 Florence Nicot1*
Florence Nicot1* Chloé Dimeglio1,2
Chloé Dimeglio1,2 Marion Migueres1,2
Marion Migueres1,2 Nicolas Jeanne1
Nicolas Jeanne1 Justine Latour1
Justine Latour1 Florence Abravanel1,2,3Noémie Ranger1Agnès Harter1Martine Dubois1Sonia Lameiras4
Florence Abravanel1,2,3Noémie Ranger1Agnès Harter1Martine Dubois1Sonia Lameiras4 Sylvain Baulande4Sabine Chapuy-Regaud1,2,3
Sylvain Baulande4Sabine Chapuy-Regaud1,2,3 Nassim Kamar2,3,5
Nassim Kamar2,3,5 Sébastien Lhomme1,2,3
Sébastien Lhomme1,2,3 Jacques Izopet1,2,3*
Jacques Izopet1,2,3*- 1CHU de Toulouse, Hôpital Purpan, Laboratoire de Virologie, Centre National de Référence du Virus de l’Hépatite E, Toulouse, France
- 2INSERM, U1043, Toulouse, France
- 3Department of Virology, Université Toulouse III Paul-Sabatier, Toulouse, France
- 4Institut Curie Genomics of Excellence Platform, Institut Curie Research Center, Paris, France
- 5CHU de Toulouse, Hôpital Rangueil, Service de Néphrologie, Dialyse et Transplantation d’Organe, Toulouse, France
Hepatitis E virus (HEV) genotype 3 is the most common genotype linked to HEV infections in Europe and America. Three major clades (HEV-3.1, HEV-3.2, and HEV-3.3) have been identified but the overlaps between intra-subtype and inter-subtype p-distances make subtype classification inconsistent. Reference sequences have been proposed to facilitate communication between researchers and new putative subtypes have been identified recently. We have used the full or near full-length HEV-3 genome sequences available in the Genbank database (April 2020; n = 503) and distance analyses of clades HEV-3.1 and HEV-3.2 to determine a p-distance cut-off (0.093 nt substitutions/site) in order to define subtypes. This could help to harmonize HEV-3 genotyping, facilitate molecular epidemiology studies and investigations of the biological and clinical differences between HEV-3 subtypes.
Introduction
The hepatitis E virus (HEV) is a significant human pathogen causing viral hepatitis worldwide. Most of the strains that infect humans belong to two species, Orthohepevirus A (8 genotypes; HEV 1–8) and Orthohepevirus C (Purdy et al., 2017; Smith and Simmonds, 2018; Sridhar et al., 2018; Primadharsini et al., 2019). The most prevalent genotype in industrialized countries at least in Europe and America is HEV genotype 3 (HEV-3). It is transmitted zoonotically by direct contact with infected animals, eating contaminated food, or via the environment. HEV-3 infection is frequently asymptomatic but it can result in severe acute hepatitis in patients with chronic liver disease and lead to chronic hepatitis and cirrhosis in immunocompromised patients (Kamar et al., 2017). Extra-hepatic manifestations have been also described in patients with acute and chronic hepatitis E (Kamar et al., 2017).
Hepatitis E virus genotype 3 variants have been assigned to one of several subtypes based on analysis of a limited number of complete genome sequences and subgenomic regions (Lu et al., 2006). Despite the increasing number of full-length or near full-length genomes deposited in the NCBI database, it is difficult to provide consistent criteria that identify viruses that are members of the same subtype due to overlaps between the intra-subtype and inter-subtype p-distances commonly used for classification (Smith et al., 2015, 2016). Nevertheless, HEV-3 viruses can be classified into three major clades based on phylogenetic grouping. Clade 3.1 includes HEV-3 subtypes a, b, c, h, i, and j; clade 3.2 includes HEV-3 subtypes e, f, and g, and clade 3.3 contains rabbit strains corresponding to the HEV-3ra subtype (Oliveira-Filho et al., 2013; Ijaz et al., 2014; Vina-Rodriguez et al., 2015). A standard reference set of genome sequences including 17 that are full-length or near full-length HEV-3 genomes was proposed in 2016 using a conservative pragmatic approach (Smith et al., 2016). Subsequently, new potential subtypes have been proposed: 3k (Miura et al., 2017), 3l (De Sabato et al., 2018), 3chi-new (Lhomme et al., 2019), and 3s (Wist et al., 2018; Sahli et al., 2019). The standard reference set of genome sequence was recently updated identifying 3k, 3l, and 3m (previously named 3chi-new) as new subtypes (Smith et al., 2020). Recently, an automated partition of phylogenetic trees constructed from 250 full-length HEV-3 genome sequences has been used to classify more than 99% of the complete genome sequences into subtypes (Nicot et al., 2018).
This study was done to determine a distance cut-off that can be used to assign HEV-3 sequences to a subtype using the new full-length HEV-3 sequences available in NCBI and the recently defined new subtypes.
Materials and Methods
Sequencing of HEV Complete Genome Sequence
Stored plasma samples from HEV-infected patients consecutively tested for HEV RNA between 2017 and 2019 in the laboratory of Virology at Toulouse University Hospital, National Reference Center for HEV, with viral load of HEV-3 >100,000 copies/mL were selected for PacBio single molecule real-time sequencing. HEV-RNA extraction and F1 and F2 amplifications were realized for 188 samples as previously described (Nicot et al., 2018). SMRT bell library was constructed by pooling 96 barcoded samples according the manufacturer instructions for SMRTbell Barcoded Adapter Prep kit. Sequencing was performed by using chemistry v3.0 on a PacBio Sequel sequencer available at ICGex, Institut Curie Research Center, Paris, France. Bioinformatics analysis and complete genome reconstruction were realized with an in-house developed pipeline. From demultiplexed.bam files provided by ICGex, CCS were constructed using min passes = 3 and min RQ 0.999 parameters. Reads were mapped to a reference sequence (Minimap 2 2.17) to retain HEV reads and remove chimeric reads. Non-identical reads were subsequently combined using a medoïd-based clustering (cluster-fast from USEARCH 11.0.667) into clusters at 99% genetic identity. A consensus sequence was generated for each cluster. F1 and F2 sequences were assembled with Megamerger (EMBOSS 6.6.0). The consensus sequence with the higher number of reads was used as complete genome sequence, annotated as previously described (Nicot et al., 2018) and submitted to Genbank with accession numbers MW355217–MW355404.
Nucleotide Sequences and Phylogenetic Analysis
The 188 complete genome sequences obtained by SMRT sequencing and all full or near full-length genome sequences of genotype 3 (n = 315) of human or animal origin available in the Genbank database on April 2020 were included (Supplementary Table 1). Duplicate sequences from a single individual and six recombinant sequences (D11092, MG783571, KJ013414, KJ013415, KT633715, and DQ450072) were removed. We also included 29 complete genome sequences of genotypes 1, 2, 4, 5, 6, 7, and 8 (Smith et al., 2020) (accession numbers HEV-1: FJ457024, MH918640; HEV-1a: M73218; HEV-1b: L088816; HEV-1c: X98292; HEV-1d: AY230202; HEV-1e: AY204877; HEV-1f: JF443721; HEV-1g: LC225387; HEV-2a: KX578717; HEV-2b: MH809516; HEV-4: MK410048, AB369688; HEV-4a: AB197673; HEV-4b: DQ279091; HEV-4c: AB074915; HEV-4d: AJ272108; HEV–4e: AY723745; HEV-4f: AB220974; HEV-4g: AB108537; HEV-4h: GU119961; HEV-4i: AB369690; HEV-5a: AB573435; HEV-6: AB856243; HEV-6a: AB602441; HEV-7: KJ496144; HEV-7a: KJ496143; and HEV-8: MH410174; HEV-8a: KX387865). The 532 sequences (503 HEV-3 and 29 HEV non-3 genotype) were aligned with MUSCLE v.3.8.31 and a bootstrapped tree (100 replicates) using the maximum likelihood (ML) method with the general time reversible model (GTR + I + G) was constructed on phyML v3.3. Interactive Tree Of Life (iTOL) v3 was used to visualize the whole large tree.
Automated Partition of Phylogenetic Tree
The ML phylogenetic tree was partitioned and strain clusters within genotype 3 were identified using a method adapted from Prosperi et al. (2011). Briefly, the topology of the ML tree was analyzed with a depth first search by considering the number of subtrees with a node reliability ≥70% and an associated number of leaves with at least two distinct patients. A subtree was identified as a cluster if the median value of the subtree distance distribution was below a t-percentile threshold of the whole-tree distance distribution. If a node satisfied this condition, the search was stopped at that node, children nodes were ignored, and other sibling nodes were analyzed. The threshold t was evaluated over the (5th, 50th) percentile range of the whole tree distance distribution with steps of 1 between the 5th and 15th and 5 between the 15th and 50th percentiles.
Distance Cut-Off for Identification of HEV Genotype 3 Subtypes
The clusters of sequences identified by automated partition of ML phylogenetic tree were used to analyze the intra- and inter-subtype nucleotide pairwise distances. The distances were estimated on MEGA X using the maximum composite likelihood (MCL) method and a gamma distribution to model evolutionary rate differences among sites (four categories). All distances were analyzed (to determine a cut-off that identified subtypes) by generating boxplots in Matlab R2018B software. Based on the algorithm described in Supplementary Figure 1, a sequence Xi can be considered to be a new subtype if all the intra-subtype and inter-subtype distances (Si,j)j ∈ [1,N] are above a cut-off α. If at least one distance d(Xi,XVj),j ∈ [1,N] between the new sequence Xi and one of the known sequences XVj is below the cut-off α, the sequence Xi is assigned to the subtype containing the sequence with the shorter distance.
Statistical Analysis
Continuous variables were tested with Student’s t-test on STATA 14.0 software. p-Values of <0.05 were considered to be significant.
Results
Identification of Clusters
We identified clusters among genotype 3 sequences using automated partition of a ML phylogenetic tree. Clusters of known subtypes were identified with a threshold of 13% (median distance <0.126 nt substitutions/site). This threshold assigns 99.1% of the 483 sequences belonging to clade 3.1 and 3.2 to one of the following known subtypes: 3aj (n = 26), 3b (n = 29), 3c (n = 117), 3h (n = 17), 3k (n = 4), 3i (n = 6), 3l (n = 6), 3m (n = 6) for clade 3.1 and 3e (n = 40), 3f (n = 228), 3g (n = 1) for clade 3.2 (Figure 1). The complete genome of HEV-3j was included in the HEV-3a subtype. The sequences proposed as HEV-3s were classified as HEV-3h subtypes. Only four sequences belonging to the group HEV-3chi (MK390370, MK390371, LC260517, and MF959765) were not classified (Figure 1). The 13% threshold also defined two clusters within clade 3.3 (HEV-3ra) composed of six (HEV-3ra1) and six sequences (HEV-3ra2) while seven sequences were outside the HEV-3ra1 and HEV-3ra2 clusters (Figure 1).
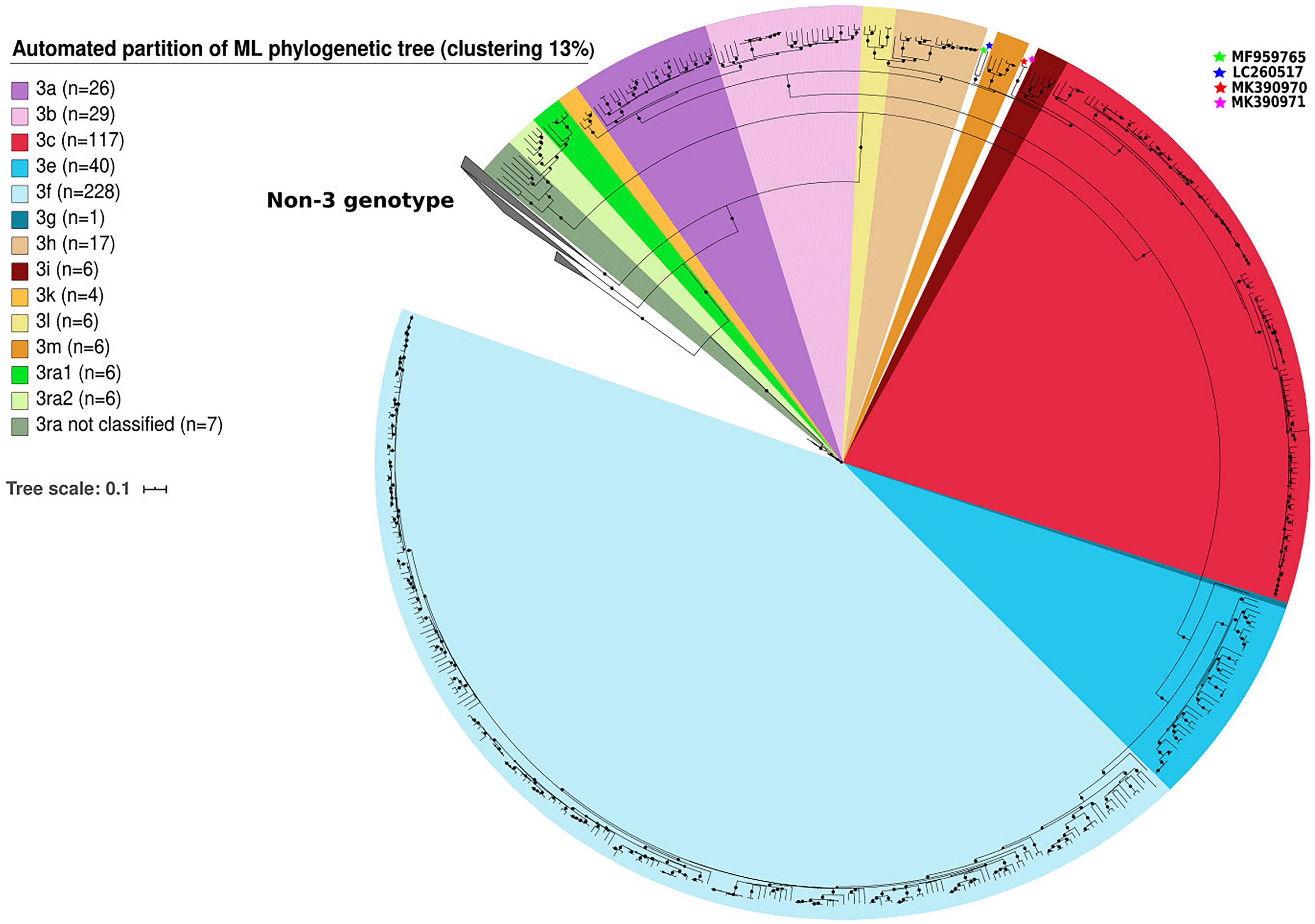
Figure 1. Automated partition of genotype 3 (n = 503) clusters within full-length or near full-length genomes. HEV-3 sequences not assigned to a cluster in clades HEV-3.1 are indicated with stars.
Determination of a Distance Cut-Off for Subtype Discrimination
The pairwise distances were estimated on MEGA X using the MCL method. Means for intra-subtype and inter-subtype distances, standard deviation, 95% confidence interval (CI) and 99% CI for each subtype are shown in Table 1. The mean intra-subtype distances obtained for HEV-3ra strains from clade 3.3 (0.116 ± 0.002 nt substitutions/site) was significantly greater than each of the mean intra-subtype distances obtained for the other HEV-3 subtypes (p < 0.01 for each subtype). Therefore, the HEV-3ra sequences are too heterogeneous to be used to determine the distance cut-off discriminating between subtypes. Analysis of sequences from clades 3.1 and 3.2 indicated that the overall mean intra-subtype distances was 0.064 (95% CI: 0.03–0.09) whereas the overall mean inter-subtype distances was 0.142 (95% CI: 0.106–0.181). The 95% CI upper limit of intra-subtype distance for each subtype was lower than the 95% CI lower limit of the inter-subtype distance (Table 1). Similarly, the 99% CI upper limit of intra-subtype distance was lower than the 99% CI lower limit of inter-subtype distance, except for subtypes 3aj and 3b. Analysis of the intra- and inter-subtype distances for each subtype indicated that 0.093 can be used as a distance cut-off for assigning a sequence to a subtype (Figure 2). We therefore designed an algorithm based on this cut-off distance that would assign sequences to a subtype (Supplementary Figure 1) and used it to assign 99.1% of the HEV-3 sequences to a defined subtype (3aj, 3b, 3c, 3e, 3f, 3h, 3i, 3k, 3l, 3m). Only four sequences were not assigned (MK390370, MK390371, LC260517, and MF959765), in agreement with the data from automated partition of the ML phylogenetic tree. The distance between sequences MK390370 and MK390371, 0.006 nt substitutions/site, assigned these sequences to the same cluster. LC260517 and MF959765 were isolated sequences (minimum intersubtype-distance: 0.099 nt substitutions/site for both sequences).
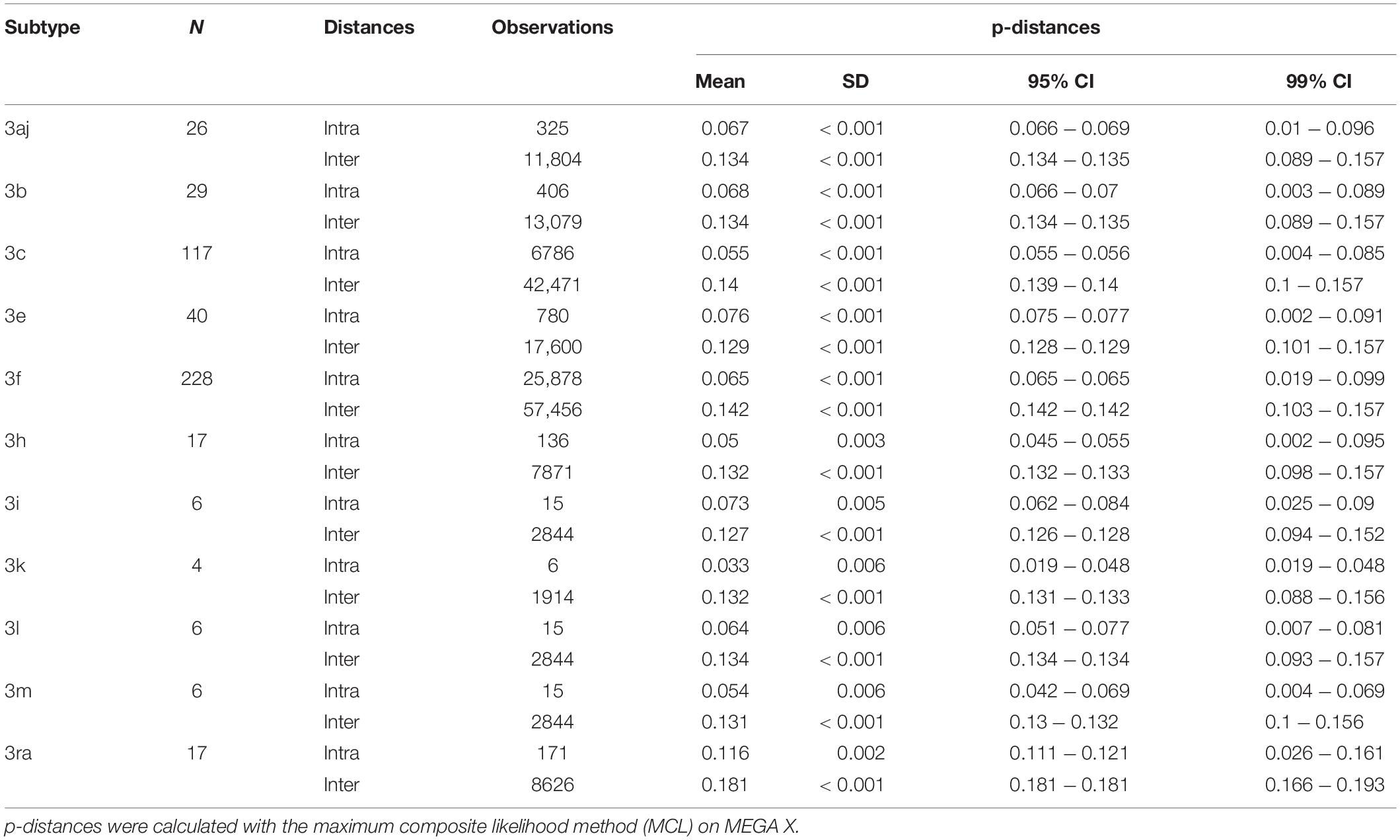
Table 1. Nucleotide p-distances calculated for each subtype (intra and inter-subtype distances).
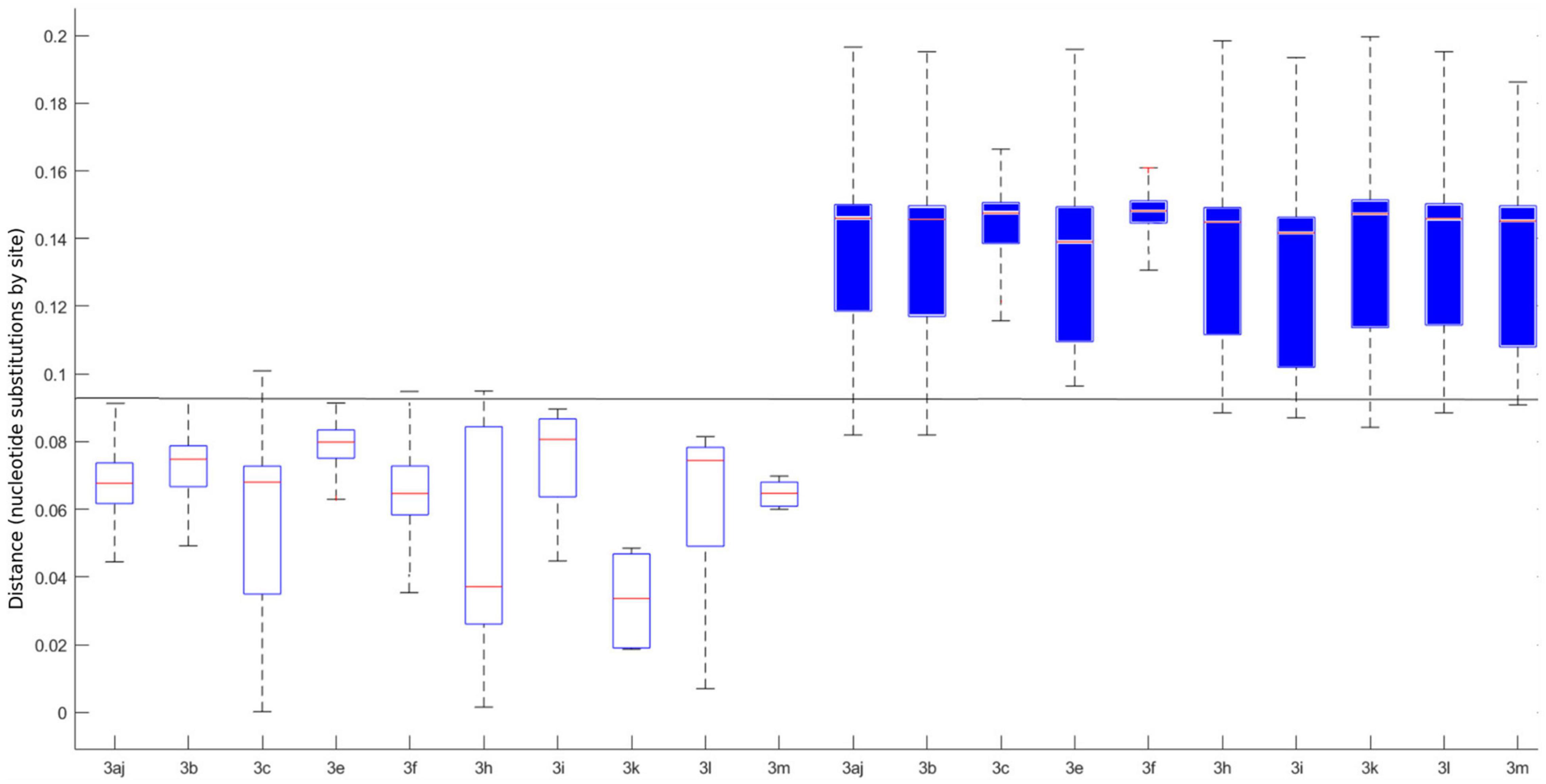
Figure 2. Individual intra (white boxplots) and inter (blue boxplots) p-distances for each determined subtype. The horizontal line represents the 0.093 cut-off. The central mark on each box indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points considered the minimum and the maximum of the distribution.
Classification of HEV-3 Sequences
We used 11 subtypes to classifying sequences in clades 3.1 (3aj, 3b, 3c, 3h, 3i, 3k, 3l, 3m) and 3.2 (3e, 3f, 3g), based on the criteria proposed by Smith et al. (2020) for subtype assignment and the results of the automated partition and distance cut-off methods (Table 2). Each complete genome sequence used in our study was assigned to a subtype (Supplementary Table 1). New complete genome sequences can be assigned to an existing subtype provided there is at least one distance less than 0.093. Otherwise, the new sequence may be a new subtype, which then needs to be confirmed (Smith et al., 2016). Complete genome sequences of subtype 3a, 3b, and 3ra were detected worldwide (Asia, Europe, and America), subtypes 3c, 3e, 3f, and 3h were detected in Asia and Europe, subtypes 3i, 3l, and 3m were detected only in Europe and subtype 3k only in Japan (Table 2 and Supplementary Table 1).
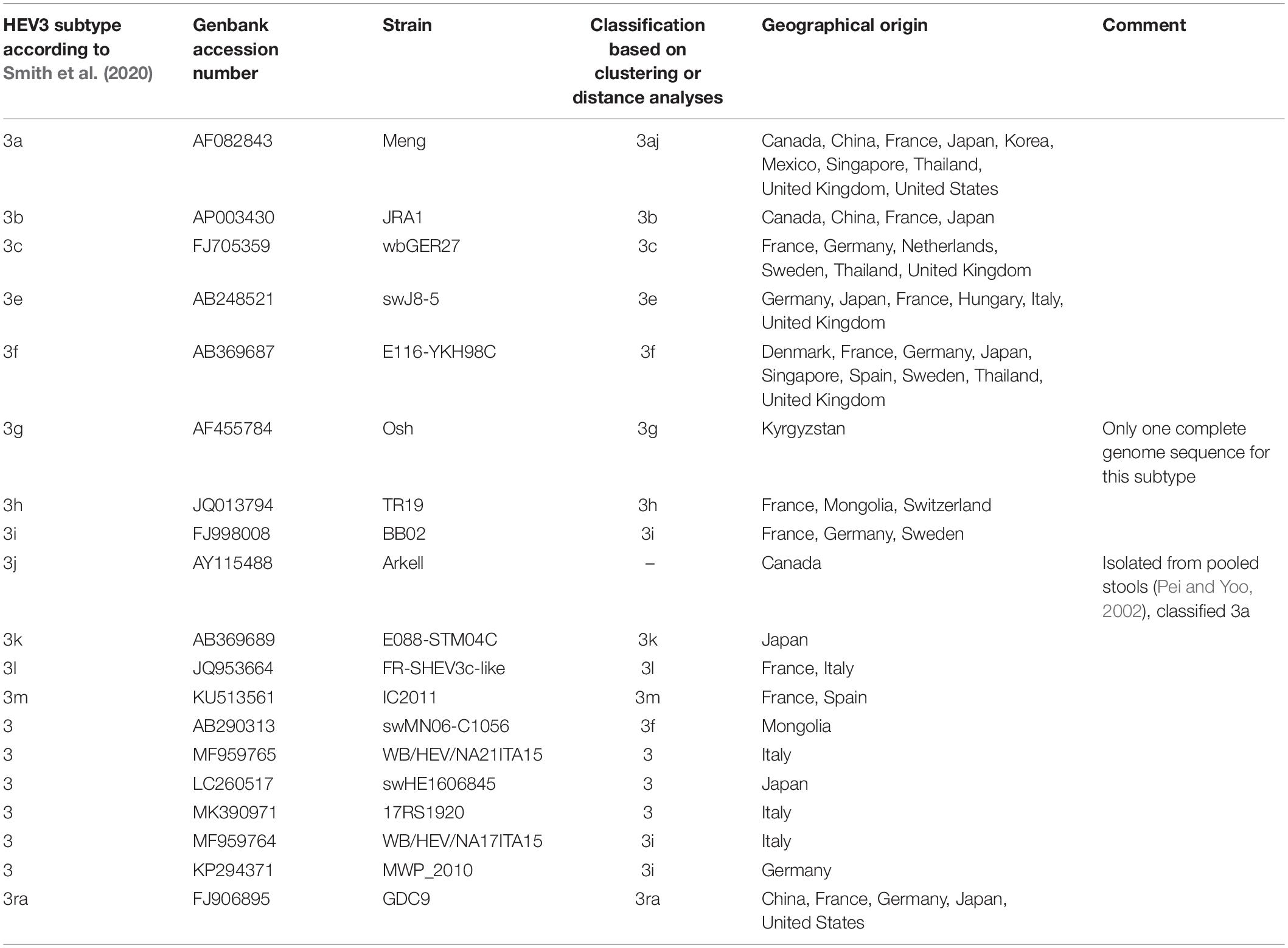
Table 2. HEV-3 subtype reference sequences based on full or near full-length genomes.
Discussion
Hepatitis E virus genotype 3 viruses display considerable diversity and have been classified into subtypes with no clear criteria based on distance and phylogenetic methods for demarcation. The set of reference sequences proposed by Smith et al. (2016) has enabled common subtypes to be assigned, but more than 10% of HEV-3 strains were not classified (Nicot et al., 2018). Automated partition of a ML phylogenetic tree using 503 HEV-3 sequences and distance analysis confirmed the classification of 250 HEV-3 sequences (Nicot et al., 2018) and supported the existence of several new post-2016 subtypes included in update classification (Smith et al., 2020). Subtype assignment using our new analysis method is automated and allow the classification of sequences not classified by Smith et al. (2020). It is important to classify a majority of sequences within a subtype and to have an objective method of classification.
The putative subtypes 3k (Miura et al., 2017), 3l (De Sabato et al., 2018), 3m (Nicot et al., 2018; Lhomme et al., 2019), and 3s (Wist et al., 2018; Sahli et al., 2019) have been described. Evidence for subtypes 3k, 3l, and 3m was provided by the automated partition of phylogenetic trees and the distance cut-off of 0.093. HEV-3k has been found in humans and pigs in Japan (Miura et al., 2017) while the HEV-3l subtype was first described in pigs in Northern Italy (De Sabato et al., 2018). Our analyses indicate that HEV-3l subtype also occurs in humans in France (sequences MF444121, MF444131, and HESQL113). The first strain of subtype 3m was detected in a Spanish patient in 2011 (Munoz-Chimeno et al., 2016) and has since been detected in France, Belgium, Netherlands and the United Kingdom (Ijaz et al., 2014; Nicot et al., 2018; Lhomme et al., 2019). A recent study showed that this subtype circulates in wild boar in Spain and also in human in Sweden, suggesting that it is transmitted via consumption of contaminated meat or water or direct contact with wild boar (Wang et al., 2019). In contrast, all the putative subtype 3s sequences were assigned to subtype 3h by the automated partition of ML phylogenetic tree, and distance analysis indicated that 3s sequences should be assigned to subtype 3h, all with distances below the cut-off of 0.093. In addition, these strains, which have been found in both humans and animals in Switzerland, form a cluster that is transmitted by the consumption of locally produced pork meat (Sahli et al., 2019). They cannot be assigned to a new subtype because they are epidemiologically related (Smith et al., 2016). Our analyses also indicate that HEV-3i, described in boar in Germany (Adlhoch et al., 2009) or human in Sweden (Norder et al., 2018), occurs in human in France (HESQL053 and HESQL059).
The sequence AY115488 classified 3j with Smith criteria (Smith et al., 2020) was obtained from the feces of pigs housed in Canada (Pei and Yoo, 2002). This sequence was classified among subtype 3a with our analysis. Indeed, the minimum intra-subtype distance observed between AY115488 and AB089824 (0.083 nucleotide subtitutions/site) in subtype 3aj is much lower than the cut-off value of 0.093 for assigning a sequence to a different subtype. The four sequences (MK390370, MK390371, LC260517, and MF95765) were not classified according Smith et al. (2020) criteria and our analysis. They could be consider as three potential new subtypes, considering MK390370 and MK390371 are assigned to the same cluster. However, in the absence of at least three complete genome sequences epidemiologically unrelated (Smith et al., 2016), these new subtypes could not be confirmed.
Hepatitis E virus genotype 3 is found worldwide and is the predominant genotype in Europe and America. The majority of Asian and North American strains of HEV-3 belong to subtypes 3a and 3b (Zehender et al., 2014). Subtype 3b is indigenous to Japan, although 3b strains have occasionally been identified in Europe (Legrand-Abravanel et al., 2009; Vina-Rodriguez et al., 2015). Subtypes 3k strains have been described only in Asia (Miura et al., 2017). The majority of European strains belong to subtypes 3c, 3f, and 3e. Changes in the distribution of variants within genotypes have been highlighted. A switch from clade 3.2 (mainly 3f and 3e) to clade 3.1 (mainly 3c) infections was observed in France and the United Kingdom after 2010 (Nicot et al., 2018; Oeser et al., 2019) and a similar switch occurred more recently in Belgium; the subtype 3f strains that were predominant before 2015 were replaced by subtype 3c strains after 2015 (Suin et al., 2019). The reason for these changes in subtype distribution is uncertain, it could reflect the distributions of HEV-3 subtypes in the pig reservoirs of different countries. Both locally produced and imported pigs or pork meat could be involved. Phylogenetic and coalescence analyses based on full-length sequences of HEV-3 from acute hepatitis patients, domestic pigs and wild boars provide evidence that HEV-3e strains were introduced from Europe into Japan through importation of pigs in the 1960s (Nakano et al., 2013). Transmission of HEV-3e strains from pigs to wild boars has been also suggested in Japan (Nakano et al., 2013). HEV-3f subtypes were recently detected in humans, domestic pigs and wild mammals in Japan, but indigenous Japanese HEV-3 strains belong to subtypes 3a, 3b, and 3e (Nakano et al., 2018). These new HEV-3f strains may have entered Japan from Europe in this way because the proportion of pork meat imported from Europe has increased in the past decade, leading to cases of hepatitis due to eating pork meat. The changes in HEV-3 subtype distribution are probably the result of changes in the origin of pork meat.
The clinical significance of infection with different HEV-3 subtypes has been discussed. Most studies have shown that asymptomatic blood donors and patients with symptomatic hepatitis E had similar genotype distributions and neither the severity of symptoms nor liver enzyme activities were significantly associated with clades 3.1 or 3.2 (Smith et al., 2015; Lhomme et al., 2019). However, two recent studies from Belgium and France found that the risk of HEV-3-infected patients being hospitalized varied with the subtype (Subissi et al., 2019; Abravanel et al., 2020). Patients infected with subtype 3c were at lower risk of hospitalization than those infected with subtypes 3f or 3e (Subissi et al., 2019; Abravanel et al., 2020). Larger studies are now needed to clarify the influence of host factors and virus diversity on HEV-3 pathogenesis.
A limitation of the present study is the relatively limited number of full length HEV-3 genome sequences available worldwide and the way diversity varies within HEV-3 subtypes. Despite very good groupings of sequences, there are outliers for most subtypes indicating that subtype assignment can be ambiguous. In addition, HEV-3ra strains are particularly heterogeneous. Nevertheless, all HEV-3ra sequences have a common signature, a 93-nucleotide insertion within the macrodomain of the HEV genome (Izopet et al., 2012).
Our findings suggest that the strains in clades 3.1 and 3.2 can be assigned to 1 of 11 subtypes, each represented by a full length or near full-length reference sequence. We have proposed a cut-off value for assigning subtypes. Update of the reference sequences (Smith et al., 2020) could help harmonize HEV-3 classification, which would be useful for comparing strains circulating in humans and the animal reservoir, for tracing the source of an individual infection and for investigating the pathogenicity of HEV-3 subtypes.
Data Availability Statement
The sequences have been submitted in Genbank database. The accession numbers corresponding the sequences data are MW355217–MW355404.
Author Contributions
FN and JI designed the project. FN, CD, and JI analyzed the results and wrote the manuscript. NJ, JL, and FN performed the bio-informatics analyses. CD performed the statistics analysis. NK provided the plasma samples. FA, SLh, SC-R, NR, AH, and MD carried out the experiments. SLa and SB performed Sequel sequencing. All authors contributed to the article and approved the submitted version.
Funding
High-throughput sequencing has been performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d’Avenir” program), by the Canceropole Ile-de-France, and by SiRIC-Curie program-SiRIC Grant “INCa-DGOS-4654.”
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The English text was checked by Owen Parkes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.634430/full#supplementary-material
References
Abravanel, F., Dimeglio, C., Castanier, M., Peron, J. M., Kamar, N., Lhomme, S., et al. (2020). Does HEV-3 subtype play a role in the severity of acute hepatitis E? Liver Int. 40, 333–337. doi: 10.1111/liv.14329
Adlhoch, C., Wolf, A., Meisel, H., Kaiser, M., Ellerbrok, H., and Pauli, G. (2009). High HEV presence in four different wild boar populations in east and west germany. Vet. Microbiol. 139, 270–278. doi: 10.1016/j.vetmic.2009.06.032
De Sabato, L., Lemey, P., Vrancken, B., Bonfanti, L., Ceglie, L., Vaccari, G., et al. (2018). Proposal for a new subtype of the zoonotic genotype 3 Hepatitis E virus: HEV-3l. Virus Res. 248, 1–4. doi: 10.1016/j.virusres.2018.02.010
Ijaz, S., Said, B., Boxall, E., Smit, E., Morgan, D., and Tedder, R. S. (2014). Indigenous hepatitis E in England and wales from 2003 to 2012: evidence of an emerging novel phylotype of viruses. J. Infect Dis. 209, 1212–1218. doi: 10.1093/infdis/jit652
Izopet, J., Dubois, M., Bertagnoli, S., Lhomme, S., Marchandeau, S., Boucher, S., et al. (2012). Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 18, 1274–1281. doi: 10.3201/eid1808.120057
Kamar, N., Izopet, J., Pavio, N., Aggarwal, R., Labrique, A., Wedemeyer, H., et al. (2017). Hepatitis E virus infection. Nat. Rev. Dis. Primers 3:17086. doi: 10.1038/nrdp.2017.86
Legrand-Abravanel, F., Mansuy, J. M., Dubois, M., Kamar, N., Peron, J. M., Rostaing, L., et al. (2009). Hepatitis E virus genotype 3 diversity, France. Emerg. Infect. Dis. 15, 110–114. doi: 10.3201/eid1501.080296
Lhomme, S., Gallian, P., Dimeglio, C., Assal, A., Abravanel, F., Tiberghein, P., et al. (2019). Viral load and clinical manifestations of hepatitis E virus genotype 3 infections. J Viral Hepat 26, 1139–1142. doi: 10.1111/jvh.13128
Lu, L., Li, C., and Hagedorn, C. H. (2006). Phylogenetic analysis of global hepatitis E virus sequences: genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 16, 5–36. doi: 10.1002/rmv.482
Miura, M., Inoue, J., Tsuruoka, M., Nishizawa, T., Nagashima, S., Takahashi, M., et al. (2017). Full-length genomic sequence analysis of new subtype 3k hepatitis E virus isolates with 99.97% nucleotide identity obtained from two consecutive acute hepatitis patients in a city in northeast Japan. J. Med. Virol. 89, 1116–1120. doi: 10.1002/jmv.24743
Munoz-Chimeno, M., Forero, J. E., Echevarria, J. M., Munoz-Bellido, J. L., Vazquez-Lopez, L., Morago, L., et al. (2016). Full coding hepatitis E virus genotype 3 genome amplification method. J. Virol. Methods 230, 18–23. doi: 10.1016/j.jviromet.2016.01.004
Nakano, T., Takahashi, K., Arai, M., Okano, H., Kato, H., Ayada, M., et al. (2013). Identification of European-type hepatitis E virus subtype 3e isolates in Japanese wild boars: molecular tracing of HEV from swine to wild boars. Infect. Genet. Evol. 18, 287–298. doi: 10.1016/j.meegid.2013.06.004
Nakano, T., Takahashi, M., Takahashi, K., Nagashima, S., Suzuki, Y., Nishigaki, Y., et al. (2018). Hepatitis E virus subtype 3f strains isolated from Japanese hepatitis patients with no history of travel to endemic areas - the origin analyzed by molecular evolution. Virology 513, 146–152. doi: 10.1016/j.virol.2017.08.008
Nicot, F., Jeanne, N., Roulet, A., Lefebvre, C., Carcenac, R., Manno, M., et al. (2018). Diversity of hepatitis E virus genotype 3. Rev. Med. Virol. 28:e1987. doi: 10.1002/rmv.1987
Norder, H., Galli, C., Magnil, E., Sikora, P., Ekvarn, E., Nystrom, K., et al. (2018). Hepatitis E virus genotype 3 genomes from RNA-positive but serologically negative plasma donors have CUG as the start Codon for ORF3. Intervirology 61, 96–103. doi: 10.1159/000491926
Oeser, C., Vaughan, A., Said, B., Ijaz, S., Tedder, R., Haywood, B., et al. (2019). Epidemiology of hepatitis E in England and wales: a 10-year retrospective surveillance study, 2008-2017. J. Infect. Dis. 220, 802–810. doi: 10.1093/infdis/jiz207
Oliveira-Filho, E. F., Konig, M., and Thiel, H. J. (2013). Genetic variability of HEV isolates: inconsistencies of current classification. Vet. Microbiol. 165, 148–154. doi: 10.1016/j.vetmic.2013.01.026
Pei, Y., and Yoo, D. (2002). Genetic characterization and sequence heterogeneity of a canadian isolate of swine hepatitis E virus. J. Clin. Microbiol. 40, 4021–4029. doi: 10.1128/jcm.40.11.4021-4029.2002
Primadharsini, P. P., Nagashima, S., and Okamoto, H. (2019). Genetic variability and evolution of hepatitis E virus. Viruses 11:27. doi: 10.3390/v11050456
Prosperi, M. C., Ciccozzi, M., Fanti, I., Saladini, F., Pecorari, M., Borghi, V., et al. (2011). A novel methodology for large-scale phylogeny partition. Nat. Commun. 2:321. doi: 10.1038/ncomms1325
Purdy, M. A., Harrison, T. J., Jameel, S., Meng, X. J., Okamoto, H., Van der Poel, W. H. M., et al. (2017). ICTV virus taxonomy profile: hepeviridae. J. Gen. Virol. 98, 2645–2646. doi: 10.1099/jgv.0.000940
Sahli, R., Fraga, M., Semela, D., Moradpour, D., and Gouttenoire, J. (2019). Rabbit HEV in immunosuppressed patients with hepatitis E acquired in Switzerland. J. Hepatol. 70, 1023–1025. doi: 10.1016/j.jhep.2019.01.025
Smith, D. B., Ijaz, S., Tedder, R. S., Hogema, B., Zaaijer, H. L., Izopet, J., et al. (2015). Variability and pathogenicity of hepatitis E virus genotype 3 variants. J. Gen. Virol. 96, 3255–3264. doi: 10.1099/jgv.0.000264
Smith, D. B., Izopet, J., Nicot, F., Simmonds, P., Jameel, S., Meng, X. J., et al. (2020). Update: proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 101, 692–698. doi: 10.1099/jgv.0.001435
Smith, D. B., and Simmonds, P. (2018). Classification and genomic diversity of enterically transmitted hepatitis viruses. Cold Spring Harb. Perspect. Med. 8:a031880. doi: 10.1101/cshperspect.a031880
Smith, D. B., Simmonds, P., Izopet, J., Oliveira-Filho, E. F., Ulrich, R. G., Johne, R., et al. (2016). Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 97, 537–542. doi: 10.1099/jgv.0.000393
Sridhar, S., Yip, C. C. Y., Wu, S., Cai, J., Zhang, A. J., Leung, K. H., et al. (2018). Rat hepatitis E virus as cause of persistent hepatitis after liver transplant. Emerg. Infect. Dis. 24, 2241–2250. doi: 10.3201/eid2412.180937
Subissi, L., Peeters, M., Lamoral, S., Klamer, S., Suin, V., and Van Gucht, S. (2019). Subtype-specific differences in the risk of hospitalisation among patients infected with hepatitis E virus genotype 3 in Belgium, 2010-2018. Epidemiol. Infect. 147:e224. doi: 10.1017/S0950268819001122
Suin, V., Klamer, S. E., Hutse, V., Wautier, M., Jacques, M., Abady, M., et al. (2019). Epidemiology and genotype 3 subtype dynamics of hepatitis E virus in belgium, 2010 to 2017. Euro. Surveill. 24:1800141. doi: 10.2807/1560-7917.ES.2019.24.10.1800141
Vina-Rodriguez, A., Schlosser, J., Becher, D., Kaden, V., Groschup, M. H., and Eiden, M. (2015). Hepatitis E virus genotype 3 diversity: phylogenetic analysis and presence of subtype 3b in wild boar in Europe. Viruses 7, 2704–2726. doi: 10.3390/v7052704
Wang, H., Castillo-Contreras, R., Saguti, F., Lopez-Olvera, J. R., Karlsson, M., Mentaberre, G., et al. (2019). Genetically similar hepatitis E virus strains infect both humans and wild boars in the Barcelona area, Spain, and Sweden. Transbound. Emerg. Dis. 66, 978–985. doi: 10.1111/tbed.13115
Wist, V., Kubacki, J., Lechmann, J., Steck, M., Fraefel, C., Stephan, R., et al. (2018). Complete genome sequence of a swiss hepatitis E virus isolate from the liver of a fattening pig. Genome Announc. 6:e113-18. doi: 10.1128/genomeA.00113-18
Keywords: HEV-3, subtype, classification, diversity, full-length genome, phylogeny
Citation: Nicot F, Dimeglio C, Migueres M, Jeanne N, Latour J, Abravanel F, Ranger N, Harter A, Dubois M, Lameiras S, Baulande S, Chapuy-Regaud S, Kamar N, Lhomme S and Izopet J (2021) Classification of the Zoonotic Hepatitis E Virus Genotype 3 Into Distinct Subgenotypes. Front. Microbiol. 11:634430. doi: 10.3389/fmicb.2020.634430
Received: 27 November 2020; Accepted: 30 December 2020;
Published: 28 January 2021.
Edited by:
Lilly Yuen, Victorian Infectious Diseases Reference Laboratory, AustraliaReviewed by:
Antonio Rivero-Juarez, Maimonides Biomedical Research Institute of Cordoba (IMIBIC), SpainAnna Rosa Garbuglia, Istituto Nazionale per le Malattie Infettive Lazzaro Spallanzani (IRCCS), Italy
Copyright © 2021 Nicot, Dimeglio, Migueres, Jeanne, Latour, Abravanel, Ranger, Harter, Dubois, Lameiras, Baulande, Chapuy-Regaud, Kamar, Lhomme and Izopet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florence Nicot, bmljb3QuZkBjaHUtdG91bG91c2UuZnI=; Jacques Izopet, aXpvcGV0LmpAY2h1LXRvdWxvdXNlLmZy