 Aleksi Husso1†
Aleksi Husso1† Leen Lietaer2†
Leen Lietaer2† Tiina Pessa-Morikawa1Thomas Grönthal3Jan Govaere2Ann Van Soom2
Tiina Pessa-Morikawa1Thomas Grönthal3Jan Govaere2Ann Van Soom2 Antti Iivanainen1‡
Antti Iivanainen1‡ Geert Opsomer2‡
Geert Opsomer2‡ Mikael Niku1*‡
Mikael Niku1*‡- 1Department of Veterinary Biosciences, Faculty of Veterinary Medicine, University of Helsinki, Helsinki, Finland
- 2Department of Reproduction, Obstetrics and Herd Health, Faculty of Veterinary Medicine, Ghent University, Merelbeke, Belgium
- 3Central Laboratory, Faculty of Veterinary Medicine, University of Helsinki, Helsinki, Finland
The development of a healthy intestinal immune system requires early microbial exposure. However, it remains unclear whether microbial exposure already begins at the prenatal stage. Analysis of such low microbial biomass environments are challenging due to contamination issues. The aims of the current study were to assess the bacterial load and characterize the bacterial composition of the amniotic fluid and meconium of full-term calves, leading to a better knowledge of prenatal bacterial seeding of the fetal intestine. Amniotic fluid and rectal meconium samples were collected during and immediately after elective cesarean section, performed in 25 Belgian Blue cow-calf couples. The samples were analyzed by qPCR, bacterial culture using GAM agar and 16S rRNA gene amplicon sequencing. To minimize the effects of contaminants, we included multiple technical controls and stringently filtered the 16S rRNA gene sequencing data to exclude putative contaminant sequences. The meconium samples contained a significantly higher amount of bacterial DNA than the negative controls and 5 of 24 samples contained culturable bacteria. In the amniotic fluid, the amount of bacterial DNA was not significantly different from the negative controls and all samples were culture negative. Bacterial sequences were identified in both sample types and were primarily of phyla Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, with some individual variation. We conclude that most calves encounter in utero maternal-fetal transmission of bacterial DNA, but the amount of bacterial DNA is low and viable bacteria are rare.
Introduction
Characterizing the very first intestinal bacteria is essential for a better understanding of the co-development of the newborn and its intestinal microbiome. Host-microbiome interactions enable early life education and maturation of the immune system during a window of opportunity. The use of prenatal and intrapartum antibiotics may disturb this process (Dierikx et al., 2020). However, the timing of the microbial colonization of the mammalian gut is still unclear (Perez-Munoz et al., 2017; Korpela and de Vos, 2018; Liu et al., 2019; Guzman et al., 2020; Blaser et al., 2021; Silverstein and Mysorekar, 2021). Vertical as well as environmental transmission of bacteria occur during and after birth and seed the neonatal gastrointestinal (GI) tract (Funkhouser and Bordenstein, 2013; Gomez de Agüero et al., 2016; Korpela and de Vos, 2018). Transmission of an orally inoculated Enterococcus faecium strain in pregnant mice to the meconium of their fetuses has been described (Jimenez et al., 2008).
Recent studies in humans, mice and cattle have reported the identification of microbial DNA or culturable bacteria in the mammalian fetal environment (Aagaard et al., 2014; Seferovic et al., 2019; Al Alam et al., 2020; Guzman et al., 2020; Rackaityte et al., 2020). Nevertheless, others interpret such observations as intrauterine infections or contamination (Lauder et al., 2016; Eisenhofer et al., 2018; de Goffau et al., 2019; Hockney et al., 2020; Olomu et al., 2020; Theis et al., 2020a,b). The newborn’s GI tract is a low microbial biomass environment, implying multiple challenges for performing reliable microbiome analyses (Glassing et al., 2016; Eisenhofer et al., 2018; Stinson et al., 2018). Besides the technical limitations inherent to DNA sequencing, there are practical and ethical concerns while collecting samples in humans. First-pass meconium has been used as a proxy to assess the fetal intestinal microbiome, although samples are often collected hours after birth and even breastfeeding, suggesting potential postnatal effects on the microbial composition of the meconium (Hansen et al., 2015; Collado et al., 2016; Liu et al., 2019; Stinson et al., 2019). Mammalian animal models can overcome several of these ethical and practical difficulties, providing insights in the fetal gastrointestinal microbiome.
In the current study, the microbial composition of the full-term fetal gut and the corresponding amniotic fluid was assessed in Belgian Blue cow-calf couples. In this double-muscled beef breed, cesarean sections (C-sections) are performed on a routine basis during the very early stages of parturition, while fetal membranes are still intact, rendering this breed a highly suitable model for full-term gestation microbiome studies.
Besides the potential role of the cow model for research, there is also a significant interest in the composition and the development of the calf’s intestinal microbiome. Gut health and growth performance during the first weeks of life are main drivers for cost effective livestock rearing (Thornton, 2010; Lorenz et al., 2011). Few studies have been conducted on the intestinal microbiome in vaginally born neonatal calves. These have reported low numbers of bacteria, mostly belonging to the phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes (Alipour et al., 2018; Klein-Jöbstl et al., 2019; Guzman et al., 2020). Shared microbiota were found between calf meconium and the maternal vaginal vestibulum (Alipour et al., 2018; Yeoman et al., 2018; Klein-Jöbstl et al., 2019). To the best of our knowledge, no microbiome studies have yet been performed on calves born by C-section, and no studies are available to assess the association between the microbial DNA signatures in meconium and the corresponding amniotic fluid.
Our aims were to assess the bacterial load and characterize the bacterial composition of the amniotic fluid and meconium of full-term neonatal calves. To this end, samples were collected during elective C-section, and analyzed by qPCR, bacterial culture under carefully controlled and validated conditions, and 16S rRNA gene amplicon sequencing. To minimize reagent and environmental bacterial DNA contaminants, we included multiple technical controls and stringently filtered the 16S rRNA gene sequencing data to exclude putative contaminant sequences.
Materials and Methods
All experimental procedures were approved by the institutional ethics and animal welfare committee of the Faculty of Veterinary Medicine (EC2018/002 - Ghent University, Belgium). The cows’ owners were informed about the study and gave their written consent.
Study Design
The sampling of this study was performed at the teaching hospital of the Department of Reproduction, Obstetrics, and Herd Health of the Faculty of Veterinary Medicine in Ghent (Belgium), where pregnant Belgian Blue beef cows of different herds (parity between 1 and 5) were housed for an elective C-section.
In total 25 Belgian Blue cows and their calves (7 males and 18 females) were sampled from November 2017 until March 2019. The cows were housed in tie-stalls at the facility for 9.5 d ± 5.8 (mean ± standard deviation) prior to C-section and had ad libitum access to hay and water. During this period, rectal temperature was measured twice daily, and calving indicators such as udder distension, teat filling, pelvic ligament relaxation, vaginal discharge, vulvar edema, and behavioral changes were monitored every 2 h by graduate veterinary students.
Prior to elective C-section, in cows that exhibited a drop in temperature, cervical dilation was assessed by manual palpation. The vulvar region was cleaned with iodine soap and water. A gloved hand was inserted vaginally and the opening of the portio vaginalis cervicis was estimated. For the present study, elective C-section was performed when the cow had a minimal cervical dilation of 8 cm, with no rupture of the fetal membranes prior to surgery. All cows were healthy according to their vital parameters (heart rate, temperature, respiratory rate) and there was no clinical evidence of intrauterine infection or contamination.
Sampling
Prior to surgery, the cows were restrained in a standing position in a surgery chute specifically designed for cattle. C-section procedure was done as described by Kolkman et al. (2007). Briefly, the surgical area (left flank) was washed and disinfected, an abdominal incision was made, and part of the uterus was exteriorized for uterotomy. The allantoic sac was opened up to expose the intact amniotic sac. Amniotic fluid was aspirated through the amniotic membrane, using a sterile 16 G needle (Agani, Terumo Europe, Hamburg, Germany) and sterile 20 ml syringe (B. Braun, Melsungen, Germany). Within 1 h after sampling, the retrieved volume was aliquoted, under a laminar-flow hood, into 2 sterile 15 ml tubes (188271, Cellstar, Greiner bio-one, Frickenhausen, Germany). In the first tube, 6 ml of amniotic fluid was dissolved in 3 ml of glycerol (≥99%, G2025, Sigma-Aldrich (Merck), Overijse, Belgium) and subsequently stored at −80°C to be used in culture experiments. In the second tube, 12 ml of amniotic fluid was stored at −80°C to be used for bacterial DNA extraction.
Meconium samples were acquired directly from the calves’ rectum, immediately after birth (no more than 30 min). Until the moment of sampling, calves laid on a clean concrete floor, with access to neither the dam, nor colostrum. The perineum of the calf was dried with a clean paper towel and disinfected with 70% ethanol. A sterile double-guarded equine uterine culture swab (Har-vet, 17705, Spring Valley, United States; or Minitube, 17214/2950, Tiefenbach, Germany) was gently introduced in the rectum and the swab was exposed. Samples were taken in duplicate, one stored immediately at −80°C with no additives, and the other stored at −80°C in a sterile 2 ml cryovial containing 1 ml of a 30% glycerol solution, prepared by diluting glycerol (≥99%, G2025, Sigma-Aldrich (Merck), Overijse, Belgium), in ultra-pure, nuclease-free water (W4502, Sigma-Aldrich (Merck), Overijse, Belgium) to a final 30% concentration.
Negative field controls were processed in the surgery room, using the same sampling procedures and disposables. In total, 16 empty, sterile double-guarded equine uterine culture swabs (10 Har-vet swabs and 6 Minitube swabs) were included for the meconium sampling, 5 of the Har-vet swabs stored in 1 ml of a 30% glycerol stock solution, the others with no additives. Additionally, 12 negative field controls were included for the amniotic fluid sampling, aspirating ultra-pure, nuclease-free water (W4502, Sigma-Aldrich (Merck), Overijse, Belgium) instead of amniotic fluid. For 8 of them, the ultra-pure, nuclease-free water was stored with no additives, and for 4 of them, 6 ml ultra-pure, nuclease-free water was dissolved in 3 ml of glycerol.
All samples were shipped on dry ice to the laboratory of the Department of Veterinary Biosciences of the Faculty of Veterinary Medicine in Helsinki (Finland) for further processing.
Culture
Validation of Culture Media
We aimed to define a single bacterial culture medium, capable of sustaining a majority of the calf intestinal core microbiota. Consequently, this single medium could be used for culturing the meconium and amniotic fluid samples, avoiding further splitting or dilution of the low biomass samples. We tested GAM “Nissui” medium (Gifu Anaerobic Medium Agar, Code 05420, HyServe, Germany) mostly used for anaerobic bacteria, YCFA medium (Yeast extract, Casitone and Fatty Acid) containing volatile fatty acids typically supporting the growth of several gut bacteria, LB medium (Lysogeny Broth, BD) as a general medium and BB medium (Trypticase Soy Agar supplemented with Bovine Blood 211043, Tammer BioLab Oy, Tampere, Finland) supporting fastidious bacteria by blood enrichment. Each medium was tested for the ability to sustain calf intestinal core microbiota, by plating each type anaerobically with feces of a healthy 7 days old calf (Lopez-Siles et al., 2012; Alipour et al., 2018). After 7 days of growth, mixed cultures were extracted from the plates, 16S rRNA gene amplicon sequenced, and compared to the core microbiota previously observed in fecal samples of young calves (Alipour et al., 2018).
Sample Culturing
Bacterial culture on GAM-agar plates was performed for 24 meconium samples (5 corresponding negative field controls) and 24 amnion samples (4 negative field controls). The frozen samples were first transferred to an anaerobic workstation (Ruskinn Concept Plus), mixed thoroughly and plated, using an aseptic technique, at +37°C. After this, the samples were transferred to a laminar flow cabinet and plated in aerobic conditions at +37°C. The culture plates were checked for growth daily for 14 days. All visible bacterial colonies were subcultured until pure isolates were obtained. Fresh cultures were then identified using MALDI-TOF (Bruker Microflex LT) at the Central laboratory of the Faculty of Veterinary Medicine (Helsinki, Finland).
Samples were prepared using MALDI Biotyper MSP Identification Standard Method v 1.1. Mass spectra were analyzed in a mass/charge range from 2,000 to 20,000 Da with MBT Compass v4.1 on flexControl v3.4 (Bruker Daltonik GmbH) using BDAL-7311 as the reference library. The Bruker Bacterial Test Standard (RUO) (Bruker Daltonik GmbH) was used for instrument calibration. If the identification confidence score was < 2.00, further identification was done with 16S rRNA gene amplicon Sanger sequencing at the Institute of Biotechnology (University of Helsinki, Finland).
DNA Extraction
DNA from the meconium samples (N = 25) and corresponding negative field controls (N = 11) were extracted using ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA, United States) according to the manufacturer’s instructions, with minor modifications to the protocol as described previously (Alipour et al., 2018; Husso et al., 2020). ZymoBIOMICSTM Microbial Community Standard and an in-house fecal standard were processed with the meconium samples in every batch.
For the amniotic fluid samples (N = 23) and the corresponding negative field controls (N = 8), 2 ml of each sample was first centrifuged in a microcentrifuge at 16,100 × g 10 min in +4°C. Most of the supernatant was removed and 750 μl of ZymoBIOMICS Lysis Solution and 19 μl of proteinase K (D3001-2-20/D3001-2-5, Zymo Research) were added to the remaining 200 μl of the samples and incubated for 30 min at +55°C. After these steps, the same protocol as applied for the meconium samples was followed.
All manipulations of the tubes during the process were performed in a laminar flow cabinet, and the workplace, instruments and pipettes were cleaned routinely with 10% bleach. Certified DNA, RNase, DNase and PCR inhibitor free tubes (STARLAB International, Germany) and Nuclease-free Water (Ambion, Thermo Fisher Scientific, United States) were used for DNA extraction and downstream analyses. All extracted DNA was stored at −80°C.
Quantitative PCR
The bacterial 16S rRNA gene copy numbers in the meconium, amnion and negative field control samples were determined using quantitative PCR. The analyses were performed as described previously (Alipour et al., 2018; Husso et al., 2020), with the exception that the PCR master mix was first treated using a dsDNAse based decontamination kit (Enzo Life Sciences, Farmingdale, New York). The amplification was performed using the Bio-Rad CFX96 instrument (Bio-Rad, Hercules, California) and universal eubacteria probe and primers (Nadkarni et al., 2002). A standard series, negative field controls and no-template controls were included in every run. The data were analyzed using the Bio-Rad CFX Maestro software.
Library Preparation and 16S rRNA Gene Amplicon Sequencing
The V3-V4 region of the 16S rRNA gene amplicons was sequenced using the Illumina MiSeq platform in the DNA core facility of the University of Helsinki, as described previously (Alipour et al., 2018; Husso et al., 2020). In total, 23 amniotic fluid samples and 23 meconium samples of the same cow-calf couple were sequenced, together with the corresponding field controls, no-template controls, a ZymoBIOMICS Microbial Community Standard (Zymo Research, United States) and an in-house adult cow fecal standard. The observed composition and abundances for the commercial standard matched the expected composition provided by the manufacturer (data not shown).
The numbers of pre-amplification PCR cycles were optimized based on the amounts of bacterial DNA template in each sample type. The ZymoBIOMICS Microbial Community Standard and the in-house adult cow fecal standard were pre-amplified with 12 cycles and all other sample types, including negative controls, with 21 cycles.
Bioinformatics
The detailed bioinformatics pipeline is described in Supplementary Materials. Briefly, the read quality was first inspected with FastQC and MultiQC (Andrews, 2010; Ewels et al., 2016). Leftover primes and spacers were then trimmed with Cutadapt v1.10 (Martin, 2011). A mapping file was created for QIIME2 and validated with Keemei (Rideout et al., 2016). The FASTQ-files were imported to QIIME2 v2019.4, where the DADA2 plugin was used to denoise and quality filter the reads, call ASVs and generate a feature table (Callahan et al., 2016; Bolyen et al., 2019). A naïve Bayes classifier was trained in QIIME2 against SILVA v132 99% database, extracted to only include the V3-V4 region and used to assign taxonomy to ASVs (Quast et al., 2013; Bokulich et al., 2018). Sequences derived from chloroplasts or mitochondria were removed and singletons were filtered out, leaving only bacteria with at least phylum-level identification.
Decontamination of the 16S rRNA Gene Amplicon Sequencing Data
The processed data was in silico filtered to remove amplicon sequence variants (ASVs) which represented probable contaminants (reagent contaminants and environmental bacterial DNA), as described previously (Husso et al., 2020).
Briefly, an ASV was removed if its prevalence in actual samples was ≤2× its prevalence in field controls (DNA extracted from empty sampling instruments exposed to the surgery room environment), and if its mean relative abundance in actual samples was ≤ 10 × its mean abundance in field controls (Supplementary Figure 1). The filtering was performed separately for meconium and amnion samples. If less than 500 reads remained after the decontamination, as was the case for six meconium samples, the sample was removed from further analyses. The original and remaining read counts are shown in Table 1.
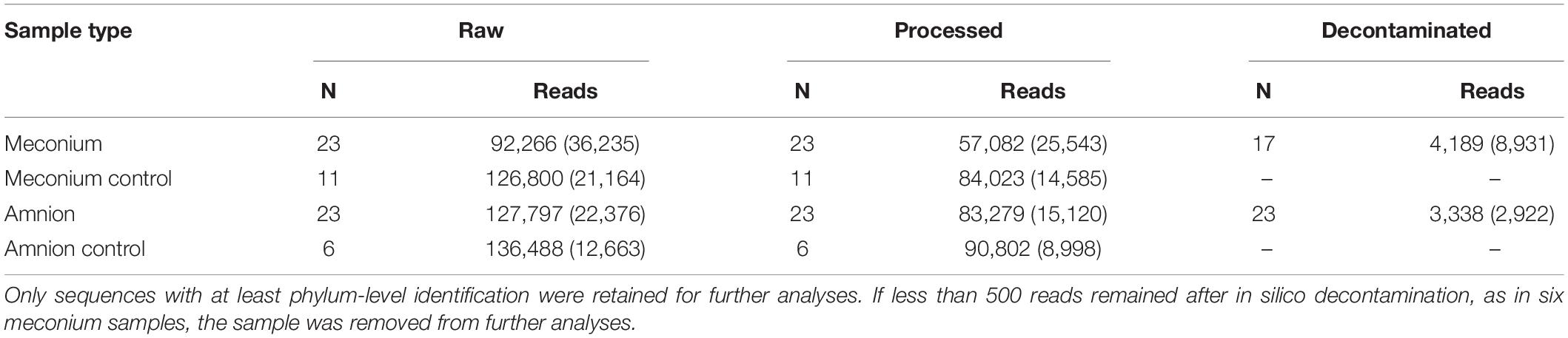
Table 1. Mean read counts (standard deviation) detected in meconium and amniotic fluid samples and their negative controls.
Statistics
The 16S rRNA gene qPCR results from samples and negative field controls were compared using two-tailed Mann-Whitney U test in IBM SPSS Statistics 25. Shannon diversity indices were calculated for genus-level data with the R package phyloseq without rarefaction (McMurdie and Holmes, 2013). The Kruskal-Wallis rank sum test was used to compare the bacterial community structure of both sample types in RStudio (R Studio Team, 2020). The PCoA figures were plotted using ASV and genus-level data and Bray-Curtis distances with the R package phyloseq (McMurdie and Holmes, 2013). Permutational analysis of variance (PERMANOVA) and multivariate homogeneity of group dispersions (betadisper) were calculated using the R package vegan, using 9,999 permutations (Oksanen et al., 2019). DESeq2 was used to explore differentially abundant ASVs by calculating differential expression between sample groups (Love et al., 2014). The LEfSe (Linear discriminant analysis Effect Size) web application was used to identify the taxons most likely explaining the differences between the sample types (Segata et al., 2011). An ecologically organized heatmap of the top 40 most abundant ASVs was created with the R package phyloseq (Rajaram and Oono, 2010; McMurdie and Holmes, 2013). Spearman correlations and their Bonferroni corrected p-values were calculated using R-package Hmisc 4.3.0 (Harrel, 2019), using the absolute counts of ASVs and genera that were present >100 times in all of the samples. Differences were considered significant at P < 0.05.
Results
Quantification of Bacterial 16S rRNA Genes in Amniotic Fluid and Meconium Samples by qPCR
The 16S rRNA gene copy numbers in amniotic fluid and meconium samples were assessed by qPCR (Figure 1). In the amniotic fluid samples, there was no significant difference (P = 0.176) between the 16S rRNA gene copy numbers of the samples (N = 23; mean = 3,170 copies per 100 μl of centrifuged amniotic fluid; SD = 2,580) and the negative field controls (N = 8; mean = 1,610; SD = 790). In the meconium samples, the 16S rRNA gene copy number (N = 25; mean = 4,350 copies per sampling swab; SD = 6,410) was significantly higher (P = 0.016) than in the negative field controls (N = 11; mean = 980; SD = 850).
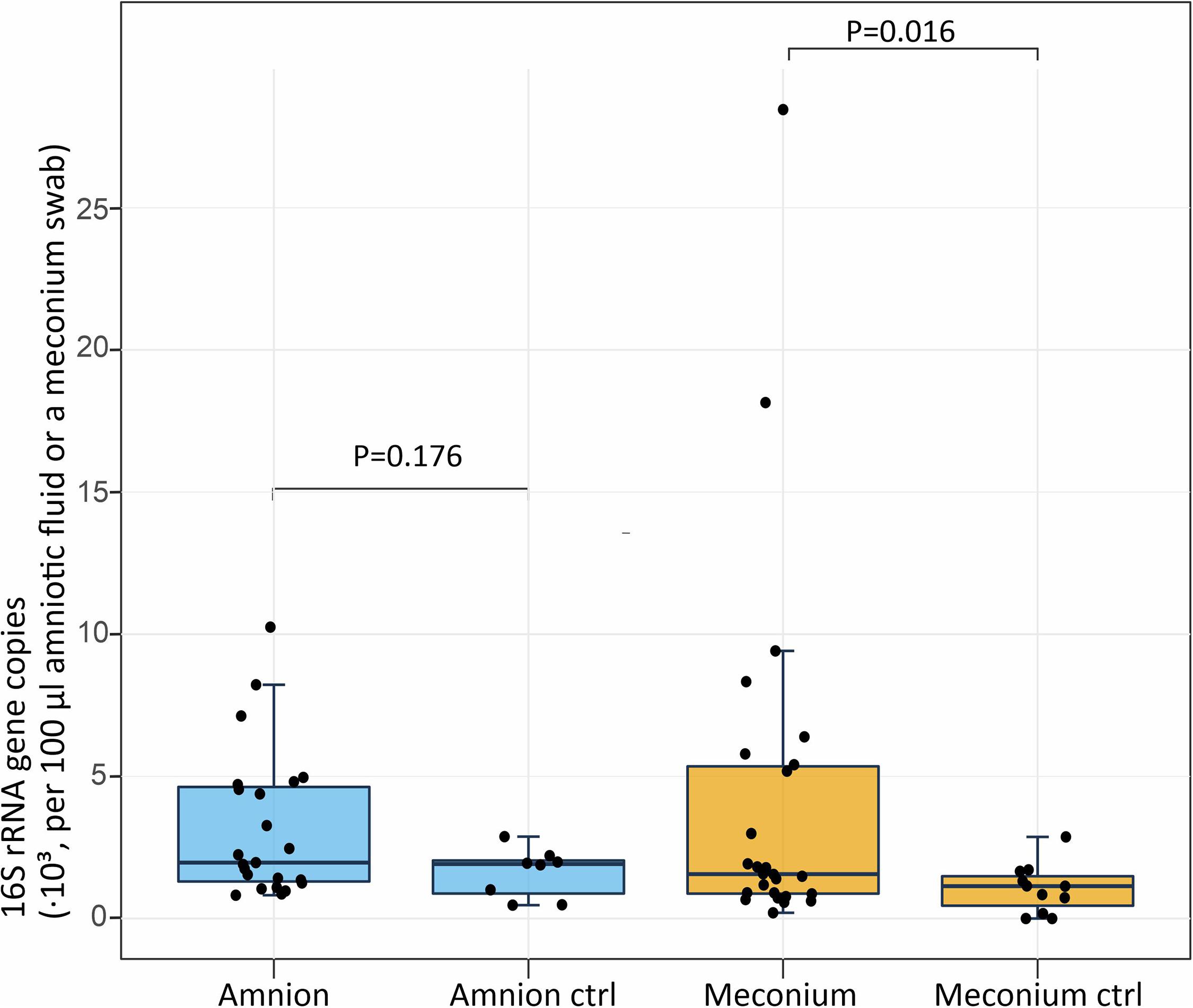
Figure 1. Absolute 16S rRNA gene copy numbers per 100 μl of centrifuged amniotic fluid (n = 23, negative controls n = 8) and per meconium sampling swab (n = 25, negative controls n = 11). The midline of the box is the median, with the upper and lower limits of the box being the third and first quartile. The whiskers extend up to 1.5 times the interquartile range from the box to the furthest data point within that distance.
Microbial Composition of Amniotic Fluid and Meconium Samples
We analyzed the microbial DNA profiles in the fetal samples using 16S rRNA gene amplicon sequencing. The microbial signature of both sample types consisted primarily of Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria phyla, with individual variation (Figure 2). A heatmap analysis of the 40 most abundant ASVs shows the difference between the sample types in more detail (Figure 3). In both sample types, the inter-individual variation increased at lower taxonomic levels (Supplementary Table 1 and Figure 3). The most abundant bacterial genera in meconium were Delftia, Staphylococcus, and Clostridium sensu stricto 1, while the most prevalent genera were Delftia, Acinetobacter, unclassified Burkholderiaceae, Staphylococcus, and Corynebacterium 1 (Supplementary Table 1). In the amniotic fluid samples, Staphylococcus, Streptococcus, Delftia, Sphingomonas, and Enterococcus were the most abundant genera, and Delftia, Streptococcus, Staphylococcus, Sphingomonas, and Acinetobacter were the most prevalent genera (Supplementary Table 1).
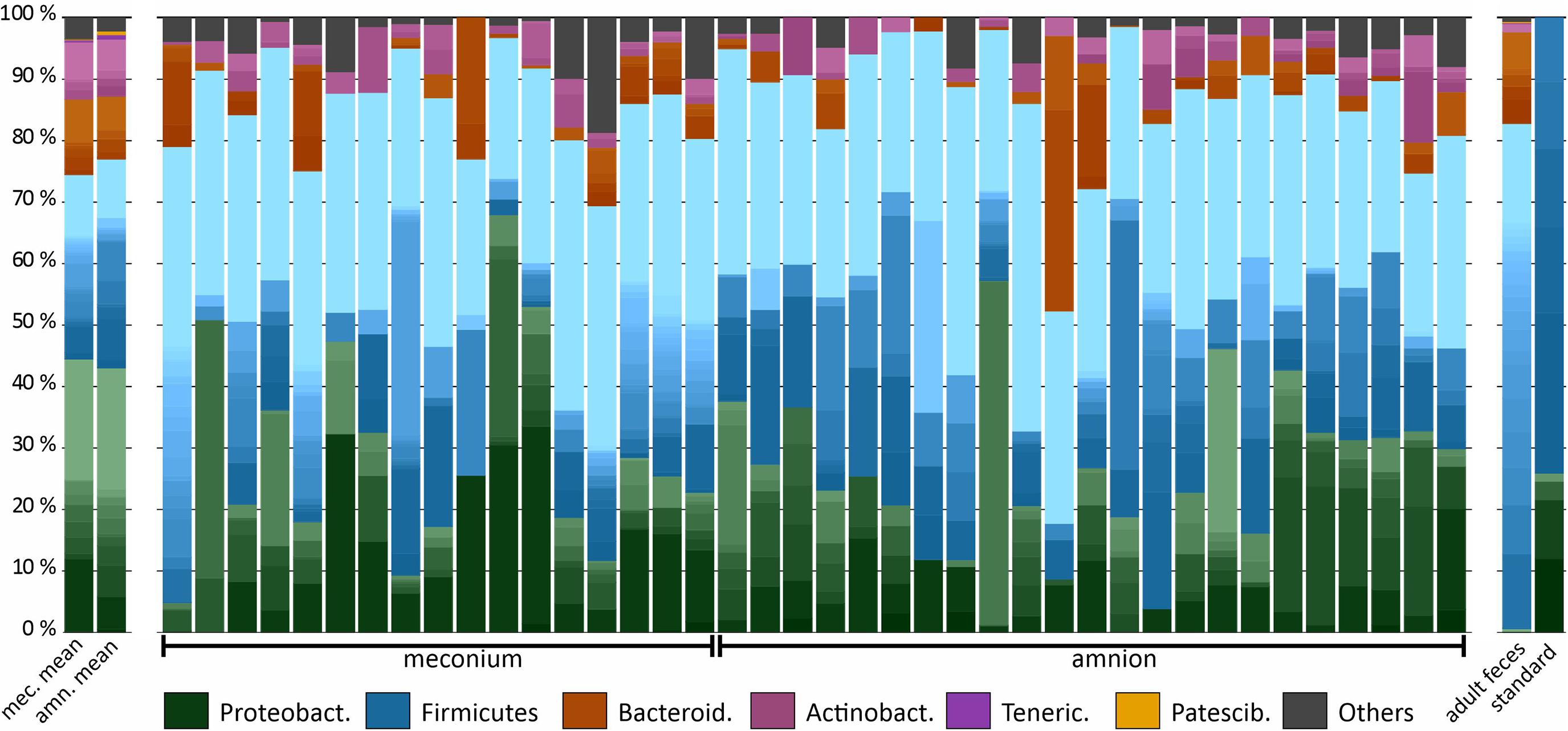
Figure 2. Microbiota composition in bovine meconium and amniotic fluid samples collected during elective C-section, adult feces, and a commercial community composition standard. The main colors indicate the bacterial phyla. Within phyla, the shades indicate bacterial genera. The lightest shade of each phylum shows the combined abundance of the least abundant genera (with a maximum of < 0.5% of total).
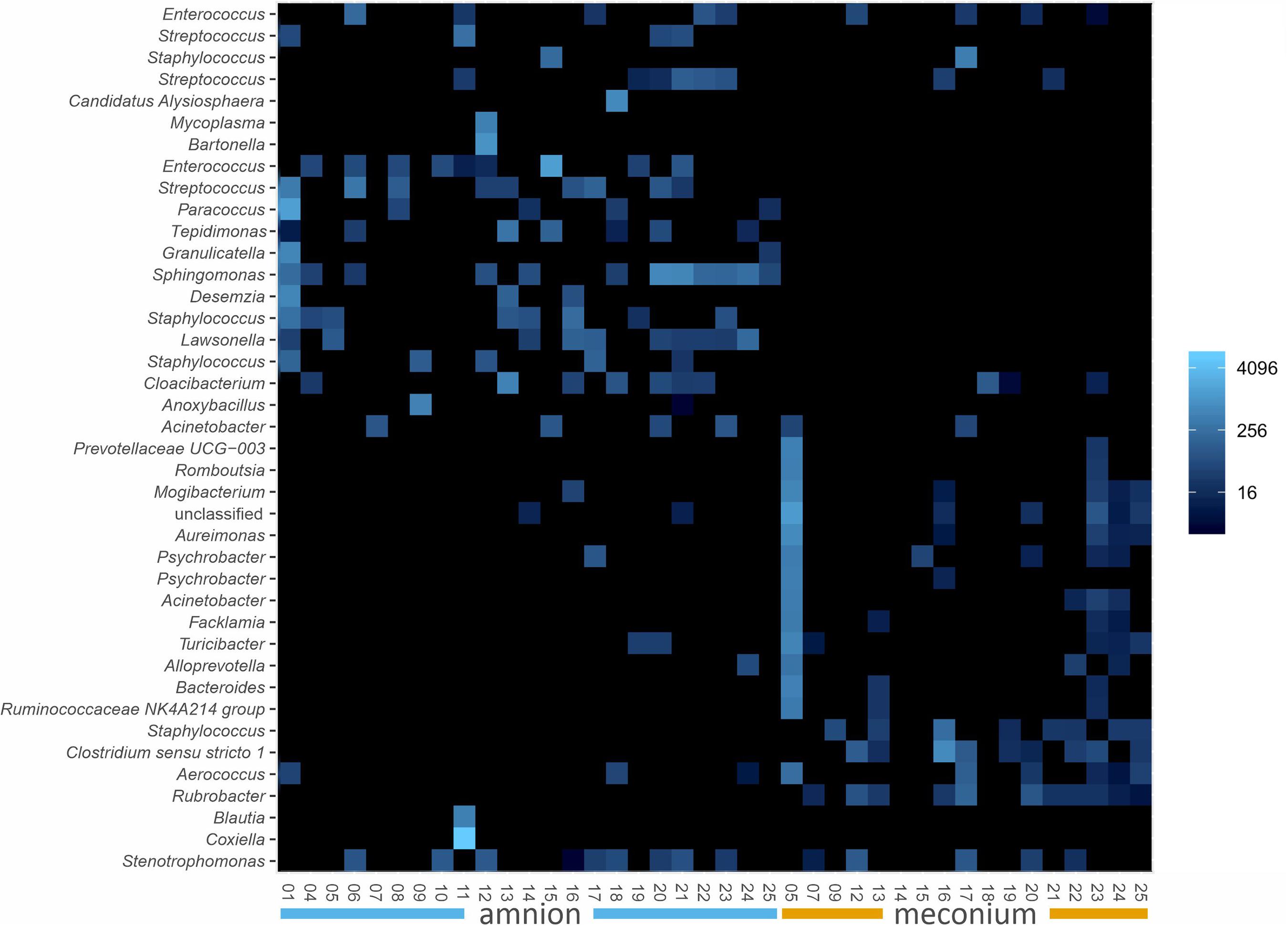
Figure 3. Ecologically organized heatmap (NMDS, Bray) of the 40 most abundant ASVs in bovine meconium and amniotic fluid samples collected during elective C-section, sorted by sample type. The taxon names are presented as genus level identifications. Color scale indicates relative abundance and is a log transformation with base 4.
The alpha diversity (Shannon index) at the genus level was not significantly different between the meconium and amniotic fluid samples (Figure 4, P = 0.889).
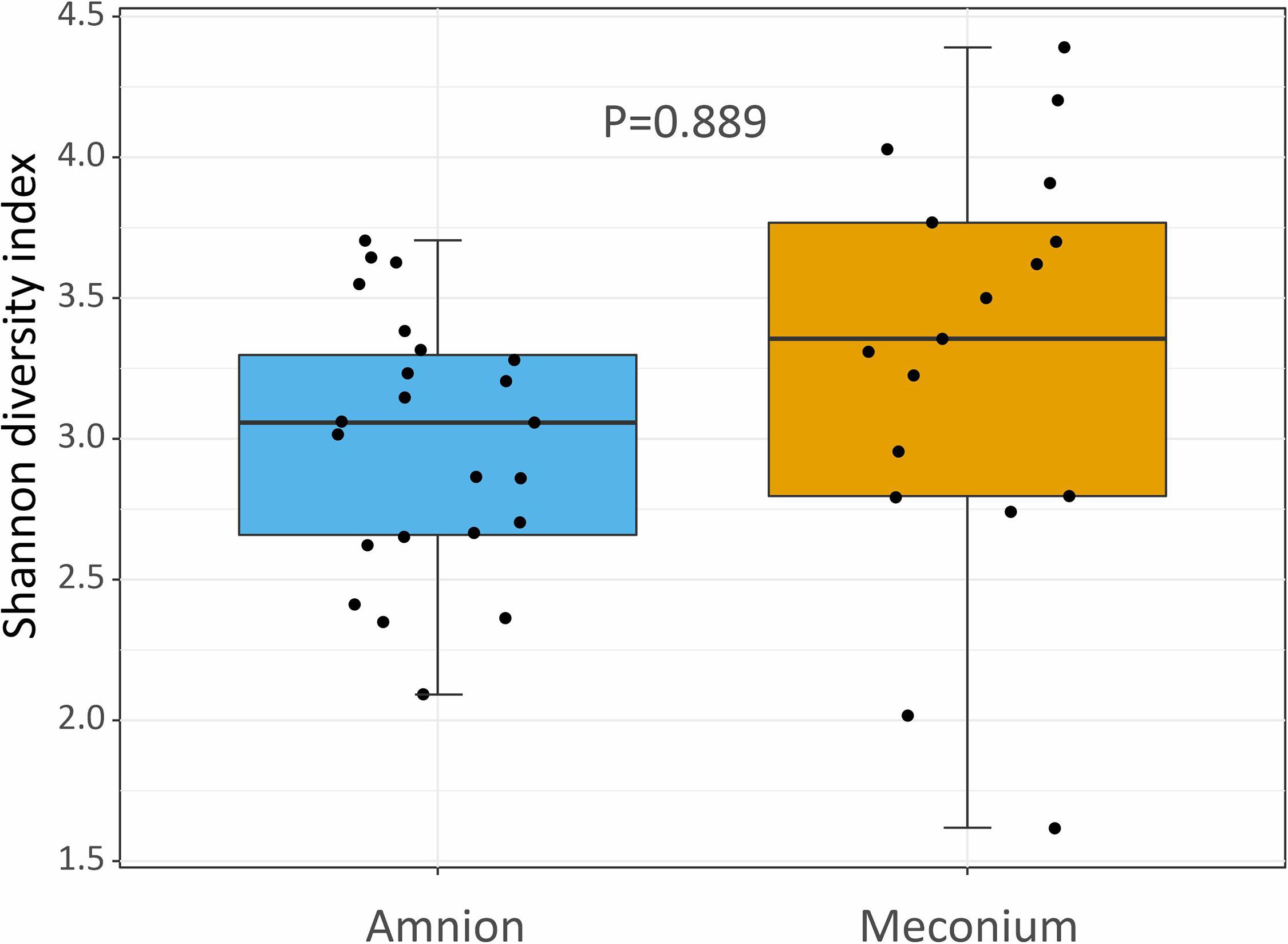
Figure 4. Shannon diversity index showing the bacterial community structure (at genus level) of amniotic fluid and meconium samples, collected during elective C-section in Belgian Blue cow-calf pairs. Boxplot as in Figure 1.
Comparison of Amniotic Fluid and Meconium Microbial DNA Signature
The microbial DNA signature was significantly different for amniotic fluid and meconium samples at the ASV level (PERMANOVA, P < 0.001, R2 = 0.049, Figure 5A), and genus level (PERMANOVA, P < 0.001, R2 = 0.062, Figure 5B).
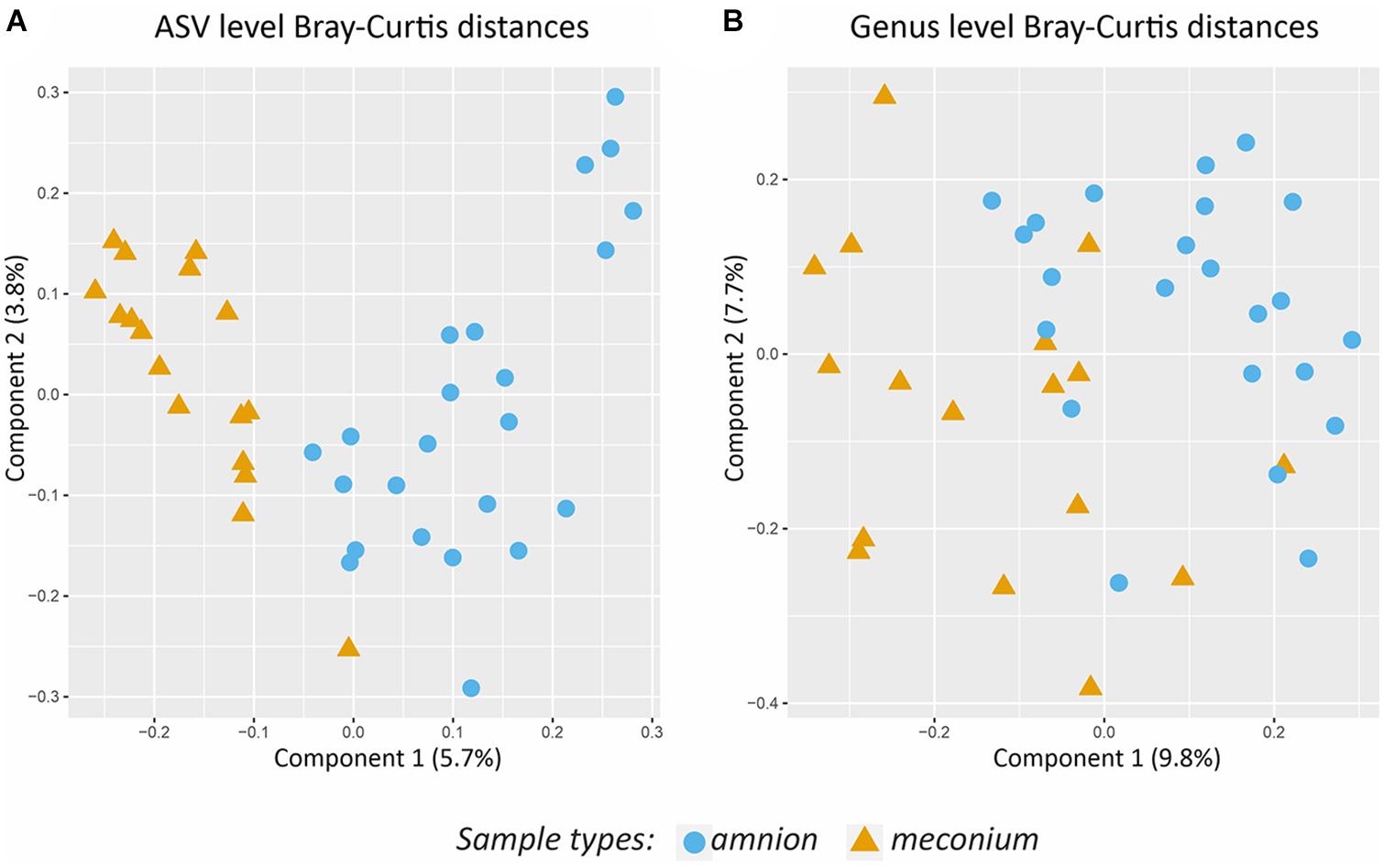
Figure 5. Comparison of the bacterial community structure of amniotic fluid and meconium. (A) PCoA on Bray-Curtis distances based on ASV level data. (B) PCoA on Bray- Curtis distances based on genus level data. Colors and shapes indicate the sample types.
According to the DESeq2 analysis, three ASVs were more abundant in meconium than in amniotic fluid: one Staphylococcus ASV (log2 Fold Change = 25.084, Padj < 0.001), one Rubrobacter ASV (log2 Fold Change = 8.337, Padj < 0.001), and one Clostridium ASV (log2 Fold Change = 8.055, Padj = 0.004). In amniotic fluid, one Sphingomonas ASV (log2 Fold Change = −8.420, Padj < 0.001) and one Staphylococcus ASV (log2 Fold Change = −23.438, Padj < 0.001) were more abundant than in meconium.
By LefSe (Linear discriminant analysis effect size) analysis including all taxonomic levels, meconium samples had a greater relative abundance of Clostridiales and Rubrobacter (LDA score (log10) > 4.0) than the amniotic fluid, but a lower relative abundance of Bacillales and Corynebacteriales (LDA score (log10) < −4.0).
Correlations Between Meconium and Amniotic Fluid From the Same Animal
To assess whether the meconium and amniotic fluid microbial DNA profiles may have common origins, we calculated Spearman rank correlations between meconium and amniotic fluid 16S rRNA gene sequencing data, using the absolute counts of ASVs and genera that were present > 100 times in all of the samples. Average correlations (ρ) at ASV (N = 17, ρavg = −0.0201, Pavg < 0.05) and genus level (N = 17, ρavg = 0.1685, Pavg < 0.05) were very weak and biologically not significant. All p-values were Bonferroni corrected for multiple testing.
Bacterial Cultures
Bacterial culture was performed to assess the presence of viable bacteria in the fetal samples. Based on our testing of multiple media, the GAM (Gifu Anaerobic Medium) agar sustained the largest number of bacterial core genera found in calf feces (Alipour et al., 2018), including some additional unique genera not found on any other plate type. Thus, we selected this medium for these experiments.
All amniotic samples and their negative field controls were culture negative, both in anaerobic and aerobic conditions. Five out of 24 meconium samples and 1 out of 5 negative field controls were culture positive on GAM agar. The results for each sample are presented in detail in Table 2. Both gram-negative and gram-positive strains were isolated from the samples.
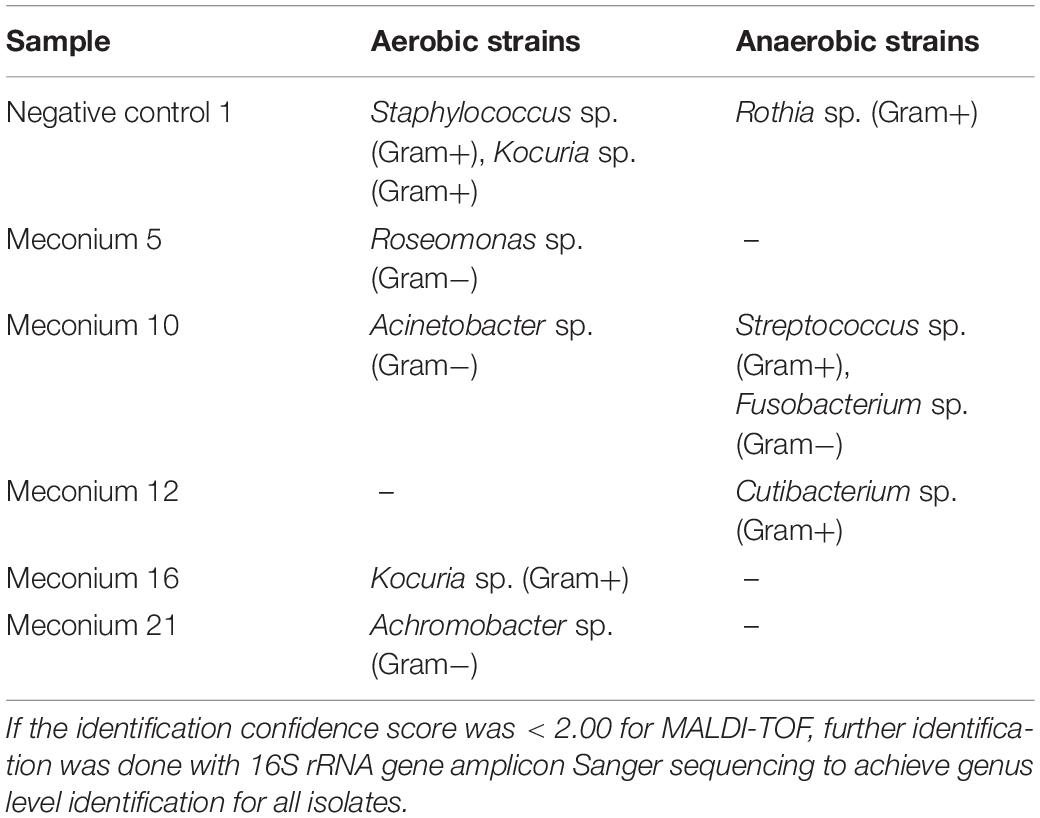
Table 2. Culture positive meconium samples and identified bacteria.
The Sanger sequence from the Roseomonas sp. isolate acquired from a meconium sample was a 100% identical match to the 16S rRNA gene amplicon sequencing data from the same sample. In addition, the Sanger sequences of Staphylococcus sp., Kocuria sp., Roseomonas sp., and Acinetobacter sp. isolates matched the 16S rRNA gene amplicon sequencing data when compared to all meconium samples. Three bacterial genera could be identified in the negative controls: Rothia sp., Staphylococcus sp., and Kocuria sp. Of these, only Kocuria sp. was also identified from the actual meconium samples.
Discussion
Vertical transmission of microbiota in utero remains controversial (Korpela and de Vos, 2018; Liu et al., 2019; Blaser et al., 2021; Silverstein and Mysorekar, 2021). In this study, we used a carefully controlled bovine elective C-section model for assessing the bacterial load and microbiota composition in full-term fetal gut and amniotic fluid.
We first examined the presence of microbial DNA in meconium and amniotic fluid by 16S rRNA gene qPCR and amplicon sequencing. Since contamination is a major challenge in the analysis of low bacterial biomass samples, we processed several types of negative controls alongside our biological samples, treated the qPCR mastermix with dsDNAse, and applied rigorous in silico filtering of potential contaminants (Glassing et al., 2016; Eisenhofer et al., 2018).
We observed a small but significant amount of bacterial DNA in the meconium samples by 16S qPCR, suggesting prenatal transmission of bacterial DNA to the fetal intestine. This is consistent with previous studies in cattle, in which vaginally born calves were sampled (Mayer et al., 2012; Alipour et al., 2018; Yeoman et al., 2018; Klein-Jöbstl et al., 2019). A profile dominated by the phyla Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria was identified in the meconium, in agreement with previous studies in newborn animals or slaughterhouse fetuses (Alipour et al., 2018; Yeoman et al., 2018; Klein-Jöbstl et al., 2019; Guzman et al., 2020; Husso et al., 2020). In contrast, Malmuthuge and Griebel (2018) concluded that fetal ovine intestinal tissue collected in C-sections were devoid of any bacterial DNA. Their PCR and agarose gel protocols were likely not sufficiently sensitive to detect small amounts of specific template as all the amplifications appeared negative, although probe-based qPCR is generally able to detect contaminating microbial DNA even in high-quality molecular biology reagents. Such technical differences may explain the different conclusions from 16S rRNA gene amplicon sequencing. Malmuthuge and Griebel (2018) obtained an extremely small number of reads also from their positive controls and compared the samples and controls only at phylum level.
In amniotic fluid, the amount of bacterial DNA was insignificant. However, we observed a 16S rRNA gene sequence profile distinguishable from the negative controls, suggesting the presence of microbial DNA at a very low abundance. Previous studies on amniotic fluid microbiota have yielded conflicting results and the study designs are diverse further complicating their comparisons. The amniotic fluid sampled by Collado et al. (2016) in women during pre-labor C-section was found to contain a distinct, low diversity, low biomass microbiome, predominated by Proteobacteria (Enterobacteriaceae). Malmuthuge and Griebel (2018) did not observe bacteria in their ovine amniotic fluid samples, within technical limits described above. Moore et al. (2017) reported a microbiome in bovine amniotic fluid by 16S rRNA gene amplicon sequencing, but did not utilize sufficient negative controls to accurately study low-abundance microbiota. Guzman et al. (2020) observed bacterial DNA in bovine amniotic fluid by qPCR and sequencing. The microbial DNA signature was similar to the one in our study at the phylum level but differed at family and genus levels.
We observed 16s rRNA genes of several intestine associated bacterial taxa in the meconium and/or amniotic fluid samples, including Bacteroides, Clostridium, Enterococcus, Lachnospiraceae, Christensenellaceae, and Ruminococcaceae. These most likely originate from the dam. Some of the other commonly mucosa-associated genera, such as Lactobacillus, Streptococcus, Staphylococcus, and Corynebacterium are also prevalent in human meconium (Chu et al., 2017). Sphingomonas, Acinetobacter, Pseudomonas, and Stenotrophomonas were also previously described in neonatal calves (Klein-Jöbstl et al., 2019). Some of the prevalent genera found in our study represent more ubiquitous taxons which can be found in both animal and environmental sources. Of these, Delftia as well as Burkholderiaceae are commonly observed reagent contaminants and were highly abundant in the raw, non-decontaminated data, suggesting that they may represent persistent contamination in the reagents (Salter et al., 2014).
The microbial composition did not correlate between meconium and amniotic fluid apart from the apparent similarity at phylum level. The genera Staphylococcus, Rubrobacter, and Clostridium were relatively more abundant in meconium samples than in amniotic fluid samples. This is in contrast with some earlier studies in humans (Collado et al., 2016; He et al., 2020), but confirms the observations by Guzman et al. (2020) who also observed different microbial communities between the bovine meconium and amniotic fluid. As the fetus swallows the amniotic fluid, which is then concentrated and retained in the intestine as meconium, together with epithelial cells and intestinal secretions, it is unexpected that the two would harbor completely different bacteria. However, the amniotic fluid microbial signature may fluctuate dynamically over time, while meconium represents a more stable collection of substances accumulated over the gestation period. Moreover, fetal excretion into the amniotic fluid is limited in cattle. Meconium is usually expelled only after birth, and fetal urine largely accumulates in the allantoic cavity, between chorion and amnion, rather than in the amniotic cavity (Bongso and Basrur, 1976). Maternal microbial components may also be translocated via the placental and umbilical blood vessels directly to the fetal internal organs. Microbial macromolecules and even live bacteria may be transported by leukocytes via these blood vessels (Perez et al., 2007). In cattle, the less permeable synepitheliochorial placenta may also restrict the translocation of bacteria and their components from the dam to the fetus, in comparison to humans.
Bacteria were successfully cultured from 5 out of 24 meconium samples, representing both gram-negative and gram-positive bacterial genera known to live in mammalian hosts. This is in general agreement with the carefully controlled study by Guzman et al. (2020), who obtained rare bacterial colonies from the rumen of bovine fetuses, but the genera were different from our data. Of the bacteria observed in our study, Fusobacteria species are typically associated with mucous membranes, especially in the oral cavity, while Cutibacterium and Kocuria species have most often been isolated from the skin (Hofstad, 1992; Savini et al., 2010; Scholz and Kilian, 2016). Various Acinetobacter and Streptococcus species have physiological functions in their mammalian hosts, but the genera also include pathogenic and/or environmental species (Krzyściak et al., 2013; Touchon et al., 2014). Species of genera Roseomonas and Achromobacter are known to act as opportunistic pathogens but they have also been isolated from a wide variety of environmental sources (Reverdy et al., 1984; Rihs et al., 1993). One of our negative controls yielded Kocuria colonies. This was the only taxon shared between the actual meconium samples and the negative field controls, suggesting low-level contamination from the sampling environment.
All amniotic fluid samples were culture negative. Taken together, live bacteria appear extremely rare in healthy fetal cattle, in contrast to the prevalent bacterial DNA. This is not likely explained by limitations in culture methods, as we validated the growth conditions for calf intestinal core microbiota. The small number of viable bacteria observed in our study may be partially explained by fetal bacteriostatic substances. Both the mucosal surface of the intestine as well as the amniotic fluid contain lactoferrin and salivary scavenger and agglutinin (SALSA), which are able to suppress bacterial growth and viability (Reichhardt and Meri, 2016; Lisowska-Myjak et al., 2019). In human meconium, SALSA amounts to 10% of all proteins, highlighting its potential role in antimicrobial defense (Reichhardt et al., 2014). Individual differences in amounts of viable bacteria retrieved from meconium were also described in a recent study on dogs. Interestingly, puppies born without detectable meconium microbiota were shown to have a slower growth rate than those in which meconium microbiota were detected (Pipan et al., 2020).
In conclusion, we detected small amounts of diverse bacterial DNA and rare culturable bacteria in the meconium of full-term calves delivered by elective cesarean section. In the amniotic fluid, bacteria were not observed by 16S qPCR or culturing, but a microbial DNA profile was distinguishable from negative controls by deep sequencing. Based on these observations, bacterial components are translocated to the bovine fetus in utero, but the prenatal acquisition of live bacteria is likely not physiologically significant.
Data Availability Statement
The dataset of the current study has been submitted to NCBI (https://www.ncbi.nlm.nih.gov) with accession PRJNA643145.
Ethics Statement
The animal study was reviewed and approved by the Institutional ethics and animal welfare committee of the Faculty of Veterinary Medicine (EC2018/002—Ghent University, Belgium). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
MN, LL, AI, and GO contributed to conception and design of the study. AH performed the bioinformatics, statistical analyses, and bacterial culture. AH and TG performed MALDI-TOF analyses. LL and JG organized the sampling and collected the samples. AH and LL wrote the manuscript. All authors contributed to manuscript preparation, read, and approved the submitted version.
Funding
The study was funded by the Finnish Veterinary Research Foundation. LL was funded by Special Research Fund (BOF) of Ghent University, under project number 01D26917.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the owners of the cows for allowing sampling of their animals, and all clinicians and graduate veterinary students for helping with the monitoring of the animals, performing the surgery and sampling. Additionally, we thank Kirsi Lahti for expert technical assistance in the laboratory.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.626421/full#supplementary-material
References
Aagaard, K., Ma, J., Antony, K. M., Ganu, R., Petrosino, J., and Versalovic, J. (2014). The placenta harbors a unique microbiome. Sci. Transl. Med. 6:237ra65. doi: 10.1126/scitranslmed.3008599
Al Alam, D., Danopoulos, S., Grubbs, B., Ali, N. A. B. M., MacAogain, M., Chotirmall, S. H., et al. (2020). Human fetal lungs harbor a microbiome signature. Am. J. Respir. Crit. Care Med. 201, 1002–1006. doi: 10.1164/rccm.201911-2127le
Alipour, M. J., Jalanka, J., Pessa-Morikawa, T., Kokkonen, T., Satokari, R., Hynönen, U., et al. (2018). The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 8:10437.
Andrews, S. (2010). FastQC: a Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed March 17, 2021).
Blaser, M. J., Devkota, S., McCoy, K. D., Relman, D. A., Yassour, M., and Young, V. B. (2021). Lessons learned from the prenatal microbiome controversy. Microbiome 9:8.
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chu, D. M., Ma, J., Prince, A. L., Antony, K. M., Seferovic, M. D., and Aagaard, K. M. (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326. doi: 10.1038/nm.4272
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E., and Salminen, S. (2016). Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6:23129.
de Goffau, M. C., Lager, S., Sovio, U., Gaccioli, F., Cook, E., Peacock, S. J., et al. (2019). Human placenta has no microbiome but can contain potential pathogens. Nature 572, 329–334. doi: 10.1038/s41586-019-1451-5
Dierikx, T. H., Visser, D. H., Benninga, M. A., van Kaam, A., de Boer, N. K. H., de Vries, R., et al. (2020). The influence of prenatal and intrapartum antibiotics on intestinal microbiota colonisation in infants: a systematic review. J. Infect. 81, 190–204. doi: 10.1016/j.jinf.2020.05.002
Eisenhofer, R., Minich, J. J., Marotz, C., Cooper, A., Knight, R., and Weyrich, L. S. (2018). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Funkhouser, L. J., and Bordenstein, S. R. (2013). Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 11:e1001631. doi: 10.1371/journal.pbio.1001631
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8:24.
Gomez de Agüero, M., Ganal-Vonarburg, S. C., Fuhrer, T., Rupp, S., Uchimura, Y., et al. (2016). The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302. doi: 10.1126/science.aad2571
Guzman, C. E., Wood, J. L., Egidi, E., White-Monsant, A. C., Semenec, L., Grommen, S. V. H., et al. (2020). A pioneer calf foetus microbiome. Sci. Rep. 10:17712.
Hansen, R., Scott, K. P., Khan, S., Martin, J. C., Berry, S. H., Stevenson, M., et al. (2015). First-pass meconium samples from healthy term vaginally-delivered neonates: an analysis of the microbiota. PLoS One 10:e0133320. doi: 10.1371/journal.pone.0133320
Harrel, F. (2019). Hmisc: Harrell Miscellaneous. Available online at: https://CRAN.R-project.org/package=Hmisc (accessed March 17, 2021).
He, Q., Kwok, L. Y., Xi, X., Zhong, Z., Ma, T., Xu, H., et al. (2020). The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes 12:1794266. doi: 10.1080/19490976.2020.1794266
Hockney, R., Waring, G. J., Taylor, G., Cummings, S. P., Robson, S. C., Orr, C. H., et al. (2020). Fetal membrane bacterial load is increased in histologically confirmed inflammatory chorioamnionitis: a retrospective cohort study. Placenta 91, 43–51. doi: 10.1016/j.placenta.2020.01.006
Hofstad, T. (1992). “The genus fusobacterium,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (Ney York, NY: Springer), 4114–4126. doi: 10.1007/978-1-4757-2191-1_75
Husso, A., Jalanka, J., Alipour, M. J., Huhti, P., Kareskoski, M., Pessa-Morikawa, T., et al. (2020). The composition of the perinatal intestinal microbiota in horse. Sci. Rep. 10:441.
Jimenez, E., Marin, M. L., Martin, R., Odriozola, J. M., Olivares, M., Xaus, J., et al. (2008). Is meconium from healthy newborns actually sterile? Res. Microbiol. 159, 187–193. doi: 10.1016/j.resmic.2007.12.007
Klein-Jöbstl, D., Quijada, N. M., Dzieciol, M., Feldbacher, B., Wagner, M., Drillich, M., et al. (2019). Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves’ gastrointestinal microbiota. PLoS One 14:e0220554. doi: 10.1371/journal.pone.0220554
Kolkman, I., De Vliegher, S., Hoflack, G., Van Aert, M., Laureyns, J., Lips, D., et al. (2007). Protocol of the caesarean section as performed in daily bovine practice in belgium. Reprod. Domest. Anim. 42, 583–589. doi: 10.1111/j.1439-0531.2006.00825.x
Korpela, K., and de Vos, W. M. (2018). Early life colonization of the human gut: microbes matter everywhere. Curr. Opin. Microbiol. 44, 70–78. doi: 10.1016/j.mib.2018.06.003
Krzyściak, W., Pluskwa, K. K., Jurczak, A., and Kościelniak, D. (2013). The pathogenicity of the streptococcus genus. Eur. J. Clin. Microbiol. Infect. Dis. 32, 1361–1376. doi: 10.1007/s10096-013-1914-9
Lauder, A. P., Roche, A. M., Sherrill-Mix, S., Bailey, A., Laughlin, A. L., Bittinger, K., et al. (2016). Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4:29.
Lisowska-Myjak, B., Skarżyñska, E., Wojdan, K., and Nasierowska-Guttmejer, A. (2019). Protein and peptide profiles in neonatal meconium. J. Obstet. Gynaecol. Res. 45, 556–564. doi: 10.1111/jog.13888
Liu, C.-J., Liang, X., Niu, Z.-Y., Jin, Q., Zeng, X.-Q., Wang, W.-X., et al. (2019). Is the delivery mode a critical factor for the microbial communities in the meconium? EBioMedicine 49, 354–363. doi: 10.1016/j.ebiom.2019.10.045
Lopez-Siles, M., Khan, T. M., Duncan, S. H., Harmsen, H. J., Garcia-Gil, L. J., and Flint, H. J. (2012). Cultured representatives of two major phylogroups of human colonic faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl. Env. Microbiol. 78, 420–428. doi: 10.1128/aem.06858-11
Lorenz, I., Fagan, J., and More, S. J. (2011). Calf health from birth to weaning. ii. management of diarrhoea in pre-weaned calves. Ir. Vet. J. 64:9.
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Malmuthuge, N., and Griebel, P. J. (2018). Fetal environment and fetal intestine are sterile during the third trimester of pregnancy. Vet. Immunol. Immunopathol. 204, 59–64. doi: 10.1016/j.vetimm.2018.09.005
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Mayer, M., Abenthum, A., Matthes, J. M., Kleeberger, D., Ege, M. J., Hölzel, C., et al. (2012). Development and genetic influence of the rectal bacterial flora of newborn calves. Vet. Microbiol. 161, 179–185. doi: 10.1016/j.vetmic.2012.07.023
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an r package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Moore, S. G., Ericsson, A. C., Poock, S. E., Melendez, P., and Lucy, M. C. (2017). Hot Topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy Sci. 100, 4953–4960. doi: 10.3168/jds.2017-12592
Nadkarni, M. A., Martin, F. E., Jacques, N. A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad-range (Universal) probe and primers set. Microbiology 148, 257–266. doi: 10.1099/00221287-148-1-257
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., and McGlinn, D. (2019). Vegan: Community Ecology Package. R package version 2.5-4. Available online at: https://CRAN.R-project.org/package=vegan (accessed March 17, 2021).
Olomu, I. N., Pena-Cortes, L. C., Long, R. A., Vyas, A., Krichevskiy, O., Luellwitz, R., et al. (2020). Elimination of “kitome” and “splashome” contamination results in lack of detection of a unique placental microbiome. BMC Microbiol. 20:157.
Perez, P. F., Doré, J., Leclerc, M., Levenez, F., Benyacoub, J., Serrant, P., et al. (2007). Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics 119, e724–e732.
Perez-Munoz, M. E., Arrieta, M. C., Ramer-Tait, A. E., and Walter, J. (2017). A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 5:48.
Pipan, M. Z., Kajdiè, L., Kalin, A., Plavec, T., and Zdovc, I. (2020). Do newborn puppies have their own microbiota at birth? influence of type of birth on newborn puppy microbiota. Theriogenology 152, 18–28. doi: 10.1016/j.theriogenology.2020.04.014
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596.
Rackaityte, E., Halkias, J., Fukui, E. M., Mendoza, V. F., Hayzelden, C., Crawford, E. D., et al. (2020). Viable bacterial colonization is highly limited in the human intestine in utero. Nat. Med. 26, 599–607. doi: 10.1038/s41591-020-0761-3
Rajaram, S., and Oono, Y. (2010). NeatMap-non-clustering heat map alternatives in R. BMC Bioinform. 11:45.
Reichhardt, M. P., and Meri, S. (2016). SALSA: a regulator of the early steps of complement activation on mucosal surfaces. Front. Immunol. 7:85.
Reichhardt, M. P., Jarva, H., de Been, M., Rodriguez, J. M., Quintana, E. J., Loimaranta, V., et al. (2014). The salivary scavenger and agglutinin in early life: diverse roles in amniotic fluid and in the infant intestine. J. Immunol. 193, 5240–5248. doi: 10.4049/jimmunol.1401631
Reverdy, M. E., Freney, J., Fleurette, J., Coulet, M., Surgot, M., Marmet, D., et al. (1984). Nosocomial colonization and infection by achromobacter xylosoxidans. J. Clin. Microbiol. 19, 140–143. doi: 10.1128/jcm.19.2.140-143.1984
Rideout, J. R., Chase, J. H., Bolyen, E., Ackermann, G., Gonzalez, A., Knight, R., et al. (2016). Keemei: cloud-based validation of tabular bioinformatics file formats in google sheets. Gigascience 5:27.
Rihs, J. D., Brenner, D. J., Weaver, R. E., Steigerwalt, A. G., Hollis, D. G., and Yu, V. L. (1993). Roseomonas, a new genus associated with bacteremia and other human infections. J. Clin. Microbiol. 31, 3275–3283. doi: 10.1128/jcm.31.12.3275-3283.1993
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:78.
Savini, V., Catavitello, C., Masciarelli, G., Astolfi, D., Balbinot, A., Bianco, A., et al. (2010). Drug sensitivity and clinical impact of members of the genus kocuria. J. Med. Microbiol. 59, 1395–1402. doi: 10.1099/jmm.0.021709-0
Scholz, C. F., and Kilian, M. (2016). The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 66, 4422–4432. doi: 10.1099/ijsem.0.001367
Seferovic, M. D., Pace, R. M., Carroll, M., Belfort, B., Major, A. M., Chu, D. M., et al. (2019). Visualization of microbes by 16S in situ hybridization in term and preterm placentas without intraamniotic infection. Am. J. Obstet. Gynecol. 221, 146.e1–146.e23.
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
Silverstein, R. B., and Mysorekar, I. U. (2021). Group therapy on in utero colonization: seeking common truths and a way forward. Microbiome 9:7.
Stinson, L. F., Boyce, M. C., Payne, M. S., and Keelan, J. A. (2019). The not-so-sterile womb: evidence that the human fetus is exposed to bacteria prior to birth. Front. Microbiol. 10:1124.
Stinson, L. F., Keelan, J. A., and Payne, M. S. (2018). Comparison of meconium DNA extraction methods for use in microbiome studies. Front. Microbiol. 9:270.
R Studio Team (2020). RStudio: Integrated Development for R. RStudio Support. Available online at: http://support.rstudio.com/hc/en-us/articles/206212048 [Accessed March 17, 2021]
Theis, K. R., Romero, R., Greenberg, J. M., Winters, A. D., Garcia-Flores, V., Motomura, K., et al. (2020a). No consistent evidence for microbiota in murine placental and fetal tissues. mSphere 5:e000933-19.
Theis, K. R., Romero, R., Winters, A. D., Jobe, A. H., and Gomez-Lopez, N. (2020b). Lack of evidence for microbiota in the placental and fetal tissues of rhesus macaques. mSphere 5:e000210-20.
Thornton, P. K. (2010). Livestock production: recent trends, future prospects. Phil. Trans. R. Soc. B 365, 2853–2867. doi: 10.1098/rstb.2010.0134
Touchon, M., Cury, J., Yoon, E.-J., Krizova, L., Cerqueira, G. C., Murphy, C., et al. (2014). The genomic diversification of the whole acinetobacter genus: origins, mechanisms, and consequences. Genome Biol. Evol. 6, 2866–2882. doi: 10.1093/gbe/evu225
Keywords: bovine, gut, amniotic fluid, meconium, fetal, cesarean section, microbiome, neonatal
Citation: Husso A, Lietaer L, Pessa-Morikawa T, Grönthal T, Govaere J, Van Soom A, Iivanainen A, Opsomer G and Niku M (2021) The Composition of the Microbiota in the Full-Term Fetal Gut and Amniotic Fluid: A Bovine Cesarean Section Study. Front. Microbiol. 12:626421. doi: 10.3389/fmicb.2021.626421
Received: 05 November 2020; Accepted: 29 March 2021;
Published: 30 April 2021.
Edited by:
Spyridon Ntougias, Democritus University of Thrace, GreeceReviewed by:
Jasenka Zubcevic, University of Florida, United StatesMariana De Paula Reis, Federal University of Minas Gerais, Brazil
Copyright © 2021 Husso, Lietaer, Pessa-Morikawa, Grönthal, Govaere, Van Soom, Iivanainen, Opsomer and Niku. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mikael Niku, bWlrYWVsLm5pa3VAaGVsc2lua2kuZmk=
†These authors have contributed equally to this work