 Tracy L. Nicholson1*
Tracy L. Nicholson1* Ursula Waack1,2
Ursula Waack1,2 Tavis K. Anderson1
Tavis K. Anderson1 Darrell O. Bayles1
Darrell O. Bayles1 Sam R. Zaia3,4
Sam R. Zaia3,4 Isaiah Goertz3,4
Isaiah Goertz3,4 Mark Eppinger3,4
Mark Eppinger3,4 Samantha J. Hau1,2
Samantha J. Hau1,2 Susan L. Brockmeier1Sarah M. Shore1
Susan L. Brockmeier1Sarah M. Shore1- 1National Animal Disease Center, Agricultural Research Service, United States Department of Agriculture, Ames, IA, United States
- 2Oak Ridge Institute for Science and Education, United States Department of Energy, Oak Ridge, TN, United States
- 3South Texas Center for Emerging Infectious Diseases, The University of Texas at San Antonio, San Antonio, TX, United States
- 4Department of Biology, University of Texas at San Antonio, San Antonio, TX, United States
Streptococcus suis is a zoonotic bacterial swine pathogen causing substantial economic and health burdens to the pork industry. Mechanisms used by S. suis to colonize and cause disease remain unknown and vaccines and/or intervention strategies currently do not exist. Studies addressing virulence mechanisms used by S. suis have been complicated because different isolates can cause a spectrum of disease outcomes ranging from lethal systemic disease to asymptomatic carriage. The objectives of this study were to evaluate the virulence capacity of nine United States S. suis isolates following intranasal challenge in swine and then perform comparative genomic analyses to identify genomic attributes associated with swine-virulent phenotypes. No correlation was found between the capacity to cause disease in swine and the functional characteristics of genome size, serotype, sequence type (ST), or in vitro virulence-associated phenotypes. A search for orthologs found in highly virulent isolates and not found in non-virulent isolates revealed numerous predicted protein coding sequences specific to each category. While none of these predicted protein coding sequences have been previously characterized as potential virulence factors, this analysis does provide a reliable one-to-one assignment of specific genes of interest that could prove useful in future allelic replacement and/or functional genomic studies. Collectively, this report provides a framework for future allelic replacement and/or functional genomic studies investigating genetic characteristics underlying the spectrum of disease outcomes caused by S. suis isolates.
Introduction
Streptococcus suis is the leading bacterial pathogen affecting swine and contributes to significant economic losses to the swine industry worldwide (Feng et al., 2014; Segura et al., 2014a,b, 2017). S. suis causes a spectrum of clinical disease outcomes in pigs including pneumonia, endocarditis, septicemia, and meningitis. In addition, and perhaps more notably, S. suis is a zoonotic pathogen capable of causing diseases in humans, mainly arthritis, sepsis, meningitis, as well as streptococcal toxic shock-like syndrome (STSS) (Wertheim et al., 2009a,b; Feng et al., 2014; Segura et al., 2014a,b). Human infections have been either sporadic, and thought to be acquired from penetrating injuries associated with occupational exposure, or epidemic, and associated with consumption of raw or undercooked pork products, primarily in southeast Asia (Dutkiewicz et al., 2018).
Colonization and virulence mechanisms used by S. suis to cause disease in swine are not comprehensively characterized. Studies addressing specific virulence mechanisms used by S. suis have been confounded because different isolates cause a spectrum of disease outcomes ranging from lethal systemic disease to asymptomatic carriage (Segura et al., 2017). Factors generally regarded to contribute the most to virulence are the capsular polysaccharide (CPS), muramidase-released protein (mrp), extracellular protein factor (epf), and suilysin (sly) (Fittipaldi et al., 2012; Segura et al., 2014a,b, 2017). Individually, these factors have not been associated with the ability to cause invasive systemic disease and therefore virulence is thought to be multifactorial (Fittipaldi et al., 2012; Segura et al., 2017). Moreover, invasive clinical isolates obtained from people and pigs often do not harbor all of these factors (Fittipaldi et al., 2012; Feng et al., 2014; Segura et al., 2014a,b, 2017).
Mobile genetic elements (MGEs) are ubiquitous in bacteria and are the single most significant driver of gene transfer resulting in intra- and interspecies dissemination of antimicrobial resistance (AMR) and virulence determinants. The 89-kb pathogenicity island (89K PAI) carried by Chinese epidemic strains is an example of a S. suis MGE harboring factors contributing to a highly invasive phenotype (Li et al., 2011; Zhao et al., 2011). S. suis MGEs carrying AMR determinants for vancomycin, tetracycline, macrolide, aminoglycoside, and chloramphenicol resistance have been identified (Takamatsu et al., 2003; Palmieri et al., 2011a,b, 2012; Huang J. et al., 2016; Huang K. et al., 2016; Huang et al., 2018).
The majority of studies investigating S. suis isolate diversity rely on evaluating isolates by pulsed-field gel electrophoresis (PFGE), molecular serotyping, PCR typing of virulence markers, and/or determination of AMR profile (De Greeff et al., 2011; Zhang et al., 2011; Zheng et al., 2014; Prufer et al., 2019). While these methods consistently identify a small collection of virulence markers and AMR that S. suis isolates may harbor, they have yet to provide a complete evaluation of the genomic diversity, and what components of that diversity contributes to the virulence potential of S. suis isolates. A few recent studies have undertaken whole-genome sequencing (WGS) in combination with comparative genomic approaches to fully evaluate the genomic diversity and pathotype identification (Weinert et al., 2015; Estrada et al., 2019; Wileman et al., 2019). Although these studies relied on accurate clinical veterinary data, they did not directly test the virulence capacity of the S. suis isolates in pigs with similar health and immune status. Isolates from pigs with clinical signs and lesions consistent with S. suis disease are presumed to be virulent, while those obtained from the upper respiratory tract of healthy pigs are generally presumed to be avirulent. Unfortunately, this is an imprecise measure of virulence since conditions such as coinfection could enhance the apparent virulence of some isolates and existing immunity may mask the true virulence potential of others. The first objective of this study was to evaluate the virulence capacity of United States S. suis field isolates following intranasal challenge in swine. We choose to include eight isolates obtained from pigs exhibiting clinical disease consistent with S. suis disease and considered to be virulent, one S. suis strain isolated from the nasal cavity of an asymptomatic pig and considered to be non-virulent, and a reference isolate (P1/7) with known virulence. We hypothesized that the isolates considered to be virulent would exhibit different capacities to cause disease in swine if tested using a reference model free of confounding factors such as age, health status, differences in maternal antibody titers toward S. suis, and coinfection with other respiratory pathogens. After accurately establishing the virulence capacity of the isolates, our second objective was to obtain the closed-whole genome of each isolate and use comparative genomic analyses to evaluate the genetic and phenotypic attributes associated with swine-virulent phenotypes to link specific genes to S. suis disease outcomes.
Materials and Methods
S. suis Isolates and Culture Conditions
Nine S. suis isolates were evaluated in this study (SRD478, ISU2414, ISU2514, ISU2614, ISU2714, ISU2812, ISU2912, ISU1606, ISU2660) alongside a reference isolate (P1/7) with known virulence (Table 1). S. suis isolates were routinely grown at 37°C in Todd-Hewitt broth (Thermo Fisher Scientific Inc., Waltham, MA, United States) supplemented with 0.2% yeast extract (MilliporeSigma, St. Louis, MO, United States) (THY) and 5% filtered heat-inactivated horse serum (MilliporeSigma, St. Louis, MO, United States) (THY+) or on tryptic soy agar (TSA) containing 5% sheep blood (Becton, Dickinson and Co., Franklin Lakes, NJ, United States).
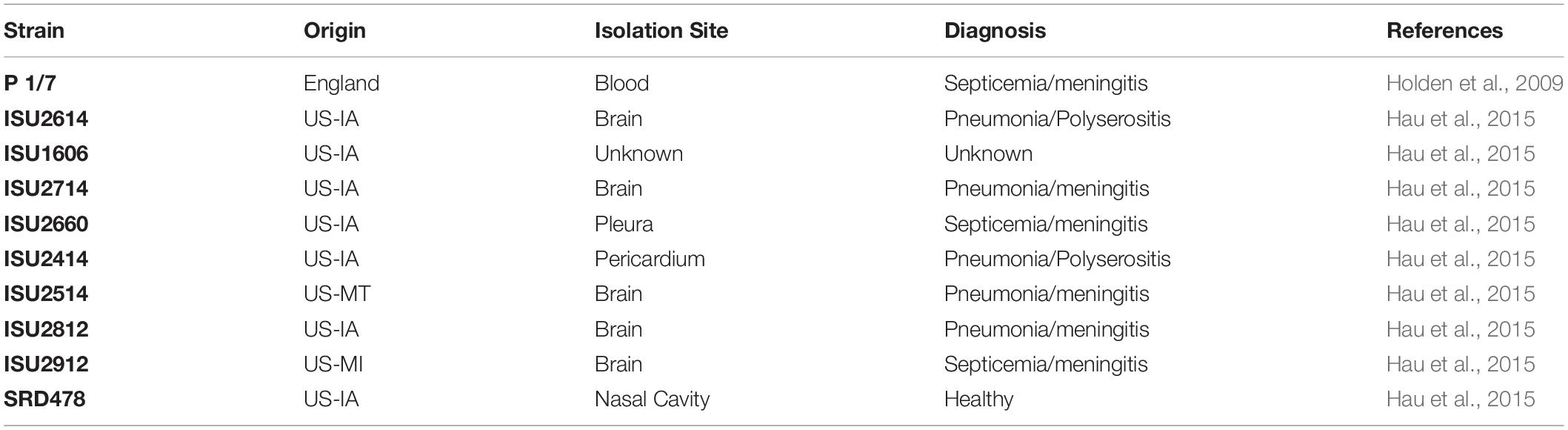
Table 1. Isolation site, clinical diagnosis, and reference for the nine United States Streptococcus suis isolates and reference isolate used in this study.
Swine Virulence Experiments
Four-week-old Cesarean-derived, colostrum-deprived (CDCD) pigs (Struve Labs International, Manning, IA, United States) were housed in agricultural biosafety level 2 (ABSL2) containment and cared for in compliance with the Institutional Animal Care and Use Committee (IACUC) of the National Animal Disease Center (NADC). Each experimental group, consisting of 4-5 pigs, was housed in a separate isolation room in ABSL2 biocontainment facilities and was challenged with a separate S. suis isolate (Table 1). Experimental group size consisting of 4-5 pigs was based on a power analysis using analysis of variance (ANOVA) analysis with a minimal significance and statistical power of 0.05 and 0.80 respectively. For inoculum, S. suis isolates were grown on TSA containing 5% sheep blood (Becton, Dickinson and Co., Franklin Lakes, NJ, United States) at 37°C overnight, scraped from the plates and resuspended in phosphate buffered saline (PBS) to contain approximately 1 × 109 colony forming units (CFU)/mL.
At approximately 8 weeks of age, nasal swabs were collected, placed in 1 mL phosphate buffered saline (PBS), and 100 μL of undiluted sample was immediately plated on TSA containing 5% sheep blood for determining the presence of S. suis and other common swine bacterial pathogens. Pigs were then intranasally challenged with 2 mL (1 mL per nostril) of approximately 1 × 109 CFU/mL of each S. suis isolate in PBS. Following challenge, pigs were examined twice daily (morning and afternoon), except for an 8-h overnight period, for clinical signs of disease including lameness, lethargy, and neurological symptoms. When clinical signs were noted, a third check at 9 pm was performed. Pigs were euthanized if severe clinical presentation was observed (dyspneic, paddling and/or did not rise upon human entry into the pen). Ten days post-challenge, pigs failing to exhibit any signs of clinical disease were then comingled along with two CDCD naïve pigs, to evaluate transmission potential. As with the primary challenge, pigs were evaluated for clinical signs of disease for the next 15 days at which time all pigs were euthanized.
At necropsy gross lesions were recorded and samples were collected for culture: tonsil swabs, swab of serosa (pericardium, thoracic cavity, and abdominal cavity), joint fluid from the hock joint (or other affected joint), cerebrospinal fluid (CSF), lung lavage, and serum. Samples were collected in 2 mL PBS, except lung lavage, where 50 mL of PBS was instilled into the lung just above the tracheal bifurcation and aspirated with a pipette. 100 μL of all of the samples were plated on TSA containing 5% sheep blood for S. suis culture, which was confirmed by colony morphology and species-specific PCR. To determine the S. suis strain isolated from necropsy samples collected from pigs post-comingling, genomic DNA was extracted from isolated colonies and used as the template in PCR amplicon sequencing targeting the aroA and mutS MLST alleles. DNA sequence analysis was performed on a 557-bp DNA fragment within the aroA gene amplified by PCR using the primers 5′-TTCCATGTGCTTGAGTCGCTA-3′ and 5′-ACGTGACCTACCTCCGTTGAC-3′ and on a 716-bp DNA fragment within the mutS gene amplified by PCR using the primers 5′-GGCACAACAGTATATCGACACACTTG-3′and 5′-CCTGACGATTCTCAATCCGCTTAA-3′.
Ethics Statement
Animal studies were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal experiments were approved by the USDA-National Animal Disease Center’s Institutional Animal Care and Use Committee (protocol #2724).
Whole-Genome Sequencing, Assembly, and Annotation
Genomic DNA from S. suis isolates (Table 1) was extracted using a MasterPure Gram-positive bacterial DNA purification kit (Lucigen Corporation, Middleton, WI, United States) with the following modifications to the manufacturer’s instructions to prevent degradation. 1 mL from overnight grown liquid cultures was pelleted (5,000 × g for 5 min), the supernatant was removed, and the pelleted cells were resuspended in 300 μL lysis buffer consisting of 2% SDS (Thermo Fisher Scientific Inc., Waltham, MA, United States), 0.25 M EDTA (Thermo Fisher Scientific Inc., Waltham, MA, United States), and 30% Proteinase K (Roche, Mannheim, Germany). The suspension was incubated for 3 h at 55°C. 300 uL of Gram Positive Lysis solution from the MasterPure Gram-positive bacterial DNA purification kit was then added and the suspension was incubated for 30 min at 70°C. Samples were then placed on ice for 5 min, 350 uL of MPC Protein Precipitation Reagent from the MasterPure Gram-positive bacterial DNA purification kit was added and the DNA precipitation instructions provided by the manufacturer’s protocol were followed.
Whole genome sequencing was performed using the Pacific Biosciences (PacBio) platform. Library preparation for PacBio sequencing was performed following the PacBio 10-kb insert library preparation protocol available online at http://www.pacb.com/wp-content/uploads/2015/09/Procedure-Checklist-10-kb-Template-Preparation-and-Sequencing.pdf. The 10 kb library for each strain was sequenced using the PacBio RSII platform with two SMRT cells for each isolate. Closed whole-genome assemblies for isolates ISU1606, ISU2660, ISU2414, ISU2515, ISU2614, ISU2714, ISU2812, and SRD478 were generated using the hybrid assembler Unicycler v. 0.4.4 (Wick et al., 2017) software along with both the PacBio sequencing reads and the Illumina MiSeq platform paired-end sequencing reads, which were previously used to obtain draft assemblies for the 9 United States isolates (Hau et al., 2015). Closed whole-genome assembly for isolate ISU2912 was generated using PacBio smrtanalysis v. 2.3.01, CANU v. 1.3 (Koren et al., 2017), and GARM v. 0.7.5 (Soto-Jimenez et al., 2014) along with PacBio sequencing reads and the Illumina MiSeq platform paired-end sequencing reads (Hau et al., 2015). Default parameters were used for all software. Final annotations were completed using NCBI’s Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016). Accession numbers and genome statistics are summarized in Table 4.

Table 4. S. suis genome sequences general summary.
Comparative Genomic Analysis
Closed whole-genomes were compared with Blast Ring Image Generator (BRIG) (Alikhan et al., 2011) and progressiveMauve (Darling et al., 2010). Prophage regions were determined using PHASTER (Arndt et al., 2016).
Genomic islands were predicted using IslandViewer 4 (Bertelli et al., 2017). Insertion elements were determined using ISEscan in Galaxy (Afgan et al., 2018). Multilocus sequence type (MLST) was determined in silico based on the PCR typing scheme developed by King et al. (2002). Serotype was determined in silico based on PCR typing schemes described by Liu et al. (2013) for the classical serotypes and by Qiu et al. (2016) for the Novel CPS Loci (NCL) serotypes. Blastn analysis of the CPS genes was employed to determine sequence similarity to four non-typeable S. suis strains available in GenBank (accession numbers KX870048, KX870054, KX870055, and KX870059). Pan-genome analysis was performed using PanACEA2 (Clarke et al., 2018). For protein alignments, gene sequences were identified by blastn, translated, and subsequently aligned using default settings in MAFFT (Katoh et al., 2002). Percent identity for virulence-associated genes was determined for each isolate relative to the P1/7 ortholog with the following exceptions: strain P5/11/88 was used as hylA reference, strain 05ZHY33 was used as revS reference, strain GZ0565 was used as stp reference, and strain ZY05719 was used as reference for vraR and vraS. These exceptions were based on choosing a reference gene sequence in which functional characterization had previously been reported (stp, vraR, and vraS) (Zhu et al., 2011; Zhong et al., 2018) or due to annotation of the P1/7 gene as a pseudogene (hylA and revS). Heatmap generation and hierarchical clustering analysis was performed using MeV 4.8.1 (Saeed et al., 2006).
Phenotypic and Genomic AMR Analysis
Phenotypic antibiotic resistance was determined using the broth microdilution method by Iowa State University Veterinary Diagnostic Laboratory following standard operating procedures. Minimum inhibitory concentrations (MICs) were determined for each isolate using the Trek BOPO6F plate (Thermo Fisher Scientific Inc., Oakwood Village, OH, United States) with Streptococcus pneumoniae ATCC 49619 and Mannheimia haemolytica ATCC 33369 (ATCC, Manassas, VA, United States) serving as the quality control strains. MICs were evaluated in accordance with Clinical Laboratory Standards Institute (CLSI) recommendations based on VET01-A4 and VET01S to give resistance interpretations (Supplementary Table 3; Clinical and Laboratory Standards Institute, 2013, 2015).
ResFinder 3.0 from the Center for Genomic Epidemiology3 and the Comprehensive Antibiotic Resistance Database (CARD)4 were employed for AMR genomic element identification. Genomes submitted to ResFinder 3.0 were evaluated for AMR determinants using standard default parameters of a threshold ID of 90% and a minimum length of 60%. To ensure that all AMR determinants had been identified, a second less stringent set of parameters of a threshold ID of 60% and a minimum length of 20% were used. The second and less stringent search did not result in identifying any additional AMR determinants. Genomes submitted to CARD were evaluated for AMR determinants using the criteria “default – perfect and strict hits only.”
Cell Surface Hydrophobicity
The relative surface hydrophobicity of S. suis isolates was determined by evaluating the change in absorbance following xylenes treatment, as previously described with modifications (Rosenberg et al., 1980). Briefly, S. suis isolates were suspended in PUM buffer (150 mM phosphate, potassium, urea, and magnesium, pH 7.1) to an absorbance at OD400 between 1.5 and 2.0. In a 10 mm round bottom test tube, 1.2 mL of bacterial suspension and 0.1 mL xylenes (Mallinckrodt Pharmaceuticals, Staines-upon-Thames, United Kingdom) were combined. The mixture was incubated at 37°C for 10 min. After incubation, the mixture was vortexed for 2 min and allowed to rest for 15 min at room temperature. Absorbance of the aqueous phase was determined at OD400 and used to calculate the change in OD400. Results are represented as a% OD400 retained after the assay. S. suis P1/7 Δcps2E, a capsule deletion mutant, was used as a control (Faulds-Pain and Wren, 2013). At least three independent experiments were performed.
Whole-Blood Sensitivity Assay
Overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL THY + and grown at 37°C to an OD600 of approximately 0.5 (range 0.4 – 0.6). Bacterial cells were pelleted by centrifugation, washed with PBS, and resuspended in PBS. 1 mL of bacterial cells was added with 9 mL whole pig blood (collected morning of assay from non-infected conventional pigs housed on-site, NADC, Ames, IA, United States) or 9 mL PBS in a sterile flask. Flasks were then incubated for 1 h at 37°C with gentle agitation. Serial dilutions in PBS were then plated on blood agar plates to determine CFU counts, which were then used to calculate the ratio of treated bacteria (whole pig blood) compared to control (PBS) and these data were reported as the percent viable bacteria. At least three independent experiments were performed.
Serum Sensitivity Assay
Overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL THY + and grown at 37°C to an OD600 of approximately 0.5 (range 0.4 – 0.6). Bacterial cells were pelleted by centrifugation, washed with PBS, and resuspended in 5 ml PBS. To each well of a 96-well flat bottom plate (Corning, Sigma-Aldrich, Darmstadt, Germany) the following was added: 10 μL bacterial cells and 90 μL guinea pig serum (Quidel Corp., San Diego, CA, United States), or 90 μL heat-inactivated guinea pig serum, or 90 μL PBS. Plates were then incubated for 1 h at 37°C with gentle agitation. Serial dilutions in PBS were then plated on blood agar plates to determine CFU counts, which were then used to calculate the ratio of bacteria in treatment wells (guinea pig serum) compared to control wells (PBS) and reported as the percent viable bacteria. A serum-sensitive Glasserella parasuis H465 isolate was used as a quality control strain. At least three independent experiments with three technical replicates in each experiment were performed.
Growth Kinetics
Kinetic growth of cultures was measured using a Bioscreen C Automated Microbiology Growth Curve Analysis System (Growth Curves USA, Piscataway, NJ, United States). Overnight cultures started from a single colony were diluted to an OD600 of 0.02 in 300 μL THY +. Plates were then incubated at 37°C for 24 h and the OD600 of each well was recorded every 15 min after 5 s of shaking. At least three independent experiments with three technical replicates were performed.
Microtiter Plate Assay for Static Biofilm Formation
A static biofilm assay was performed using the standard crystal violet method as previously reported (Nicholson et al., 2013). Briefly, overnight cultures started from a single colony were diluted to an OD600 of 0.1 in THY+. Cultures (100 μL) were added to each well of a flat-bottomed 96-well plate (Corning, Sigma-Aldrich, Darmstadt, Germany) and incubated statically for 24 hrs at 37°C. After incubation, the OD600 was measured in all wells to determine growth. The supernatant and any unadhered bacteria were aspirated from all cultures and then the wells were washed three times with 200 μL PBS. The wells were stained with 150 μL 0.1% crystal violet for 10 min. Crystal violet dye was then removed and wells were washed three times with 200 μL PBS. After the plate had dried, 150 μL of 100% ethanol was added to the wells and allowed to incubate for 15 min. To determine biofilm levels, 125 μL was then transferred to a new plate and the absorbance was measured at OD538. At least three independent experiments with three technical replicates in each experiment were performed.
Oxidative Stress Assay
Overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL THY + and grown at 37°C to an OD600 of approximately 0.5 (range 0.4 – 0.6). Cultures were then divided into two 2.5 mL cultures. Hydrogen peroxide was added to the treated cultures to 10 mM final concentration and an equal volume of water was added to the untreated cultures. Cultures were incubated at 37°C for 15 min in a shaking incubator (250 rpm). After incubation, catalase was added to 10 μg/mL final concentration. Serial dilutions in PBS + 10 μg/mL catalase were then plated on blood agar plates to determine CFU counts, which were then used to calculate the ratio of bacteria in treated cultures (10 mM H2O2) and untreated cultures (H2O), and the results were reported as the percent viable bacteria. At least three independent experiments with three technical replicates in each experiment were performed.
Hemolysis Assay
Overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL complex media containing pullulan (CM-P) (Ferrando et al., 2014). Bacteria were pelleted by centrifugation, washed with PBS, resuspended in 5 mL PBS, and pelleted again to collect the supernatant. DTT was added to a final concentration of 5 mM and two-fold dilutions were prepared in CM-P + 5 mM DTT. 100 μL of the supernatant dilution was added to 100 μL 2% RBCs and incubated for 2 h at 37°C. Unlysed RBCs were pelleted by centrifugation (1500 × g, 10 min) and 100 μL of the supernatant transferred to a flat-bottomed 96-well plate (Corning, Sigma-Aldrich, Darmstadt, Germany) plate and absorbance was read at 540 nm. Lysis by 1% Triton X-100 was used as a reference for 100% lysis. Hemolytic activity was calculated as the highest dilution that induced at least 50% hemolysis. One Hemolytic unit was defined as the reciprocal of the highest dilution that induced at least 50% lysis of RBCs. At least three independent experiments with three technical replicates in each experiment were performed.
Adherence to BEAS2B or J774.16 Cells
BEAS-2B (ATCC CRL-9609; human bronchial epithelial cell line) and J774A.1 (ATCC TIB-67; murine monocyte/macrophage cell line) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) broth supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids and 1% sodium pyruvate to 85% confluency or 2 × 105 cells/well at 37°C with 5% CO2. Overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL THY + and grown at 37°C to an OD600 of approximately 0.5 (range 0.4 – 0.6). Cultures were then pelleted by centrifugation and resuspended in DMEM at approximately 2 × 107 CFU/mL (MOI = 100) in DMEM. Adhesion assays were carried out in triplicate and performed by removing growth medium from BEAS-2B or J774A.1 cells and then adding either medium alone or 1 mL of bacterial inoculum. Plates were centrifuged at 800 × g for 10 min and incubated at 37°C with 5% CO2 for 2 h. Wells were washed four times with 1 mL of the growth medium to remove non-adherent bacteria. BEAS-2B or J774A.1 cells were trypsinized using 0.5 mL of 0.125% trypsin and incubated for 10 min at 37°C. The total volume of each well was brought up to 1 mL with DMEM and cells were homogenized by pipetting. Serial dilutions were then plated on blood agar plates to determine CFU counts, which were then used to calculate the proportion of adherent bacteria, expressed as a percentage of the original inoculum. At least three independent experiments with three technical replicates in each experiment were performed.
Nuclease Activity
Both cell-associated and secreted nuclease assays were performed using a previously reported method (Kiedrowski et al., 2011). Briefly, overnight cultures started from a single colony were diluted to an OD600 of 0.05 in 5 mL THY + and grown at 37°C to an OD600 of approximately 0.5 (range 0.4 – 0.6). Cultures were normalized to ensure an equivalent OD600 and pelleted by centrifugation to remove the supernatant. Bacterial cells were resuspended in fresh THY + and an equivalent volume of bacterial sample was incubated with 2 μM FRET substrate for 10 min. Nuclease activity was then quantified by measuring the difference in fluorescence (ex. 552 nm/em. 580 nm). Secreted nuclease activity assays were performed similarly except, after the bacterial cultures were pelleted by centrifugation, the supernatant was collected and used for measuring nuclease activity by incubation with 2 μM FRET substrate for 10 min, followed by measuring the difference in fluorescence (ex. 552 nm/em. 580 nm). At least three independent experiments with three technical replicates in each experiment were performed.
Statistical Analysis
For each assay, we applied a one-way ANOVA with Tukey’s multiple comparisons test to assess the differences between groups performed using GraphPad Prism software (GraphPad, La Jolla, CA, United States). A P-value less than 0.05 was considered significant.
Results and Discussion
Swine Virulence Assessment
The virulence capacity of nine United States S. suis field isolates, along with a highly virulent reference strain, P1/7, was assessed by evaluating morbidity and mortality following intranasal challenge of Cesarean-derived, colostrum-deprived (CDCD) pigs, a reference model free from many confounding factors such as health and immune status inherent in other model systems. S. suis isolates ISU2614 and ISU1606 exhibited high virulence, similar to strain P1/7, with all pigs in each of these groups developing systemic clinical disease within 8 days post-challenge (dpc) (Figure 1). Specifically, three of the pigs challenged with P1/7 exhibited clinical signs 1 day post-challenge. Two were euthanized 2 dpc, and the third was euthanized 4 dpc. One pig from this group began to exhibit clinical signs 2 dpc and was euthanized on the same day due to severe dyspnea. All pigs infected with P1/7 exhibited lethargy, while three pigs exhibited lameness on front and/or hind limbs, two pigs exhibited respiratory signs, and one pig exhibited neurological signs of disease (Table 2). At necropsy, S. suis was isolated from the CSF in all pigs infected with P1/7 (Table 3). S. suis was additionally isolated from a joint, or the serosa, or from serum for the majority, and from the lung for half of the pigs within this group (Table 3). One pig challenged with ISU2614 exhibited clinical signs 1 day post-challenge and was euthanized due to severe neurological signs (Figure 1). Three pigs infected with ISU2614 began to exhibit similar clinical signs 2 dpc and were euthanized on the same day due to either severe neurological signs or lameness due to swollen hocks (Figure 1). One pig challenged with ISU2614 exhibited clinical signs 3 dpc and was euthanized on day 4 post-challenge (Figure 1). The majority of pigs within this group exhibited lethargy and lameness on front and/or hind limbs, and neurological signs of disease, while 2 out of 5 pigs exhibited respiratory signs of disease (Table 2). At necropsy, S. suis was isolated from the serum and joint in all pigs infected with ISU2614, and from the serosa and CSF for the majority, and from the lung for 2 out of 5 pigs within this group (Table 3). Two of the pigs challenged with ISU1606 exhibited clinical signs 3 dpc and were euthanized, while the other pigs began to exhibit similar clinical signs 7 dpc and were euthanized on day 8 (Figure 1). All pigs within this group exhibited lethargy and lameness on front and/or hind limbs, two pigs exhibited neurological signs of disease, and one pig exhibited respiratory signs (Table 2). At necropsy, S. suis was isolated from the serum and serosa in all pigs infected with ISU1606, and from a joint for the majority, and from the lung and CSF for half of the pigs within this group (Table 3). Given that all pigs within these three groups rapidly developed clinical signs of systemic disease consistent with S. suis infection and were subsequently euthanized leaving no survivors, S. suis isolates ISU2614 and ISU1606 were categorized as highly virulent, along with strain P1/7.
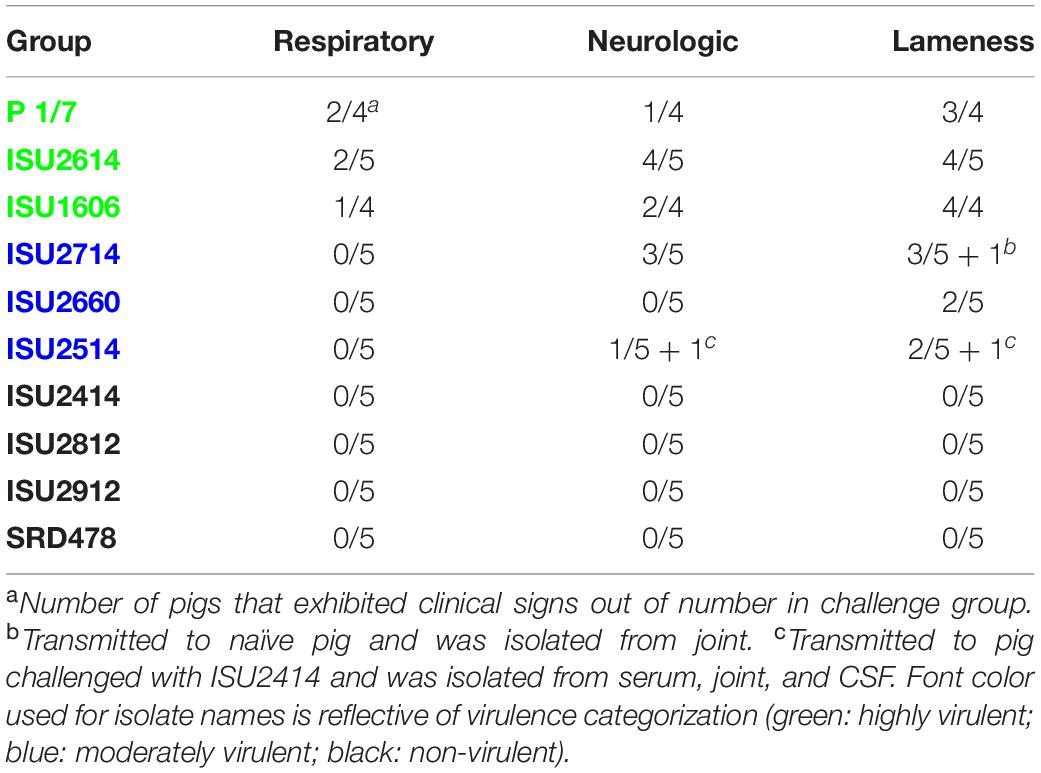
Table 2. Frequency of clinical signs in S. suis infected pigs.
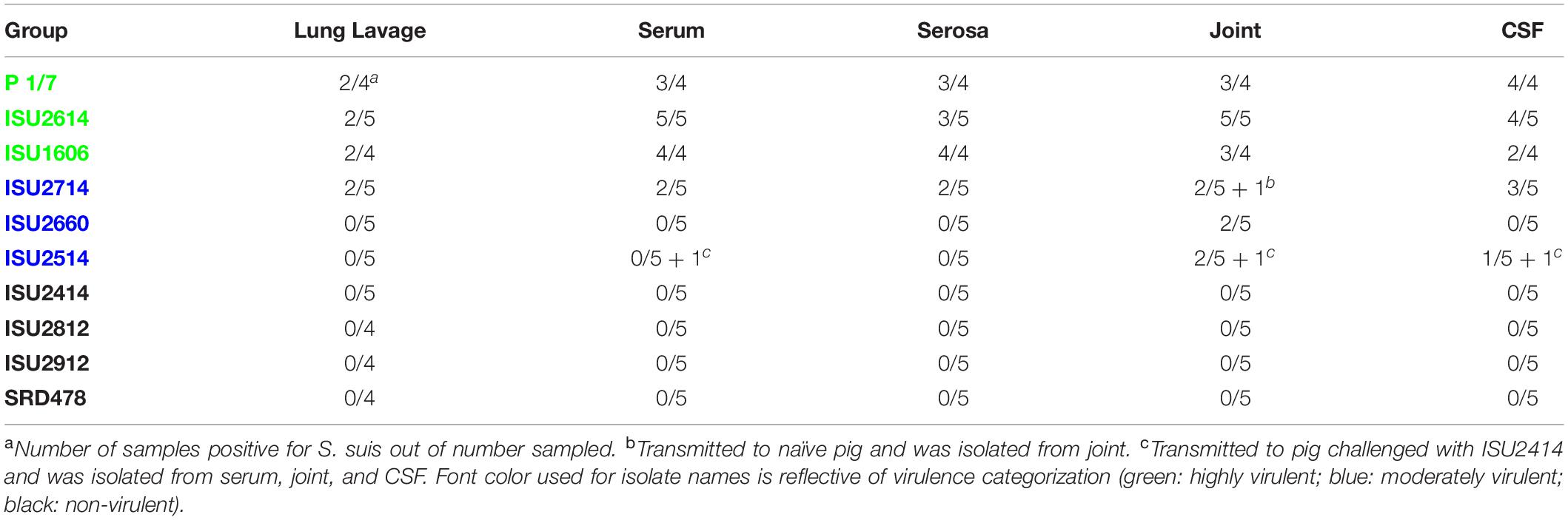
Table 3. Frequency of S. suis culture from various sites from pigs that died or were euthanized after demonstrating clinical signs.
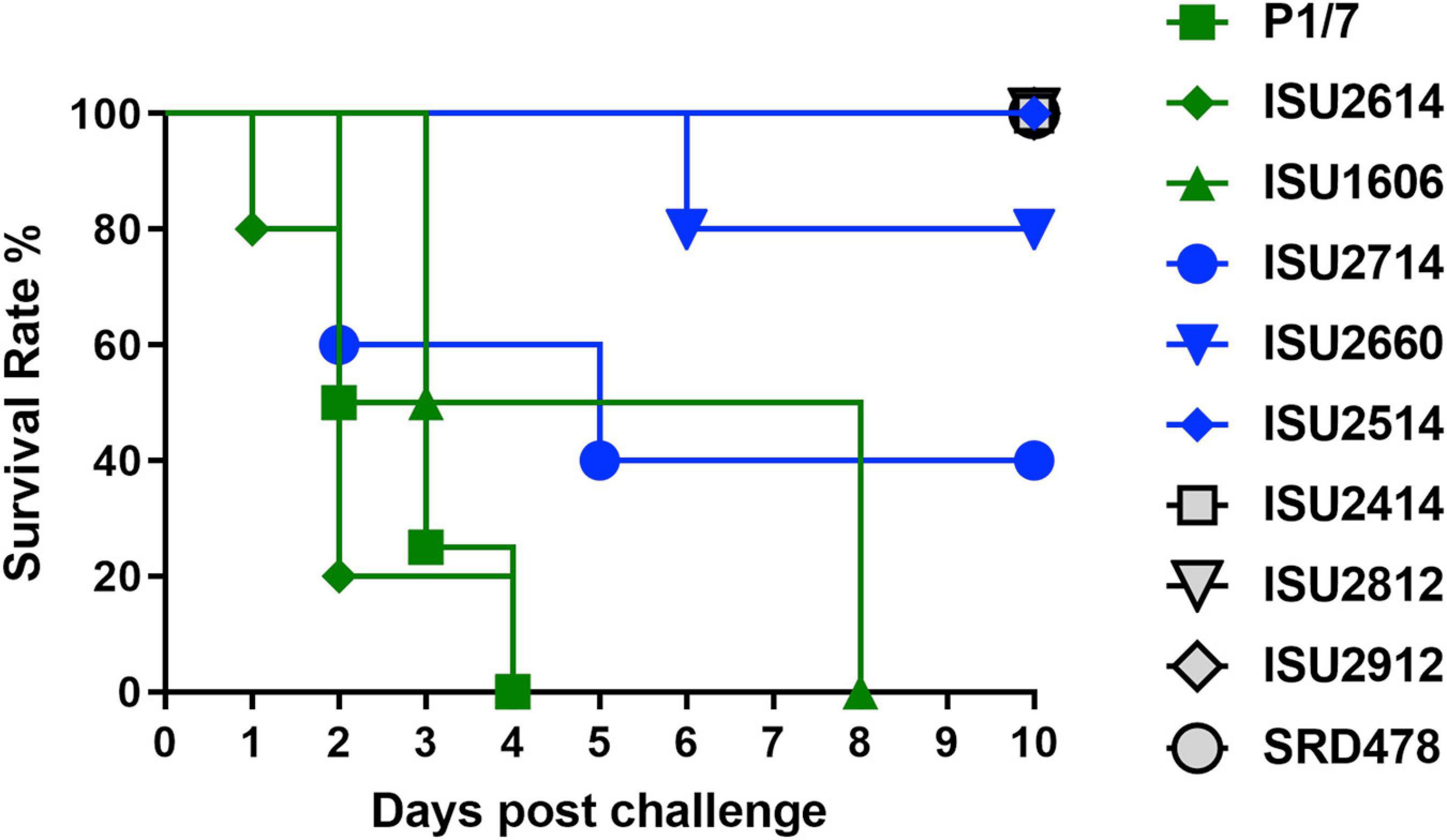
Figure 1. Survival rates of pigs post intranasal challenge. On day 0 groups of 4-5, 8-week-old CDCD pigs were intranasally inoculated with 2 mL (1 mL per nostril) of approximately 1 × 109 CFU/mL of each S. suis isolate. The x-axis indicates days post-challenge, and the y-axis indicates percent survival.
Streptococcus suis isolates ISU2714, ISU2660, and ISU2514 were moderately virulent following intranasal challenge. Three out of five pigs challenged with ISU2714 exhibited neurologic signs and lameness 2-4 dpc and were euthanized, while the other two pigs remained healthy and exhibited no signs of clinical disease (Figure 1 and Table 2). At necropsy, S. suis was isolated from the CSF in 3 out of 5 pigs infected with ISU2714, and from the joint, serosa, serum, and lung in 2 out of 5 pigs within this group (Table 3). Following challenge with ISU2660, one pig developed severe lameness 5 dpc and was euthanized on day 6 post-challenge (Figure 1 and Table 2). A second pig from this group also developed lameness 6 dpc, but subsequently recovered and remained healthy (Figure 1 and Table 2). The other 3 pigs challenge with ISU2660 remained healthy and exhibited no signs of clinical disease (Figure 1 and Table 2). At necropsy, S. suis was isolated from a joint in 2 out of 5 pigs infected with ISU2660 (Table 3). One pig challenged with ISU2514 developed lameness 8 dpc, but subsequently recovered and remained healthy (Figure 1 and Table 2). A second pig from this group exhibited delayed clinical signs of disease, which included lethargy and severe neurological signs and lameness, 13 dpc. The other 3 pigs challenged with ISU2514 remained healthy and exhibited no signs of clinical disease (Figure 1 and Table 2). At necropsy, S. suis was isolated from a joint in 2 out of 5 pigs infected with ISU2514 and from the CSF of a pig that exhibited delayed clinical disease (Table 3).
Following 10 dpc, pigs failing to exhibit any signs of clinical disease were subsequently comingled along with two CDCD naïve pigs, to evaluate transmission potential. During this period, a pig initially challenged with ISU2714 transmitted the bacterium to a naïve pig and ISU2714 was subsequently isolated from the joint of the contact pig. Additionally, a pig initially challenged with ISU2514 transmitted the bacterium to a pig initially challenged with ISU2414. ISU2514 was subsequently isolated from the joint, serum, and the CSF of the pig initially challenged with ISU2414 demonstrating transmission of isolate ISU2514. Collectively, the majority of the pigs initially challenged with isolates ISU2714, ISU2660, or ISU2514 remained healthy and exhibited no signs of clinical disease. Additionally, isolates ISU2714 and ISU2514 transmitted to cohorts leading to clinical signs of disease consistent with S. suis infection and were subsequently isolated from systemic sites. Because these isolates were capable of causing severe systemic disease but they were not uniformly lethal, isolates ISU2714, ISU2660, or ISU2514 were categorized as moderately virulent.
Following intranasal challenge with S. suis isolates ISU2414, ISU2812, ISU2912, or SRD478, all pigs in these groups remained healthy and exhibited no signs of clinical disease (Figure 1 and Tables 2, 3). Additionally, comingling with cohorts including naïve pigs failed to result in a transmission event leading to clinical signs of disease consistent with S. suis infection (Table 3). Based on these results, isolates ISU2414, ISU2812, ISU2912, and SRD478 were categorized as non-virulent.
While isolates ISU2414, ISU2812, and ISU2912 were categorized as non-virulent after internasal challenge of CDCD pigs, there were among the eight isolates obtained from pigs exhibiting clinical disease consistent with S. suis disease and were originally considered to be virulent (Table 1). In fact, ISU2414 was isolated from the pericardium and ISU2812 and ISU2912 were isolated from the brain (Table 1). It is possible that these isolates may have the capacity to cause systemic disease under different experimental conditions, such as circumventing the normal route of infection and using an intravenous challenge instead of an internasal challenge. Taken together, these results highlight the importance of utilizing a challenge method that closely resembles the normal route of infection along with a model free of confounding factors such as coinfections, which could enhance the apparent virulence of some isolates and existing immunity, which may mask the virulence potential of other isolates.
Genome Assemblies, Features, and Comparisons
The complete genome assembly and annotation of all the S. suis isolates included in this study, along with a reference strain P1/7, is summarized in Table 4. The isolates harbored a variety of MLST sequence types (ST) with only ISU1606 and ISU2714 harboring the same ST as P1/7 (ST-1).
The chromosome size, total number of predicted protein coding sequences (CDSs), and G + C content of all the isolates was similar to P1/7, with the exception of isolates ISU2812 and ISU2912. ISU2812 contained a 2,563,863-bp chromosome, encoding 2,434 predicted CDSs, ISU2912 contained a 2,720,381-bp chromosome, encoding 2,667 predicted CDSs: the reference P1/7 contained a 2,007,491-bp chromosome, encoding 1,908 predicted CDSs (Table 4). These isolates were also observed to encode a greater number of predicted pseudogenes and functional CDSs compared to other isolates (Table 4). Previously, it has been hypothesized that virulence and zoonotic potential are correlated with a smaller genome size (Weinert et al., 2015). This correlative observation is consistent when comparing the genomic features of isolates ISU2812 and ISU2912 to P1/7, however, it is not consistent when comparing the genomic features of SRD478 to P1/7. Specifically, SRD478 contained a 2,063,454-bp chromosome, encoding 1,990 predicted CDSs, 96 predicted pseudogenes, and 1,894 functional CDSs, and is very similar to P1/7 genome features (Table 4).
The genomes for the 9 United States isolates were aligned and compared to the genome of P1/7 to examine genome architecture, chromosomal sequences and gene inventories, including mobile elements such as genomic islands, prophage regions, and insertion elements (Figure 2). A high degree of global synteny was observed among the ST-1 isolates P1/7, ISU1606, and ISU2714 (Figure 2). Genome synteny was further explored by comparing the linear organization of the chromosome of each S. suis isolate to P1/7. The highest degree of synteny was observed for isolate ISU1606 (Figure 3). A high degree of synteny within contiguously assembled regions of the genome was additionally observed for isolates SRD478 and ISU2714, with three and four, respectively, collinear regions of sequence alignment compared to P1/7 (Figure 3). An increased number of genome re-arrangements and inversions were observed for isolates ISU2414, ISU2660, and ISU2614 with nine, ten, and eleven, respectively, collinear regions of sequence alignment compared to P1/7 (Figure 3). Isolates ISU2514, ISU2812, and ISU2912 were found to contain a higher number of genome re-arrangements and inversions compared to P1/7 with 22, 29, and 38 respectively, collinear regions of sequence alignment (Supplementary Figure 1). Extensive genetic recombination among streptococcal species, including S. suis has been previously reported (Hanage et al., 2009; De Greeff et al., 2011; Zhang et al., 2011; Mostowy et al., 2014; Weinert et al., 2015). Therefore, the high number of genome re-arrangements and inversions compared to P1/7 observed for isolates ISU2514, ISU2812, and ISU2912 were not surprising. While ISU1606 and ISU2714 are both ST-1 isolates, the high degree of collinearity observed for SRD478 was unexpected given that it is a ST-785 isolate, which differs from ST-1 isolates at every allele.
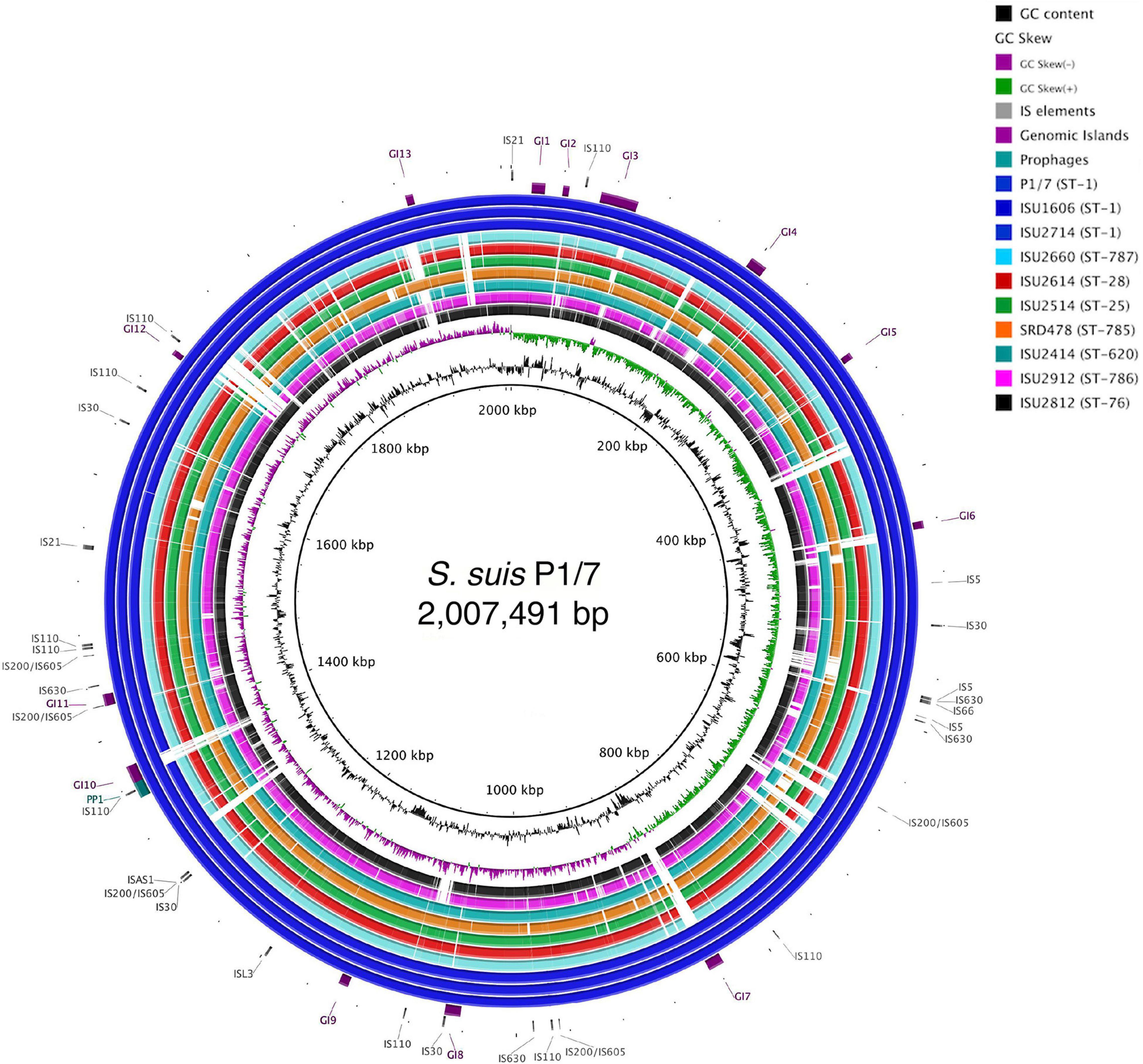
Figure 2. Genome visualization of S. suis chromosomes. Circular comparison of the of nine closed S. suis chromosomes referenced to the genome of ST-1 strain P1/7. Query genomes are and plotted from outermost to innermost ring and shown top to bottom in the legend. GC-content and -skew of the reference chromosome are depicted in the two innermost circles, respectively. Identified MGEs are shown as follows Genomic Islands (GI), ProPhage regions (PP), and Insertion elements (IS).

Figure 3. Comparison of the linear organization of S. suis chromosomes. Linear comparison of each closed S. suis genome to P1/7 (top), reference sequence. Locally collinear blocks (LCBs) representing regions of sequence alignment are shown as colored rectangles connected by lines. LCBs placed above the center line are in the same orientation as the reference; LCBs placed below the center line are in reverse orientation. Blank sections are regions of low sequence conservation. (A) P1/7 compared to ISU2614. (B) P1/7 compared to ISU1606. (C) P1/7 compared to ISU2714. (D) P1/7 compared to ISU2660. (E) P1/7 compared to ISU2514. (F) P1/7 compared to ISU2414. (G) P1/7 compared to ISU2812. (H) P1/7 compared to ISU2912. (I) P1/7 compared to SRD478.
To begin examining genomic differences that could have influenced the swine virulence capacity of the S. suis isolates, pan-genome analysis was used to investigate the genomic content among the S. suis genomes. A complete list of all the protein coding sequences (CDSs) identified within the S. suis genomes are summarized in Supplementary Table 1. Comparative analyses based on groups of orthologous proteins revealed 1,352 core CDSs that were shared among all ten isolates, 2,514 accessory CDSs that were present in 2-9 isolates, and 1,468 CDSs unique to single isolates (Supplementary Table 1). In addition to the high number of unique CDSs compared to the number of core CDSs (1,468 versus 1,352), wide variation in the number of unique CDSs harbored by single isolates was also observed. No unique CDSs were found in isolate ISU2714. Isolates P1/7 and ISU1606 both contained five unique CDSs (Supplementary Table 1). In contrast, isolate ISU2912 was found to contain 693 unique CDSs and isolate ISU2812 was found to contain 362 unique CDSs. These isolates also had a larger genome size with a higher number of functional CDSs compared to P1/7. SRD478, which was found to contain a similar number of functional CDSs compared to P1/7, was observed to contain 185 unique CDSs (Supplementary Table 1). No correlation between the number of unique genes harbored by a single isolate and ST or serotype was observed (Supplementary Table 1). The large number of unique genes and the wide variation in the number of unique genes harbored by single isolates is consistent with previous reports describing an open pan-genome characterized by a frequent gene transfer and significant genomic diversity among S. suis isolates (De Greeff et al., 2011; Zhang et al., 2011; Weinert et al., 2015).
Given that AMR is highly prevalent among S. suis isolates, the isolates in this study were screened for the phenotypic resistance and the genomes were screened for chromosomal mutations and genes conferring resistance (Supplementary Table 3). Isolates ISU2812 and ISU2912 exhibited phenotypic intermediate beta-lactam (penicillin) resistance, however no chromosomal mutations or genes conferring resistance were found (Supplementary Table 3). All isolates except P1/7 exhibited phenotypic tetracycline resistance (Supplementary Table 3). The tetracycline resistance gene tetM was found in SRD478, while tetO was found in the other tetracycline resistant isolates (Supplementary Table 3). With the exception of P1/7, ISU2660, and SRD478, all isolates exhibited phenotypic macrolide, lincosamide, and streptogramin (MLS) resistance (Supplementary Table 3). The MLS resistance gene ermB was found in ISU2614, ISU1606, ISU2714, ISU2514, ISU2812, and ISU2912 and both lnuB and lsaE were found in ISU2414 (Supplementary Table 3). All isolates except ISU2812 and ISU2912 exhibited phenotypic aminoglycoside resistance (Supplementary Table 3). The aminoglycoside resistance gene ANT(9) was identified in ISU2414. No other chromosomal mutations or genes conferring aminoglycoside resistance were found (Supplementary Table 2). ISU2414 was the only isolate to exhibit pleuromutilin resistance (tiamulin), however no genes conferring pleuromutilin resistance were found (Supplementary Table 3). With the exception of ISU2660, ISU2414, and SRD478, all isolates were phenotypically resistant to sulfonamide antibiotics (sulphadimethoxine); however, no chromosomal mutations or genes conferring resistance were found (Supplementary Table 3). No isolates were found to exhibit phenotypic resistance to phenicol, fluoroquinolone, or cephalosporin antibiotics and no chromosomal mutations or genes conferring resistance to these antibiotic classes were found (Supplementary Table 3).
Mobile genetic elements substantially contribute to evolution, adaption, and genomic diversification through horizontal gene transfer. Numerous MGEs including genomic islands and prophage insertions were observed among the genomes of the S. suis isolates in this study, appearing to contribute to genome plasticity and the disruption of genome synteny and plasticity (Figure 2). A complete list of all MGEs identified in the S. suis genomes are summarized in Supplementary Table 2. The number of prophage regions within a given genome varied from one, for isolates P1/7 and ISU2812 up to nine for ISU2912 (Table 5). Sequence analysis was used to determine if any AMR elements were located within any MGEs. Both tetO (DK235_04640) and ermB (DK235_04660) were found to be located within a genomic island for ISU1606 (Supplementary Tables 2, 3). Similarly, tetO (DK876_03195) and ermB (DK876_03150) from ISU2714 were found to be located within a genomic island (Supplementary Tables 2, 3). The ANT(9) (DK877_09725), lnu(B) (DK877_09745), and lsaE (DK877_09750) genes from ISU2414 are collocated within the same chromosomal region, resembling an operon structure. This region encompasses six CDSs and is not located within any of the prophage or other MGEs identified in ISU2414.
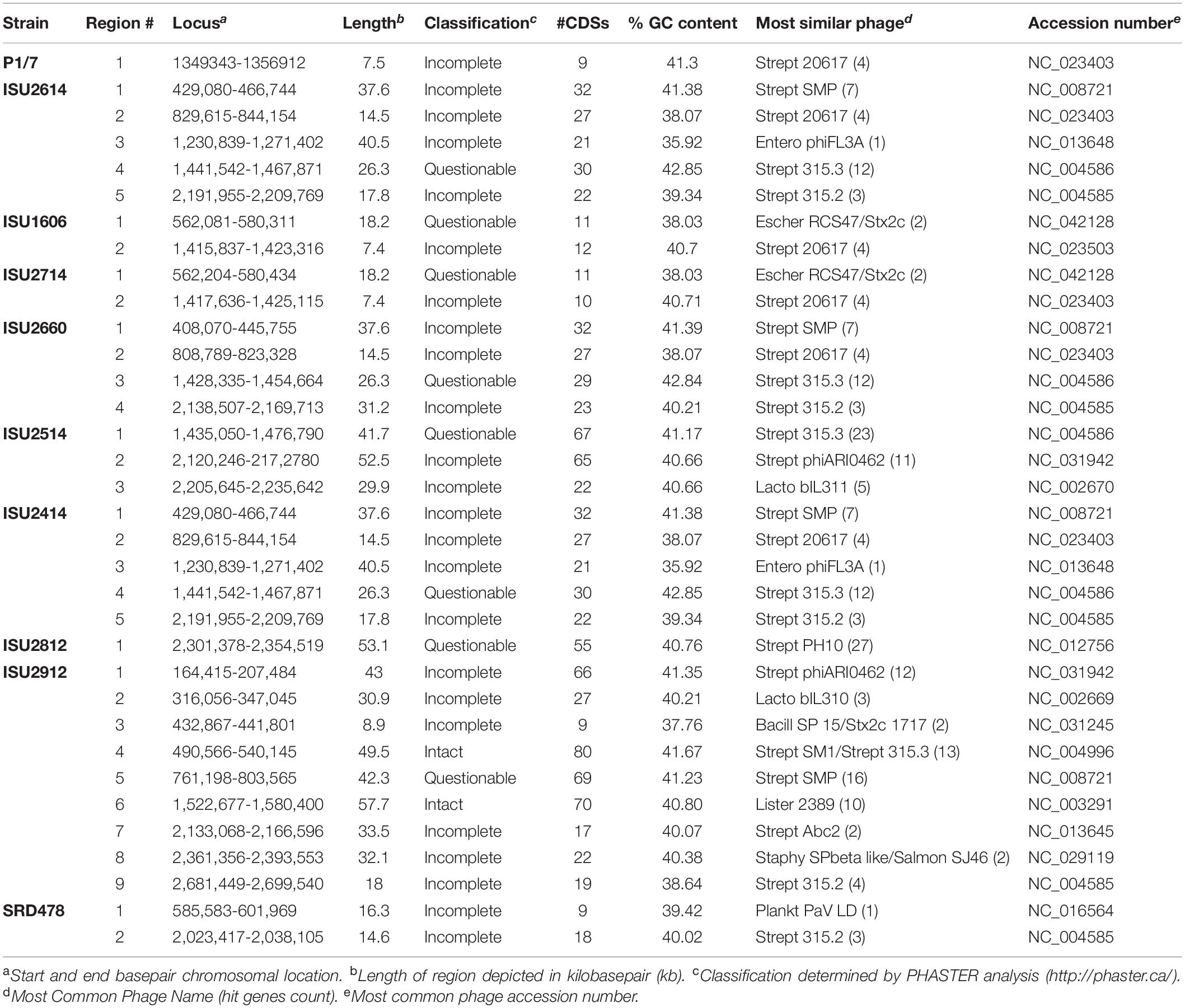
Table 5. Prophage regions identified in S. suis genome sequences.
Isolates ISU2614, ISU2514, and ISU2812 were observed to contain a single plasmid and SRD478 was observed to harbor four plasmids (Table 4). The plasmid harbored by ISU2614 is 4,984-bp and contains seven CDSs including rec, repB, a predicted transcriptional regulator, and four CDSs of unknown function. ISU2514 contains a 5,581-bp plasmid that encodes six CDSs including rec, rep, and four CDSs of unknown function and ISU2812 contains a similar size plasmid, 5,586-bp, which also encodes six CDSs including rep, and five CDSs of unknown function (Supplementary Figure 1). The first plasmid harbored by SRD478 is 15,154-bp and encodes fifteen CDSs including parA, repR, a recombinase, a kinase, a transcriptional regulator, two transporter proteins, an ATP-binding cassette-domain containing protein, a type II toxin and antitoxin (TA) system, and five additional CDSs of unknown function. A second plasmid harbored by SRD478 is 4,723-bp and contains a predicted type II toxin and antitoxin system, rep, and four CDSs of unknown function (Supplementary Figure 1). The third plasmid is 3,972-bp and encodes a rep, a bacteriocin, a bacteriocin immunity protein, and three CDSs of unknown function (Supplementary Figure 1). The fourth plasmid and smallest plasmid is 2,945-bp and contains a rep and two CDSs of unknown function (Supplementary Figure 1). TA systems are small genetic elements comprised of two components, a stable protein toxin and a more labile antagonistic antitoxin, and typically provide a variety of functions, such as stabilization of genomic regions, anti-addiction against similar plasmid-borne toxins, defense against phage infection, biofilm formation, control of the stress response, and bacterial persistence (Yamaguchi and Inouye, 2011; Yamaguchi et al., 2011). With the exception of bacteriocin, no other chromosomal mutations and genes conferring resistance were identified in any of the plasmids in these three strains. Bacteriocins comprise a group of structurally diverse small antimicrobial peptides, which are ribosomally synthesized by a wide range of bacteria and archaea (Dobson et al., 2012). Bacteriocin production has been considered an important and desirable trait in the selection of probiotic strains (Dobson et al., 2012; O’Connor et al., 2020). However, a bacteriocin locus was recently found encoded within an ICE of the S. suis prophylactic candidate strain 90-133 (Sun et al., 2019). The authors cautioned against the use of strain 90-133 due to potential transfer of bacteriocin resistance given the bacteriocin locus was found encoded within a mobile genetic element. The finding of a bacteriocin locus encoded within a relatively small mobile plasmid reported here supports the idea that bacteriocin resistance may be easily transferred and more prevalent than originally assumed.
Capsule Loci Comparisons
Four types of capsule polysaccharide (CPS) loci were observed among the isolates in this study with the majority harboring a type 2 capsule (P1/7, ISU2614, ISU1606, ISU2714, ISU2660, ISU2514, and ISU2414) (Supplementary Figure 2). The serotype 2 strains are highly similar to each other and to reference strain P1/7, but were not identical. Sequence conservation is very high in the region spanning from the 5′ regulatory genes to the N-acetylneuraminic acid synthesis genes located near the 3′ end of the CPS locus. Within this region, identical annotated gene content compared to P1/7 was observed except for the region predicted to encode several small CDSs of unknown function between the glycosyltransferase and sialyltransferase genes (Supplementary Figure 2). While similar nucleotide sequence was observed, the annotation of P1/7 differs compared to the other serotype 2 strains. The greatest variation within the capsule region of the serotype 2 strains was seen in the region near the 3′ end containing genes predicted to encode transposases or integrases located between the N-acetylneuraminic acid synthesis and the aroA genes (Supplementary Figure 2). ISU2812 was identified as serotype 19 and is highly similar but not identical to the CPS locus of strain 42A, another serotype 19 strain (Supplementary Figure 2; Liu et al., 2013). As with the serotype 2 strains, gene content is conserved compared to strain 42A and the region of greatest nucleotide sequence divergence is within the mobile-element related genes near the 3′ end (Supplementary Figure 2). The CPS locus of ISU2912 was identified as a NCL serotype 1-3, originally described by Zheng et al. (2015) (Supplementary Figure 2). Sequence comparisons with other NCL1-3 strains showed similarity in gene content, with the exception that ISU2912 contains transposases not present in the NCL1-3 strains from Zheng et al. (2015) (Supplementary Figure 2). These are the transposases located between the sugar epimerase gene and the sugar dehydrogenase gene, and the 4 genes immediately downstream of the nucleotidyl transferase gene (1 unknown function, 3 transposase/integrase genes) (Supplementary Figure 2; Zheng et al., 2015). The CPS locus of SRD478 does not match any published serotypes; however, a BLASTn search revealed a high similarity to CPS loci from four non-typeable S. suis isolates (accession numbers KX870048, KX870054, KX870055, and KX870059) (Supplementary Figure 2).
While a variety of virulence factors have been described for S. suis, CPS is perhaps considered the most important (Charland et al., 1998; Fittipaldi et al., 2012; Roy et al., 2015; Zhao et al., 2015; Segura et al., 2017). Therefore, the capsule production ability among the S. suis isolates was evaluated using in vitro assays, which have been previously used to demonstrate CPS production (Bonifait et al., 2010; Roy et al., 2015; Auger et al., 2018). All of the isolates harboring a type 2 capsule (P1/7, ISU2614, ISU1606, ISU2714, ISU2660, ISU2514, and ISU2414) exhibited a low surface hydrophobicity (higher percent retained in the aqueous phase), reflective of high capsule production, and significantly different compared to the P1/7 capsule mutant Δcps2E (Figure 4A). In contrast, ISU2812 (serotype 19), ISU2912 (serotype 1-3), and SRD478 (undefined/non-typeable) exhibited a high surface hydrophobicity, indicating reduced CPS production, compared to the type 2 capsule isolates (Figure 4A). ISU2912 was the only isolate that exhibited no significant difference in surface hydrophobicity compared to the P1/7 capsule mutant Δcps2E (Figure 4A). The ability to survive incubation in whole blood was tested for one isolate from each swine virulence category. Out of the isolates that exhibited high surface hydrophobicity, ISU2912 would have been the best choice to further test for the ability to survive in both whole blood and serum. However, ISU2912 was observed to have a variety of traits, including genomic size, sequence diversity, and growth medium conditions, which differed from the other isolates and was therefore excluded from consideration in an effort to minimize confounders. Out of the isolates that exhibited high surface hydrophobicity, SRD478 was chosen to test for the ability to survive in both whole blood and serum. No significant difference in the ability to survive incubation in whole blood was observed for ISU2614 (highly virulent), ISU2660 (moderately virulent), and SRD478 (non-virulent) (Figure 4B). While a reduction in the percent survival was observed after incubation in serum compared to heat-inactivated serum, no significant difference in the percent survival between P1/7 (highly virulent) and SRD478 (non-virulent) was observed (Figure 4C).
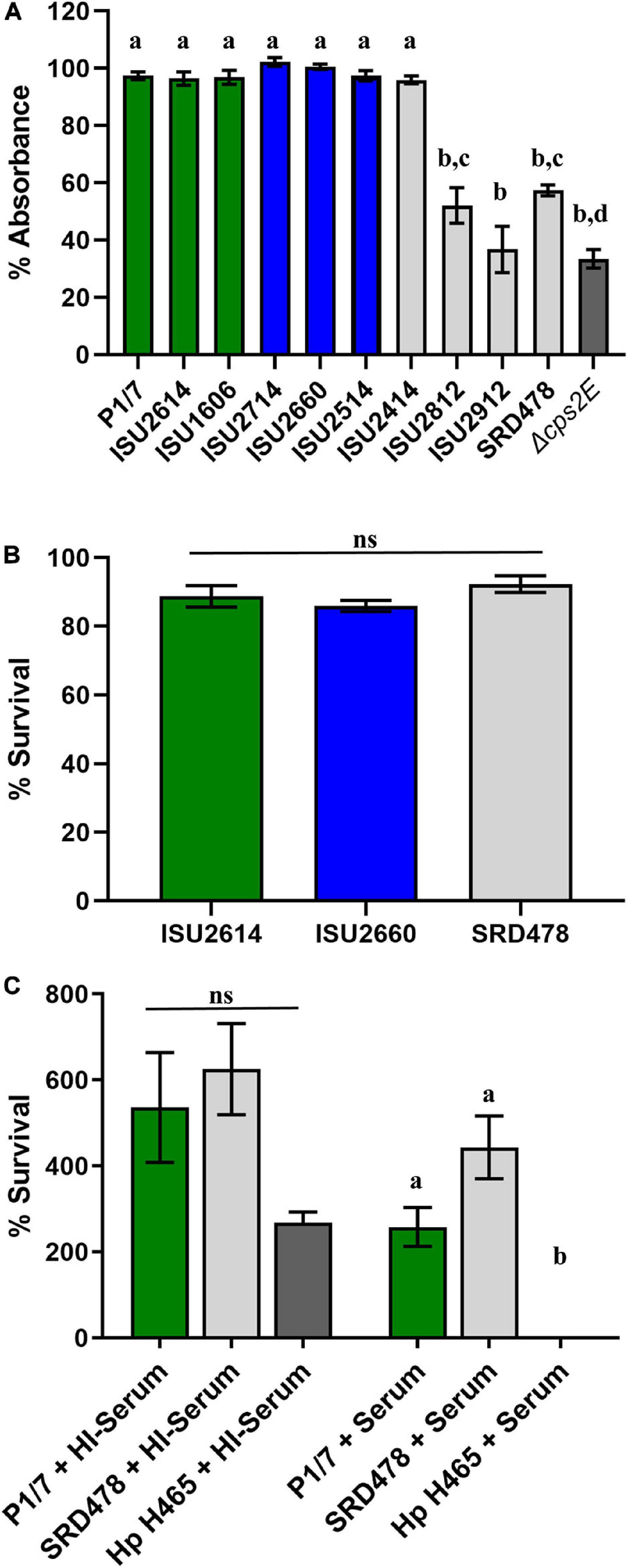
Figure 4. Comparison of phenotypes related to capsule production. (A) Bacterial hydrophobicity assay. Relative hydrophobicity of S. suis isolates were determined by measuring their absorption (y-axis) to n-hexadecane. S. suis P1/7 Δcps2E, a capsule deletion mutant, was used as a control. (B) Whole blood sensitivity assay. S. suis isolates were incubated with swine whole blood for 1 h and enumerated to determine the percent viable bacteria (y-axis) expressed. (C) Serum sensitivity assay. Bacteria were incubated with guinea pig serum (+Serum) or heat-inactivated guinea pig serum (HI + Serum) and enumerated to determine the percent survival (y-axis) calculated as the proportion of treated samples to untreated samples. Serum-sensitive Glasserella parasuis H465 (Hp H465) was used as a control. Bars represent means ± SEM from three independent experiments. Bar color reflective of virulence categorization (green: highly virulent; blue: moderately virulent; light gray: non-virulent) or control (dark gray: S. suis P1/7 Δcps2E). Data was analyzed using a one-way ANOVA with a Tukey’s post-test (GraphPad Prism 8.3.0). Groups with different letter designations were significantly different (p < 0.05); ns, not statistically significantly different.
It has been previously demonstrated that CPS production results in low surface hydrophobicity (Bonifait et al., 2010; Roy et al., 2015; Auger et al., 2018). While the high surface hydrophobicity exhibited by ISU2812, ISU2912, and SRD478 could suggest a decreased ability to produce a capsule, the ability of SRD478 to survive in both whole blood and serum suggest that these isolates may have the ability to produce a capsule, albeit with different functional characteristics. The lack of significant difference between P1/7 (highly virulent) and SRD478 (non-virulent) in the ability to survive in both whole blood and serum suggest that any differences or functional characteristics between their capsules does not correlate with their respective capacity to cause disease in swine.
Comparison of in vitro Growth and Virulence-Associated Phenotypes
Given the lack of correlation between capsule type and the capacity to cause disease in swine, we further tested the S. suis isolates in a variety of in vitro assays routinely used to measure virulence capacity. Differences in growth rate dynamics of S. suis isolates were measured and revealed a wide range of growth rates among the isolates (Figure 5A). Overall, no correlation between the growth rate dynamics of the S. suis isolates and their capacity to cause disease in swine was observed (Figure 5A). It has been reported that biofilm formation in S. suis causes a reduction in virulence due to the downregulation of virulence factors, such as CPS (Wang et al., 2011). Biofilm formation among the S. suis isolates was quantified by standard microtiter crystal violet assays (Figure 5B). While significant differences in biofilm formation were observed among some of the isolates, no correlation between biofilm formation by the S. suis isolates and their capacity to cause disease in swine was observed (Figure 5B). Moreover, no correlation between biofilm formation by the S. suis isolates and their surface hydrophobicity was observed (Figures 4A, 5B). Next, the ability to survive oxidative stress, to produce hemolytic activity, to adhere to either BEAS-2B or J774A.1 cells, as well as the ability to produce cell-associated or secreted nuclease activity among the S. suis isolates was measured (Figures 5C–H). Similar to the growth rate dynamics and biofilm formation capacity among the S. suis isolates, significant differences in each of these virulence-associated phenotypes were observed among some of the isolates; however, there was no correlation between in vitro virulence-associated phenotypes by the S. suis isolates and their capacity to cause disease in swine (Figures 5C–H).
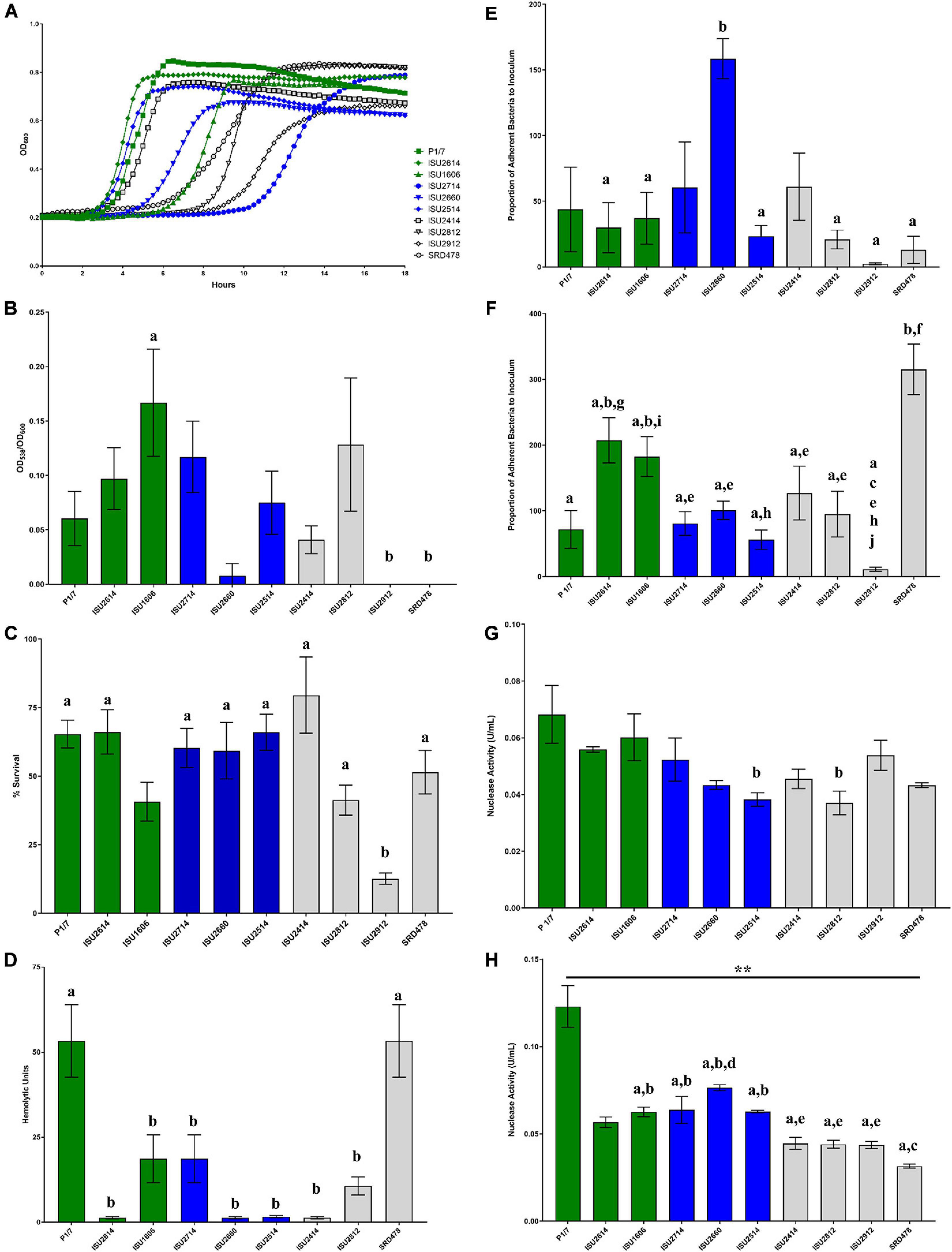
Figure 5. Comparison of in vitro growth and virulence-associated phenotypes. (A) Growth dynamics of S. suis isolates. Isolates were cultivated at 37°C for 24 h. Growth was measured by OD600 every 15 min. All data points represent averages obtained from three independent experiments. (B) Biofilm formation by S. suis isolates. S. suis isolates were cultivated statically for 24 h in microtiter plates. Growth was then measured by OD600 followed by quantification crystal violet staining by OD538. Biofilm mass (y-axis) is expressed as the OD538 normalized to OD600. (C) Oxidative stress assay. S. suis isolates were cultivated until exponential phase and divided into treated (10 mM H2O2) and untreated cultures (H20), incubated at 37°C for 15 min followed by addition of 10 μg/mL catalase. Bacteria were enumerated to determine the percent survival (y-axis) calculated as the proportion of treated samples to untreated samples. (D) Hemolysis assay. Hemolytic activity of supernatants collected from S. suis isolates. (E) Adherence of S. suis isolates to BEAS-2B cells. Adherence (y-axis) to BEAS-2B cells (human lung/bronchus epithelial cell line) is expressed as the proportion of bacteria in the original inoculum found to be adherent after a 2-h incubation period. (F) Adherence of S. suis isolates to macrophages. Adherence (y-axis) to J774A.1 cells (murine macrophage-like cell line cell line) is expressed as the proportion of bacteria in the original inoculum found to be adherent after a 2-h incubation period. (G) Cell-Associated Nuclease Activity. Cell-associated nuclease activity for S. suis isolates was measured using whole-cell FRET assay. (H) Secreted Nuclease Activity. Secreted nuclease activity for S. suis isolates was measured using extracellular FRET assay. Bars (B–H) represent means ± SEM from three independent experiments. Bar color reflective of virulence categorization (green: highly virulent; blue: moderately virulent; light gray: non-virulent). Data (B–H) was analyzed using a one-way ANOVA with a Tukey’s post-test (GraphPad Prism 8.3.0). Groups with connecting lines were significantly different (p < 0.0003). Groups with different letter designations were significantly different (p < 0.05).
Virulence-Associated Gene Comparisons
To further examine genomic differences that could have influenced the swine virulence capacity of the S. suis isolates, we compared the nucleotide sequences of fifty-one virulence-associated genes, which were included in a comprehensive review (Fittipaldi et al., 2012). The percent identity for each gene was determined for each isolate relative to the P1/7 ortholog with the following exceptions: hylA, revS, stp, vraR, and vraS. These exceptions were based on choosing a reference gene sequence in which functional characterization had previously been reported or due to annotation of the P1/7 gene as a pseudogene. Hierarchical clustering analysis of the nucleotide percent identity of analyzed virulence genes revealed two general observations. First, a correlation between ST and the presence of genes encoding known virulence factors, and the nucleotide identity among those genes, was observed (Figure 6A). The highest sequence identity among the analyzed genes was observed in the genes from isolates ISU1606 and ISU2714. Similar to P1/7, both of these isolates are ST-1 (Figure 6A). Second, no clear correlation between the capacity to cause disease in swine and the presence of genes encoding known virulence factors, and the nucleotide identity among those genes, was found (Figure 6A). For example, while the highest sequence divergence in the genes analyzed was observed from the non-virulent isolate ISU2912, similarly high sequence divergence was not noted for the other non-virulent isolates (ISU2414, ISU2812, and SRD478) (Figure 6A). While a high sequence identity among the analyzed genes was observed for the highly virulent ISU1606, high sequence identity among the analyzed genes was not observed for ISU2614, also categorized as highly virulent. Some of the analyzed ISU2614 genes with the lowest sequence identity include sadP with 77% identity, ideS with 87.5% identity, and mrp with 87.8% identity compared to the corresponding P1/7 orthologs (Figures 6A,B). ISU2614 genes ofs, with 75.9% identity, and zmpC, with 75.9% identity, are both predicted pseudogenes Figures 6A,B. Suilysin gene sly from ISU2714 and ISU1606 was 100% identical to sly from P1/7 and sly from the non-virulent isolates ISU2812 and SRD478 were 99.6% and 99.2% identical to sly from P1/7. However, sly was absent from all other isolates (ISU2660, ISU2414 ISU2614, ISU2514, and ISU2912) (Figure 6A).
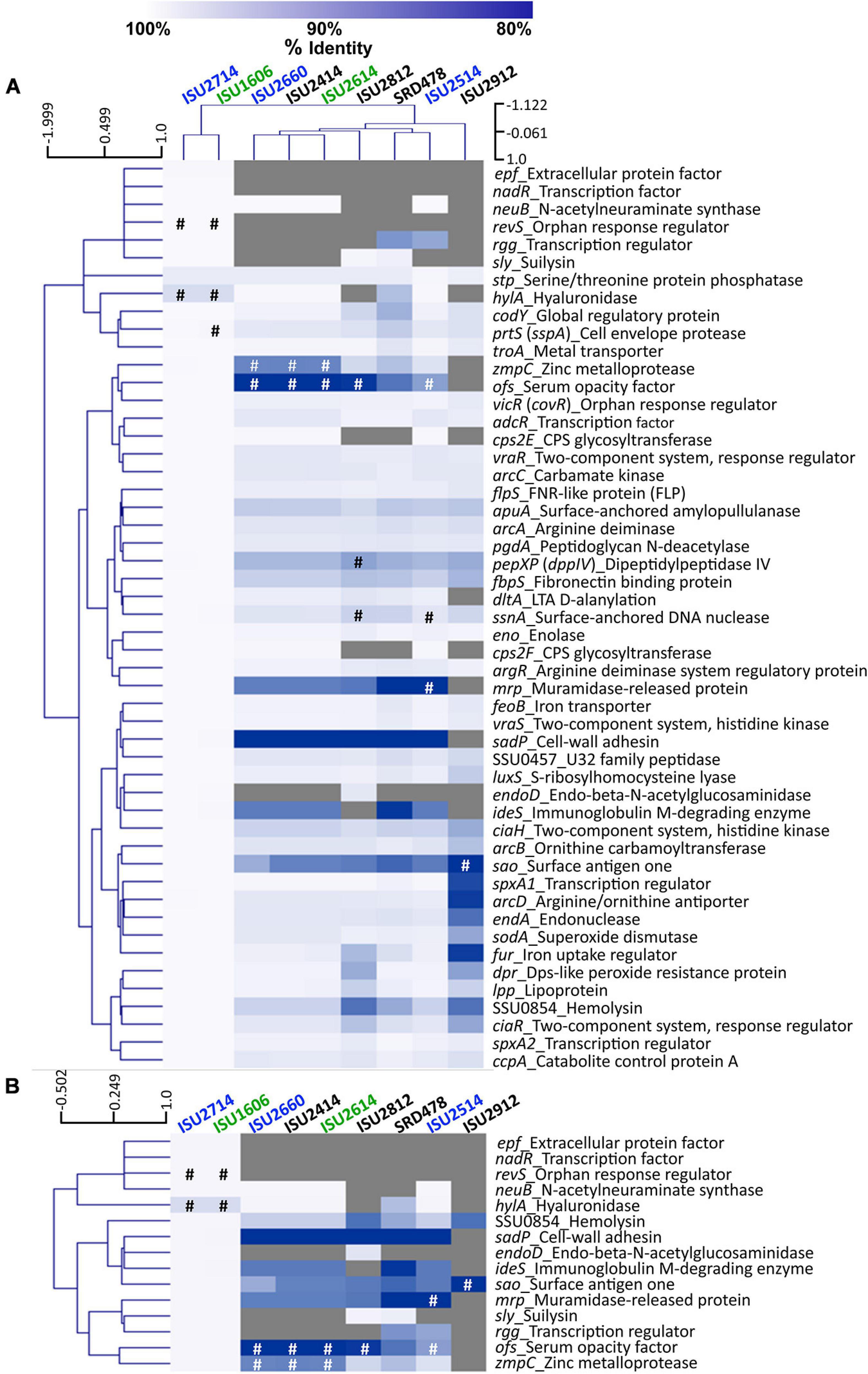
Figure 6. Hierarchical cluster heatmap displaying the relatedness of S. suis isolates based on the nucleotide percent identity of analyzed virulence genes. (A) Heatmap based on nucleotide percentage identity for all analyzed virulence genes. Heatmap generated from nucleotide percentage identity was converted into a distance matrix and clustered by means of complete hierarchical clustering based on Pearson correlation distance for both genes and isolates. (B) Heatmap based on nucleotide percentage identity for selected genes with high sequence divergence. Heatmap generated from nucleotide percentage identity was converted into a distance matrix and clustered by means of complete hierarchical clustering based on Pearson correlation distance for genes only. Isolates names are provided at the top of the heat map and gene names are provided at the right side of heat map. Font color used for isolate names color reflective of virulence categorization (green: highly virulent; blue: moderately virulent; black: non-virulent). The percent identity of analyzed genes (rows) from each isolate (columns) is represented using the color scale at top, while genes not present within a strain are indicated by gray. Annotated pseudogenes are indicated by # (white or black). Dendrograms are on the left side and on top of the heat map.
The closest correlation between the capacity to cause disease in swine and the presence of genes encoding known virulence factors, or their nucleotide identity, was observed for the genes neuB, ciaR, codY, and SSU0854 encoding a hemolysin. Specifically, neuB was present in ISU2414, but absent from the other non-virulent isolates ISU2812, ISU2912, and SRD478 (Figure 6A). A similar sequence identity was observed between P1/7 and ISU2414 for ciaR, codY, and SSU0854 encoding a hemolysin, while a low sequence identity was observed between P1/7 and the other non-virulent isolates ISU2812, ISU2912, and SRD478 (Figure 6A). Therefore, no clear correlation between the presence of genes encoding known virulence factors, or their nucleotide identity, and the capacity to cause disease in swine was identified.
Overall the highest sequence identity among the analyzed genes was observed in the genes from isolates ISU1606, categorized as highly virulent, and ISU2714, categorized as moderately virulent (Figure 6A). As mentioned, ISU1606 and ISU2714 harbor the same ST as P1/7 (ST-1). The hylA gene encoding hyaluronidase from both ISU1606 and ISU2714 was found to have the most sequence divergence with 97.3% identity and is a predicted pseudogene in ISU1606 and ISU2714 (Figure 6A). The prtS (sspA) gene encoding a cell envelope protease is also a predicted pseudogene in ISU1606 (Figure 6A). Another predicted pseudogene in ISU1606 and ISU2714 is revS, which is also annotated as a pseudogene in P1/7 (Figures 6A,B). Since revS is annotated as a pseudogene in P1/7, the revS gene from S. suis 05ZHY33 (Wu et al., 2009) was used as the comparison reference. Previous studies reported revS to be a 519-bp gene encoding a 173-amino acid protein (De Greeff et al., 2002; Wu et al., 2009). The revS gene for ISU1606 and ISU2714 is predicted to be 821-bp and 815-bp for P1/7. The 5′ end of revS for all three isolates is 100% identical to the 519-bp revS gene from S. suis 05ZHY33, which was used as reference. Therefore, it is possible that the first 519-bp of the revS gene from ISU1606, ISU2714, and P1/7, could encode a functional RevS protein. revS orthologs were not identified in any of the other isolates analyzed (Figures 6A,B). Similarly, nadR and epf from ISU1606 and ISU2714 were 99.9% and 100% identical to their respective P1/7 orthologs and were not identified in any of the other isolates (Figures 6A,B).
Due to the high degree of nucleotide divergence observed for some of the genes listed in Figure 6B, protein sequence alignments were generated to explore the amino acid variation. IdeS is a S. suis immunoglobulin M-degrading enzyme, first described by Seele et al. (2013). The predicted amino acid sequence contains a Mac-1 domain and is similar to the IdeS, also known as Mac-1, a S. pyogenes endopeptidase with specificity for IgG (Agniswamy et al., 2004, 2006; Wenig et al., 2004). IdeS from P1/7, ISU1606, and ISU2714 is identical and is 1,141 amino acids (AA) in length (Figure 7A). IdeS from ISU2614, ISU2660, and ISU2514 and ISU2414 is shorter with a predicted 1,027 AA length, mainly due to a 114 AA deletion corresponding to amino acid residues 717-830 of P1/7 IdeS (Figure 7A). There are 151 AA differences between IdeS from ISU2614, ISU2660, and ISU2414 compared to P1/7 IdeS and 150 AA differences between IdeS from ISU2514 and P1/7 (Figure 7A). These changes are observed throughout the protein sequences (Figure 7A). The greatest amino acid divergence was observed in IdeS from SRD478, predicted to be 1092 AA in length and contain 170 differences compared to P1/7 IdeS (Figure 7A). Many of these differences are located with the N-terminus region as well two small deletion regions of 25 AA and 29 AA that correspond to 806-830 AA and 86-891 AA in P1/7 IdeS (Figure 7A).

Figure 7. Protein sequence alignments. Gene sequences were identified by BLASTn, translated, and multiple alignments of the protein sequences were created using MAFFT. Gray regions indicate identical amino acids, black regions indicate amino acid sequence differences compared to the reference (top) sequence. (A) Multiple alignment of IdeS (immunoglobulin M-degrading enzyme); P1/7 IdeS used as reference sequence. (B) Multiple alignment of MRP (muramidase-released protein); P1/7 MRP used as reference sequence. (C) Multiple alignment of OFS (serum opacity factor); Strain 10 OFS used as reference sequence. (D) Multiple alignment of SadP (cell wall adhesin); P1/7 SadP used as reference sequence. (E) Multiple alignment of SAO (surface antigen one); P1/7 SAO used as reference sequence.
While a search failed to identify a corresponding ideS ortholog in ISU2812 and ISU2912, further inspection of the genomic region within these isolates corresponding to the ideS location in the P1/7 genome revealed a CDS annotated as a hypothetical protein containing a Mac-1 domain in both ISU2812 (A7J09_05595) and ISU2912 (A7J10_03470). Both CDSs are 2,727-bp and are 99.4% identical to each other. The predicted amino acid sequence for both is 908 AA and has 99.1% similarity to each other. The predicted amino acid sequence for the CDS from ISU2812 (A7J09_05595) only has 28.7% similarity to P1/7 IdeS, with the highest level of similarity located within the cysteine proteinase/Mac-1 domain region.
Muramidase-released protein is a muraminidase-released protein that has been characterized as associated with the cell wall of S. suis via an LPxTG motif as well as released into culture supernatants (Vecht et al., 1991). MRP is often reported as an epidemic infection marker for highly virulent strains in Europe and Asia (Vecht et al., 1996; Silva et al., 2006; Rehm et al., 2007; Wei et al., 2009; Onishi et al., 2012; Maneerat et al., 2013; Li et al., 2017). The amino acid sequence of MRP from P1/7, ISU1606, and ISU2714 were identical, 1,256 AA in length, and contained a signal peptide domain followed by several repeat MucBP (Mucin-Binding Protein) domains, followed by a LPXTG cell wall anchor domain (Figure 7B). MRP from ISU2614 and ISU2660, and ISU2414 was predicted to be 1,268 AA in length and the protein sequence from these isolates differed from P1/7 MRP by 177 AA. MRP from ISU2812 was predicted to be 1,242 AA in length and contained 183 differences compared to P1/7 MRP (Figure 7B). The predicted AA sequence for MRP from SRD478 was 1,391 AA and differed from P1/7 MRP by 338 AA. MRP from SRD478 contained a large 136 AA insert within a MucBP domain corresponding to 970-971 AA in P1/7 MRP (Figure 7B). Many of the AA differences in the MRP from isolates ISU2614, ISU2660, ISU2414, ISU2812, as well as in SRD478 compared to P1/7 MRP were located upstream of the first MucBP domain (Figure 7B). The observed variation in size of MRP from the isolates analyzed was not surprising given that MRP size variants have been previously described (Silva et al., 2006). MRP from ISU2514 contained a frameshift mutation and was annotated as a pseudogene.
The ofs gene encodes for the S. suis serum opacity factor (OFS) protein. The predicted OFS protein structure has been described to contain several structural features of a MSCRAMMs (microbial surface components recognizing adhesive matrix molecules), such as a putative N-terminal signal sequence, a large N-terminal domain including a proline-rich region, repetitive sequence elements, and a C-terminal LPXTG anchor motif (Baums et al., 2006). Several allelic variants of OFS have been previously reported (Takamatsu et al., 2008). Due to frameshift mutations in P1/7, ISU2414, ISU2614, ISU2812, and ISU2660, and a truncation in ISU2514, the ofs gene was annotated as a pseudogene.
Protein alignments were generated using OFS from S. suis Strain 10 as the reference since it has been functionally characterized and demonstrated to serve a role in virulence (Baums et al., 2006). OFS from Strain 10 is predicted to be 938 AA in length, while OFS from ISU1606 and ISU2714 is predicted to be 930 AA (Figure 7C). OFS from ISU1606 and ISU2714 differs from Strain 10 OFS by 1 AA, which happens to be the start codon for ISU1606 and ISU2714. The difference in predicted length is due to annotation differences in which the annotation for Strain 10 uses an upstream start codon compared to the start codon used in the annotation of ISU1606 and ISU2714. The nucleotide sequence upstream of the annotated ofs CDS for ISU1606 and ISU2714 is identical to the corresponding region in Strain 10 and the corresponding start codon used in the annotation of Strain 10 is inframe with the annotated ofs CDS for ISU1606 and ISU2714. Therefore, despite the predicted length difference, OFS from Strain 10, ISU1606, and ISU2714 is likely to be highly similar. OFS from SRD478 is predicted to be 989 AA in length and was observed to contain the greatest amino acid divergence with 183 differences compared to Strain 10 OFS (Figure 7C). Many of these differences are located in two regions. First, several small insertions are located within the first 22 AA of the N-terminal signal peptide and the second location is a 37 AA insertion corresponding to 759 and 760 of Strain 10 OFS (Figure 7C).
SadP (streptococcal adhesin P) has been demonstrated to mediate the binding of S. suis to galactosyl-α1–4-galactose (Galα1–4Gal)-containing host receptors (Kouki et al., 2011). Substantial nucleotide sequence variation in the sadP gene and structural variants for SadP have been described (Ferrando et al., 2017). SadP from P1/7, ISU1606, and ISU2714 is 765 amino acids (AA) in length and SadP from P1/7 and ISU2714 is identical, while SadP from ISU1606 differs from P1/7 SadP by 1 AA (Figure 7D). A high degree of amino acid divergence was observed for SadP from the other isolates (Figure 7D). SadP from ISU2614, ISU2660, and ISU2414 is shorter with a predicted 714 AA in length (Figure 7D). There are 241 AA differences between SadP from ISU2614 and ISU2414 compared to P1/7 SadP and 242 AA differences between SadP from ISU2660 and P1/7 (Figure 7D). SadP from ISU2514 is predicted to be 747 AA in length and contain 254 differences compared to P1/7 SadP (Figure 7D). The predicted amino acid sequence for SadP from ISU2812 is 728 AA and differs from P1/7 SadP by 235 AA. SRD478, predicted to be 769 AA in length and differs from P1/7 SadP by 422 AA, which includes a 26 AA insert corresponding to 524-525 of P1/7 SadP (Figure 7D). Kouki et al. (2011) reported that the central portion of the protein, particularly amino acids 31-328, are critical for ligand binding to Galα1–4Gal-containing host receptors. Given that the majority of the numerous AA changes observed in SadP from ISU2614, ISU2660, ISU2514, ISU2414, ISU2812, and SRD478 are located within this region, it is possible that the variation could impact binding affinity, ligand septicity, or both.
Surface antigen one (SAO) was originally identified by a phage display library screen utilizing convalescent swine sera and has been reported to contain a LPXTG anchor motif and is consequently anchored to the cell-wall peptidoglycan by sortase A (Li et al., 2007; Zhang et al., 2008). While it has not previously been associated with virulence, it has been evaluated as a protective immunogen (Li et al., 2007; Zhang et al., 2008; Hsueh et al., 2014, 2017; Roy et al., 2014). To date, three allelic variants have been reported with sao-M, encoding an amino acid sequence 580 AA in length, as the most prevalent (Feng et al., 2007). SAO from P1/7, ISU1606, and ISU2714 are identical and are 580 AA in length (Figure 7D). The amino acid sequence contains an FctA domain, found in the major pilin of from Streptococcus pyogenes and other fibronectin- and collagen-binding proteins, located between AA524-525 of P1/7 SAO. SAO from ISU2614, ISU2514, ISU2414, and ISU2812 is longer with a predicted 639 AA length (Figure 7E). There are 92 AA differences between SAO from ISU2614 compared to P1/7 SAO, 99 AA differences between SAO from ISU2514 and P1/7 SAO, and 91 AA differences between SAO from ISU2414 and P1/7 SAO (Figure 7E). The greatest amino acid divergence was observed in SAO from ISU2812, which differs from P1/7 SAO by110 AA (Figure 7E). SAO from SRD478 is predicted to be 670 AA in length and contain 105 differences compared to P1/7 SAO (Figure 7E). The sao gene in ISU2912 (A7J10_07360, A7J10_07370) is interrupted by a transposase (A7J10_07365). The alignment revealed two areas of insertions downstream of the FctA domain that are different lengths for different isolates (Figure 7E). The first insertion location corresponds to AA363-364 in P1/7 SAO (Figure 7E). ISU2614, ISU2514, ISU2414, ISU2812, and SRD478 contain a 30 AA insertion and ISU2660 contains an 11 AA insertion at this location (Figure 7E). The second insertion location corresponds to positions 414-415 in P1/7 SAO (Figure 7E). ISU2614, ISU2660, ISU2514, ISU2414, and ISU2812 contain a 30 AA insertion and SRD478 contains a 60 AA insertion at this location (Figure 7E).
Due to the high degree of nucleotide divergence observed for the fifty-one previously reported virulence-associated genes (Figure 6A), we performed an additional search for orthologs that were found in any one of the three isolates categorized as highly virulent (P1/7, ISU2614, or ISU1606) and not found in any of the isolates categorized as non-virulent (ISU2414, ISU2812, ISU2912, and SRD478). This analysis identified 42 CDSs including one predicted DNA binding protein and three predicted transcriptional regulators (Supplementary Table 1). Unfortunately, the majority of the other CDSs were predicted to be uncharacterized hypothetical proteins (Supplementary Table 1). A reciprocal search for orthologs was then performed in which a search for orthologs found in any one of the isolates categorized as non-virulent (ISU2414, ISU2812, ISU2912, or SRD478) and not found in any of the isolates categorized as highly virulent (P1/7, ISU2614, and ISU1606). This analysis identified 1,425 CDSs including 13 predicted DNA binding proteins and 63 predicted transcriptional regulators (Supplementary Table 1). The much larger number of CDSs identified from this search is likely reflective of the larger genome size of isolates ISU2812 and ISU2912. While this analysis failed to pinpoint any specific CDSs that likely function as potential virulence factors, it does provide a reliable one-to-one assignment of specific genes of interest that could prove useful in future allelic replacement and/or functional genomic studies.
Conclusion
In summary, a spectrum of virulence phenotypes was observed among the nine United States S. suis isolates following intranasal challenge in pigs. Comparative genomic analysis of the genomes revealed a high degree of similarity among isolates P1/7, ISU1606, and ISU2714 from a variety of different assessments, while extensive genetic variation was observed among the other isolates. Genomic analysis revealed a bacteriocin locus encoded within a relatively small mobile plasmid, as well as numerous other chromosomal MGEs within these S. suis genomes, some of which contained AMR genes. While virulence mechanisms utilized by S. suis are not fully understood, a variety of different functional characteristics of S. suis isolates have been hypothesized to serve a role in virulence. In this study, no correlation between the characteristics evaluated here, such as genome size, serotype, ST, in vitro virulence-associated phenotypes and the capacity to cause disease in swine was observed. Focusing in on the in vitro virulence-associated phenotypes, results from cell-surface hydrophobicity, biofilm capacity, and the ability to survive in both whole-blood and serum, suggest that any differences or functional characteristics between S. suis capsules does not correlate with their respective capacity to cause disease in swine. It is important to note that while no correlation between the capacity to cause disease in swine and the presence of genes encoding known virulence factors was observed, the differential expression of any one or a combination of the genes encoding known virulence factors could strongly influence the capacity of an isolate to cause disease in swine. Despite not observing a correlation between the capacity to cause disease in swine and the presence of genes encoding known virulence factors, numerous CDSs were identified from the search for orthologs present in any one of the three isolates categorized as highly virulent and not found in any of the isolates categorized as non-virulent as well as the reciprocal ortholog search. Our hope is that the assembly and annotation of these genomes, coupled with the comparative genomic analyses reported in this study provides a framework for future allelic replacement and/or functional genomic studies investigating genetic characteristics that underlie and influence the phenotypic differences among these isolates.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal studies were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal experiments were approved by the USDA-National Animal Disease Center’s Institutional Animal Care and Use Committee (protocol #2724).
Author Contributions
TN conceived and designed the experiment. TN, UW, SH, SB, and SS performed the experiments. UW, TA, SZ, IG, ME, SH, SB, DB, and SS analyzed the data. TN, UW, TA, ME, SH, SB, DB, and SS contributed reagents, materials, and analysis tools and wrote the manuscript. All authors gave approval of the final version to be published and agreed to be accountable for all aspects of the work.
Funding
The funding was provided by the United States Department of Agriculture, Agriculture Research Service (Project #5030-32000-119-00-D) and by the National Pork Board (Project #17-084). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. UW and SH were supported in part by an appointment to the USDA-ARS Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the United States Department of Energy (DOE) and USDA under contract number DE-AC05-06OR23100. This research used resources provided by the SCINet project of the USDA, ARS. The contributions of SZ, IG, and ME were funded by the National Institute of Allergy and Infectious Diseases (NIAID) (SC2AI120941) and the United States Department of Homeland Security (2014-ST-062-000058) to ME. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA, DOE, or ORISE. USDA is an equal opportunity provider and employer.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.620843/full#supplementary-material
Supplementary Figure 1 | Map of plasmids detected in S. suis isolates. Plasmid name, accession number, and size (bp) is indicated for each plasmid (center). Arrows indicate orientation of annotated CDSs; blue arrows represent CDSs with predicted functions based on sequence homology along with indicated names. Gray arrows represent predicted CDSs of unknown function. (A) pISU2614. (B) pISU2514. (C) pISU2812. (D) pSRD487_1. (E) pSRD487_2. (F) pSRD487_3. (G) pSRD487_4.
Supplementary Figure 2 | Comparison of capsule loci. Colored arrows represent the genes present within the CPS locus of the representative serotypes and S. suis isolates (left). Predicted function is indicated by the arrow color, as shown at the bottom. ISU2614 was chosen as a representative of the serotype 2 strains, ISU2812 is serotype 19, ISU2912 is serotype NCL1-3 and the serotype of SRD478 is undefined. Specific regions of greatest nucleotide sequence divergence observed within a specific serotype is indicated by brackets and letters.
Footnotes
- ^ https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/
- ^ https://github.com/JCVenterInstitute/PanACEA
- ^ http://www.genomicepidemiology.org/
- ^ https://card.mcmaster.ca/home
References
Afgan, E., Baker, D., Batut, B., Van Den Beek, M., Bouvier, D., Cech, M., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544.
Agniswamy, J., Lei, B., Musser, J. M., and Sun, P. D. (2004). Insight of host immune evasion mediated by two variants of group a Streptococcus Mac protein. J. Biol. Chem. 279, 52789–52796. doi: 10.1074/jbc.m410698200
Agniswamy, J., Nagiec, M. J., Liu, M., Schuck, P., Musser, J. M., and Sun, P. D. (2006). Crystal structure of group A Streptococcus Mac-1: insight into dimer-mediated specificity for recognition of human IgG. Structure 14, 225–235. doi: 10.1016/j.str.2005.10.012
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21.
Auger, J. P., Dolbec, D., Roy, D., Segura, M., and Gottschalk, M. (2018). Role of the Streptococcus suis serotype 2 capsular polysaccharide in the interactions with dendritic cells is strain-dependent but remains critical for virulence. PLoS One 13:e0200453. doi: 10.1371/journal.pone.0200453
Baums, C. G., Kaim, U., Fulde, M., Ramachandran, G., Goethe, R., and Valentin-Weigand, P. (2006). Identification of a novel virulence determinant with serum opacification activity in Streptococcus suis. Infect. Immun. 74, 6154–6162. doi: 10.1128/iai.00359-06
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35.
Bonifait, L., Gottschalk, M., and Grenier, D. (2010). Cell surface characteristics of nontypeable isolates of Streptococcus suis. FEMS Microbiol. Lett. 311, 160–166. doi: 10.1111/j.1574-6968.2010.02086.x
Charland, N., Harel, J., Kobisch, M., Lacasse, S., and Gottschalk, M. (1998). Streptococcus suis serotype 2 mutants deficient in capsular expression. Microbiology 144(Pt 2), 325–332. doi: 10.1099/00221287-144-2-325
Clarke, T. H., Brinkac, L. M., Inman, J. M., Sutton, G., and Fouts, D. E. (2018). PanACEA: a bioinformatics tool for the exploration and visualization of bacterial pan-chromosomes. BMC Bioinformatics 19:246.
Clinical and Laboratory Standards Institute (2013). Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals, Approved Standard, 4th Edn. Wayne, PA: Clinical and Laboratory Standards Institute. CLSI document VET01-A4.
Clinical and Laboratory Standards Institute (2015). Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals, Approved Standard, 3th Edn. Wayne, PA: Clinical and Laboratory Standards Institute. CLSI supplement VET01S.
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
De Greeff, A., Buys, H., Van Alphen, L., and Smith, H. E. (2002). Response regulator important in pathogenesis of Streptococcus suis serotype 2. Microb. Pathog. 33, 185–192. doi: 10.1016/s0882-4010(02)90526-7
De Greeff, A., Wisselink, H. J., De Bree, F. M., Schultsz, C., Baums, C. G., Thi, H. N., et al. (2011). Genetic diversity of Streptococcus suis isolates as determined by comparative genome hybridization. BMC Microbiol. 11:161. doi: 10.1186/1471-2180-11-161
Dobson, A., Cotter, P. D., Ross, R. P., and Hill, C. (2012). Bacteriocin production: a probiotic trait? Appl. Environ. Microbiol. 78, 1–6. doi: 10.1128/aem.05576-11
Dutkiewicz, J., Zajac, V., Sroka, J., Wasinski, B., Cisak, E., Sawczyn, A., et al. (2018). Streptococcus suis: a re-emerging pathogen associated with occupational exposure to pigs or pork products. Part II - Pathogenesis. Ann. Agric. Environ. Med. 25, 186–203. doi: 10.26444/aaem/85651
Estrada, A. A., Gottschalk, M., Rossow, S., Rendahl, A., Gebhart, C., and Marthaler, D. G. (2019). Serotype and genotype (multilocus sequence type) of Streptococcus suis isolates from the United States serve as predictors of pathotype. J. Clin. Microbiol. 57:e00377-19.
Faulds-Pain, A., and Wren, B. W. (2013). Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in clostridium difficile and Streptococcus suis. Appl. Environ. Microbiol. 79, 4768–4771. doi: 10.1128/aem.01195-13
Feng, Y., Zheng, F., Pan, X., Sun, W., Wang, C., Dong, Y., et al. (2007). Existence and characterization of allelic variants of Sao, a newly identified surface protein from Streptococcus suis. FEMS Microbiol. Lett. 275, 80–88. doi: 10.1111/j.1574-6968.2007.00859.x
Feng, Y., Zhang, H., Wu, Z., Wang, S., Cao, M., Hu, D., et al. (2014). Streptococcus suis infection: an emerging/reemerging challenge of bacterial infectious diseases? Virulence 5, 477–497. doi: 10.4161/viru.28595
Ferrando, M. L., Van Baarlen, P., Orru, G., Piga, R., Bongers, R. S., Wels, M., et al. (2014). Carbohydrate availability regulates virulence gene expression in Streptococcus suis. PLoS One 9:e89334. doi: 10.1371/journal.pone.0089334
Ferrando, M. L., Willemse, N., Zaccaria, E., Pannekoek, Y., Van Der Ende, A., and Schultsz, C. (2017). Streptococcal Adhesin P (SadP) contributes to Streptococcus suis adhesion to the human intestinal epithelium. PLoS One 12:e0175639. doi: 10.1371/journal.pone.0175639
Fittipaldi, N., Segura, M., Grenier, D., and Gottschalk, M. (2012). Virulence factors involved in the pathogenesis of the infection caused by the swine pathogen and zoonotic agent Streptococcus suis. Future Microbiol. 7, 259–279. doi: 10.2217/fmb.11.149
Hanage, W. P., Fraser, C., Tang, J., Connor, T. R., and Corander, J. (2009). Hyper-recombination, diversity, and antibiotic resistance in pneumococcus. Science 324, 1454–1457. doi: 10.1126/science.1171908
Hau, S. J., Bayles, D. O., Alt, D. P., Brockmeier, S. L., Frana, T. S., and Nicholson, T. L. (2015). Draft genome sequences of nine Streptococcus suis strains isolated in the United States. Genome Announc. 3:e01301-15.
Holden, M. T., Hauser, H., Sanders, M., Ngo, T. H., Cherevach, I., Cronin, A., et al. (2009). Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS One 4:e6072.
Hsueh, K. J., Cheng, L. T., Lee, J. W., Chung, Y. C., Chung, W. B., and Chu, C. Y. (2017). Immunization with Streptococcus suis bacterin plus recombinant Sao protein in sows conveys passive immunity to their piglets. BMC Vet. Res. 13:15.
Hsueh, K. J., Lee, J. W., Hou, S. M., Chen, H. S., Chang, T. C., and Chu, C. Y. (2014). Evaluation on a Streptococcus suis vaccine using recombinant sao-l protein manufactured by bioreactors as the antigen in pigs. Transbound Emerg. Dis. 61, e35–e43.
Huang, J., Chen, L., Li, D., Wang, M., Du, F., Gao, Y., et al. (2018). Emergence of a vanG-carrying and multidrug resistant ICE in zoonotic pathogen Streptococccus suis. Vet. Microbiol. 222, 109–113. doi: 10.1016/j.vetmic.2018.07.008
Huang, J., Ma, J., Shang, K., Hu, X., Liang, Y., Li, D., et al. (2016). Evolution and diversity of the antimicrobial resistance associated mobilome in Streptococcus suis: a probable mobile genetic elements reservoir for other Streptococci. Front. Cell Infect. Microbiol. 6:118.
Huang, K., Song, Y., Zhang, Q., Zhang, A., and Jin, M. (2016). Characterisation of a novel integrative and conjugative element ICESsD9 carrying erm(B) and tet(O) resistance determinants in Streptococcus suis, and the distribution of ICESsD9-like elements in clinical isolates. J. Glob. Antimicrob. Resist. 7, 13–18. doi: 10.1016/j.jgar.2016.05.008
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kiedrowski, M. R., Kavanaugh, J. S., Malone, C. L., Mootz, J. M., Voyich, J. M., Smeltzer, M. S., et al. (2011). Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS One 6:e26714. doi: 10.1371/journal.pone.0026714
King, S. J., Leigh, J. A., Heath, P. J., Luque, I., Tarradas, C., Dowson, C. G., et al. (2002). Development of a multilocus sequence typing scheme for the pig pathogen Streptococcus suis: identification of virulent clones and potential capsular serotype exchange. J. Clin. Microbiol. 40, 3671–3680. doi: 10.1128/jcm.40.10.3671-3680.2002
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kouki, A., Haataja, S., Loimaranta, V., Pulliainen, A. T., Nilsson, U. J., and Finne, J. (2011). Identification of a novel streptococcal adhesin P (SadP) protein recognizing galactosyl-alpha1-4-galactose-containing glycoconjugates: convergent evolution of bacterial pathogens to binding of the same host receptor. J. Biol. Chem. 286, 38854–38864. doi: 10.1074/jbc.m111.260992
Li, M., Shen, X., Yan, J., Han, H., Zheng, B., Liu, D., et al. (2011). GI-type T4SS-mediated horizontal transfer of the 89K pathogenicity island in epidemic Streptococcus suis serotype 2. Mol. Microbiol. 79, 1670–1683. doi: 10.1111/j.1365-2958.2011.07553.x
Li, Q., Fu, Y., Ma, C., He, Y., Yu, Y., Du, D., et al. (2017). The non-conserved region of MRP is involved in the virulence of Streptococcus suis serotype 2. Virulence 8, 1274–1289. doi: 10.1080/21505594.2017.1313373
Li, Y., Gottschalk, M., Esgleas, M., Lacouture, S., Dubreuil, J. D., Willson, P., et al. (2007). Immunization with recombinant Sao protein confers protection against Streptococcus suis infection. Clin. Vaccine Immunol. 14, 937–943. doi: 10.1128/cvi.00046-07
Liu, Z., Zheng, H., Gottschalk, M., Bai, X., Lan, R., Ji, S., et al. (2013). Development of multiplex PCR assays for the identification of the 33 serotypes of Streptococcus suis. PLoS One 8:e72070. doi: 10.1371/journal.pone.0072070
Maneerat, K., Yongkiettrakul, S., Kramomtong, I., Tongtawe, P., Tapchaisri, P., Luangsuk, P., et al. (2013). Virulence genes and genetic diversity of Streptococcus suis serotype 2 isolates from Thailand. Transbound Emerg. Dis. 60, (Suppl. 2), 69–79.
Mostowy, R., Croucher, N. J., Hanage, W. P., Harris, S. R., Bentley, S., and Fraser, C. (2014). Heterogeneity in the frequency and characteristics of homologous recombination in pneumococcal evolution. PLoS Genet. 10:e1004300. doi: 10.1371/journal.pgen.1004300
Nicholson, T. L., Shore, S. M., Smith, T. C., and Frana, T. S. (2013). Livestock-associated methicillin-resistant Staphylococcus aureus (LA-MRSA) isolates of swine origin form robust biofilms. PLoS One 8:e73376. doi: 10.1371/journal.pone.0073376
O’Connor, P. M., Kuniyoshi, T. M., Oliveira, R. P., Hill, C., Ross, R. P., and Cotter, P. D. (2020). Antimicrobials for food and feed; a bacteriocin perspective. Curr. Opin. Biotechnol. 61, 160–167.
Onishi, H., Sugawara, M., Okura, M., Osaki, M., and Takamatsu, D. (2012). Prevalence of Streptococcus suis genotypes in isolates from porcine endocarditis in East Japan. J. Vet. Med. Sci. 74, 1681–1684. doi: 10.1292/jvms.12-0301
Palmieri, C., Magi, G., Mingoia, M., Bagnarelli, P., Ripa, S., Varaldo, P. E., et al. (2012). Characterization of a Streptococcus suis tet(O/W/32/O)-carrying element transferable to major streptococcal pathogens. Antimicrob. Agents Chemother. 56, 4697–4702. doi: 10.1128/aac.00629-12
Palmieri, C., Princivalli, M. S., Brenciani, A., Varaldo, P. E., and Facinelli, B. (2011a). Different genetic elements carrying the tet(W) gene in two human clinical isolates of Streptococcus suis. Antimicrob. Agents Chemother. 55, 631–636. doi: 10.1128/aac.00965-10
Palmieri, C., Varaldo, P. E., and Facinelli, B. (2011b). Streptococcus suis, an emerging drug-resistant animal and human pathogen. Front. Microbiol. 2:235.
Prufer, T. L., Rohde, J., Verspohl, J., Rohde, M., De Greeff, A., Willenborg, J., et al. (2019). Molecular typing of Streptococcus suis strains isolated from diseased and healthy pigs between 1996-2016. PLoS One 14:e0210801. doi: 10.1371/journal.pone.0210801
Qiu, X., Bai, X., Lan, R., Zheng, H., and Xu, J. (2016). Novel capsular polysaccharide loci and new diagnostic tools for high-throughput capsular gene typing in Streptococcus suis. Appl. Environ. Microbiol. 82, 7102–7112. doi: 10.1128/aem.02102-16
Rehm, T., Baums, C. G., Strommenger, B., Beyerbach, M., Valentin-Weigand, P., and Goethe, R. (2007). Amplified fragment length polymorphism of Streptococcus suis strains correlates with their profile of virulence-associated genes and clinical background. J. Med. Microbiol. 56, 102–109. doi: 10.1099/jmm.0.46616-0
Rosenberg, M., Gutnick, D., and Rosenberg, E. (1980). Adherence of bacteria to hydrocarbons: a simple method for measuring cell-surface hydrophobicity. FEMS Microbiol. Lett. 9, 29–33. doi: 10.1111/j.1574-6968.1980.tb05599.x
Roy, D., Auger, J. P., Segura, M., Fittipaldi, N., Takamatsu, D., Okura, M., et al. (2015). Role of the capsular polysaccharide as a virulence factor for Streptococcus suis serotype 14. Can. J. Vet. Res. 79, 141–146.
Roy, D., Fittipaldi, N., Dumesnil, A., Lacouture, S., and Gottschalk, M. (2014). The protective protein Sao (surface antigen one) is not a critical virulence factor for Streptococcus suis serotype 2. Microb. Pathog. 67-68, 31–35. doi: 10.1016/j.micpath.2014.02.002
Saeed, A. I., Bhagabati, N. K., Braisted, J. C., Liang, W., Sharov, V., Howe, E. A., et al. (2006). TM4 microarray software suite. Methods Enzymol. 411, 134–193. doi: 10.1016/s0076-6879(06)11009-5
Seele, J., Singpiel, A., Spoerry, C., Von Pawel-Rammingen, U., Valentin-Weigand, P., and Baums, C. G. (2013). Identification of a novel host-specific IgM protease in Streptococcus suis. J. Bacteriol. 195, 930–940. doi: 10.1128/jb.01875-12
Segura, M., Fittipaldi, N., Calzas, C., and Gottschalk, M. (2017). Critical Streptococcus suis virulence factors: are they all really critical? Trends Microbiol. 25, 585–599. doi: 10.1016/j.tim.2017.02.005
Segura, M., Zheng, H., De Greeff, A., Gao, G. F., Grenier, D., Jiang, Y., et al. (2014a). Latest developments on Streptococcus suis: an emerging zoonotic pathogen: part 1. Future Microbiol. 9, 441–444.
Segura, M., Zheng, H., De Greeff, A., Gao, G. F., Grenier, D., Jiang, Y., et al. (2014b). Latest developments on Streptococcus suis: an emerging zoonotic pathogen: part 2. Future Microbiol. 9, 587–591.
Silva, L. M., Baums, C. G., Rehm, T., Wisselink, H. J., Goethe, R., and Valentin-Weigand, P. (2006). Virulence-associated gene profiling of Streptococcus suis isolates by PCR. Vet. Microbiol. 115, 117–127. doi: 10.1016/j.vetmic.2005.12.013
Soto-Jimenez, L. M., Estrada, K., and Sanchez-Flores, A. (2014). GARM: genome assembly, reconciliation and merging pipeline. Curr. Top. Med. Chem. 14, 418–424. doi: 10.2174/1568026613666131204110628
Sun, Y., Veseli, I. A., Vaillancourt, K., Frenette, M., Grenier, D., and Pombert, J. F. (2019). The bacteriocin from the prophylactic candidate Streptococcus suis 90-1330 is widely distributed across S. suis isolates and appears encoded in an integrative and conjugative element. PLoS One 14:e0216002. doi: 10.1371/journal.pone.0216002
Takamatsu, D., Osaki, M., and Sekizaki, T. (2003). Chloramphenicol resistance transposable element TnSs1 of Streptococcus suis, a transposon flanked by IS6-family elements. Plasmid 49, 143–151. doi: 10.1016/s0147-619x(02)00149-x
Takamatsu, D., Osaki, M., Tharavichitkul, P., Takai, S., and Sekizaki, T. (2008). Allelic variation and prevalence of serum opacity factor among the Streptococcus suis population. J. Med. Microbiol. 57, 488–494. doi: 10.1099/jmm.0.47755-0
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vecht, U., Wisselink, H. J., Jellema, M. L., and Smith, H. E. (1991). Identification of two proteins associated with virulence of Streptococcus suis type 2. Infect. Immun. 59, 3156–3162. doi: 10.1128/iai.59.9.3156-3162.1991
Vecht, U., Wisselink, H. J., Stockhofe-Zurwieden, N., and Smith, H. E. (1996). Characterization of virulence of the Streptococcus suis serotype 2 reference strain Henrichsen S 735 in newborn gnotobiotic pigs. Vet. Microbiol. 51, 125–136. doi: 10.1016/0378-1135(96)00028-4
Wang, Y., Zhang, W., Wu, Z., and Lu, C. (2011). Reduced virulence is an important characteristic of biofilm infection of Streptococcus suis. FEMS Microbiol. Lett. 316, 36–43. doi: 10.1111/j.1574-6968.2010.02189.x
Wei, Z., Li, R., Zhang, A., He, H., Hua, Y., Xia, J., et al. (2009). Characterization of Streptococcus suis isolates from the diseased pigs in China between 2003 and 2007. Vet. Microbiol. 137, 196–201. doi: 10.1016/j.vetmic.2008.12.015
Weinert, L. A., Chaudhuri, R. R., Wang, J., Peters, S. E., Corander, J., Jombart, T., et al. (2015). Genomic signatures of human and animal disease in the zoonotic pathogen Streptococcus suis. Nat. Commun. 6:6740.
Wenig, K., Chatwell, L., Von Pawel-Rammingen, U., Bjorck, L., Huber, R., and Sondermann, P. (2004). Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc. Natl. Acad. Sci. U.S.A. 101, 17371–17376. doi: 10.1073/pnas.0407965101
Wertheim, H. F., Nghia, H. D., Taylor, W., and Schultsz, C. (2009a). Streptococcus suis: an emerging human pathogen. Clin. Infect. Dis. 48, 617–625.
Wertheim, H. F., Nguyen, H. N., Taylor, W., Lien, T. T., Ngo, H. T., Nguyen, T. Q., et al. (2009b). Streptococcus suis, an important cause of adult bacterial meningitis in northern Vietnam. PLoS One 4:e5973. doi: 10.1371/journal.pone.0005973
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wileman, T. M., Weinert, L. A., Howell, K. J., Wang, J., Peters, S. E., Williamson, S. M., et al. (2019). Pathotyping the zoonotic pathogen Streptococcus suis: novel genetic markers to differentiate invasive disease-associated isolates from non-disease-associated isolates from england and wales. J. Clin. Microbiol. 57:e01712-18.
Wu, T., Chang, H., Tan, C., Bei, W., and Chen, H. (2009). The orphan response regulator RevSC21 controls the attachment of Streptococcus suis serotype-2 to human laryngeal epithelial cells and the expression of virulence genes. FEMS Microbiol. Lett. 292, 170–181. doi: 10.1111/j.1574-6968.2008.01486.x
Yamaguchi, Y., and Inouye, M. (2011). Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 9, 779–790. doi: 10.1038/nrmicro2651
Yamaguchi, Y., Park, J. H., and Inouye, M. (2011). Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61–79. doi: 10.1146/annurev-genet-110410-132412
Zhang, A., Xie, C., Chen, H., and Jin, M. (2008). Identification of immunogenic cell wall-associated proteins of Streptococcus suis serotype 2. Proteomics 8, 3506–3515. doi: 10.1002/pmic.200800007
Zhang, A., Yang, M., Hu, P., Wu, J., Chen, B., Hua, Y., et al. (2011). Comparative genomic analysis of Streptococcus suis reveals significant genomic diversity among different serotypes. BMC Genomics 12:523.
Zhao, J., Pan, S., Lin, L., Fu, L., Yang, C., Xu, Z., et al. (2015). Streptococcus suis serotype 2 strains can induce the formation of neutrophil extracellular traps and evade trapping. FEMS Microbiol. Lett. 362:fnv022.
Zhao, Y., Liu, G., Li, S., Wang, M., Song, J., Wang, J., et al. (2011). Role of a type IV-like secretion system of Streptococcus suis 2 in the development of streptococcal toxic shock syndrome. J. Infect. Dis. 204, 274–281. doi: 10.1093/infdis/jir261
Zheng, H., Ji, S., Lan, R., Liu, Z., Bai, X., Zhang, W., et al. (2014). Population analysis of Streptococcus suis isolates from slaughtered swine by use of minimum core genome sequence typing. J. Clin. Microbiol. 52, 3568–3572. doi: 10.1128/jcm.00536-14
Zheng, H., Ji, S., Liu, Z., Lan, R., Huang, Y., Bai, X., et al. (2015). Eight novel capsular polysaccharide synthesis gene loci identified in nontypeable Streptococcus suis isolates. Appl. Environ. Microbiol. 81, 4111–4119. doi: 10.1128/aem.00315-15
Zhong, X., Zhang, Y., Zhu, Y., Dong, W., Ma, J., Pan, Z., et al. (2018). The two-component signaling system VraSRss is critical for multidrug resistance and full virulence in Streptococcus suis serotype 2. Infect. Immun. 86:e00096-18.
Keywords: Streptococcus suis, whole-genome sequencing, comparative genomics, virulence, mobile genetic elements, antimicrobial resistance, swine
Citation: Nicholson TL, Waack U, Anderson TK, Bayles DO, Zaia SR, Goertz I, Eppinger M, Hau SJ, Brockmeier SL and Shore SM (2021) Comparative Virulence and Genomic Analysis of Streptococcus suis Isolates. Front. Microbiol. 11:620843. doi: 10.3389/fmicb.2020.620843
Received: 23 October 2020; Accepted: 21 December 2020;
Published: 26 January 2021.
Edited by:
Chih-Horng Kuo, Institute of Plant and Microbial Biology, Academia Sinica, TaiwanReviewed by:
Anusak Kerdsin, Kasetsart University, ThailandAstrid De Greeff, Wageningen University and Research, Netherlands
Sophie Payot, INRA Centre Nancy-Lorraine, France
Copyright © 2021 Nicholson, Waack, Anderson, Bayles, Zaia, Goertz, Eppinger, Hau, Brockmeier and Shore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy L. Nicholson, VHJhY3kuTmljaG9sc29uQHVzZGEuZ292; dHJhY3kubmljaG9sc29uQGFycy51c2RhLmdvdg==