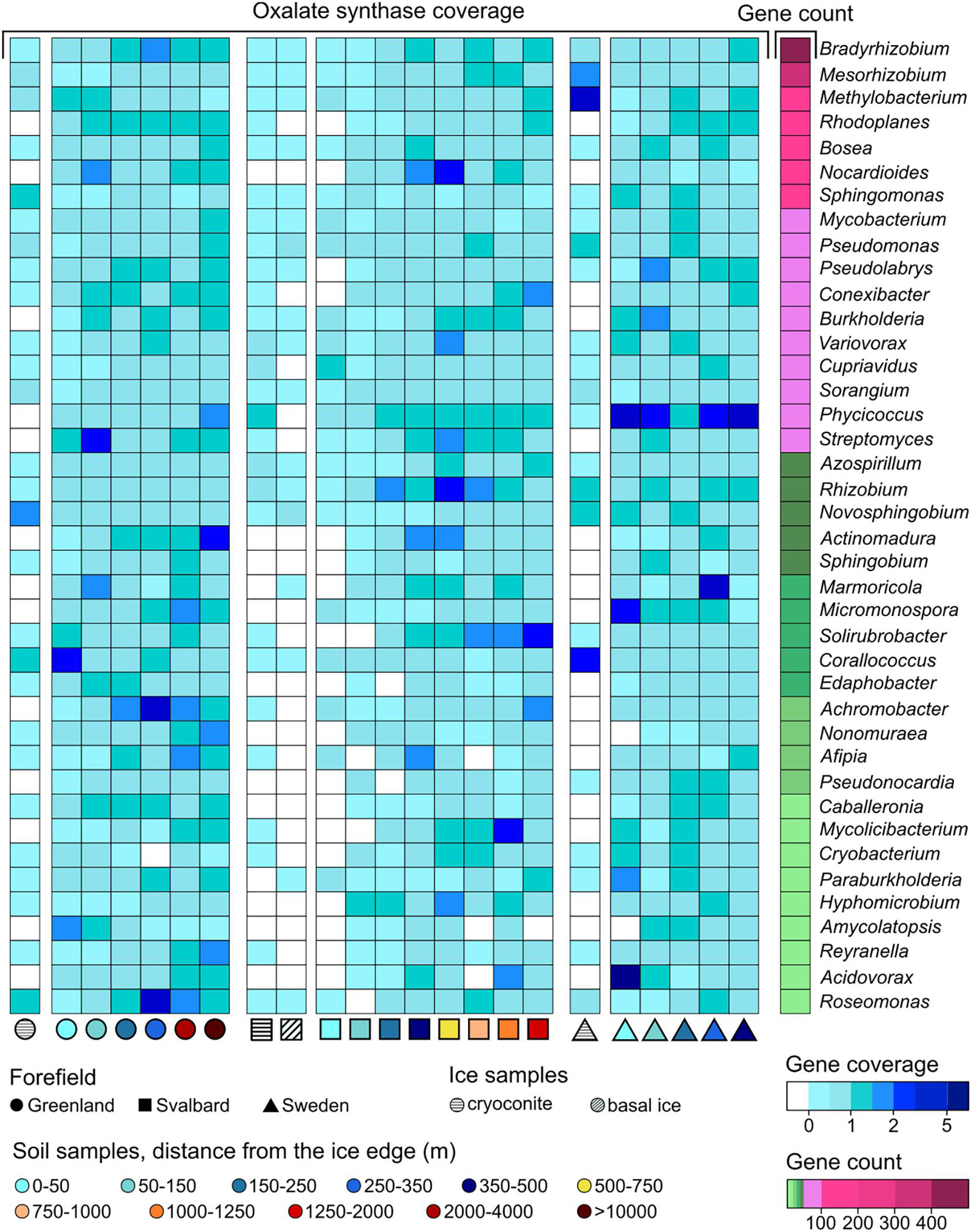
Figure 8. obcA gene (involved in oxalate biosynthesis) trends at the genus level along the microbial successions. Only genera with more than 10 coding regions associated with obcA genes are reported.
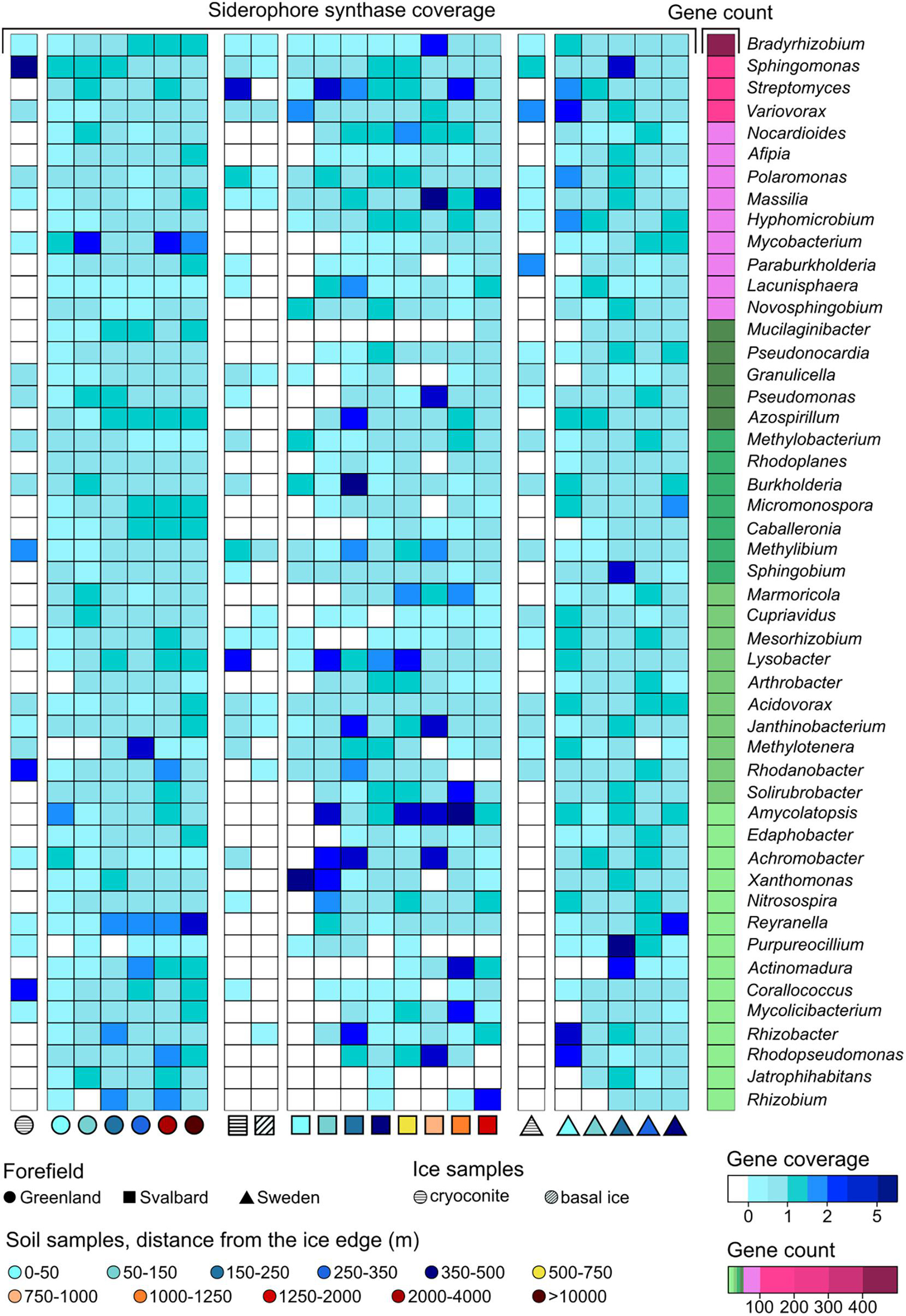
Figure 9. Siderophore-related gene trends at the genus level along the microbial successions. Only genera with more than 10 coding regions associated with siderophore-related genes are reported.
Sixty-five genera had at least one obcA gene assigned to their contigs. The genera Bradyrhizobium, Mesorhizobium, Methylobacterium, Rhodoplanes, Bosea, Nocardiales, and Sphingomonas had the highest content with 456, 320, 157, 127, 117, 117, and 105 genes, respectively (Figure 8). Some of the other genera showed fewer associated contig areas (i.e., fewer genes), but a high coverage of this gene was involved in oxalate biosynthesis (i.e., obcA) at different soil stages. Cyanide synthase genes were less abundant in the three datasets, and only 16 genera were associated with these. The genus Gemmata had most of the genes with 18, followed by Microbacterium (7) and Singulisphaera (5). These genera were also the most ubiquitous in the successions compared to the other cyanide synthase-related genera (Supplementary Figure 3). The three genera that, in particular, showed a higher base coverage in some sites of the microbial succession stages were Streptomyces, Pseudomonas, and Variovorax. More genera were associated with genes involved in siderophore synthesis and transport (81). Bradyrhizobium, Sphingomonas, Streptomyces, and Variovorax had most of the genes, and they had overall a higher abundance in the early stages of the SV and SW successions (Figure 9).
The obcA gene was also associated with 2,234 unclassified contigs at the genus level, the cyanide synthase gene to 49, and the siderophore genes to 1,919 contig regions. These unclassified contigs mainly belonged to the phyla Proteobacteria, Acidobacteria, Actinobacteria, and Planctomycetes (Supplementary Figures 2B–D).
Microbial successions have been widely studied with the aim of understanding microbial-driven environmental processes and how communities are shaped by environmental factors (Fierer et al., 2010). Glacial forefields constitute ideal systems to study microbial successions, as they are characterized by different environmental gradients, such as an increase in TOC, TN, and vegetation coverage and a decrease in pH; these vary in different soil genesis stages from the bedrocks close to the ice edge to the more developed soil. Many physiochemical factors (e.g., pH, TOC, TN, and water availability) have been shown to influence and shape the microbial diversity and structure at biogeographic scales (Malard and Pearce, 2018) and in forefield specifically (Bradley et al., 2014, 2016; Wang et al., 2020). In our study, we report only two environmental variables (i.e., TOC and TN). Two main biological-driven processes that help create forefield geochemical and microbial trends are nitrogen fixation and rock weathering. We investigated microbial successions from three different proglacial systems with the aims of exploring glacial forefield microbial trends, investigating nitrogen-fixing and rock weathering processes, and exploring the taxonomy associated with these processes.
Whereas the Svalbard and Sweden communities were mainly shaped by their distance from the ice edge, the Greenland communities were more shaped by TN and TOC gradients (Table 3F). This could be due to the nature of its complex dynamics where the GrIS does not retreat linearly and it retreats slower compared to the other systems, leading to more complex dynamics for microbial development (Nienow et al., 2017). The same result was observed when looking at the functional dataset (Table 3G). However, this dataset (not divided into different forefields) also showed a significant correlation with the distance from the ice edge, suggesting that, in this case, certain functional profiles such as photosynthesis and response to starvation or to antimicrobial compounds were conserved across the different forefield gradients (Supplementary Table 4). The variance observed in the gene dataset was also better explained by the forefield differentiation compared to the taxon dataset (Table 3). This indicates how gene and trait-based data could be used in biogeographic studies instead of the more widely used taxonomy data (Green et al., 2008).
In this study, along the microbial succession, microbial communities increased in diversity between ice and soil samples and also with soil complexity (i.e., soil age since deglaciation), where the biggest increase was after the early soil stages of the succession (Figure 3). This was previously observed in Jiang et al. (2018) where bacterial diversity was higher in later soil stages compared to early stages. The first succession stages are characterized by challenging environmental conditions (e.g., nutrient depletion and hydrological disturbances) and are deeply influenced by the glacier environment (characterized by a lower microbial diversity). For these reasons, this soil has a lower community diversity compared to the more developed soil where there is the formation of differentiated ecological niches due to the nutrient increase and plant development (Reynolds et al., 2003; Dumbrell et al., 2010).
In all three proglacial systems, the ice microbial communities comprised of organisms typically found in the cryosphere (Figure 5C). Cyanobacteria, Firmicutes, Bacteroidetes, and Armatimonadetes showed a high abundance in the first stages of the succession where the soil is highly affected by glacial water discharges (Boyd et al., 2014; Bradley et al., 2014; Dieser et al., 2014) and environmental characteristics favor the development of communities adapted to nutrient-depleted conditions. Cyanobacteria and Firmicutes (spore-forming organisms) are widely found in the glacial environment (Galperin, 2016; Segawa et al., 2017). Organisms belonging to the Bacteroidetes phylum have been found in many glacial habitats (e.g., Zhang et al., 2009; Wilhelm et al., 2013; Smith et al., 2016). The diffused presence of these non-spore-forming heterotrophic organisms has been proposed to be related to their ability to assimilate and use recalcitrant substances (Zeng et al., 2013) abundant in the glacier environment (Kmezik et al., 2020). Organisms belonging to Armatimonadetes have been found in this environment (e.g., Bajerski and Wagner, 2013; Zhang et al., 2015; Gokul et al., 2016). This phylum is not well characterized, but its few isolates have been characterized as chemoheterotrophs (Tamaki et al., 2011; Lee et al., 2014).
Whereas in the first soil stages of the succession, microbial diversity showed a presence of autotrophic organisms (i.e., Cyanobacteria), spore-forming organisms (i.e., Firmicutes), and organisms specialized to use recalcitrant compounds (i.e., Bacteroidetes), processes and organisms associated with heterotrophy were shown to increase in later stages of the microbial succession. In the three proglacial systems, microbial succession was characterized by the increase of Acidobacteria (Figure 5C). Organisms belonging to this phylum are heterotrophs and adapted to use a variety of organic carbon sources. Furthermore, many of these organisms are acidophiles (Kielak et al., 2016) and therefore more adapted to live in the later stages of the succession where the soil is enriched of organic carbon and pH is lower (Zumsteg et al., 2012). Other phyla such as Candidatus Rokubacteria, Elusimicrobia, Nitrospirae, and Planctomycetes showed an increased abundance in the more developed soil of the later stages. Elusimicrobia have been isolated mainly from soil and are principally insect symbionts (Méheust et al., 2019). Candidatus Rokubacteria and Verrucomicrobia are characterized by organisms able to perform sulfate reduction (Anantharaman et al., 2018). Nitrospirae metabolism relies on nitrite oxidation (Daims et al., 2016). These microorganisms are favored in the later stages of the succession where nitrogen and sulfur stocks have been built by diazotrophs and rock weathering organisms. Actinobacteria and Proteobacteria (Alphaproteobacteria in particular) showed oscillating trends along the three proglacial environments (Figure 5A and Supplementary Table 3). Actinobacteria and Proteobacteria are two ubiquitous phyla and are both characterized by a wide metabolic range (Barka et al., 2016; Taylor et al., 2019). However, a change in the relative abundance of these two phyla has previously been observed in relation to different soil matrices and mineralogy that can be encountered both at small-scale distances and along forefield successions in soil (Mitchell et al., 2013; Uroz et al., 2015).
In the soil successions, the main CO2-fixing organisms in the early successional soil stages belonged to the phylum Cyanobacteria, where Nostoc was the most abundant genus. In later soil stages, autotrophic organisms were ascribed to several microbial phyla, such as Planctomycetes, Nitrospirae, Chloroflexi, Proteobacteria, and Verrucomicrobia (Xu et al., 2017; Chen et al., 2018; Li et al., 2019). Most of these phyla showed an increase along the soil successions, in particular in the SV and SW forefields where a more linear soil chronosequence was present (Figures 1, 5C). This suggests phototrophs to be substituted by chemolithoautotrophic organisms along forefield microbial successions, where more nutrients are present in later soil stages to sustain chemolithoautotrophic metabolisms.
Although ice-related/soil-related organism trends were observed in all the proglacial systems, the latter showed differences in relative abundances of several taxa (e.g., Nitrospirae, Candidatus division AD3, Chloroflexi, and Gammatimonadetes), showing how local patterns and characteristics could influence the microbial distribution and the biogeography of different soil microbial communities. Environmental selection to specific conditions (e.g., DOC, TN, and pH) has previously been observed to strongly shape microbial communities at large biogeographic spatial scales (Hanson et al., 2012; Malard and Pearce, 2018). Highlighting this aspect, no correlation between distance from the ice edge and the entire forefield dataset was observed; a high correlation was obtained only when looking at the microbial distribution across the separate forefields (Table 3F), showing how the forefield local factors had a role in shaping the microbial communities.
We investigated the taxonomy of the organisms performing two important microbially driven environmental processes: nitrogen fixation and rock weathering. To do so, we focused on four different sets of genes: (i) nitrogenase genes, (ii) obcA genes involved in the first step of the oxalate biosynthesis, (iii) genes involved in cyanide synthesis, and (iv) genes involved in siderophore synthesis and transport. Whereas the first one is involved in nitrogen fixation, the other three are involved in rock weathering processes where oxalate and cyanide have been observed to increase rock and mineral dissolution and siderophore is a molecule used to facilitate iron uptake inside the cells. These processes have previously been shown to be prevalent closer to the ice edge where microorganisms that are able to produce metabolic energy from atmospheric nitrogen and inorganic compounds (e.g., minerals) are favored (Brankatschk et al., 2011; Fernández-Martínez et al., 2016).
In all three systems, genes associated with these processes were shared by the lowest percentage of taxa in the early stages and by more in later succession stages (Figures 6–9). Interestingly, nitrogenase genes showed a correlation with TN concentration values, highlighting the pivotal role of diazotrophs in the creation of a nitrogen pool in proglacial systems (Nash et al., 2018). Gene coverages observed at specific genus levels showed a variety of trends and distributions, suggesting that different organisms drive the same ecological processes (i.e., nitrogen fixation and rock weathering) but at different stages of microbial succession.
Nitrogenases are key enzymes in the fixation of atmospheric nitrogen to ammonium. These enzymes are coded by a wide gene class (e.g., nifK, nifH, and nifD genes) and have been widely used to detect biological nitrogen fixation potential in a variety of environmental settings (Hoffman et al., 2014). Further, diazotrophic organisms have been found spanning a wide range of different bacterial phyla (Addo and Dos Santos, 2020).
Most of the 30 genera associated with nitrogenase genes in our dataset belonged to the phyla Proteobacteria (17) and Actinobacteria (6). The only cyanobacterial organism to which nitrogenase was assigned in our dataset was Nostoc, which also showed a higher abundance in the ice realm compared to the proglacial soil and has previously been detected by Fernandez et al. (2020) in the Arctic region. The other cyanobacterial organisms present in our dataset were present in a lower percentage (1%) compared to Nostoc (4%); between them, there are, for example, Phormidesmis and Chamaesiphon. These organisms are typical of the cold environments and do not perform nitrogen fixation (Jungblut and Vincent, 2017; Segawa et al., 2017; Uetake et al., 2019). In the soil, diazotrophic organisms belonging to Proteobacteria and Actinobacteria were more abundant (Figure 7). These phyla are usually associated with soil environment and root symbiosis (Rudnick et al., 1997; Gtari et al., 2012). In addition to Nostoc, the nitrogen fixers Bradyrhizobium, Geobacter, Paraburkholderia, and Frankia (Sellstedt and Richau, 2013; Calderoli et al., 2017; Thangaraj et al., 2017; Choi and Im, 2018; Hara et al., 2019) were abundant in all our studied systems (Figure 7). The same dominant nitrogen fixers were found in Nash et al. (2018) where Nostoc, Geobacter, Rhizobium, Polaromonas, and Frankia were found dominant in all three forefield systems. In our study, Streptomyces was also found associated with nitrogenase genes. This genus has only recently been shown to perform nitrogen fixation by isolation and sequencing from environmental soil (Dahal et al., 2017). This result represents the first confirmation of the presence of nitrogenase genes in Streptomyces. Furthermore, the genus Fimbriiglobus was also associated with these genes in our dataset. The only organism previously sequenced belonging to this genus (Fimbriiglobus ruber) does not have nitrogen-fixing genes (Ravin et al., 2018). However, our result could suggest that other members of the genus do.
Compared to biological nitrogen fixation, less is known about biological rock weathering. Whereas some microbial processes involved in the rock weathering have already been identified, the protagonists of these processes still remain unclear (Samuels et al., 2020). We explored the taxonomy associated with three different rock weathering-associated genes: the obcA gene, cyanide synthase genes, and genes involved in siderophore transport and synthesis. Whereas oxalate, cyanide, and siderophore syntheses have been shown to be correlated with rock weathering (Welch et al., 1999; Frey et al., 2010; Ferreira et al., 2019), these three molecules are involved in other processes. For instance, the release of oxalate has been shown to play an important role in pathogenicity and metal tolerance (Palmieri et al., 2019) and cyanide release plays a role in microbial competition (Blumer and Haas, 2000). Siderophores are molecules commonly used by microorganisms for the uptake of environmental iron but also to sequester iron from hosts (Krewulak and Vogel, 2008; Braun and Hantke, 2011). Even if all of these processes are present in organisms not involved in rock weathering, we expect a higher abundance of these organisms in early succession samples where the rock weathering is predominant. However, the genera associated with rock-weathering genes did not show common and continuous trends along the chronosequence, suggesting that different organisms can conduct a certain environmental function (e.g., rock dissolution) at different succession stages and soil conditions (Figures 8, 9 and Supplementary Figure 3).
The genus that showed most of the genes involved in oxalate and siderophore biosynthesis was Bradyrhizobium. Organisms belonging to this genus are mainly recognized as plant symbionts but can also be present as free-living organisms (Kulkarni et al., 2015; Čuklina et al., 2016). The high content and diversity of genes involved in siderophore metabolism relate to the fact that high production of siderophore is needed to uptake iron released from the rock dissolution performed by oxalate release. These organisms are also diazotrophs and require iron as a nitrogenase cofactor (Rubio and Ludden, 2008). Mesorhizobium and Paraburkholderia are also common diazotrophic components of the rhizosphere but have been detected as free-living organisms (Ahmad et al., 2008), and they showed high presence of both these genes. Paraburkholderia was found to have chemotactic sensitivity for oxalate even if it was not directly shown that it can produce it (Haq et al., 2018). Micromonospora also had high oxalate-related gene abundance especially in the first stages of the SW dataset. Organisms related to this genus have been found in soil by different studies (Thawai et al., 2016; Malisorn et al., 2020) and have previously been found to increase phosphate and iron solubilization in soil due to organic acid release, such as oxalate (El-Tarabily et al., 2008). Overall, Burkholderia had a higher presence of both siderophore and oxalate-related genes in early stages compared to later soil stages. These organisms have been identified by many different studies as oxalate producers (Nakata, 2011; Oh et al., 2014) and often have been found in proglacial soil (Frey et al., 2010). Pseudomonas was shown to produce oxalic acids (Hamel et al., 1999), which concords with the trends in our datasets. Additionally, contigs associated with Pseudomonas were found to contain cyanide synthase-coding regions. Streptomyces was previously identified as a phosphate solubilizer, but mechanisms were not identified (Liu et al., 2016). Other than the oxalate genes, this genus was also shown to have cyanide synthase genes in our dataset. Finally, Fimbriiglobus, Corallococcus, Variovorax, and Phycicoccus are organisms typically found in soil (Yoon et al., 2008; Zhang et al., 2011; Singh et al., 2015) and were associated with oxalate, cyanide, and siderophore genes for the first time in this study.
Microbial successions in the three proglacial systems showed early soil stages enriched with autotrophic, spore-forming, and recalcitrant compound degraders showing a community influenced by the common ice microbiome, whereas later soil stages showed a higher heterotrophic microbial component. Although there were common taxonomic trends among the proglacial systems, taxon contribution to the different proglacial microbial communities showed differences, suggesting the presence of biogeographic differences between proglacial systems. Furthermore, rock weathering and nitrogenase gene distributions peaked at different succession soil stages in different proglacial systems. Different genera showed different gene coverages at different stages of the microbial successions, indicating a community constituted by a plurality of organisms involved in these processes, and where the latter were driven by different organisms in different soil succession stages. Whereas we confirmed the presence of nitrogenase and rock weathering genes in known nitrogen-fixing and rock weathering organisms, in this study, we also present organisms that, even if often found in soil and proglacial systems, have never been related to these processes before. These results represent a further step toward a more comprehensive understanding of nitrogen-fixing and biotic rock weathering processes and highlight how a better understanding of these and other microbially driven environmental processes (e.g., CO2 fixation) is necessary for a better comprehension of microbial successions in a warming Arctic.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ebi.ac.uk/ena/browser/view/PRJEB41174.
All authors wrote the manuscript. GV analyzed the data and performed the statistical analysis.
Funding support was provided through NERC grants NE/J02399X/1 awarded to AA and NE/J022365/1 awarded to GB. Funding support for GV came from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 675546–MicroArctic.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors GB.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Maisie Nash for sharing the sequencing and geochemical data.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.627437/full#supplementary-material
Abdi, H., and Williams, L. J. (2010). Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2, 433–459. doi: 10.1002/wics.101
Addo, M. A., and Dos Santos, P. C. (2020). Distribution of nitrogen-fixation genes in prokaryotes containing alternative nitrogenases. ChemBioChem 21, 1749–1759. doi: 10.1002/cbic.202000022
Ahmad, F., Ahmad, I., and Khan, M. S. (2008). Screening of free-living rhizospheric bacteria for their multiple plant growth promoting activities. Microbiol. Res. 163, 173–181. doi: 10.1016/j.micres.2006.04.001
Anantharaman, K., Hausmann, B., Jungbluth, S. P., Kantor, R. S., Lavy, A., Warren, L. A., et al. (2018). Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 12, 1715–1728. doi: 10.1038/s41396-018-0078-0
Anderson, M. J. (2017). “Permutational multivariate analysis of variance (PERMANOVA).” in Wiley StatsRef Statistical Reference Online, eds N. Balakrishnan, T. Colton and B. Everitr. Hoboken, NJ: John Wiley & Sons doi: 10.1002/9781118445112.stat07841
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bajerski, F., and Wagner, D. (2013). Bacterial succession in Antarctic soils of two glacier forefields on Larsemann Hills, East Antarctica. FEMS Microbiol. Ecol. 85, 128–142. doi: 10.1111/1574-6941.12105
Barka, E. A., Vatsa, P., Sanchez, L., Nathalie Gaveau-Vaillant, C. J., Klenk, H.-P., Clément, C., et al. (2016). Taxonomy, physiology, and natural products of Actinobacteria. Am. Soc. Microbiol. 80, 1–43. doi: 10.1128/MMBR.00019-15
Blumer, C., and Haas, D. (2000). Mechanism, regulation, and ecological role of bacterial cyanide biosynthesis. Arch. Microbiol. 173, 170–177. doi: 10.1007/s002039900127
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borin, S., Ventura, S., Tambone, F., Mapelli, F., Schubotz, F., Brusetti, L., et al. (2010). Rock weathering creates oases of life in a high Arctic desert. Environ. Microbiol. 12, 293–303. doi: 10.1111/j.1462-2920.2009.02059.x
Boyd, E. S., Hamilton, T. L., Havig, J. R., Skidmore, M. L., and Shock, E. L. (2014). Chemolithotrophic primary production in a subglacial ecosystem. Appl. Environ. Microbiol. 80, 6146–6153. doi: 10.1128/AEM.01956-14
Bradley, J. A., Arndt, S., Šabacká, M., Benning, L. G., Barker, G. L., Blacker, J. J., et al. (2016). Microbial dynamics in a high Arctic glacier forefield: a combined field, laboratory, and modelling approach. Biogeosciences 13, 5677–5696. doi: 10.5194/bg-13-5677-2016
Bradley, J. A., Singarayer, J. S., and Anesio, A. M. (2014). Microbial community dynamics in the forefield of glaciers. Proc. R. Soc. B Biol. Sci. 281:20140882. doi: 10.1098/rspb.2014.0882
Brankatschk, R., Töwe, S., Kleineidam, K., Schloter, M., and Zeyer, J. (2011). Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J. 5, 1025–1037. doi: 10.1038/ismej.2010.184
Braun, V., and Hantke, K. (2011). Recent insights into iron import by bacteria. Curr. Opin. Chem. Biol. 15, 328–334. doi: 10.1016/j.cbpa.2011.01.005
Brunner, I., Plötze, M., Rieder, S., Zumsteg, A., Furrer, G., and Frey, B. (2011). Pioneering fungi from the Damma glacier forefield in the Swiss Alps can promote granite weathering. Geobiology 9, 266–279. doi: 10.1111/j.1472-4669.2011.00274.x
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Calderoli, P. A., Collavino, M. M., Behrends Kraemer, F., Morrás, H. J. M., and Aguilar, O. M. (2017). Analysis of nifH-RNA reveals phylotypes related to Geobacter and Cyanobacteria as important functional components of the N2-fixing community depending on depth and agricultural use of soil. Microbiologyopen 6:e00502. doi: 10.1002/mbo3.502
Carbon, S., Douglass, E., Dunn, N., Good, B., Harris, N. L., Lewis, S. E., et al. (2019). The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
Chen, D., Wang, H., Yang, K., and Ma, F. (2018). Performance and microbial communities in a combined bioelectrochemical and sulfur autotrophic denitrification system at low temperature. Chemosphere 193, 337–342. doi: 10.1016/j.chemosphere.2017.11.017
Choi, G. M., and Im, W. T. (2018). Paraburkholderia azotifigens sp. nov., a nitrogen-fixing bacterium isolated from paddy soil. Int. J. Syst. Evol. Microbiol. 68, 310–316. doi: 10.1099/ijsem.0.002505
Čuklina, J., Hahn, J., Imakaev, M., Omasits, U., Förstner, K. U., Ljubimov, N., et al. (2016). Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genomics 17:302. doi: 10.1186/s12864-016-2602-9
Dahal, B., NandaKafle, G., Perkins, L., and Brözel, V. S. (2017). Diversity of free-living nitrogen fixing Streptomyces in soils of the badlands of South Dakota. Microbiol. Res. 195, 31–39. doi: 10.1016/j.micres.2016.11.004
Daims, H., Lücker, S., and Wagner, M. (2016). A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol. 24, 699–712. doi: 10.1016/j.tim.2016.05.004
Dieser, M., Broemsen, E. L. J. E., Cameron, K. A., King, G. M., Achberger, A., Choquette, K., et al. (2014). Molecular and biogeochemical evidence for methane cycling beneath the western margin of the Greenland Ice Sheet. ISME J. 8, 2305–2316. doi: 10.1038/ismej.2014.59
Duc, L., Noll, M., Meier, B. E., Bürgmann, H., and Zeyer, J. (2009). High diversity of diazotrophs in the forefield of a receding alpine glacier. Microb. Ecol. 57, 179–190. doi: 10.1007/s00248-008-9408-5
Dumbrell, A. J., Nelson, M., Helgason, T., Dytham, C., and Fitter, A. H. (2010). Relative roles of niche and neutral processes in structuring a soil microbial community. ISME J. 4, 337–345. doi: 10.1038/ismej.2009.122
Edwards, A., and Cook, S. (2015). Microbial dynamics in glacier forefield soils show succession is not just skin deep. Mol. Ecol. 24, 963–966. doi: 10.1111/mec.13098
El-Tarabily, K. A., Nassar, A. H., and Sivasithamparam, K. (2008). Promotion of growth of bean (Phaseolus vulgaris L.) in a calcareous soil by a phosphate-solubilizing, rhizosphere-competent isolate of Micromonospora endolithica. Appl. Soil Ecol. 39, 161–171. doi: 10.1016/j.apsoil.2007.12.005
Fernandez, L., Bertilsson, S., and Peura, S. (2020). Non-cyanobacterial diazotrophs dominate nitrogen-fixing communities in permafrost thaw ponds. Limnol. Oceanogr. 65, S180–S193. doi: 10.1002/lno.11243
Fernández-Martínez, M. A., Pérez-Ortega, S., Pointing, S. B., Allan Green, T. G., Pintado, A., Rozzi, R., et al. (2017). Microbial succession dynamics along glacier forefield chronosequences in Tierra del Fuego (Chile). Polar Biol. 40, 1939–1957. doi: 10.1007/s00300-017-2110-7
Fernández-Martínez, M. A., Pointing, S. B., Pérez-Ortega, S., Arróniz-Crespo, M., Allan Green, T. G., Rozzi, R., et al. (2016). Functional ecology of soil microbial communities along a glacier forefield in Tierra del Fuego (Chile). Int. Microbiol. 19, 161–173.
Ferreira, C. M. H., Vilas-Boas, Â, Sousa, C. A., Soares, H. M. V. M., and Soares, E. V. (2019). Comparison of five bacterial strains producing siderophores with ability to chelate iron under alkaline conditions. AMB Express 9:78. doi: 10.1186/s13568-019-0796-3
Fierer, N., Nemergut, D., Knight, R., and Craine, J. M. (2010). Changes through time: integrating microorganisms into the study of succession. Res. Microbiol. 161, 635–642. doi: 10.1016/j.resmic.2010.06.002
Fountain, A. G., Campbell, J. L., Schuur, E. A. G., Stammerjohn, S. E., Williams, M. W., and Ducklow, H. W. (2012). The disappearing cryosphere: impacts and ecosystem responses to rapid cryosphere loss. Bioscience 62, 405–415. doi: 10.1525/bio.2012.62.4.11
Frey, B., Rieder, S. R., Brunner, I., Plötze, M., Koetzsch, S., Lapanje, A., et al. (2010). Weathering-associated bacteria from the damma glacier forefield: physiological capabilities and impact on granite dissolution. Appl. Environ. Microbiol. 76, 4788–4796. doi: 10.1128/AEM.00657-10
Galperin, M. Y. (2016). “Genome diversity of spore-forming firmicute,” in The Bacterial Spore, eds A. Driks and P. Eiechenberger. Hoboken, NJ: Wiley doi: 10.1128/9781555819323.ch1
Gokul, J. K., Hodson, A. J., Saetnan, E. R., Irvine-Fynn, T. D. L., Westall, P. J., Detheridge, A. P., et al. (2016). Taxon interactions control the distributions of cryoconite bacteria colonizing a high Arctic ice cap. Mol. Ecol. 25, 3752–3767. doi: 10.1111/mec.13715
Green, J. L., Bohannan, B. J. M., and Whitaker, R. J. (2008). Microbial biogeography: from taxonomy to traits. Science 320, 1039–1043. doi: 10.1126/science.1153475
Gtari, M., Ghodhbane-Gtari, F., Nouioui, I., Beauchemin, N., and Tisa, L. S. (2012). Phylogenetic perspectives of nitrogen-fixing actinobacteria. Arch. Microbiol. 194, 3–11. doi: 10.1007/s00203-011-0733-6
Hamel, R., Levasseur, R., and Appanna, V. D. (1999). Oxalic acid production and aluminum tolerance in Pseudomonas fluorescens. J. Inorg. Biochem. 76, 99–104. doi: 10.1016/S0162-0134(99)00120-8
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Haq, I. U., Zwahlen, R. D., Yang, P., and van Elsas, J. D. (2018). The response of Paraburkholderia terrae strains to two soil fungi and the potential role of oxalate. Front. Microbiol. 9:989. doi: 10.3389/fmicb.2018.00989
Hara, S., Morikawa, T., Wasai, S., Kasahara, Y., Koshiba, T., Yamazaki, K., et al. (2019). Identification of nitrogen-fixing bradyrhizobium associated with roots of field-grown sorghum by metagenome and proteome analyses. Front. Microbiol. 10:407. doi: 10.3389/fmicb.2019.00407
Heckmann, T., Mccoll, S., and Morche, D. (2016). Retreating ice: research in pro-glacial areas matters. Earth Surf. Process. Landforms 41, 271–276. doi: 10.1002/esp.3858
Hoffman, B. M., Lukoyanov, D., Yang, Z. Y., Dean, D. R., and Seefeldt, L. C. (2014). Mechanism of nitrogen fixation by nitrogenase: The next stage. Chem. Rev. 114, 4041–4062. doi: 10.1021/cr400641x
Hotaling, S., Hood, E., and Hamilton, T. L. (2017). Microbial ecology of mountain glacier ecosystems: biodiversity, ecological connections and implications of a warming climate. Environ. Microbiol. 19, 2935–2948. doi: 10.1111/1462-2920.13766
Jiang, Y., Lei, Y., Qin, W., Korpelainen, H., and Li, C. (2019a). Revealing microbial processes and nutrient limitation in soil through ecoenzymatic stoichiometry and glomalin-related soil proteins in a retreating glacier forefield. Geoderma 338, 313–324. doi: 10.1016/j.geoderma.2018.12.023
Jiang, Y., Lei, Y., Yang, Y., Korpelainen, H., Niinemets, Ü, and Li, C. (2018). Divergent assemblage patterns and driving forces for bacterial and fungal communities along a glacier forefield chronosequence. Soil Biol. Biochem. 118, 207–216. doi: 10.1016/j.soilbio.2017.12.019
Jiang, Y., Song, H., Lei, Y., Korpelainen, H., and Li, C. (2019b). Distinct co-occurrence patterns and driving forces of rare and abundant bacterial subcommunities following a glacial retreat in the eastern Tibetan Plateau. Biol. Fertil. Soils 55, 351–364. doi: 10.1007/s00374-019-01355-w
Jungblut, A. D., and Vincent, W. F. (2017). “Cyanobacteria in polar and alpine ecosystems,” in Psychrophiles: From Biodiversity to Biotechnology, 2nd edn, eds R. Margesin, F. Schinner, and J. C. Marx (Berlin: Springer). doi: 10.1007/978-3-319-57057-0_9
Kelly, E. F., Chadwick, O. A., and Hilinski, T. E. (1998). The effect of plants on mineral weathering. Biogeochemistry 42, 21–53. doi: 10.1023/A:1005919306687
Kielak, A. M., Barreto, C. C., Kowalchuk, G. A., van Veen, J. A., and Kuramae, E. E. (2016). The ecology of acidobacteria: moving beyond genes and genomes. Front. Microbiol. 7:744. doi: 10.3389/fmicb.2016.00744
Kmezik, C., Bonzom, C., Olsson, L., Mazurkewich, S., and Larsbrink, J. (2020). Multimodular fused acetyl-feruloyl esterases from soil and gut Bacteroidetes improve xylanase depolymerization of recalcitrant biomass. Biotechnol. Biofuels 13, 1–14. doi: 10.1186/s13068-020-01698-9
Knelman, J. E., Legg, T. M., O’Neill, S. P., Washenberger, C. L., González, A., Cleveland, C. C., et al. (2012). Bacterial community structure and function change in association with colonizer plants during early primary succession in a glacier forefield. Soil Biol. Biochem. 46, 172–180. doi: 10.1016/j.soilbio.2011.12.001
Koshila Ravi, R., Anusuya, S., Balachandar, M., and Muthukumar, T. (2019). Microbial Interactions in Soil Formation and Nutrient Cycling. Berlin: Springer doi: 10.1007/978-981-13-6480-8_21
Krewulak, K. D., and Vogel, H. J. (2008). Structural biology of bacterial iron uptake. Biochim. Biophys. Acta Biomembr. 1778, 1781–1804. doi: 10.1016/j.bbamem.2007.07.026
Kulkarni, G., Busset, N., Molinaro, A., Gargani, D., Chaintreuil, C., Silipo, A., et al. (2015). Specific hopanoid Ccasses differentially affect free-living and symbiotic states of Bradyrhizobium diazoefficiens. MBio 6, 1–9. doi: 10.1128/mBio.01251-15
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, K. C. Y., Morgan, X. C., Dunfield, P. F., Tamas, I., McDonald, I. R., and Stott, M. B. (2014). Genomic analysis of Chthonomonas calidirosea, the first sequenced isolate of the phylum Armatimonadetes. ISME J. 8, 1522–1533. doi: 10.1038/ismej.2013.251
Lepleux, C., Turpault, M. P., Oger, P., Frey-Klett, P., and Uroz, S. (2012). Correlation of the abundance of betaproteobacteria on mineral surfaces with mineral weathering in forest soils. Appl. Environ. Microbiol. 78, 7114–7119. doi: 10.1128/AEM.00996-12
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, Q., Liu, Y., Gu, Y., Guo, L., Huang, Y., Zhang, J., et al. (2020). Ecoenzymatic stoichiometry and microbial nutrient limitations in rhizosphere soil along the Hailuogou glacier forefield chronosequence. Sci. Total Environ. 704:135413. doi: 10.1016/j.scitotenv.2019.135413
Li, Y., Wu, Z., Dong, X., Jia, Z., and Sun, Q. (2019). Variance in bacterial communities, potential bacterial carbon sequestration and nitrogen fixation between light and dark conditions under elevated CO2 in mine tailings. Sci. Total Environ. 652, 234–242. doi: 10.1016/j.scitotenv.2018.10.253
Liu, D. F., Lian, B., and Wang, B. (2016). Solubilization of potassium containing minerals by high temperature resistant Streptomyces sp. isolated from earthworm’s gut. Acta Geochim. 35, 262–270. doi: 10.1007/s11631-016-0106-6
Liu, G. X., Hu, P., Zhang, W., Wu, X., Yang, X., Chen, T., et al. (2012). Variations in soil culturable bacteria communities and biochemical characteristics in the Dongkemadi glacier forefield along a chronosequence. Folia Microbiol. 57, 485–494. doi: 10.1007/s12223-012-0159-9
Malard, L. A., and Pearce, D. A. (2018). Microbial diversity and biogeography in Arctic soils. Environ. Microbiol. Rep. 10, 611–625. doi: 10.1111/1758-2229.12680
Malisorn, K., Embaen, S., Sribun, A., Saeng-in, P., Phongsopitanun, W., and Tanasupawat, S. (2020). Identification and antimicrobial activities of Streptomyces, Micromonospora, and Kitasatospora strains from rhizosphere soils. J. Appl. Pharm. Sci. 10, 123–128. doi: 10.7324/JAPS.2020.102018
Maurer, J. M., Schaefer, J. M., Rupper, S., and Corley, A. (2019). Acceleration of ice loss across the Himalayas over the past 40 years. Sci. Adv. 5:aav7266. doi: 10.1126/sciadv.aav7266
Méheust, R., Castelle, C. J., Carnevali, P. B. M., Farag, I. F., He, C., Chen, L.-X., et al. (2019). Aquatic Elusimicrobia are metabolically diverse compared to gut microbiome Elusimicrobia and some have novel nitrogenase-like gene clusters. bioRxiv [Preprint] doi: 10.1101/765248
Mitchell, A. C., Lafrenière, M. J., Skidmore, M. L., and Boyd, E. S. (2013). Influence of bedrock mineral composition on microbial diversity in a subglacial environment. Geology 41, 855–858. doi: 10.1130/G34194.1
Moon, T., Ahlstrøm, A., Goelzer, H., Lipscomb, W., and Nowicki, S. (2018). Rising oceans guaranteed: Arctic land ice loss and sea level rise. Curr. Clim. Chang. Rep. 4, 211–222. doi: 10.1007/s40641-018-0107-0
Nakata, P. A. (2011). The oxalic acid biosynthetic activity of Burkholderia mallei is encoded by a single locus. Microbiol. Res. 166, 531–538. doi: 10.1016/j.micres.2010.11.002
Nash, M. V., Anesio, A. M., Barker, G., Tranter, M., Varliero, G., Eloe-Fadrosh, E. A., et al. (2018). Metagenomic insights into diazotrophic communities across Arctic glacier forefields. FEMS Microbiol. Ecol. 94:fiy114. doi: 10.1093/femsec/fiy114
Nienow, P. W., Sole, A. J., Slater, D. A., and Cowton, T. R. (2017). Recent advances in our understanding of the role of meltwater in the Greenland ice sheet system. Curr. Clim. Chang. Rep. 3, 330–344. doi: 10.1007/s40641-017-0083-9
Oh, J., Goo, E., Hwang, I., and Rhee, S. (2014). Structural basis for bacterial quorum sensing-mediated oxalogenesis. J. Biol. Chem. 289, 11465–11475. doi: 10.1074/jbc.M113.543462
Olsson-Francis, K., Boardman, C. P., Pearson, V. K., Schofield, P. F., Oliver, A., and Summers, S. (2015). A culture-independent and culture-dependent study of the bacterial community from the bedrock soil interface. Adv. Microbiol. 05, 842–857. doi: 10.4236/aim.2015.513089
Palmieri, F., Estoppey, A., House, G. L., Lohberger, A., Bindschedler, S., Chain, P. S. G., et al. (2019). Oxalic Acid, a Molecule at the Crossroads of Bacterial-Fungal Interactions, 1st Edn. Amsterdam: Elsevier. doi: 10.1016/bs.aambs.2018.10.001
Porder, S. (2019). How plants enhance weathering and how weathering is important to plants. Elements 15, 241–246. doi: 10.2138/gselements.15.4.241
R Core Team 2019 (2019). R: a Language and Environment for Statistical Computing. Vienna: R Found. Statistical Computing.
Ravin, N. V., Rakitin, A. L., Ivanova, A. A., Beletsky, A. V., Kulichevskaya, I. S., Mardanov, A. V., et al. (2018). Genome analysis of Fimbriiglobus ruber SP5T, a planctomycete with confirmed chitinolytic capability. Appl. Environ. Microbiol. 84:e2645-17. doi: 10.1128/AEM.02645-17
Reynolds, H. L., Packer, A., Bever, J. D., and Clay, K. (2003). Grassroots ecology: plant-microbe-soil interactions as drivers of plant community structure and dynamics. Ecology 84, 2281–2291. doi: 10.1890/02-0298
Rime, T., Hartmann, M., and Frey, B. (2016). Potential sources of microbial colonizers in an initial soil ecosystem after retreat of an alpine glacier. ISME J. 10, 1625–1641. doi: 10.1038/ismej.2015.238
Rousk, J., and Bengtson, P. (2014). Microbial regulation of global biogeochemical cycles. Front. Microbiol. 5:103. doi: 10.3389/978-2-88919-297-7
Rubio, L. M., and Ludden, P. W. (2008). Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 62, 93–111. doi: 10.1146/annurev.micro.62.081307.162737
Rudnick, P., Meletzus, D., Green, A., He, L., and Kennedy, C. (1997). Regulation of nitrogen fixation by ammonium in diazotrophic species of proteobacteria. Soil Biol. Biochem. 29, 831–841. doi: 10.1016/S0038-0717(96)00238-6
Samuels, T., Bryce, C., Landenmark, H., Marie-Loudon, C., Nicholson, N., Stevens, A. H., et al. (2020). Microbial Weathering of Minerals and Rocks in Natural Environments. Washington, DC: AGU. doi: 10.1002/9781119413332.ch3
Sayers, E. W., Beck, J., Brister, J. R., Bolton, E. E., Canese, K., Comeau, D. C., et al. (2020). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 48, D9–D16. doi: 10.1093/nar/gkz899
Schmalenberger, A., and Noll, M. (2010). Shifts in desulfonating bacterial communities along a soil chronosequence in the forefield of a receding glacier. FEMS Microbiol. Ecol. 71, 208–217. doi: 10.1111/j.1574-6941.2009.00799.x
Schmidt, S. K., Reed, S. C., Nemergut, D. R., Grandy, A. S., Cleveland, C. C., Weintraub, M. N., et al. (2008). The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc. R. Soc. B Biol. Sci. 275, 2793–2802. doi: 10.1098/rspb.2008.0808
Segawa, T., Yonezawa, T., Edwards, A., Akiyoshi, A., Tanaka, S., Uetake, J., et al. (2017). Biogeography of cryoconite forming cyanobacteria on polar and Asian glaciers. J. Biogeogr. 44, 2849–2861. doi: 10.1111/jbi.13089
Sellstedt, A., and Richau, K. H. (2013). Aspects of nitrogen-fixing actinobacteria, in particular free-living and symbiotic frankia. FEMS Microbiol. Lett. 342, 179–186. doi: 10.1111/1574-6968.12116
Singh, H., Won, K., Ngo, H. T. T., Du, J., Kook, M., and Yi, T. H. (2015). Phycicoccus soli sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 65, 2351–2356. doi: 10.1099/ijs.0.000265
Smith, H. J., Schmit, A., Foster, R., Littman, S., Kuypers, M. M. M., and Foreman, C. M. (2016). Biofilms on glacial surfaces: hotspots for biological activity. npj Biofilms Microbiomes 2:16008. doi: 10.1038/npjbiofilms.2016.8
Smouse, P. E., Long, J. C., and Sokal, R. R. (1986). Society of systematic biologists multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst. Zool. 35, 627–632. doi: 10.2307/2413122
Tamaki, H., Tanaka, Y., Matsuzawa, H., Muramatsu, M., Meng, X. Y., Hanada, S., et al. (2011). Armatimonas rosea gen. nov., sp. nov., of a novel bacterial phylum, Armatimonadetes phyl. nov., formally called the candidate phylum OP10. Int. J. Syst. Evol. Microbiol. 61, 1442–1447. doi: 10.1099/ijs.0.025643-0
Taylor, J. A., Sichel, S. R., and Salama, N. R. (2019). Bent bacteria: a comparison of cell shape mechanisms in Proteobacteria. Annu. Rev. Microbiol. 73, 457–480. doi: 10.1146/annurev-micro-020518-115919
Thangaraj, B., Rajasekar, D. P., Vijayaraghavan, R., Garlapati, D., Devanesan, A. A., Lakshmanan, U., et al. (2017). Cytomorphological and nitrogen metabolic enzyme analysis of psychrophilic and mesophilic Nostoc sp.: a comparative outlook. 3 Biotech 7:107. doi: 10.1007/s13205-017-0724-7
Thawai, C., Kittiwongwattana, C., Thanaboripat, D., Laosinwattana, C., Koohakan, P., and Parinthawong, N. (2016). Micromonospora soli sp. nov., isolated from rice rhizosphere soil. Antonie van Leeuwenhoek. Int. J. Gen. Mol. Microbiol. 109, 449–456. doi: 10.1007/s10482-016-0651-3
Uetake, J., Nagatsuka, N., Onuma, Y., Takeuchi, N., Motoyama, H., and Aoki, T. (2019). Bacterial community changes with granule size in cryoconite and their susceptibility to exogenous nutrients on NW Greenland glaciers. FEMS Microbiol. Ecol. 95, 1–8. doi: 10.1093/femsec/fiz075
Uroz, S., Calvaruso, C., Turpault, M. P., and Frey-Klett, P. (2009). Mineral weathering by bacteria: ecology, actors and mechanisms. Trends Microbiol. 17, 378–387. doi: 10.1016/j.tim.2009.05.004
Uroz, S., Kelly, L. C., Turpault, M. P., Lepleux, C., and Frey-Klett, P. (2015). The mineralosphere concept: mineralogical control of the distribution and function of mineral-associated bacterial communities. Trends Microbiol. 23, 751–762. doi: 10.1016/j.tim.2015.10.004
Wang, Y. L., Wang, Q., Yuan, R., Sheng, X. F., and He, L. Y. (2019). Isolation and characterization of mineral-dissolving bacteria from different levels of altered mica schist surfaces and the adjacent soil. World J. Microbiol. Biotechnol. 35, 1–13. doi: 10.1007/s11274-018-2573-x
Wang, Y., Ma, A., Liu, G., Ma, J., Wei, J., Zhou, H., et al. (2020). Potential feedback mediated by soil microbiome response to warming in a glacier forefield. Glob. Chang. Biol. 26, 697–708. doi: 10.1111/gcb.14936
Warnes, G. R. (2012). gplots: various R programming tools for plotting data. J. Phycol. 33, 569–575.
Welch, S. A., Barker, W. W., and Banfield, J. F. (1999). Microbial extracellular polysaccharides and plagioclase dissolution. Geochim. Cosmochim. Acta 63, 1405–1419. doi: 10.1016/S0016-7037(99)00031-9
Wickham, H. (2007). Reshaping data with the reshape package. J. Stat. Softw. 21, 1–20. doi: 10.18637/jss.v021.i12
Wickham, H. (2016). ggplot2 Elegant Graphics for Data Analysis. Berlin: Springer. doi: 10.1007/978-3-319-24277-4
Wilhelm, L., Singer, G. A., Fasching, C., Battin, T. J., and Besemer, K. (2013). Microbial biodiversity in glacier-fed streams. ISME J. 7, 1651–1660. doi: 10.1038/ismej.2013.44
Wongfun, N., Plötze, M., Furrer, G., and Brandl, H. (2014). Weathering of granite from the Damma glacier area: the contribution of Cyanogenic bacteria. Geomicrobiol. J. 31, 93–100. doi: 10.1080/01490451.2013.802396
Xi, J., Wei, M., and Tang, B. (2018). Differences in weathering pattern, stress resistance and community structure of culturable rock-weathering bacteria between altered rocks and soils. RSC Adv. 8, 14201–14211. doi: 10.1039/C8RA01268G
Xu, D., Xiao, E., Xu, P., Zhou, Y., He, F., Zhou, Q., et al. (2017). Performance and microbial communities of completely autotrophic denitrification in a bioelectrochemically-assisted constructed wetland system for nitrate removal. Bioresour. Technol. 228, 39–46. doi: 10.1016/j.biortech.2016.12.065
Yoon, J. H., Lee, S. Y., Kang, S. J., and Oh, T. K. (2008). Phycicoccus dokdonensis sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 58, 597–600. doi: 10.1099/ijs.0.65284-0
Zeng, Y. X., Yan, M., Yu, Y., Li, H. R., He, J. F., Sun, K., et al. (2013). Diversity of bacteria in surface ice of Austre Lovénbreen glacier, Svalbard. Arch. Microbiol. 195, 313–322. doi: 10.1007/s00203-013-0880-z
Zhang, J. Y., Liu, X. Y., and Liu, S. J. (2011). Phycicoccus cremeus sp. nov., isolated from forest soil, and emended description of the genus Phycicoccus. Int. J. Syst. Evol. Microbiol. 61, 71–75. doi: 10.1099/ijs.0.020842-0
Zhang, S., Hou, S., Qin, X., Du, W., Liang, F., and Li, Z. (2015). Preliminary study on effects of glacial retreat on the dominant glacial snow bacteria in Laohugou glacier no. 12. Geomicrobiol. J. 32, 113–118. doi: 10.1080/01490451.2014.929761
Zhang, X., Ma, X., Wang, N., and Yao, T. (2009). New subgroup of Bacteroidetes and diverse microorganisms in Tibetan plateau glacial ice provide a biological record of environmental conditions. FEMS Microbiol. Ecol. 67, 21–29. doi: 10.1111/j.1574-6941.2008.00604.x
Keywords: forefield, soil, microbial succession, rock weathering, nitrogen fixation, functional profiles, microbial diversity
Citation: Varliero G, Anesio AM and Barker GLA (2021) A Taxon-Wise Insight Into Rock Weathering and Nitrogen Fixation Functional Profiles of Proglacial Systems. Front. Microbiol. 12:627437. doi: 10.3389/fmicb.2021.627437
Received: 09 November 2020; Accepted: 05 August 2021;
Published: 16 September 2021.
Edited by:
Anne D. Jungblut, Natural History Museum, United KingdomReviewed by:
Weidong Kong, Institute of Tibetan Plateau Research (CAS), ChinaCopyright © 2021 Varliero, Anesio and Barker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilda Varliero, Z2lsZGEudmFybGllcm9AYnJpc3RvbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.