
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 25 March 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.614957
 Camila Gazolla Volpiano1
Camila Gazolla Volpiano1 Fernando Hayashi Sant’Anna1
Fernando Hayashi Sant’Anna1 Adriana Ambrosini1
Adriana Ambrosini1 Jackson Freitas Brilhante de São José2
Jackson Freitas Brilhante de São José2 Anelise Beneduzi2
Anelise Beneduzi2 William B. Whitman3
William B. Whitman3 Emanuel Maltempi de Souza4
Emanuel Maltempi de Souza4 Bruno Brito Lisboa2
Bruno Brito Lisboa2 Luciano Kayser Vargas2
Luciano Kayser Vargas2 Luciane Maria Pereira Passaglia1*
Luciane Maria Pereira Passaglia1*Taxonomic decisions within the order Rhizobiales have relied heavily on the interpretations of highly conserved 16S rRNA sequences and DNA–DNA hybridizations (DDH). Currently, bacterial species are defined as including strains that present 95–96% of average nucleotide identity (ANI) and 70% of digital DDH (dDDH). Thus, ANI values from 520 genome sequences of type strains from species of Rhizobiales order were computed. From the resulting 270,400 comparisons, a ≥95% cut-off was used to extract high identity genome clusters through enumerating maximal cliques. Coupling this graph-based approach with dDDH from clusters of interest, it was found that: (i) there are synonymy between Aminobacter lissarensis and Aminobacter carboxidus, Aurantimonas manganoxydans and Aurantimonas coralicida, “Bartonella mastomydis,” and Bartonella elizabethae, Chelativorans oligotrophicus, and Chelativorans multitrophicus, Rhizobium azibense, and Rhizobium gallicum, Rhizobium fabae, and Rhizobium pisi, and Rhodoplanes piscinae and Rhodoplanes serenus; (ii) Chelatobacter heintzii is not a synonym of Aminobacter aminovorans; (iii) “Bartonella vinsonii” subsp. arupensis and “B. vinsonii” subsp. berkhoffii represent members of different species; (iv) the genome accessions GCF_003024615.1 (“Mesorhizobium loti LMG 6125T”), GCF_003024595.1 (“Mesorhizobium plurifarium LMG 11892T”), GCF_003096615.1 (“Methylobacterium organophilum DSM 760T”), and GCF_000373025.1 (“R. gallicum R-602 spT”) are not from the genuine type strains used for the respective species descriptions; and v) “Xanthobacter autotrophicus” Py2 and “Aminobacter aminovorans” KCTC 2477T represent cases of misuse of the term “type strain”. Aminobacter heintzii comb. nov. and the reclassification of Aminobacter ciceronei as A. heintzii is also proposed. To facilitate the downstream analysis of large ANI matrices, we introduce here ProKlust (“Prokaryotic Clusters”), an R package that uses a graph-based approach to obtain, filter, and visualize clusters on identity/similarity matrices, with settable cut-off points and the possibility of multiple matrices entries.
The classification of Rhizobiales species is complex and has undergone many changes over the years. Frank (1889) described Rhizobium, the type genus of the order, to accommodate different symbiotic nitrogen-fixing bacteria based only on their selective interaction with legume plants. In 1991, this classification was abandoned after extensive criticism (Wilson, 1944) and the discovery that genes required for symbiosis are often located on transmissible plasmids (Nuti et al., 1979; Brewin et al., 1980; Prakash et al., 1981). The minimal standards for species descriptions have usually incorporated DNA–DNA hybridization (DDH) and the 16S rRNA sequence analyses, as well as morphological, physiological, and biochemical features (Graham et al., 1991).
In the pre-genomic era, the DDH was considered the gold standard for prokaryotic species circumscriptions. The DDH measures the fraction of DNA that hybridizes under optimal conditions, and a threshold of at least 70% between two strains was widely recognized as the species boundary (Tindall et al., 2010). In the first formal species definition based on the DDH proposed by Wayne et al. (1987), the change in the melting temperature (ΔTm) was recommended as a second measure of genetic relatedness to be considered with DDH, and a ΔTm value of 5°C or less was established as the cut-off for prokaryotic species. The ΔTm represents the difference of the Tm of the heteroduplex of the two strains being tested, reflecting primarily the sequence identity (Johnson and Whitman, 2007). However, because DDH is technically easier to measure, ΔTm is usually not determined in most species’ descriptions (Li et al., 2015).
With the advent of genomics, taxonomy has relied heavily on comparative measurements that calculate surrogates of both the ΔTm and DDH in silico directly between genomes sequences. These comparisons, as forms of similarity or distance, have been coined as overall genome-related indices (OGRI). Among them, average nucleotide identity (ANI) using the BLASTn algorithm to perform alignments (ANIb) and the Genome BLAST Distance Phylogeny (GBDP)-based digital DDH (dDDH) methods have been most widely used (Konstantinidis and Tiedje, 2005; Richter and Rosselló-Móra, 2009; Chun et al., 2018; Sant’Anna et al., 2019). The ANI is considered a good surrogate for the ΔTm because it only compares homologous DNA fragments that meet sequence identity and coverage criteria. In its turn, the dDDH using formula d0 and d6 seems to most closely approximate the properties expected for the experimentally determined DDH values (Auch et al., 2010a; Li et al., 2015).
The ANIb was originally proposed on the basis of benchmarking with respect to DDH values by Goris et al. (2007). In this method, the genomic sequence from one of the genomes in a pair (“query”) is cut in silico into consecutive 1,020 nt fragments, mimicking the DNA fragmentation step during the DDH experiments. The fragments are then used to search against the whole genomic sequence of the other genome in the pair (“reference”) using BLASTn. The ANI between the query and the reference genomes is calculated as the mean identity of all BLASTn matches that show more than 30% overall sequence identity (recalculated to an identity along the entire sequence) over an alignable region of at least 70% of their length. The classical cut-off point of 70% DDH for species delineation corresponded to an ANIb of 95%. Later, Richter and Rosselló-Móra (2009) recommended applying an ANI boundary of 95–96% after correlating ANIm (ANI with MUMmer ultra-rapid aligning tool) and DDH values between strains putatively representing single species. In its turn, Meier-Kolthoff et al. (2013) established a 70% dDDH for species boundaries based on the comparisons of the dDDH predictions with in vitro DDH.
The application of OGRI to prokaryote taxonomy has the advantage of being objective and proved to unravel a microbial phylogenetic novelty at an unprecedented pace (Sant’Anna et al., 2019; Sanford et al., 2021), however, it is still being subject to controversies. The ANI and dDDH cut-offs were calibrated to yield the species previously determined by the DDH and ΔTm, which were based on a threshold that was calibrated upon previous bacterial species definitions that were based entirely on phenotypic properties, mostly in the Enterobacteriaceae (Gevers et al., 2006). Finally, some authors consider that a universal metric or species cut-off for in silico genome comparisons should be interpreted only as a guideline considering that all species have evolved via independent evolutionary trajectories which could result in cohesion among individuals occurring at different levels of similarity (Palmer et al., 2020).
In the analysis of genomic sequence data from prokaryotes with OGRI values, new algorithms with optimized search parameters (Lee I. et al., 2016; Rodriguez-R and Konstantinidis, 2016) or to speeding up the search process (Richter and Rosselló-Móra, 2009; Varghese et al., 2015; Yoon et al., 2017b; Jain et al., 2018) are continuously being developed. However, effective algorithms for mining the output data from large OGRI matrices are still in high demand. A diverse set of “traditional” hierarchical clustering approaches (i.e., average linkage, complete linkage, Ward, UPGMA, and neighbor-joining) are already available. These approaches can create clusters by grouping genes/genomes with high identity measures together and returning tree-shaped diagrams. Such diagrams do not allow overlapping clusters and are difficult to extract information in complex cases where not all the members of a cluster share sufficient identity to cluster with all the other members. Moreover, it is usually not possible to combine different matrices in such approaches. This is important considering that ANI values should be used in conjunction with complementary measures of the minimum amount that genomes must overlap, such as the alignment coverage (pyANI), AF (Alignment Fraction; Varghese et al., 2015), or dDDH. If the homologous regions are short with respect to the total length of the genomes, as might be seen following an horizontal gene transfer (HGT), then ANI values may be high even though the bacteria are distantly related.
The present study aims to evaluate the taxonomic structure at the species level within the Rhizobiales order using genome-scale comparisons with ANIb and dDDH from a collection of 520 genome assemblies identified as belonging to type strains. We note, however, that Hördt et al. (2020) recently proposed an emended description of Hyphomicrobiales (Douglas, 1957) to replace Rhizobiales (Kuykendall, 2005). Our secondary objective is to introduce ProKlust, an R package developed to facilitate the downstream analysis of large identity matrices.
Ensifer terangae SEMIA 6460T (USDA 4894T = ORS 1009T = LMG 7834T = ATCC 51692T = DSM 11282T) and Rhizobium gallicum SEMIA 4085T (USDA 2918T = R-602 spT = EMBRAPA Soja 172T), previously isolated from common bean nodules and maintained at SEMIA Culture Collection (World Data Center on Microorganisms no. 443), were re-hydrated from lyophilized cultures and grown on yeast mannitol (YM) agar medium (Somasegaran and Hoben, 2012) at 28°C. DNA from late log phase SEMIA 6460T cultures was extracted using the PureLinkTM Microbiome DNA Purification Kit (Thermo Fisher Scientific). A phenol: chloroform method adapted from Sambrook and Russell (2006) was used to obtain the genomic DNA from SEMIA 4085T.
Genomic Encyclopedia of Bacteria and Archaea (GEBA) KMG Phase III project from the United States Department of Energy (DOE) Joint Genome Institute (JGI) provided a draft genome sequence for SEMIA 6460T. Genomic libraries for SEMIA 4085T were prepared at the Department of Biochemistry and Molecular Biology (UFPR, Brazil) using the Nextera prep kit (Illumina). The sequencing was performed on an Illumina MiSeq platform with a 250 paired-end protocol. SPAdes v.3.11.1 (Bankevich et al., 2012) was used to assemble the reads, and Blobtools (Laetsch and Blaxter, 2017) was used to identify and remove contaminated contigs.
Genomic sequences for 518 assemblies were retrieved from the NCBI assembly database upon searching for “Rhizobiales” with filters “latest RefSeq,” and “assembly from any type” on April 29th, 2020. Additional 21 assemblies were retrieved from the database upon searching for “Aminobacter” with filters “latest RefSeq” on November 26th, 2020.
Pairwise comparisons among the genomes were calculated using the ANIb method from pyANI v 0.2.10 Python3 module1 and FastANI v 1.3 (Jain et al., 2018). GGDC (Genome-to-Genome Distance Calculator) 2.1 with the recommended BLAST+ aligner were used to compute dDDH values and confidence intervals (C.I.) using GBDP formula d0 (GGDC formula 1) and d6 (GGDC formula 3) at http://ggdc.dsmz.de.
ProKlust uses a graph-based approach implemented in R language for the downstream analysis of large identity/similarity matrices. First, the input pairwise matrix is formatted into a triangular matrix using the average of each pair. Then, the matrix is formatted using the cut-off values chosen by the user. To obtain the Boolean matrix, values were replaced according to the criterion chosen. If more than one matrix is used as input, the generated logical matrices are also multiplied to obtain a consensus. Afterward, the graph is formed by connecting the nodes (i.e., genomes or genes) using a modified Bron–Kerbosch algorithm from the “igraph” R package (Csardi and Nepusz, 2006) to find the maximal cliques, which is superior in performance (Eppstein et al., 2010). The following filters were implemented to filter the data (i) “filterRemoveIsolated,” to remove isolated nodes i.g., nodes that do not form groups/clusters; (ii) “filterRemoveLargerComponent” and “filterOnlyLargerComponent” to remove or retain only the component containing the highest number of nodes); (iii) “filterDifferentNamesConnected” to retain groups of connected nodes containing more than one binomial species name; and (iv) “filterSameNamesNotConnected” to retain groups of unconnected nodes containing the same species names. Four types of outputs were implemented on ProKlust: (i) “maxCliques,” the maximal clique is the largest subset of nodes in which each node is directly connected to every other node in the subset; (ii) “components” that contains the isolated nodes or groups formed of complete graphs; (iii) “graph,” an igraph object graph, that can be further handled by the user; and (iv) the “plot,” where the final graph could be promptly visualized with forceNetwork function from the “networkD3” R package (Allaire et al., 2017).
The data generated in the previous topic was clustered with ProKlust to extract groups of genospecies. For both pyANI and FastANI identity matrices, the cut-off criterion chosen was ANI ≥95%. Additionally, the pyANI alignment coverage matrix, representing the fraction of each genome that was aligned, was combined with ANIb, with an arbitrary cut-off point of ≥50%. The data were filtered using the parameters “filterDifferentNamesConnected” and “filterSameNamesNotConnected”.
For quality check, miComplete (Hugoson et al., 2019) was employed to infer weighted completeness and redundancy of genomes using the precalculated weights associated with the inbuilt marker sets “Bact105.” In miComplete, completeness is calculated based on the presence/absence of a set of marker genes.
Additionally, we analyzed the 16S rRNA gene copies present on our genome set. The 16S rRNA genes were collected from RNA sequences by genomic FASTA. Barrnap2 was employed to predict the location of the 16S rRNA genes in E. terangae SEMIA 6460T. Taxonomic assignments from order to genus ranks were made for the extracted sequences with the IdTaxa function available via the “DECIPHER” v2.14.0 R package (Murali et al., 2018) using the SILVA SSU r138 trained classifier (Yilmaz et al., 2014; link to the full license:3).
To guarantee the identity of the genomes present on the RefSeq database, we choose to further analyze the 16S rRNA sequences extracted in a subset of genomes that were selected based on the results from the clustering step or/and were assigned to different genera using the SILVA SSU r138 trained classifier. To provide reliable comparisons, we removed seven sequences with ≤400 nucleotides. The 16S rRNA reference sequences determined by Sanger method from type strains were then retrieved according to the sequence accessions provided on LPSN – List of Prokaryotic names with Standing in Nomenclature (as available at4). An additional 16S rRNA sequence for Methylobacterium organophilum ATCC 27886T (NR_041027) sequenced by Kato et al. (2005) was added as a reference. The 16S rRNA for “Bartonella mastomydis” (KY555064) was retrieved from Dahmani et al. (2018). We performed a profile-to-profile alignment considering RNA secondary structure using “AlignSeqs” function from “DECIPHER.” The “seqinr” v.3.6.1 R package (Charif and Lobry, 2007) was employed to compute pairwise distances from aligned sequences with no gaps.
In this work, we first checked the general quality of 520 genomes obtained from the type strains of species from the order Rhizobiales. To obtain genomic groups with high identity, the values computed with ANIb and FastANI were clustered using ProKlust. An additional authenticity check was performed to support our proposals of changes in the actual taxonomic classification. To apply Wayne et al. (1987) recommendations, we also computed dDDH using GGDC formula d0 and d6 among closely related strains. A diagram capturing the steps performed in this work is shown in Figure 1.
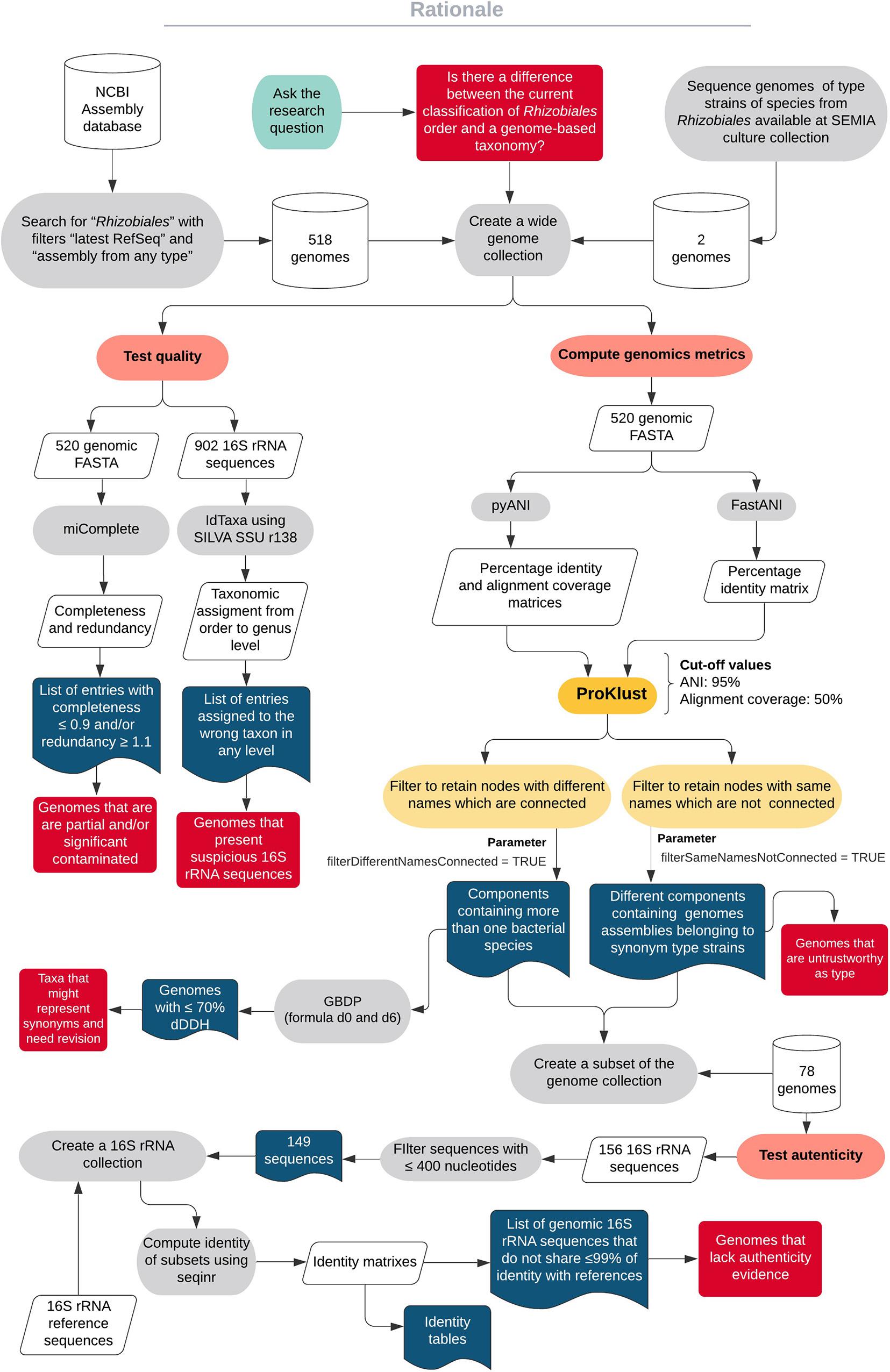
Figure 1. A diagram showing the workflow of the study.
The genomic sequence generated for E. terangae SEMIA 6460T can be found according to the following identifiers: 2838074704 (IMG Taxon OID, Integrated Microbial Genomes platform), Ga0394399 (GOLD Id, Genomes Online Database), and PRJNA581033 (NCBI BioProject Accession). The genomic sequence generated for R. gallicum SEMIA 4085T can be found according to the RefSeq accession GCF_013004495.1. The genomes retrieved from NCBI can be found using the RefSeq accessions provided in Supplementary Table 1. ProKlust is available at https://github.com/camilagazolla/ProKlust.
Quality-related statistics obtained from the genome collection are presented in Supplementary Figure 1 and Table 1. The genomes presented a range of 1.44–10.11 Mb in length, with an average weighted completeness and redundancy parameters of 0.9814 and 1.026, respectively. From the 520 genomes present in the set, 12 were detected with weighted completeness ≤0.9 and/or weighted redundancy ≥1.1 (Table 1). This low sequence quality must be considered by taxonomists dealing with these specific accessions.
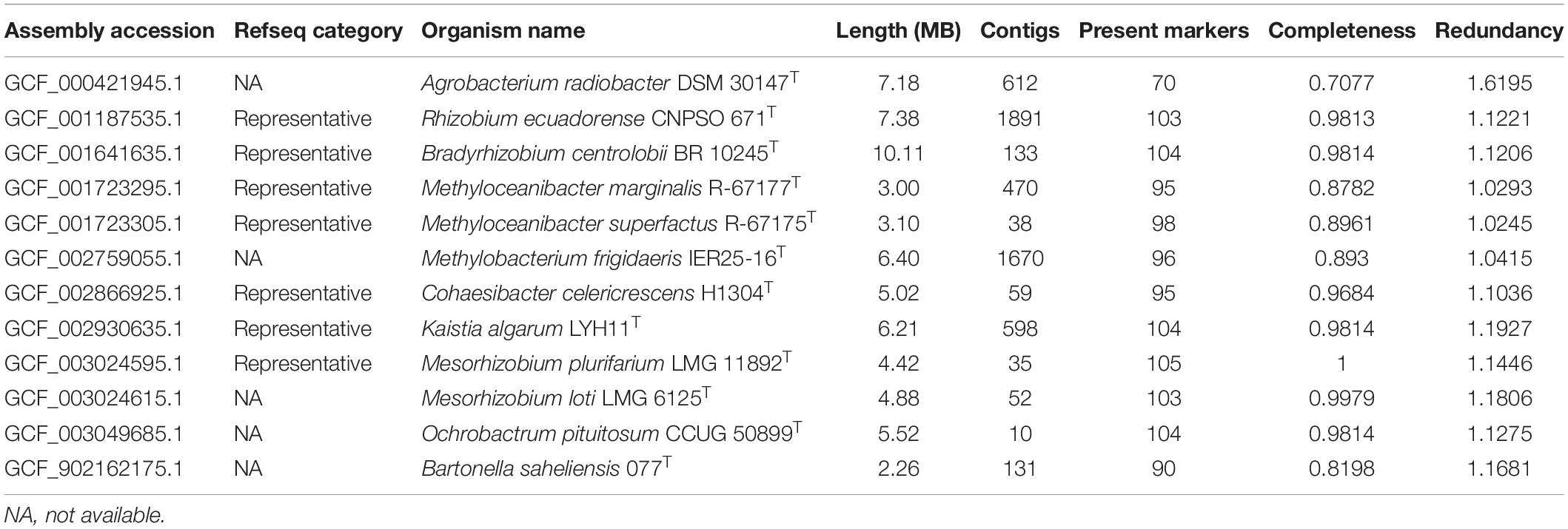
Table 1. Genomes detected with high contamination and/or low completeness.
We also analyzed the taxonomic assignments of the 16S rRNA genes present in the genome sequences. A total of 902 genes were found among 513 genomes, varying from 1 to 13 copies per genome. We then compared the taxonomic assignments of these copies using SILVA SSU r138 (Yilmaz et al., 2014) to those of the genomes. A total of 715 gene copies were correctly assigned to the genus, 160 gene copies were not taxonomically assigned, and 28 copies were assigned to suspicious taxa (Supplementary Table 2). After the removal of sequences belonging to recently described genera that were absent in the classifier, a total of 17 genomes remained assigned to suspicious copies of the 16S rRNA (Table 2). These were further evaluated to determine if they represented misclassified genome sequences.
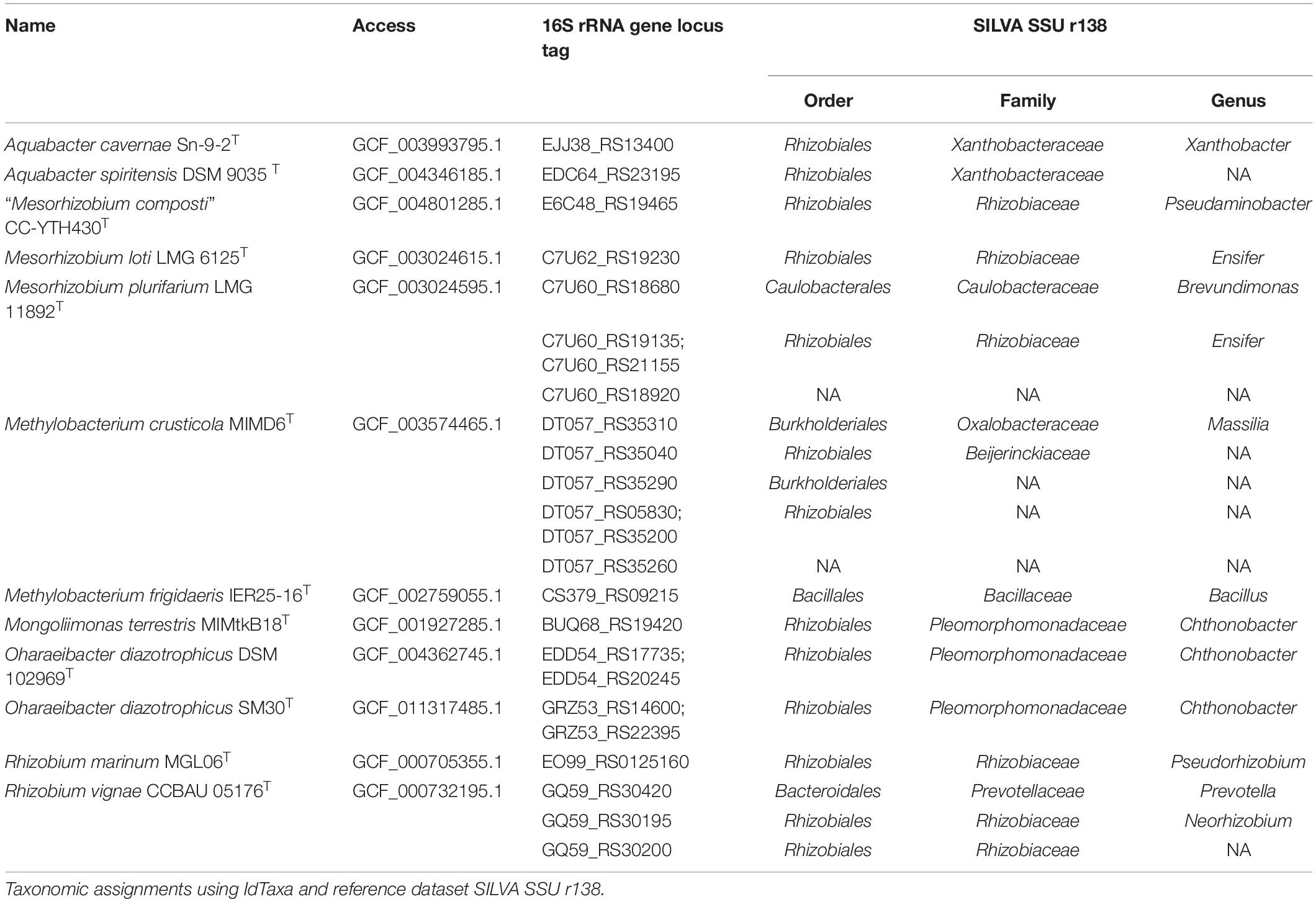
Table 2. 16S rRNA copies extracted from genomes and incorrectly taxonomically assigned.
The 16S rRNA genes extracted from Aquabacter cavernae Sn-9-2T and A. spiritensis DSM 9035T genomes were both assigned to Xanthobacteraceae. However, these sequences shared a high identity of 99.8 and 100%, respectively, with the 16S rRNA gene references MF958452 and FR733686 (Supplementary Table 3). Xanthobacter autotrophicus DSM 432T and X. tagetidis ATCC 700314T were reported by Duo et al. (2019) as the nearest phylogenetic neighbors of A. cavernae Sn-9-2T. The unexpected affiliation of the A. spiritensis DSM 9035T 16S rRNA gene sequence within the family Xanthobacteraceae was also reported previously by Yarza et al. (2013). Recently, Hördt et al. (2020) proposed including Aquabacter into Xanthobacteraceae. The authors reported that Aquabacter, Xanthobacter, and Azorhizobium formed a highly supported clade in a GBDP d3 tree and that those genera are difficult to discern as currently circumscribed with 16S rRNA gene analyses. Given that the genome sequences from all the type strains of this genus are not yet available, it remains to be elucidated if A. cavernae and A. spiritensis represent species of Xanthobacter.
“Mesorhizobium composti” was proposed by Lin et al. (2019), although the name is not validly published. The assembly employed here was based upon the WGS accession given for the type strain CC-YTH430T, and the 16S rRNA sequence extracted from the genome and the reference KX988315 presented 100% identity. In a 16S rRNA gene tree, “M. composti” CC-YTH430T was reported to form a cluster with Mesorhizobium and Pseudaminobacter species (Lin et al., 2019), which could explain its wrong assignment to Pseudaminobacter in SILVA SSU r138 (Table 2). Besides, several Mesorhizobium species have been described as intermixed with Pseudaminobacter in 16S rRNA gene (Lin et al., 2019) and GBDP d5 phylogenies Hördt et al. (2020). Taken into consideration, the genome available for “M. composti” CC-YTH430T seems to be authentic.
Methylobacterium crusticola MIMD6T was recently described by Jia et al. (2020) isolated from biological soil crusts. The assembly employed here holds the WGS accession given in the M. crusticola description. The M. crusticola MIMD6T genome presents six 16S rRNA sequences, of which only the locus tag DT057_RS35040 assigned to Beijerinckiaceae shared a high identity of 99.1% with the reference KT346425 sequence, while the identity of the remaining five sequences ranged from 56.1 to 75.8%. In a core-proteome dendrogram constructed using the neighbor-joining method (Supplementary Figure 2), M. crusticola MIMD6T was placed along other Methylobacterium strains instead of the ones belonging to Beijerinckiaceae. Considering this, the M. crusticola MIMD6T genome is authentic but wrongly designated in SILVA SSU r138 classifier. It remains to be elucidated if those suspiciously assigned copies represent contamination by foreign DNA or even a HGT event. Similarly, Methylobacterium frigidaeris IER25-16T presented only one 16S rRNA sequence, and it was assigned to Bacillus (Table 2). The WGS accession provided from its description corresponded to the genome employed here (Lee and Jeon, 2018), and despite that, it presented only 86.2% of identity with the reference (KY864396). In the core-proteome dendrogram (Supplementary Figure 2), M. frigidaeris IER25-16T was also found along with other Methylobacterium instead of Bacillus strains. Considering this, the 16S rRNA copy of M. frigidaeris IER25-16T is more likely to represent a contamination or HGT event than a reliable taxonomic marker.
The family Pleomorphomonadaceae currently comprises the genera Chthonobacter, Hartmannibacter, Methylobrevis, Mongoliimonas, Oharaeibacter, and Pleomorphomonas (Hördt et al., 2020). The 16S rRNA genes extracted from Mongoliimonas terrestris MIMtkB18T and two assemblies belonging to Oharaeibacter diazotrophicus (DSM 102969T and SM30T) were assigned to the Chthonobacter genus. However, the WGS for M. terrestris MIMtkB18T genome given by Xi et al. (2017) is comprised within the assembly employed here. Also, the 16S rRNA gene present in the genome is identical to the reference (KP993300), confirming its authenticity. Regarding O. diazotrophicus, the four 16S rRNA sequences extracted from its assemblies shared 99.9–100% of identity with the reference for O. diazotrophicus SM30T (LC153750; Lv et al., 2017), confirming their authenticity. Remarkably, the 16S rRNA genes of M. terrestris and O. diazotrophicus share a 16S rRNA sequence identity of 98.4–98.3% and 97.3–97.9%, respectively, with the type species of Chthonobacter, C. albigriseus ED7T (KP289282). Once the genome of C. albigriseus ED7T becomes available, the placement of Mongoliimonas, Oharaeibacter, and Chthonobacter as separate genera should be reevaluated.
The Rhizobium vignae CCBAU 05176T genome sequence possessed three different 16S rRNA gene copies, which were ultimately assigned to Prevotella, Neorhizobium, and an unidentified genus of Rhizobiaceae. The 16S rRNA copy which was assigned to Neorhizobium (GQ59_RS30195) presented a high identity of 99.9% with the reference GU128881 given on the R. vignae description (Ren et al., 2011), while the remaining copies shared less than 76% identity. Considering this, it seems that GCF_000732195.1 represents an authentic R. vignae CCBAU 05176T genome, but contamination or HGT occurred. Recently, Hördt et al. (2020) proposed that R. vignae should be assigned to Neorhizobium because R. vignae was placed as a sister group of Neorhizobium galegae (Mousavi et al., 2014) with strong support in a GBDP d5 tree.
The genomes attributed to Mesorhizobium plurifarium LMG 11892T and Mesorhizobium loti LMG 6125T indeed represented unauthentic genome sequences. The Rhizobium marinum MGL06T is a synonym of Pseudorhizobium pelagicum. These issues are explored further in the next section below.
Here, we describe ProKlust, an R package for the downstream analysis of large identity matrices using maximal cliques enumeration (Figure 2). ProKlust is open source and not computationally intensive. Due to its flexibility, ProKlust could be employed to analyze any identity/similarity matrix, such as barcoding gene identity. Additionally, it contains useful filter options to deal with taxonomical data.
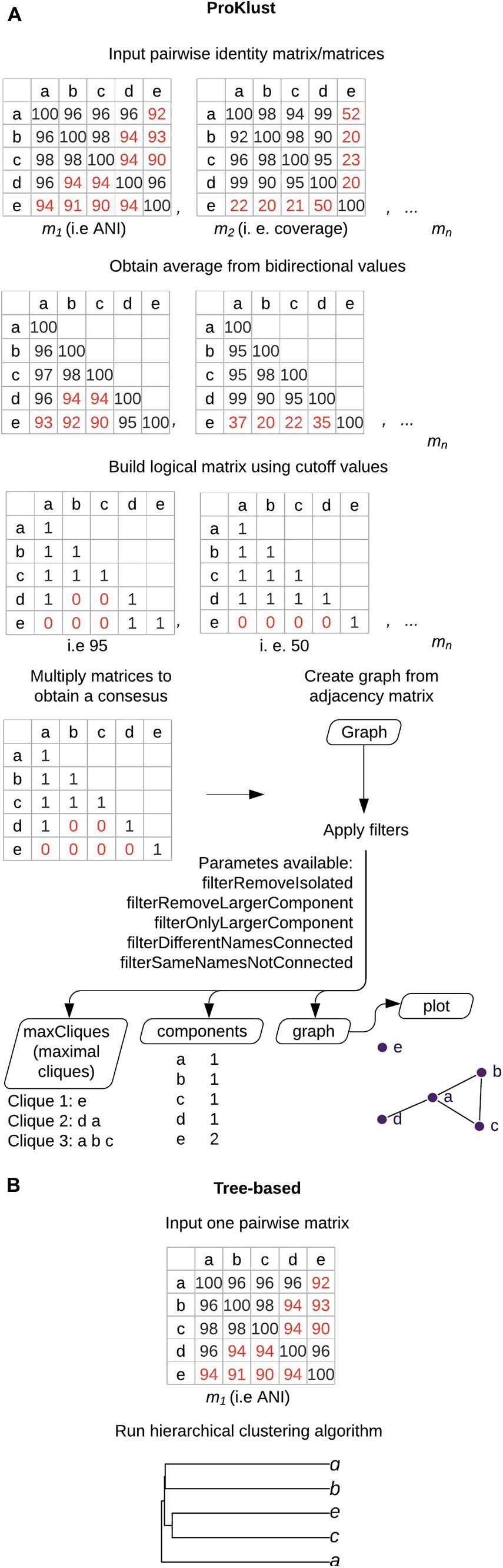
Figure 2. Graph-based clustering using ProKlust compared to hierarchical clustering. (A) The average of each pair from the pairwise input. matrix/matrices is/are obtained. A Boolean matrix/matrices is/are obtained according to the cut-off values chosen by the user. If more than one matrix is used as input, the final generated matrix is obtained by multiplying the elements of the matrices. A graph is formed by connecting the nodes which present the positive values. In this example, nodes correspond to genomes and edges correspond to ANI ≥95% with coverage alignment ≥50%. The data could be filtered to retain components containing more than one species name or unconnected nodes containing the same species names. In addition, filters to remove isolated nodes (“filterRemoveIsolated”) or the largest component (“filterOnlyLargerComponent”) are also available. The tool generates four types of outputs: (i) the maximal cliques on “maxCliques,” which is the largest subset of nodes in which each node is directly connected to every other node in the subset i.e., all the possible species groups that could be delimited in the graph, which could result in groups having genomes in common; (ii) “components” that contains the isolated nodes or groups formed of complete graphs; (iii) “graph,” an igraph object graph, that can be further handled by the user; and (iv) the “plot,” where the final graph could be visualized. (B) Overview of the hierarchical-based clustering approach. These approaches return tree-shaped diagrams with non-overlapping clusters.
A total of 415 components were obtained using ProKlust to analyze the input of 270,400 ANI values computed for the 520-genome set. Components are the groups formed by linking the genomes together according to the chosen criteria, i.e., ≥95% ANI. A genome that is part of a component does not necessarily share ANI values above the established cut-off with all the other genomes of that component, but it must share an ANI value above the cut-off for at least one other genome. Cliques, instead, are formed by genomes that all share ANI values above the chosen criteria. A genome could belong at the same time to different cliques within the same component.
All components detected were congruent between the ANIb and FastANI methods. Employing a filter step to retain genomes clusters containing more than one bacterial species name, we were able to easily identify genomes clusters containing heterotypic synonyms on our genome set (Figure 3). Some of these heterotypic synonyms had already been identified by other authors (Table 3). We discuss some of these proposals in addition to the heterotypic synonyms identified. Additionally, we found strains incorrectly assigned as members of the same inspecting the clustering behavior of “supposedly” synonym type strains.

Figure 3. Genomic clusters detected using pairwise ANI values from 520 Rhizobiales genomes. Here, only a subset is shown following filtering to (i) retain genomes clusters containing more than one bacterial species name and (ii) the unconnected genomes containing the same species names (synonym strains). In the graph, the clusters have nodes corresponding to genomes and edges corresponding to ANI values above the cut-off for species delineation. In the case of ANIb from pyANI, where an alignment coverage matrix was also generated, the edges additionally correspond to a reliable alignment of between the set of genomes. The same graph structure was obtained using FastANI values. Different colors were used whenever possible to indicate different species names.
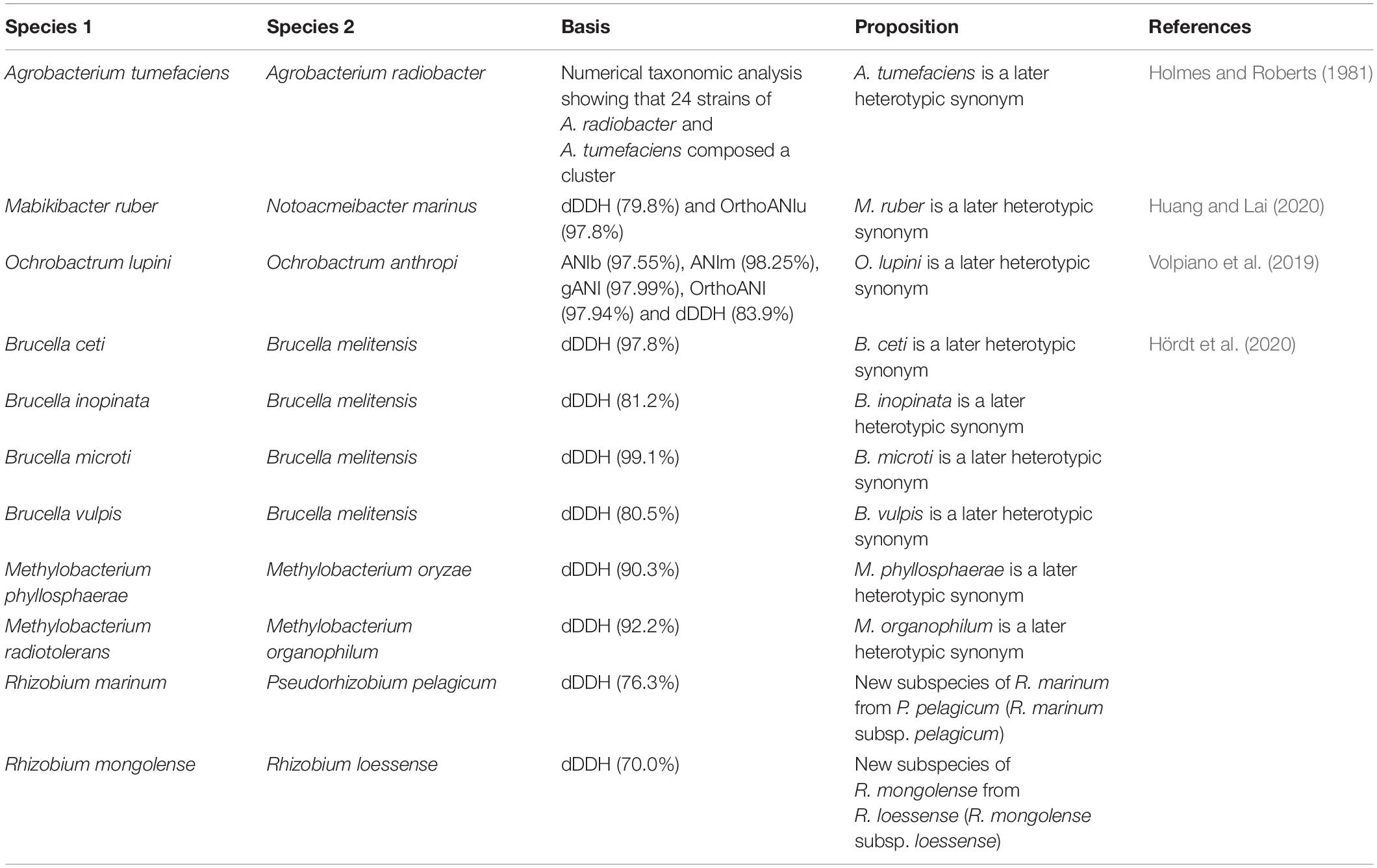
Table 3. Heterotypic synonyms found here that have already been detected by other authors.
The minimal standards for the use of genome data for the taxonomy of prokaryotes recommend the utilization of 16S rRNA sequences to confirm the authenticity of genome data (Chun et al., 2018). Thus, the 16S rRNA genes found in the 78 genomes presented in Figure 3 were also examined to ensure their authenticity. When the 16S rRNA sequences shared ≥99% identity, it confirmed their assignments to the same operational taxonomic unit (Stackebrandt and Ebers, 2006; Kim et al., 2014). The 16S rRNA distance matrices for each taxon can be found in Supplementary Table 3. The dDDHs using formulas d0 and d6 were also computed (Supplementary Table 4) considering the proposal of two measures of genetic relatedness to set the boundary for prokaryotic species established by Wayne et al. (1987).
The genomic clusters formed by Rhizobium favelukesii LPU83T and Rhizobium tibeticum CCBAU 85039T and CGMCC 1.7071T assemblies possessed an ANI value of 95.9% and a dDDH up to 64.7%. Similarly, the genomes of R. flavum YW14T and “Rhizobium halotolerans” AB21 shared 95.8% of ANIb and 65.4% of dDDH. These strains should remain classified in separate species according to Wayne et al. (1987).
Aurantimonas manganoxydans SI85-9A1T and “Aurantimonas litoralis” HTCC 2156T are described as Mn(II)-oxidizing bacteria isolated from the oxic/anoxic interface of a stratified Canadian fjord and the surface waters of the Oregon coast, respectively (Anderson et al., 2009). Despite sharing nearly identical 16S rRNA gene sequences with the previously described A. coralicida WP1T (Denner et al., 2003), the measured DDH similarity between A. manganoxydans SI85-9A1T and “A. litoralis” HTCC 2156T with WP1T was only 21.8 and 9.45%, respectively.
Aurantimonas coralicida DSM 14790T and A. manganoxydans SI85-9A1T each have two genome assemblies in the RefSeq database, while a genome sequence for an “A. litoralis” type strain is still not available. We found a total of four and two 16S rRNA gene copies in the A. manganoxydans SI85-9A1T and A. coralicida DSM 14790T genomes, respectively, which shared a high identity with the reference sequences (AJ786361 and AJ786360). Also, these genomes share 95.25–95.24% of ANIb and at least 78.7% of dDDH, greatly differing from the genome relatedness values reported by Anderson et al. (2009).
Thus, A. manganoxydans corrig. Anderson et al. (2011) should be considered a later heterotypic synonym of A. coralicida Denner et al. (2003) considering the bacterial species threshold of 95% of ANIb originally proposed by Goris et al. (2007).
Doronina et al. (2010) proposed the genus Chelativorans to accommodate C. multitrophicus DSM 9103T isolated from samples taken from industrial wastewater treatment plants and C. oligotrophicus LPM-4T, isolated from sewage sludge. The two EDTA-degrading strains shared a high similarity of 99.3% between their 16S rRNA gene sequences.
We found that the C. multitrophicus DSM 9103T and C. oligotrophicus LPM-4T genomes shared 99.86% of ANIb and at least 98.9% of dDDH. Additionally, the 16S rRNA gene sequences extracted from them presented ≥99% identity to the references EF457243 and EF457242, respectively.
Considering these findings, C. multitrophicus Doronina et al. 2010 and C. oligotrophicus Doronina et al. 2010 represent members of a single species. The former name should be retained because it represents the type species of Chelativorans genus.
After Frank’s (1889) description of Rhizobium genus, Jordan (1984) described R. meliloti, R. loti, and three biovars of R. leguminosarum (uniting the former species of R. leguminosarum, R. phaseoli, and R. trifolii). Subsequently, Chen et al. (1988) proposed creation of a separate genus, Sinorhizobium, to include the previously described R. fredii (Scholla and Elkan, 1984). Rhizobium meliloti was later transferred to Sinorhizobium (de Lajudie et al., 1994), and S. arboris and S. kostiense were proposed as new Sinorhizobium species (Nick et al., 1999). Jarvis et al. (1997) proposed the genus Mesorhizobium for encompassing R. loti and R. huakuii, after observing that the fatty acid profiles, additional physiological characteristics, and the 16S rRNA genes from these species were distinct from those of members of the genera Agrobacterium, Rhizobium, and Sinorhizobium. Later, de Lajudie et al. (1998) described M. plurifarium ORS 1032T, isolated from root nodules of Acacia species.
Other significant revisions have been made by Willems et al. (2003). These authors utilized several characterization methods, including 16S rRNA and recA sequence analyses, and showed that Ensifer and Sinorhizobium formed a single group in neighbor-joining dendrograms, leading to the conclusion that Ensifer and Sinorhizobium were synonyms and a proposal that the name Sinorhizobium should be preferred to Ensifer. However, Young (2003) considered that Ensifer has priority over Sinorhizobium because it was validly published earlier.
Up to date, there are two deposits in RefSeq for genomes of the M. loti type strains: “LMG 6125T” (GCF_003024615.1) and DSM 2626T (GCF_003148495.1), both listed as the type strains. However, we found that both genomes unexpectedly share only 74.6% of ANIb and 14.1% of dDDH, indicating that they do not represent the genome sequences of the same species. The “M. loti LMG 6125T” genome shared 62.5 (d0) to 70.1% (d6) of dDDH and 99.5% of ANIb with Ensifer meliloti USDA1002T, which suggested that “M. loti LMG 6125T” was a strain of E. meliloti. The 16S rRNA gene sequence extracted from “M. loti LMG 6125T” present only 97.6% of identity with the reference (AB680660). On the other hand, the M. loti DSM 2626T genome shared only 74.6% of ANIb with E. meliloti USDA1002T. The 16S rRNA sequences extracted from this second M. loti genome presented ≥99% of identity with the reference. In conclusion, “LMG 6125T” (GCF_003024615.1) represents an unauthentic genome sequence for the type strain of M. loti. The genome of M. loti DSM 2626T (GCF_003148495.1) should be used as a reference for this species.
Similarly, “M. plurifarium LMG 11892T” genome (GCF_003024595.1) shared an ANI of 99.6% and up to 65.6% of dDDH with the genome of E. arboris LMG 14919T. None of the three 16S rRNA gene copies found in the “M. plurifarium LMG 11892T” genome presented sufficient identity with the reference sequence (AB681835) to confirm its identity (Supplementary Table 3).
“Mesorhizobium plurifarium LMG 11892T” genome accession GCF_003024595.1 and “M. loti LMG 6125T” genome accession GCF_003024615.1 shared high values of 1.15 and 1.18 for weighted redundancy, respectively (Table 1). Moreover, the 16S rRNA gene copies extracted from these genomes were assigned to suspicious taxa (Table 2). Researchers must utilize these assemblies with caution considering the inconsistencies found here. This observation is especially important for the accession GCF_003024595.1, once it is categorized as the representative genome for M. plurifarium on RefSeq database.
The genus Methylobacterium was first proposed by Patt et al. (1976), and it was defined by the type species M. organophilum, a Gram-stain-negative, methane-utilizing bacterium. Thereafter, Green and Bousfield (1983) demonstrated that M. organophilum was phenotypically highly similar to the pink-pigmented, facultatively methylotrophic bacteria that do not utilize methane. Consequently, methane assimilation was omitted as an essential feature in the emended description of the genus. As a result, Pseudomonas rhodos Heumann (1962), renamed Methylobacterium rhodinum; Pseudomonas mesophilica Austin and Goodfellow (1979), renamed Methylobacterium mesophilicum; and Pseudomonas radiora Ito and Iizuka (1971), renamed Methylobacterium radiotolerans, were placed in the emended genus Methylobacterium. More recently, based on 16S rRNA gene and multi-locus sequence analyses, genomic, and phenotypic data, Green and Ardley (2018) proposed the Methylorubrum genus to accommodate the 11 species previously classified in Methylobacterium.
There are two deposits on RefSeq for genomes of M. radiotolerans type strains: NBRC 15690T (GCF_007991055.1) and JCM 2831T (GCF_000019725.1). Both genomes share ANIb of 99% and at least 84.4% of dDDH with the “M. organophilum DSM 760T” genome (GCF_003096615.1), thus indicating that the three genomes belong to the same species. The eight 16S rRNA gene copies from M. radiotolerans assemblies possess ≥99% identity with the reference (D32227), confirming their authenticity. However, the 16S rRNA gene from the “M. organophilum DSM 760T” genome shared a low identity of only 96.1% with the reference (AJ400920).
Kato et al. (2005) has already reported that strain DSM 760T was different from the other M. organophilum type strain (JCM 2833T) in physiological and biochemical characteristics, although their origins were reported to be the same and they were expected to possess the same properties. After conducting an extensive investigation of M. organophilum strains, Kato et al. (2005) concluded that DSM 760T had been mislabeled.
We thus recommend that the GCF_003096615.1 assembly should not be used as a reference for the type strain of M. organophilum because it would lead to erroneous conclusions. As an example, the recent report of Hördt et al. (2020) used GCF_003096615.1 to propose M. organophilum as a later heterotypic synonym of M. radiotolerans.
Rhizobium leguminosarum is the nomenclatural type of the genus Rhizobium (Frank, 1889), and the type strain, which was isolated from nodules of pea (Pisum sativum), has the original designation of 3Hoq18T. After the proposal of Jordan (1984) for the reclassification of R. trifolii and R. phaseoli as two biovars of R. leguminosarum, the description of these species of R. leguminosarum was included in the second edition of Bergey’s Manual; however, its re-examination was also recommended by Kuykendall et al. (2005). Ramírez-Bahena et al. (2008) in a paper published on 01 November 2008, analyzed the taxonomic status of these species employing DDH and 16S–23S ITS, rrs, recA, and atpD sequence analyses, in addition to phenotypic characteristics. The 16S rRNA gene sequence of R. leguminosarum ATCC 10004T (held in the author’s lab since 1990) was compared with the sequence of R. leguminosarum USDA 2370T, surprisingly sharing only 99.2% of similarity. The authors then compared the sequences of the recA and atpD genes from strain ATCC 10004T with those of R. leguminosarum USDA 2370T and the 16S–23S ITS region of ATCC 10004T with the sequence of R. leguminosarum LMG 14904T. The results suggested that the strain ATCC 10004T did not belong to the same species of USDA 2370T and LMG 14904T. According to the information recorded from culture collections and molecular analyses from 3Hoq18T and additional R. leguminosarum type strains, concluded that the ATCC collection distributed different strains with the same accession number. The results obtained from sequence analysis confirmed that the strain ATCC 10004T received in 1,990 was identical to strains DSM 30132 and NCIMB 11478 and was different from strains LMG 14904T and USDA 2370T (which were identical to each other). Also, the strain that was being provided in 2008 by the ATCC under the designation of ATCC 10004T was identical to strains LMG 14904T and USDA 2370T. Since strain USDA 2370T was the original deposit corresponding to strain 3Hoq18T, it retained the name R. leguminosarum. The strain DSM 30132 = NCIMB 11478 (old strain ATCC 10004T, incorrectly distributed) shared DDH of 57% with R. leguminosarum USDA 2370T and was thus described as R. pisi. Finally, regarding the decision about the status of the names R. trifolii and R. phaseoli, Ramírez-Bahena et al. (2008) suggested that R. trifolii should be considered as a later synonym of R. leguminosarum.
Later, on 01 December 2008, R. fabae was described by Tian et al. (2008). The type strain CCBAU 33202T was isolated from root nodules of Vicia faba. According to the 16S rRNA gene analysis, the closest relative of R. fabae CCBAU 33202T was reported to be Rhizobium etli CFN42T (99.5% similarity), followed by “R. leguminosarum bv. trifolii” T24 (99.3%), “R. leguminosarum bv. viciae” USDA2370T (99.1%), and “R. leguminosarum bv. phaseoli” USDA 2671 (99.1%). The DDH value described from the comparison of R. fabae CCBAU 33202T with R. etli CFN 42T and strains of the three biovars of R. leguminosarum were 19 and 14–43%, respectively.
Here, the genomes of R. fabae CCBAU 33202T and R. pisi DSM 30132T shared 97.5% of ANIb and at least 87.8% of dDDH. The 16S rRNA gene sequences extracted from them shared high identity with the reference sequences (DQ835306 and AY509899). Consequently, we propose that R. fabae Tian et al. 2008 should be considered as a later heterotypic synonym of R. pisi Ramírez-Bahena et al. 2008.
Rhizobium gallicum was described by Amarger et al. (1997). The type strain R-602 spT had been isolated from root nodules of field-grown Phaseolus vulgaris sampled in France (Geniaux et al., 1993).
We found that the genome available on RefSeq for R. gallicum R-602 spT (GCF_000373025.1) shared an ANIb ≥98% and a dDDH ≥79.2% with the two genomic assemblies available for R. leguminosarum USDA 2370T. However, the 16S rRNA sequences extracted from R-602 spT genome shared only 98.6% of identity with the reference (AF008130), suggesting that the assembly was misclassified.
The genome for another representative of the R. gallicum type strain (SEMIA 4085T) was sequenced in this study. The 16S rRNA sequence extracted from this genome shared 99.8% identity to the reference AF008130, confirming that it was correctly identified. Moreover, SEMIA 4085T shared 80.3–80.4% of ANIb and 21.2–22.3% of dDDH with the R-602 spT genome and the type strains of R. leguminosarum. Thus, the genome of strain SEMIA 4085T (GCF_013004495.1) was different from those of R-602 spT (GCF_000373025.1) and R. leguminosarum USDA 2370T (GCF_002008365.1 and GCF_003058385.1). According to these findings, we recommend that the genome accession of SEMIA 4085T should be used as a reference for R. gallicum species. The genome GCF_000373025.1 is not from the genuine type strain R-602 spT used for the R. gallicum species description.
We also detected that R. gallicum SEMIA 4085T shared 99.2% of ANIb and at least 83.3% of dDDH with R. azibense 23C2T. Importantly, the genome of R. azibense 23C2T also contained a 16S rRNA sequence that possessed a high identity of 99.13% with the reference sequence (JN624691) and, thus, appeared to be correctly identified. R. azibense was described by Mnasri et al. (2014) as representing a genomic group closely related to R. gallicum isolated from root nodules of P. vulgaris. Considering the genome relatedness found here, R. azibense Mnasri et al. 2014 should be considered as a later heterotypic synonym of R. gallicum Amarger et al. 1997.
Rhizobium loessense was described by Wei et al. (2003) based upon the type strain CCBAU 7190BT (= CGMCC 1.3401T) isolated from Astragalus complanatus nodules. The 16S rRNA sequence similarities between R. loessense CCBAU 7190BT and the most closely related strains described, Rhizobium galegae HAMBI 540T and Rhizobium huautlense SO2T, were 96.8 and 97.5 %, respectively. The DDH similarity between R. galegae HAMBI 540T and R. huautlense SO2T with R. loessense CCBAU 7190BT, were reported to be 40.1 and 9.3%, respectively.
Rhizobium mongolense USDA 1844T was isolated from Medicago ruthenica nodules as described by Van Berkum et al. (1998). Although Wei et al. (2003) cited previous reports where R. galegae and R. huautlense were grouped with R. mongolense, and R. gallicum (Wang et al., 1998; Peng et al., 2002), the type-strains from these species were not included in DDH experiments.
Here, R. loessense CCBAU 7190BT and two genomes assemblies from R. mongolense USDA 1844T share 96.03% of ANIb and 66.3% (C.I. 62.9 – 69.5%, formula d6) to 63.3 (C.I. 59.6 – 66.9%, formula d0) of dDDH. All the four 16S rRNA sequences extracted from R. mongolense assemblies and the one sequence extracted from R. loessense CCBAU 7190BT presented high identities with the reference sequences (U89817 and AF364069) confirming their authenticity.
To be coherent with Wayne et al. (1987), those strains should be placed in separate species. However, this case is open for alternative interpretations. Hördt et al. (2020) obtained 70% of dDDH between R. mongolense USDA 1844T and R. loessense CGMCC 1.3401T and proposed the new subspecies R. mongolense subsp. loessense from R. loessense based on a threshold of <79% for subspecies according to Meier-Kolthoff et al. (2014).
Rhodoplanes piscinae was described with the type strain JA266T isolated from a surface water sample from a freshwater fishpond (Chakravarthy et al., 2012). According to the 16S rRNA gene phylogeny, R. piscinae JA266T (= DSM 19946T) was closely related to R. serenus TUT3530T (= DSM 18633T) isolated from pond water and described by Okamura et al. (2009). To differentiate R. piscinae JA266T from its closest relative, Chakravarthy et al. (2012) conducted a DDH experiment between both strains, which yielded a value of less than 65%.
In this work, R. piscinae DSM 19946T and R. serenus DSM 18633T genomes shared 97.6% of ANIb and 88.5% of dDDH. The 16S rRNA gene sequences extracted from these genomes were compatible with the reference sequences (LC178579 and AB087717) and confirmed the identity of the assemblies. We thus propose that R. piscinae Chakravarthy et al. 2012 should be considered as a later heterotypic synonym of Rhodoplanes serenus Okamura et al. 2009.
“Bartonella mastomydis” was proposed by Dahmani et al. (2018), with the type strain 008T being isolated from Mastomys erythroleucus rodents. Based on a phylogeny reconstructed from concatenated gltA, rpoB, 16S RNA, and ftsZ sequences, B. elizabethae F9251T (Brenner et al., 1993) was recognized as the closest relative to strain “B. mastomydis” 008T, with a dDDH value of 60.3 ± 2.8%.
Here, “B. mastomydis” 008T and two genome assemblies from B. elizabethae F9251T were found to share an ANIb of 95.1% and at least 89.5% of dDDH. The 16S rRNA sequences extracted from “B. mastomydis” 008T and B. elizabethae F9251T genomes shared identity values (≥99%) with reference sequences KY555064 and L01260, respectively, confirming their identity. These results suggest that “B. mastomydis” represents a synonym of B. elizabethae (Daly et al. 1993) Brenner et al. 1993.
Bartonella vinsonii was proposed by Brenner et al. (1993), with the type strain ATCC VR-152T isolated from voles by Baker, 1946. Later, Kordick et al. (1996) isolated the strain 93-C01T from the blood of a dog with valvular endocarditis. According to the 16S rRNA gene analysis, 93-C01T was closely related to the type strain of B. vinsonii. According to DDH tests, 93-C01T and B. vinsonii ATCC VR-152T genomes shared 81% (hybridization at 55°C) to 70% (hybridization at 70°C) of DDH and 5°C of ΔTm. Kordick et al. (1996) then proposed B. vinsonii subsp. berkhoffii to accommodate 93-C01T, considering that: (i) a second isolate (G7464) was significantly more closely related to 93-C01T than both were related to B. vinsonii; (ii) 93-C01T and G7464 shared a unique insertion in their 16S rRNAs; and (iii) both were isolated from dogs.
In 1999, Welch et al. (1999) compared an isolate from human blood culture (OK 94-513T) to Bartonella species. They reported that its reciprocal DNA relatedness to B. vinsonii subsp. vinsonii and B. vinsonii subsp. berkhoffii was 65 to 85% at 55°C. Based on these and other results, proposed creating a third subspecies within B. vinsonii, B. vinsonii subsp. arupensis with the type strain OK 94-513T.
We found that the genomes of B. vinsonii subsp. berkhoffii ATCC 51672T and B. vinsonii subsp. arupensis OK-94-513T were not within the same genome cluster, sharing ANIb values of only 91.3%. The dDDH values present between them were 74.5 (d0) and 81.5% (d6). To be consistent with the Wayne et al. (1987) recommendation, these strains should be placed in separate species. The 16S rRNA sequences extracted from B. vinsonii subsp. arupensis and subsp. berkhoffii genomes presented high identity with the reference sequences of HF558389 and L35052, respectively, confirming their identity. The taxonomic status of the three B. vinsonii subspecies may need to be revised after sequencing the type-strain of B. vinsonii subsp. vinsonii.
So far, we have focused on classification and authenticity errors. Now we deal with the third type of error where genomes are erroneously assigned as type strains in the databases. These errors were discovered by the identification of clusters containing more than one described bacterial species as well as different genomes assemblies supposedly belonging to the same strains that were not clustered together.
The genomes of X. autotrophicus DSM 432T (GCF_005871085.1) and “X. autotrophicus” Py2 (GCF_000017645.1) presented only 91% of ANIb and up to 57.6% of dDDH, consequently, the strains were not found forming a cluster. In 1986, Van Ginkel and De Bont, 1986 isolated bacterial strains from soil and water samples that were able to grow in an atmosphere of 5% alkene in the air. Based on physiological, morphological, and GC content data, the yellow-pigmented strain “X. autotrophicus” Py2 was assigned to the genus Xanthobacter. Meanwhile, the original type strain of X. autotrophicus was isolated from a black pool sludge 8 years before (Wiegel et al., 1978). Considering this, “X. autotrophicus” Py2 was incorrectly identified as a type strain in the RefSeq database.
Genomes of Aminobacter aminovorans DSM 7048T (GCF_004341645.1) and NCTC10684T (GCF_900445235.1) shared ANIb and dDDH of 100%. A third type strain, “A. aminovorans” KCTC 2477T (GCF_001605015.1), was not clustered with the other two strains. DSM 7048T and NCTC 10684T indeed represented A. aminovorans type strain, while KCTC 2477T (= ATCC 29600T = DSM 10368T) does not. KCTC 2477T was classified as the type strain of Chelatobacter heintzii before being reclassified as A. aminovorans (Kämpfer et al., 2002). In the KCTC 2477T genome announcement (Lee S.H. et al., 2016), the strain is wrongly assigned as A. aminovorans type strain, which can explain its erroneous assignment in the NCBI metadata.
“Pseudomonas” spp. ATCC 29600T was isolated by successive enrichment in a medium with nitrilotriacetate (NTA) from a soil surrounding a dry well that had received septic tank effluent (Tiedje et al., 1973). In a study of NTA-utilizing organisms, Auling et al. (1993) described Chelatobacter genus with ATCC 29600T representing C. heintzii as the only species. Later, C. heintzii was reclassified based on (i) similarities above 99 ± 6% between the 16S rRNA gene sequences from C. heintzii DSM 10368T with A. aminovorans DSM 7048T, A. aganoensis DSM 7051T, and A. niigataensis DSM 7050T (Kämpfer et al., 1999); and (ii) a DDH similarity study that showed that C. heintzii DSM 10368T strain shared DDH values of at least 70% with A. aminovorans DSM 7048T (Kämpfer et al., 2002).
Here, the C. heintzii KCTC 2477T genome shared an ANIb of only 90.8% and a dDHH value up to 65.2% with the two assemblies of A. aminovorans DSM 7048T and NCTC 10684T. The 16S rRNA gene sequences extracted from C. heintzii KCTC 2477T and A. aminovorans assemblies were compatible with their reference sequences (LT984904 and AJ011759, respectively) and confirmed their identities. Considering this, C. heintzii KCTC 2477T and A. aminovorans DSM 7048T represent members of different species.
To identify additional strains belonging to the same species as C. heintzii KCTC 2477T, 21 genome sequences for Aminobacter were obtained, including a recently deposited genome for DSM 10368T. They included the genomes for A. aganoensis DSM 7051T, A. carboxidus DSM 1086T, A. ciceronei DSM 17455T and DSM 15910T, A. lissarensis DSM 17454T, and A. niigataensis DSM 7050. The quality of these genomes was checked (Supplementary Table 5), and the comparisons were calculated using ANIb, dDDH, and ProKlust as described before.
In addition to the expected C. heintzii DSM 10368T genome, the cluster containing C. heintzii KCTC 2477T (Figure 4) also included Aminobacter sp. SR38, Aminobacter sp. MDW-2 (accessions GCF_009674635.1 and GCF_014250155.1), and A. ciceronei DSM 17455T (GCF_014138635.1) and DSM 15910T (GCF_014138625.1). Importantly, all the genomes forming this cluster shared ANIb values above 98%. The dDDH calculated (Supplementary Table 6) with formula d6 between these strains ranged from 74.1 to 100%. All comparisons presented a dDDH average of ≥70% for formula d0, except between A. ciceronei DSM 17455T/DSM 15910T with Aminobacter sp. SR38 (69.8, CI: 65.8 – 73.4%), and C. heintzii DSM 10368T and Aminobacter sp. SR38 (69.9, CI: 66 – 73.5%). The 16S rRNA sequences extracted from C. heintzii DSM 10368T and A. ciceronei DSM 17455T/DSM 15910T genomes were compatible with the reference sequences LT984904 and AF034798, respectively.
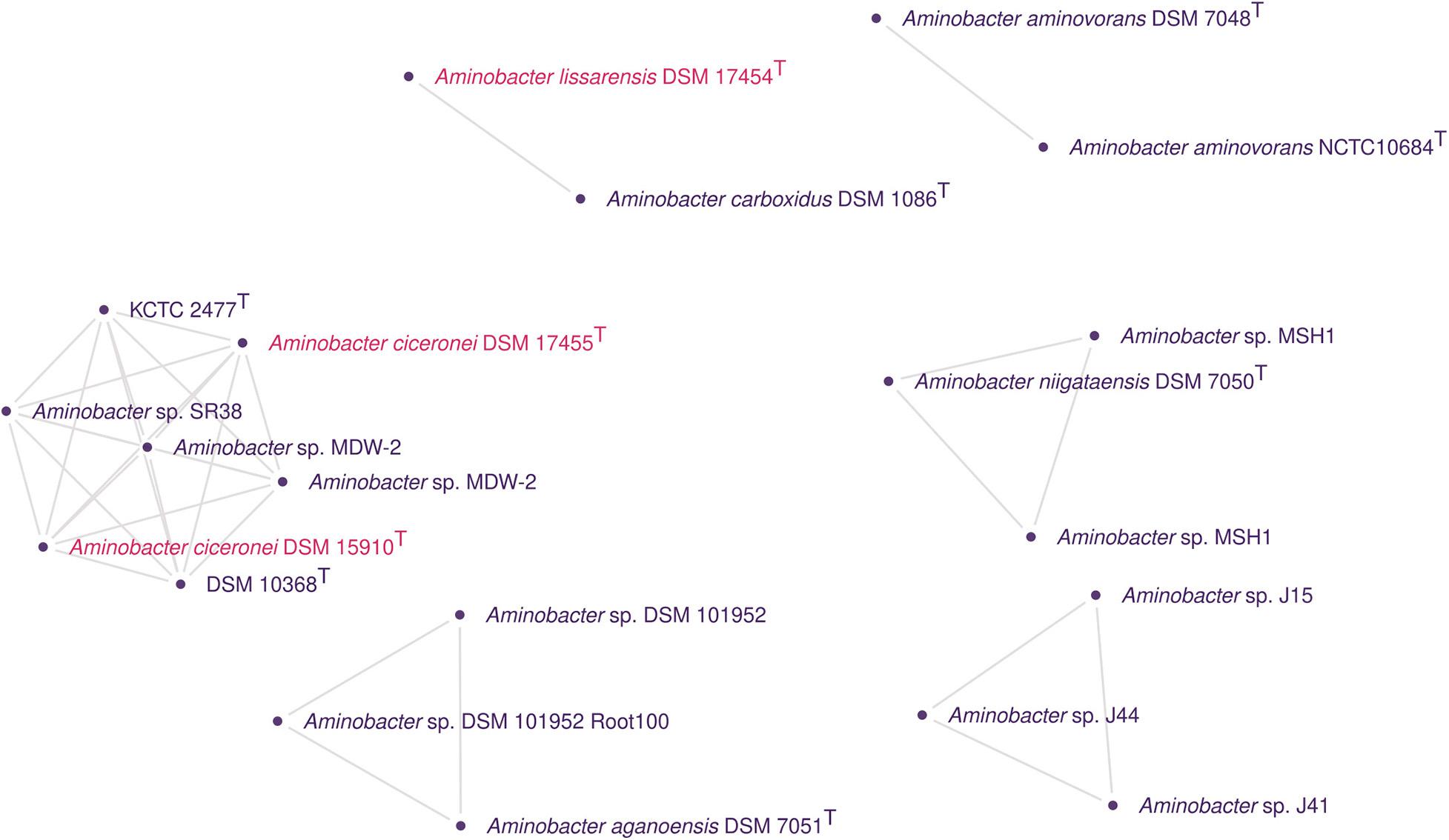
Figure 4. Genomic clusters detected using pairwise ANI values from Aminobacter genomes. In the graph, the clusters have nodes corresponding to genomes and edges corresponding to ANI values above the cut-off for species delineation with a reliable alignment of between the genomes. Isolated nodes were removed. Different colors were used whenever possible to indicate different species names.
All the strains present in the C. heintzii KCTC 2477T/DSM 10368T cluster were pesticide-/herbicide-degrading bacteria. Rousseaux et al. (2001) isolated Aminobacter sp. SR38 (= LR3-3), an atrazine-degrading bacterium, from agricultural French soil with a history of treatment with the herbicide. The nearest neighbor of SR38 was C. heintzii (AJ011762) according to the 16S rRNA gene analysis conducted. MDW-2 was isolated from sludge from the wastewater-treating system of a pesticide manufacturer (Zhang et al., 2017). The strain can degrade methomyl completely in biochemical cooperation with Afipia sp. MDW-3 (Zhang et al., 2017). MDW−2 was also reported to show the highest similarity with A. aganoensis DSM 7051T (AJ011760).
IMB-1T (= DSM 17455T = DSM 15910T) was isolated from CH3Br-fumigated soil in California. It has been reported to be capable of growth on CH3Cl, CH3Br, CH3I, and methylated amines as sole carbon and energy sources (Miller et al., 1997; Connell Hancock et al., 1998). After, McDonald et al. (2005) proposed the species A. ciceronei to accommodate IMB-1T based on DDH comparisons with A. aminovorans DSM 7048T (47.7%), A. aganoensis DSM 7051T (37.2%), and A. niigataensis DSM 7050T (17.9%). In the same paper, A. lissarensis was also proposed to accommodate CC495T (= DSM 17454T), a strain isolated from unpolluted beech woodland soil in Northern Ireland by enrichment culture using CH3Cl (Coulter et al., 1999). A. lissarensis CC495T was able to grow on methylamine as sole carbon and energy source, as well as with CH3Cl and CH3Br with cyanocobalamin supplementation. The DDH analysis of A. lissarensis CC495T indicated that it shared 20.8, 49.5, and 31.8% hybridization with A. aminovorans DSM 7048T, A. aganoensis DSM 7051T, and A. niigataensis DSM 7050T, respectively. The authors also reported that replicate DDH experiments between A. aminovorans DSM 7048T and strain A. lissarensis CC495T gave widely differing values (20.8–72.0%) in reciprocal hybridizations. Here, we found that A. lissarensis DSM 17454T shared 98.1% of ANIb and at least 81.7% of dDDH with A. carboxidus DSM 1086T.
Strain Z-1171T (CIP 105722T = DSM 1086T) was isolated from soil in Moscow (Russia) and first described as Achromobacter carboxydus (Nozhevnikova and Zavarzin, 1974). Z-1171T was assigned to the physiological group of carboxydobacteria due to its ability to grow aerobically on carbon monoxide as the sole carbon and energy source (Zavarzin and Nozhevnikova, 1977; Meyer and Schlegel, 1983). Meyer et al. (1993) transferred A. carboxydus to a new genus as Carbophilus carboxidus based on 16S rRNA similarities and phenotypic characteristics. Recently, Hördt et al. (2020) reported a 16S rRNA gene tree where C. carboxidus CIP 105722T was nested within Aminobacter, leading to the transferring Carbophilus to Aminobacter.
Considering the genome relatedness found here, we propose A. heintzii, comb. nov. as a new combination for C. heintzii Auling et al. 1993. We also propose the reclassification of A. ciceronei McDonald et al. 2005 as A. heintzii. Finally, A. lissarensis McDonald et al. 2005 should be considered as a later heterotypic synonym of A. carboxidus (Meyer et al. 1994) Hördt et al. 2020.
The results of the OGRI and ProKlust analyses revealed several inaccuracies with the taxonomic scheme with the type strains of species from the order Rhizobiales. This came as no surprise considering that the rapid expansion of sequenced bacterial and archaeal genomes in the past decade (Garrity, 2016; Hugenholtz et al., 2016; Yoon et al., 2017a) was accompanied with numerous reclassification and name changes (Sant’Anna et al., 2017; Nouioui et al., 2018; García-López et al., 2019; Volpiano et al., 2019; Hördt et al., 2020). The most likely reason for this is that traditional DDH and ΔTm measurements are more imprecise than the application of ANI and dDDH in silico surrogates (Auch et al., 2010b; Meier-Kolthoff et al., 2013).
The genome metrics approach is based on computationally intensive pairwise genomic alignments and calculations, which are a disadvantage in large-scale studies. FastANI uses fast approximate read mapping with Mapmash, based on MinHash alignment identity estimates, being reported to be 50-4608 times faster than ANIb (Jain et al., 2018). We found that the same groups of genospecies in our set of 520 genomes were obtained using both ANIb and FastANI algorithms. To guarantee accuracy and to reduce the computing cost for species delineation, FastANI could be employed to sieve a lower total number of genomes for subsequent alignment and calculation of identity with ANIb and dDDH.
ProKlust demonstrated to be a useful graph-based approach to extract genomic groups from large OGRI matrices, which can also be applied to other identity/similarity matrices. Our tool has the advantage of settable cut-off points, the possibility of multiple matrices entries, besides useful functions to filter and visualize the obtained clusters. Graph-based approaches depend upon finding cliques or completely connected subgraphs utilizing a threshold value. Higher threshold values generally result in a less connected graph and therefore smaller cluster sizes (Jay et al., 2012). To date, we have found two graph-based tools that were useful with OGRI data. The first is the “Genome Clustering” web-based tool from MicroScope (Microbial Genome Annotation and Analysis Platform) described by Vallenet et al. (2009). The interface allows the user to define a set of 3 to 500 genomes from the current 5,033 genomes available at the platform. The user can also add their own data. The platform estimates the genomic similarity using Mash distances, which are reported to be well correlated to ANI, especially in the range of 90–100% (Ondov et al., 2016). To obtain genome clusters, “Genome Clustering” i) connects all nodes using the pairwise Mash distances; ii) removes edges representing distances above 0.06 (which is expected to correspond to 94% of ANI); (iii) removes incomplete or contaminated genomes with CheckM (Parks et al., 2015); and (iv) extracts communities from the network with the Louvain community detection algorithm (Blondel et al., 2008). The second approach that we have found is described by Varghese et al. (2015), which also employs an approach based on MCE to analyze the genome-wide ANI (gANI) metric and AF between the matrices generated from 13,151 genomes. They employed a PERL script that uses the C++ Bron–Kerbosch module to construct maximal cliques using the criteria of a minimum pairwise AF of at least 0.6 and a minimum pairwise gANI of at least 96.5%. However, the script used by the authors was not made available with the publication, and no filter parameter seems to be available.
When dealing with genome assemblies from public databases, taxonomists should be aware of the level of contamination and the presence of misidentified type strains on their genome sets (Salvà-Serra et al., 2019). In our study, a series of erroneous conclusions could have been made if the noteworthy presence of misidentified type strains and unauthentic genomes found in our genome set was not previously detected. The 16S rRNA gene analysis and a careful check on genome associated metadata have proved to be an important sanity check step on wide genome-based taxonomy surveys, which could prevent, for example, the recent report of Hördt et al. (2020) to propose the reclassification of M. radiotolerans using comparisons with an unauthentic M. organophilum genome.
Genomic and phenotypic approaches have unique contributions to microbial taxonomy, but the integration of both worlds is challenging (Sanford et al., 2021), especially in studies dealing with hundreds of strains. Phenotypic evaluation is important to characterize a taxon of interest, but its accuracy is affected by differential gene expression and interpretation bias (Petti et al., 2005; Vital et al., 2015; Sant’Anna et al., 2017). Nevertheless, an evaluation of phenotypic and morphologic properties of problematic type-strains found here, along with a comparison with the species’ descriptions, would corroborate that these strains are misidentified, but was not within the scope of this study.
The results of this study are considered to contribute to improving the taxonomic classification of species within Rhizobiales order. The genome clustering approach and the tool described here are proven as useful to detect species and to provide identity information from large genome sets.
Basonym: Chelatobacter heitzii Auling et al. 1993.
Aminobacter heintzii (hein.tzii, N. L. gen. masc. n. heintzii of Heintz’s, named after the chemist W. Heintz, an honor proposed by Auling et al., 1993).
The description for the type strain KCTC 2477T = ATCC 29600T = DSM 10368T is as given for C. heitzii (Auling et al., 1993). Phenotypes for strain IMB-1 = DSM 17455 = DSM 15910 are present by Connell Hancock et al. (1998) and McDonald et al. (2005). Strains SR38 = LR3-3 and MDW-2 are characterized by Rousseaux et al. (2001) and Zhang et al. (2017).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
CV, FS, and AA elaborated the conception of the study. CV wrote ProKlust, collected and analyzed the data, and wrote the manuscript. JS and AB provided the strain SEMIA 6460T. ES and WW provided sequence for SEMIA 4085T and SEMIA 6460T genomes, respectively. FS, AA, JS, AB, BL, LV, WW, ES, and LP revised the manuscript critically. All authors read and approved the final manuscript.
The work conducted by the United States Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, was supported by the Office of Science of the United States Department of Energy under Contract No. DE-AC02-05CH11231. FS and AA received scholarships from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil). CV received a scholarship from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Sequence data for SEMIA 6460T were produced by the United States Department of Energy Joint Genome Institute https://www.jgi.doe.gov., in collaboration with the user community. JGI and IMG resource was used for analysis. Sequence data for SEMIA 4085T was produced by the Department of Biochemistry and Molecular Biology, Federal University of Paraná. We gratefully acknowledge the support of the following agencies: CNPq, CAPES, and FAPERGS (Fundação de Amparo à Pesquisa do Estado do RS). We would like to thank the Computer Engineering undergraduate student Pedro Durayski Saccilotto for the assistance on the ProKlust code.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.614957/full#supplementary-material
Allaire, J. J., Gandrud, C., Russell, K., and Yetman, C. J. (2017). networkD3: D3 JavaScript Network Graphs from R. R package version 0.4. Vienna: R Core Team.
Amarger, N., Macheret, V., and Laguerre, G. (1997). Rhizobium gallicum sp. nov. and Rhizobium giardinii sp. nov., from Phaseolus vulgaris nodules. Int. J. Syst. Evol. Microbiol. 47, 996–1006. doi: 10.1099/00207713-47-4-996
Anderson, C. R., Dick, G. J., Chu, M. L., Cho, J. C., Davis, R. E., Brauer, S. L., et al. (2009). Aurantimonas manganoxydans, sp. nov. and Aurantimonas litoralis, sp. nov.: Mn (II) oxidizing representatives of a globally distributed clade of alpha-Proteobacteria from the order Rhizobiales. Geomicrobiol. J. 26, 189–198. doi: 10.1080/01490450902724840
Auch, A. F., Hans-Peter, K., and Göker, M. (2010a). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic. Sci. 2, 142–148. doi: 10.4056/sigs.541628
Auch, A. F., von Jan, M., Klenk, H. P., and Göker, M. (2010b). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic. Sci. 2, 117–134. doi: 10.4056/sigs.531120
Auling, G., Busse, H. J., Egli, T., El-Banna, T., and Stackebrandt, E. (1993). Description of the Gram-negative, obligately aerobic, nitrilotriacetate (NTA)-utilizing bacteria as Chelatobacter heintzii, gen. nov., sp. nov., and Chelatococcus asaccharovorans, gen. nov., sp. nov. Syst. Appl. Microbiol. 16, 104–112. doi: 10.1016/S0723-2020(11)80254-7
Austin, B., and Goodfellow, M. (1979). Pseudomonas mesophilica, a new species of pink bacteria isolated from leaf surfaces. Int. J. Syst. Evol. Microbiol. 29, 373–378. doi: 10.1099/00207713-29-4-373
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blondel, V. D., Guillaume, J. L., Lambiotte, R., and Lefebvre, E. (2008). Fast unfolding of communities in large networks. J. Stat. Mech. 2008:10008. doi: 10.1088/1742-5468/2008/10/P10008
Brenner, D. J., O’Connor, S. P., Winkler, H. H., and Steigerwalt, A. G. (1993). Proposals to unify the genera Bartonella and Rochalimaea, with descriptions of Bartonella quintana comb. nov., Bartonella vinsonii comb. nov., Bartonella henselae comb. nov., and Bartonella elizabethae comb. nov., and to remove the family Bartonellaceae from the order Rickettsiales. Int. J. Syst. Evol. Microbiol. 43, 777–786. doi: 10.1099/00207713-43-4-777
Brewin, N., Beringer, J., and Johnston, A. (1980). Plasmid-mediated transfer of host-range specificity between two strains of Rhizobium leguminosarum. Microbiology 120, 413–420. doi: 10.1099/00221287-120-2-413
Chakravarthy, S. K., Ramaprasad, E., Shobha, E., Sasikala, C., and Ramana, C. V. (2012). Rhodoplanes piscinae sp. nov. isolated from pond water. Int. J. Syst. Evol. Microbiol. 62, 2828–2834. doi: 10.1099/ijs.0.037663-0
Charif, D., and Lobry, J. R. (2007). “SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis,” in The Structural Approaches to Sequence Evolution, eds U. Bastolla, M. Porto, H. E. Roman, and M. Vendruscolo (Berlin: Springer), 207–232.
Chen, W., Yan, G., and Li, J. (1988). Numerical taxonomic study of fast-growing soybean rhizobia and a proposal that Rhizobium fredii be assigned to Sinorhizobium gen. nov. Int. J. Syst. Evol. Microbiol. 38, 392–397. doi: 10.1099/00207713-38-4-392
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Connell Hancock, T. L., Costello, A. M., Lidstrom, M. E., and Oremland, R. S. (1998). Strain IMB-1, a novel bacterium for the removal of methyl bromide in fumigated agricultural soils. Appl. Environ. Microbiol. 64, 2899–2905. doi: 10.1128/AEM.64.8.2899-2905.1998
Coulter, C., Hamilton, J. T., McRoberts, W. C., Kulakov, L., Larkin, M. J., and Harper, D. B. (1999). Halomethane: bisulfide/halide ion methyltransferase, an unusual corrinoid enzyme of environmental significance isolated from an aerobic methylotroph using chloromethane as the sole carbon source. Appl. Environ. Microbiol. 65, 4301–4312. doi: 10.1128/AEM.65.10.4301-4312.1999
Csardi, G., and Nepusz, T. (2006). The Igraph Software Package for Complex Network Research. Kalamazoo, MI: Kalamazoo College.
Dahmani, M., Diatta, G., Labas, N., Diop, A., Bassene, H., Raoult, D., et al. (2018). Noncontiguous finished genome sequence and description of Bartonella mastomydis sp. nov. N. Microbes N. Infect. 25, 60–70. doi: 10.1016/j.nmni.2018.03.005
Daly, J. S., Worthington, M. G., Brenner, D. J., Moss, C. W., Hollis, D. G., Weyant, R. S., et al. (1993). Rochalimaea elizabethae sp. nov. isolated from a patient with endocarditis. J. Clin. Microbiol. 31, 872–881. doi: 10.1128/JCM.31.4.872-881.1993
de Lajudie, P., Willems, A., Nick, G., Moreira, F., Molouba, F., Hoste, B., et al. (1998). Characterization of tropical tree rhizobia and description of Mesorhizobium plurifarium sp. nov. Int. J. Syst. Evol. Microbiol. 48, 369–382. doi: 10.1099/00207713-48-2-369
de Lajudie, P., Willems, A., Pot, B., Dewettinck, D., Maestrojuan, G., Neyra, M., et al. (1994). Polyphasic taxonomy of rhizobia: Emendation of the genus Sinorhizobium and description of Sinorhizobium meliloti comb. nov., Sinorhizobium saheli sp. nov., and Sinorhizobium teranga sp. nov. Int. J. Syst. Evol. Microbiol. 44, 715–733. doi: 10.1099/00207713-44-4-715
Denner, E. B. M., Smith, G. W., Busse, H. J., Schumann, P., Narzt, T., Polson, S. W., et al. (2003). Aurantimonas coralicida gen. nov., sp. nov., the causative agent of white plague type II on Caribbean scleractinian corals. Int. J. Syst. Evol. Microbiol. 53, 1115–1122. doi: 10.1099/ijs.0.02359-0
Doronina, N. V., Kaparullina, E. N., Trotsenko, Y. A., Nörtemann, B., Bucheli-Witschel, M., Weilenmann, H. U., et al. (2010). Chelativorans multitrophicus gen. nov., sp. nov. and Chelativorans oligotrophicus sp. nov., aerobic EDTA-degrading bacteria. Int. J. Syst. Evol. Microbiol. 60, 1044–1051. doi: 10.1099/ijs.0.003152-0
Douglas, H. C. (1957). “Order III. Hyphomicrobiales Douglas, ordo nov,” in Bergey’s Manual of Determinative Bacteriology, 7th Edn, eds R. S. Breed, E. G. D. Murray, and N. R. Smith (Baltimore, MD: The Williams and Wilkins Co), 276.
Duo, J. L., Cha, Q. Y., Zhou, X. K., Zhang, T. K., Qin, S. C., Yang, P. X., et al. (2019). Aquabacter cavernae sp. nov., a bacterium isolated from cave soil. Int. J. Syst. Evol. Microbiol. 69, 3716–3722. doi: 10.1099/ijsem.0.003585
Eppstein, D., Löffler, M., and Strash, D. (2010). “Listing all maximal cliques in sparse graphs in near-optimal time,” in Algorithms and Computation, eds O. Cheong, K. Y. Chwa, and K. Park (Berlin: Springer), 403–414.
Frank, B. (1889). Über die Pilzsymbiose der Leguminosen. Ber. Dtsch. Bot. Ges. 7, 332–346. doi: 10.1111/j.1438-8677.1889.tb05711.x
García-López, M., Meier-Kolthoff, J. P., Brian, J. T., Sabine, G., Tanja, W., Nikos, C. K., et al. (2019). Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front. Microbiol. 10:2083. doi: 10.3389/fmicb.2019.02083
Garrity, G. M. (2016). A new genomics-driven taxonomy of Bacteria and Archaea: Are we there yet? J. Clin. Microbiol. 54, 1956–1963. doi: 10.1128/jcm.00200-16
Geniaux, E., Laguerre, G., and Amarger, N. (1993). Comparison of geographically distant populations of Rhizobium isolated from root nodules of Phaseolus vulgaris. Mol. Ecol. 2, 295–302. doi: 10.1111/j.1365-294X.1993.tb00022.x
Gevers, D., Dawyndt, P., Vandamme, P., Willems, A., Vancanneyt, M., Swings, J., et al. (2006). Stepping stones towards a new prokaryotic taxonomy. Phil. Trans. R. Soc. B 361, 911–1916. doi: 10.1098/rstb.2006.1915
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Graham, P., Sadowsky, M., Keyser, H., Barnet, Y., Bradley, R., Cooper, J., et al. (1991). Proposed minimal standards for the description of new genera and species of root-and stem-nodulating bacteria. Int. J. Syst. Evol. Microbiol. 41, 582–587. doi: 10.1099/00207713-41-4-582
Green, P. N., and Ardley, J. K. (2018). Review of the genus Methylobacterium and closely related organisms: a proposal that some Methylobacterium species be reclassified into a new genus, Methylorubrum gen. nov. Int. J. Syst. Evol. Microbiol. 68, 2727–2748. doi: 10.1099/ijsem.0.002856
Green, P. N., and Bousfield, I. J. (1983). Emendation of Methylobacterium Patt, Cole, and Hanson 1976; Methylobacterium rhodinum (Heumann, 1962) comb. nov. corrig.; Methylobacterium radiotolerans (Ito and Iizuka, 1971) comb. nov. corrig.; and Methylobacterium mesophilicum (Austin and Goodfellow, 1979). Int. J. Syst. Evol. Microbiol. 33, 875–877. doi: 10.1099/00207713-33-4-875
Heumann, W. (1962). Die methodik der kreuzung sternbildender bakterien. Biol. Zentralbl. 81, 341–354. doi: 10.1007/BF00888802
Holmes, B., and Roberts, P. (1981). The classification, identification and nomenclature of agrobacteria. J. Appl. Microbiol. 50, 443–467. doi: 10.1111/j.1365-2672.1981.tb04248.x
Hördt, A., López, M. G., Meier-Kolthoff, J. P., Schleuning, M., Weinhold, L. M., Tindall, B. J., et al. (2020). Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 11:468. doi: 10.3389/fmicb.2020.00468
Huang, Z., and Lai, Q. (2020). Mabikibacter ruber Choi et al. 2017 is a later heterotypic synonym of Notoacmeibacter marinus Huang et al. 2017. Int. J. Syst. Evol. Microbiol. 70, 439–441. doi: 10.1099/ijsem.0.003771
Hugenholtz, P., Skarshewski, A., and Parks, D. H. (2016). Genome-based microbial taxonomy coming of age. Cold Spring Harb. Perspect. Biol. 8:a018085. doi: 10.1101/cshperspect.a018085
Hugoson, E., Lam, W. T., and Guy, L. (2019). miComplete: weighted quality evaluation of assembled microbial genomes. Bioinformatics 36, 936–937. doi: 10.1093/bioinformatics/btz664
Ito, H., and Iizuka, H. (1971). Taxonomic studies on a radio-resistant Pseudomonas: part XII. Studies on the microorganisms of cereal grain. Agric. Biol. Chem. 35, 1566–1571. doi: 10.1080/00021369.1971.10860119
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jarvis, B., Van Berkum, P., Chen, W., Nour, S., Fernandez, M., Cleyet-Marel, J., et al. (1997). Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium ciceri, Rhizobium mediterraneum, and Rhizobium tianshanense to Mesorhizobium gen. nov. Int. J. Syst. Evol. Microbiol. 47, 895–898. doi: 10.1099/00207713-47-3-895
Jay, J. J., Eblen, J. D., Zhang, Y., Benson, M., Perkins, A. D., Saxton, A. M., et al. (2012). A systematic comparison of genome-scale clustering algorithms. BMC Bioinform. 13:S7. doi: 10.1186/1471-2105-13-S10-S7
Jia, J. L., Zhang, S. K., Tang, K., Meng, Y. J., Zheng, C., and Feng, Y. F. (2020). Methylobacterium crusticola sp. nov., isolated from biological soil crusts. Int. J. Syst. Evol. Microbiol. 70, 2089–2095. doi: 10.1099/ijsem.0.004020
Johnson, J. L., and Whitman, W. B. (2007). “Similarity analysis of DNAs,” in Methods for General and Molecular Microbiology, 3th Edn, ed. C. A. Reddy (Washington, DC: American Society of Microbiology Press), 624–652. doi: 10.1128/9781555817497.ch26
Jordan, D. (1984). “Family III. Rhizobiaceae Conn 1938,” in The Bergey’s Manual of Systematic Bacteriology, eds D. H. Bergey, N. R. Krieg, and J. G. Holt (Baltimore, MD: Williams & Wilkins), 234–256.
Kämpfer, P., Müller, C., Mau, M., Neef, A., Auling, G., Busse, H. J., et al. (1999). Description of Pseudaminobacter gen. nov. with two new species, Pseudaminobacter salicylatoxidans sp. nov. and Pseudaminobacter defluvii sp. nov. Int. J. Syst. Evol. Microbiol. 49, 887–897. doi: 10.1099/00207713-49-2-887
Kämpfer, P., Neef, A., Salkinoja-Salonen, M. S., and Buss, H. J. (2002). Chelatobacter heintzii (Auling et al., 1993) is a later subjective synonym of Aminobacter aminovorans (Urakami et al. 1992). Int. J. Syst. Evol. Microbiol. 52, 835–839. doi: 10.1099/00207713-52-3-835
Kato, Y., Asahara, M., Arai, D., Goto, K., and Yokota, A. (2005). Reclassification of Methylobacterium chloromethanicum and Methylobacterium dichloromethanicum as later subjective synonyms of Methylobacterium extorquens and of Methylobacterium lusitanum as a later subjective synonym of Methylobacterium rhodesianum. J. Gen. Appl. Microbiol. 51, 287–299. doi: 10.2323/jgam.51.287
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U S A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kordick, D. L., Swaminathan, B., Greene, C. E., Wilson, K. H., Whitney, A. M., O’Connor, S., et al. (1996). Bartonella vinsonii subsp. berkhoffii subsp. nov., isolated from dogs; Bartonella vinsonii subsp. vinsonii; and emended description of Bartonella vinsonii. Int. J. Syst. Evol. Microbiol. 46, 704–709. doi: 10.1099/00207713-46-3-704
Kuykendall, L. D. (2005). “Order VI, Rhizobiales ord. nov,” in Bergey’s Manual of Systematic Bacteriology: The Proteobacteria, eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 324.
Kuykendall, L., Young, J., Martínez-Romero, E., Kerr, A., and Sawada, H. (2005). “Genus Rhizobium, a highly divergent genus in a revised family, the Rhizobiaceae,” in The Bergey’s Manual of Systematic Bacteriology, eds G. Garrity, D. J. Brenner, N. R. Krieg, and J. T. Staley (Berlin: Springer), 324–340.
Laetsch, D. R., and Blaxter, M. L. (2017). BlobTools: Interrogation of genome assemblies. F1000research 6:1287. doi: 10.12688/f1000research.12232.1
Lee, I., Ouk, K. Y., Park, S. C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Lee, S. H., Choe, H., Nasir, A., Park, D. S., and Kim, K. M. (2016). Complete genome sequence of nitrilotriacetate-degrading Aminobacter aminovorans KCTC 2477T. Genome Announc. 4, 1363–1316e. doi: 10.1128/genomeA.01363-16
Lee, Y., and Jeon, C. O. (2018). Methylobacterium frigidaeris sp. nov., isolated from an air conditioning system. Int. J. Syst. Evol. Microbiol. 68, 299–304. doi: 10.1099/ijsem.0.002500
Li, X., Huang, Y., and Whitman, W. B. (2015). The relationship of the whole genome sequence identity to DNA hybridization varies between genera of prokaryotes. Antonie Van Leeuwenhoek 107, 241–249. doi: 10.1007/s10482-014-0322-1
Lin, S. Y., Hameed, A., Hsieh, Y. T., and Young, C. C. (2019). Mesorhizobium composti sp. nov., isolated from compost. Antonie Van Leeuwenhoek 112, 1387–1398. doi: 10.1007/s10482-019-01270-y
Lv, H., Masuda, S., Fujitani, Y., Sahin, N., and Tani, A. (2017). Oharaeibacter diazotrophicus gen. nov., sp. nov., a diazotrophic and facultatively methylotrophic bacterium, isolated from rice rhizosphere. Int. J. Syst. Evol. Microbiol. 67, 576–582. doi: 10.1099/ijsem.0.001660
McDonald, I. R., Kämpfer, P., Topp, E., Warner, K. L., Cox, M. J., Hancock, T. L. C., et al. (2005). Aminobacter ciceronei sp. nov. and Aminobacter lissarensis sp. nov., isolated from various terrestrial environments. Int. J. Syst. Evol. Microbiol. 55, 1827–1832. doi: 10.1099/ijs.0.63716-0
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 21:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Hahnke, R. L., Petersen, J., Scheuner, C., Michael, V., Fiebig, A., et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic. Sci. 9:2. doi: 10.1186/1944-3277-9-2
Meyer, O., and Schlegel, H. G. (1983). Biology of aerobic carbon monoxide oxidizing bacteria. Annu. Rev. Microbiol. 37, 277–310. doi: 10.1146/annurev.mi.37.100183.001425
Meyer, O., Stackebrandt, E., and Auling, G. (1993). Reclassification of ubiquinone Q-10 containing carboxidotrophic bacteria: transfer of “[Pseudomonas] carboxydovorans” OM5T to Oligotropha, gen. nov., as Oligotropha carboxidovorans, comb. nov., transfer of “[Alcaligenes] carboxydus” DSM 1086T to Carbophilus, gen. nov., as Carbophilus carboxidus, comb. nov., transfer of “[Pseudomonas] compransoris” DSM 1231T to Zavarzinia, gen. nov., as Zavarzinia compransoris, comb. nov., and amended descriptions of the new genera. Syst. Appl. Microbiol. 16, 390–395. doi: 10.1016/S0723-2020(11)80271-7
Miller, L. G., Connell, T. L., Guidetti, J. R., and Oremland, R. S. (1997). Bacterial oxidation of methyl bromide in fumigated agricultural soils. Appl. Environ. Microbiol. 63, 4346–4354.
Mnasri, B., Liu, T. Y., Saidi, S., Chen, W. F., Chen, W. X., Zhang, X. X., et al. (2014). Rhizobium azibense sp. nov., a nitrogen fixing bacterium isolated from root-nodules of Phaseolus vulgaris. Int. J. Syst. Evol. Microbiol. 64, 1501–1506. doi: 10.1099/ijs.0.058651-0
Mousavi, S. A., Osterman, J., Wahlberg, N., Nesme, X., Lavire, C., Vial, L., et al. (2014). Phylogeny of the Rhizobium-Allorhizobium-Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst. Appl. Microbiol. 37, 208–215. doi: 10.1016/j.syapm.2013.12.007
Murali, A., Bhargava, A., and Wright, E. S. (2018). IDTAXA: a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 6, 1–14. doi: 10.1186/s40168-018-0521-5
Nick, G., De Lajudie, P., Eardly, B. D., Suomalainen, S., Paulin, L., Zhang, X., et al. (1999). Sinorhizobium arboris sp. nov. and Sinorhizobium kostiense sp. nov., isolated from leguminous trees in Sudan and Kenya. Int. J. Syst. Evol. Microbiol. 49, 1359–1368. doi: 10.1099/00207713-49-4-1359
Nouioui, I., Lorena, C., García-López, M., Meier-Kolthoff, J. P., Woyke, T., Kyrpides, N. C., et al. (2018). Genome-based taxonomic classification of the phylum Actinobacteria. Front. Microbiol. 9:2007. doi: 10.3389/fmicb.2018.02007
Nozhevnikova, A. N., and Zavarzin, G. A. (1974). Taxonomy of co-oxidizing gram-negative bacteria. Izv. Akad. Nauk. SSSR Biol. 436–440.
Nuti, M., Lepidi, A., Prakash, R., Schilperoort, R., and Cannon, F. (1979). Evidence for nitrogen fixation (nif) genes on indigenous Rhizobium plasmids. Nature 282, 533–535. doi: 10.1038/282533a0
Okamura, K., Kanbe, T., and Hiraishi, A. (2009). Rhodoplanes serenus sp. nov., a purple non-sulfur bacterium isolated from pond water. Int. J. Syst. Evol. Microbiol. 59, 531–535. doi: 10.1099/ijs.0.000174-0
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Palmer, M., Steenkamp, E. T., Blom, J., Hedlund, B. P., and Venter, S. N. (2020). All ANIs are not created equal: implications for prokaryotic species boundaries and integration of ANIs into polyphasic taxonomy. Int. J. Syst. Evol. Microbiol. 70, 2937–2948. doi: 10.1099/ijsem.0.004124
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patt, T. E., Cole, G. C., and Hanson, R. S. (1976). Methylobacterium, a new genus of facultatively methylotrophic bacteria. Int. J. Syst. Evol. Microbiol. 26, 226–229. doi: 10.1099/00207713-26-2-226
Peng, G. X., Tan, Z. Y., Wang, E. T., Reinhold-Hurek, B., Chen, W. F., and Chen, W. X. (2002). Identification of isolates from soybean nodules in Xinjiang Region as Sinorhizobium xinjiangense and genetic differentiation of S. xinjiangense from Sinorhizobium fredii. Int. J. Syst. Evol. Microbiol. 52, 457–462. doi: 10.1099/00207713-52-2-457
Petti, C. A., Polage, C. R., and Schreckenberger, P. (2005). The role of 16S rRNA gene sequencing in identification of microorganisms misidentified by conventional methods. J. Clin. Microbiol. 43, 6123–6125. doi: 10.1128/JCM.43.12.6123-6125.2005
Prakash, R., Schilperoort, R., and Nuti, M. (1981). Large plasmids of fast-growing rhizobia: homology studies and location of structural nitrogen fixation (nif) genes. J. Bacteriol. 145, 1129–1136.
Ramírez-Bahena, M. H., García-Fraile, P., Peix, A., Valverde, A., Rivas, R., Igual, J. M., et al. (2008). Revision of the taxonomic status of the species Rhizobium leguminosarum (Frank 1879) Frank, 1889AL, Rhizobium phaseoli Dangeard 1926AL and Rhizobium trifolii Dangeard 1926AL. R. trifolii is a later synonym of R. leguminosarum. Reclassification of the strain R. leguminosarum DSM 30132 (=NCIMB 11478) as Rhizobium pisi sp. nov. Int. J. Syst. Evol. Microbiol. 58, 2484–2490. doi: 10.1099/ijs.0.65621-0
Ren, D. W., Chen, W. F., Sui, X. H., Wang, E. T., and Chen, W. X. (2011). Rhizobium vignae sp. nov., a symbiotic bacterium isolated from multiple legume species. Int. J. Syst. Evol. Microbiol. 61, 580–586. doi: 10.1099/ijs.0.023143-0
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U S A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Rousseaux, S., Hartmann, A., and Guy, S. (2001). Isolation and characterisation of new Gram-negative and Gram-positive atrazine degrading bacteria from different French soils. FEMS Microbiol. Ecol. 36, 211–222. doi: 10.1111/j.1574-6941.2001.tb00842.x
Salvà-Serra, F., Jaén-Luchoro, D., Karlsson, R., Bennasar-Figueras, A., Jakobsson, H. E., and Moore, E. R. (2019). Beware of false “type strain” genome sequences. Microbiol. Resour. Announc. 8, 369–319e. doi: 10.1128/MRA.00369-19
Sambrook, J., and Russell, D. (2006). “Purification of nucleic acids by extraction with phenol: chloroform,” in Molecular Cloning, eds J. Sambrook and D. W. Russell (New York, NY: Cold Spring Harbor Laboratory Press), doi: 10.1101/pdb.prot4455
Sanford, R. A., Lloyd, K. G., Konstantinidis, K. T., and Löffler, F. E. (2021). Microbial Taxonomy Run Amok. Trends Microbiol. Preprint. doi: 10.1016/j.tim.2020.12.010
Sant’Anna, F. H., Ambrosini, A., Souza, R., Fernandes, G. C., Bach, E., Balsanelli, E., et al. (2017). Reclassification of Paenibacillus riograndensis as a genomovar of Paenibacillus sonchi: Genome-based metrics improve bacterial taxonomic classification. Front. Microbiol. 8:1849. doi: 10.3389/fmicb.2017.01849
Sant’Anna, F. H., Bach, E., Porto, R. Z., Guella, F., Sant’Anna, E. H., and Passaglia, L. M. (2019). Genomic metrics made easy: what to do and where to go in the new era of bacterial taxonomy. Crit. Rev. Microbiol. 45, 182–200. doi: 10.1080/1040841X.2019.1569587
Scholla, M. H., and Elkan, G. H. (1984). Rhizobium fredii sp. nov., a fast-growing species that effectively nodulates soybeans. Int. J. Syst. Evol. Microbiol. 34, 484–486. doi: 10.1099/00207713-34-4-484
Somasegaran, P., and Hoben, H. J. (2012). “Appendix 3,” in The Handbook for Rhizobia: Methods in Legume–Rhizobium Technology, eds P. Somasegaran and H. J. Hoben (New York, NY: Springer), 382–383. doi: 10.1007/978-1-4613-8375-8
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today 33, 152–155.
Tian, C. F., Wang, E. T., Wu, L. J., Han, T. X., Chen, W. F., Gu, C. T., et al. (2008). Rhizobium fabae sp. nov., a bacterium that nodulates Vicia faba. Int. J. Syst. Evol. Microbiol. 58, 2871–2875. doi: 10.1099/ijs.0.2008/000703-0
Tiedje, J. M., Mason, B. B., Warren, C. B., and Malec, E. J. (1973). Metabolism of nitrilotriacetate by cells of Pseudomonas species. Appl. Microbiol. 25, 811–818.
Tindall, B. J., Rosselló-Móra, R., Busse, H. J., Ludwig, W., and Kämpfer, P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60, 249–266. doi: 10.1099/ijs.0.016949-0
Vallenet, D., Engelen, S., Mornico, D., Cruveiller, S., Fleury, L., Lajus, A., et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database 2009:ba021. doi: 10.1093/database/bap021
Van Berkum, P., Beyene, D., Bao, G., Campbell, T. A., and Eardly, B. D. (1998). Rhizobium mongolense sp. nov. is one of three rhizobial genotypes identified which nodulate and form nitrogen-fixing symbioses with Medicago ruthenica [(L.) Ledebour]. Int. J. Syst. Evol. Microbiol. 48, 13–22. doi: 10.1099/00207713-48-1-13
Van Ginkel, C. G., and De Bont, J. A. M. (1986). Isolation and characterization of alkene-utilizing Xanthobacter spp. Arch. Microbiol. 145, 403–407. doi: 10.1007/BF00470879
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. doi: 10.1093/nar/gkv657
Vital, M., Chai, B., Østman, B., Cole, J., Konstantinidis, K. T., and Tiedje, J. M. (2015). Gene expression analysis of E. coli strains provides insights into the role of gene regulation in diversification. ISME J. 9, 1130–1140. doi: 10.1038/ismej.2014.204
Volpiano, C. G., Sant’Anna, F. H., Ambrosini, A., Lisboa, B. B., Vargas, L. K., and Passaglia, L. M. P. (2019). Reclassification of Ochrobactrum lupini as a later heterotypic synonym of Ochrobactrum anthropi based on whole-genome sequence analysis. Int. J. Syst. Evol. Microbiol. 69, 2312–2314. doi: 10.1099/ijsem.0.003465
Wang, E. T., Van Berkum, P., Beyene, D., Sui, X. H., Dorado, O., Chen, W. X., et al. (1998). Rhizobium huautlense sp. nov., a symbiont of Sesbania herbacea that has a close phylogenetic relationship with Rhizobium galegae. Int. J. Syst. Evol. Microbiol. 48, 687–699. doi: 10.1099/00207713-48-3-687
Wayne, L. G., Moore, W. E. C., Stackebrandt, E., Kandler, O., Colwell, R. R., Grimont, P. A. D., et al. (1987). Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Evol. Microbiol. 37, 463–464. doi: 10.1099/00207713-37-4-463
Wei, G. H., Tan, Z. Y., Zhu, M. E., Wang, E. T., Han, S. Z., and Chen, W. X. (2003). Characterization of rhizobia isolated from legume species within the genera Astragalus and Lespedeza grown in the Loess Plateau of China and description of Rhizobium loessense sp. nov. Int. J. Syst. Evol. Microbiol. 53, 1575–1583. doi: 10.1099/ijs.0.02031-0
Welch, D. F., Carroll, K. C., Hofmeister, E. K., Persing, D. H., Robison, D. A., Steigerwalt, A. G., et al. (1999). Isolation of a new subspecies, Bartonella vinsonii subsp. arupensis, from a cattle rancher: Identity with isolates found in conjunction with Borrelia burgdorferi and Babesia microti among naturally infected mice. J. Clin. Microbiol. 37, 2598–2601.
Wiegel, J., Wilke, D., Baumgarten, J., Opitz, R., and Schlegel, H. G. (1978). Transfer of the nitrogen-fixing hydrogen bacterium Corynebacterium autotrophicum Baumgarten et al. to Xanthobacter gen. nov. Int. J. Syst. Evol. Microbiol. 28, 573–581. doi: 10.1099/00207713-28-4-573
Willems, A., Fernández-López, M., Muñoz-Adelantado, E., Goris, J., De Vos, P., Martínez-Romero, E., et al. (2003). Description of new Ensifer strains from nodules and proposal to transfer Ensifer adhaerens Casida 1982 to Sinorhizobium as Sinorhizobium adhaerens comb. nov. Request for an opinion. Int. J. Syst. Evol. Microbiol. 53, 1207–1217. doi: 10.1099/ijs.0.02264-0
Wilson, J. (1944). Over five hundred reasons for abandoning the cross inoculation groups of the legumes. Soil Sci. 58, 61–70.
Xi, J., Wang, Y., Yang, X., Tao, Y., Shao, Y., and Feng, F. (2017). Mongoliimonas terrestris gen. nov., sp. nov., isolated from desert soil. Int. J. Syst. Evol. Microbiol. 67, 3010–3014. doi: 10.1099/ijsem.0.002067
Yarza, P., Spröer, C., Swiderski, J., Mrotzek, N., Spring, S., Tindall, B. J., et al. (2013). Sequencing orphan species initiative (SOS): filling the gaps in the 16S rRNA gene sequence database for all species with validly published names. Syst. Appl. Microbiol. 36, 69–73. doi: 10.1016/j.syapm.2012.12.006
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Young, J. (2003). The genus name Ensifer Casida 1982 takes priority over Sinorhizobium Chen et al., 1988, and Sinorhizobium morelense Wang et al. 2002 is a later synonym of Ensifer adhaerens Casida 1982. Is the combination “Sinorhizobium adhaerens” (Casida 1982) Willems et al., 2003 legitimate? Request for an Opinion. Int. J. Syst. Evol. Microbiol. 53, 2107–2110. doi: 10.1099/ijs.0.02665-0
Zavarzin, G. A., and Nozhevnikova, A. N. (1977). Aerobic carboxydobacteria. Microb. Ecol. 3, 305–326. doi: 10.1007/BF02010738
Keywords: ANI, dDDH, species-cluster, genome clustering, Rhizobium
Citation: Volpiano CG, Sant’Anna FH, Ambrosini A, de São José JFB, Beneduzi A, Whitman WB, de Souza EM, Lisboa BB, Vargas LK and Passaglia LMP (2021) Genomic Metrics Applied to Rhizobiales (Hyphomicrobiales): Species Reclassification, Identification of Unauthentic Genomes and False Type Strains. Front. Microbiol. 12:614957. doi: 10.3389/fmicb.2021.614957
Received: 07 October 2020; Accepted: 04 March 2021;
Published: 25 March 2021.
Edited by:
Denis Grouzdev, Federal Center Research Fundamentals of Biotechnology (RAS), RussiaReviewed by:
José David Flores Félix, Universidade da Beira Interior, PortugalCopyright © 2021 Volpiano, Sant’Anna, Ambrosini, de São José, Beneduzi, Whitman, de Souza, Lisboa, Vargas and Passaglia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciane Maria Pereira Passaglia, bHVjaWFuZS5wYXNzYWdsaWFAdWZyZ3MuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.