 Terje Klemetsen
Terje Klemetsen Christian R. Karlsen
Christian R. Karlsen Nils P. Willassen
Nils P. Willassen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 February 2021
Sec. Aquatic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.626759
Genus Aliivibrio is known to harbor species exhibiting bioluminescence as well as pathogenic behavior affecting the fish farming industry. Current phylogenetic understanding of Aliivibrio has largely remained dormant after reclassification disentangled it from the Vibrio genus in 2007. There is growing evidence of wider diversity, but until now the lack of genomes and selective use of type strains have limited the ability to compare and classify strains firmly. In this study, a total of 143 bacterial strains, including 51 novel sequenced strains, were used to strengthen phylogenetic relationships in Aliivibrio by exploring intra-species and inter-species relations. Multilocus sequence analysis (MLSA), applying the six housekeeping genes 16S ribosomal RNA (rRNA), gapA, gyrB, pyrH, recA, and rpoA, inferred 12 clades and a singular branch in Aliivibrio. Along with four new phylogenetic clades, the MLSA resolved prior inconsistencies circumscribing Aliivibrio wodanis and formed a unique clade we propose as the novel species Aliivibrio sp. “friggae.” Furthermore, phylogenetic assessment of individual marker genes showed gyrB, pyrH, and recA superior to the 16S rRNA gene, resolving accurately for most species clades in Aliivibrio. In this study, we provide a robust phylogenetic groundwork for Aliivibrio as a reference point to classification of species.
The family of Vibrionaceae contains a large number of bacterial species, many of which are described from marine habitats (Thompson et al., 2004). A comprehensive study of the family and its evolutionary history suggested a common ancestor dating back to the Devonian era some 600 million years ago (Sawabe et al., 2007). Vibrionaceae is versatile, delineated and holds 22 distinct phylogenetic clades with highly diverse species of which several are harbored within the genus Aliivibrio (Sawabe et al., 2007, 2013). Aliivibrio is a firmly established genus, separate from Vibrio (Ast et al., 2009; Boyd et al., 2015). The genus harbors bioluminescent bacteria that have symbiotic relationships with aquatic organisms (Bongrand and Ruby, 2019), but also includes pathogens of marine animals (Hjerde et al., 2015; Kashulin et al., 2017).
Aliivibrio fischeri is studied extensively for its bioluminescence and symbiotic capability with marine squids and fishes (Visick, 2009). Exploration of A. fischeri has focused on revealing and understanding mechanisms of host adaptation, biofilm formation, flagellar function, quorum sensing and subsequent pathways to express its observed phenotypes (Visick, 2009; Verma and Miyashiro, 2013). The ability to form bioluminescent symbiosis with marine hosts has additionally been observed in Aliivibrio logei and Aliivibrio sp. “thorii” (Ast et al., 2009), while Aliivibrio sifiae is capable of forming independent bioluminescent colonies on marine agar (Yoshizawa et al., 2010). Aliivibrio logei is associated to skin of farmed fish (Benediktsdóttir et al., 1998), but also found in shellfish (Bang et al., 1978) and in the intestine of fish residing in the seas of Bering and Okhotsk (Bazhenov et al., 2019). Aliivibrio finisterrensis has been isolated from free living clams (Ruditapes philippinarum) and shown to be seasonally present in the hindgut of Tasmanian farmed Atlantic salmon (Beaz-Hidalgo et al., 2010; Hatje et al., 2014). Aliivibrio wodanis is associated to ulcerative skin problems of farmed fish (Karlsen et al., 2014), and Aliivibrio salmonicida is the causative agent of the seasonal cold-water vibrosis (Egidius et al., 1986). Strains of Aliivibrio sp. “thorii,” A. sifiae and the non-luminescent A. finisterrensis have been given less attention. With the description of A. finisterrensis (Beaz-Hidalgo et al., 2010) and A. sifiae (Yoshizawa et al., 2010), the number of Aliivibrio species is currently six (Parte, 2018).
Utilization of the 16S ribosomal RNA (rRNA) gene marker-sequence is the prevalent method of inferring evolutionary relationships between taxa. However, 16S rRNA gene sequences may not provide interspecies resolution, and it is deemed a poor marker for resolving phylogenetically distinct species of Vibrionaceae (Sawabe et al., 2013; Ashok Kumar et al., 2020). Alternative marker genes are used to improve the resolution of species and the accuracy of classification in PCR based methods. Markers with a low degree of sequence conservation are favorable (Lan et al., 2016). The same study additionally found the coaE marker gene to phylogenetically mimic the genome wide amino acid identity in Bacillus. Other studies have applied the fur gene to further increase discriminatory power within Vibrionaceae (Machado and Gram, 2015), while glnAI has been proposed as an improvement for Bifidobacteriaceae compared to the 16S rRNA marker (Killer et al., 2020). Although single markers can provide phylogenetic resolution of species, a combination is often necessary to increase discriminatory power and expose monophyletic groups. This became evident following the use of multilocus sequence analysis (MLSA) reclassifying Aliivibrio (Urbanczyk et al., 2007), and in the identification of Aliivibrio sp. “thorii” (Ast et al., 2009). To further improve the accuracy of species identification, genome sequencing and genome-wide analysis were introduced (Konstantinidis and Tiedje, 2005). Indeed, genomic taxonomy suggests a correction of the whole Vibrionaceae family to change its parent order from the Vibrionales to the Enterobacterales (Parks et al., 2018).
As methods for more accurate classification has advanced, corrections of strains representative for species within Aliivibrio (formerly Photobacterium and later Vibrio) have occurred several times. For example, strain ATCC 15382, formerly classified as Vibrio logei, has later been suggested as a representative of A. wodanis (Ast et al., 2009). The same study additionally linked luminescent strain SR6 and SA12 to A. wodanis. However, bioluminescence in A. wodanis has not been described (Lunder et al., 2000; Hjerde et al., 2015). Furthermore, A. sifiae was published by introducing strain H1-1T and H1-2 (Yoshizawa et al., 2010), while a set of 11 strains by Ast et al. (2009) were informally named as the “sifiae” clade. Without comparison to the described type strains there is no evidence for this classification.
Understanding the ecology and evolution of Aliivibrio requires robust and accurate genus- and species-level taxa. The present taxonomic classification results from representative type strains of selected species, eluding the species concepts of genomically coherent groups of microorganisms (Rosselló-Mora and Amann, 2001). The aim of the study was to firmly establish phylogenetic knowledge about the Aliivibrio genus with new sequenced data and to analyze marker genes for accurate classification of Aliivibrio species.
In this study, marker genes from 143 bacterial strains obtained from in-house sequenced genomes and GenBank (Benson et al., 2017; RRID:SCR_004860; Supplementary Table 1) were used to inferring the evolutionary relationships. Among these, 134 were represented by Aliivibrio strains and for comparison with neighboring genera, four strains of Vibrio and Photobacterium were included. The outgroup was represented by Photorhabdus luminescens subsp. laumondii TT01T in line with prior phylogenetic studies (Urbanczyk et al., 2007; Ast et al., 2009). The type strains of A. finisterrensis DSM 23419T and A. logei ATCC 29985T, and 51 additional Aliivibrio isolates were genome-sequenced for this study. Strains and isolates sequenced in this study are available from the authors upon request.
For sequencing, Aliivibrio isolates, were revived from cryopreserved glycerol stocks and cultured in Luria-Bertani (LB) broth supplied with 3.5% w/v sodium chloride at 12°C. Genomic DNA was extracted using the Qiagen DNeasy blood and tissue kit protocol for Gram-negative bacteria. Sequencing libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina) according to the manufacturer’s protocol. The fragment size distribution was verified to 500–1,000 bp using the Agilent 2100 Bioanalyzer System. Libraries were multiplexed and sequenced on an Illumina MiSeq instrument (RRID:SCR_016379), using either MiSeq Reagent kits v2 (500 cycles) or v3 (600 cycles), yielding an average of 3.8 million reads per bacterial isolate (Supplementary Table 2).
All sequence reads were quality controlled and each genome was de novo assembled using the CLC Genomics Workbench (RRID:SCR_011853) version 8.0.3. Briefly, paired-end reads were imported using the built-inn CLC pipeline removing failed reads. Reads were further quality trimmed with an ambiguous limit of 2 and a quality limit of 0.05 while reads shorter than 15 bases were removed. De novo assembly was performed with default parameters, auto-detecting paired distances and performing scaffolding. A cutoff for minimum contig length was set to 500 bp. On average, the de novo assembly gave 343 scaffolds (N50 of 62,646) with total assembly lengths between 3.7 and 5.2 Mb, and an average coverage of 247.43x (Supplementary Table 2).
The assembled genomes were annotated using Prokka (Seemann, 2014; RRID:SCR_014732) version 1.13 on the Galaxy platform (Afgan et al., 2016; RRID:SCR_006281) with a default parameter setting. Annotated genomes were screened for the 16S rRNA, gapA (P0A9B2), gyrB (P0A2I3), pyrH (P65933), recA (P65977), and rpoA (Q664U6) genes, and identified sequences extracted. Public sequence data under the Aliivibrio taxa (taxonomy ID 511678) which contained all six genes were gathered from GenBank. Locus tag identifiers from the 90 public strains used in this study are listed in Supplementary Table 1.
Sequences in each gene locus were aligned individually using MUSCLE (Edgar, 2004; RRID:SCR_011812) version 3.8.31 with default parameters for nucleotide sequences. Gene regions were selected according to Sawabe et al. (2007). However, flanking ends in each alignment were recursively trimmed. Briefly, flanking positions with gaps occurring in more than 5% of the alignment sequences were trimmed using Aliview (Larsson, 2014; RRID:SCR_002780) version 1.2.6. Vibrio cholerae strain N16961 (ungapped gene numbering) was used as reference to the trimmed alignments with the following gene name, gene position range, and reference locus tag: 16S rRNA, 252-1422, and VCr001; gapA, 89-862, and VC2000; gyrB, 308-1496, and VC0015; pyrH, 21-624, and VC2258, recA, 69-865 VC0543; and rpoA, 20-950, and VC2571. The concatenated sequence of the six trimmed fragments (16S rRNA-gapA-gyrB-pyrH-recA-rpoA) produced a multilocus sequence alignment (MLSA) of 5,473 positions.
Phylogenetic relationships between bacterial strains included in this study were constructed on the basis of the concatenated MLSA. A network graph was created in SplitsTree4 (Huson and Bryant, 2005; RRID:SCR_014734) version 4.13.1 by applying the Jukes-Cantor (JC69) distance correction between sequences while NeighborNet was used as network model.
To analyze the evolutionary variance within species and the average evolutionary distance between species, strains were assigned to designated groups based on the network model (Figure 1). Briefly, the concatenated gene loci dataset was imported as a nucleotide dataset into MEGA X (Kumar et al., 2018; RRID:SCR_000667) version 10.1.7. Fourteen groups were assigned by the Sequence Data Explorer in MEGA using P. luminescens subsp. laumondii TT01T as outgroup and Aliivibrio sp. appey-12 as a singular group. Distance estimation with SE was calculated for within and between groups of species under the conditions: uniform rates among sites and pairwise deletion was used while applying the JC69 as substitution model. All estimations were statistically tested with 1,000 bootstrap replications. MEGA was further used to construct a Neighbor-joining tree using the MLSA, 16S rRNA, gapA, gyrB, pyrH, recA, rpoA, concatenated recA-rpoA, gyrB-rpoH, and gyrB-recA with equivalent parameters as given for the distance measurements. Resulting newick files from each inferred tree was compared topologically against the MLSA using the MutualClusteringInfo algorithm in the TreeDist R package (Smith, 2020). Due to conflicting overlap between sequences in the 16S rRNA alignment, Photobacterium angustum strains ATCC 33977 and S14 were removed from the datasets only prior to tree construction and topological comparison.
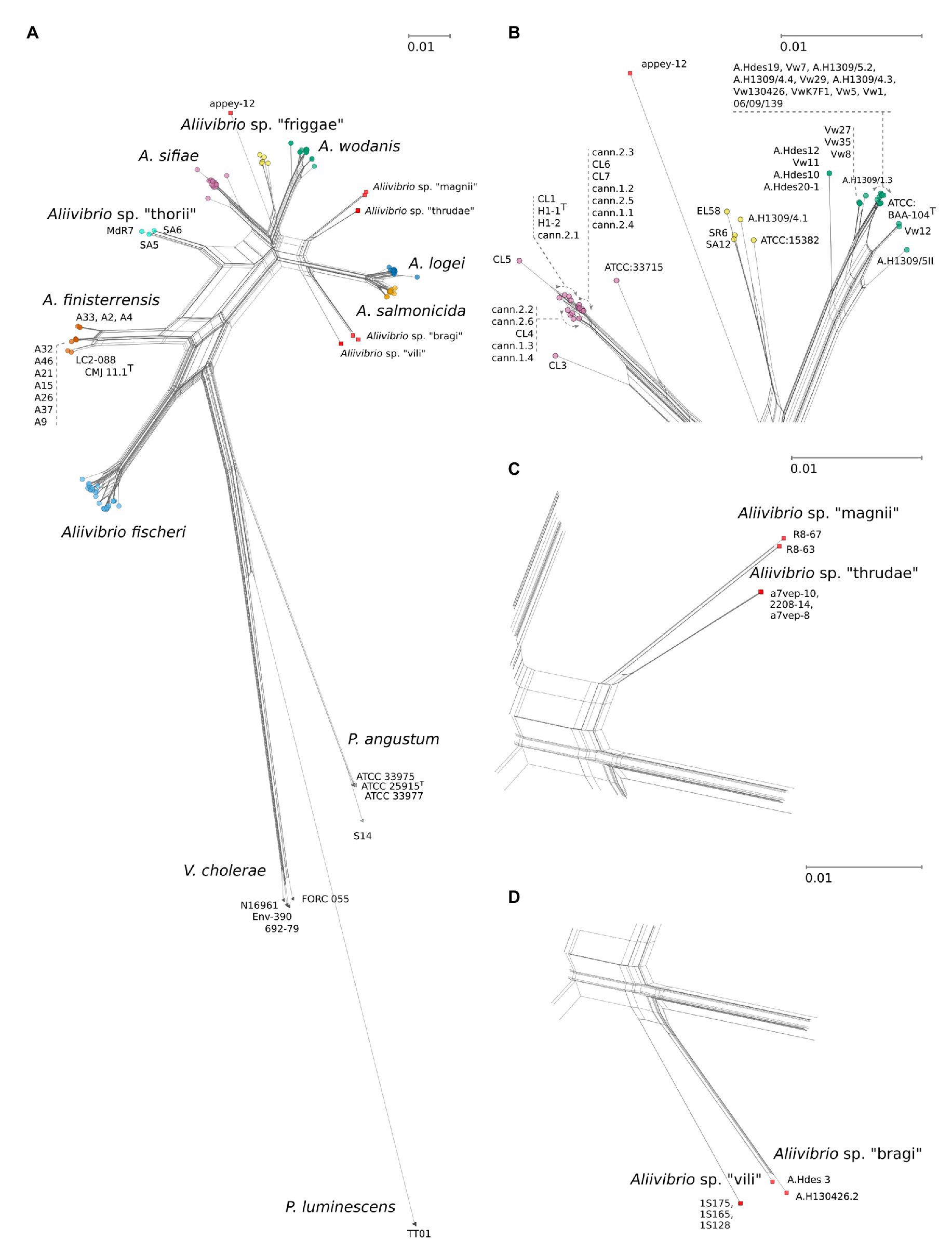
Figure 1. (A) JC69 corrected NeighborNet comprising 143 strains including Aliivibrio, Vibrio cholerae, Photobacterium angustum, and with Pluminescens luminescens acting as outgroup. Network is based on a concatenated alignment of the 16S ribosomal RNA (rRNA) gene, gapA, gyrB, pyrH, recA, and rpoA spanning 5,473 nt positions. Arrows indicate relative placements according to listed strains. Scale bars are relative to the given panels and represent the number of base substitutions per site. (B) Details on Aliivibrio sifiae, Aliivibrio sp. “friggae,” and Aliivibrio wodanis groups. (C) Detailed view of “magnii” and “thrudae” clades. (D) Strain details for the “bragi” and “vili” clades. See Figure 3, for details on Aliivibrio fischeri, Aliivibrio logei, and Aliivibrio salmonicida.
Sequence identities between and within designated groups were estimated as described for evolutionary distances using single genes as well as the MLSA and a 5-gene concatemer excluding the 16S rRNA gene. The python script identity.py1 was created to calculate sequence identities. In short, the script evaluates all sequences pairwise, removing any gapped positions before calculating the identity as a percentage. For each dataset (single genes and MLSA) the identity values were enlisted in either of two subsets; those associated with the same genus (within-genera) or any different genera (between-genera). The same procedure was repeated to differentiate subsets of intra- and inter- species identity values. Values not within and between hitherto described species were filtered. Distributions were plotted as log10 transformed histograms to simplify identification of overlapping data. Subsets became colored based on their affiliation as intra- or inter- subsets. GC content was calculated using Biopython GC (Cock et al., 2009; RRID:SCR_007173).
In this study, a MLSA scheme based on six concatenated genes (16S rRNA gene, gapA, gyrB, pyrH, recA, and rpoA) were used to infer the phylogeny and evolutionary relationships in the Aliivibrio genus. Based on the MLSA sequence data both the inferred phylogenetic network (Figure 1) and phylogenetic tree (Supplementary Figure 1) remained congruent with only minor topology inconsistencies. Twelve individual clades were identified of which seven corresponded to clades described in earlier studies: A. fischeri (Urbanczyk et al., 2007), A. finisterrensis (Beaz-Hidalgo et al., 2010), Aliivibrio sp. “thorii” (Ast et al., 2009), A. sifiae (Yoshizawa et al., 2010), A. wodanis (Lunder et al., 2000), A. logei (Bang et al., 1978), and A. salmonicida (Egidius et al., 1986). The results corroborate the reclassification by Urbanczyk et al. (2007), the wider description of Aliivibrio (Ast et al., 2009) and confirm the presence of Aliivibrio sp. “thorii,” A. finisterrensis and A. sifiae to the genus. Sixteen strains could not be affiliated to any of the described clades. These strains gave rise to one singular branch (appey-12) and five clades in which we suggest the provisionally names “friggae,” “magnii,” “thrudae,” “bragi,” and “vili” (Figure 1) in order to provide working names in line with other species within Aliivibrio genus that have derived their names after Norse mythology gods (A. wodanis, A. logei, A. sifiae, and Aliivibrio sp. “thorii”). Clades inferred by the neighbor-joining approach (Supplementary Figure 1) had, except for Aliivibrio sp. “magni” and Aliivibrio sp. “thrudae,” robust support values. The friggae clade consists of five strains and includes the SR6, SA12 and ATCC 15382, previous classified as wodanis (Ast et al., 2009), EL58, and A.H1309/4.1. These strains were isolated from different hosts such as fishes (Atlantic salmon and Pacific cod), gorgonian coral and bobtail squids. The “thrudae” clade represent three strains isolated from lumpfish while the “magnii” clade represent two isolates from amphipods. Filtered seawater is the source of all three strains composing the “vili” clade. Lastly, strains in the “bragi” clade originate from skin ulcer and head kidney samples isolated from Atlantic salmon.
This study utilizes several strains from each species group in Aliivibrio rather than a single representative type strain. This approach provides a more robust statistical measure of group affiliations (intra-species), their circumference, and observed interrelations between groups (inter-species). The results showed fluctuating evolutionary variances in Aliivibrio, Vibrio, and Photobacterium (Figure 2) that closely reflect the circumference of clades in the phylogenetic tree (Supplementary Figure 1). Aliivibrio species ranged from the narrow intra-species variance of A. salmonicida (0.0012) to the nine times wider variance of A. fischeri (0.0109). Still, these measurements are expected to be inaccurate for clades represented by a low number of strains such as Aliivibrio sp. “vili” and Aliivibrio sp. “thrudae.”
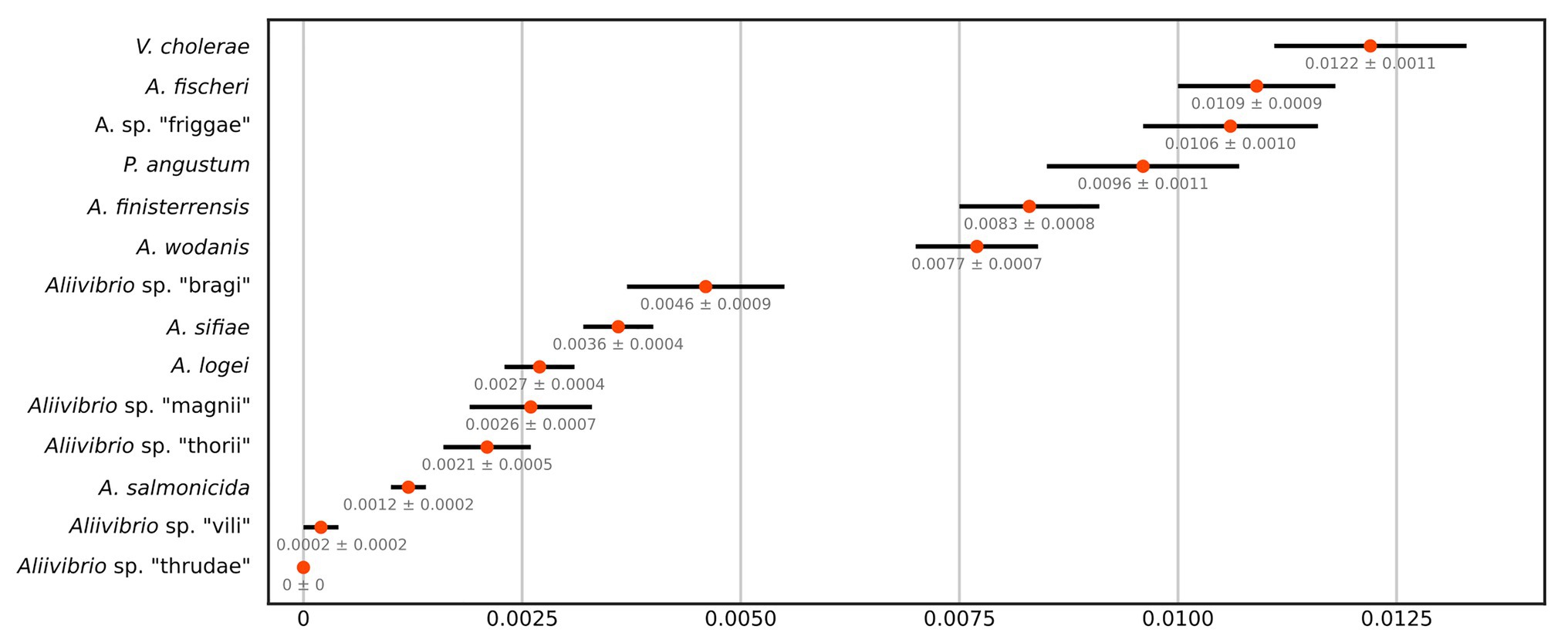
Figure 2. JC69 corrected intra-species distances of the MLSA dataset as number of base substitutions per site. Orange centroids reflect the average over all sequence pairs compared within each group and are drawn with SE values from 1,000 bootstrap replicates.
Analysis of inter-species distances using the JC69 substitution model is shown in Table 1. These values reflect the distance and error value deviation between species groups as they appear in the phylogenetic tree diagram (Supplementary Figure 1). In Aliivibrio, the average evolutionary inter-species distance was 0.060 where the general defined species groups diverged by ≥0.041. The most distant species groups were between A. fischeri and Aliivibrio sp. “bragi” (0.104), while the smallest distance was between A. salmonicida and A. logei (0.013), which make them the closest neighboring species in Aliivibrio (Figure 3A). It is noteworthy that the low sequence variances of A. salmonicida and A. logei contrast the number of different host species. Extended sampling of environments might result in the emergence of one interchangeable species rather than two, as a full genome ANI approach has reported representatives of both A. salmonicida and A. logei as “s__Aliivibrio salmonicida_A” (Parks et al., 2020). This is intriguing as A. salmonicida causes cold-water vibriosis and A. wodanis causes wodanosis and/or winter ulcer in Atlantic salmon, while A. logei is not known to be a salmon pathogen. The phylogenetic structure of A. fischeri reflects differences in the host species, but also colonization behavior (Bongrand et al., 2016). Comparable to the A. fischeri phylogenetic clades, A. salmonicida, A. wodanis, and A. logei similarities may relate to colonization effectiveness while their differences may be related to behavioral specialization and dependent on environmental factors such as temperature (Nishiguchi, 2000; Hatje et al., 2014; Sunagawa et al., 2015). When considering the 5-gene average GC content (Table 2), higher values (42.5–43.1%) were measured compared to the reported genome average of A. salmonicida (39.8%), A. wodanis (39.4%) and A. fischeri (39.0%; Hjerde et al., 2008, 2015; Califano et al., 2015). Indeed, the genome traits of A. salmonicida are suggestive of host specificity adaptation (Hjerde et al., 2008), which do not rule out that other lineages of Aliivibrio may have evolved by similar ecological strategies. Genetic drifts for specialization of Aliivibrios could involve genes with lower GC content than the house keeping genes, such as the MLSA scheme used in this study.
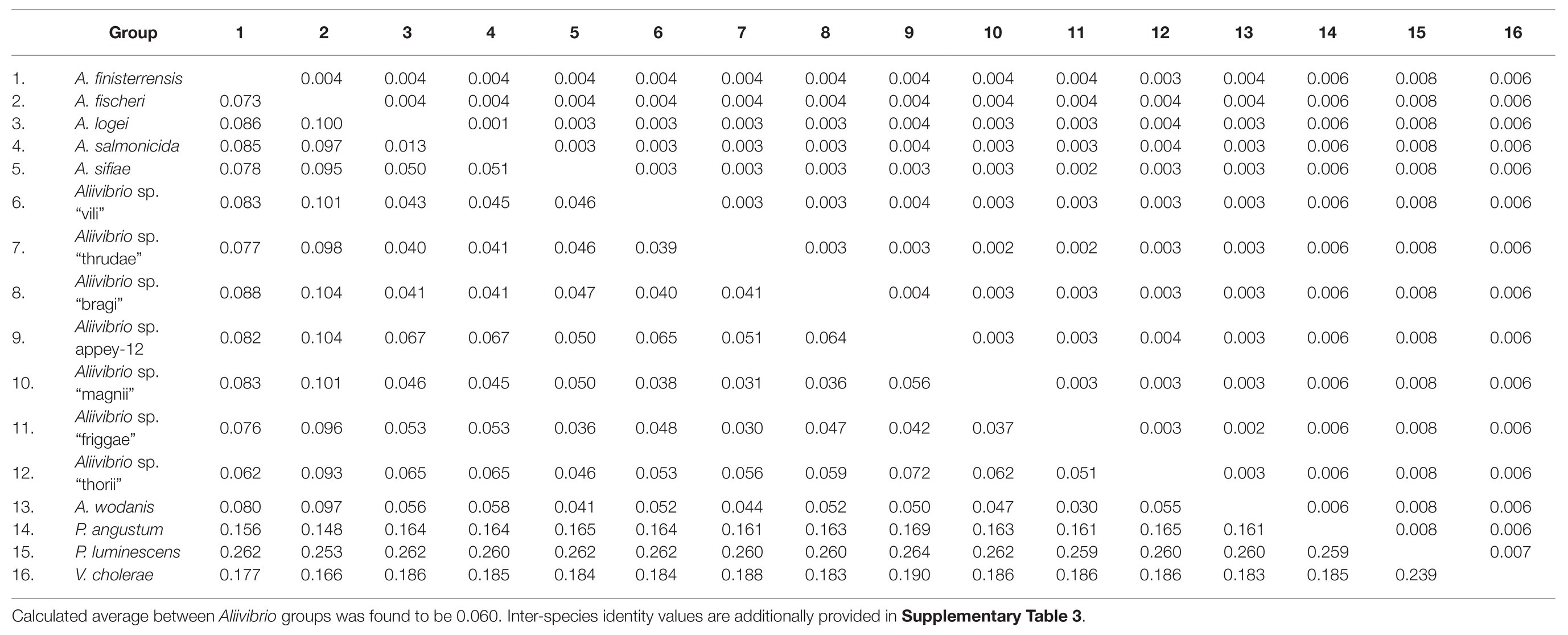
Table 1. JC69 corrected inter-species distances (lower) of the MLSA dataset as number of base substitutions per site with SE values (upper) from 1,000 bootstrap replicates.
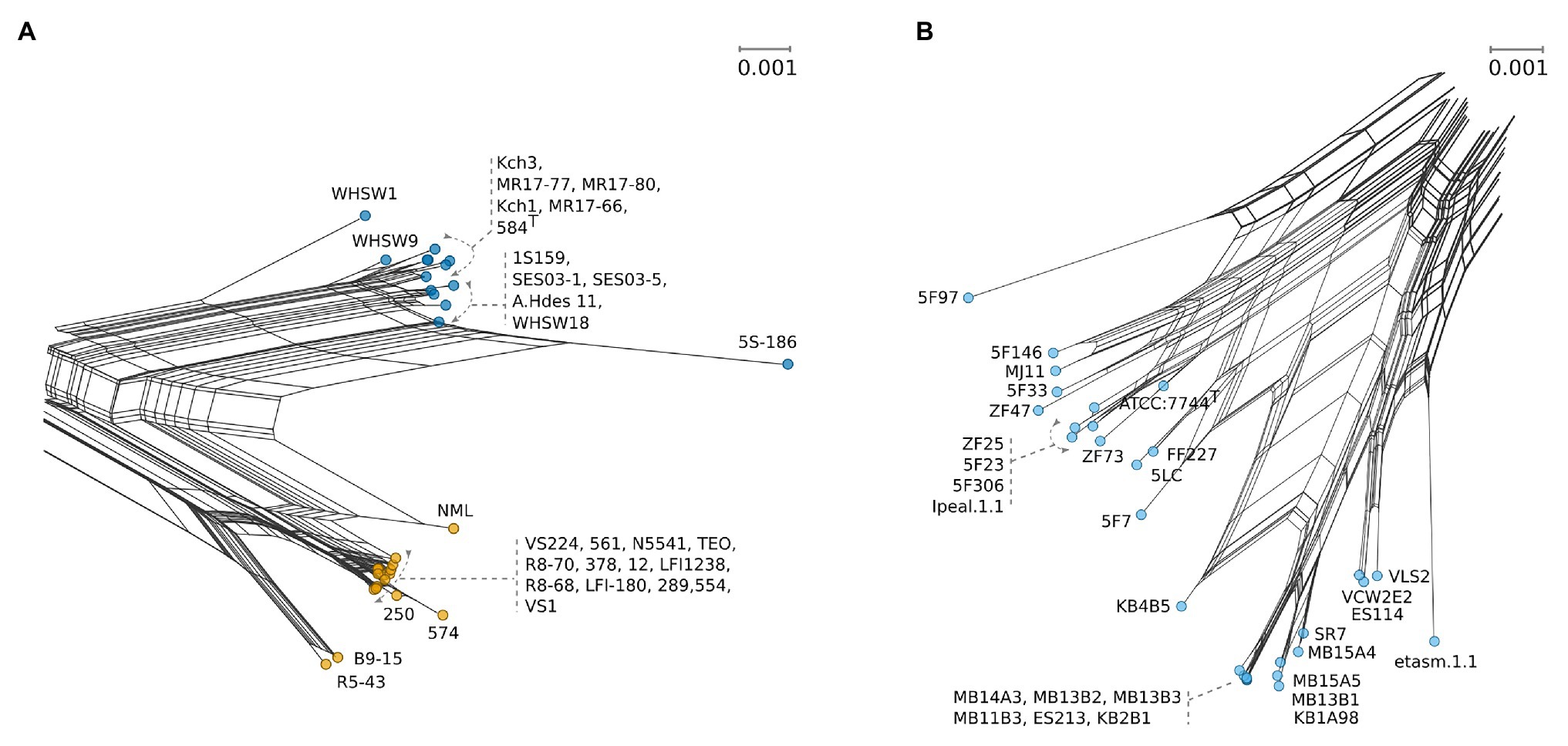
Figure 3. Detailed selections from NeighborNet Figure 1 with uniform scales showing number of base substitutions per site. (A) Strain details for A. logei (blue) and A. salmonicida (yellow). (B) Detailed view for the A. fischeri clade (pale blue).
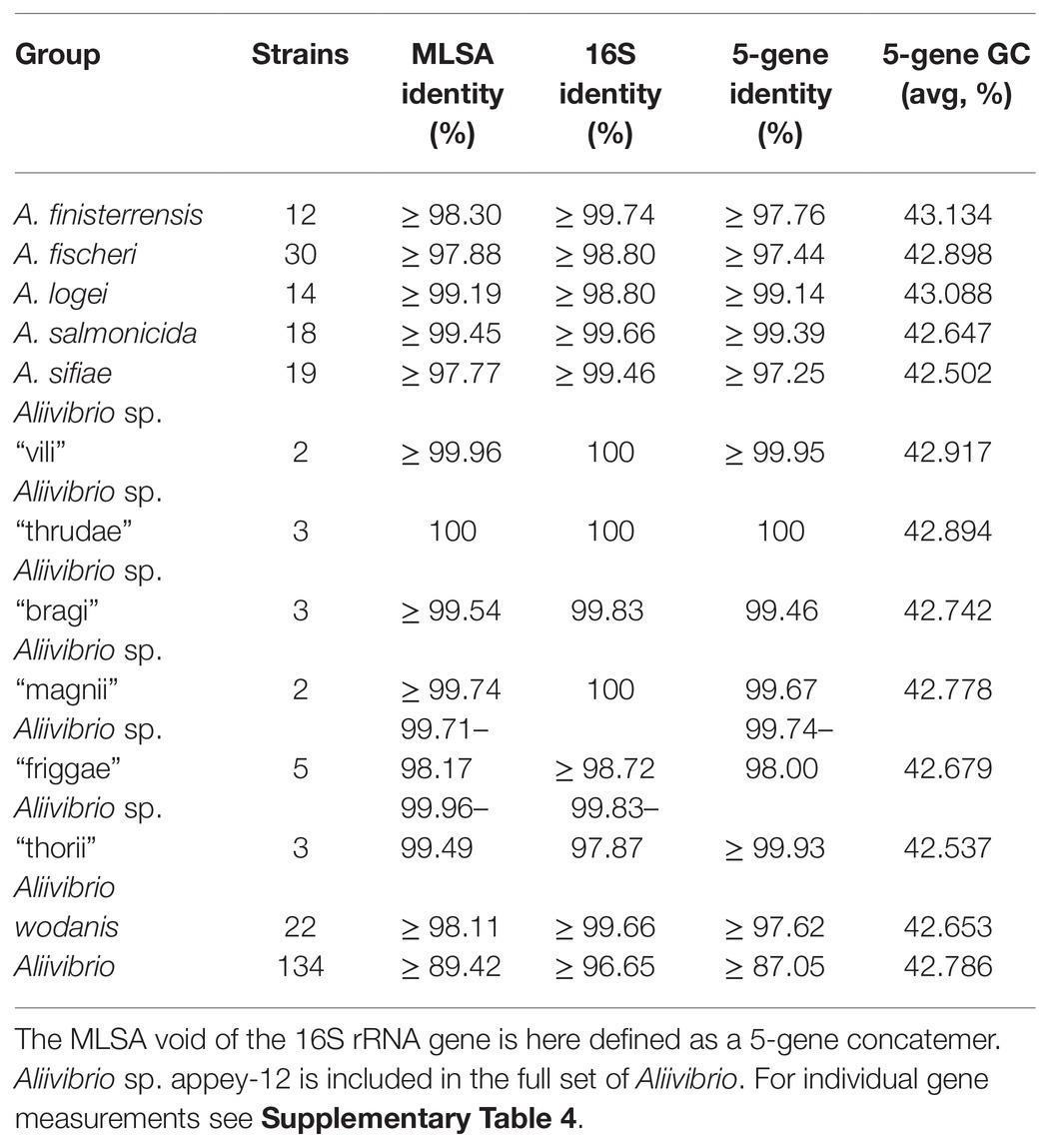
Table 2. Minimum sequence identities for Aliivibrio from trimmed and ungapped sequence pairs within intra-species groups.
Gene sequence identity is frequently used as distance measurements in homolog comparisons and for clustering operational taxonomic units (OTUs; Edgar, 2017). Here, the inferred phylogeny of Aliivibrio was measured interchangeably for species and genera. Estimates based on the 16S rRNA gene sequences show ≥98.80% intra-species identity and ≥96.65% for Aliivibrio (Table 2). Similarly, Urbanczyk et al. (2007) reported ≥97.4 intra-species identity among four representative Aliivibrio species, illustrating the limited resolving power that corroborate previous assessments by Sawabe et al. (2013). Sequence identity showed no overlap between the within-genera and between-genera subset for the genes rpoA, pyrH, and the concatenated MLSA (Figure 4A). Gaps between subsets of rpoA, pyrH, and MLSA were 5.31, 2.45, and 2.72%, respectively. The two extremes of rpoA; P. angustum ATCC 25915T and A. sifiae H1-2 (between genera) shared 90.67% identity, while Aliivibrio sp. ATCC 15382 and A. fischeri 5LC (between species in same genera) shared 96.01% identity. Hence, the rpoA, pyrH, and MLSA datasets have capability to discriminating genera, but dataset overlaps are unreliable at species level differentiation (Figure 4B). Genus-level overlap was observed for the 16S rRNA gene (1171 bp, covering hyper variable regions v3–v8). This causes potentially inaccurate or erroneous classification and OTU clustering of Aliivibrios using near full length 16S rRNA gene sequences. Utilization of the MLSA dataset resulted in lower intra-species identity (≥97.77%) and considerably reduced identity for the whole Aliivibrio genus (≥89.42%), indicating improved resolution. Also, the 5-gene concatemer, without the 16S rRNA gene, resulted in low identity values (Table 2). However, estimates for A. logei and Aliivibrio sp. “thorii” were found contradicting, suggesting the 16S rRNA gene to be favorable for classification of some species.
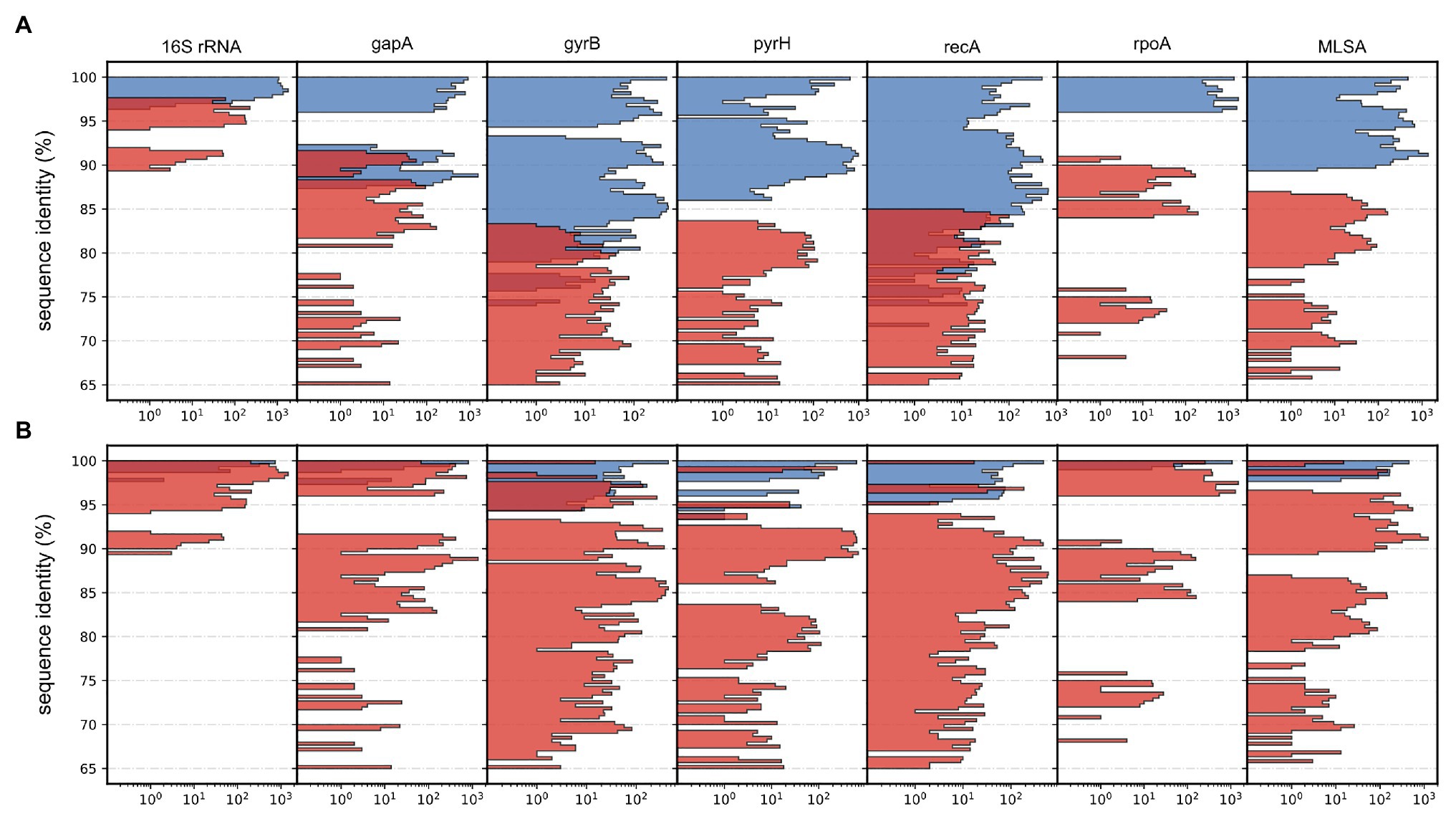
Figure 4. Histograms depicting distributions of pairwise sequence identities for each applied gene and the multilocus sequence analysis (MLSA). Data counts (x-axis) are log10 transformed for improved visualization and presented as colored subsets based on relative affiliation. (A) Between-genera (red) and within-genera (blue). (B) Intra-species (red) and inter-species (blue).
The ability of individual gene markers to classify monophyletic groups with shared topology to the MLSA scheme was assessed. Marker gene recA produced a polyphyletic group of Aliivibrio sp. “thorii” and Aliivibrio sp. “friggae” (indicated in Figure 5A) that shared 67% of the MLSA topology with a misplacement of Aliivibrio sp. appey-12. Similar ability to resolve species was observed by pyrH (sharing 56.66% of the MLSA topology) in which individual strains of Aliivibrio sp. “friggae,” A. sifiae and A. salmonicida mixed with neighboring clades (Figure 5B) – similar to previously reported pyrH discrepancies in the Vibrio group (Pascual et al., 2010). Marker gyrB shared 73.51% of the MLSA topology. Manual assessment of the tree revealed A. wodanis and Aliivibrio sp. “friggae” as polyphyletic (Figure 5C) while Aliivibrio sp. appey-12 interfere with the A. sifiae clade. Still, gyrB, pyrH, and recA markers show significant improvement for Aliivibrio classification compared to the 16S rRNA, gapA, and rpoA which had 54.66, 53.73, and 53.73% topological similarity to the MLSA, respectively. Visual inspection of the resulting trees from 16S rRNA and gapA (Supplementary Figures 2, 3) show concerns in resolving A. sifiae, A. logei and smaller clades like Aliivibrio sp. “thorii.” Furthermore, the relative wide identity gap between genera described earlier becomes apparent for the rpoA marker tree (Supplementary Figure 4). It firmly discriminates Aliivibrio from Vibrio and Photobacterium, but show similar conserved nature as 16S rRNA for highly similar strains like A. salmonicida and A. logei.
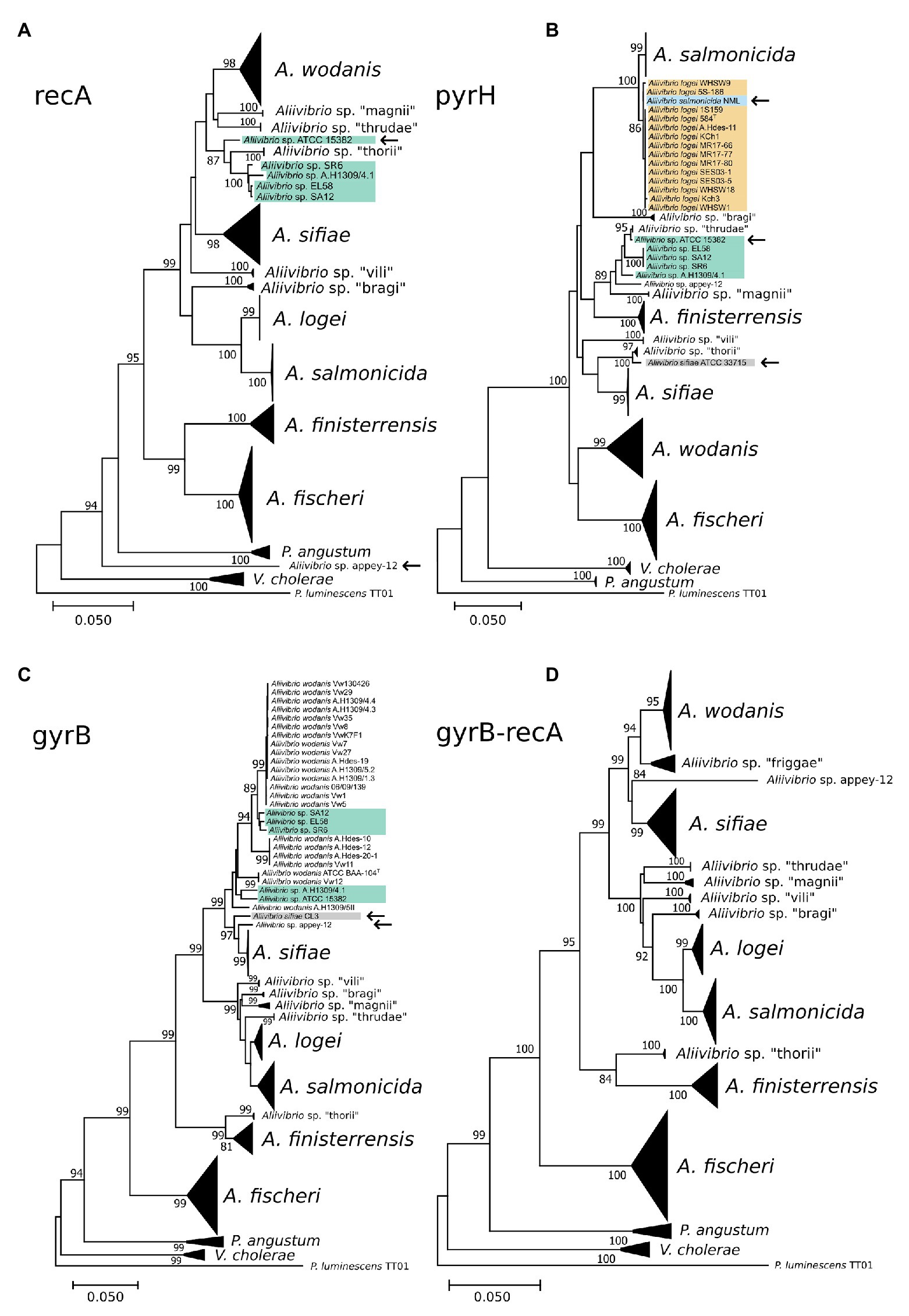
Figure 5. Phylogenetic reconstruction of the marker genes recA (A), pyrH (B), gyrB (C), and gyrB-recA (D). Collapsed clades represent conserved classification similarly as in the full MLSA tree. Strains interfering in neighboring or other clades are marked by an arrow and strains of Aliivibrio sp. “friggae” has teal background color.
Although, none of the discussed markers showed discriminatory power equal to that of the MLSA, paired combinations of concatenated gyrB, pyrH, recA, and rpoA marker sets were tested. Comparable classification to the MLSA was found in recA-rpoA (data not shown), gyrB-pyrH (data not shown), and gyrB-recA (Figure 5D). Based on mutual clustering information trees from these markers shared 68.17, 77.45, and 80.46% of the MLSA topology, respectively. High topological similarity by gyrB produced a discriminating power of gyrB-recA that best resembled the MLSA phylogeny for Aliivibrio.
In this study, using 143 strains we have applied MLSA to gain new insight into the evolutionary structure and relationships in the Aliivibrio genus. Five new clades (friggae, vili, magnii, thrudae, and bragi) and one singular branch was identified in addition to the seven earlier described clades. These presented clades can be illustrated as a snapshot of current knowledge using available data. Future sampling will likely be expanding the complexity and number of clades in genus Aliivibrio.
In this study the discrepancy in intra-species variance for some clades, highly identical sequences may be attributed to a bias toward singular sampling origins or repetitive sampling of target species. The different Aliivibrio clades, independent of host range, show different inter-species sequence variances but may also be globally distributed with little sequence variations. This underline the need to include a sufficient number of strains that represent the population for each species, and not only type strains in taxonomic studies.
Host-associated microbiomes can influence their host’s welfare and health and there is an ongoing effort to identify individual members and their contributions. To help, insight into the evolutionary structure of a genus would be beneficial. Here, the MSLA scheme generated is successfully used to infer a significant insight into the evolutionary relationships in the Aliivibrio genus. As a major member of the Vibrionaceae family in marine environments (Sunagawa et al., 2015; Machado and Gram, 2017), it includes members pathogenic to aquaculture species. Therefore, awareness is required to reinforce phylogenetic relationship of strains within the genus Aliivibrio. Continuing the MLSA approach would be of great value to further investigate the distribution and prevalence in the marine environment and to improve the accuracy of clinical diagnosis when Aliivibrio is detected from farmed aquatic animals. The identification of Aliivibrio species determined by the gyrB-recA approach could be an alternative and even more cost-effective way for a rapid and informative molecular method down to the taxonomic level of species within the Aliivibrio.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJEB34882.
TK conducted and performed the bioinformatic analyses. NW coordinated the work. TK and CK drafted the manuscript. TK, CK, and NW authors provided critical feedback and helped shape the research, analysis, and manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by UiT The Arctic University of Norway and through the Research Council of Norway (RCN) by the Nofima basic grant (194050/F40). The publication charges for this article have been funded by a grant from the publication fund of UiT The Arctic University of Norway.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Concetta De Santi, Seila Pandur and Juan Fu at the Center for Bioinformatics, UiT The Arctic University of Norway who revived the cultures, extracted DNA and performed the sequencing. Additional thanks to Erik Hjerde for giving helpful feedback on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.626759/full#supplementary-material
Afgan, E., Baker, D., van den Beek, M., Blankenberg, D., Bouvier, D., Čech, M., et al. (2016). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10. doi: 10.1093/nar/gkw343
Ashok Kumar, J., Vinaya Kumar, K., Avunje, S., Akhil, V., Ashok, S., Kumar, S., et al. (2020). Phylogenetic relationship among Brackishwater vibrio species. Evol. Bioinforma. 16:117693432090328. doi: 10.1177/1176934320903288
Ast, J. C., Urbanczyk, H., and Dunlap, P. V. (2009). Multi-gene analysis reveals previously unrecognized phylogenetic diversity in Aliivibrio. Syst. Appl. Microbiol. 32, 379–386. doi: 10.1016/j.syapm.2009.04.005
Bang, S. S., Baumann, P., and Nealson, K. H. (1978). Phenotypic characterization of Photobacterium logei (sp. nov.), a species related to P. fischeri. Curr. Microbiol. 1, 285–288. doi: 10.1007/BF02601683
Bazhenov, S. V., Khrulnova, S. A., Konopleva, M. N., and Manukhov, I. V. (2019). Seasonal changes in luminescent intestinal microflora of the fish inhabiting the Bering and Okhotsk seas. FEMS Microbiol. Lett. 366:fnz040. doi: 10.1093/femsle/fnz040
Beaz-Hidalgo, R., Doce, A., Balboa, S., Barja, J. L., and Romalde, J. L. (2010). Aliivibrio finisterrensis sp. nov., isolated from Manila clam, Ruditapes philippinarum and emended description of the genus Aliivibrio. Int. J. Syst. Evol. Microbiol. 60, 223–228. doi: 10.1099/ijs.0.010710-0
Benediktsdóttir, E., Helgason, S., and Sigurjónsdóttir, H. (1998). Vibrio spp. isolated from salmonids with shallow skin lesions and reared at low temperature. J. Fish Dis. 21, 19–28. doi: 10.1046/j.1365-2761.1998.00065.x
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2017). GenBank. Nucleic Acids Res. 45, D37–D42. doi: 10.1093/nar/gkw1070
Bongrand, C., Koch, E. J., Moriano-Gutierrez, S., Cordero, O. X., McFall-Ngai, M., Polz, M. F., et al. (2016). A genomic comparison of 13 symbiotic Vibrio fischeri isolates from the perspective of their host source and colonization behavior. ISME J. 10, 2907–2917. doi: 10.1038/ismej.2016.69
Bongrand, C., and Ruby, E. G. (2019). The impact of Vibrio fischeri strain variation on host colonization. Curr. Opin. Microbiol. 50, 15–19. doi: 10.1016/j.mib.2019.09.002
Boyd, E. F., Carpenter, M. R., Chowdhury, N., Cohen, A. L., Haines-Menges, B. L., and Kalburge, S. S., et al. (2015). Post-genomic analysis of members of the family Vibrionaceae. Microbiol. Spectr. 3:10. doi: 10.1128/microbiolspec.VE-0009-2014
Califano, G., Franco, T., Gonçalves, A. C. S., Castanho, S., Soares, F., Ribeiro, L., et al. (2015). Draft genome sequence of Aliivibrio fischeri strain 5LC, a bacterium retrieved from gilthead sea bream (Sparus aurata) larvae reared in aquaculture. Genome Announc. 3, e00593–e00615. doi: 10.1128/genomeA.00593-15
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. (2017). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. bioRxiv [Preprint]. doi: 10.1101/192211
Egidius, E., Wiik, R., Andersen, K., Hoff, K. A., and Hjeltnes, B. (1986). Vibrio salmonicida sp. nov., a new fish pathogen. Int. J. Syst. Bacteriol. 36, 518–520. doi: 10.1099/00207713-36-4-518
Hatje, E., Neuman, C., Stevenson, H., Bowman, J. P., and Katouli, M. (2014). Population dynamics of Vibrio and Pseudomonas species isolated from farmed Tasmanian Atlantic Salmon (Salmo salar L.): a seasonal study. Microb. Ecol. 68, 679–687. doi: 10.1007/s00248-014-0462-x
Hjerde, E., Karlsen, C., Sørum, H., Parkhill, J., Willassen, N. P., and Thomson, N. R. (2015). Co-cultivation and transcriptome sequencing of two co-existing fish pathogens Moritella viscosa and Aliivibrio wodanis. BMC Genomics 16:447. doi: 10.1186/s12864-015-1669-z
Hjerde, E., Lorentzen, M., Holden, M. T., Seeger, K., Paulsen, S., Bason, N., et al. (2008). The genome sequence of the fish pathogen Aliivibrio salmonicida strain LFI1238 shows extensive evidence of gene decay. BMC Genomics 9:616. doi: 10.1186/1471-2164-9-616
Huson, D. H., and Bryant, D. (2005). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Karlsen, C., Vanberg, C., Mikkelsen, H., and Sørum, H. (2014). Co-infection of Atlantic salmon (Salmo salar), by Moritella viscosa and Aliivibrio wodanis, development of disease and host colonization. Vet. Microbiol. 171, 112–121. doi: 10.1016/j.vetmic.2014.03.011
Kashulin, A., Seredkina, N., and Sørum, H. (2017). Cold-water vibriosis. The current status of knowledge. J. Fish Dis. 40, 119–126. doi: 10.1111/jfd.12465
Killer, J., Mekadim, C., Bunešová, V., Mrázek, J., Hroncová, Z., and Vlková, E. (2020). Glutamine synthetase type I (glnAI) represents a rewarding molecular marker in the classification of bifidobacteria and related genera. Folia Microbiol. 65, 143–151. doi: 10.1007/s12223-019-00716-0
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lan, Y., Rosen, G., and Hershberg, R. (2016). Marker genes that are less conserved in their sequences are useful for predicting genome-wide similarity levels between closely related prokaryotic strains. Microbiome 4:18. doi: 10.1186/s40168-016-0162-5
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. doi: 10.1093/bioinformatics/btu531
Lunder, T., Sorum, H., Holstad, G., Steigerwalt, A. G., Mowinckel, P., and Brenner, D. J. (2000). Phenotypic and genotypic characterization of Vibrio viscosus sp. nov. and Vibrio wodanis sp. nov. isolated from Atlantic salmon (Salmo salar) with “winter ulcer”. Int. J. Syst. Evol. Microbiol. 50, 427–450. doi: 10.1099/00207713-50-2-427
Machado, H., and Gram, L. (2015). The fur gene as a new phylogenetic marker for Vibrionaceae species identification. Appl. Environ. Microbiol. 81, 2745–2752. doi: 10.1128/AEM.00058-15
Machado, H., and Gram, L. (2017). Comparative genomics reveals high genomic diversity in the genus Photobacterium. Front. Microbiol. 8:1204. doi: 10.3389/fmicb.2017.01204
Nishiguchi, M. K. (2000). Temperature affects species distribution in symbiotic populations of Vibrio spp. Appl. Environ. Microbiol. 66, 3550–3555. doi: 10.1128/AEM.66.8.3550-3555.2000
Parks, D. H., Chuvochina, M., Chaumeil, P. A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. -A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parte, A. C. (2018). LPSN-list of prokaryotic names with standing in nomenclature (Bacterio.net), 20 years on. Int. J. Syst. Evol. Microbiol. 68, 1825–1829. doi: 10.1099/ijsem.0.002786
Pascual, J., Macián, M. C., Arahal, D. R., Garay, E., and Pujalte, M. J. (2010). Multilocus sequence analysis of the central clade of the genus vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int. J. Syst. Evol. Microbiol. 60, 154–165. doi: 10.1099/ijs.0.010702-0
Rosselló-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Sawabe, T., Kita-Tsukamoto, K., and Thompson, F. L. (2007). Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
Sawabe, T., Ogura, Y., Matsumura, Y., Feng, G., Amin, A. R., Mino, S., et al. (2013). Updating the vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 4:414. doi: 10.3389/fmicb.2013.00414
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smith, M. R. (2020). Information theoretic generalized Robinson–Foulds metrics for comparing phylogenetic trees. Bioinformatics 36, 5007–5013. doi: 10.1093/bioinformatics/btaa614
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359
Thompson, F. L., Iida, T., and Swings, J. (2004). Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 68, 403–431. doi: 10.1128/mmbr.68.3.403-431.2004
Urbanczyk, H., Ast, J. C., Higgins, M. J., Carson, J., and Dunlap, P. V. (2007). Reclassification of Vibrio fischeri, Vibrio logei, Vibrio salmonicida and Vibrio wodanis as Aliivibrio fischeri gen. nov., comb. nov., Aliivibrio logei comb. nov., Aliivibrio salmonicida comb. nov. and Aliivibrio wodanis comb. nov. Int. J. Syst. Evol. Microbiol. 57, 2823–2829. doi: 10.1099/ijs.0.65081-0
Verma, S. C., and Miyashiro, T. (2013). Quorum sensing in the squid-vibrio symbiosis. Int. J. Mol. Sci. 14, 16386–16401. doi: 10.3390/ijms140816386
Visick, K. L. (2009). An intricate network of regulators controls biofilm formation and colonization by Vibrio fischeri: MicroReview. Mol. Microbiol. 74, 782–789. doi: 10.1111/j.1365-2958.2009.06899.x
Keywords: Vibrionaceae, Aliivibrio, phylogeny, multilocus sequence analysis, marine bacteria, species group coherence, marker gene
Citation: Klemetsen T, Karlsen CR and Willassen NP (2021) Phylogenetic Revision of the Genus Aliivibrio: Intra- and Inter-Species Variance Among Clusters Suggest a Wider Diversity of Species. Front. Microbiol. 12:626759. doi: 10.3389/fmicb.2021.626759
Edited by:
Marcelino T. Suzuki, Sorbonne Universités, FranceReviewed by:
Maxime Bruto, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2021 Klemetsen, Karlsen and Willassen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terje Klemetsen, dGVyamUua2xlbWV0c2VuQHVpdC5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.