 Shasha Wang1,2,3,4†
Shasha Wang1,2,3,4† Lijing Jiang2,3,4†
Lijing Jiang2,3,4† Qitao Hu2,3,4
Qitao Hu2,3,4 Liang Cui1
Liang Cui1 Bitong Zhu1
Bitong Zhu1 Xiaoteng Fu2,3,4
Xiaoteng Fu2,3,4 Qiliang Lai2,3,4
Qiliang Lai2,3,4 Zongze Shao2,3,4,5*
Zongze Shao2,3,4,5* Suping Yang1*
Suping Yang1*- 1Department of Bioengineering and Biotechnology, Huaqiao University, Xiamen, China
- 2Key Laboratory of Marine Genetic Resources, Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
- 3State Key Laboratory Breeding Base of Marine Genetic Resources, Xiamen, China
- 4Fujian Key Laboratory of Marine Genetic Resources, Xiamen, China
- 5Southern Marine Science and Engineering Guangdong Laboratory, Zhuhai, China
Bacteria of the genus Sulfurimonas within the class Campylobacteria are predominant in global deep-sea hydrothermal environments and widespread in global oceans. However, only few bacteria of this group have been isolated, and their adaptations for these extreme environments remain poorly understood. Here, we report a novel mesophilic, hydrogen- and sulfur-oxidizing bacterium, strain NW10T, isolated from a deep-sea sulfide chimney of Northwest Indian Ocean.16S rRNA gene sequence analysis showed that strain NW10T was most closely related to the vent species Sulfurimonas paralvinellae GO25T with 95.8% similarity, but ANI and DDH values between two strains were only 19.20 and 24.70%, respectively, indicating that strain NW10 represents a novel species. Phenotypic characterization showed strain NW10T is an obligate chemolithoautotroph utilizing thiosulfate, sulfide, elemental sulfur, or molecular hydrogen as energy sources, and molecular oxygen, nitrate, or elemental sulfur as electron acceptors. Moreover, hydrogen supported a better growth than reduced sulfur compounds. During thiosulfate oxidation, the strain can produce extracellular sulfur of elemental α-S8 with an unknown mechanism. Polyphasic taxonomy results support that strain NW10T represents a novel species of the genus Sulfurimonas, and named as Sulfurimonas hydrogeniphila sp. nov. Genome analyses revealed its diverse energy metabolisms driving carbon fixation via rTCA cycling, including pathways of sulfur/hydrogen oxidation, coupled oxygen/sulfur respiration and denitrification. Comparative analysis of the 11 available genomes from Sulfurimonas species revealed that vent bacteria, compared to marine non-vent strains, possess unique genes encoding Type V Sqr, Group II, and Coo hydrogenase, and are selectively enriched in genes related to signal transduction and inorganic ion transporters. These phenotypic and genotypic features of vent Sulfurimonas may explain their thriving in hydrothermal environments and help to understand the ecological role of Sulfurimonas bacteria in hydrothermal ecosystems.
Introduction
Deep-sea hydrothermal vent is one of the most extreme environments on earth and provides unique and diverse habitats for various microorganisms (Zeng et al., 2020). However, the sharp physical and chemical gradients across the vent chimneys and their surroundings impose great challenges to bacterial survival (Sievert et al., 2008a). In the vent ecosystems, biomass production is mainly energized by oxidation of reduced sulfur compounds and hydrogen driving carbon fixation via chemolithoautotrophic microorganisms, which constitute a dominant bacterial group in situ (Nakagawa and Takai, 2008).
Among these chemolithoautotrophs, members of the genus Sulfurimonas (class Campylobacteria) represent one of the most widespread and preponderant mesophilic bacteria in global deep-sea hydrothermal environments. They have been described as strictly chemolithoautotrophic, metabolically versatile sulfur, and/or hydrogen oxidizers and widely distribute in various hydrothermal habitats, including chimneys, sediments, plumes and diffuse-flow vent fluids (Nakagawa et al., 2005; Campbell et al., 2006; Mino et al., 2017; Dick, 2019). For example, the genus Sulfurimonas comprised 70 and 20%, respectively, of the bacterial abundance in plume waters and diffuse fluids of deep-sea vents (Akerman et al., 2013; Perner et al., 2013; Han and Perner, 2015). Obviously, bacteria of this genus play the important role in the biogeochemical cycles of hydrothermal vent systems.
The Sulfurimonas genus currently contains eight species with validly published names. Only two strains, Sulfurimonas autotrophica OK10T and Sulfurimonas paralvinellae GO25T, were isolated from deep-sea hydrothermal environments, respectively, from sediments and polychaete nests in Mid-Okinawa Trough hydrothermal field (Inagaki et al., 2003; Takai et al., 2006). Four species were isolated from coastal marine sediments, including Sulfurimonas denitrificans DSM1251T, Sulfurimonas hongkongensis AST-10, Sulfurimonas xiamenensis 1-1NT and Sulfurimonas lithotrophica GYSZ_1T (Timmer-ten, 1975; Cai et al., 2014; Wang et al., 2020b). Another marine isolate representing Sulfurimonas gotlandica GD1T was from the pelagic redox cline of the Baltic Sea (Labrenz et al., 2013). In addition, Sulfurimonas crateris SN118T was from a terrestrial mud volcano (Ratnikova et al., 2019).
Of the two deep-sea hydrothermal vent strains, only S. autotrophica OK10T has genome sequence publicly available (Sikorski et al., 2010). Genomic analysis indicated that strain OK10T possess all genes essential for carbon fixation via the reductive tricarboxylic acid (rTCA) cycle. Oxidation of reduced sulfur compounds by strain OK10T proceeds via the Sox pathway and sulfide: quinone oxidoreductase (Sqr) (Sikorski et al., 2010). Furthermore, metagenomic and metatranscriptomic analyses revealed the processes and activities involving sulfur/hydrogen oxidation, oxygen respiration and denitrification as well as carbon fixation in genus Sulfurimonas inhabiting in vent fluids of Axial Seamount (Fortunato and Huber, 2016). These metabolic pathways were also observed in other hydrothermal samples such as actively venting chimney of East Pacific Rise and hydrothermal chimneys from the Roman Ruins vent field based on metagenomic and metaproteomic analyses (Pjevac et al., 2018; Hou et al., 2020).
To define the ecological roles of Sulfurimonas in deep-sea hydrothermal environments, culturable bacteria representing the predominant member in situ are required. Six potential novel species of genus Sulfurimonas were recently isolated from different marine environments, including three from deep-sea hydrothermal vents, one from deep-sea sediment and two from coastal marine sediments (Wang et al., 2020a). Two isolates from coastal environments, S. xiamenensis 1-1NT and S. lithotrophica GYSZ_1T, have been just assigned as novel species (Wang et al., 2020b). Bacteria of novel species, represented by strain NW10, were recently reported to predominate the bacterial population in in situ deep-sea hydrothermal vents globally (Wang et al., 2020a). To understand its environment adaptation and ecological role in hydrothermal ecosystems, we firstly characterized it via a polyphasic taxonomic approach as a novel species. Further, comparative genomic analyses were carried out between vent and non-vent marine Sulfurimonas to reveal the unique genotypic features that help to clarify their adaptation to deep-sea hydrothermal environments.
Results
Enrichment of Chemolithoautotrophic Sulfur-Oxidizing Bacteria (CSOB) and Isolation of Sulfurimonas Species
To obtain CSOB from a newly discovered hydrothermal vent on the Carlsberg Ridge, active vent chimney samples were ground on board and inoculated into sealed bottles filled with MMJHS medium with thiosulfate and hydrogen as the energy sources. After about one and half month incubation on board, enriched bacterial cultures were transferred into 10 ml MMJHS medium, and incubated at 28°C in the laboratory. After 2 days of incubation, bacterial growth was obvious with cells in form of short rods. The bacterium was subsequently purified with the dilution-to-extinction method. The culture in the serum bottle showing growth at the highest dilution was designated as strain NW10T. The purity of the culture was further confirmed by microscopic examination and 16S rRNA gene sequencing. Interestingly, the bacterium can produce large amount of elemental sulfur in the form of extracellular granules as determined below.
Morphology of Strain Sulfurimonas sp. NW10T
Morphological observations by phase-contrast light microscopy and transmission electron microscope showed that cells of strain NW10T were Gram-negative, motile and short rod-shaped with a size of 0.4–0.8 μm wide and 0.8–3.5 μm long (Supplementary Figure S1). Spore formation was not found during the culture incubation. Cells in older cultures tended to form aggregates. These morphological features are shared with other vent species S. autotrophica OK10T and S. paralvinellae GO25T (Table 1). In addition, on solid MMJHS medium agar plates, strain NW10T formed small, white, round-shaped colonies with smooth boundaries.
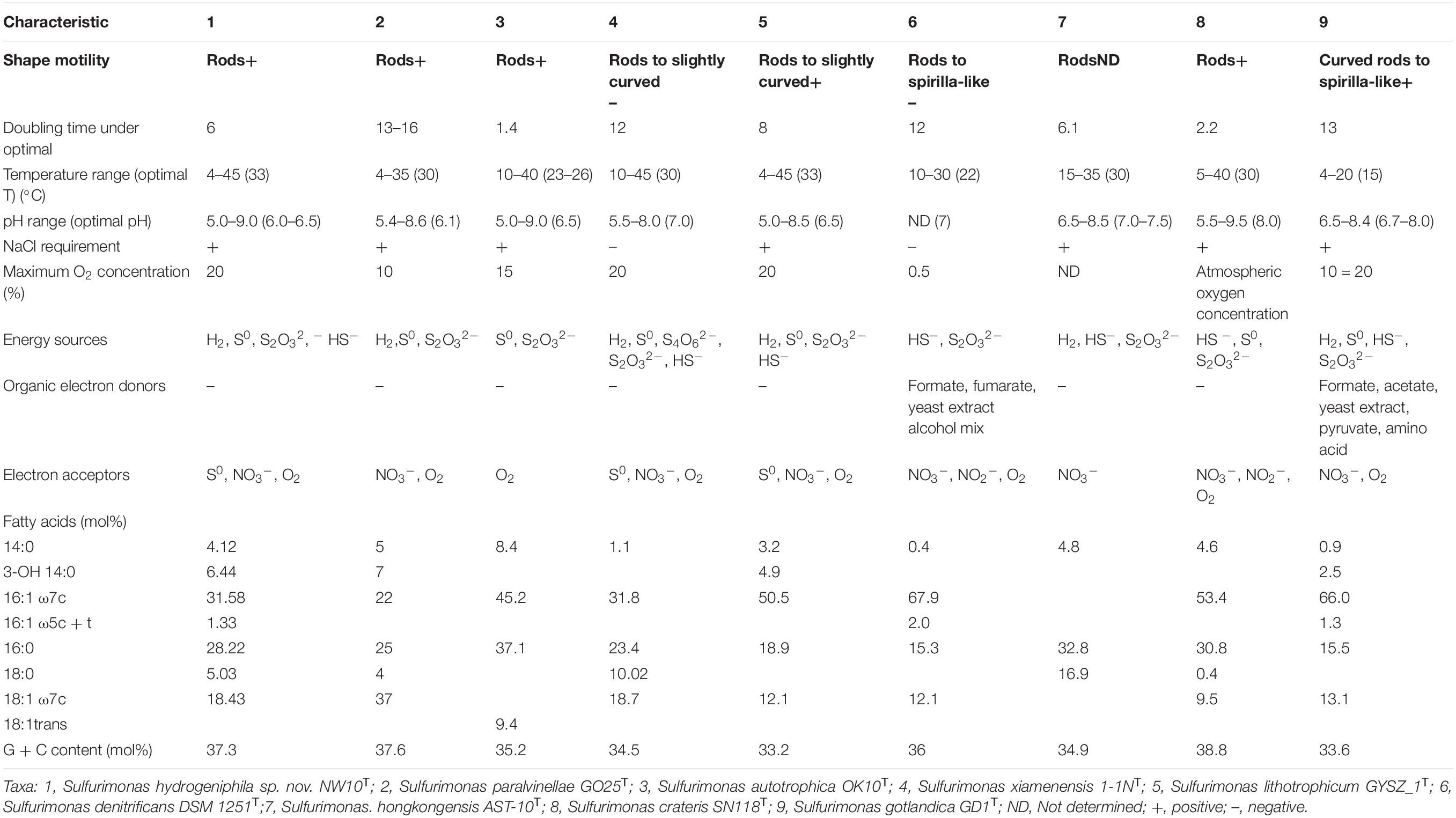
Table 1. Comparison of characteristics of Sulfurimonas hydrogeniphila NW10T sp. nov. with related species of the genus Sulfurimonas.
Phylogenetic Analysis Based on 16S rRNA and Core Gene Sequences
Sequence comparison of the 16S rRNA gene sequences obtained from PCR amplification (1,447 bp) showed that strain NW10T is closest to the only two deep-sea vent bacterial species S. paralvinellae GO25T and S. autotrophica OK10T, with 95.8 and 95.2% sequence identities, respectively. The maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences showed that strain NW10T clusters with S. paralvinellae GO25T within the genus Sulfurimonas (Figure 1). This was further confirmed through the phylogenomic trees constructed with the neighbor-joining and minimum evolution methods (Supplementary Figures S2, S3). The phylogenomic tree based on the up-to-date bacterial core gene sequences showed that strain NW10T forms a branch with S. autotrophica OK10T and S. paralvinellae GO25T (Figure 2), supporting further that strain NW10T should belong to the Sulfurimonas genus and likely represents a novel species.
Figure 1. Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences showing the relationship of Sulfurimonas hydrogeniphila NW10T with other members within the genus Sulfurimonas. Bootstrap values based on 1,000 replicates are shown at branch nodes. Branch node values below 50% are not shown. Bar = 0.02 substitutions per nucleotide position.
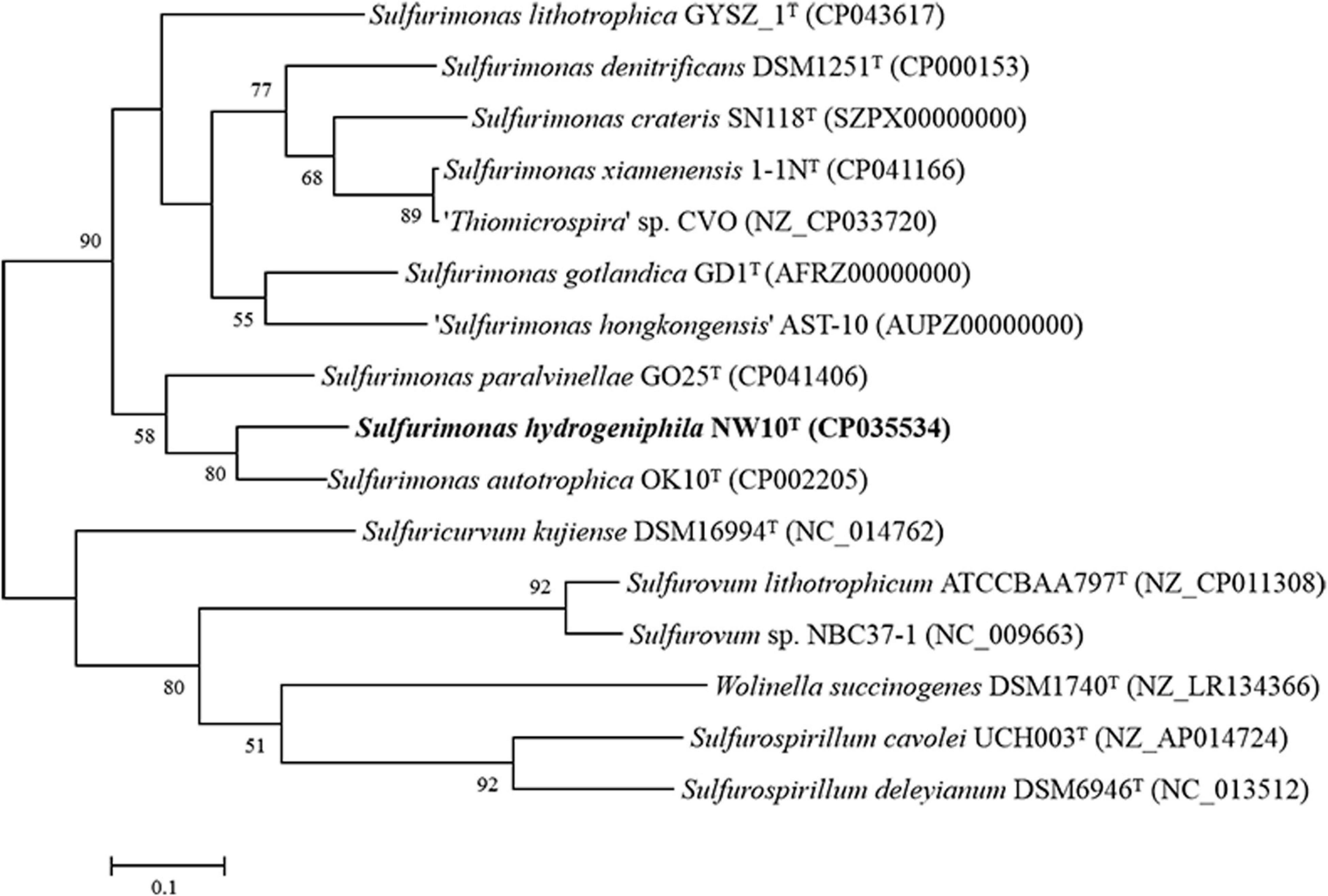
Figure 2. Phylogenetic tree inferred using UBCGs showing the position of Sulfurimonas hydrogeniphila NW10T and closely related taxa within the genus Sulfurimonas using the maximum-likelihood algorithm. The node is labeled with Gene Support Index (GSI) values. Branch node values below 50% are not shown. The accession numbers of the genomes are shown in parentheses. Bar, 0.1 substitutions per position.
Physiological Characteristics of the Putative Novel Species
Growth experiment showed that strain NW10T could grow in the range of temperatures (4–45°C), salinities (2–4% (w/v) NaCl), pH (5.0–9.0) and oxygen concentrations (1–20%). Strain NW10T also could use nitrate as sole electron acceptor in the absence of oxygen. The optimal growth occurred at 33°C, 6% O2, pH 6.0–6.5 and 3% (w/v) NaCl (Table 1). Under optimal growth conditions, the maximum cell concentration was approximately 5.0 × 108 cells/ml and the doubling time was approximately 6 h. Chemoautotrophic growth showed that strain NW10T could grow with thiosulfate, sulfide, elemental sulfur, and hydrogen as energy sources, but not with sulfite and tetrathionate. Strain NW10T grew better with hydrogen as the sole energy source, which brought about the highest cell concentration far more than other energy sources. Thus, hydrogen is possibly the preferred energy source for this bacterium. Similarly, hydrogen also supported the best growth of S. paralvinellae GO25T (Takai et al., 2006). When hydrogen was used as the energy source, strain NW10T could respire element sulfur. The product of sulfur reduction was sulfide, which reached up to 42 μM at the late exponential phase in the medium (Wang et al., 2020a). As a chemolithoautotroph, strain NW10T could not grow using any of the tested organic compounds as the carbon source. In addition, none of these organic compounds could be used as an energy source.
The predominant cellular fatty acids of strain NW10T were C16:1 ω7c (31.6%), C16:0 (28.2%), C18:1 ω7c (18.4%) and 3-OH C14:0 (6.4%), which are similar to those of S. paralvinellae GO25T, S. lithotrophicum GYSZ_1T and S. gotlandica GD1T, and distinctly different from those of other Sulfurimonas species. The significant difference between strain NW10T and its closest relative S. paralvinellae GO25T was that its prominent fatty acid in strain NW10T was C16:1 ω7c, representing 31.6%, whereas in S. paralvinellae GO25T, C18:1 ω7c was prominent, accounting for 37.0%. In addition, C16:1 ω5c + t (1.3%) was observed in strain NW10T, but not in S. paralvinellae GO25T. The fatty acid profile of strain NW10T and the related type strains are detailed in Table 1.
Generation of Extracellular Biogenic Sulfur and Characterization With SEM, EDS, and Raman Spectroscopy
When strain NW10T was incubated with hydrogen and thiosulfate as mixed electron donors and oxygen as the sole electron acceptor, elemental sulfur occurred in the culture at the mid-exponential growth phase, and accumulated in the late exponential phase and during the stationary phase (Figure 3A). When grown in MMJHS medium with neutral pH such as 7.0 (unbuffered) or weakly acidic pH such as 5.5 (in buffered medium), strain NW10T produced large amount of naked-eye elemental sulfur in the culture. However, there was no accumulation of elemental sulfur observed in alkaline conditions, such as at pH 8.0. Elemental sulfur production was also affected by the oxygen concentration in the gaseous phase and the result showed the maximum accumulation of elemental sulfur occurred at 6% oxygen concentration. Morphological observations under SEM showed that extracellular sulfur was mainly shaped as crystalline bars (Figures 3B,C), with length varying from 10 to 100 μm. EDS analysis revealed that these crystals mainly contained elemental sulfur (Figure 3D). Raman spectroscopy further showed the presence of α-S8 when compared against a standard chemical product of S80 (Figure 3E). This confirmed that the extracellular aggregates produced by strain NW10T were biogenic sulfur and composed of crystalline α-S8. These findings suggested that strain NW10T performs incomplete oxidation of thiosulfate with elemental sulfur as an oxidation intermediate accumulated outside of the cells.
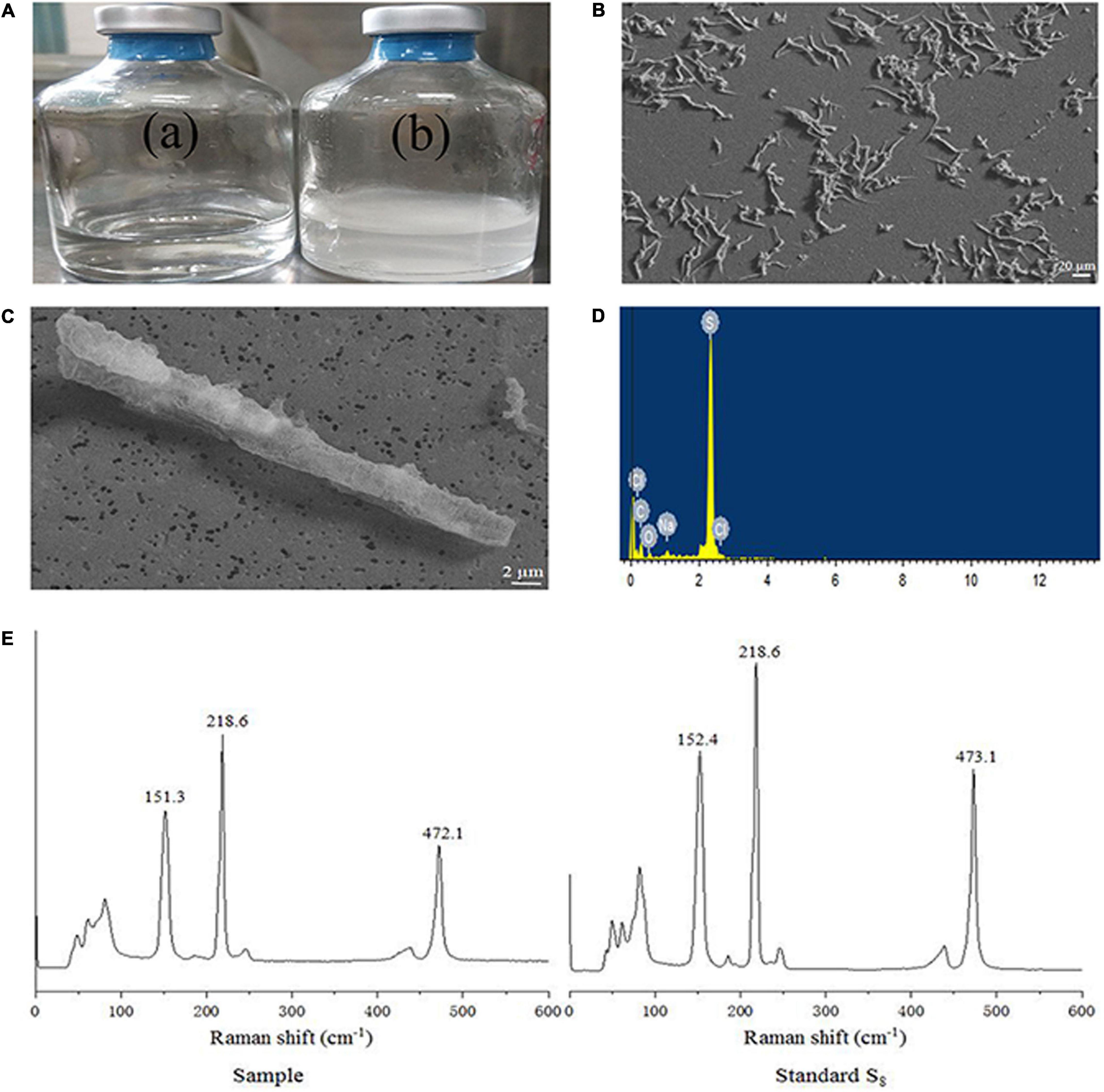
Figure 3. S. hydrogeniphila NW10T produces extracellular sulfur when cultured in MMJHS medium. (A) Strain NW10T cultured in MMJHS medium without (a) or with 10 mM thiosulfate (b). (B,C) SEM observation of S0 particles produced by strain NW10T. (D) Energy dispersive spectrum analysis of extracellular S0. (E) Raman spectra of extracellular S0 produced by strain NW10T and standard S8.
Genomic Properties and Genetic Relatedness
Strain NW10T has a single circular chromosome (Supplementary Figure S4) with complete genome size of 2,432,011 bp with GC content of 37.3%, which is similar to that of S. paralvinellae GO25T (Table 1). No plasmid was detected in the genome of this bacterium. Total 2,472 genes were predicted, which contained 2,367 protein coding genes and 57 RNA genes. The RNA genes cover 45 tRNAs and 12 rRNAs. ANI and DDH were calculated to identify the genomic similarities of strain NW10T with other species of the genus Sulfurimonas. Pairwise ANI values between strain NW10T and its closest relatives, S. paralvinellae GO25T and S. autotrophica OK10T, were 74.50 and 81.15%, respectively. The predicted DDH value between strain NW10T and S. paralvinellae GO25T was 19.20% and the value between strain NW10T and S. autotrophica OK10T was 24.70%. These findings supported the classification of strain NW10T as one distinct species according to the cut-off thresholds of ANI (95–96%) and DDH (70%) for delineation of prokaryotic species (Chun et al., 2018).
Comparative Genomic Analysis of the Sulfurimonas Genus
Pan-genomic studies were carried out to investigate the genotypic features of 11 Sulfurimonas species, including five vent bacteria-S. hydrogeniphila NW10T, Sulfurimonas sp. NW8N, Sulfurimonas sp. S2-6, S. paralvinellae GO25T and S. autotrophica OK10T, and six non-vent marine bacteria-Sulfurimonas sp. B2, S. xiamenensis 1-1NT, S. lithotrophica GYSZ_1T, S. denitrificans DSM1251T, S. hongkongensis AST-10 and S. gotlandica GD1T (Table 2). Comparative analyses based on orthologous groups of proteins revealed 886 core genes shared by all the eleven Sulfurimonas genomes (Figure 4A). The percentages of core genes in each genome ranged from 32.7 to 47.6%, indicating that the 11 strains of Sulfurimonas shared a low percentage of common functional proteins. The percentages of unique genes in each genome varied from 10.8 to 23.0%, however, a relative high proportion of distributed genes in each genome was detected, ranging from 36.2 to 51.0% (Figure 4B). The distributed genes usually offer species diversity, environmental adaptation and other characteristics for bacteria (Innamorati et al., 2020). In addition, considering that the definition of unique genes is subject to change, a singleton today may be reclassified into a multi-gene cluster when new genome data is included in the future (Zhang and Sievert, 2014), we decided to focus on the distributed genes to investigate common adaptation characteristics of the Sulfurimonas genus to deep-sea hydrothermal vent environments.
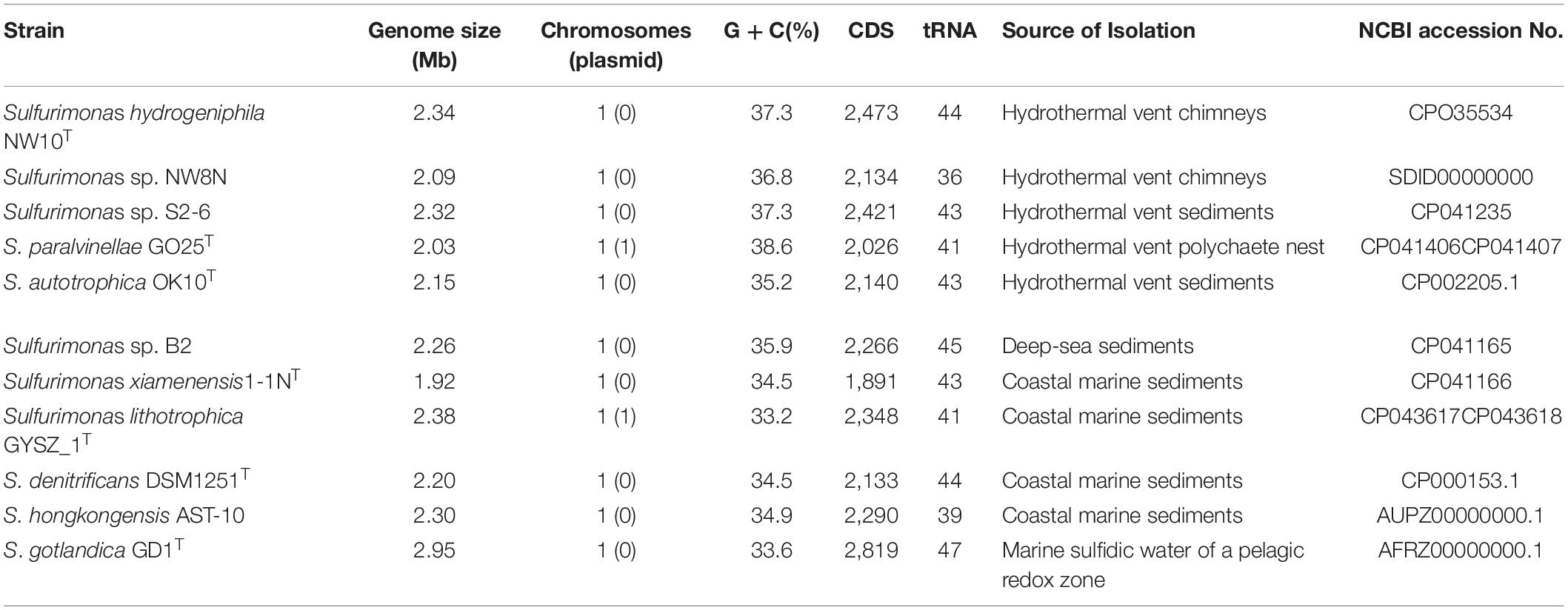
Table 2. General genome features of Sulfurimonas species used in this study.
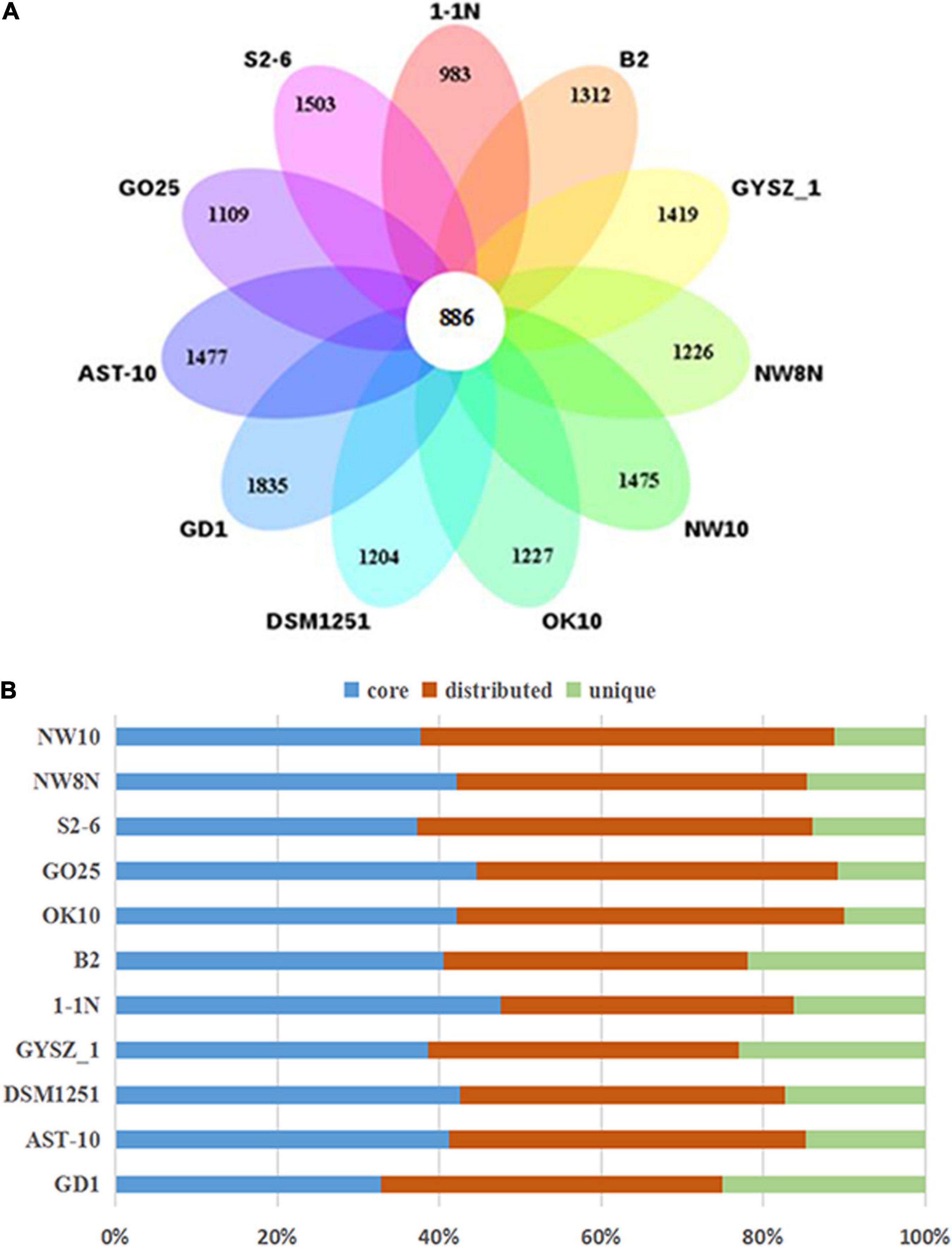
Figure 4. Comparisons of Sulfurimonas orthologous protein groups in 11 Sulfurimonas genomes. (A) Venn diagram displaying the numbers of core gene families and flexible genes for each of the 11 Sulfurimonas strains. (B) Percentage of core, distributed, and unique genes in each of the 11 genomes.
Central Metabolisms Within Genus Sulfurimonas
Sulfur Metabolism
Genomic analysis revealed that strain NW10T contains two Sox gene clusters (soxABXY1Z1 and soxCDY2Z2) forming a complete Sox multi-enzyme system (Table 3 and Supplementary Table S1). Furthermore, comparative analysis showed that soxCDY2Z2 is present in all Sulfurimonas species, indicating that it was conserved among Sulfurimonas spp. The gene cluster SoxABXY1Z1 was absent in the non-vent strains S. xiamenensis 1-1NT and S. lithotrophica GYSZ-1T. Despite this absence, growth experiments showed that both strains still oxidize thiosulfate to sulfur. Strain NW10T contained three homologs of sqr gene, usually responsible for oxidizing sulfide to elemental sulfur. Comparative analysis showed that Sulfurimonas species harbor diverse types of Sqr, including Type II, III, IV, V, and VI (Table 3 and Figure 5). As shown in Table 3, except for Type VI Sqr that was absent in S. denitrificans DSM1251T, both Type IV and VI Sqrs were conserved among all 11 sequenced Sulfurimonas species. Phylogenetic analysis showed that Type IV Sqr of hydrothermal vent Sulfurimonas clustered together and those from non-vent Sulfurimonas species formed another cluster, indicating a differential evolution of Type IV Sqr was in accordance to the environmental origins of the hosts (Figure 5). All non-vent strains harbored Type II Sqr, and among them strains B2, S. hongkongensis AST-10 and S. gotlandica GD1T had two copies. In contrast, strains NW8N and S. autotrophica OK10T from hydrothermal vents only had one copy of type II Sqr. Interestingly, Type III Sqr only occurred in non-vent strains, S. denitrificans DSM1251T, S. hongkongensis AST-10 and S. gotlandica GD1T. However, Type V Sqr only presented in the three vent strains NW10T, S. autotrophica OK10T and S. paralvinellae GO25T. The Type III Sqrs were phylogenetically close to the cluster of Type II (Figure 5), indicating that they may have similar functions, as previously reported by Han and Perner (2015). Phylogenetic analysis also showed that Type V Sqr from strains NW10T, S. autotrophica OK10T and S. paralvinellae GO25T clustered with those of thermophilic bacteria and archaea as well as green sulfur bacteria (Figure 5).
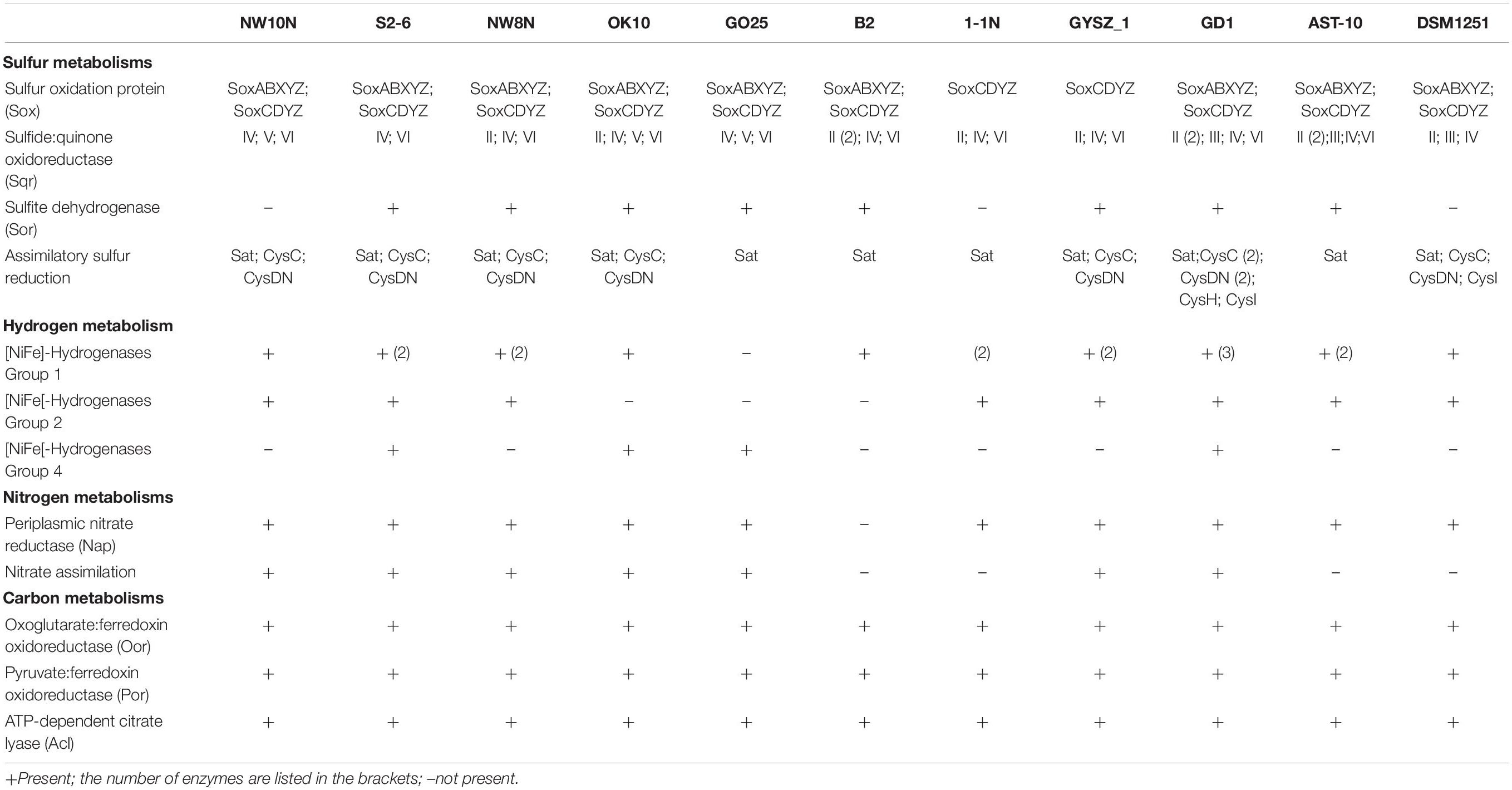
Table 3. Comparison of key enzymes for sulfur, nitrogen, hydrogen, and carbon metabolisms in Sulfurimonas species based on RAST annotations in this study.
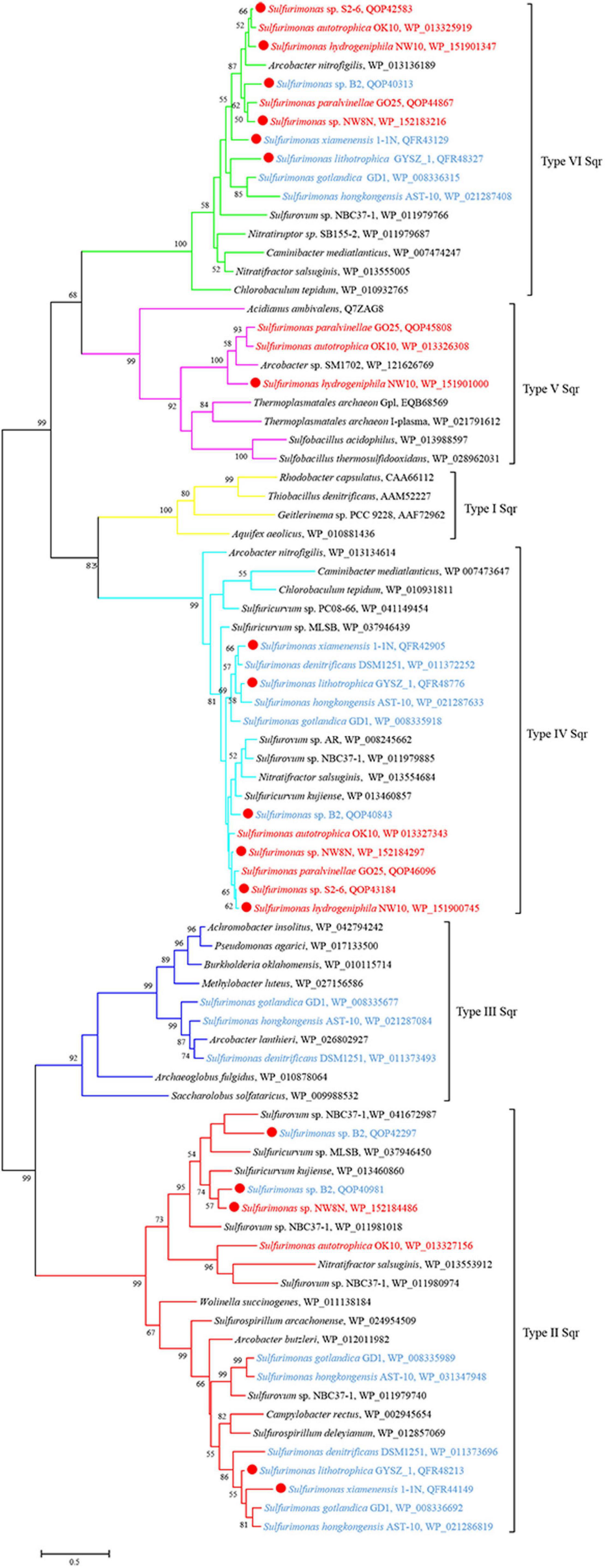
Figure 5. Maximum likelihood phylogenetic tree of Sqr protein sequences derived from Sulfurimonas strains and other representative species within the class Campylobacteria. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branch node values below 50% are not shown. The solid red circles represent our own isolated strains. Branches representing different type of Sqr are different color. Isolation sources of Sulfurimonas species are indicated in different font colors: red, hydrothermal environments; blue, marine non-vent system.
Besides Sox multienzyme complex and Sqr, sorAB genes responsible for sulfite oxidation were detected in most Sulfurimonas genomes, but absent in strains NW10T, 1-1NT and S. denitrificans DSM1251T (Table 3). In addition, strain NW10T had an incomplete assimilation sulfate reduction pathway, only containing genes encoding sulfate adenylyltransferase (Sat), adenylylsulfate kinase (CysC) and sulfate adenylyltransferase (CysDN), which were also found in all other Sulfurimonas bacteria except strain S. gotlandica GD1T (Table 3).
Hydrogen Metabolism
Genomic analysis revealed that Sulfurimonas spp. contain three types of hydrogenases: [NiFe]-Hydrogenases Group 1 (Hyd and Hyp), Group 2b (Hup) and Group 4 (Hyc, Coo, and Ech) (Table 3). Except for S. paralvinellae GO25T, all Sulfurimonas species contained the Group 1 hydrogenases (Table 3), suggesting that this group might be essential for growth. Strains S2-6, NW8N, 1-1NT, GYSZ_1T, and S. hongkongensis AST-10 had two Group I hydrogenases, and strain S. gotlandica GD1T had three. Phylogenetic analysis showed that Group I hydrogenases of Sulfurimonas grouped into different clusters with diverse Campylobacteria (Figure 6). Group II hydrogenases existed in most of Sulfurimonas species (Table 3) and surprisingly, three vent strains, i.e., NW10T, NW8N, and S2-6, also harbored this type of hydrogenase. It is the first observation that Group II hydrogenases are present in vent Sulfurimonas species. Type IV hydrogenases were only found in four Sulfurimonas species, strains S2-6, S. autotrophica OK10T, S. paralvinellae GO25T, and S. gotlandica GD1T (Table 3). Phylogenetic analysis showed that group IV hydrogenase can be further classified into three types: Hyc, Coo and Ech. Hyc proteins from S. autotrophica OK10T and S. paralvinellae GO25T were clustered with those from other vent Campylobacteria (Figure 6). Ech was only found in strain S. gotlandica GD1T, and Coo was only found in vent strain S2–6.
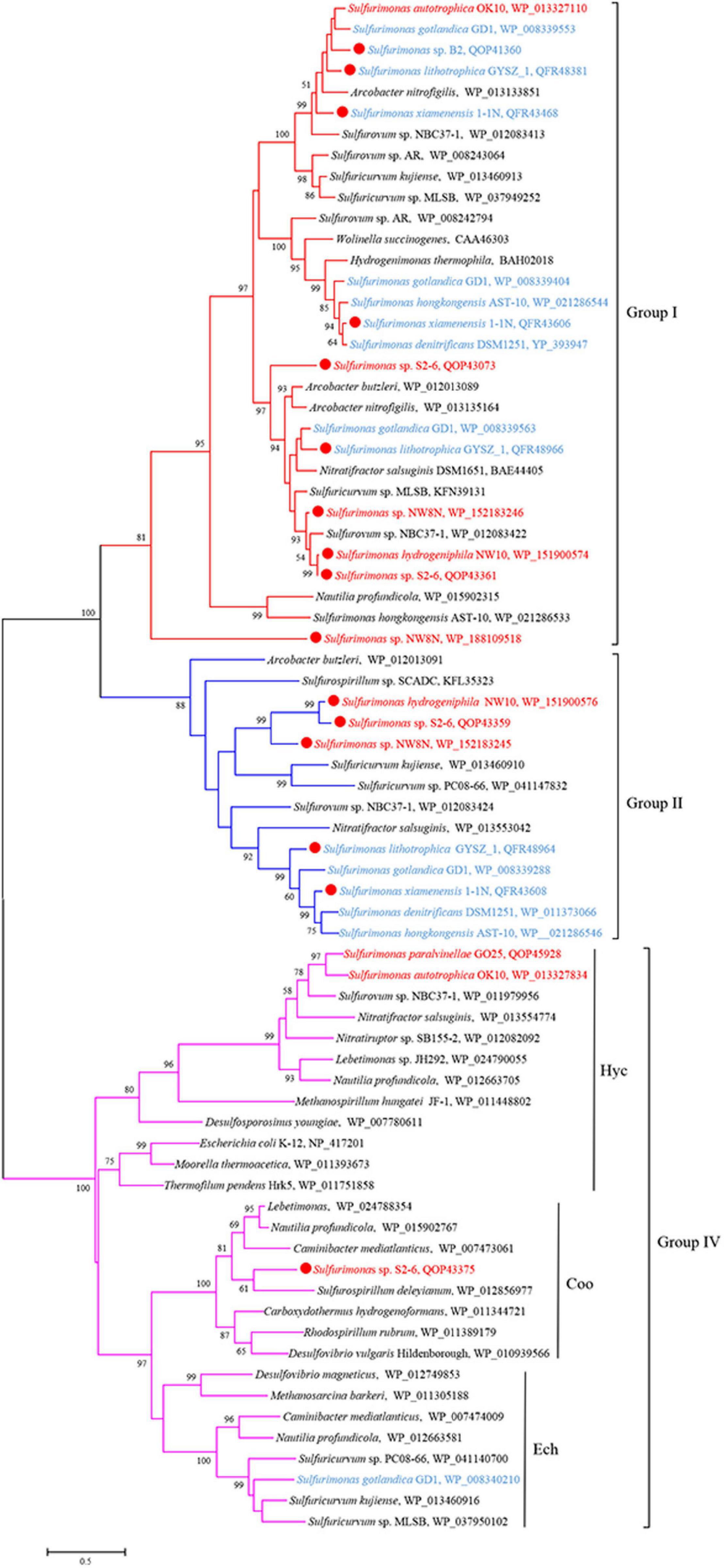
Figure 6. Maximum likelihood phylogenetic tree of hydrogenase large subunit sequences derived from Sulfurimonas strains and other representative species within the class Campylobacteria. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branch node values below 50% are not shown. The solid red circles represent our own isolated strains. Branches representing different hydrogenase group are different color. Isolation sources of Sulfurimonas species are indicated in different font colors: red, hydrothermal environments; blue, marine non-vent system.
Nitrogen Metabolism
Except for strain B2, genes encoding all components required for the complete reduction of nitrate to nitrogen gas, i.e., nitrate reductases (nap), nitrite reductases (nir), nitric oxide reductases (nor) and nitrous oxide reductases (nos), were found in all Sulfurimonas species (Table 3). However, even though the genomes of strains NW8N and S. autotrophica OK10T contained the napAGHBFLD operon, both strains were incapable of growing with nitrate as the sole electron acceptor under the tested conditions. We speculated that strains NW8N and S. autotrophica OK10T may use nitrate as the electron acceptor under certain unidentified environmental conditions. Phylogenetic analysis showed that NapAs from vent Sulfurimonas species clustered together, while those from non-vent but marine habitats clustered apart and were close to those from Arcobacter species (Figure 7). In addition, strain NW10T had the complete assimilatory nitrate reduction pathway, containing the genes encoded nitrate transporter (nasAB) and ferredoxin-nitrite reductase (nirA). This gene cluster was also found in all other hydrothermal vent isolates, but absent in most non-vent strains (4/6) (Table 3).
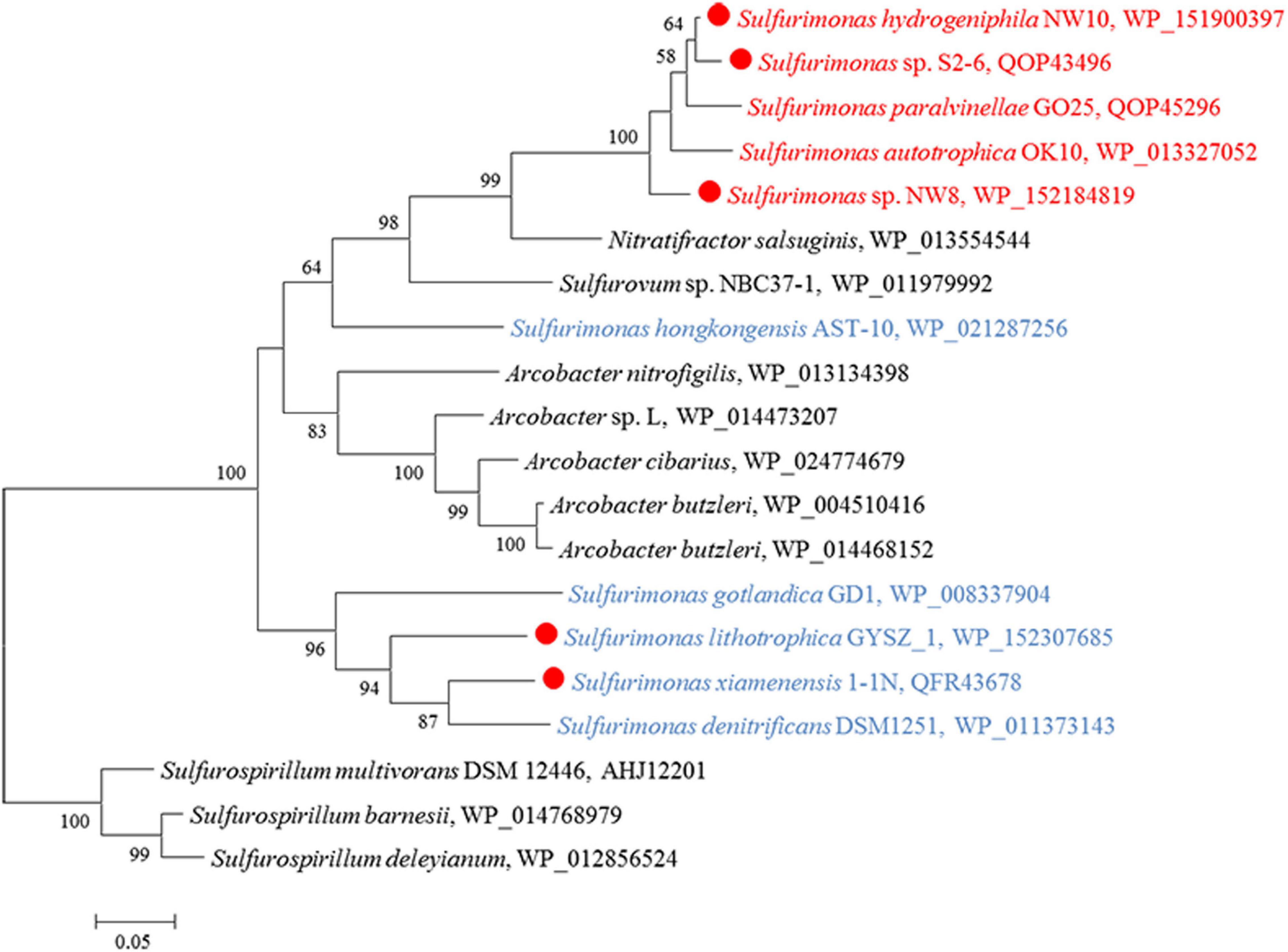
Figure 7. Maximum likelihood phylogenetic tree of the NapA protein sequences derived from Sulfurimonas strains and other representative species within the class Campylobacteria. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branch node values below 50% are not shown. The solid red circles represent our own isolated strains. Isolation sources of Sulfurimonas species are indicated in different font colors: red, hydrothermal environments; blue, marine non-vent system.
Central Carbon Metabolism
CO2 fixation coupled to oxidation of reduced sulfur compounds and hydrogen is a typical characteristic in Campylobacterial species for biomass production (Waite et al., 2017). Strain NW10T was capable of chemoautotrophic growth with CO2/HCO3–. All of the enzymes essential for rTCA cycle were encoded in strain NW10T, including ATP-dependent citrate lyase (Acl), 2-oxoglutarate: ferredoxin oxidoreductase (Oor) and pyruvate:ferredoxin oxidoreductase (Por) (Table 3). These key enzymes for CO2 fixation were also found in all other Sulfurimonas species (Table 3). Phylogenetic analysis showed that the AclBs from Sulfurimonas species formed a separate clade, with the exception of strain GYSZ_1T (Figure 8), suggesting this pathway is conserved in Sulfurimonas species. In addition, AclBs from vent isolates clustered together, while non-vent-AclBs formed a distinct cluster.
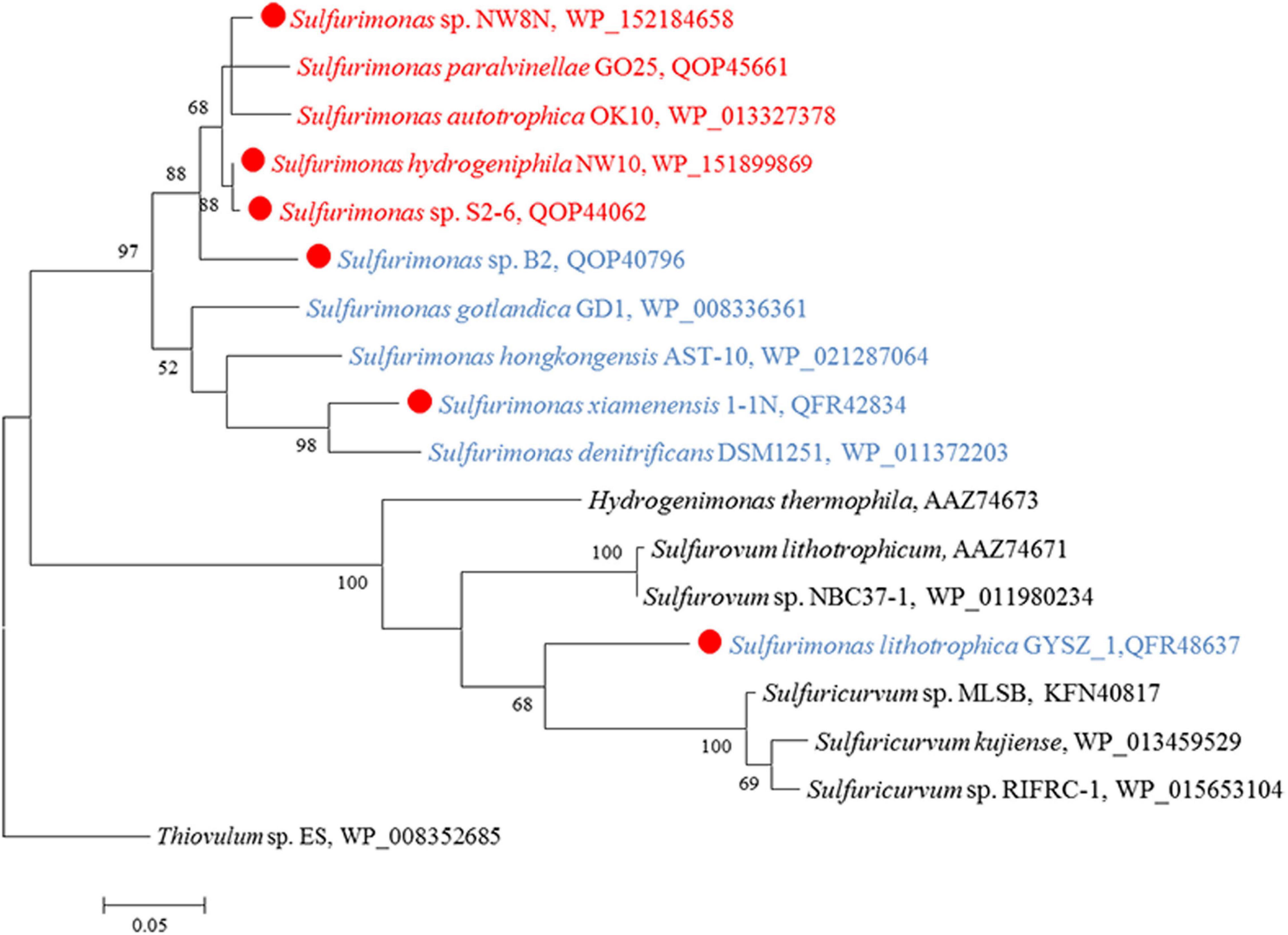
Figure 8. Maximum likelihood phylogenetic tree of the AclB protein sequences derived from Sulfurimonas strains and other representative species within the class Campylobacteria. Bootstrap values indicated at each node are based on a total of 1,000 bootstrap replicates. Branch node values below 50% are not shown. The solid red circles represent our own isolated strains. Isolation sources of Sulfurimonas species are indicated in different font colors: red, hydrothermal environments; blue, marine non-vent system.
Niche-Specific Genes at the Deep-Sea Hydrothermal Vents
The niche-specific genes were retrieved based on comparative genome analysis. We compared the genes common in five hydrothermal vent strains with those shared by non-hydrothermal vent strains. Overall, 57 unique genes specific of the hydrothermal vent isolates were found (Supplementary Table S2). The hydrothermal vent-specific genes encoded four major functions, including signal transduction, energy production and conversion, inorganic ion transport and cell wall/membrane/envelope biogenesis (Supplementary Table S2).
As for signal transduction, some transcriptional regulators belonging to the Per-ARNT-Sim (PAS) family were shared by the hydrothermal vent strains (Supplementary Table S2). Two histidine kinases (clusters 1,373 and 1,406), as the sensing component of the two-component signal transduction system, and one response regulator (cluster 1,293) were also found across the five vent strains (Supplementary Table S2), which may be responsible for sensing certain hydrothermal vent environmental conditions. In addition, the genomes of vent strains encoded relatively high numbers of signaling proteins (Supplementary Table S2), and particularly the genes encoding proteins with EAL and GGDEF domains, likely involved in the synthesis and hydrolysis of the intracellular signaling compound cyclic diguanylate. Regarding energy conservation, some genes involved in energy metabolism such as pyruvate dehydrogenase complex, which converts pyruvate to acetyl-CoA, NADH and CO2, were also shared by vent strains (Supplementary Table S2). We also found three rhodanese-related sulfur transferase (clusters 1,452, 1,501, and 1,507), involved in sulfur metabolism, uniquely present in the genomes of all vent strains. In addition, the vent strains shared multiple transporters, including the ABC transporter systems (clusters 1,349, 1,362, and 1,399) and some metal ion transporters such as Zn2+, Mg2+, Cu2+ and K+ (Supplementary Table S2). Finally, we found three outer membrane proteins TolC (clusters 1,471, 1,475, and 1,511), previously associated with different efflux systems and type I protein secretion (Pérez-Llarena and Bou, 2016), shared by all vent strains.
Discussion
Chemoautotrophic bacteria of the genus Sulfurimonas in the class Campylobacteria are ubiquitous and numerically dominant in global deep-sea hydrothermal vents (Mino et al., 2017; Dick, 2019). It is clear that Sulfurimonas species likely play an important role in the biogeochemical cycles of carbon, nitrogen and sulfur. However, yet little is known about their adaptations to vent environments. In this study, a novel species designated strain NW10T was characterized, which was isolated from a sulfide chimney on the Carlsberg Ridge of Northwestern Indian Ocean. In addition, we carried out a comparative genomic analysis including core and distributed genes to gain insights into the adaptation mechanisms of Sulfurimonas species to the deep-sea hydrothermal vents.
A New Species of Sulfurimonas Genus Representing a Predominant Bacterium in situ
Strain NW10T shared the highest 16S rRNA gene sequence similarity (95.8%) with S. paralvinellae GO25T and formed a phylogenetic subcluster within the genus Sulfurimonas, indicating that this strain should belong to the Sulfurimonas genus. Furthermore, ANI and DDH analyses clearly showed that strain NW10T could be differentiated genetically from other previously described Sulfurimonas species. Phenotypically, strain NW10T was different from its closest relative S. paralvinellae GO25T in many characteristics (Table 1), such as the growth conditions and maximum growth rate. The utilization patterns of electron donor were also different, in that strain NW10T could oxidize sulfide and elemental sulfur, whereas S. paralvinellae GO25T could not (Table 1). The combined evidence in phenotypic, chemotaxonomic and phylogenetic features conclusively demonstrates that strain NW10T represents a novel species in the genus Sulfurimonas, for which the name Sulfurimonas hydrogeniphila sp. nov. is proposed.
In deep-sea hydrothermal environments, bacteria of this species are widely spread, as supported by ecological distribution search in different environments through the Integrated Microbial Next Generation Sequencing (IMNGS) (Lagkouvardos et al., 2016). Across the 274,621 deep-sea hydrothermal vent samples gathered in the IMNGS platform, the relative abundance of Sulfurimonas sp. NW10-like sequences (>97% similarity of 16S rRNA gene in length 1,447 bps) was more than 1% total sequence reads in 49% of the vent samples and more than 0.1–1% in 39% of the vent samples (Wang et al., 2020a). The highest relative abundance was found in a deep-sea hydrothermal vent fluid sample from the Pacific Ocean, accounting for 22.4% of the total 16S sequences (Wang et al., 2020a). The relative high abundance and wide distribution of the new species highlight its importance in the biogeochemical cycles of sulfur in in situ hydrothermal environments.
Incomplete Thiosulfate Oxidation and Extracellular Biogenic S0
In sulfur-oxidizing bacteria, thiosulfate is usually oxidized by a Sox multi-enzyme system (SoxABCDXYZ) located in the periplasm. Two kinds of Sox pathways have been described according to the presence or absence of SoxCD. When SoxCD is present, it acts as a sulfur dehydrogenase and completely oxidizes thiosulfate to sulfate without formation of sulfur globule. Otherwise, sulfur is formed as an intermediate without SoxCD (Frigaard and Dahl, 2008). In this study, we found the soxCD genes were present in all Sulfurimonas genomes (Table 3). In addition, previous study showed that Sulfurimonas species did perform a complete thiosulfate oxidation without elemental sulfur as an intermediate (Inagaki et al., 2003; Sievert et al., 2008a; Labrenz et al., 2013). However, strain NW10T seems to be an exception in genus Sulfurimonas. Despite the presence of SoxCD, it performed incomplete oxidation of thiosulfate, with elemental sulfur accumulation outside of the cells (Figure 3).
The accumulation of extracellular S0 was significantly influenced by culture conditions, such as pH. At alkaline pH, strain NW10T completely oxidized thiosulfate to sulfate, while under neutral and acidic conditions, thiosulfate was oxidized to sulfate, with formation of elemental sulfur as an intermediate. The phenomenon was also observed in different strains of Hydrogenovibrio genus (Javor et al., 1990; Houghton et al., 2016; Jiang et al., 2017). Considering that the vent fluids of black chimneys are typically acidic, it is likely that the Sulfurimonas species inhabiting on vent chimneys can generate extracellular S0 in situ. In addition, oxygen concentrations can also significantly influence the elemental sulfur generation, with a maximum accumulation of extracellular S0 occurred at 6% oxygen.
Furthermore, the structures of extracellular S0 produced by NW10T were mainly in the form of crystalline bars with clear edges composed of α-S8, which was significantly different from the sulfur globules usually resulting from most chemotrophic and phototrophic bacteria activity (Dahl and Prange, 2006; Jiang et al., 2017; Cron et al., 2019). α-S8 is the thermodynamically most stable form of elemental sulfur at ambient temperature and pressure, and has been found in very diverse environments, such as marine sediments, water columns, euxinic lakes, sulfidic caves, hydrothermal vents, as well as cold or hot springs (Roy and Trudinger, 1970; Taylor et al., 1999; Steudel, 2000; Gleeson et al., 2012; Findlay et al., 2014; Hamilton et al., 2015). This crystalline sulfur could serve as an important intermediate in the biogeochemical sulfur cycle, and be further consumed by a wide diversity of microorganisms, such as sulfur oxidizer, sulfur reducer, or sulfur disproportionator. Our results highlight the potential ecological significance of extracellular S0 produced by Sulfurimonas in deep-sea hydrothermal ecosystems.
The mechanisms involved in the formation of extracellular S0 by SOB are still enigmatic compared with those of intracellular S0, although a variety of chemotrophic and phototrophic SOB and archaea can produce extracellular S0 (Dahl and Friedrich, 2008). So far, a large number of studies focused on the analysis of chemical form and structure of microbial extra-and intra-cellular S0 (Pickering et al., 2001; George et al., 2008; Berg et al., 2014). To our knowledge, the enzymes catalyzing the oxidation of sulfide or thiosulfate to S0 are usually located in the bacterial periplasm, but sulfur globules are observed outside of the cells, and sometimes even keeping the cells at a distance (Gregersen et al., 2011; Marnocha et al., 2016; Cron et al., 2019). Therefore, it has been proposed that reduced sulfur compounds could be initially oxidized to soluble polysulfide intermediates in the periplasm, and then be transported outside the cells to form sulfur globules. It is still not clear how extracellular S0 can accumulate outside of the cells (Cron et al., 2019). Recently, increasing number of studies indicate that SOB could excrete soluble organics to help form and stabilize S0 in the environment (Cosmidis et al., 2019; Cron et al., 2019; Marnocha et al., 2019). This could explain many of the puzzling fact observed in microbial extracellular S0, such as coated by organics on the surface, and growing extracellularly at a distance from the cells (Hanson et al., 2016; Marnocha et al., 2016, 2019). Here, the extracellular S0 produced by strain NW10T mainly contained elemental sulfur, with extremely low amounts of carbon and oxygen. In addition, there were no known homologs of sulfur globule proteins (SGP) genes in its genome, and comparative genome analysis showed no significant difference in Sox pathways between strain NW10T and other Sulfurimonas species and the only difference is that SoxC protein of strain NW10T lacks 18 bases at the N-terminal. The mechanism of extracellular S0 production requires further investigations by means of multiple omics in future.
Environmental Adaptations of Sulfurimonas Revealed by Comparative Genomic Analyses
Bacteria of the Sulfurimonas genus have been found to colonize a broad range of natural habitats from terrestrial, coastal sediment, shallow waters to deep-sea hydrothermal vents globally (Grote et al., 2008; Dahle et al., 2013; Meier et al., 2017). In this report to understand their environmental adaptation, we carried out comparative genomic analyses between vent and non-vent marine Sulfurimonas strains. Regarding energy conservation, all Sulfurimonas genomes contained the broad suite of genes encoding the enzymes capable of oxidizing thiosulfate, sulfite, sulfide and hydrogen (Table 3). In relation to sulfide oxidation, Sulfurimonas species contained genes encoding diverse types of Sqrs, allocated into Types II, III, IV, V, and VI. The variation and diversification of these Sqrs are presumed to play important roles in sulfide oxidation, sulfide assimilation, energy generation, heavy metal tolerance, detoxification and sulfide signaling (Marcia et al., 2010). Type IV and VI Sqrs were relatively conserved in genus Sulfurimonas (Table 3), likely to cope with different concentrations of sulfide, such as free sulfide in hydrothermal vents and metal sulfide in marine sediments (Han and Perner, 2015). Additionally, Sulfurimonas bacteria had one or more copies of Type II Sqrs (Table 3), which may compensate for the function of other Sqrs under specific environmental conditions (Han and Perner, 2015). Distinct roles of Type II Sqr have been proposed in different microorganisms. For example, it may be involved in heavy metal tolerance in the yeast Saccharomyces pombe (Vande-Weghe and Ow, 1999), sulfur assimilation in the non-pathogenic bacterium Pseudomonas putida KT2440 (Shibata and Kobayashi, 2006), and sulfide signaling in mammalian cells (Shahak and Hauska, 2008). In addition, some Sulfurimonas species harbored Type III and V Sqrs besides Type II, IV and VI Sqrs (Table 3), which may function in sulfide oxidation to enhance energy generation or detoxification and sulfide signaling (Han and Perner, 2015). It is interesting to find that Type V Sqr is only present in vent strains, such as NW10T, S2-6 and S. autotrophica OK10T. Type V Sqrs are a group of archaeal proteins, predominantly found in Creanarcheaota and Euryarchaeota (Sousa et al., 2018). It was only characterized in the archaeon Acidianus ambivalens and has been confirmed to possess Sqr activity with high substrate affinity (Brito et al., 2009). In addition, this type of Sqr in A. ambivalens exhibited the highest activity at 70°C, and only 3% activity remained at 25°C (Brito et al., 2009). This indicates that Type V Sqr may tolerate elevated temperatures. Phylogenetic relationship showed that Sulfurimonas has likely acquired its Type V Sqr gene from archaea through horizontal gene transfer (Figure 5; Han and Perner, 2015). Further experiments will be required to confirm the temperature tolerance attributed to Type V Sqr. In addition, genes encoding rhodanese-related sulfurtransferase were significantly enriched in vent strains (Supplementary Table S1). Previous study has indicated that the rhodanese-related sulfurtransferase serves as a polysulfide-sulfur transferase at lower polysulfide concentration in Wolinella succinogenes (Klimmek et al., 1991) and was possibly involved in polysulfide reduction in Desulfurella amilsii (Florentino et al., 2019). Here, we hypothesize that these sulfur transferases from vent strains may play a key role in the sulfur/polysulfide respiration process, where elemental sulfur or polysulfides are abundant.
Hydrogen oxidation is popular in bacteria of the Sulfurimonas genus. Members of this genus harbor a relatively broad suite of hydrogenases (Table 3), which can catalyze the hydrogen oxidation coupled to various electron acceptors reduction, including oxygen, nitrate and elemental sulfur. Almost all Sulfurimonas members contained multiple copies of Group I hydrogenases for hydrogen uptake, likely exhibiting different hydrogen affinities (Grote et al., 2012; Labrenz et al., 2013). Group II hydrogenases likely participate in hydrogen sensing or energy conversion at a low concentration of hydrogen (Campbell et al., 2009; Sievert et al., 2008b; Han and Perner, 2015). Previous study showed that Group II hydrogenases were not found in the hydrothermal vent Sulfurimonas isolates, but present in the marine water and sediments, thus speculated that in these habitats hydrogen concentration is relatively low than in hydrothermal vents, and that this group of hydrogenases may be more important under low hydrogen concentrations (Han and Perner, 2015). Yet, in this study, Group II hydrogenase was found in vent Sulfurimonas species including strains NW10T, NW8N, and S2-6. Hence, it is unlikely that Group II hydrogenase in vent Sulfurimonas is specialized to function at low hydrogen concentrations, and the elucidation of the role of Group II hydrogenase in Sulfurimonas species will require examination of its activity under different hydrogen concentrations.
Additionally, four Sulfurimonas species harbored Group IV hydrogenases, possibly involved in hydrogen evolution or energy conversion (Vignais and Billoud, 2007). In addition to Hyc and Ech, Coo subtype of group IV hydrogenase was first found in vent strain Sulfurimonas sp. S2-6, containing the cluster CooLUHF (Table 3). This energy-converting hydrogenases can couple CO and H2 metabolism with energy conservation. The Coo hydrogenase cluster has been identified in 30 bacterial representatives, at particularly high frequency in Deltaproteobacteria (many sulfate reducers, e.g., Desulfovibrio vulgaris), Alpha- (e.g., Rhodospirillum rubrum) and Campylobacteria (Nautilia profundicola), Firmicutes (e.g., Carboxydothermus hydrogenoformans), Betaproteobacteria and Gammaproteobacteria (Schoelmerich and Müller, 2019). Phylogenetic analysis based on the large subunit CooH indicated that this hydrogenase was clustered together with those of R. rubrum, D. vulgaris and C. hydrogenoformans (Figure 6), which have been experimentally demonstrated to oxidize CO, producing carbon dioxide and hydrogen as products, through CooMKLXUHF complexes (Bonam and Ludden, 1987; Wu et al., 2005; Caffrey et al., 2007). Further, protein domain analyses by DELTA-BLAST and PSI-BLAST in NCBI showed that CooH contains three functional domains: two respiratory-chain NADH dehydrogenases and one nickel-dependent hydrogenase. The Coo hydrogenases could be involved in H2 production from CO according to the overall equation CO + H2O→CO2 + H2 (Heidelberg et al., 2004). In any case, this is the first report of Coo hydrogenase found in the genus Sulfurimonas and the function of H2 production from Coo hydrogenase in Sulfurimonas species needs further experimental confirmation.
In a word, the evolution and functions of different types of Sqr and hydrogenases within one host remain enigmatic and need further investigations, especially to elucidate their relevance in relation to host adaptation to hydrothermal environments. In addition to energy metabolism, comparative genome analysis revealed other vent-specific gene signatures related to signal transduction mechanisms and inorganic ion transporter mechanisms, including unique two-component signal transduction system and a relative abundance of signaling proteins, the ABC transporter system and metal ion transporter (such as Zn2+, Mg2+, Cu2+ and K+ transporter), and outer membrane protein TolC. Overall, this versatile energy metabolism, environmental sensing systems, and multiple transporter mechanisms could contribute to the wide spreading and high adaptability of these organisms to different hydrothermal vent fields globally.
Conclusion
Strain NW10T represents a novel species named as S. hydrogeniphila, which is abundant (≥1%) in nearly half of deep-sea hydrothermal vent environments globally. It differs from other established species of this genus in that it can produce a large amount of extracellular sulfur during thiosulfate oxidation. This discovery highlights that the role in hydrothermal vent ecosystems worth further investigations. Strain NW10T can grow with a variety of electron donors (various sulfur compounds and hydrogen) and acceptors (nitrate, oxygen, and elemental sulfur), with a preference for hydrogen utilization. Correspondingly, its genome encodes very diverse energy metabolisms for carbon fixation, with sulfur oxidation coupled nitrate/oxygen reduction, or hydrogen oxidation coupled the reduction of nitrate, oxygen, and even elemental sulfur, showing its adaptability to fluctuating hydrothermal vent environments. Comparative genomics revealed unique genes encoding Type V Sqr, Group II and Coo hydrogenases that may facilitate vent Sulfurimonas survival in deep-sea hydrothermal environments.
Description of Sulfurimonas hydrogeniphila sp. nov.
Sulfurimonas hydrogeniphila (hy.dro.ge.ni.phi’la. N.L. neut. n. hydrogenum hydrogen; Gr. adj. philos loving; N.L. fem. adj. hydrogeniphila hydrogen-loving, because growth prefers hydrogen).
Cells are Gram-negative short rods (0.8–3.5 × 0.4–0.8 μm) that are motile by means of a polar flagellum. Anaerobic to aerobic, even with the air in the headspace (optimum 6%). Growth occurs at 4-45°C (optimum 33°C), pH 5.0–9.0 (optimum pH 6.0–6.5), and with 2.0–4.0% (w/v) NaCl concentration (optimum 3.0%). Obligate chemolithoautotrophic growth occurs with H2, thiosulfate, sulfide, elemental sulfur as electron donors, and oxygen, nitrate, and S0 can be utilized as an electron acceptor. Organic substrates are not utilized as carbon sources and energy sources. Major cellular fatty acids are C16:1 ω7c (31.6%), C16:0 (28.2%) and C18:1 ω7c (18.4%).
The type strain, NW10T (=MCCC 1A13987T = KTCC 15781T) was isolated from the deep-sea hydrothermal sulfide chimneys in the Carlsberg Ridge of Northwest Indian Ocean. The G + C content of its genomic DNA is 37.3 mol%.
Materials and Methods
Sample Collection and Enrichment
The chimney samples were collected near an active hydrothermal vent on the Carlsberg Ridge (60°31′E, 6°21′N), at a depth of 2936 m, by a human operated vehicle “Jiao-long” during COMRA DY 38 oceanic scientific cruise in March 2017. Aboard the research vessel Xiang-Yang-Hong No. 9, chimney samples were immediately transferred into MMJHS medium under a gas phase mixture of 80% H2/18% CO2/2% O2 (200 kPa) and then incubated at 28°C according to the previous description (Inagaki et al., 2003). After successful enrichment with MMJHS medium, the well-grown culture was further purified using the dilution-to-extinction technique with the same medium. MMJS medium consisted of NaCl (30 g l–1), KCl (0.33 g l–1), NH4Cl (0.25 g l–1), MgCl2⋅6H2O (4.18 g l–1), CaCl2⋅2H2O (0.14 g l–1), K2HPO4 (0.14 g l–1), NaHCO3 (1 g l–1), Na2S2O3⋅5H2O (10 mM), Wolfe’s vitamins (1 ml l–1) and trace element solution (10 ml l–1).
Phylogenomic Analysis
The genomic DNA was prepared according to the method described previously (Jiang et al., 2010) and the 16S rRNA gene was amplified by PCR primers described previously (Lane, 1991). The sequence was compared with those of other type strains using the EzTaxon-e server (Yoon et al., 2017). The 16S rRNA gene sequences of the related taxa were obtained from the GenBank database. Phylogenetic trees were constructed by using MEGA 6.0 (Tamura et al., 2013) using the neighbor-joining (Saitou and Nei, 1987), maximum-likelihood (Felsenstein, 1981) and minimum evolution methods (Rzhetsky and Nei, 1992) after multiple alignments of the data by CLUSTAL_W. Evolutionary distances were calculated using Kimura’s two-parameter model and bootstrap values were determined based on 1,000 replications. The phylogenomic tree was constructed based on an up-to-date 92 bacterial core gene sets by UBCG version 3.0 (Na et al., 2018). Genome sequences of reference taxa were retrieved from the NCBI database and the 92 concatenated core genes were extracted, aligned and concatenated using default parameters. The tree topology was supported by the maximum-likelihood method for 100 bootstrap replications using RAxML version 8.2.11 (Stamatakis, 2014) with the GTR + CAT model.
Phenotypic and Chemotaxonomic Characterization
Cell morphology was observed under a transmission electron microscopy (Model JEM-1230, JEOL, Japan) with cultures grown in MMJHS medium at 28°C for 1 day. The physiological characterization of the isolate was tested on MMJHS medium (Inagaki et al., 2003). After autoclaving, the medium (10 ml) was dispensed into 50 ml serum bottles, then sealed with a butyl-rubber stopper under a gas phase of 80% H2/18% CO2/2% O2 (200 kPa). All cultivation experiments were performed in triplicate, unless otherwise specified. The growth was measured by direct cell counting using a phase contrast microscope (Eclipse 80i, Nikon, Japan). Growth at different temperatures was examined in MMJHS at 4, 10, 15, 20, 25, 28, 30, 33, 35, 37, 45, 50, and 60°C. The growth salinity range was examined by adjusting the concentrations of NaCl between 0 and 9.0% (w/v), at 0.5 (w/v) intervals. To determine the effect of pH on growth, the pH of MMJHS medium was adjusted from 4.5 to 9.0 with a 0.5 pH unit interval by using 30 mM acetate/acetic acid buffer (pH 4.0–5.0), MES (pH 5.0–6.0), PIPES (pH 6.0–7.0), HEPES (pH 7.0–7.5), Tris and CAPSO (pH 8.0 and above). The effect of O2 on growth was examined by adjusting the oxygen concentration (0, 1, 2, 4, 6, 8, 10% at 200 kPa and 20% at 100 kPa) in the headspace gas. To test the anaerobic growth, 10 mM nitrate was added as an alternative electron acceptor.
The ability of sulfur oxidation of strain NW10T was tested using the following sulfur compounds as the sole energy source in the MMJS medium, including thiosulfate (10 mM), sulfite (5 mM), thiocyanate (5 mM), tetrathionate (5 mM), elemental sulfur (1% w/v), or sodium sulfide (50, 100, 500 μM, and 1 mM) with a gas phase of 74% N2/20% CO2/6% O2 (200 kPa). Molecular hydrogen was also tested in MMJH medium in the absence of thiosulfate under a gas phase of 74% H2/20% CO2/6% O2 (200 kPa). To determine the utilization of other electron acceptors, each of the potential electron acceptors, such as thiosulfate (10 mM), tetrathionate (10 mM), sulfite (2 mM and 10 mM), elemental sulfur (1%, w/v), nitrate (10 mM), nitrite (1 mM and 5 mM), selenate (5 mM), arsenate (5 mM), fumarate (10 mM), and ferric citrate (20 mM) was examined with MMJHS medium under 80% H2/20% CO2 (200 kPa). Heterotrophic growth was examined in MMJHS medium in the absence of NaHCO3 under a gas phase of 94% N2/6% O2 (200 kPa). Each of the following potential organic carbon compounds was tested: 0.1% (w/v) peptone, starch, tryptone, yeast extract, casein, and casamino acids, 5 mM formate, acetate, propionate, citrate, tartrate, fumarate, succinate, malate, and pyruvate, 5 mM of each of 20 amino acids, 0.02% (w/v) glucose, galactose, sucrose, fructose, lactose, maltose, and trehalose. In an attempt to determine the alternative energy source, these organic compounds were used as an energy source in MMJ medium to replace thiosulfate under a gas phase of 74% N2/20% CO2/6% O2 (200 kPa).
For analyses of fatty acids, cells grown on MMJHS medium at 33°C for 24 h were saponified, methylated, and extracted following the standard MIDI protocol (Sherlock Microbial Identification System, version 6.0B). The fatty acids were analyzed by gas chromatography (Agilent Technologies 6850) and then the result was identified using the TSBA6.0 database of the Microbial Identification System.
Scanning Electron Microscopy and Raman Spectromicroscopy of Extracellular Sulfur
Scanning electron microscope (SEM) (S-3400N; Hitachi, Japan) and Raman spectra (XploRA; Horiba JY, France) were used to identify the shape, components and structure of extracellular sulfur produced by strain NW10T. For SEM analysis, a milky white suspension was collected using polycarbonate filters (Merck Millipore, pore size 3.0 μm), rinsed three times with deionized water and observed by SEM at 5 kV. Energy-Dispersive Spectrum (EDS) (model 550i, IXRF systems, United States) equipment with SEM was employed at an accelerating voltage of 5 keV for 30 s. For Raman analysis, about 5 ml samples were concentrated by centrifugation and rinsed three times with deionized water. The supernatant was decanted and the pellet was lyophilized overnight. A small amount of powder was put on the glass slide and observed by microconfocal method. Raman spectra were collected using a Horiba XploRA Raman spectrometer coupled with a 532 nm laser source. For α-S8 standard, commercial precipitated sulfur (purity > 99%, Thermo Fisher Scientific) was used.
Whole Genome Sequence Analysis
The complete genome of strain NW10T was sequenced by Tianjin Biochip Corporation (Tianjin, PR China), using the single molecule real-time (SMRT) technology on the Pacific Biosciences (PacBio) sequencing platform. The sequenced reads were filtered, and high quality paired-end reads were assembled to construct a circular genome with SOAPdenovo (version 2.04)1. The G + C content of the chromosomal DNA was determined according to the genome sequence. Gene prediction was carried out by Glimmer program (Delcher et al., 2007). rRNA identification was performed with the RNAmmer 1.2 software (Lagesen et al., 2007), and tRNAscan-SE (version 1.21) was used to identify the tRNA genes (Schattner et al., 2005). Gene prediction and annotation were carried out using NCBI Prokaryotic Genomes Annotation Pipeline (PGAP) and the Rapid Annotation using Subsystem Technology (RAST) pipeline2 (Aziz et al., 2008). The functional annotation and metabolic pathways were analyzed by searching against KEGG and COG databases. To further clarify the genetic relatedness between strain NW10T and related species of the genus Sulfurimonas, the average nucleotide identity (ANI) value between two genomes was calculated using the web service of EZGenome3 (Richter and Rosselló-Móra, 2009). The predicted in silico DNA-DNA hybridization (DDH) values were determined online4 using the Genome-to-Genome Distance Calculator (GGDC) (Auch et al., 2010).
Comparative Genomics Analyses
To avoid the possible deviations due to different annotation methods, we used RAST server for re-annotation. A pan-genome for the eleven genomes was determined by BPGA (Chaudhari et al., 2016) pipeline to identify orthologous groups among Sulfurimonas strains and to extrapolate the pan-genome models of applying default parameters. Orthologous clusters were assigned by grouping all protein sequences in the 11 genomes using USEARCH based on their sequence similarity (E-value < 10–5, >50% coverage) and each protein was assigned to one protein family. The pan genome analysis complied the set of core genes shared among all strains, a set of distributed genes shared with more than two but not all strains, and unique genes only found in a single strain. COG and KEGG distributions of the core, distributed and unique gene families were calculated based on representative sequences.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
SW, LJ, SY, and ZS designed the study. ZS contributed to deep sea sampling and conducted on board experiments. SW and QH conducted the experiments and analyzed the data. SW, LJ, and LC drafted the manuscript. BZ, XF, QL, and ZS revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was financially supported by the National Key R&D Program of China (No. 2018YFC0310701 to ZS), the National Natural Science Foundation of China (No. 41672333 to LJ), COMRA program (No. DY135-B2-01 to ZS), and the Subsidized Project for Cultivating Postgraduates Innovative Ability in Scientific Research of Huaqiao University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the whole team of the cruise DY38-leg1 conducted by R/V Xiang-Yang-Hong No.9 for samples collecting. We thank Dr. Chuan Liu (Xiamen University) for Raman spectrum analysis, and Dr. Li Gu from the Third Institute of Oceanography for the scanning electron microscopy pictures. We thank Dr. Zhaobin Huang (Quanzhou Normal University) for helpful discussion and advices.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.626705/full#supplementary-material
Supplementary Figure 1 | Transmission electron micrograph of cells of S. hydrogeniphila NW10T. Bar, 2.0 μm.
Supplementary Figure 2 | Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the relationship of strain NW10T with other members within the genus Sulfurimonas. Bootstrap values based on 1,000 replicates are shown at branch nodes. Bar, 0.01 substitutions per nucleotide position.
Supplementary Figure 3 | Minimum evolution phylogenetic tree based on 16S rRNA gene sequences showing the relationship of strain NW10T with other members within the genus Sulfurimonas. Bootstrap values based on 1,000 replicates are shown at branch nodes. Bar, 0.01 substitutions per nucleotide position.
Supplementary Figure 4 | Circular diagrams of the S. hydrogeniphila NW10 chromosomes displaying relevant genome features. The following information is provided from outside to inside: The first circle and the fourth circle are protein coding sequences on the forward and reverse strand; The second and third circles are CDS, tRNA and rRNA on the forward and reverse stand, respectively. The fifth circle is the GC content (swell outward/inward indicates higher/lower G + C compared with the average G + C content); The sixth circle is the GC-Skew value (purple/yellow indicate positive/negative values) and the innermost circle identifies the genome size.
Supplementary Table 1 | The specific information of key enzymes involved in sulfur, nitrogen, hydrogen, and carbon metabolisms in 11 Sulfurimonas species based on RAST annotations.
Supplementary Table 2 | Vent-specific genes and their functional annotations. Annotation is the functional annotation based on the SEED database. COG_sutype, shows the functional classification based on COG family assignments.
Abbreviations
MCCC, Marine Culture Collection of China; KCTC, Korean Collection for Type Cultures; ANI, average nucleotide identity; DDH, DNA-DNA hybridization.
Footnotes
- ^ http://soap.genomics.org.cn/
- ^ http://rast.nmpdr.org/
- ^ www.ezbiocloud.net/tools
- ^ http://ggdc.dsmzde/distcalc2.php
References
Akerman, N. H., Butterfield, D. A., and Huber, J. A. (2013). Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front. Microbiol. 4:185. doi: 10.3389/fmicb.2013.00185
Auch, A. F., Von, J. M., Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Berg, J. S., Schwedt, A., Kreutzmann, A. C., Kuypers, M. M. M., and Milucka, J. (2014). Polysulfides as intermediates in the oxidation of sulfide to sulfate by Beggiatoa spp. Appl. Environ. Microbiol. 80, 629–636. doi: 10.1128/AEM 02852-13
Bonam, D., and Ludden, P. W. (1987). Purification and characterization of carbon monoxide dehydrogenase, a nickel, zinc, iron-sulfur protein, from rhodospirillum rubrum. J. Biol. Chem. 262, 2980–2987. doi: 10.1016/S0021-9258(18)61456-5
Brito, J. A., Sousa, F. L., Stelter, M., Bandeiras, T. M., Vonrhein, C., Teixeira, M., et al. (2009). Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 48, 5613–5622. doi: 10.1021/bi9003827
Caffrey, S. M., Park, H. S., Voordouw, J. K., He, Z. L., Zhou, J. Z., and Voordouw, G. (2007). Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 189, 6159–6167. doi: 10.1128/JB.00747-07
Cai, L., Shao, M., and Zhang, T. (2014). Non-contiguous finished genome sequence and description of Sulfurimonas hongkongensis sp. nov., a strictly anaerobic denitrifying, hydrogen- and sulfur-oxidizing chemolithoautotroph isolated from marine sediment. Stand. Genomic Sci. 9, 1302–1310. doi: 10.1007/s00248-015-0722-4
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile epsilon-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468. doi: 10.1038/nrmicro1414
Campbell, B. J., Smith, J. L., Hanson, T. E., Klotz, M. G., Stein, L. Y., Lee, C. K., et al. (2009). Adaptations to submarine hydrothermal environments exemplified by the genome of Nautilia profundicola. PLoS Genet. 5:e1000362. doi: 10.1371/journal.pgen.1000362
Chaudhari, N. M., Kumar, G. V., and Chitra, D. (2016). BPGA an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Cosmidis, J., Nims, C. W., Diercks, D., and Templeton, A. S. (2019). Formation and stabilization of elemental sulfur through organomineralization. Geochim. Cosmochim. Acta 247, 59–82. doi: 10.1016/j.gca.2018.12.025
Cron, B., Henri, P., Chan, C. S., Macalady, J. L., and Cosmidis, J. (2019). Elemental sulfur formation by Sulfuricurvum kujiense is mediated by extracellular organic compounds. Front. Microbiol. 10:2710. doi: 10.3389/fmicb.2019.02710
Dahl, C., and Prange, A. (2006). Bacterial sulfur globules: occurrence, structure and metabolism. Microbiol. Monogr. 26, 21–51. doi: 10.1007/3-540-33774-1_2
Dahle, H., Roalkvam, I., Thorseth, I. H., Pedersen, R. B., and Steen, I. H. (2013). The Versatile in situ gene expression of an Epsilonproteobacteria-dominated biofilm from a hydrothermal chimney. Environ. Microbiol. Rep. 5, 282–290. doi: 10.1111/1758-2229.12016
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinform 23, 673–679. doi: 10.1093/bioinformatics/btm009
Dick, G. J. (2019). The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat. Rev. Microbiol. 17, 271–283. doi: 10.1038/s41579-019-0160-2
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Findlay, A. J., Gartman, A., MacDonald, D. J., Hanson, T. E., Shaw, T. J., and Luther, G. W. (2014). Distribution and size fractionation of elemental sulfurin aqueous environments: the Chesapeake Bay and mid-Atlantic ridge. Geochim. Cosmochim. Acta 142, 334–348. doi: 10.1016/j.gca.2014.07.032
Florentino, A. P., Inês, A. C., Boeren, S., Born, M. V. D., Stams, A. J., and Sánchez-Andrea, I. (2019). Insight into the sulfur metabolism of Desulfurella amilsii by differential proteomics. Environ. Microbiol. 21, 209–225. doi: 10.1111/1462-2920.14442
Fortunato, C., and Huber, J. (2016). Coupled RNA-SIP and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. ISME J. 10, 1925–1938. doi: 10.1038/ismej.2015.258
Frigaard, N. U., and Dahl, C. (2008). “Sulfur metabolism in phototrophic sulfur bacteria,” in Advances in Microbial Physiology, ed. K. P. Robert (Waltham, MA: Academic Press), 103–200.
George, G. N., Gnida, M., Bazylinski, D. A., Prince, R. C., and Pickering, I. J. (2008). X-ray absorption spectroscopy as a probe of microbial sulfur biochemistry: the nature of bacterial sulfur globules revisited. J. Bacteriol. 190, 6376–6383. doi: 10.1128/JB.00539-08
Gleeson, D. F., Pappalardo, R. T., Anderson, M. S., Grasby, S. E., Mielke, R. E., Wright, K. E., et al. (2012). Biosignature detection at an Arctic analog to Europa. Astrobiology 12, 135–150. doi: 10.1089/ast.2010.0579
Gregersen, L. H., Bryant, D. A., and Frigaard, N. U. (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front. Microbiol. 2:116. doi: 10.3389/fmicb.2011.00116
Grote, J., Jost, G., Labrenz, M., Herndl, G. J., and Jurgens, K. (2008). Epsilonproteobacteria represent the major portion of chemoautotrophic bacteria in sulfidic waters of pelagic redoxclines of the Baltic and Black Seas. Appl. Environ. Microbiol. 74, 7546–7551. doi: 10.1128/AEM.01186-08
Grote, J., Schott, T., Bruckner, C. G., Glockner, F. O., Jost, G., Teeling, H., et al. (2012). Genome and physiology of a model Epsilonproteobacterium responsible for sulfide detoxification in marine oxygen depletion zones. Proc. Natl. Acad. Sci. U S A. 109, 506–510. doi: 10.1073/pnas.1111262109
Hamilton, T. L., Jones, D. S., Schaperdoth, I., and Macalady, J. L. (2015). Metagenomic insights into S(0) precipitation in a terrestrial subsurface lithoautotrophic ecosystem. Front. Microbiol. 5:756. doi: 10.3389/fmicb.2014.00756
Han, Y., and Perner, M. (2015). The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 6:989. doi: 10.3389/fmicb.2015.00989
Hanson, T. E., Bonsu, E., Tuerk, A., Marnocha, C. L., Powell, D. H., and Chan, C. S. (2016). Chlorobaculum tepidum growth on biogenic S(0) as the sole photosynthetic electron donor. Environ. Microbiol. 18, 2856–2867. doi: 10.1111/1462-2920.12995
Heidelberg, J. F., Seshadri, R., Haveman, S. A., Hemme, C. L., Paulsen, I. T., Kolonay, J. K., et al. (2004). The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22, 554–559. doi: 10.1038/nbt959
Hou, J., Sievert, S. M., Wang, Y., Seewald, J. S., Natarajan, V. P., Wang, F., et al. (2020). Microbial succession during the transition from active to inactive stages of deep-sea hydrothermal vent sulfide chimneys. Microbiome 8:102. doi: 10.21203/rs.3.rs-16462/v2
Houghton, J., Foustoukos, D., Flynn, T., Vetriani, C., Bradley, A. S., and Fike, D. (2016). Thiosulfate oxidation by Thiomicrospira thermophilus: metabolic flexibility in response to ambient geochemistry. Environ. Microbiol. 9, 3057–3072. doi: 10.1111/1462-2920.13232
Inagaki, F., Takai, K., Kobayashi, H., Nealson, K. H., and Horikoshi, K. (2003). Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing ε-Proteobacteria isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 53, 1801–1805. doi: 10.1099/ijs.0.02682-0
Innamorati, K. A., Earl, J. P., Aggarwal, S. D., Ehrlich, G. D., and Hiller, N. L. (2020). “The bacterial guide to designing a diversified gene portfolio,” in The Pangenome, eds H. Tettelin and D. Medini (Cham: Springer), 51–87.
Javor, B. J., Wilmot, D. B., and Vetter, R. D. (1990). pH-Dependent metabolism of thiosulfate and sulfur globules in the chemolithotrophic marine bacterium Thiomicrospira crunogenus. Arch. Microbiol. 154, 231–238. doi: 10.1007/BF00248960
Jiang, L., Lyu, J., and Shao, Z. (2017). Sulfur metabolism of Hydrogenovibrio thermophilus strain S5 and its adaptations to deep-sea hydrothermal vent environment. Front. Microbiol. 8:2513. doi: 10.3389/fmicb.2017.02513
Jiang, L., Zheng, Y., Peng, X., Zhou, H., Zhang, C., Xiao, X., et al. (2010). Vertical distribution and diversity of sulfate-reducing prokaryotes in the Pearl River estuarine sediments, Southern China. FEMS Microbiol. Ecol. 70, 93–106. doi: 10.1111/j.1574-6941.2009.00758.x
Klimmek, O., Kröger, A., Steudel, R., and Holdt, G. (1991). Growth of Wolinella succinogenes with polysulphide as terminal acceptor of phosphorylative electron transport. Arch. Microbiol. 155, 177–182. doi: 10.1007/BF00248614
Labrenz, M., Grote, J., Mammitzsch, K., Boschker, H. T., Laue, M., Jost, G., et al. (2013). Sulfurimonas gotlandica sp. nov., a chemoautotrophic and psychrotolerant epsilonproteobacterium isolated from a pelagic redoxcline, and an emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 63, 4141–4148. doi: 10.1099/ijs.0.048827-0
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lagkouvardos, I., Joseph, D., Kapfhammer, M., Giritli, S., Horn, M., Haller, D., et al. (2016). IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6:33721. doi: 10.1038/srep33721
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackbrandt and M. Goodfellow (New York: Wiley), 115–176.
Marcia, M., Ermler, U., Peng, G., and Michel, H. (2010). A new structure-based classification of sulfide:quinone oxidoreductases. Proteins 78, 1073–1083. doi: 10.1002/prot.22665
Marnocha, C. L., Levy, A. T., Powell, D. H., Hanson, T. E., and Chan, C. S. (2016). Mechanisms of extracellular S0 globule production and degradation in Chlorobaculum tepidum via dynamic cell–globule interactions. Microbiol 162, 1125–1134. doi: 10.1099/mic.0.000294
Marnocha, C. L., Sabanayagam, C. R., Modla, S., Powell, D. H., Henri, P. A., Steele, A. S., et al. (2019). Insights into the mineralogy and surface chemistry of extracellular biogenic S(0) globules produced by Chlorobaculum tepidum. Front. Microbiol. 10:271. doi: 10.3389/fmicb.2019.00271
Meier, D. V., Pjevac, P., Bach, W., Hourdez, S., Girguis, P. R., Vidoudez, C., et al. (2017). Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 11, 1545–1558. doi: 10.1038/ismej.2017.37
Mino, S., Nakagawa, S., Makita, H., Toki, T., Miyazak, J., and Sievert, M. S. (2017). Endemicity of the cosmopolitan mesophilic chemolithoautotroph Sulfurimonas at deep-sea hydrothermal vents. ISME J. 11, 909–919. doi: 10.1038/ismej.2016.178
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Nakagawa, S., and Takai, K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65, 1–14. doi: 10.1111/j.1574-6941.2008.00502.x
Nakagawa, S., Takai, K., Inagaki, F., Hirayama, H., Nunoura, T., Horikoshi, K., et al. (2005). Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ. Microbiol. 7, 1619–1632. doi: 10.1111/j.1462-2920.2005.00856.x
Pérez-Llarena, F. J., and Bou, G. (2016). Proteomics as a tool for studying bacterial virulence and antimicrobial resistance. Front. Microbiol. 7:410. doi: 10.3389/fmicb.2016.00410
Perner, M., Gonnella, G., Hourdez, S., Böhnke, S., Kurtz, S., and Girguis, P. (2013). In situ chemistry and microbial community compositions in five deep-sea hydrothermal fluid samples from Irina II in the Logatchev field. Environ. Microbiol. 15, 1551–1560. doi: 10.1111/1462-2920.12038
Pickering, I. J., George, G. N., Yu, E. Y., Brune, D. C., Tuschak, C., Overmann, J., et al. (2001). Analysis of sulfur biochemistry of sulfur bacteria using X-ray absorption spectroscopy. Biochem 40, 8138–8145. doi: 10.1021/bi0105532
Pjevac, P., Meier, D. V., Markert, S., Hentschker, C., Schweder, T., Becher, D., et al. (2018). Metaproteogenomic profiling of microbial communities colonizing actively venting hydrothermal chimneys. Front. Microbiol. 9:680. doi: 10.3389/fmicb.2018.00680
Ratnikova, N. M., Slobodkin, A. I., Merkel, A. Y., Kopitsyn, D. S., Kevbrin, V. V., Bonch-Osmolovskaya, E. A., et al. (2019). Sulfurimonas crateris sp. nov., a facultative anaerobic sulfur-oxidizing chemolithoautotrophic bacterium isolated from a terrestrial mud volcano. Int. J. Syst. Evol. Microbiol. 70, 487–492. doi: 10.1099/ijsem.0.003779
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U S A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roy, A. B., and Trudinger, P. A. (1970). The Biochemistry of Inorganic Compounds of Sulfur. London: Cambridge University Press.
Rzhetsky, A., and Nei, M. (1992). Statistical properties of the ordinary least-squares, generalized least-squares, and minimum-evolution methods of phylogenetic inference. J. Mol. Evol. 35, 367–375. doi: 10.1007/BF00161174
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schoelmerich, M. C., and Müller, V. (2019). Energy conservation by a hydrogenase-dependent chemiosmotic mechanism in an ancient metabolic pathway. Proc. Natl. Acad. Sci. U S A. 116, 6329–6334. doi: 10.1073/pnas.1818580116
Shahak, Y., and Hauska, G. (2008). “Sulfide oxidation from cyanobacteria to humans: sulfide-quinone oxidoreductase (SQR),” in Sulfur Metabolism in Phototrophic Organisms, Vol. 27, eds C. Dahl, D. Knaff, and T. Leustek (Amsterdam: Springer), 319–335.
Shibata, H., and Kobayashi, S. (2006). Characterization of a HMT2-likeenzyme for sulfide oxidation from Pseudomonas putida. Can. J. Microbiol. 52, 724–730. doi: 10.1139/W06-022
Sievert, S. M., Hügler, M., Taylor, C. D., and Wirsen, C. O. (2008a). “Sulfur oxidation at deep-sea hydrothermal vents,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (New York: Springer), 238–258.
Sievert, S. M., Scott, K. M., Klotz, M. G., Chain, P. S., Hauser, L. J., Hemp, J., et al. (2008b). Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 74, 1145–1156. doi: 10.1128/AEM.01844-07
Sikorski, J., Munk, C., Lapidus, A., Ngatchou, D. O., Lucas, S., Glavina, D. R., et al. (2010). Complete genome sequence of Sulfurimonas autotrophica type strain (OK10T). Stand. Genomic Sci. 3, 194–202. doi: 10.4056/sigs.1173118
Sousa, F. M., Pereira, J. G., Marreiros, B. C., and Pereira, M. M. (2018). Taxonomic distribution, structure/function relationship and metabolic context of the two families of sulfide dehydrogenases: SQR and FCSD. Biochim. Biophys. Acta 1859, 742–753. doi: 10.1016/j.bbabio.2018.04.004
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinform 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steudel, R. (2000). “The chemical sulfur cycle,” in Environmental Technologies to Treat Sulfur Pollution, eds P. Lens and W. H. Pol (London: IWA Publishing), 1–31.
Takai, K., Suzuki, M., Nakagawa, S., Miyazaki, M., Suzuki, Y., Inagaki, F., et al. (2006). Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen- and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 56, 1725–1733. doi: 10.1099/ijs.0.64255-0
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Taylor, C. D., Wirsen, C. O., and Gaill, F. (1999). Rapid microbial production of filamentous sulfur mats at hydrothermal vents. Appl. Environ. Microbiol. 65, 2253–2255.
Timmer-ten, H. T. (1975). A new type of thiosulphate oxidizing, nitrate reducing microorganism: Thiomicrospira denitrificans sp. nov. Neth. J. Sea. Res. 9, 344–350. doi: 10.1016/0077-7579(75)90008-3
Vande-Weghe, J. G., and Ow, D. W. (1999). A fission yeast gene for mitochondrial sulfide oxidation. J. Biol. Chem. 274, 13250–13257. doi: 10.1074/jbc.274.19.13250
Vignais, P. M., and Billoud, B. (2007). Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272. doi: 10.1021/cr050196r
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2018.00772
Wang, S., Jiang, L., Hu, Q., Liu, X., Yang, S., and Shao, Z. (2020a). Elemental sulfur reduction by a deep-sea hydrothermal vent Campylobacterium Sulfurimonas sp. NW10. Environ. Microbiol. Preprint. doi: 10.1111/1462-2920.15247
Wang, S., Jiang, L., Liu, X., Yang, S., and Shao, Z. (2020b). Sulfurimonas xiamenensis sp. nov. And Sulfurimonas lithotrophica sp. nov., hydrogen-and sulfur-oxidizing chemolithoautotrophs within the Epsilonproteobacteria isolated from coastal sediments, and an emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 70, 2657–2663. doi: 10.1099/ijsem.0.004087
Wu, M., Ren, Q., Durkin, A. S., Daugherty, S. C., and Eisen, J. A. (2005). Life in hot carbon monoxide: the complete genome sequence of carboxydothermus hydrogenoformans z-2901. PLoS Genet. 1:e65. doi: 10.1371/journal.pgen.0010065
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zeng, X., Zhang, X., and Shao, Z. (2020). Metabolic Adaptation to Sulfur of Hyperthermophilic Palaeococcus pacificus DY20341T from Deep-Sea Hydrothermal Sediments. Int. J. Mol. Sci. 21:368. doi: 10.3390/ijms21010368
Keywords: Sulfurimonas hydrogeniphila, hydrogen oxidation, sulfur oxidation, hydrothermal vent, environmental adaptation
Citation: Wang S, Jiang L, Hu Q, Cui L, Zhu B, Fu X, Lai Q, Shao Z and Yang S (2021) Characterization of Sulfurimonas hydrogeniphila sp. nov., a Novel Bacterium Predominant in Deep-Sea Hydrothermal Vents and Comparative Genomic Analyses of the Genus Sulfurimonas. Front. Microbiol. 12:626705. doi: 10.3389/fmicb.2021.626705
Received: 06 November 2020; Accepted: 11 February 2021;
Published: 26 February 2021.
Edited by:
Osvaldo Ulloa, University of Concepcion, ChileReviewed by:
James F. Holden, University of Massachusetts Amherst, United StatesKathleen Scott, University of South Florida, United States
Copyright © 2021 Wang, Jiang, Hu, Cui, Zhu, Fu, Lai, Shao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongze Shao, c2hhb3p6QDE2My5jb20=; Suping Yang, eWFuZ3N1cGluZ0BocXUuZWR1LmNu
†These authors have contributed equally to this work