Abstract
Three out of the seven ribosomal RNA operons in Escherichia coli end in dual terminator structures. Between the two terminators of each operon is a short sequence that we report here to be an sRNA gene, transcribed as part of the ribosomal RNA primary transcript by read-through of the first terminator. The sRNA genes (rrA, rrB and rrF) from the three operons (rrnA, rrnB and rrnD) are more than 98% identical, and pull-down experiments show that their transcripts interact with Hfq and CsrA. Deletion of rrA, B, F, as well as overexpression of rrB, only modestly affect known CsrA-regulated phenotypes like biofilm formation, pgaA translation and glgC translation, and the role of the sRNAs in vivo may not yet be fully understood. Since RrA, B, F are short-lived and transcribed along with the ribosomal RNA components, their concentration reflect growth-rate regulation at the ribosomal RNA promoters and they could function to fine-tune other growth-phase-dependent processes in the cell. The primary and secondary structure of these small RNAs are conserved among species belonging to different genera of Enterobacteriales.
Introduction
Bacterial small regulatory RNAs (sRNA) are major post-transcriptional regulators of gene expression. Mechanistically, the majority of these sRNAs act by base paring to complementary sequences in mRNA targets, thereby altering translation initiation rates and/or mRNA stability (Wagner and Romby, 2015). Association rates between sRNAs and their target RNAs are often strongly increased by the presence of the homohexameric RNA chaperone Hfq, which binds both RNAs and facilitates base pairing (Santiago-Frangos and Woodson, 2018). Many sRNAs are involved in the rapid reorganization of bacterial gene expression as a response to various types of stresses (recently reviewed in Holmqvist and Wagner, 2017). However, sRNAs that are expressed in the absence of an acute stress have also been described, including anti-toxin sRNAs (reviewed in Brantl and Jahn, 2015), the sRNAs ChiX and Spot 42 which regulate different aspects of carbohydrate metabolism (Møller et al., 2002; Rasmussen et al., 2009; Beisel and Storz, 2011) and MgrR (Moon and Gottesman, 2009), a regulator of lipopolysaccharide composition. The sRNAs CsrB and CsrC are expressed in response to the accumulation of end-metabolism products at the entry to stationary phase (Lawhon et al., 2002; Gonzalez Chavez et al., 2010). In contrast to base-pairing sRNAs, CsrB/C act by sequestering a single protein target, the global regulator CsrA. This small (7 kDa) homodimeric RNA-binding protein acts by binding at or close to ribosome binding sites (RBS) in a myriad of different mRNAs (Potts et al., 2017). Targets of CsrA include mRNAs encoding proteins involved in carbon metabolism (Liu et al., 1995), biofilm formation (Jackson et al., 2002), motility (Wei et al., 2001), quorum sensing, and virulence (Altier et al., 2002). CsrB and CsrC function by mimicking CsrA targets and carry ∼18 and nine motifs for CsrA binding, respectively (Liu et al., 1997; Weilbacher et al., 2003). As a consequence, they antagonize CsrA by sequestering it away from its lower-affinity mRNAs targets, thereby decreasing the effective concentration of CsrA. More recent, similar activities on CsrA have been described for two additional sRNAs in E. coli; McaS (Jorgensen et al., 2013) and GadY (Parker et al., 2017), both of which are believed to contain two binding sites for CsrA. Unlike CsrB/C, both McaS and GadY also regulate gene expression independent of CsrA (Opdyke et al., 2004; Jørgensen et al., 2012; Thomason et al., 2012).
While the majority of characterized sRNAs from E. coli are transcribed from intergenic regions (IGRs) under the control of a dedicated promoter, several reports suggest that a substantial number of sRNAs are generated from 5′ or 3′ untranslated regions (UTRs) by RNase-dependent mRNA processing (Kawano et al., 2005; Chao et al., 2012; Miyakoshi et al., 2015). Additionally, the glyW-cysT-leuZ transcript, which is processed to give rise to tRNAglyW, tRNAcysT and tRNAleuZ, also generates the sRNA 3′ETSleuZ (Lalaouna et al., 2015). This sRNA, which is generated by RNase E-dependent processing, base-pairs to two other sRNAs, RyhB and RybB. The pairing neutralizes transcriptional noise from the ryhB and rybB genes and counteracts potential regulatory outcomes of inadvertent expression of the corresponding sRNAs. The 3′-ETSleuZ is the first functional tRNA-derived fragment (tRF) described in bacteria. However, numerous tRFs have been reported in eukaryotes, where they control multiple different cellular processes, including genome stability (Martinez et al., 2017; Schorn et al., 2017), cell-cell signaling (Baglio et al., 2015), response to viral infection (Yeung et al., 2009) and stress responses (Emara et al., 2010; Saikia et al., 2014).
In the present study, we have investigated the family of so-called tRNA-linked repeats (TLRs) from E. coli. The TLRs are a class of sequences located in tRNA or ribosomal RNA (rRNA) operons. Since the first description of TLRs in 1978 (Egan and Landy, 1978), a total of 22 TLR genes have been identified (Rudd, 1999), which are distributed between ten different loci on the E. coli K-12 chromosome, each locus harboring one to five TLRs. A striking feature common to all TLRs is that 18–19 bp of their 3′-end is identical to the 3′-end of the tRNA or rRNA gene that is located immediately upstream of the TLR (Figure 1). Regarding TLR functionality, one of the TLRs found in the pre-tRNA transcript tyrT-tyrV was initially reported to be involved in recovery from amino acid starvation (Bösl and Kersten, 1991). However, this claim was later retracted as the phenotype was shown to originate from a nearby open reading frame (Bosl and Kersten, 1994). Thus far, the TLRs have no known function. In the following, we present evidence that the three TLRs located downstream from rRNA operon A (rrA), B (rrB) and D (rrF), respectively, are transcribed, processed, and bind the post-transcriptional regulators Hfq and CsrA. We present evidence that these novel sRNAs may act as regulators to fine-tune CsrA activity.
FIGURE 1
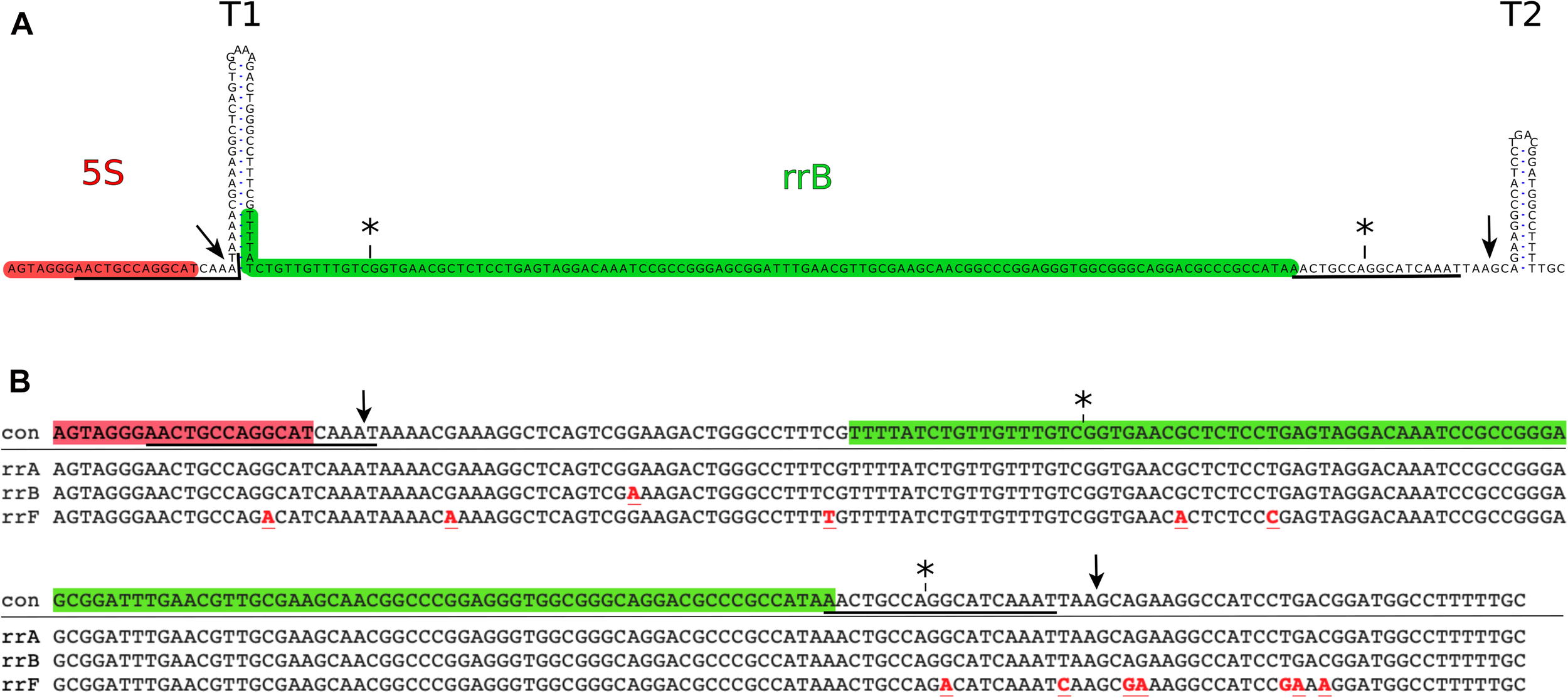
Genomic position of the rrB sequence and alignment of rrA rrB and rrF. (A) The rrB sequence found in rrnB. The 3′-end of the mature 5S transcript (highlighted in red) is followed by two transcriptional terminators (T1 and T2). An 18 base pair sequence (underlined) overlapping the 3′-end of the 5S gene, is found twice in the sequence with a spacing of 150 bp. The predominant form of RrB found on Hfq (RrBshort) is highlighted in green (see also Figures 2, 3). The asterisks (*) above the sequence indicate the ends of the annotated version of rrB (Ecogene; Zhou and Rudd, 2012) and the arrows indicate the ends found by S1 mapping of the longer transcript (Figure 3). (B) Alignment of the sequences found in the 3′-end of three out of seven rRNA operons in E coli.: rrnA (rrA), rrnB (rrB) and rrnD (rrF). con: consensus. Symbols are as in Figure 1A, red font indicates sequence differences compared to the consensus sequence.
Materials and Methods
Culture Growth and Media
The study was carried out in E. coli K-12 MAS1081 (MG1655 rph+ gatC+ glpR+). All strains used in the study are listed in Supplementary Table 1. Unless otherwise noted, all cultures were grown in MOPS minimal medium (Neidhardt et al., 1974) at 37°C shaking at 160 rpm and were grown exponentially for at least ten generations before start of the experiment to obtain balanced growth. Antibiotics were added as described for each experiment.
RNA Purification and Northern Blotting
Culture aliquots were harvested into 1/4 vol ice-cold stop solution (95% ethanol, 5% phenol) (Bernstein et al., 2002). Subsequently RNA was purified using hot phenol and flash freezing in liquid nitrogen as in Fessler et al. (2020). Briefly: Stopped culture aliquots were centrifuged 2 min at 20.000 g and resuspended in 0.1 vol cold 0.3 M sucrose, 0.01 M NaOAc pH 4.5 followed by addition of 0.1 vol 2% SDS 0.01 M NaOAc pH 4.5. Phenol (saturated with water) was added to the liquid phase at a 1:1 ratio, the tubes were vortexed and incubated 3 min at 65°C. After freezing 15 sec in liquid N2 and centrifugation at 20.000 g for 5 min, the water phase was transferred to new tubes and the phenol extraction was repeated. If the RNA was used in an enzymatic reaction after purification, a chloroform extraction step was included. The RNA was ethanol-precipitated, washed by 96% ethanol, air dried at room temperature and dissolved in 10 mM NaOHAc, 1 mM EDTA. For northern blots, RNA was mixed 1:1 with loading buffer (0.1 M NaOAc (pH 5.0), 8 M urea, 0.05% (w/v) bromophenol blue and 0.05% (w/v) xylene cyanol) and size-separated on denaturing 0.4 mm thick polyacrylamide gels using 1 × TBE buffer (90 mM Tris, 90 mM boric acid and 2 mM EDTA). The RNA was electroblotted onto Hybond-N membranes (GE Healthcare) (1.5 V/cm, 1.5 h) in 40 mM Tris-acetate (pH 8.1), 2 mM EDTA. After UV-crosslinking (0.12 J/cm2) the membranes were pre-hybridized (1 h, rotating at 42°C) in hybridization solution (0.9 M NaCl, 0.05 M NaH2PO4 (pH7.7), 5 mM EDTA, 5 × Denhardt’s solution (0.1% BSA, 0.1% Ficoll 400, 0.1% polyvinylpyrrolidone), 0.5% (w/v) SDS and 100 mg/ml sheared, denatured salmon sperm DNA). Probe hybridization was done by adding 30 pmol of oligo-DNA, 5′-end labeled with 32P (overnight, rotating at 42°C). Subsequently, membranes were washed several times with 0.3 M NaCl, 30 mM sodium citrate, 0.1% SDS at 42°C. Radioactive signals were quantified on a PhosphorImager (Typhoon-GE Healthcare) using ImageQuant software as previously described (Sørensen, 2001; Stenum et al., 2017) and in case of very low signals (e.g., Figure 2C) the signal found in the estimated position of a band was used. Before re-probing, membranes were stripped by washing several times with 98°C, 15 mM NaCl, 1.5 mM sodium citrate, 0.1% SDS, until no more radioactive signal could be detected by a Geiger-Müller tube. Probe sequences used in this study are listed in Supplementary Table 4.
FIGURE 2
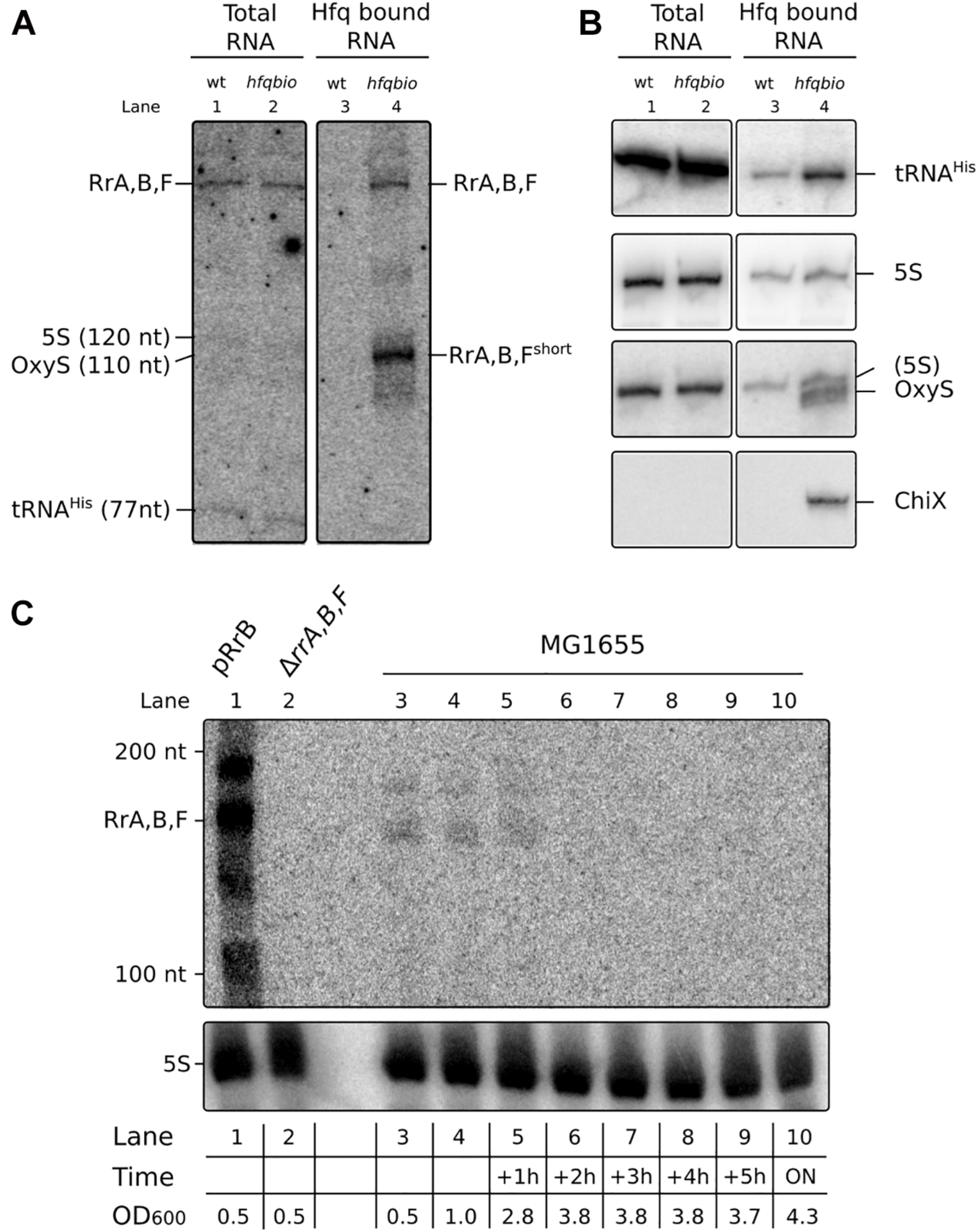
RrA, B, F is present as a distinct band in total RNA and a shorter version of RrA, B, F is enriched on Hfq. (A) Northern blot of a denaturing 10% polyacrylamide gel with RNA harvested from cultures of MG1655 (wt) or the isogenic Hfq-tagged derivative (hfq_bio). E. coli cultures were grown exponentially in MOPS minimal media supplemented with 15 mg/l chloramphenicol, 0.2% glucose, 10 mg/l uracil, 50 μM biotin. Both strains contain the plasmid pBirA. RNA was harvested directly from the cultures (lane 1 & 2) or from pull-down assays with Hfq (lane 3 & 4) as described in Materials and Methods. Equal volumes of the co-purified RNA were loaded in each lane to reflect equal numbers of input cells. (B) The same Northern blot membrane was probed and re-probed for tRNAhis, 5S RNA, OxyS and ChiX to obtain the size markers indicated in panel (A) and as controls for the pull-down assay. The probe for OxyS was used after the 5S probe, and carryover signal from the 5S probe is clearly visible. Due to the low amount of RNA loaded in lane 1 and 2 (0.7 μg/lane) we did not detect OxyS or ChiX in the total RNA fraction. Lane 4 has 2.0 μg Hfq co-purified RNA, here we detect OxyS and ChiX but not in the control reaction with an untagged hfq, as expected. (C) Northern blot of a denaturing 6% polyacrylamide gel with RNA harvested from cultures of MG1655 + pRrB (lane 1), ΔrrA, B, F (lane 2) and MG1655 (lanes 3–10) as indicated. The cultures were grown exponentially in MOPS minimal media supplemented with 0.2% glucose. The first two samples of MG1655 were collected at OD600 values of 0.5 and 1, then samples were collected every hour for 5 h and the last sample was harvested after overnight (ON) incubation. The OD600 values for the culture at different harvest time points are shown in the table below the graph. The OD600 value for the pRrB and ΔrrA, B, F strains were both 0.5 at the time of harvest.
Hfq Pull-Down Assay
RNAs that bind Hfq were isolated using an E. coli strain, where the hfq allele was tagged with a biotinylation sequence (Hfq_bio). We chose this tag as it is not positively charged, to reduce the risk that the tag might unspecifically bind negatively charged RNA. Hfq_bio was biotinylated by the biotin ligase BirA, and biotin’s high-affinity binding to avidin was used for purifying Hfq_bio (Kay et al., 2009). To ensure full biotinylation of Hfq_bio we introduced the plasmid pBirA where birA is under the control of an IPTG-inducible promoter. We found that production of sufficient BirA was achieved without induction of transcription by IPTG.
Wildtype and Hfq_bio cells both containing the pBirA plasmid were grown exponentially in MOPS medium supplemented with; 15 μg/ml chloramphenicol, 0.2% glucose, 10 μg/ml uracil, 50 μM biotin, at 37°C for at least 10 generations. At OD436 = 0.8 the cells were pelleted and washed in medium without biotin, re-suspended in 2 ml lysis buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5% glycerol), lysed by sonication and centrifuged (20,000 g, 60 min). Total RNA was prepared from an aliquot of the cleared lysate, and the remaining lysate was transferred to fresh tubes containing 300 μl equilibrated SoftLinkTM Avidin Resin (Promega) and left overnight rotating at 4°C. Then, the resin was washed four times in lysis buffer as above and RNA was harvested by phenol extraction and ethanol precipitation. For total RNA and the Hfq-bound RNA, RNA corresponding to 0.05 and 10% of the total culture volume respectively (approximately 2,000 ng for Hfq-bound RNA from the Hfq_Bio strain and 700 ng for total RNA samples), was used for analysis by northern blotting.
S1 Nuclease Mapping
Briefly, a [γ-32P]-ATP end-labeled DNA oligo antisense to the TLR area of interest was hybridized to total RNA and single-stranded overhangs were removed by addition of S1 nuclease. The resulting fragments were visualized on a denaturing polyacrylamide gel. If the synthetic oligo extends beyond the end of the target RNA, the position of the end can be determined by the number of nucleotides removed from the DNA oligo. S1 nuclease was used to map the RrB transcript of a strain harboring the plasmid pTSS1. RNA was harvested by hot phenol 1 h after IPTG induction. One pmol of 5′-end [32P] -labeled probe was hybridized to 30 μg of total RNA from the strain of interest. Hybridization was done in 50% formamide, 20 mM HEPES, 0.5 mM EDTA, 0.2 M NaCl, 0.05% (w/v) SDS and performed overnight in a thermocycler (68°C for 10 min, then the temperature was lowered to 54°C and decreased 1°C every 30 min until reaching 20°C). Digestion was performed by adding 300 μl 0.28 M NaCl, 50 mM NaOAc pH 4.6, 4.5 mM ZnSO4 along with 300 U/ml S1 nuclease (Thermo Fisher Scientific) and incubating at RT for 60 min. Samples were phenol/CHCl3 extracted, ethanol precipitated, size separated by electrophoresis on 7 M urea, 10% polyacrylamide sequencing gels and detected by autoradiography.
Circular RACE Mapping
Circular RACE was used to map the isoform of RrB enriched on Hfq. Briefly, RNA purified from the Hfq_bio purification experiment was circularized using RNA ligase, reverse transcribed using random hexamer primers, PCR amplified twice using nested sets of RrA, B, F-specific primers, and subjected to deep sequencing. The site of circularization thus reveals both ends of the transcript. Mapping was carried out essentially as described by McGrath (2011) omitting the TAP treatment: 500 ng of RNA co-purified with Hfq was circularized in 1× buffer by adding T4 RNA ligase. Reverse transcription was carried out using Super Script III RT (Thermo Fischer) and primed by random hexamer oligos. The area of interest was amplified twice by PCR with two different sets of specific primers (cRACE rrB-2 1F + cRACE rrB-2 1R and cRACE rrB-2 2F + cRACE rrB-2 2R, see Supplementary Table 3). The PCR-library was sequenced on an Illumina Mi-seq by 300 bp paired-end sequencing. The resulting sequences were merged and subsequently listed by abundance. All reads that could not be merged were left out of the analysis.
RNA Stability During Rifampicin Treatment
Cultures were grown exponentially at 37°C, shaking at 160 rpm for at least 10 generations in MOPS medium supplemented with 0.2% glucose and 10 μg/ml uracil. At an OD436 of 0.7, 3 × 15 ml culture aliquots were collected (time 0 min) and rifampicin was added to the remaining culture to a final concentration of 100 μg/ml. Aliquots of 15 ml were collected at 2.5, 5, 10, and 20 min post rifampicin treatment. Five percent spike-in culture overexpressing tRNAselC was added to each sample aliquot as in Stenum et al. (2017). RNA was harvested using TRI-reagent (Sigma), as described by the manufacturer. The RNA was size-separated on polyacrylamide gels, northern blotted and probed as described above. RrBshort transcript levels were normalized to the tRNAselC level for each sample.
Structure Probing
The structure of RrB was investigated in vitro with and without Hfq.
Reactions (10 μl) containing 0.1 pmol of 5′-end [γ-32P] ATP-labeled transcript and 50 nM unlabeled E. coli tRNA were incubated with different concentrations of hexameric Hfq (Hfq6) (3, 1.5, and 0 μM) at 37°C for 100 min along with the relevant cleavage buffer. For Pb2+ cleavage: 1 × Structural Probing Buffer (Ambion AM2237), Pb2+ was added to a final concentration of 10 mM and samples incubated 1 min at 37°C. Control (C1) was without Hfq and Pb2+. RNaseIII cleavage: 1 × Short Cut MnCl2 buffer (New England Biolabs) and 0.002 units Short Cut RNaseIII (New England Biolabs), samples were incubated 20 min at 37°C. Control (C2) was without Hfq and RNaseIII. Control T1: 1 × Structural Probing Buffer (Ambion AM2237), sample was incubated at 95°C for 1 min, transferred to 37°C for 1 min. After addition of 0.05 U RNase T1 (Ambion AM2237) the sample was incubated at 37°C for 5 min. OH ladder: 1 × Alkaline Hydrolysis Buffer (Ambion AM2237), sample was incubated at 95°C for 5 min. All samples were cooled by addition of 200 μl ice-cold H2O and transferred to ice. The digested RNA was phenol extracted, ethanol precipitated, resuspended in 1 × loading buffer II (Ambion AM2237), and separated on an 8% polyacrylamide/urea/TBE gel. Radioactive signal from the dried gel was visualized on a PhosphorImager (Typhoon -GE Healthcare). Hfq protein used for all in vitro experiments was a kind gift from Anders Boysen.
MS2 Affinity Purification, RNA-Seq and MS-MS
Affinity purification of MS2-tagged RNAs was done either with in vivo expressed RNA or in vitro transcripts that were added to the cell lysate. The 115 bp RrBshort sequence, mapped as the most abundant variant pulled down with Hfq (Figure 3) was cloned into the plasmid pNS21 using PCR amplifying RrBshort with either NheI or XbaI restriction sites on the ends. Purified PCR fragments were restriction digested with either NheI or XbaI and ligated into the pNS21 plasmid cut with the corresponding restriction enzyme, resulting in RrBshort 3′-fused to MS2 (NheI digestion) or RrBshort 5′-fused to MS2 (XbaI digestion), respectively. The transcripts from the resulting plasmids are terminated by the vrrA terminator originally found in Vibrio species. Transcription from the plasmids is under control of the PLlacO–1 promoter. As the strains used for affinity purification harbor only the chromosomal copy of lacI, expression from these plasmids should be constitutive. This was verified by northern blotting (Supplementary Figure 4). Cell lysates of the strain hfq-FLAG (JVS814) harboring the different MS2-aptamer expressing plasmids, were prepared by growing cells in LB with 100 μg/ml ampicillin, to an OD600 of 1.0. Cell pellets corresponding to 50 OD600 units were resuspended in 800 μl of buffer A (20 mM Tris-HCl pH 8.0, 150 mM KCl, 1 mM MgCl2, 1 mM DTT) and lysed by addition of glass beads (0.1 mm), flash freezing in liquid N2 and shaking at 30 Hz for 10 min. Lysates were cleared by centrifugation (30 min 16,000 g, 4°C). Lysates corresponding to 2 and 0.5 OD600 units of cell culture were used to prepare RNA and protein, respectively. Before addition of lysate, the affinity chromatography columns (Bio-Spin #732-6008, Bio-Rad) were prepared by adding 100 μl amylose resin (New England Biolabs, #E8021S), washing three times with 2 ml buffer A and subsequently adding 200 pmol of MS2-MBP recombinant protein (a gift from the Jörg Vogel group). All steps of the affinity chromatography were done at 4°C. Following addition of the lysate, the columns were washed four times with 2 ml buffer A, and the RNA-protein complexes were eluted with 900 μl buffer A containing 12 mM maltose. RNA was purified using phenol-chloroform, followed by ethanol precipitation with the addition of 1 μl GlycoBlue (Thermo Fisher). Protein was harvested from the organic phase by acetone precipitation. RNA was subjected to next generation sequencing on the Illumina platform at the University of Würzburg. Proteins were detected and quantified by mass spec at the mass spec facility at University of Würzburg, see Supplementary Methods.
FIGURE 3
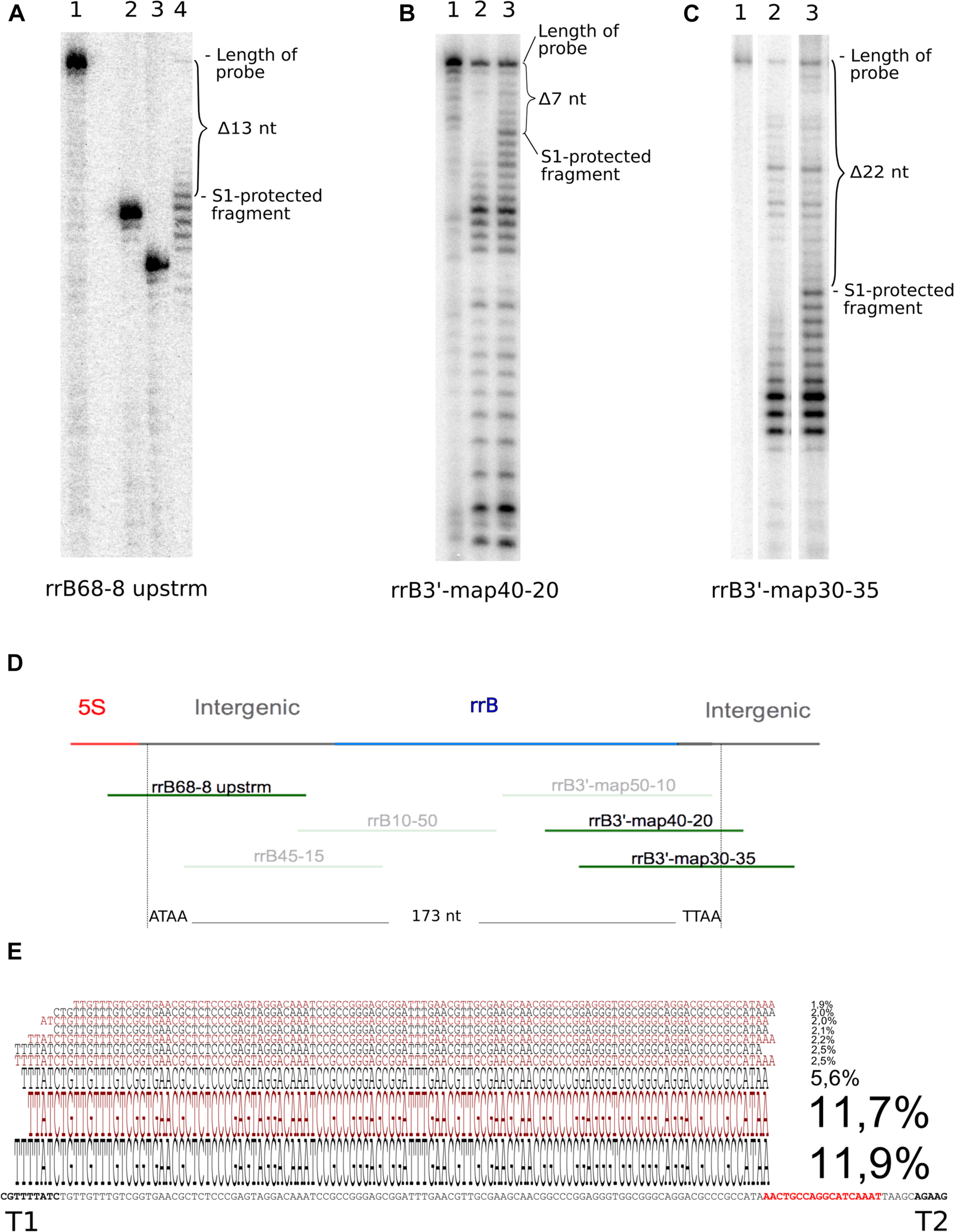
Determination of 5′ and 3′ ends of RrB by S1 nuclease analysis and of RrBshort by circular RACE. In the S1 nuclease mapping analysis (A–C) the 32P-labeled probe DNA was visualized by autoradiography of 10% poly-acrylamide sequencing gels. (A) mapping of the 5′-end using probe rrB68-8upstrm [shown in panel (D)] and total RNA from an IPTG-induced culture of MG1655 + pRrB over-expressing truncated ‘rrfB and rrB. The S1-protected fragment (lane 4) is 13 nt shorter than the untreated probe (lane 1). The exact number of nucleotides removed was determined by loading two labeled oligos as size markers [lane 2 (rrB54-8 upstrm) and 3 (rrB50-8 upstrm), probe rrB68- 8upstrm truncated by 14 and 18 nt respectively]. (B) probe rrB3′-map40-20 was hybridized to total RNA from an IPTG-induced culture of Δ22TLR + pRrfB-RrB over-expressing rrfB and rrB. The S1 protected band was shortened by 7 nt (lane 3) compared to the untreated probe (lane 1). Lane 2 shows a similar experiment using total RNA from the IPTG-induced strain Δ22TLR harboring empty vector (pJFR1). (C) Probe rrB3′-map30-35 was hybridized to total RNA from MG1655 + pRrfB-RrB over-expressing rrfB and rrB. The S1 protected band was shortened by 22 nt (lane 3) compared to the untreated probe (lane 1). Lane 2 shows a similar experiment using total RNA from the strain Δ22TLR harboring empty plasmid. (D) Map of the probes used for the experiments shown in panels (A–C). Top line represents the genomic map; rrfB (5S) is shown in red, the intergenic sequences in gray and the annotated version of rrB in blue. Probe sequences are presented as green lines and probes shown in transparent colors were also used for mapping but detected no ends. The vertical broken lines denote the two ends detected in this study. (E) Circular RACE mapping of the RrB sequences that co-precipitated with Hfq. The ten most abundant RrBshort sequences detected by circular RACE and deep sequencing are aligned to the genomic sequence. The size of the characters correlates with their relative abundance, which is also stated as a percentage of total merged reads (n = 4722). Bold characters highlight the two terminators T1 and T2 and the direct repeat, which is also found at the 3′-end of the mature 5S RNA is highlighted in red (see Figure 1).
Input Into Invenire CsrB/C Family RNA Prediction Algorithm
The web version of Invenire sRNA found at http://markov.math.umb.edu/inveniresrna/was used for the analysis. The dataset included the 85 sRNAs, 22 TLR sequences, and 2217 intergenic regions (<1,000 bp) annotated in the Ecogene database1 on November 24, 2017. In addition, 578 sequence peaks identified experimentally as CsrA binding sites by CLIP-seq (Potts et al., 2017) were included in the analysis.
In vitro Transcription and Electrophoretic Mobility Shift Assay
Binding of RNAs to CsrA was examined using electrophoretic mobility shift assay (EMSA) with recombinant CsrA-3xFLAG (Supplementary Tables 1, 2) and in vitro transcribed RNA. DNA templates for in vitro transcription were made by PCR using pRrB, pRrB-3GGA and pCrsB (see Supplementary Tables 1, 2) as template for RrB, RrB-3GGA and CsrB, respectively. All primers used in the study are listed in Supplementary Table 3. Transcription was performed overnight at room temperature using the MEGAscript T7 transcription kit (ThermoFisher). The resulting RNA was treated with TurboDNase (ThermoFischer) (0.1 unit/μl, 30 min at 37°C), gel-purified from denaturing polyacrylamide gels, dephosphorylated using calf intestinal alkaline phosphatase (30 min at 37°C) and 5′ radiolabeled using T4 polynucleotide kinase and [γ-32P] ATP. After each step the RNA was phenol extracted once, chloroform extracted twice and ethanol precipitated. Binding reactions contained 100 mM KCl, 10 mM MgCl2, 2 mM DTT, 7.5% glycerol, 0.1 U SUPERase-IN RNase inhibitor (ThermoFisher), 2 ng total yeast RNA, 120 pM labeled RNA and 0–1,600 nM CsrA-3xFLAG in a 10 μl reaction. Samples were incubated 10 min at 37°C and separated on 8% native polyacrylamide gels using 1x TBE as buffer. Radioactive signals were detected on a PhosphorImager (Typhoon -GE Healthcare). The CsrA-3xFLAG was purified from the strain CsrA-3xFLAG carrying the plasmid pBAD-RBS-csrA:3xFLAG as described by Jorgensen et al. (2013).
Construction of Deletion Mutants
For λ Red recombineering, we used a MAS1081-derivative strain (MAS1080) harboring the λ RED prophage [λ cI857 Δ(cro-bioA)] imported from strain HME68 (Sawitzke et al., 2007) by P1 transduction. Deletion mutations were constructed in this strain as described (Sawitzke et al., 2007). For each deletion, a cat-sacB cassette was first inserted at the desired genomic region by selecting for chloramphenicol resistance and confirming by PCR. The cassette was then replaced by a DNA fragment designed to yield deletion of 107 bp, 103 bp and 103 bp for rrnA, rrnB and rrnD, respectively, by counter-selection of the cassette by sucrose tolerance and confirmation by PCR and DNA sequencing. The deletions roughly match the annotated TLR sequences, including all GGA motifs found in RrA, B, F. For reasons outside the scope of this study, the DNA fragments were constructed so that the TLR sequence was replaced with a tRNA gene in two out of the three deletion sites (see Supplementary Table 1).
To obtain an E. coli mutant where the λ Red recombination enzymes had not been expressed, and thereby reduce the risk of undesired genome mutations, each mutated locus was then moved to an otherwise wildtype strain by P1 transduction first of each cat-sacB cassette and next of the locus carrying a deletion. P1 transduction was done as described by Miller (1972).
Biofilm Measurements
Biofilm was measured in microtiter plates using peg-lids (Nunc-TSP, cat. no. 445497) and crystal violet staining. Cultures were grown in 96-well flat-bottom microtiter plates. Ten microliter outgrown culture was used to inoculate each well containing 150 μl YT media (per liter: 8 g tryptone, 5 g yeast extract and 5 g NaCl) supplemented with 100 μl/ml ampicillin and 1 mM IPTG for strains carrying plasmids. The outer-most wells on the plates were not used, as the results from these were found to fluctuate more than average. Plates were incubated at 37°C for 48 h (no shaking) to ensure all cultures were completely outgrown. After incubation the pegs were washed once in wash buffer (25 mM Tris pH 7.5, 100 mM NaCl) and placed in 0.01% crystal violet for 15 min. Then, the pegs were washed three times in wash buffer and transferred to a fresh microtiter plate containing 180 μl 96% ethanol in each well. When the crystal violet was completely dissolved, the A590 of each well was measured along with the OD600 of each well of the growth plate. The optical densities from the growth plate were used to normalize the absorbance signals from the stained biofilm.
Motility Measurements
Mobility was measured on soft agar plates. One microliter outgrown culture was used to inoculate each plate by injection of 1 μl culture halfway into the agar in the center of the plate. Plates contained YT media (per liter: 8 g tryptone, 5 g yeast extract and 5 g NaCl) + 0.3% agar, and were supplemented with 100 μl/ml ampicillin and 1 mM IPTG for strains carrying plasmids. The plates were incubated approx. 16 h at 37°C before the spread of the bacteria was measured. Measurements were repeated several times with approximately 2 h in between to compensate for differences in the growth rates of different strains.
Translational Reporters
The glgC-gfp in-frame fusion was constructed by replacing the NsiI-NheI fragment of pXG10-SF (Supplementary Table 2) with the 5′UTR and first 30 nucleotides of the glgC coding sequence by restriction digestion and ligation. The pgaA-lacZ in-frame fusion was made by cloning the promoter of lacI along with a NheI restriction site into pGH253-kan (Supplementary Table 2) by replacing the EcoRI-BamHI fragment. Subsequently, the 5′UTR and first 30 nucleotides of the pgaA coding sequence was inserted using BamHI-NheI digestion. All primers used in the study are listed in Supplementary Table 3.
Beta-Galactosidase Measurement
Beta-Galactosidase (β-gal) activity was measured from overnight cultures harboring the translational fusion pgaA-lacZ, grown in test tubes in YT media (per liter: 8 g tryptone, 5 g yeast extract and 5 g NaCl) supplemented with 15 μg/ml kanamycin. The β-gal activity was measured using the fluorescent substrate 4-methylumbelliferone b-D-galactopyranoside (MUG), as described (Li et al., 2018). OD600 of the cultures was measured and used to normalize the corresponding β-gal values.
GFP Measurements
Fluorescence of GFP was measured from strains harboring the glgC-gfp translational fusion. Cultures were grown in MOPS minimal media supplemented with 0.2% glucose, 15 μg/ml chloramphenicol and 100 μg/ml ampicillin when needed, at 37°C shaking at 300 rpm in 96-well black microtiter plates with clear, flat bottoms (Costar) in a plate reader (Synergy H1, Biotek). OD600 and GFP fluorescence (excitation 470 nm and emission 510 nm) was measured simultaneously every 10 min. The values for GFP were normalized to the corresponding values for OD600. The normalized data was plotted against time and the area under the curve was calculated as a measure of relative GFP expression. The same amount of data points were used for each strain and only data where cultures were growing exponentially was used in the analysis.
Results
Expression of RrA, B, F and Interaction With Hfq
Three out of seven ribosomal RNA (rRNA) operons from E. coli contain a sequence from the TLR-family downstream from the 5S gene (Supplementary Figure 1). These three sequences, named rrA, rrB and rrF respectively, are very similar in sequence (Figure 1). Furthermore, their location downstream of 5S, and their sequence, is highly conserved in many species of the Enterobacteriales, suggesting a specific role for these sequences and structures (Supplementary Figure 2).
We hypothesized that the sequences may function as sRNAs. To begin testing this hypothesis we first examined whether RNA molecules of defined sizes could be detected after processing of the primary rrn transcripts. Total RNA harvested from exponentially growing E. coli K-12 cultures was subjected to northern blot analysis. To detect RrA, RrB and RrF, here collectively referred to as RrA, B, F, we used a single probe, which is expected to detect all three sequences since they only differ at a few positions. As shown in Figure 2A (lane 1 and 2), a distinct band of 150–200 nt was observed, confirming that RrA, B, F can be detected after processing of the primary transcript. The size of the RrA, B, F RNA was estimated based on re-probing of the membrane for transcripts of known sizes (Figures 2A,B) and comparison to an RNA ladder (Figure 2C). Next, we asked whether the RrA, B, F RNAs interact with the RNA chaperone Hfq, like many well-characterized E. coli sRNAs (Bilusic et al., 2014). To enable pull-down of Hfq and analysis of co-purified RNAs, a biotinylation sequence (Beckett et al., 2008) was inserted in the chromosomal hfq gene. The C-terminally tagged Hfq protein (Hfq_bio) is functional, as shown by intact repression of an mRNA target by an Hfq-dependent sRNA in the tagged strain (Supplementary Figure 3). After affinity purification of Hfq_bio using a streptavidin resin, co-precipitated RNA was analyzed by northern blot analysis. Detection of the known Hfq-binding sRNAs OxyS and ChiX only in the co-precipitated RNA verified that Hfq-binding RNAs had been enriched in the Hfq pull-down assay (Figure 2B). Interestingly, the pull-down revealed that a shorter form of RrA, B, F (RrA, B, Fshort) was highly enriched on Hfq in addition to the band detected in total RNA (Figure 2A, lane 4). Further, to verify the identity of the bands on our northern blots found by the RrA, B, F, probe, we made a blot containing RNA from a strain overexpressing RrB from pRrB, a strain deleted for rrA, B, F, and several samples from the wildtype strain harvested at different stages of growth (Figure 2C). The blot verifies the identity of the bands and shows that RrA, B, F are only detected during exponential growth, where the rRNA operons are actively transcribed. During stationary phase, the transcriptional activity of rRNA operons is absent or very low (Baracchini and Bremer, 1991) but the 5S, 16S and 23S RNAs are stable molecules. Therefore the 5S rRNA detected during stationary phase in Figure 2C was transcribed during growth and serves as a qualitative loading control.
The ends of the longer RNA species were mapped using S1 nuclease protection assay (Berk and Sharp, 1977). As this assay was not sensitive enough to detect chromosomally expressed RrA, B, F, we used a strain overexpressing the 5S gene rrfB along with rrB from a plasmid. The S1 protected fragments are displayed in Figures 3A–C. Several bands spaced with one nucleotide intervals are seen, which is a common observation when using S1 nuclease transcript mapping (Green and Roeder, 1980; Aiba et al., 1981; Brosius et al., 1982) and is probably due to “end-nibbling” (Shenk et al., 1975). Thus, we define the transcript ends based on the longest protected bands. For the 5′ end the probe was shortened by 13 nt (Figure 3A). For the 3′ end the two different probes were shortened by 7 nt (Figure 3B) and 22 nt (Figure 3C), respectively. They both predict the same end. The 5′end of RrB was detected 3 nt downstream of the mature 3′end of the 5S transcript (see Figures 1, 3A). This site has been described as the initial RNase E processing site of the pre-5S RNA (Roy et al., 1983; Li and Deutscher, 1995) suggesting that the 5′ end of RrB is generated by RNase E cleavage. The 3′end of RrB was detected 13 nt downstream of its annotated 3′end (Figure 1). This 3′end was verified using two different probes (Figures 3B,C). These results predict that the most abundant RrA, B, F transcript in total RNA has a length of 173 nt, which is in good agreement with the northern blots shown in Figures 2A,C. We also carried out S1 analysis with probes antisense to the area between the two observed ends to detect any alternative transcript ends, but could not detect any (data not shown, the probes are shown in Figure 3D).
To determine the ends of the Hfq-enriched shorter version of RrA, B, F (RrA, B, Fshort) we used the circular RACE method (McGrath, 2011), coupled with next generation sequencing. As this method is PCR-based it requires substantially less input RNA than the S1 nuclease assay. Circular RACE yielded a variety of sequences mapping to rrA, B, F, of which the ten most abundant are shown in Figure 3E. The most abundant reads predict the Hfq-associated RrA, B, Fshort transcript to be 113–115 nt in length. This is in accordance with our observations from northern blots (Figure 2).
To detect the RrA, B, Fshort fragment in the total RNA fraction, we carried out another northern blot analysis using substantially more total RNA per lane than the one presented in Figure 2A. This blot showed three different species of the RrA, B, F RNA (Figure 4A): the 115 nt RrA, B, Fshort, the 173 nt fragment, and a longer version. We suspect that the longer version represents a processing intermediate that includes the T2 terminator sequence. Thus, the RrA, B, Fshort is not uniquely seen in the RNA fraction co-precipitating with Hfq but can also be detected in the total RNA fraction.
FIGURE 4
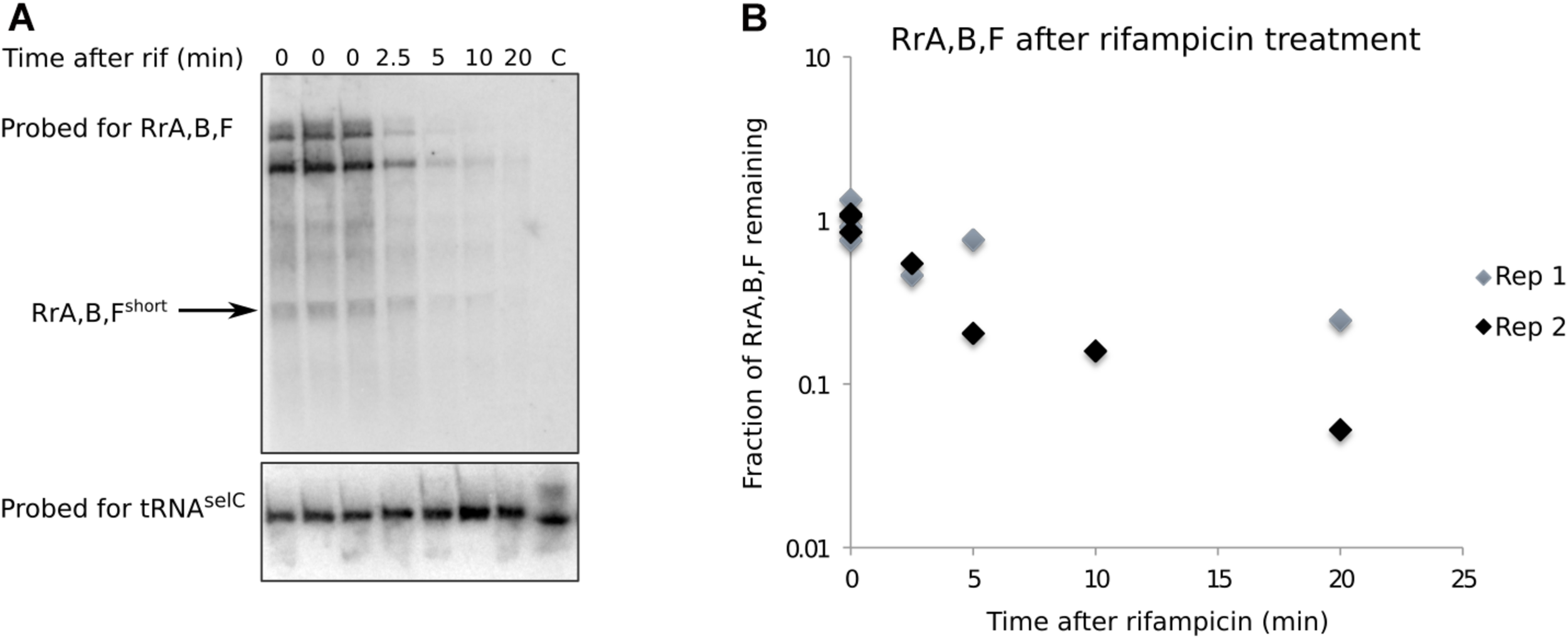
RrA, B, Fshort is a shortlived transcript. (A) Northern blot made from total RNA harvested from a culture of MG1655. The culture was grown in MOPS minimal media supplemented with 0.2% glucose, 10 mg/l uracil. Rifampicin was added at 0 min to a concentration of 100 μg/ml. RNA was harvested at the indicated timepoints, right before and after rifampicin addition. Before RNA purification, spike-in cells overexpressing the tRNA gene selC were added to each experimental sample (see section “Materials and Methods”). 8–10 μg RNA was loaded in each lane. The blot was probed for RrA, B, F and tRNAselC as indicated. The arrow indicates the band used for quantification. The lane marked “C” only contain spike-in cells. (B) Quantification of northern blots. The half-life of RrA, B, F was estimated as the average half-life calculated from two independent experiments (Rep 1, Rep 2). Rep 2 is shown in panel (A). Independent biological replicates were obtained for all sample points except for the 10 min time point post rifampicin addition.
A short half-life of the RrA, B, F RNA could explain its low abundance relative to the rRNA transcripts expressed from the same operons and the absence of signal from stationary phase cells (Figure 2C). We measured the half-life of RrA, B, Fshort by monitoring the levels of RrA, B, Fshort upon transcription initiation blocking by addition of rifampicin to a culture in balanced growth (Figure 4). Indeed, we found that the half-life of RrA, B, Fshort was ∼2.5 min, which is very short relative to the rRNA transcripts that are stable on the time scale of hours during exponential growth (Piir et al., 2011).
Taken together, we conclude that the RrA, B, F RNAs can be detected in exponentially growing cells as at least two transcripts of ∼173 and ∼115 nt that appear to interact with Hfq and that the shorter variant RrA, B, Fshort has a half-life of about 2–3 min. The presence of RrA, B, F is therefore dependent on active rRNA transcription.
The Structure of RrA, B, F Is Conserved Within the Order of Enterobacteriales
In order to assess the conservation of RrA, B, F between species we did a BLAST search2 with rrB and the sequences surrounding the locus as input (see Supplementary Figure 2 and Supplementary Material). Both the sequence and the location of the rrB downstream of the gene encoding 5S was found to be conserved in at least 31 species from 12 genera, all belonging to the order of Enterobacteriales (Supplementary Figure 2). To evaluate the structural conservation of the RrA, B, F homologs we used the locARNA algorithm (Will et al., 2007, 2012; Raden et al., 2018) to predict a consensus structure (Figure 5). The consensus structure shows a high degree of structural conservation, particularly in the three stem loop structures named 2-4 in the figure, which could indicate a conserved function of the RNA.
FIGURE 5
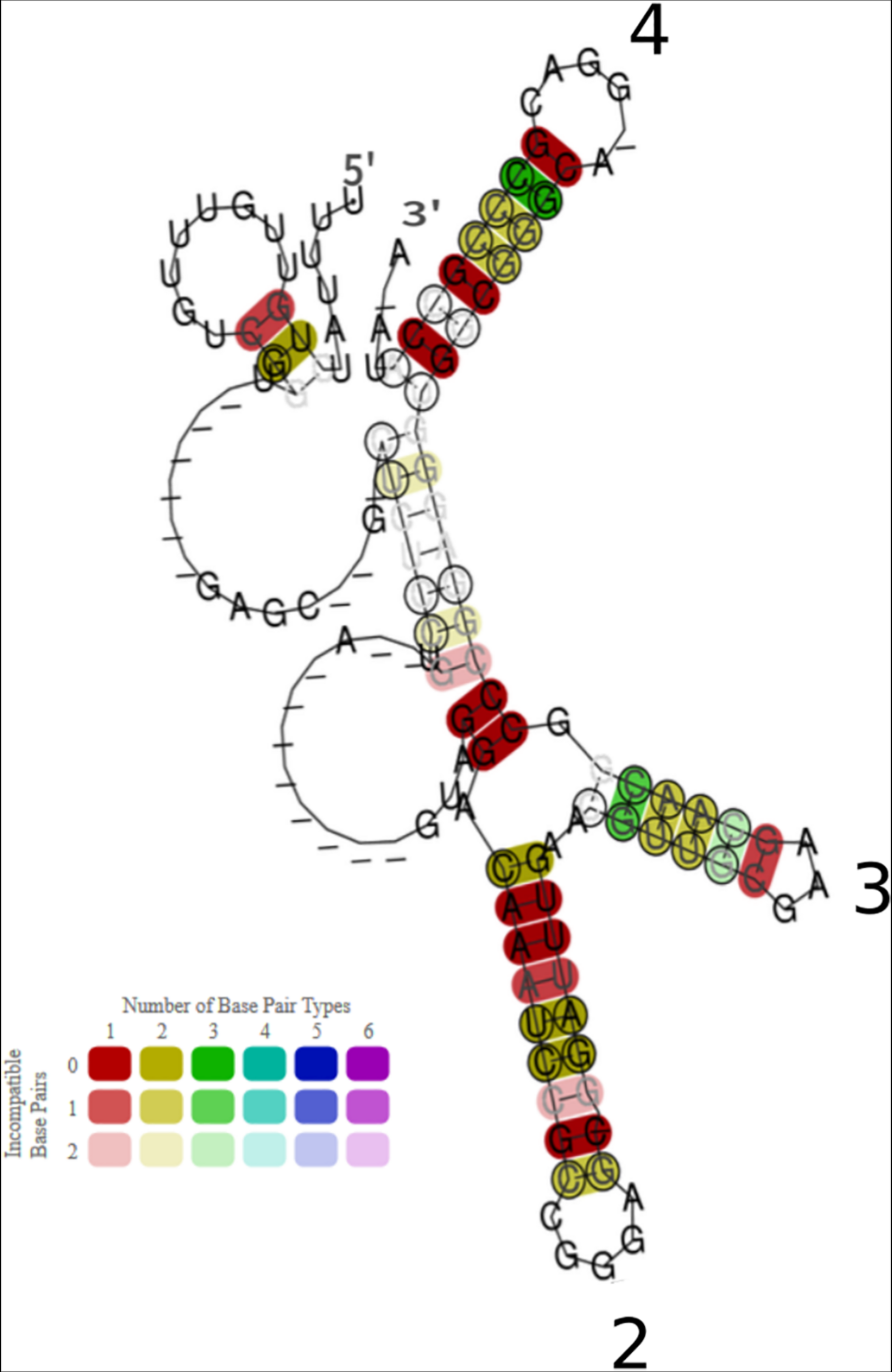
Conservation of sequence and structure of 30 RrB homologs found in Enterobacteria. The presented consensus structure was made using the algorithm LocARNA (Will et al., 2007, 2012; Raden et al., 2018) It is based on an alignment of 30 RrBshort homologs, from 30 different bacterial species, identified by BLAST search (see Supplementary Figure 2 and Supplementary Material to Figure 5). The base pairs of the locARNA structure are colored according to the level of structure and sequence conservation, as specified in the color legend. The hue indicates sequence conservation of the base pair (red: most conserved, purple: least conserved). The color saturation indicates the structural conservation of the base pair (High saturation: most conserved, low saturation: least conserved). Stem-loops are numbered as in Figure 6, Stem-loop 1 is not predicted to form in this consensus structure. Genera of the organisms included in the analysis are: Salmonella, Shigella, Klebsiella, Escherichia, Pectobacterium, Brenneria; the more distant: Citrobacter, Enterobacter, Providencia, Raoultella; and the most distant: Cedecea, Serratia and Hafnia.
We speculated that the enrichment of RrA, B, Fshort in RNA that had co-precipitated with Hfq could be due to a role for Hfq in chaperoning the correct folding of RrA, B, F, as shown for other RNAs (Geissmann and Touati, 2004; Soper and Woodson, 2008; Bordeau and Felden, 2014; Hoekzema et al., 2019). In this case, we would expect to see differences in the RrA, B, Fshort structure with and without Hfq. To experimentally investigate the structure of the 115 nt RrBshort RNA, we performed RNA structure probing in the presence or absence of Hfq (Figure 6). As shown in Figure 1B, RrAshort and RrBshort are identical in sequence, and differ by only two nucleotides from RrFshort. We therefore expect RrA, B, Fshort to serve identical functions, and arbitrarily chose the 115 nt long RrBshort from the rrnB operon for these experiments. An in vitro transcript of the RNA was incubated with increasing concentrations of Hfq, followed by exposure to the RNA cleavage agent Pb2+ that primarily hydrolyzes single-stranded RNA (Ciesiołka et al., 1998), or the endoribonuclease RNase III that predominantly hydrolyzes double-stranded RNA with a strong preference for helices of sufficient length (Robertson, 1990). Fragmented RNA was size-separated on a polyacrylamide gel and visualized by autoradiography (Figure 6A) In general, only subtle changes in the fragment pattern were observed upon Hfq addition (Figure 6A). The most notable Hfq-induced change was found in the Pb2+-treated samples at the apparent single-stranded region at nt 63-64, 66-68. This sequence displayed reduced hydrolysis by Pb2+ in the presence of Hfq (Figure 6A). In contrast, nt 53-56 showed slightly enhanced Pb2+ cleavage, upon addition of Hfq. Finally, nt 48-50 displayed slightly enhanced RNase III cleavage after incubation with Hfq (Figure 6A). The change in cleavage pattern observed at nt 63-68 as a consequence of Hfq addition could be interpreted as direct binding of Hfq in the area, leading to protection from Pb2+-induced cleavage. Alternatively, the main outcome of interaction with Hfq may be a structural rearrangement that allows the region around nt 63-68 to engage in intramolecular base-pairing. The structure probing data do not allow us to distinguish between these two potential effects of Hfq. To pursue the hypothesis that Hfq may facilitate a structural rearrangement of the region, we used the algorithm Mfold (Zuker, 2003)3 to predict the most thermodynamically favorable RrBshort structure with (Figure 6B) and without (Figure 6C) the constraint of a single-stranded region from nt 63-68 (open red triangles). The predicted structure for RrBshort in the absence of the constraint (Figure 6C) forms stem loop #2 predicted by the consensus structure (Figure 5), whereas the predicted structure given the structural constraint of a single-stranded region from nt 63-68, does not (Figure 6B). The structure shown in Figure 6B is not the energetically most favorable one. This leads us to suggest that Hfq could be involved in folding of the RrA, B, F in order for the RNA to attain its most thermodynamically favorable structure, namely that shown in Figure 6C, which shows the consensus stem-loop structure (stem loop #2) predicted by locARNA.
FIGURE 6

Structure probing of RrBshort with and without Hfq. The RrBshortin vitro transcript was incubated with 0, 1.5, or 3 μM of Hfq6 before addition of the cleavage agent. The partly digested RNA was size-separated on a denaturing 8% polyacrylamide gel. An autoradiogram of the gel is shown in panel (A). C1 indicates the transcript in Pb2 + -buffer without addition of Pb2 +, C2 indicates the transcript in RNase III-buffer without addition of RNase III. T1 indicates the heated transcript treated with RNase T1 and OH indicates the transcript heated in alkaline buffer. Gray bars on the left side of the gel mark areas of Pb2 + protection, Pb2 + enhancement and RNase III enhancement. The nucleotide number with respect to the 5′ end is indicated at the right side of the gel. Intense bands at position 39 and 43 in all lanes can probably be attributed to background hydrolysis of the purified transcript before the analysis since they are abundant in all lanes. The structure of RrBshort was predicted by mfold with the constraint of a single-stranded region at nt 63-68 (B) or without any constraints (C). The predicted free energies of the structures are indicated. Triangles and stars mark Hfq-dependent structural changes as indicated. GGA sequence motifs are marked by green ellipses; see section “RrB binds to CsrA in vitro.”
RrB Binds to CsrA in vitro
In order to identify potential interaction partners of RrA, B, F besides Hfq, we used MS2 affinity purification coupled with either mass spectrometry or RNA sequencing (Said et al., 2009; Corcoran et al., 2012; Lalaouna et al., 2015). Here, an RNA of interest is expressed as a fusion with an MS2-aptamer sequence, to allow purification of in vivo formed RNA-RNA or RNA-protein complexes using immobilized MS2 protein (Bardwell and Wickens, 1990; Said et al., 2009). We constructed plasmid-borne versions of RrBshort, MS2-tagged at either the 5′-end or the 3′-end and affinity-purified the RNAs. Expression and purification of the tagged RNAs was verified by northern blotting (Supplementary Figure 4). RNAs co-purifying with RrBshort were identified by deep RNA sequencing (RNA-seq) and co-purifying proteins were identified by mass spectrometry. The results were compared to results for affinity purification of the MS2 RNA alone. While no specific enrichment was reproducibly detected in the RNA-seq analysis (data not shown), several proteins were specifically enriched in pull-downs with MS2-tagged RrBshort, the most strongly enriched protein being the RNA-binding post-transcriptional regulator CsrA (Table 1). Somewhat surprising, Hfq was not found among the enriched proteins. We verified that RrBshort binds Hfq in vitro by electrophoretic mobility shift assay (EMSA). The EMSA analysis showed that RrBshort bound Hfq in vitro but with substantially lower affinity than the established Hfq-binding sRNA OxyS (Supplementary Figure 5). We will return to this point in section “Discussion.”
TABLE 1
 |
Proteins identified by MS2-affinity purification to co-purify with RrBshort.
The canonical CsrA binding motif identified both in S. typhimurium (Holmqvist et al., 2016) and E. coli (Dubey et al., 2005; Potts et al., 2017) contains a GGA sequence located in the loop of a stem-loop structure. Interestingly, the RrA, B, Fshort sequences have five GGA sequences, two of which are predicted to be located in the loops of stem-loop structures by the mfold and LocaRNA structure prediction algorithms (Figures 5, 6). Intriguingly, the structure probing experiments shown in Figure 6A suggest that the potential CsrA binding motif in loop 2 is only formed after interaction with Hfq.
The machine-learning-based algorithm InvenireSRNA (Fakhry et al., 2017) is designed to predict sRNAs of the CsrB/C family. To gauge how RrA, B, F rank in this algorithm compared to other E. coli non-coding RNAs, we provided the algorithm with a total of 2902 sequences, including all annotated E. coli sRNA sequences, the 22 TLR sequences, all sequences identified as CsrA-binding peaks from CLIP-seq data (Potts et al., 2017), and finally all intergenic regions of E. coli shorter than 1,000 bp. The results show that CsrB and CsrC both score high as expected (Table 2), while McaS and GadY both have low probability scores, suggesting that the features that make the latter acceptable binding partners for CsrA are not picked up by the algorithm. In accordance with the MS2-purification and structure prediction, RrAshort, RrBshort and RrFshort are predicted to bind CsrA with high probability and, remarkably, rank among the 16 highest scoring sequences in the analysis (Table 2). Notably, the remaining 19 members of the TLR family located in tRNA operons all obtained a probability score of <0.01, suggesting RrA, B, F are unique among the TLR family in their affinity for CsrA.
TABLE 2
| RNA | Probability score |
| CsrB sRNA | 0.9997 |
| csrB chip-SEQ | 0.9977 |
| speA chip-SEQ | 0.9507 |
| glyQ_ysaB | 0.9505 |
| setA_leuD | 0,8794 |
| OmpX chip-SEQ | 0.872 |
| lhr_grxD | 0.8634 |
| tatE_lipA | 0,8518 |
| CsrC sRNA | 0.8435 |
| zapB chip-SEQ | 0.8308 |
| csrC chip-SEQ | 0.7979 |
| ybhH_ybhI | 0.7439 |
| gltD chip-SEQ | 0.7013 |
| yqfE_argP | 0.6407 |
| RrA, Bshort | 0.6245 |
| RrFshort | 0.5833 |
| McaS | 0.005 |
| GadY | 0.0003 |
Transcripts predicted to regulate CsrA by the algorithm InvenireSRNA (Fakhry et al., 2017).
The input into InvenireSRNA was all annotated sRNAs, the 22 TLR sequences, all sequences identified as peaks in a comprehensive CsrA CHIP-seq experiment (Potts et al., 2017) and all intergenic regions of E. coli shorter than 1,000 bp (2902 sequences in total). The table displays the 16 highest-scoring transcripts from the analysis, and the sRNAs McaS and GadY, along with the associated probability scores. Intergenic regions are named by the two genes flaking them, separated by “_” and CHIP-seq peaks by addition of “chip-SEQ” to the gene name. Note that CsrB/C are present twice as they are included as full-length sequences from the list of sRNAs and as peak regions from the CHIP-seq data.
To further validate the binding between RrBshort and CsrA we conducted EMSAs. In vitro transcribed RrBshort was incubated with increasing amounts of purified CsrA-3xFLAG and separated on non-denaturing gels. As seen in Figure 7A, an RrBshort-CsrA complex is observed from a concentration of 50 nM CsrA, and a complete shift is seen at 100–200 nM CsrA. At higher concentrations of CsrA, several complexes with higher molecular weights are visible. The number of different band sizes suggests that RrBshort has multiple binding sites for CsrA, which is expected based on our RNA structure predictions (Figures 5, 6). The binding relationship between CsrA and RrBshort is therefore complex but a first approximation of an overall KD from our data in Figure 7A is around 100 nM. We repeated the binding experiment using a mutant version of RrBshort, where three GGA sequences have been changed to “UUU” (number 1, 2 and 5 on Figure 6C), which includes the two motifs found in loops of stem structures. By only mutating the three GGA motifs found in single stranded regions, we do not expect this to have any consequences for the secondary structure of the RNA. The affinity for CsrA is clearly lower for this mutant compared to the wildtype RrBshort as the mutant does not show any shift within the tested range of CsrA concentrations. Note that part of the RNA is degraded at the highest CsrA concentration. We attribute this to residual activity of the RNase that was added during CsrA purification, see materials and methods. To further probe the specificity of RrBshort to CsrA we did competition binding experiments where a preformed CsrA-RrBshort complex (CsrA at 200 nM, RrBshort at 120 pM) was challenged with increasing concentrations of the unlabeled competitors RrBshort, RrBshort-3GGA and CsrB (Figure 7B). CsrB is believed to have ∼18 CsrA binding sites (Liu et al., 1997), a KD around 1 nM of binding to CsrA (Weilbacher et al., 2003) and thus we expect it to be a more efficient competitor than RrBshort. This is indeed what we observe, CsrB fully competes off the labeled RrBshort at a concentration of 3.13 nM, whereas a concentration of 50 nM is needed in the self-competition with RrBshort. RrBshort-3GGA show significantly less competition and is not able to fully compete the labeled RrBshort off CsrA within the range of concentrations tested.
FIGURE 7
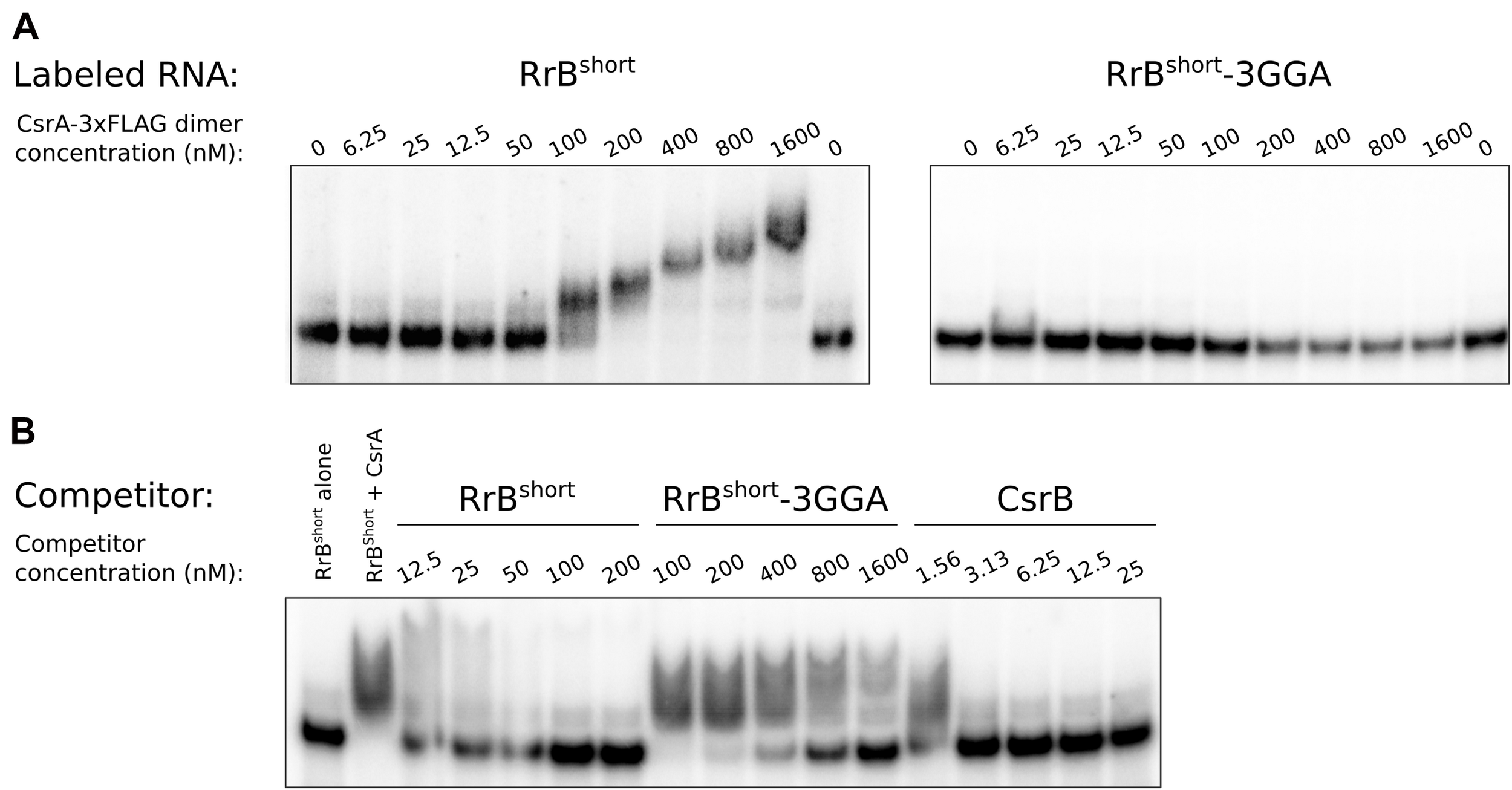
Electrophoretic mobility shift assays (EMSA) of RrBshort with CsrA. EMSA with 120 pM in vitro transcribed RNA and increasing concentration of CsrA-3xFLAG, as indicated. (A) EMSA of 32P-labeled RrBshort and an RrBshort mutant (RrBshort-3GGA) where three out of five GGA motifs where replaced with ‘UUU’, mixed with increasing concentration of CsrA-3xFLAG, as indicated. (B) A competition assay with 32P-labeled RrBshort (120 pM), CsrA (200 nM) and increasing concentration of the in vitro transcribed unlabeled RNAs: RrBshort, RrBshort-3GGA and the well-known CsrA-binding RNA CsrB. Note the differences in concentrations between the different competitor RNAs. The first lane contains no CsrA in any of the experiments. The stated CsrA concentrations refer to the concentrations of the homodimer.
With these binding assays we clearly show that RrBshort binds CsrA in vitro and that this interaction relies on the predicted binding sites located in single-stranded regions. The two remaining GGA motifs in the mutant RNA RrBshort-3GGA do not efficiently bind CsrA on their own. We suspect that these low affinity GGA sites may require nearby high affinity sites in order to bind CsrA, this type of binding has previously been described for the CsrA homolog RsmE (Duss et al., 2014).
In combination, our results from MS2 affinity purification, structure prediction, the InvenireSRNA algorithm, and the EMSAs strongly suggest that RrBshort, and likely RrA, B, Fshort, specifically interact with the post-transcriptional regulator CsrA.
Phenotypic Effects of Altered RrA, B, F Levels
We next asked whether RrA, B, F might act by sequestering CsrA, similarly to the effects of CsrB, CsrC, GadY and McaS. To this end, we constructed an E. coli mutant deleted for the chromosomal copies of rrA, B, F (see Materials and Methods). Additionally, to overexpress rrB in a fashion that would presumably allow normal processing of RrBshort, we cloned the rrB sequence including the 3′-half of the adjacent 5S gene and both terminators (T1 and T2) into a modified pUC18 plasmid (pJFR1, see Supplementary Table 2) to make pRrB.
Putative effects of RrB expression on the activity of CsrA were investigated by monitoring several CsrA-regulated phenotypes: growth rate, biofilm formation, motility, and post-transcriptional repression of the CsrA-regulated genes pgaA (Wang et al., 2005) and glgC (Baker et al., 2002). As a positive control, we moved a previously characterized csrA::kan allele (Romeo et al., 1993) into our wildtype strain (which is otherwise isogenic to the ΔrrA, B, F strain). The csrA::kan allele encodes a truncated version of CsrA which is known to have strongly reduced CsrA activity (Romeo et al., 1993). The results of these analyses are presented in Figure 8. In general, if RrA, B, F regulate CsrA activity in a manner similar to CsrB, we expect the phenotype of overexpression of rrB to display a similar tendency to that of the csrA::kan allele. Neither deletion nor overexpression of the rrA, B, F significantly affected the growth rate in minimal glucose medium (Figure 8A). For comparison, E. coli harboring a truncated CsrA have been reported to grow at approximately half the growth rate of the wildtype strain when glucose is the carbon source (Morin et al., 2016). The mutants lacking or overexpressing the rrA, B, F were also tested for motility on soft agar plates. Neither overexpression nor deletion of the rrA, B, F affected motility (Figure 8B). In contrast, the csrA::kan strain was severely impaired for motility as previously described (Romeo et al., 1993), although we note that this phenotype in our hands was very variable between replicate experiments.
FIGURE 8
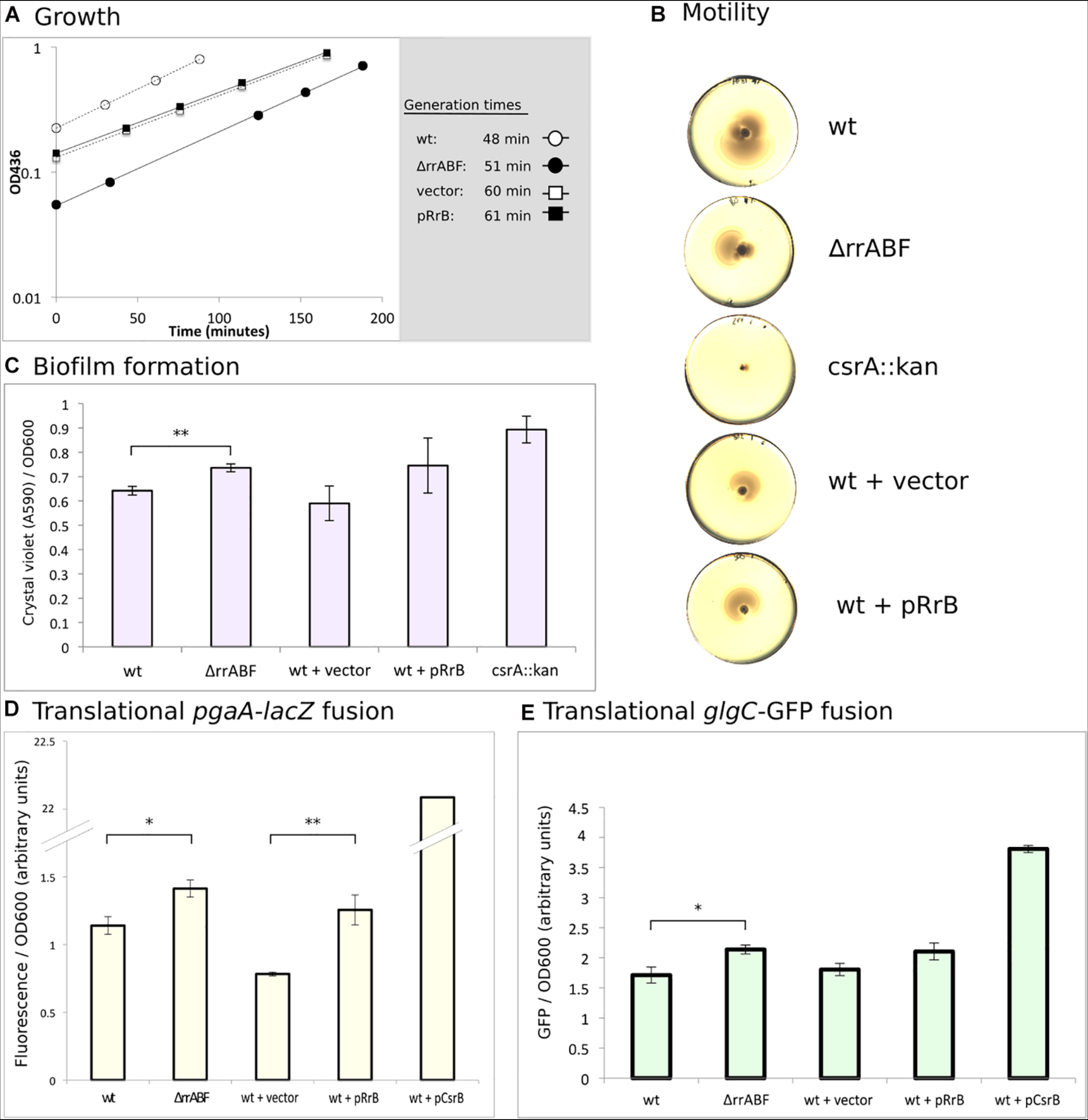
Phenotypes of deletion and overexpression of the rrA, B, F genes. (A) Growth of the four indicated strains. Cultures were grown in MOPS minimal media supplemented with 0.2% glucose and 100 μg/ml ampicillin for strains harboring plasmids. The indicated generation times were calculated as the average of two independent replicates. (B) Motility of the indicated strains was assayed on soft YT agar (0.3%) plates. Color and contrast of the photo has been manipulated to more clearly visualize the zone of bacterial growth. (C) Amount of biofilm formed on “peg-lids” by the indicated strains was quantified using crystal violet staining and absorbance measurements. (D) Quantification of the fluorescence produced by cleavage of 4-methylumbelliferyl-β-D-galactopyranoside (MUG) by β-galactosidase for outgrown cultures of the indicated strains. All strains carried a plasmid with a translational fusion of the pgaA leader to lacZ (pTSS36). (E) Quantification of GFP-signal for strains growing in exponential phase. All stains carried the translational fusion of the glgC leader to sf-gfp on a plasmid (pTSS16). In all cases, statistical probability was calculated using a two-sided student’s t-test and is indicated (∗P < 0.05, ∗∗P < 0.01).
CsrA is a negative regulator of PGA (poly-β-1,6-N-acetyl-d-glucosamine)-mediated biofilm formation (Wang et al., 2005). In agreement with this, we found that the csrA::kan strain produces more biofilm than wildtype as measured by crystal violet staining (Figure 8C). The ΔrrA, B, F strain also formed significantly more biofilm than the wildtype. However, overexpression of RrB also showed somewhat increased biofilm formation, although the difference from the strain carrying the empty vector control was not statistically significant.
In connection with biofilm, translation of the pgaABCD mRNA, encoding the machinery for synthesis and transport of PGA, is known to be strongly repressed by CsrA (Wang et al., 2005). Indeed, we found > 20-fold higher activity of β-galactosidase from a pgaA-lacZ fusion in the presence of CsrB overexpression than in the wildtype strain (Figure 8D). A modest increase in β-galactosidase activity was also observed in the ΔrrA, B, F mutant. Again, overexpression of RrB resulted in a phenotype similar to that of the Δrr,AB,F mutant, pointing to a surprisingly similar effect on the pgaA-lacZ fusion of decreasing and increasing the RrA, B, F levels. There is good correlation between our data on biofilm formation and our data on the pgaA-lacZ fusion for all the strains (Figures 8C,D), as would be expected for CsrA-dependent regulation of biofilm (Wang et al., 2005).
Lastly, we tested the expression of GFP from a plasmid-borne translational glgC-gfp fusion during exponential growth, the growth phase where RrA, B, F accumulated (Figure 2C). The glgCAP operon encodes enzymes involved in glycogen metabolism, and is known to be repressed by CsrA (Baker et al., 2002). CsrB overexpression increases glgC-gfp expression substantially, while we observed slightly more GFP signal both upon deletion and overexpression of rrA, B, F (Figure 8E). In summary, we only observed minor phenotypic effects of changes in the expression of RrA, B, F. The RrA, B, F RNAs appear to modestly influence biofilm formation and expression from the CsrA-controlled pgaA and glgC mRNAs, but all three assays showed the same curious trend, namely that both deletion of rrA, B, F and overexpression of rrB resulted in a change consistent with modestly reduced CsrA activity. A plausible explanation for this unexpected similarity of phenotypes would be if RrB RNA expressed from the vector was processed into a form that exerted a different action on the tested properties than chromosomally expressed RrB. To address this hypothesis, we also tested expression of the glgC-gfp fusion in the presence of the pKK3535 plasmid that contains the entire rrnB operon, including rrB. Although the rRNA operon promoters are feedback-controlled and thus difficult to overexpress, this plasmid was expected to cause a relative increase in RrB levels, because the pKK3535 plasmid is responsible for approximately 50% of total rRNA synthesis even in the presence of the seven chromosomal rRNA operons (Steen et al., 1986). As shown in Supplementary Figure 5, overexpression of the rrnB operon resulted in increased glgC-gfp expression, and was thus consistent with reduced CsrA activity under this condition. In our hands, however, the strains containing pKK3535 had impaired growth rates (see Supplementary Figure 6A) and thus we did not pursue additional experiments with this plasmid.
The Concentration of RrA, B, F Increases Upon Translational Halt
The very modest effects of RrA, B, F on CsrA-controlled phenotypes suggest that if there is a physiological consequence of RrA, B, F interaction with CsrA then we might not have examined it under the proper growth conditions. In E. coli, rRNA expression is controlled by the second messenger ppGpp (Zhang and Bremer, 1995). A decrease in ppGpp levels leads to increased rRNA expression (as observed upon treatment with antibiotics specifically blocking translation (Kurland and Maaløe, 1962; Muto et al., 1975)). To test whether the ppGpp effect also applies to rrA, B, F, we induced translational arrest, either by chloramphenicol addition, or by expression of the toxin MazF, and investigated the level of RrA, B, F by northern blot analysis at several time points after induction. Chloramphenicol blocks translation by binding to ribosomes and inhibiting the peptidyl transferase activity (Das et al., 1966) while MazF leads to translational arrest by disrupting ribosome biogenesis and cleaving mRNAs (Culviner and Laub, 2018), thereby removing the template for translation. In both cases of translational arrest, we detect a strong increase in RrA, B, F transcript levels 20–80 min after the treatment (Figure 9). As an independent indicator of transcription from the rRNA operons, we also investigated the level of tRNAGlu. This tRNA is exclusively expressed from genes located in four rRNA operons (rrnB, rrnC, rrnE, rrnG), so we expected its expression pattern to resemble that of RrA, B, F. Indeed, the level of tRNAGlu also increases during translational arrest, albeit not nearly to the same extent as RrA, B, F (Figure 9).
FIGURE 9
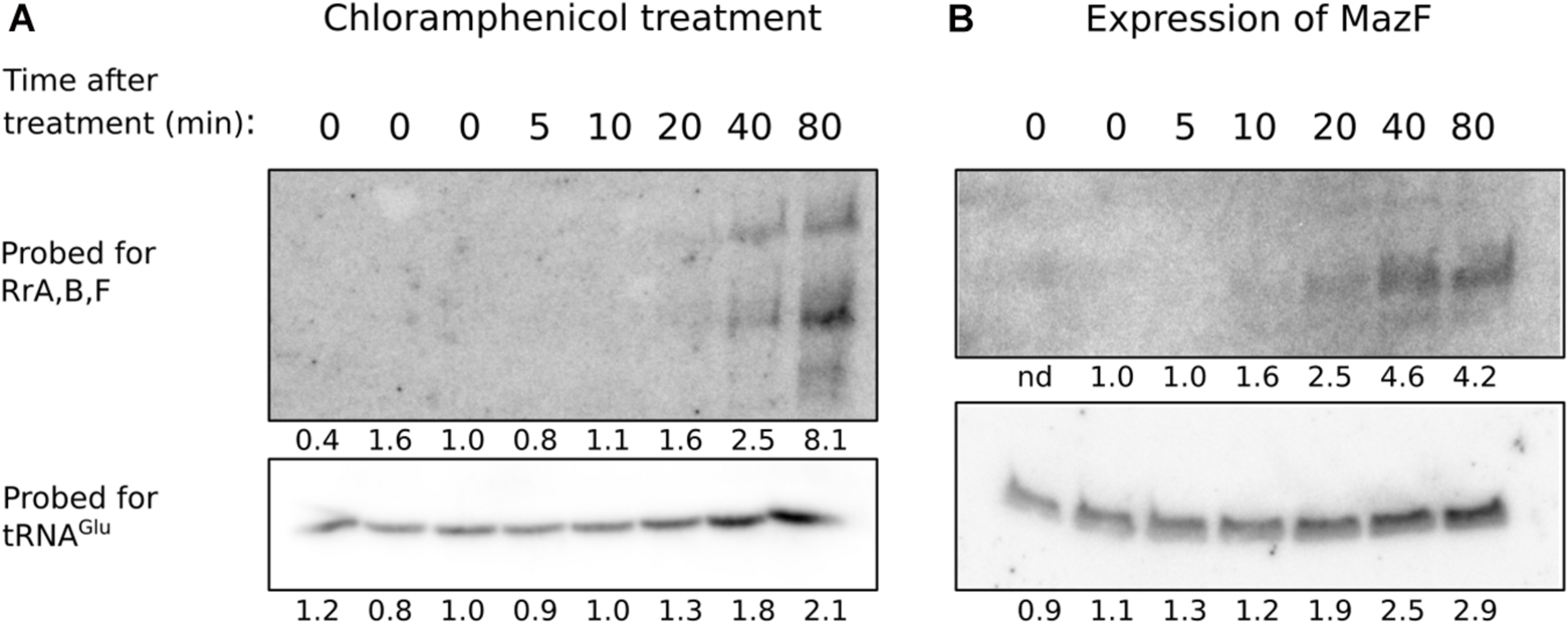
Levels of RrA, B, F after addition of chloramphenicol or induction of mazF expression. The strains MAS1081 (A) and TSS261 (MAS1081 + pMazF) (B) were grown in MOPS minimal media supplemented with 0.2% glucose or 0.4% glycerol, respectively. Treatment was induced by addition of either chloramphenicol (20 μg/ml) (A) or arabinose (0.1%) (B). RNA was harvested at the indicated times after treatment. Equal volumes of RNA (6–15 μg/lane) were separated on 6% denaturing polyacrylamide gels and northern blotted. The blots were probed for tRNAGlu and RrA, B, F as indicated on the figure. Quantification of the radioactivity signal in each lane is given relative to the average value of the steady-state levels and is shown below each lane. nd: not determined due to high background.
Discussion
We identify three small genes that are co-transcribed from rRNA operons and processed to transcripts of defined lengths that bind the post-transcriptional regulator CsrA both in vitro and in vivo. As the transcription rate of rRNA is strictly correlated with the growth rate of E. coli (Potrykus et al., 2011), it is tempting to speculate that such short-lived sRNAs transcribed along with rRNA could function to align growth-rate-regulated transcription rates with other growth-phase-dependent processes in the cell, such as those regulated by CsrA. The relatively short half-life of RrA, B, F compared to other sRNAs makes biological sense if their abundance should reflect the transcriptional activity of the rRNA operons in real time.
We find a modest effect of overexpression of rrB on three of the CsrA-regulated phenotypes tested (Figure 8). The phenotypes observed upon overexpression of rrB agree with those expected from a mutant with modestly reduced CsrA activity. Such an apparent inhibition of CsrA upon overexpression of rrB could be due to binding of RrB to CsrA, resulting in regulation by titration in the classical way first described for CsrB/C. Direct binding between CsrA and RrBshort is experimentally supported by the results of our RNA affinity purification and EMSA analyses as well as theoretically supported by the InvenireSRNA prediction and the presence of consensus CsrA-binding motifs in the RrBshort structure. The fact that deletion of the rrA, B, F genes showed similar phenotypes to RrB overexpression is then counterintuitive (see Figure 8). Potentially, the chromosomally encoded form of RrB could fulfill a function for which the version of RrB expressed from our multicopy plasmid has a dominant negative effect (overriding the phenotype of the wildtype allele), in which case similar phenotypes of the chromosomal rrA, B, F deletions and the RrB overexpression could be expected. The curious effect or RrB overexpression was investigated further by introducing a plasmid expressing the entire rrnB operon (Supplementary Figure 6), which confirmed the results obtained by specific overexpression of RrB (Figure 8E). Since overexpression of rrB by two different cloning tactics affected the glgC-gfp fusion similarly, we conclude that deletion and overexpression of RrB both result in phenotypes consistent with reduced CsrA activity, but cannot currently provide a mechanistic explanation for this curious observation.
While we have demonstrated an interaction between the RrA, B, F RNAs and CsrA, deletion and overexpression of rrA, B, F show only modest changes of the CsrA-related phenotypes tested in this study. We want to note that CsrA is the major hub in a regulatory network with many inputs (Romeo and Babitzke, 2018). For that reason, one might not expect that the absence, or presence in excess, of RrA, B, F would lead to prominent phenotypes, but merely fine tuning of the activity of CsrA. Further, EMSA analyses (Figure 7) showed that CsrB totally competed RrBshort off CsrA at a ten-fold lower concentration than RrBshort itself (Figure 7B). This shows that CsrB is a higher-affinity CsrA binding partner than RrA, B, F and that we therefore might expect small effects, if any, of changes to the RrA, B, F levels in situations where CsrA-regulation by CsrB/C is at play.
Our northern analysis shows that RrA, B, F are most abundant when the rRNA operons are expressed, namely during exponential growth (Figure 2C), and especially upon treatments that lower ppGpp production by arresting translation (Figure 9). During translational halt, RrA, B, F levels increased more than the tRNAglu, which is also expressed solely from rRNA operons. This difference in the extent of induction could either mean that there is differential expression of the genes in the rRNA operons upon translational arrest, or, more likely, translational arrest may result in increased stability of the RrA, B, F sRNAs relative to tRNAglu by an unknown mechanism. Our data do not allow us to distinguish between these possibilities. Nevertheless, the experiments show that conditions exist in which the abundance of the RrA, B, F RNAs increase substantially. If they play a greater role in cell physiology than the fine-tuning of CsrA-regulated phenotypes shown in Figure 8, their impact should maybe be sought under such conditions, or at least under conditions where CsrB/C concentrations are low but the transcription rate of rRNA is high.
We find that rrA, B, F is well conserved in various different genera of bacteria including numerous pathogenic species. It is well known that growth-phase-dependent regulation is important for bacterial virulence (Dalebroux and Swanson, 2012; Kitamoto et al., 2016). Thus, if RrA, B, F is used as an indicator of growth rate in the cell, it could also potentially have a regulatory effect on the pathogenesis of these virulent stains.
A hallmark of all the members of the TLR family found in E. coli is the presence of a short (18–19 bp) repeated sequence identical to the 3′-end of the mature tRNA or rRNA from the same operon. For the tRNA or rRNA this 3′-end sequence is important for processing (reviewed in Mackie, 2013). The 3′-end of the 173-nt form of RrB was mapped 3 nt downstream of the repeated sequence that is identical to the final 18 bp of mature 5S RNA. As this repeated sequence and structure is known to be important for the RNase E cleavage immediately downstream of 5S, we suggest that the repeat in the RrB 3′-end is also recognized by RNase E, and that this explains the function of the repeated sequence.
RrBshort was enriched on Hfq in the co-purification experiment (Figure 2), and showed specific binding to Hfq by EMSA analysis (Supplementary Figure 5), which could suggest a role for Hfq in folding or processing of RrB. Hfq could affect folding and the kinetics of the processing but is not essential for processing since RrBshort was also observed in a Δhfq mutant (Supplementary Figure 7). Alternatively, the enrichment could reflect that RrBshort functions as a base-pairing sRNA to regulate one or more target RNAs, chaperoned by Hfq. GadY (Parker et al., 2017) and McaS (Jorgensen et al., 2013) are two recent examples of dual function sRNAs that can both function to bind and titrate out CsrA, and function as generic Hfq-dependent base-pairing sRNAs to regulate target mRNAs. Although we cannot rule out either option, we favor the former option, namely that Hfq participates in the folding or processing of RrB, which is also supported by our structure probing with and without Hfq (Figure 6). If Hfq assisted with pairing of RrBshort to a target RNA(s), we would have expected to detect the binding between RrBshort and Hfq in the mass spectrometry analysis of the proteins co-purifying with RrBshort, and the RNA-seq analysis could have revealed the target RNA(s). On the other hand, if the role of Hfq is to facilitate the refolding or processing of RrB, then the affinity of Hfq for the refolded product RrBshort need not be very high, why we speculate that competing binders with higher affinity, like the abundant CsrA protein, may have excluded binding of Hfq to RrBshort in the MS2-RNA affinity purification experiments.
The rrA, B, F genes are found downstream of the gene encoding 5S in three out of seven rRNA operons of E. coli. These three operons are furthermore the only three that contain two terminator sequences (T1 and T2), whereas the remaining four rRNA operons have a single terminator (Supplementary Figure 1; Lesnik et al., 2001). The significance of the dual terminators is not clear and has been puzzling since its discovery (Brosius et al., 1981; Brosius, 1984; Ghosh et al., 1991; Orosz et al., 1991). The termination efficiencies of each of the terminators from the rrnB operon have been measured. T1 was measured to terminate 87% of transcripts while T2 terminated 100% of transcripts (Orosz et al., 1991). As the rrA, B, F genes are located between the two terminators we suggest that this dual terminator arrangement could serve the purpose of regulating the expression of rrA, B, F. In this way, the expression of rrA, B, F will be directly linked to expression of rRNA and growth rate, but they would be expressed at a lower level than the rRNA, which are among the most highly transcribed genes in the cell. We point out that the dual terminators of ribosomal operon B, including the rrB gene, are among the most highly used transcription terminator sequences on plasmids for cloning and protein expression (Denèfle et al., 1987; Andrews et al., 1996; Rogers et al., 2015; Wille et al., 2015; Engstrom and Pfleger, 2017). Our demonstration that the RrA, B, F molecules can interact with at least two pleiotropic regulatory proteins in E. coli (Hfq and CsrA), warrants careful consideration of whether expression of such putative sRNAs from a high copy number plasmid is desirable for a given application.
Several studies on eukaryotic organisms have identified potential miRNAs transcribed as part of rRNA primary transcripts (Wei et al., 2013; Chak et al., 2015; Asha and Soniya, 2017), and the murine-specific miR-712, transcribed as a part of a spacer element in the pre-rRNA transcript, has been shown to be involved in endothelial inflammation and atherosclerosis (Son et al., 2013). To our knowledge, the RrA, B, F sRNAs described here represent the first report of sRNAs transcribed as part of rRNA primary transcripts in a prokaryote. In conclusion, three out of seven rRNA operons in E. coli contains an arrangement of dual terminators with small defined RNAs encoded in between, and these RNAs are conserved both in structure and sequence among species belonging to different genera of Enterobacteriales. We have found that these RNAs interact with both Hfq and CsrA but has been unable to demonstrate clear phenotypes of both deletion and overexpression mutants. We presume that the conservation among species indicate a function for the RNAs but that this function is still not fully understood.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.ebi.ac.uk/pride/archive/projects/PXD013749.
Author contributions
TS, MK, SS, and MS wrote the manuscript, conceived and designed the research. TS, MK, BK, EH, SS, and MS designed the experiments. MK (Hfq) and TS (CsrA) performed the experiments. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Lundbeck Foundation [R108-A10583 to MS], the Danish council for Independent Research | Natural Sciences [1323-00343B to SS] and the Danish National Research Foundation [DNRF120 to SS; MS].
Acknowledgments
The authors thank Marit Warrer, Bertil Gummessson, and Pilar Menéndez Gil for excellent technical assistance. The authors thank Jörg Vogel for help and support to TS during his visit in Würzburg. The authors also thank Anders Boysen for the kind supply of purified Hfq.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.625585/full#supplementary-material
Footnotes
References
1
AibaH.AdhyaS.de CrombruggheB. (1981). Evidence for two functional gal promoters in intact Escherichia coli cells.J. Biol. Chem.25611905–11910.
2
AltierC.SuyemotoM.RuizA. I.BurnhamK. D.MaurerR. (2002). Characterization of two novel regulatory genes affecting Salmonella invasion gene expression.Mol. Microbiol.35635–646. 10.1046/j.1365-2958.2000.01734.x
3
AndrewsB.AdariH.HannigG.LahueE.GosselinM.MartinS.et al (1996). A tightly regulated high level expression vector that utilizes a thermosensitive lac repressor: production of the human T cell receptor Vβ5.3 in Escherichia coli.Gene182101–109. 10.1016/S0378-1119(96)00523-9
4
AshaS.SoniyaE. V. (2017). The sRNAome mining revealed existence of unique signature small RNAs derived from 5.8SrRNA from Piper nigrum and other plant lineages.Sci. Rep.7:41052. 10.1038/srep41052
5
BaglioS. R.RooijersK.Koppers-LalicD.VerweijF. J.Pérez LanzónM.ZiniN.et al (2015). Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species.Stem Cell Res. Ther.6:127. 10.1186/s13287-015-0116-z
6
BakerC. S.MorozovI.SuzukiK.RomeoT.BabitzkeP. (2002). CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli.Mol. Microbiol.441599–1610. 10.1046/j.1365-2958.2002.02982.x
7
BaracchiniE.BremerH. (1991). Control of rRNA synthesis in Escherichia coli at increased RRN gene dosage, Role of guanosine tetraphosphate and ribosome feedback.J. Biol. Chem.26611753–11760. 10.1016/S0021-9258(18)99021-6
8
BardwellV. J.WickensM. (1990). Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method.Nucleic Acids Res.186587–6594.
9
BeckettD.KovalevaE.SchatzP. J. (2008). A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation.Protein Sci.8921–929. 10.1110/ps.8.4.921
10
BeiselC. L.StorzG. (2011). The base-pairing RNA Spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli.Mol. Cell41286–297. 10.1016/j.molcel.2010.12.027
11
BerkA. J.SharpP. A. (1977). Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of S1 endonuclease-digested hybrids.Cell12721–732. 10.1016/0092-8674(77)90272-0
12
BernsteinJ. A.KhodurskyA. B.LinP.-H.Lin-ChaoS.CohenS. N. (2002). Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays.Proc. Natl. Acad. Sci. U.S.A.999697–9702. 10.1073/pnas.112318199
13
BilusicI.PopitschN.ReschenederP.SchroederR.LybeckerM. (2014). Revisiting the coding potential of the E. coli genome through Hfq co-immunoprecipitation.RNA Biol.11641–654. 10.4161/rna.29299
14
BordeauV.FeldenB. (2014). Curli synthesis and biofilm formation in enteric bacteria are controlled by a dynamic small RNA module made up of a pseudoknot assisted by an RNA chaperone.Nucleic Acids Res.424682–4696. 10.1093/nar/gku098
15
BöslM.KerstenH. (1991). A novel RNA product of the tyrT operon of Escherichia Coli.Nucleic Acids Res.195863–5870. 10.1093/nar/19.21.5863
16
BoslM.KerstenH. (1994). Organization and functions of genes in the upstream region of tyrT of Escherichia coli: phenotypes of mutants with partial deletion of a new gene (tgs).J. Bacteriol.176221–231. 10.1128/jb.176.1.221-231.1994
17
BrantlS.JahnN. (2015). sRNAs in bacterial type I and type III toxin-antitoxin systems.FEMS Microbiol. Rev.39413–427.
18
BrosiusJ. (1984). Toxicity of an overproduced foreign gene product in Escherichia coli and its use in plasmid vectors for the selection of transcription terminators.Gene27161–172. 10.1016/0378-1119(84)90137-9
19
BrosiusJ.CateR. L.PerlmutterA. P. (1982). Precise location of two promoters for the beta-lactamase gene of pBR322. S1 mapping of ribonucleic acid isolated from Escherichia coli or synthesized in vitro.J. Biol. Chem.2579205–9210.
20
BrosiusJ.DullT. J.SleeterD. D.NollerH. F. (1981). Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli.J. Mol. Biol.148107–127. 10.1016/0022-2836(81)90508-8
21
ChakL. L.MohammedJ.LaiE. C.Tucker-KelloggG.OkamuraK. (2015). A deeply conserved, noncanonical miRNA hosted by ribosomal DNA.RNA21375–384. 10.1261/rna.049098.114
22
ChaoY.PapenfortK.ReinhardtR.SharmaC. M.VogelJ. (2012). An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs.EMBO J.314005–4019. 10.1038/emboj.2012.229
23
CiesiołkaJ.MichałowskiD.WrzesinskiJ.KrajewskiJ.KrzyżosiakW. J. (1998). Patterns of cleavages induced by lead ions in defined RNA secondary structure motifs 1 1Edited by I. Tinoco.J. Mol. Biol.275211–220. 10.1006/jmbi.1997.1462
24
CorcoranC. P.RiederR.PodkaminskiD.HofmannB.VogelJ. (2012). “Use of aptamer tagging to identify in vivo protein binding partners of small regulatory RNAs,” inBacterial Regulatory RNA, ed.KeilerK. C. (Totowa, NJ: Humana Press), 177–200. 10.1007/978-1-61779-949-5_11
25
CulvinerP. H.LaubM. T. (2018). Global analysis of the E. coli Toxin MazF reveals widespread cleavage of mRNA and the inhibition of rRNA maturation and ribosome biogenesis.Mol. Cell70868–880.e10. 10.1016/j.molcel.2018.04.026
26
DalebrouxZ. D.SwansonM. S. (2012). ppGpp: magic beyond RNA polymerase.Nat. Rev. Microbiol.10203–212. 10.1038/nrmicro2720
27
DasH. K.GoldsteinA.KannerL. C. (1966). Inhibition by chlorampenicol of the growth of nascent protein chains in Escherichia coli.Mol. Pharmacol.2158–170.
28
DenèfleP.KovarikS.GuittonJ.-D.CartwrightT.MayauxJ.-F. (1987). Chemical synthesis of a gene coding for human angiogenin, its expression in Escherichia coli and conversion of the product into its active form.Gene5661–70. 10.1016/0378-1119(87)90158-2
29
DubeyA. K.BakerC. S.RomeoT.BabitzkeP. (2005). RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction.RNA111579–1587. 10.1261/rna.2990205
30
DussO.MichelE.Diarra dit KontéN.SchubertM.AllainF. H.-T. (2014). Molecular basis for the wide range of affinity found in Csr/Rsm protein–RNA recognition.Nucleic Acids Res.425332–5346. 10.1093/nar/gku141
31
EganJ.LandyA. (1978). Structural analysis of the tRNA1Tyr gene of Escherichia coli. A 178 base pair sequence that is repeated 3.14 times.J. Biol. Chem.2533607–3622.
32
EmaraM. M.IvanovP.HickmanT.DawraN.TisdaleS.KedershaN.et al (2010). Angiogenin-induced tRNA-derived Stress-induced RNAs promote stress-induced stress granule assembly.J. Biol. Chem.28510959–10968. 10.1074/jbc.M109.077560
33
EngstromM. D.PflegerB. F. (2017). Transcription control engineering and applications in synthetic biology.Synth. Syst. Biotechnol.2176–191. 10.1016/j.synbio.2017.09.003
34
FakhryC. T.KulkarniP.ChenP.KulkarniR.ZarringhalamK. (2017). Prediction of bacterial small RNAs in the RsmA (CsrA) and ToxT pathways: a machine learning approach.BMC Genomics18:645. 10.1186/s12864-017-4057-z
35
FesslerM.GummessonB.CharbonG.SvenningsenS. L.SørensenM. A. (2020). Short-term kinetics of rRNA degradation in Escherichia coli upon starvation for carbon, amino acid or phosphate.Mol. Microbiol.113951–963. 10.1111/mmi.14462
36
GeissmannT. A.TouatiD. (2004). Hfq, a new chaperoning role: binding to messenger RNA determines access for small RNA regulator.EMBO J.23396–405. 10.1038/sj.emboj.7600058
37
GhoshB.GrzadzielskaE.BhattacharyaP.PeraltaE.DeVitoJ.DasA. (1991). Specificity of antitermination mechanisms.J. Mol. Biol.22259–66. 10.1016/0022-2836(91)90737-Q
38
Gonzalez ChavezR.AlvarezA. F.RomeoT.GeorgellisD. (2010). The physiological stimulus for the BarA Sensor Kinase.J. Bacteriol.1922009–2012. 10.1128/JB.01685-09
39
GreenM. R.RoederR. G. (1980). Definition of a novel promoter for the major adenovirus-associated virus mRNA.Cell22231–242. 10.1016/0092-8674(80)90171-3
40
HoekzemaM.RomillyC.HolmqvistE.WagnerE. G. H. (2019). Hfq-dependent mRNA unfolding promotes sRNA-based inhibition of translation.EMBO J.38:e101199. 10.15252/embj.2018101199
41
HolmqvistE.WagnerE. G. H. (2017). Impact of bacterial sRNAs in stress responses.Biochem. Soc. Trans.451203–1212. 10.1042/BST20160363
42
HolmqvistE.WrightP. R.LiL.BischlerT.BarquistL.ReinhardtR.et al (2016). Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo.EMBO J.35991–1011. 10.15252/embj.201593360
43
JacksonD. W.SuzukiK.OakfordL.SimeckaJ. W.HartM. E.RomeoT. (2002). Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli.J. Bacteriol.184290–301. 10.1128/JB.184.1.290-301.2002
44
JørgensenM. G.NielsenJ. S.BoysenA.FranchT.Møller-JensenJ.Valentin-HansenP. (2012). Small regulatory RNAs control the multi-cellular adhesive lifestyle of Escherichia coli.Mol. Microbiol.8436–50. 10.1111/j.1365-2958.2012.07976.x
45
JorgensenM. G.ThomasonM. K.HavelundJ.Valentin-HansenP.StorzG. (2013). Dual function of the McaS small RNA in controlling biofilm formation.Genes Dev.271132–1145. 10.1101/gad.214734.113
46
KawanoM.ReynoldsA. A.Miranda-RiosJ.StorzG. (2005). Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli.Nucleic Acids Res.331040–1050. 10.1093/nar/gki256
47
KayB. K.ThaiS.VolginaV. V. (2009). High-throughput biotinylation of proteins.Methods Mol. Biol.498185–198. 10.1007/978-1-59745-196-3_13
48
KitamotoS.Nagao-KitamotoH.KuffaP.KamadaN. (2016). Regulation of virulence: the rise and fall of gastrointestinal pathogens.J. Gastroenterol.51195–205. 10.1007/s00535-015-1141-5
49
KurlandC. G.MaaløeO. (1962). Regulation of ribosomal and transfer RNA synthesis.J. Mol. Biol.4193–210. 10.1016/S0022-2836(62)80051-5
50
LalaounaD.CarrierM. C.SemseyS.BrouardJ. S.WangJ.WadeJ. T.et al (2015). A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise.Mol. Cell58393–405. 10.1016/j.molcel.2015.03.013
51
LawhonS. D.MaurerR.SuyemotoM.AltierC. (2002). Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA.Mol. Microbiol.461451–1464. 10.1046/j.1365-2958.2002.03268.x
52
LesnikE. A.SampathR.LeveneH. B.HendersonT. J.McNeilJ. A.EckerD. J. (2001). Prediction of rho-independent transcriptional terminators in Escherichia coli.Nucleic Acids Res.293583–3594. 10.1093/nar/29.17.3583
53
LiS. H.-J.LiZ.ParkJ. O.KingC. G.RabinowitzJ. D.WingreenN. S.et al (2018). Escherichia coli translation strategies differ across carbon, nitrogen and phosphorus limitation conditions.Nat. Microbiol.3939–947. 10.1038/s41564-018-0199-2
54
LiZ.DeutscherM. P. (1995). The tRNA processing enzyme RNase T is essential for maturation of 5S RNA.Proc. Natl. Acad. Sci. U.S.A.926883–6886. 10.1073/pnas.92.15.6883
55
LiuM. Y.GuiG.WeiB.PrestonJ. F.OakfordL.YükselÜ.et al (1997). The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli.J. Biol. Chem.27217502–17510. 10.1074/jbc.272.28.17502
56
LiuM. Y.YangH.RomeoT. (1995). The product of the pleiotropic Escherichia coli gene csrA modulates glycogen biosynthesis via effects on mRNA stability.J. Bacteriol.1772663–2672. 10.1128/jb.177.10.2663-2672.1995
57
MackieG. A. (2013). RNase E: at the interface of bacterial RNA processing and decay.Nat. Rev. Microbiol.1145–57. 10.1038/nrmicro2930
58
MartinezG.ChouduryS. G.SlotkinR. K. (2017). tRNA-derived small RNAs target transposable element transcripts.Nucleic Acids Res.455142–5152.
59
McGrathP. T. (2011). Characterizing cDNA Ends by Circular RACE.Methods Mol. Biol.772257–265. 10.1007/978-1-61779-228-1_15
60
MillerJ. H. (1972). Experiments in Molecular Genetics.New York, NY: Plenum Press.
61
MiyakoshiM.ChaoY.VogelJ. (2015). Regulatory small RNAs from the 3′ regions of bacterial mRNAs.Curr. Opin. Microbiol.24132–139. 10.1016/j.mib.2015.01.013
62
MøllerT.FranchT.UdesenC.GerdesK.Valentin-HansenP. (2002). Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon.Genes Dev.161696–1706. 10.1101/gad.231702
63
MoonK.GottesmanS. (2009). A PhoQ/P-regulated small RNA regulates sensitivity of Escherichia coli to antimicrobial peptides.Mol. Microbiol.741314–1330. 10.1111/j.1365-2958.2009.06944.x
64
MorinM.RopersD.LetisseF.LaguerreS.PortaisJ.-C.Cocaign-BousquetM.et al (2016). The post-transcriptional regulatory system CSR controls the balance of metabolic pools in upper glycolysis of Escherichia coli.Mol. Microbiol.100686–700. 10.1111/mmi.13343
65
MutoA.KimuraA.OsawaS. (1975). Effects of some antibiotics on the stringent control of RNA synthesis in Escherichia coli.Mol. Gen. Genet.139321–327. 10.1007/BF00267972
66
NeidhardtF. C.BlochP. L.SmithD. F. (1974). Culture medium for enterobacteria.J. Bacteriol.119736–747.
67
OpdykeJ. A.KangJ.-G.StorzG. (2004). GadY, a Small-RNA regulator of acid response genes in Escherichia coli.J. Bacteriol.1866698–6705. 10.1128/JB.186.20.6698-6705.2004
68
OroszA.BorosI.VenetianerP. (1991). Analysis of the complex transcription termination region of the Escherichia coli rrn B gene.Eur. J. Biochem.201653–659. 10.1111/j.1432-1033.1991.tb16326.x
69
ParkerA.CureogluS.De LayN.MajdalaniN.GottesmanS. (2017). Alternative pathways for Escherichia coli biofilm formation revealed by sRNA overproduction.Mol. Microbiol.105309–325. 10.1111/mmi.13702
70
PiirK.PaierA.LiivA.TensonT.MaiväliÜ. (2011). Ribosome degradation in growing bacteria.EMBO Rep.12458–462. 10.1038/embor.2011.47
71
PotrykusK.MurphyH.PhilippeN.CashelM. (2011). ppGpp is the major source of growth rate control in E. coli.Environ. Microbiol.13563–575. 10.1111/j.1462-2920.2010.02357.x
72
PottsA. H.VakulskasC. A.PannuriA.YakhninH.BabitzkeP.RomeoT. (2017). Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics.Nat. Commun.8:1596. 10.1038/s41467-017-01613-1
73
RadenM.AliS. M.AlkhnbashiO. S.BuschA.CostaF.DavisJ. A.et al (2018). Freiburg RNA tools: a central online resource for RNA-focused research and teaching.Nucleic Acids Res.4625–29. 10.1093/nar/gky329
74
RasmussenA. A.JohansenJ.NielsenJ. S.OvergaardM.KallipolitisB.Valentin-HansenP. (2009). A conserved small RNA promotes silencing of the outer membrane protein YbfM.Mol. Microbiol.72566–577. 10.1111/j.1365-2958.2009.06688.x
75
RobertsonH. D. (1990). Escherichia coli ribonuclease III.Methods Enzymol.181189–202. 10.1016/0076-6879(90)81121-A
76
RogersJ. K.GuzmanC. D.TaylorN. D.RamanS.AndersonK.ChurchG. M. (2015). Synthetic biosensors for precise gene control and real-time monitoring of metabolites.Nucleic Acids Res.437648–7660. 10.1093/nar/gkv616
77
RomeoT.BabitzkeP. (2018). “Global regulation by CsrA and its RNA antagonists,” inRegulating with RNA in Bacteria and Archaea, edsStorzG.PapenfortK. (Washington, DC: ASM Press), 339–354. 10.1128/9781683670247.ch19
78
RomeoT.GongM.LiuM. Y.Brun-ZinkernagelA.-M. (1993). Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties.J. Bacteriol.1754744–4755. 10.1128/JB.175.15.4744-4755.1993
79
RoyM. K.SinghB.RayB. K.ApirionD. (1983). Maturation of 5S RNA : ribonuclease E cleavage and their dependence on precursor sequences.Eur. J. Biochem.131119–127.
80
RuddK. E. (1999). Novel intergenic repeats of Escherichia coli K-12.Res. Microbiol.150653–664.
81
SaidN.RiederR.HurwitzR.DeckertJ.UrlaubH.VogelJ. (2009). In vivo expression and purification of aptamer-tagged small RNA regulators.Nucleic Acids Res.37:e133. 10.1093/nar/gkp719
82
SaikiaM.JobavaR.ParisienM.PutnamA.KrokowskiD.GaoX.-H.et al (2014). Angiogenin-Cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress.Mol. Cell. Biol.342450–2463. 10.1128/MCB.00136-14
83
Santiago-FrangosA.WoodsonS. A. (2018). Hfq chaperone brings speed dating to bacterial sRNA.Wiley Interdiscip. Rev. RNA9:e1475. 10.1002/wrna.1475
84
SawitzkeJ. A.ThomasonL. C.CostantinoN.BubunenkoM.DattaS.CourtD. L. (2007). Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond.Methods Enzymol.421171–199. 10.1016/S0076-6879(06)21015-2
85
SchornA. J.GutbrodM. J.LeBlancC.MartienssenR. (2017). LTR-retrotransposon control by tRNA-derived small RNAs.Cell17061–71.e11. 10.1016/J.CELL.2017.06.013
86
ShenkT. E.RhodesC.RigbyP. W.BergP. (1975). Biochemical method for mapping mutational alterations in DNA with S1 nuclease: the location of deletions and temperature-sensitive mutations in simian virus 40.Proc. Natl. Acad. Sci. U.S.A.72989–993. 10.1073/pnas.72.3.989
87
SonD. J.KumarS.TakabeW.Woo KimC.NiC.-W.Alberts-GrillN.et al (2013). The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis.Nat. Commun.4:3000. 10.1038/ncomms4000
88
SoperT. J.WoodsonS. A. (2008). The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA.RNA141907–1917. 10.1261/rna.1110608
89
SørensenM. A. (2001). Charging levels of four tRNA species in Escherichia coli Rel+ and Rel- strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy11Edited by D. E. Draper.J. Mol. Biol.307785–798. 10.1006/jmbi.2001.4525
90
SteenR.JemioloD. K.SkinnerR. H.DunnJ. J.DahlbergA. E. (1986). Expression of plasmid-coded mutant ribosomal RNA in E coli: choice of plasmid vectors and gene expression systems.Prog. Nucleic Acid Res. Mol. Biol.331–18. 10.1016/S0079-6603(08)60018-5.
91
StenumT. S.SørensenM. A.SvenningsenS. L. (2017). Quantification of the abundance and charging levels of transfer RNAs in Escherichia coli.J. Vis. Exp.126:e56212. 10.3791/56212
92
ThomasonM. K.FontaineF.De LayN.StorzG. (2012). A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli.Mol. Microbiol.8417–35. 10.1111/j.1365-2958.2012.07965.x
93
WagnerE. G. H.RombyP. (2015). Small RNAs in Bacteria and Archaea: who they are, what they do, and how they do it.Adv. Genet.90133–208. 10.1016/bs.adgen.2015.05.001
94
WangX.DubeyA. K.SuzukiK.BakerC. S.BabitzkeP.RomeoT. (2005). CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli.Mol. Microbiol.561648–1663. 10.1111/j.1365-2958.2005.04648.x
95
WeiB. L.Brun-ZinkernagelA.-M.SimeckaJ. W.PrüßB. M.BabitzkeP.RomeoT. (2001). Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli.Mol. Microbiol.40245–256. 10.1046/j.1365-2958.2001.02380.x
96
WeiH.ZhouB.ZhangF.TuY.HuY.ZhangB.et al (2013). Profiling and identification of Small rDNA-Derived RNAs and their potential biological functions.PLoS One8:e56842. 10.1371/journal.pone.0056842
97
WeilbacherT.SuzukiK.DubeyA. K.WangX.GudapatyS.MorozovI.et al (2003). A novel sRNA component of the carbon storage regulatory system of Escherichia coli.Mol. Microbiol.48657–670. 10.1046/j.1365-2958.2003.03459.x
98
WillS.JoshiT.HofackerI. L.StadlerP. F.BackofenR. (2012). LocARNA-P: accurate boundary prediction and improved detection of structural RNAs.RNA18900–914. 10.1261/rna.029041.111
99
WillS.ReicheK.HofackerI. L.StadlerP. F.BackofenR. (2007). Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering.PLoS Comput. Biol.3:e65. 10.1371/journal.pcbi.0030065
100
WilleT.BarlagB.JakovljevicV.HenselM.SourjikV.GerlachR. G. (2015). A gateway-based system for fast evaluation of protein-protein interactions in bacteria.PLoS One10:e0123646. 10.1371/journal.pone.0123646
101
YeungM. L.BennasserY.WatashiK.LeS.-Y.HouzetL.JeangK.-T. (2009). Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: evidence for the processing of a viral-cellular double-stranded RNA hybrid.Nucleic Acids Res.376575–6586. 10.1093/nar/gkp707
102
ZhangX.BremerH. (1995). Control of the Escherichia coli rrnB P1 promoter strength by ppGpp.J. Biol. Chem.27011181–11189.
103
ZhouJ.RuddK. E. (2012). EcoGene 3.0.Nucleic Acids Res.41D613–D624. 10.1093/nar/gks1235
104
ZukerM. (2003). Mfold web server for nucleic acid folding and hybridization prediction.Nucleic Acids Res.313406–3415.
Summary
Keywords
sRNA, CsrA, Hfq, ribosomal RNA operon, dual terminators
Citation
Stenum TS, Kongstad M, Holmqvist E, Kallipolitis B, Svenningsen SL and Sørensen MA (2021) Three Ribosomal Operons of Escherichia coli Contain Genes Encoding Small RNAs That Interact With Hfq and CsrA in vitro. Front. Microbiol. 12:625585. doi: 10.3389/fmicb.2021.625585
Received
04 November 2020
Accepted
09 April 2021
Published
11 May 2021
Volume
12 - 2021
Edited by
Olga Soutourina, UMR 9198 Institut de Biologie Intégrative de la Cellule (I2BC), France
Reviewed by
Branislav Vecerek, Institute of Microbiology, Academy of Sciences of the Czech Republic (ASCR), Czechia; Franz Narberhaus, Ruhr University Bochum, Germany
Updates
Copyright
© 2021 Stenum, Kongstad, Holmqvist, Kallipolitis, Svenningsen and Sørensen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Askvad Sørensen, MAS@bio.ku.dkSine Lo Svenningsen, SLS@bio.ku.dk
†Present address: Erik Holmqvist, Department of Cell and Molecular Biology, Uppsala University, Uppsala, Sweden
This article was submitted to Microbial Physiology and Metabolism, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.