 Shaohua Wen
Shaohua Wen Guoping Wang
Guoping Wang Zuokun Yang1
Zuokun Yang1 Ni Hong
Ni Hong- 1Key Lab of Plant Pathology of Hubei Province, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 2Key Laboratory of Horticultural Crop (Fruit Trees) Biology and Germplasm Creation of the Ministry of Agriculture, Wuhan, China
Kiwifruit (Actinidia spp.) is native to China. Viral disease–like symptoms are common on kiwifruit plants. In this study, six libraries prepared from total RNA of leaf samples from 69 kiwifruit plants were subjected to next-generation sequencing (NGS). Actinidia virus 1 (AcV-1), a tentative species in the family Closteroviridae, was discovered in the six libraries. Two full-length and two near-full genome sequences of AcV-1 variants were determined by Sanger sequencing. The genome structure of these Chinese AcV-1 variants was identical to that of isolate K75 and consisted of 12 open reading frames (ORFs). Analyses of these sequences together with the NGS-derived contig sequences revealed high molecular diversity in AcV-1 populations, with the highest sequence variation occurring at ORF1a, ORF2, and ORF3, and the available variants clustered into three phylogenetic clades. For the first time, our study revealed different domain compositions in the viral ORF1a and molecular recombination events among AcV-1 variants. Specific reverse transcriptase–polymerase chain reaction assays disclosed the presence of AcV-1 in plants of four kiwifruit species and unknown Actinidia spp. in seven provinces and one city.
Introduction
The family Closteroviridae contains many plant viruses causing different diseases of economically important crops (Martelli et al., 2012). Viruses in the family Closteroviridae are characterized by having 650–2,200 nm, flexuous filamentous virions consisting of a positive-sense, single-stranded (+ss) RNA with size ranging from 13 to 19.3 kb (Agranovsky et al., 1995; Rubio et al., 2013). According to the International Committee on Taxonomy of Viruses (ICTV) report posted on August 2018 (ICTV Master Species List, 2018 v1), the family Closteroviridae comprises more than 50 recognized species, which are classified into four genera, Closterovirus, Ampelovirus, Crinivirus, and Velarivirus, and seven unassigned members, based on their vectors and phylogenetic relationships (Karasev, 2000; Dolja et al., 2006). Members of the family Closteroviridae share a common genome organization, containing a replication-related module encoded by open reading frame (ORF) 1a and 1b, and a five-gene module encoding a small hydrophobic protein, a homolog of the plant heat shock protein HSP70 (HSP70h), a ∼60 kDa protein, a major coat protein (CP), and a minor coat protein (CPm) (Martelli et al., 2012). A notable exception is that viruses in the genus Ampelovirus have smaller genomes, which do not possess CPm and have unique ORFs. Previous studies showed that viruses in the family Closteroviridae have great genetic variability, and molecular evolution might be necessary for adaptation to new environments (Holland et al., 1982; Kreuze et al., 2002; Cuellar et al., 2008; Bertazzon et al., 2010; García-Arenal and Fraile, 2011; Esteves et al., 2012). Homologous recombination has been described for CP and P20 genes of citrus tristeza virus (CTV) (Rubio et al., 2001, 2013; Bar-Joseph and Mawassi, 2013), a member in the genus Closterovirus, for CP genes of ampeloviruses grapevine leafroll-associated virus 3 (GLRaV-3) (Turturo et al., 2005; Farooq et al., 2012), GLRaV-1 (Fan et al., 2015), and GLRaV-11 (Boulila, 2010), for HSP70h gene of plum bark necrosis stem pitting-associated virus (PBNSPaV) (Qu et al., 2014), and for ORF1a of GLRaV-4 (Adiputra et al., 2019).
Kiwifruit (Actinidia spp.) is an economically important fruit crop cultivated worldwide. Since the first report of apple stem grooving virus (ASGV) (Clover et al., 2003) naturally infecting kiwifruit, currently at least 19 viruses infecting kiwifruit have been reported (Pearson et al., 2011; Chavan et al., 2012, 2013; Blouin et al., 2013, 2018; Wang et al., 2016, 2020; Zheng et al., 2017; Veerakone et al., 2018; Wang D. et al., 2018; Wang Y. et al., 2018; Zhao et al., 2019, 2020). Among these viruses, Actinidia virus 1 (AcV-1), a tentative member of the family Closteroviridae, was first characterized from Actinidia chinensis in New Zealand (Blouin et al., 2018). The reported genome of AcV-1 isolate K75 (GenBank accession no. KX857665) consists of an 18,848 nt + ss RNA containing at least 12 ORFs. ORF1a encodes a multifunctional protein with two papain-like leader protease (L-Pro) domains (L1 and L2), one methyltransferase (Mtr) domain, and one helicase (Hel) domain. ORF 1b, expressed by a + 1 ribosomal frameshift, encodes an RNA-dependent RNA polymerase (RdRp). ORF2 and ORF3 encode two hypothetical proteins with unknown functions and predicted molecular masses of 13.6 and 25.4 kDa, respectively. ORFs 4–8 consist of a five-gene module, in which, instead of CPm in the module of most viruses in the family Closteroviridae, ORF7 encodes a 30 kDa protein (p30) with a conserved domain of thaumatin-like proteins. ORFs 9–11 encode three small proteins, whose functions are currently unknown. The virus was reported in China in 2018, the second country with AcV-1 infecting kiwifruit (Peng et al., 2018; Wen et al., 2018).
Until now, only one complete genome sequence of AcV-1 isolate K75 has been documented (Blouin et al., 2018), and a few sequences of the viral CP and HSP70h genes are available in GenBank. These sequences are valuable for designing molecular diagnostic methods. The relationship between the infection with the virus and occurrence of kiwifruit disease is still unknown, and the viral populations remain poorly understood. In this study, for the first time, the genome-wide genetic diversity of AcV-1 variants from kiwifruit plants grown in China was determined by using next-generation sequencing (NGS) combined with conventional Sanger sequencing for reverse transcriptase–polymerase chain reaction (RT-PCR) products. This study provides useful information for understanding the molecular evolution within AcV-1 populations and developing reliable molecular detection methods of the virus, which is necessary for epidemiological investigation of the viral disease.
Materials and Methods
Plant Materials
During the growing season of kiwifruit plants in 2016–2019, leaf samples of 245 kiwifruit plants, including 104 plants of A. chinensis, 39 Actinidia delicious plants, nine Actinidia arguta plants, 36 Actinidia eriantha plants, one Actinidia rufa plant, and 56 plants of unknown species, were collected in Hubei, Yunnan, Jiangxi, Shandong, Zhejiang, Henan, Fujian, and Shanxi provinces and Chongqing and Shanghai cities, covering the major kiwifruit production areas in China. Of these plants, 231 plants showed virus disease–like symptoms, including uneven chlorosis between veins, mosaic, ringspot, chlorotic spot, and/or deformation, whereas 14 plants were asymptomatic. Leaves from each plant were pooled as one sample named with geographic origin followed by a plant number. The presence of six known viruses AcV-1, Actinidia virus A (AcVA), Actinidia virus B (AcVB), ASGV, Actinidia chlorotic ringspot-associated emaravirus (AcCRaV), and citrus leaf blotch virus (CLBV), in these samples were tested by RT-PCR using specific primers reported previously (James, 1999; Harper et al., 2008; Blouin et al., 2012; Zheng et al., 2017; Supplementary Table S1). A leaf sample of a virus-free seedling of A. chinensis was used as a negative control.
According to the origins and symptoms of individually collected leaf samples as mentioned above, six samples were prepared for RNA-seq analyses (Supplementary Table S2). One leaf sample (ID: Z2) from an A. delicious plant showing severe chlorotic spots, leaf mottle, and deformation, grown at a field in Henan province, and one sample (ID: JX5) from an A. chinensis plant showing leaf yellowing from Jiangxi province were individually used for RNA-seq analyses. Additionally, in order to have a full view of kiwifruit virome, four pooled samples JS, ZZ, CQ, and JX were also subjected to RNA-seq analyses. The sample JS was a mixture of four leaf samples (JS27, JS29, JS30, and JS45) from Hubei province. The sample ZZ consisted of leaf samples from 30 kiwifruit plants (ZZ1–ZZ11, ZZ13–ZZ31) grown at a field in Henan province. The sample CQ consisted of leaf samples from 28 kiwifruit plants (CQ1–CQ28) grown at a field in Chongqing city. The sample JX consisted of leaf samples from five kiwifruit plants (JX1–JX4 and JX6) grown at a field in Jiangxi province. Most of these RNA-seq–analyzed samples (except for two asymptomatic samples JX6 and CQ18) showed viral disease–like symptoms as listed in Supplementary Table S4.
Additionally, one leaf sample (ID: WH4) from an A. arguta plant showing severe leaf mottle and deformation, grown at a field in Hubei province, was used for Sanger sequencing of AcV-1 genome.
RNA-Seq
Total RNAs were extracted from the six samples prepared for RNA-seq analyses as mentioned above, and ribosomal RNAs (rRNAs) were removed using an Epicenter Ribo-Zero rRNA removal kit (Epicenter, Madison, WI, United States). The rRNA-depleted RNA extracts were used for cDNA library construction with a TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA, United States) and sequenced on an Illumina HiSeq XTen sequencing machine with a paired-end 150 bp setup (Biomarker Biology Technology Ltd., Company, Beijing, China).
Raw reads were trimmed of adaptor sequences and filtered for low-quality reads with more than 5% ambiguous bases (Ns) or with more than 10% of the bases below Q20 quality as described previously (Liu et al., 2020). Qualified reads were de novo assembled into contigs using the CLC Genomics Workbench 11.0 (Qiagen, Valencia, CA, United States) platform. Contigs were subsequently screened for sequence identity against the NCBI databases1 using BlastX and BlastN programs.
Determination of Complete Genomic Sequences of AcV-1 Isolates
The complete genomic sequence of one AcV-1 variant (JS27) from sample JS27 and the near-full-length genomic sequence of one AcV-1 variant (Z2) from sample Z2 were individually determined by RT-PCR using primers (Supplementary Table S3) designed on the basis of the contig sequences derived from the RNA-seq and/or the sequence of the reported isolate K75. The complete genomic sequence of AcV-1 variant WH4-2 and the near-full-length genomic sequence of AcV-1 variant WH4-1 from sample WH4 were individually determined by RT-PCR using primers (Supplementary Table S3) designed on the basis of available AcV-1 sequences. Most of the adjacent fragments overlapped by more than 100 bp. The 5′ and 3′ terminal sequences of the viral genomic RNA were determined by a RACE strategy using a commercial kit (Invitrogen GeneRacer Kit, United States) according to the manufacturer’s instructions and specific reverse primers (Supplementary Table S3). RT-PCR solutions and conditions were like those reported previously (Zheng et al., 2017), except that annealing temperature and extension time varied, depending on the primer sets used in each reaction and the sizes of PCR products. Sequences of RT-PCR fragments from each sample were assembled into a continuous sequence using DNAMAN6.0 (Lynnon, Quebec, Canada).
RT-PCR Detection of AcV-1
The primer set cp-F/cp-R (5′-TGAGCTRGGRATAGATGTTGC-3′/5′-TCTCTCAGGGTTMGGATGAGT-3′) (Wen et al., 2018) designed on the basis of the conserved sequence of the CP gene (positions 16,607–16,627 nt/16,962–16,982 nt of isolate K75 genome) of AcV-1 was used for the specific detection of AcV-1 in kiwifruit leaf samples. Total RNA was extracted from the collected leaf materials (each about 0.5 g) using a CTAB method (Fan et al., 2015). The RNA was diluted in 40 μL RNase-free water. The 20-μL cDNA synthesis solution contained 9 μL RNA template, 1 μL p(N)6 random primer, 0.5 μL Moloney murine leukemia virus (M-MLV) reverse transcriptase, 0.5 μL recombinant RNase inhibitor (Takara, Dalian, China), 4 μL 5 × M-MLV buffer, 1 μL 2.5 mM dNTP, and 4 μL RNase-free water. A 25 μL PCR reaction solution contained 2.0 μL of the cDNA template, 2.5 μL of 10 × reaction buffer, 1.5 μL of 2.5 mM dNTP, 0.5 μL each of 10 mM primers, 0.25 μL rTaq (5 U/μl) (TakaRa, Dalian, China), and 17.75 μL ddH2O. The cycling parameters were set as follows: denaturation at 95°C for 5 min, followed by 35 cycles of denaturation (30 s at 95°C), annealing (30 s at 54°C), and extension (45 s at 72°C). The PCR products were separated on 1.2% agarose gel, stained with ethidium bromide, and visualized under UV light.
Cloning and Sequencing of RT-PCR Products
PCR products were gel purified, ligated into the pMD18-T vector (Takara, Dalian, China), and subsequently transformed into cells of Escherichia coli strain Top10. Positive clones were identified by PCR using the E. coli cultures as templates. At least three positive clones of each PCR product were sequenced at Shanghai Sangon Biological Engineering & Technology and Service Co., Ltd., Shanghai, China.
Sequence Analyses
ORFs were predicted using the Open Reading Frame Finder at https://www.ncbi.nlm.nih.gov/orffinder/. Conserved domains in predicted proteins were identified by using the Conserved Domain Database (CDD)2 websites in NCBI. Multiple alignments and identity analyses of nucleotide and amino acid sequences were performed using the ClustalW2 program3. Phylogenetic trees were constructed using the neighbor-joining method with 1,000 bootstrap in MEGA 7.0.
Recombination Analysis
Genomic sequences of AcV-1 variants were examined for potential recombination events using the Recombination Detection Program (RDP) version RDP4.94 with default parameter settings (Martin et al., 2015). The RDP software includes the RDP, GENECONV, BOOTSCAN, MaxChi, Chimaera, and SISCAN methods. AcV-1 genomic sequences were aligned in MEGA v.7.0.1 software and exported to the RDP program to generate evidence of recombination. At a Bonferroni-corrected p-value cutoff ≤ 0.05, recombinant sites detected by four or more algorithms in the RDP were considered as recombination events.
Results
Viruses Identified by RNA-Seq Analysis
In total, 67,640,546, 92,477,928, 73,387,306, 82,730,664, 79,782,996, and 73,419,336 clean reads were obtained and 177,848, 315,308, 165,312, 246,566, 221,290, and 174,235 unique contigs were generated from RNA-seq samples JS, ZZ, Z2, CQ, JX, and JX5, respectively (Supplementary Table S2). BlastX searches against the NCBI NR database revealed that 254, 154, 137, 620, 73, and 18 contigs (accounting for 0.01–0.25% of total contigs) from the six samples respectively matched with viral proteins (Supplementary Table S2). AcV-1 was recovered from all these RNA-seq samples, and 121 contigs were assigned to this virus, accounting for 9.63% of the total viral contigs (Table 1). Meanwhile, five other well-documented kiwifruit viruses AcCRaV, AcVA, AcVB, CLBV, and Actinidia seed-borne latent virus (ASbLV) (Veerakone et al., 2018) were identified in sample JS. Four other viruses, including ASGV, Actinidia emaravirus 2 (AcEV-2) (Wang et al., 2020), and two unreported kiwifruit-infecting viruses, belonging to families Betaflexiviridae and Secoviridae, were identified in samples ZZ and Z2. In sample CQ, eight viruses, including AcV-1, AcCRaV, ASbLV, AcVA, AcVB, CLBV, ASGV, and an unreported virus in the family Secoviridae, were identified. In sample JX, five known viruses, including AcV-1, ASbLV, AcVA, AcVB, and CLBV, were identified. In sample JX5, only one known virus AcV-1 was identified. Additionally, a virus potentially belonging to the family Bromoviridae was identified in samples JX and JX5. Here, only AcV-1 sequences were considered for further analyses.
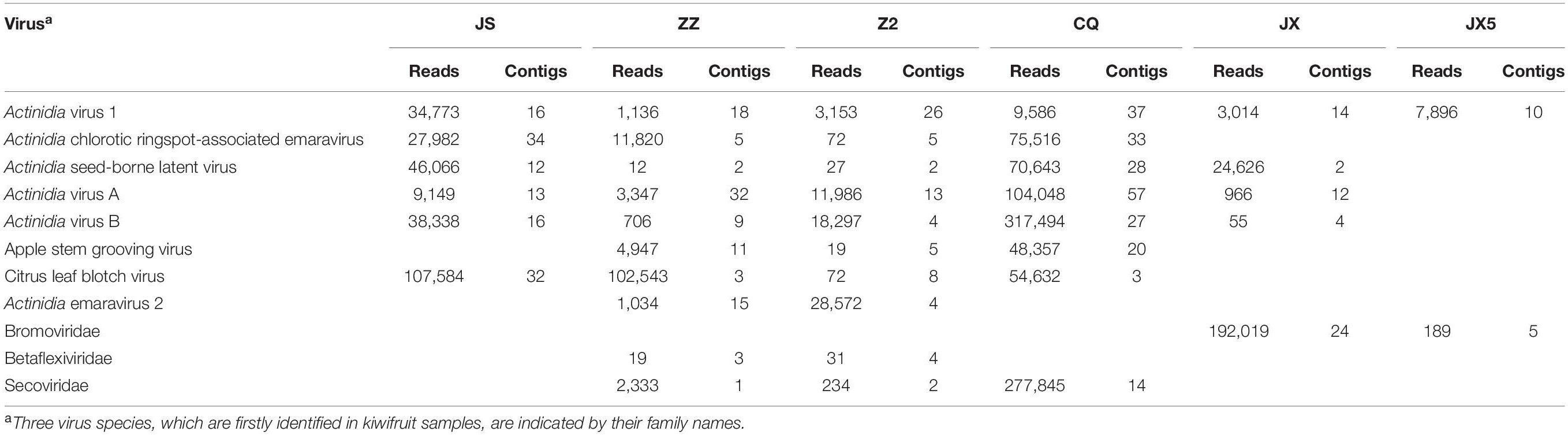
Table 1 | Viruses and their sequences identified from six kiwifruit samples by using RNA-seq analysis.
AcV-1 Sequences Identified by RNA-Seq Analysis
From sample JS, 16 contigs matched the genomic sequence of AcV-1. One contig of 18,779 nt was near the full length of AcV-1 genome sequence, while the remaining contigs were 395–1,467 nt long. These contigs shared 58.0–90.0% nt sequence identity with corresponding sequences of isolate K75 (Figure 1). Comparison of these short contigs and the 18,779 nt contig revealed 59.6–92.9% nt sequence identity with each other.
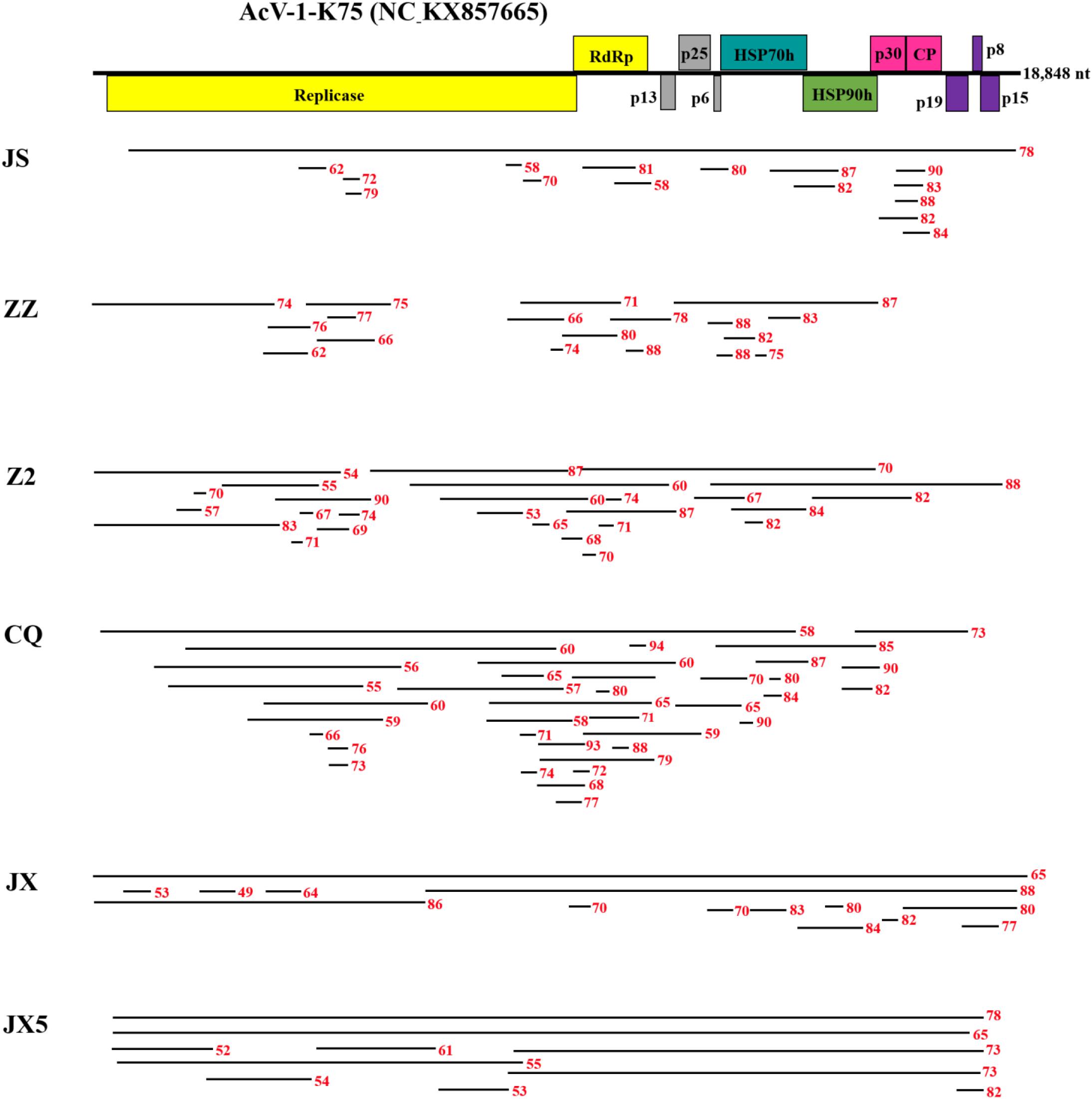
Figure 1 | Contigs with significant nucleotide sequence identity to Actinidia virus 1 (AcV-1) isolate K75. A schematic diagram of the genome organization of AcV-1 isolate K75 was used as reference showing the contig positions. Six samples JS, ZZ, Z2, CQ, JX, and JX5 were named with the abbreviation of their geographic origins. Black lines represent AcV-1 contigs and their position at the genome of isolate K75. Red numbers indicate the nucleotide identity between each contig and the corresponding sequence of AcV-1 isolate K75.
From sample ZZ, 18 AcV-1 contigs with sizes ranging from 224 to 4,132 nt covered 71.5% of the viral genome and displayed 62.1–88.2% nt sequence identity with corresponding sequences of AcV-1 isolate K75 (Figure 1).
From sample Z2, 26 contigs ranging from 248 to 6,041 nt in length matched the genomic sequence of AcV-1, with a total coverage of 97.9%. These contig sequences shared 53.1–88.2% nt identity with corresponding sequences of isolate K75 (Figure 1).
From sample CQ, 37 contigs matched the genomic sequence of AcV-1. One contig (named CQ-c13707) was 13,707 nt in length and covered the ORF1a-ORF4 (position at 248–14,118 nt) of the viral genomic sequence. The remaining contigs were 212–7,590 nt in length. These contigs shared 55.2–94.3% nt sequence identity with corresponding sequences of isolate K75 (Figure 1).
From sample JX, 14 contigs matched the genomic sequence of AcV-1. One large contig (named JX-c18869) had a size of 18,869 nt, nearly covering the full-length of AcV-1 genome sequence, and another contig (named JX-c12007) was 12,007 nt (position at 6,772–18,873 nt) covering about 64% of the viral genomic sequence. The remaining contigs were 311–6,776 nt in length. All these contigs shared 49.0–88.3% nt sequence identity with corresponding sequences of isolate K75 (Figure 1).
From sample JX5, 10 contigs matched the genomic sequence of AcV-1. Two contigs (named JX5-c18818 and JX5-c18410) were 18,818 and 18,410 nt in length, respectively, and covered nearly the complete genome of AcV-1. Meanwhile, another two contigs (named JX5-c10087 and JX5-c10084) matched approximately in the same position (8,710–18,800 nt) of the viral genome. The four contigs shared 63.3–86.4% nt sequence identity with each other and 65.2–78.1% nt sequence identity with corresponding sequences of isolate K75. The remaining six contigs were 667–8,857 nt and shared 54.4–78.3% nt sequence identity with corresponding sequences of AcV-1 isolate K75 (Figure 1).
For the following analyses, each contig derived from RNA-seq analyses was named as a sample name followed by the contig length, and each sequence was considered as a molecular variant.
Genomic Characteristics and Sequence Diversity of Chinese AcV-1 Variants
A complete genomic sequence (JS27, 18,896 nt, GenBank accession no. MT936297) of AcV-1 from the sample JS27 was reconstructed by Sanger sequencing of RT-PCR and 3′ and 5′ RACE products using primers designed on the basis of sequence of a large contig (18,779 nt) derived from RNA-seq. In addition, a near-full-length genomic sequence (Z2, 18,685 nt, excluding about 300 nt at the 3′ terminus) (GenBank accession no. MT936306) of AcV-1 infecting an A. delicious plant (ID: Z2) was reconstructed by RT-PCR using primers designed on the basis of sequences in the RNA-seq contigs. The two reconstructed genomic sequences showed greater than 98.4% identity at nt level with corresponding contig sequences generated from RNA assembly, indicating that the assembled contig sequences from RNA-seq data were reliable. Meanwhile, a complete genomic sequence of 18,851 nt (WH4-2) (GenBank accession no. MT936305) and a near-full-length genomic sequence (WH4-1, 16,871 nt, excluding about 2,000 nt at the 5′ terminus) (GenBank accession no. MT936304) of AcV-1 infecting an A. arguta plant (ID: WH4) were determined by Sanger sequencing of RT-PCR and 3′ and 5′ RACE products using primers designed on the basis of all available sequences.
These sequences together with near-full-length genomic sequences of three contigs (JX-c18869, JX5-c18818, and JX5-c18410) (GenBank accession nos. MT936303, MT936301, and MT936300), four contigs (CQ-c13707, JX-c12007, JX5-c10087, and JX5-c10084) (GenBank accession nos. MT936296, MT936302, MT936299, and MT936298) with sizes > 10,000 nt obtained from RNA-seq, and the genomic sequence of K75 were compared with each other (Table 2). The genome-wide nt sequence identity among the Chinese AcV-1 variants was about 60–87%. All these sequences showed 68.3–79.3% nt identity with that of isolate K75 reported from New Zealand. Moreover, high diversity was present in the AcV-1 sequences derived from the same sample. The two variants WH4-1 and WH4-2 from sample WH4 showed 62.6% nt sequence identity with each other, and the four contigs JX5-c18818, JX5-c18410, JX5-c10087, and JX5-c10084 from sample JX5 showed 63.3–86.5% nt sequence identity with each other. The genome structure of these Chinese AcV-1 variants was identical to that of K75 and consisted of 12 ORFs. There was variation in the length of the 5′ untranslated region (UTR) of genomic RNA among the Chinese AcV-1 variants and isolate K75. The 5′-UTR lengths of three AcV-1 variants Z2, JS27, and WH4-2 were 416, 290, and 288 nt, with 39.7, 42.9, and 78.3% nt sequence identities between Z2 and WH4-2, Z2 and JS27, and WH4-2 and JS27. These sequences shared 64.3–79.3% identity with 304-nt 5′-UTR of K75. The 3′-UTR lengths of AcV-1 variants JS27, WH4-2, and WH4-1 were 253, 252, and 252 nt, which were about 152 nt shorter than the 404-nt 3′-UTR of K75. The 3′-UTR of these Chinese AcV-1 variants shared 74.1–92.1% nt sequence identity with each other and 80.0–88.1% nt sequence identity with that of K75.
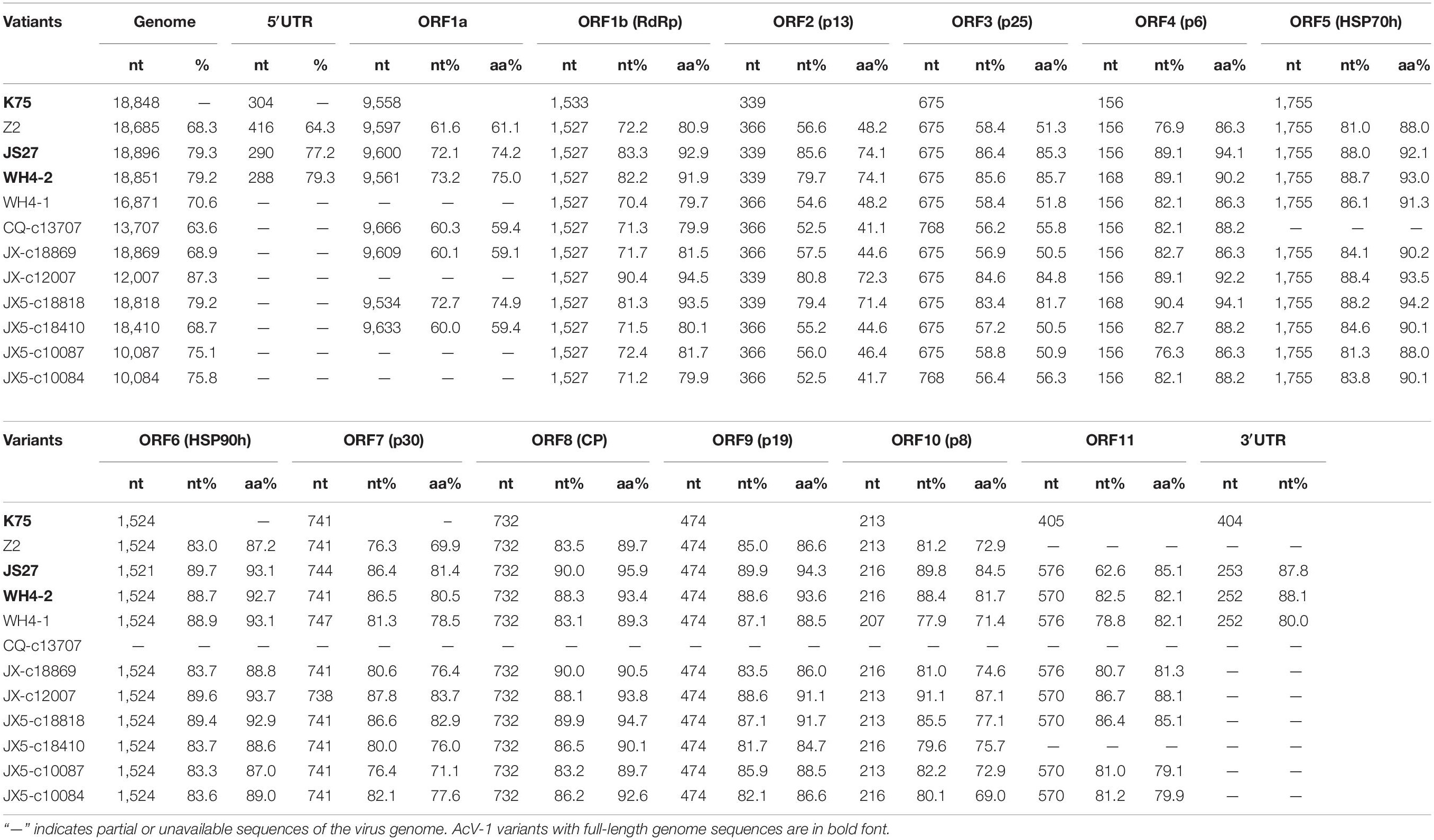
Table 2 | Nucleotide and amino acid sequence comparison of the genome, ORFs, and UTRs of 11 Actinidia virus 1 (AcV-1) variants determined in this study with the corresponding sequences of isolate K75.
Sequence alignment for each ORF (Table 2) encoded in the above genomic and contig sequences showed that sizes of ORF5, ORF6, ORF8, and ORF9 of these Chinese AcV-1 variants were the same as the corresponding ORFs of K75, except for the ORF6 in variant JS27. The ORF1b size of these Chinese AcV-1 variants was the same (1,527 nt), but 6-nt shorter than that of K75. The sizes of other ORFs were variable among these sequences. The ORF1a sizes showed the highest variation, ranging from 9,543 to 9,666 nt. ORF7 and ORF10 had three different sizes, and ORFs 2, 3, 4, and 11 had two sizes. Moreover, ORF11 sequences determined in this study were 165 nt or 171 nt larger than the ORF11 (405 nt) of K75 due to 6-nt and 164-nt deletions located near the 5′ and 3′ termini of ORF11 of K75, respectively (Supplementary Figure S1).
High nucleotide and amino acid sequence diversity occurred in each ORF of the Chinese AcV-1 populations (Table 2). Generally, ORF1a, ORF2, and ORF3 near the 5′ terminus of AcV-1 genome were more variable. ORF1a of Chinese AcV-1 variants shared 59.5–86.1% nt sequence identity with each other and 60.0–73.2% nt sequence identity with the ORF1a of K75. RdRp encoded in ORF1b of all AcV-1 variants had an AcV-1 specific 26-amino acid (aa) insert located immediately after the GDD motif, when it was compared to RdRp encoded by other viruses in the family Closteroviridae. Multiple alignments showed that the 26-aa insertion sequences were highly variable among AcV-1 variants (Figure 2A) and formed two phylogenetic clades (Figure 2B). The ORF2 sequences of AcV-1 variants determined in this study were even more variable and showed the highest divergence up to 47.5% at nt sequence level and 58.9% at aa sequence level compared to isolate K75. Nucleotide variations distributed across the entire ORF2 (Supplementary Figure S2A) and amino acid variations were mainly present at the C-terminal half of the encoded protein (p13) (Supplementary Figure S2B). Similarly, the ORF3 of these Chinese AcV-1 variants shared 56.2–86.4% nt and 50.5–85.7% aa sequence identity with that of K75. ORFs 5, 6, 8, and 9 were relatively conserved among AcV-1 variants by showing greater than 80% nt sequence identity with the corresponding sequences of K75 (Table 2).
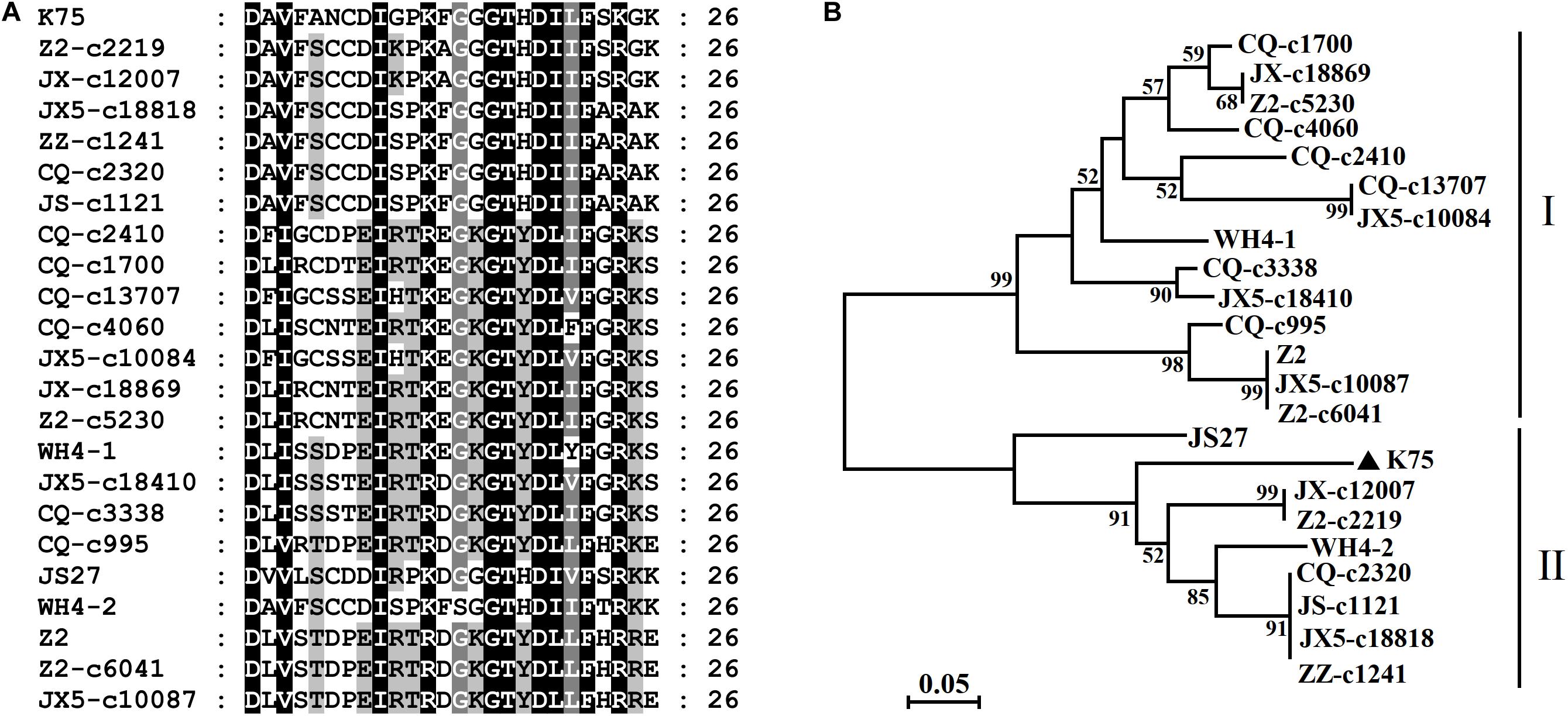
Figure 2 | Multiple alignment of the 26-aa insert in RdRp of Actinidia virus 1 (AcV-1) variants (A) and a neighbor-joining phylogenetic tree inferred from the 26–amino acid (aa) insert sequences (B).
Phylogenetic analyses for the complete aa sequences of proteins RdRp, HSP70h, and CP, for which aa sequences are specified by ICTV for genus demarcation within the family Closteroviridae, showed that all Chinese AcV-1 variants together with isolate K75 clustered into a clade, distantly related to persimmon virus B (PeVB), an unassigned closterovirus species (Ito et al., 2015) (Figure 3). In the AcV-1 clade, Chinese AcV-1 variants formed three subclades represented by JX5-c18410, Z2, and K75. In the HSP90h-, p30-, and p19-based trees, AcV-1 variants also clustered into three clades. It was found that the positions of some AcV-1 variants changed in these phylogenetic trees. For example, in the RdRp- and HSP70h-based trees, WH4-1 was in the subclade represented by JX5-c18410, but clustered in the subclade represented by K75 in the HSP90h-based tree and in the subclade represented by Z2 in the CP-, p30-, and p19-based trees.
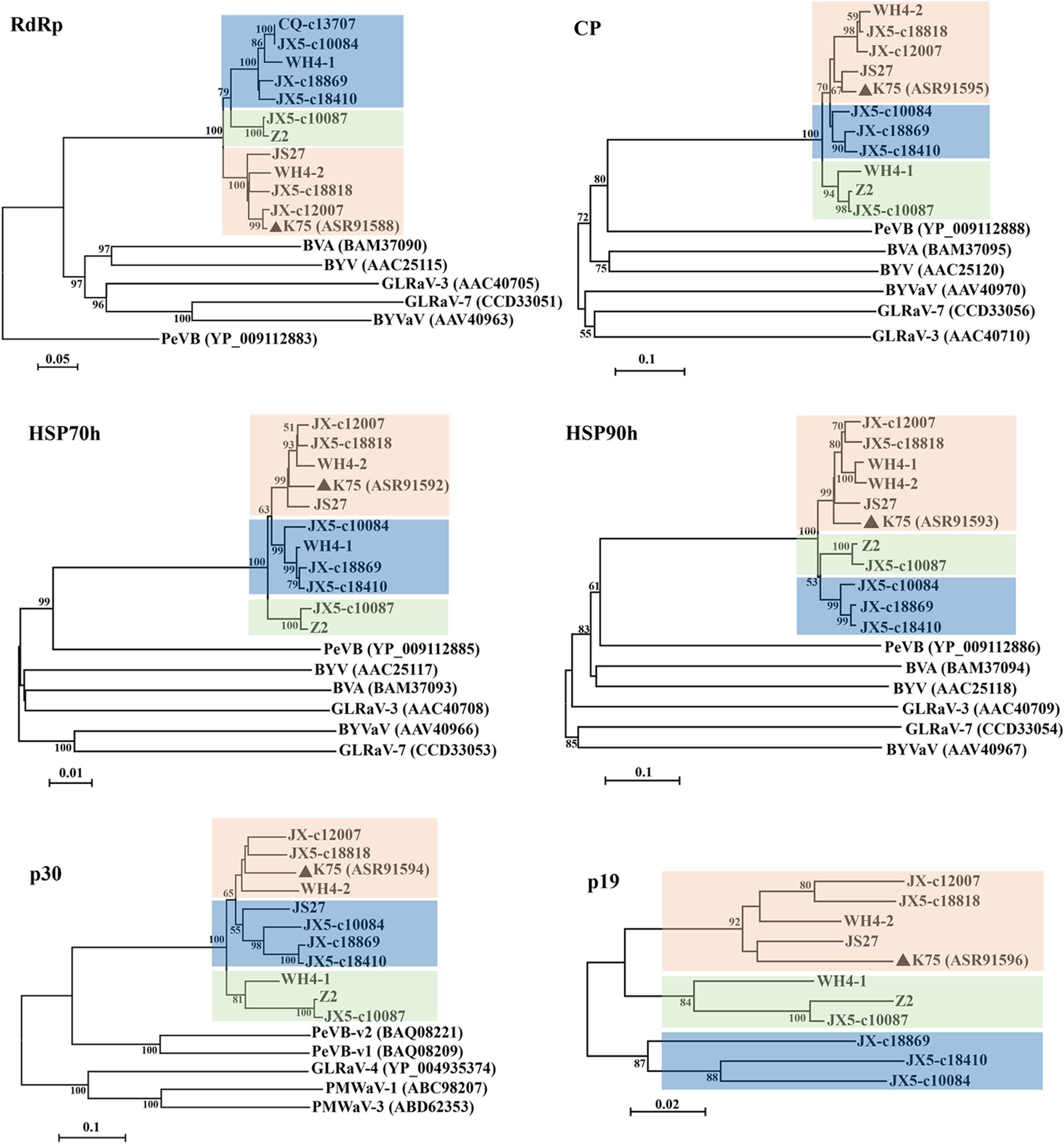
Figure 3 | Phylogenetic trees constructed using amino acid (aa) sequences of putative RdRp, CP, HSP70h, HSP90h, p30, and p19 of Actinidia virus 1 (AcV-1) variants. The genomic sequence of AcV-1 isolate K75, two complete (JS27 and WH4-2) and two near-complete (Z2 and WH4-1) genomic sequences determined in this study, and seven RNA-seq derived contigs with sizes > 10,000 nt, were included in the analysis. The contig CQ-c13707 covering the ORF1a-ORF4 (position 248–14,118 nt) of the viral genome was only included in the RdRp-based tree. Bootstrap values (1,000 replicates) > 50% were shown at branch nodes. The reported AcV-1 isolate K75 was marked by a black triangle. GenBank accession numbers of viruses referred for phylogenetic analyses were indicated following the abbreviation of each virus name. PeVB, Persimmon virus B (AB923924); BVA, blueberry virus A (AB733585); BYV, beet yellows virus (AF056575); BYVaV, blackberry yellow vein-associated virus (AY776334); GLRaV-3, grapevine leafroll-associated virus 3 (AF037268); GLRaV-7, grapevine leafroll-associated virus 7 (HE588185).
Identification of Domains of AcV-1 Proteins
Putative domains of the proteins predicted from AcV-1 genomic sequences (including assembled contig sequences) were identified by using CDD at https://www.ncbi.nlm.nih.gov/Structure/cdd. It was found that motifs of papain-like leader protease (L-Pro), methyltransferase (MTR, PF01660), and helicase (HEL, PF01443), necessary for virus replication, were present in the multifunctional protein encoded in ORF1a of all AcV-1 variants (Figure 4). However, the number of L-Pro domains varied among AcV-1 variants. Notably, the ORF1a of Z2, JX5-c8857, CQ-c7590, and ZZ-c3649 had a single L-Pro domain (L1 or L2), and the ORF1a of other AcV-1 variants had two L-Pro domains (L1 and L2). Sequence analyses revealed the presence of a sec-independent translocase domain (cl35116) in variant Z2 and an ATP-dependent RNA helicase RhiB (cl35267) in variants CQ-c13707 and JX5-c2170. These domains were not identified and documented in the reported AcV-1 isolate K75. Further sequence alignments showed that the sequences covering the two domains were highly variable in other AcV-1 variants (Supplementary Figures S3A,B). Additionally, according to the CDD prediction, ORF1a of the AcV-1 variants JS27, CQ-c5050, CQ-c3990, and JX-c6776 harbored a domain (56 or 57 aa) of 2OG-Fe (II) oxygenase superfamily (cl21496) (Aravind and Koonin, 2001; Erwin et al., 2008) upstream of the MET domain. Further alignment showed that all AcV-1 variants had the core sequence of AlkB domain (pfam 03171) (Maree et al., 2008; Ghanem-Sabanadzovic et al., 2012; Donda et al., 2017), belonging to 2OG-Fe (II) oxygenase superfamily, as in some closteroviruses (Supplementary Figure S3C).
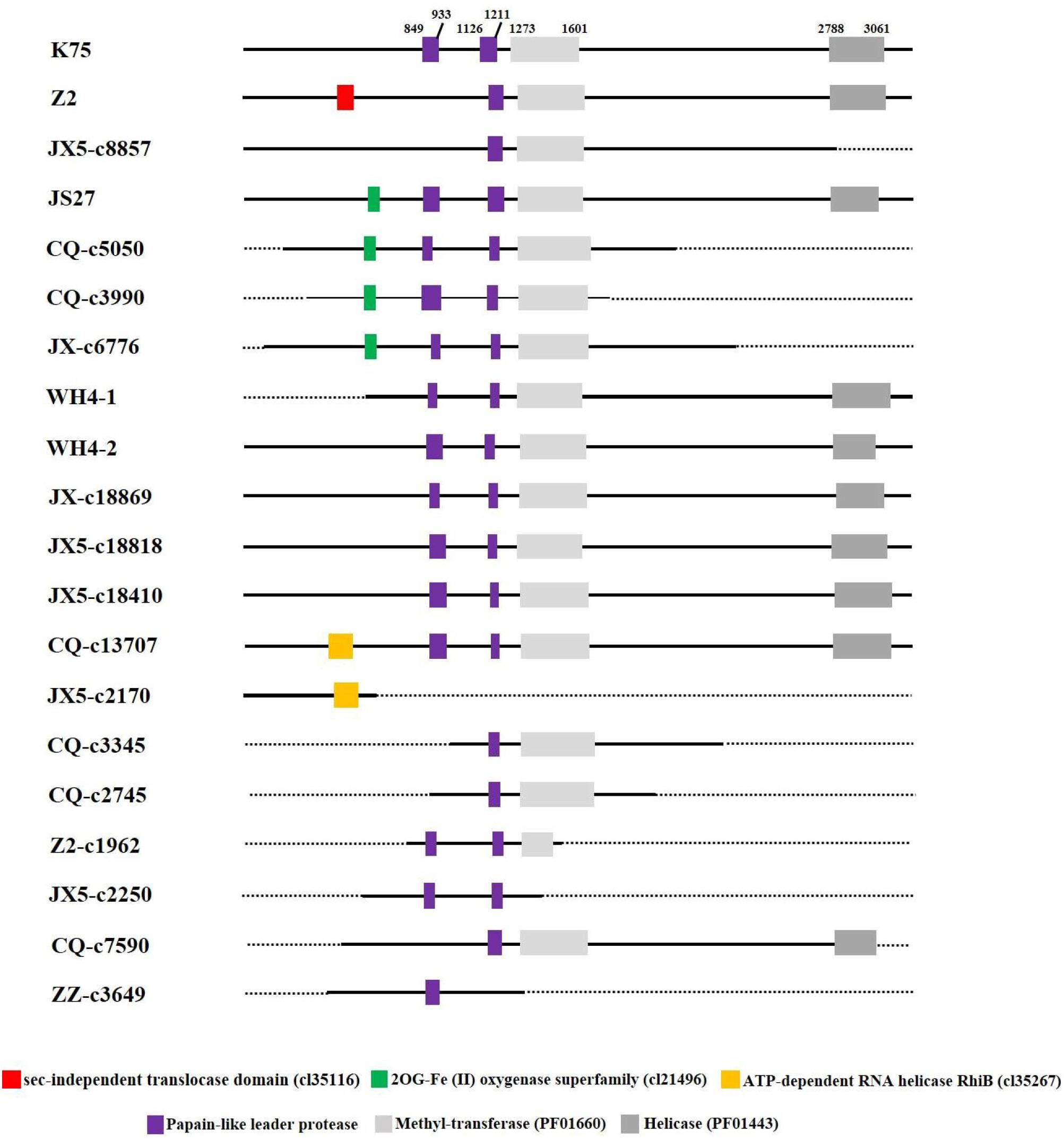
Figure 4 | Schematic diagram of domains contained in ORF1a of Actinidia virus 1 (AcV-1) variants. The starting and ending sites of each domain in the ORF1a of AcV-1 isolate K75 are indicated by black numbers. The sec-independent translocase domain (cl35116), domain belonging to the 2OG-Fe (II) oxygenase superfamily (cl21496), and ATP-dependent RNA helicase RhiB (cl35267) are marked as red, green, and orange boxes, respectively. MET and HEL are shown as light gray and dark gray boxes, respectively. Black solid lines represent available sequences, and black dotted lines indicate unavailable sequences.
A thaumatin-like domain was identified in protein p30 encoded by ORF7 of all available AcV-1 variants. The thaumatin-like domain of all available AcV-1 variants was 109 or 115 aa (Supplementary Figure S4) and shared 69.9–96.3% aa sequence identity with each other.
Recombination Analysis
The incongruent positions of some variants in phylogenetic trees based on the sequences of different proteins indicated possible recombination events occurring in the Chinese AcV-1 variants. Recombination analysis performed on an alignment of 11 AcV-1 genomic sequences revealed that 11 recombination events were present in eight variants (Table 3). All the detected recombination regions located at the 3′ half of the viral genome. Three very close recombination events at positions 13,029–14,361, 14,881–16,595, and 16,614–18,703 nt were detected in WH4-1 with JX5-c18410 as a possible major parent in two recombination events (events 2 and 3) and as a possible minor parent in one recombination event (event 1). The variant WH4-2 from the same sample as WH4-1 had two recombination events (events 4 and 5) with parents different from those of WH4-1. It was found that three recombination events (events 4, 8, and 10) in variants WH4-2, Z2, and JX5-c10087 had a common major parent JX5-c18818. Additionally, two variants JX-c12007 and JS27, for which the start sites of recombination were not detected, had the same parents WH4-2 and K75. AcV-1 variants JX5-c18410, JX5-c10087, and JX5-c10084 were from the same sample (ID: JX5), but had different parents.
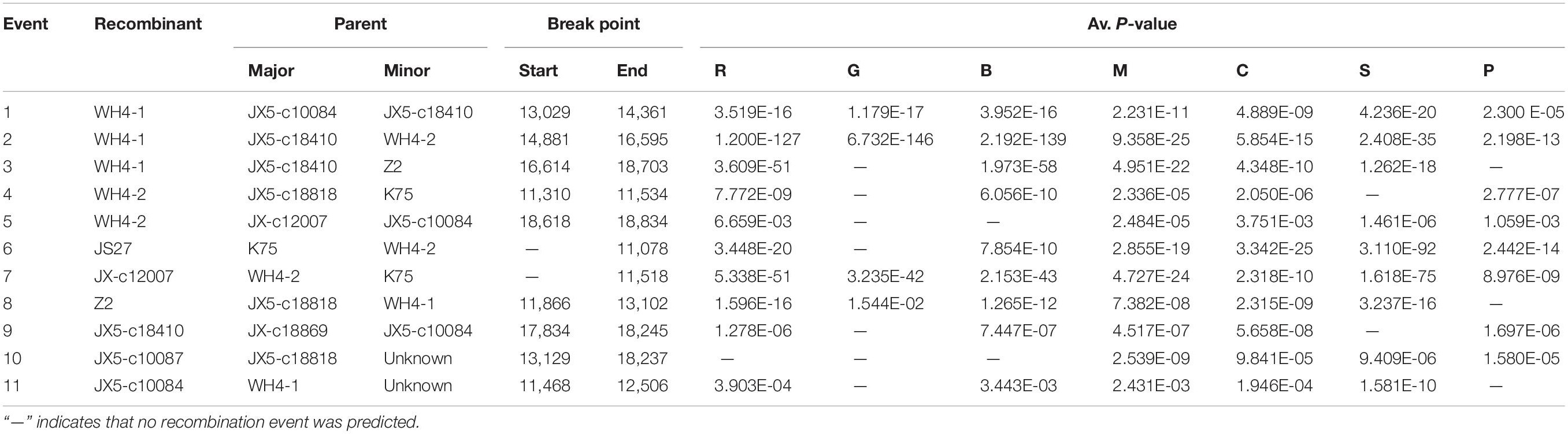
Table 3 | Putative recombination events identified by using RDP4 in Actinidia virus 1 (AcV-1) genomic sequences.
RT-PCR Detection of AcV-1 in Kiwifruit Plants Grown in China
To disclose the infection status of AcV-1, leaf samples collected from 245 kiwifruit plants grown in 10 provinces and cities in China were subjected to RT-PCR detection of AcV-1 using the primer set cp-F/cp-R. Results showed that 75 samples, including 70 symptomatic samples and five asymptomatic samples, were positive for AcV-1 (Table 4), accounting for 30.6% of total samples. The virus was detected in four analyzed species, including A. chinensis, A. delicious, A. arguta, and A. eriantha, and in some other kiwifruit plants with species unknown. Of these AcV-1–positive samples, two samples ZZ4 and CQ11 were negative for other detected viruses, and 73 samples were coinfected by AcV-1 with one or more other viruses, including AcVA, AcVB, ASGV, CLBV, and AcCRaV (Supplementary Table S4), and/or unreported kiwifruit-infecting viruses (data not shown). Except for samples from Yunnan Province and Shanghai city, where all collected samples were negative for AcV-1 in this test, the virus was detected in samples from seven provinces and one city, with relative high infection rates for samples from Hubei and Shandong provinces and Chongqing city (Table 4).
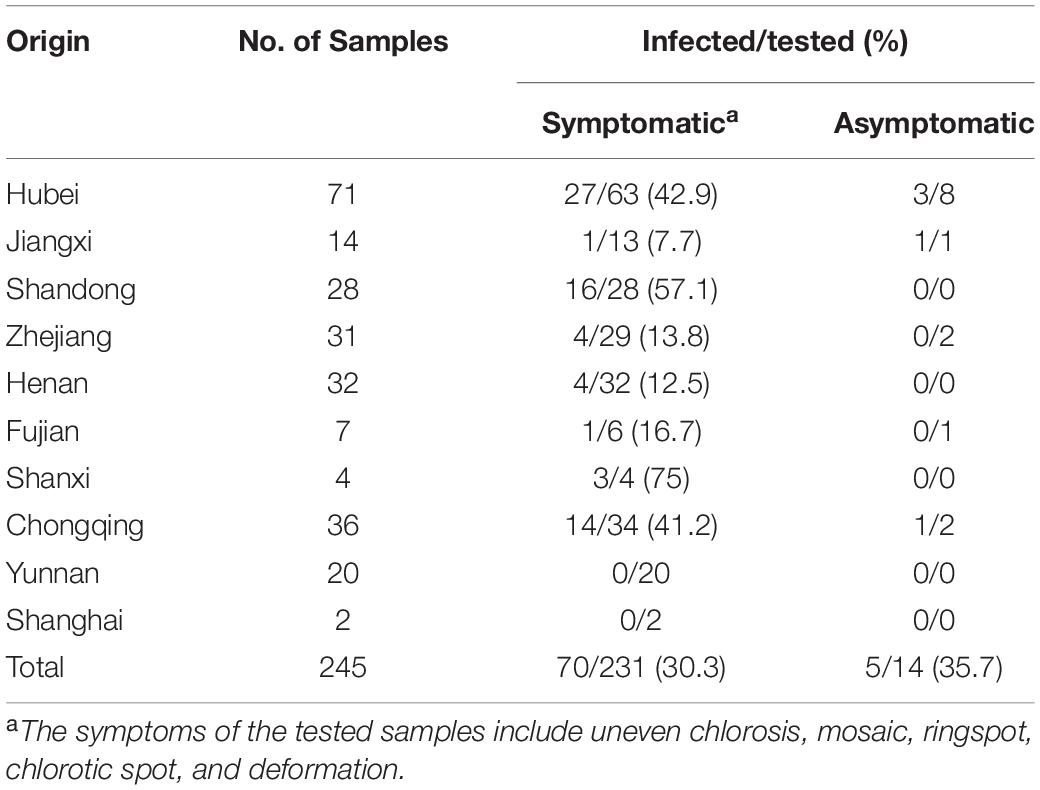
Table 4 | Incidence of Actinidia virus 1 (AcV-1) in the kiwifruit samples collected from eight provinces and two cities in China.
The 376 bp PCR products obtained using the primer set cp-F/cp-R from 75 AcV-1–positive samples were sequenced. These sequences together with the corresponding sequences derived from RNA-seq and 13 sequences of CP gene referred from GenBank database were used for phylogenetic analysis. The 376-bp sequences of all Chinese AcV-1 variants showed 78.5–100% nt identity with the equivalent sequence of isolate K75 and clustered into three distinct clades, respectively, represented by the variants K75, WH4-1, and Z2, except for one variant YT5, which was distant from the three clades (Supplementary Figure S5). All eight AcV-1 variants, previously reported from Sichuan province (Peng et al., 2018), clustered in the same clade (clade I). It was found that variants from each of samples Z2 and JX5 were distributed in different clades, confirming the sequence variation within the AcV-1 populations.
Discussion
NGS combined with bioinformatics analysis has become a routine technology for the rapid discovery and characterization of known or novel plant viruses (Adams et al., 2009; Kreuze et al., 2009; Barba et al., 2014; Wu Q. et al., 2015; Hadidi et al., 2016; Cretazzo and Velasco, 2017; Villamor et al., 2019; Zhao et al., 2020; Wang et al., 2020; Zhang et al., 2020). NGS-based metagenomic analyses have revealed the natural biodiversity of plant viruses (Roossinck, 2011; Roossinck et al., 2015). In this study, the RNA-seq analyses of two samples individually collected from two kiwifruit plants and four pooled kiwifruit samples revealed the presence of the eight kiwifruit-infecting viruses AcV-1, AcCRaV, ASbLV, AcVA, AcVB, CLBV, AcEV-2, and ASGV, and three viruses related to the families Bromoviridae, Betaflexiviridae, and Secoviridae. These data indicate that NGS technology is useful for identifying viruses infecting kiwifruit plants.
AcV-1 was identified from all six samples analyzed by RNA-seq. Except for the sample ZZ, from which AcV-1 contigs with size of 224–4,132 nt covering 71.5% of the viral genome were recovered, large contig sequences (>5,000 nt) of AcV-1 genome were obtained from the remaining five RNA-seq samples, indicating that the RNA-seq analyses were efficient for the genomic and molecular characterization of the virus. The RNA-seq sample ZZ was a pool of leaf samples from 30 kiwifruit plants, of which only two were positive for AcV-1. The relatively low AcV-1 titer in the mixed sample ZZ and molecular diversity of the virus in the two sampled plants might contribute to the absence of large contig sequences from the sample. The Sanger sequencing for the complete genome of AcV-1 variants JS27 and Z2 also confirmed the reliability of NGS sequence assembly for the viral genome. RNA-seq analyses also revealed mixed infection of AcV-1 variants in its natural host. Two contigs covering nearly the complete genome of AcV-1 were assembled from the sample JX5 and shared about 63% nt sequence identity, and the remaining contigs from the sample shared less than 85% nt sequence identity with each other. The constitution of AcV-1 contigs from sample Z2 was even more complicated. From the sample, 26 AcV-1 contigs were derived, but could not be assembled as contiguous sequences, indicating high sequence heterogeneity of these AcV-1 contigs, which was also the case in the pooled sample CQ. Consistent with RNA-seq analyses, Sanger sequencing for RT-PCR products revealed at least two divergent AcV-1 variants (WH4-1 and WH4-2) in the sample WH4. Like viruses infecting other perennial woody plants, viruses can be transmitted among kiwifruit plants through vegetative propagation. Vectors play important roles in the natural transmission of viruses in the family Closteroviridae, which might increase the virus diversity in a single host plant (Iglesias et al., 2008; Roy and Brlansky, 2009; Rubio et al., 2013).
The complete genomic sequences of variants JS27 and WH4-2 determined here were 18,896 and 18,851 nt, respectively, close to the 18,848 nt of reported isolate K75 (Blouin et al., 2018). However, the 3′-UTR of JS27, WH4-2, and a partially sequenced variant WH4-1 was about 150 nt shorter than that of K75. Analyses of the two complete genomic sequences of AcV-1 variants JS27 and WH4-2 and nine near-complete genomic sequences of AcV-1 variants Z2, WH4-1, CQ-c13707, JX-c18869, JX-c12007, JX5-c18818, JX5-c18410, JX5-c10087, and JX5-c10084 derived from different kiwifruit plants and geographical areas in China revealed high genetic divergence across the viral genome, especially in the 5′-UTR and the ORFs (ORF1a, ORF2, and ORF3) located in the 5′ half of the viral genome. The sequence variation might impede primer design to amplify the viral 5′ terminal sequence of variant WH4-1. The 5′ UTR could play a regulatory role in virus replication (Mawassi et al., 1996; Liveratos et al., 2004; Mongkolsiriwattana et al., 2016; Jarugula et al., 2018). The 5′ UTR of GLRaV-3 contains critical elements required for virus replication (Jarugula et al., 2010; Jarugula et al., 2018; Adiputra et al., 2019). Similarly, the 5′ UTR of CTV is also highly variable (Lopez et al., 1998), but can form stable SL (stem-loop) structures, which are essential for viral replication and assembly (Tatineni et al., 2002; Gowda et al., 2003).
The ORF1a of viruses in the family Closteroviridae encodes a multifunctional protein (Rubio et al., 2013). The size of ORF1a differed among all available AcV-1 variants with divergence up to 40% at nt sequence level. Previously, the ORF1a size variation was also found in GLRaV-2 (Meng et al., 2005) and GLRaV-5 (Thompson et al., 2012). Overall, the organization of ORF1a of the AcV-1 genome is similar to that of members in the family Closteroviridae (Agranovsky et al., 1995; Karasev, 2000; Dolja et al., 2006). In this study, we found that AcV-1 variants might have one or two L-Pro domains (L1 and L2). Some viruses in the family Closteroviridae possess a tandem of papain-like cysteine proteases with two divergent functional domains L1 and L2, which might have evolved via gene duplication (Karasev, 2000; Peng et al., 2001; Dolja et al., 2006; Liu et al., 2009). The absence of domain L1 in some AcV-1 variants indicated that the domain L2 might be indispensable, which was different from the case that domain L1 plays a crucial role in the establishment of infection and accumulation of some viruses in the family Closteroviridae (Liu et al., 2009; Kang et al., 2018). Except for motifs L-Pro, MTR, and HEL, necessary for viral replication, the ORF1a of some AcV-1 variants harbors a sec-independent translocase domain, or an ATP-dependent RNA helicase RhiB, or an 2OG-Fe (II) oxygenase domain, suggesting that these domains might have been acquired relatively recently via horizontal gene transfer (Bratlie and Drablos, 2005). The AlkB domain is present in the polyproteins of some viruses in the family Closteroviridae, as well as in alpha-like plant viruses belonging to genera Allexivirus, Carlavirus, Foveavirus, Potexvirus, Trichovirus, and Vitivirus (Erwin et al., 2008). The AlkB domain usually locates between MET and HEL domains, whereas the AlkB domain of AcV-1 variants identified in this study locates upstream of the MET domain. Certain cellular AlkBs are involved in RNA repair via methylation reversal (Aravind and Koonin, 2001). However, the function of AlkB in plant virus infection is unknown. The variation of L-Pro numbers and other domains in the multifunctional protein might show not only the diversity of the AcV-1 populations, but also different biological features (Liu et al., 2009; Atallah et al., 2016; Kang et al., 2018). The p30 encoded in AcV-1 ORF7 has a thaumatin-like domain like that in the p29 of PeVB-v1 (GenBank accession no. AB923924) and the p28 of PeVB-v2 (GenBank accession no. AB923925). Although the location of p30 in the AcV-1 genome is similar to the CPm of some closteroviruses, the p30 did not show aa sequence identity with the CPm encoded by other viruses in the family Closteroviridae. The function of the protein remains to be unknown.
The ORF-by-ORF comparisons also showed that the sequences of ORF2 and ORF3 were highly variable among the AcV-1 variants, with the highest divergence up to 58 and 49% at aa sequence level, respectively. Similar diversities are observed among divergent variants of some closteroviruses, such as GLRaV-3 (Maree et al., 2008), GLRaV-4 (Adiputra et al., 2019), and PeVB (Ito et al., 2015). However, other ORFs were much more conserved in AcV-1 populations. Among the viral variants, the aa divergence values of 10–20% for CP, HSP70h and RdRp are less than the species discriminating threshold of 25% approved by ICTV. Therefore, we contend that all AcV-1 sequences represent divergent variants of the virus, which often occurred in a single kiwifruit plant as illustrated by RNA-seq analyses. Phylogenetic analyses for the complete aa sequences of proteins RdRp, HSP70h and CP showed that all Chinese AcV-1 variants together with isolate K75 clustered into a clade, distantly related to PeVB. In the AcV-1 clade, Chinese AcV-1 variants formed three subclades. The results further support that the sequences determined from kiwifruit plants grown in China belong to AcV-1 variants, which are highly divergent in molecular composition.
Recombination in RNA viruses has been extensively documented as a powerful driving force for generating new variants (Karasev, 2000; Chare and Holmes, 2006; Farooq et al., 2012; Katsiani et al., 2015; James and Phelan, 2016; Ruiz et al., 2018). Frequent homologous recombination events have been reported for viruses in the family Closteroviridae (Karasev, 2000; Turturo et al., 2005; Sztuba-Solińska et al., 2011; Rubio et al., 2013). The incongruent positions of some AcV-1 variants in the phylograms based on proteins coded near the 3′ half of AcV-1 genome suggested occurrences of recombination events. Although the parents for these recombinants were identified, some parents were also potential recombinants. The exact parents for these recombinants need to be clarified as more genomic sequences available from a wide host source. The data will help to understand whether an ancient recombination event occurred before the recombinants spread worldwide, as previously suggested for CTV (Roy et al., 2005; Rubio et al., 2013; Wu G. et al., 2015). Most of the recombination events occurred in the region following the viral ORF1b. In CTV, the genes encoded in the region play important roles in the determination of pathogenicity, movement, and host range (Folimonova, 2020). The frequent recombination events in the region of AcV-1 genome might be necessary to meet possible selection pressure (Roy and Brlansky, 2009).
Finally, RT-PCR detection showed that AcV-1 occurred in four Actinidia species and some kiwifruit plants of unknown species grown in seven provinces and one city in China. Previous investigation showed that mixed infection of several viruses in a single kiwifruit plant was very common (Zheng et al., 2014, 2017; Blouin et al., 2018; Veerakone et al., 2018; Wang et al., 2020). Most of the plants tested in this study showed viral disease–like symptoms, including uneven chlorosis between veins, mosaic, ringspots, chlorotic spots, and/or deformation, which were indicated previously to be associated with some virus infections. However, some asymptomatic plants were also positive for the virus. Although two AcV-1–infected samples ZZ4 and CQ11 exhibited interveinal chlorosis and were negative for other kiwifruit viruses known in China, we could not conclude that the disease symptom was caused by the virus infection due to the potential infection of unknown viruses. Therefore, the association of AcV-1 infection with diseases of kiwifruit plants is unclear at present. The biological properties of AcV-1 need to be determined in future studies.
All nucleotide sequences obtained in this study supported high molecular diversity in AcV-1 populations from kiwifruit plants grown in China. This represents the first report for the complete genome sequences of AcV-1 in China and molecular diversity of AcV-1. The reported AcV-1 sequences would be helpful for further detailed taxonomic study and assignment of efficient molecular diagnostic techniques of AcV-1 to improve the sanitary status of kiwifruit planting materials in China.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
NH and GW supervised the project, conceived and designed the experiments. SW, ZY, MR, QL, and YW performed the experiments and analyzed the data. SW and NH wrote the manuscript, prepared the tables and figures, revised and approved final version of the manuscript. This is the first submission of the manuscript and it is not being considered for publication elsewhere in part or in whole. All authors approved the submission of this manuscript.
Funding
This work was financially supported by the Key National Project (Grant No. 2019YFD1001800); the program for Key International S&T Cooperation Proposal (Grant No. 2017YFE0110900); the earmarked fund for the China Agriculture Research System (Grant No. CARS-28-15). The funders did not influence the study design, data collection and analysis, decision to publish, or the preparation of this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Ahmed Hadidi for his linguistic assistance and suggestions during the preparation of this manuscript. We thank the team of kiwifruit research at Zhengzhou Fruit Research Institute, Academy of Agricultural Sciences, Zhengzhou, China, and Dr. Hong-Wei Yi at Fruit Research Institute, Chongqing Academy of Agricultural Sciences, Chongqing, China, for kind helps during sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.602039/full#supplementary-material
Supplementary Figure 1 | Multiple alignment of nucleotide sequences of ORF11 of Chinese Actinidia virus 1 (AcV-1) variants and reported isolate K75.
Supplementary Figure 2 | Multiple alignment of nucleotide (A) and amino acid (B) sequences of ORF2 of Actinidia virus 1 (AcV-1) variants.
Supplementary Figure 3 | Amino acid sequence alignments of sec-independent translocase domain (A), ATP-dependent RNA helicase RhiB domain (B) and AlkB domain belonging to the 2OG-Fe (II) oxygenase superfamily (C) of Actinidia virus 1 (AcV-1) variants or viruses. Black triangle indicates the AcV-1 variants containing predicated domains. A solid line above aligned sequences marks the core of the AlkB domain and arrows indicate the conserved residues in the AlkB. Accession numbers of sequences used in the analysis include: grapevine leafroll-associated virus 1 (GLRaV-1) isolates WA-CH (KU674796), WA-PN (KU674797) and Canada (NC_016509), GLRaV-3 (GU983863), GLRaV-4 (FJ467503), GLRaV-5 (JX559640), GLRaV-6 (NC_016417), GLRaV-9 (AY297819), little cherry virus 2 (LChV-2; AF531505).
Supplementary Figure 4 | Multiple sequence alignment of thaumatin-like domain in ORF7 (p30) of the Actinidia virus 1 (AcV-1) variants.
Supplementary Figure 5 | Neighbor joining (NJ) phylogenetic tree generated from the nucleotide sequences of partial CP of Actinidia virus 1 (AcV-1) variants. Bootstrap values (1,000 replicates) > 50% are shown at branch nodes. The reported AcV-1 isolate K75 is marked by a black triangle. The sequences referred from GenBank are identified by their GenBank accession numbers. The scale bar is 0.01 substitutions per site.
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/
- ^ https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^ http://www.ebi.ac.uk/Tools/msa/clustalw2/
References
Adams, I. P., Glover, R. H., Monger, W. A., Mumford, R., Jackeviciene, E., Navalinskiene, M., et al. (2009). Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol. Plant Pathol. 10, 537–545. doi: 10.1111/j.1364-3703.2009.00545.x
Adiputra, J., Jarugula, S., and Naidu, R. A. (2019). Intra-species recombination among strains of the ampelovirus grapevine leafroll-associated virus 4. Virol. J. 16, 139. doi: 10.1186/s12985-019-1243-4
Agranovsky, A. A., Lesemann, D. E., Maiss, E., Hull, R., and Atabekov, J. G. (1995). “Rattlesnake” structure of a filamentous plant RNA virus built of two capsid proteins. Proc. Natl. Acad. Sci. U.S.A. 92, 2470–2473. doi: 10.1073/pnas.92.7.2470
Aravind, L., and Koonin, E. V. (2001). The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2:research0007.1. doi: 10.1186/gb-2001-2-3-research0007
Atallah, O. O., Kang, S., El-Mohtar, C. A., Shilts, T., María, B., and Folimonova, S. Y. (2016). A 5’-proximal region of the citrus tristeza virus genome encoding two leader proteases is involved in virus superinfection exclusion. Virology 489, 108–115. doi: 10.1016/j.virol.2015.12.008
Barba, M., Czosnek, H., and Hadidi, A. (2014). Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 6, 106–136. doi: 10.3390/v6010106
Bar-Joseph, M., and Mawassi, M. (2013). The defective RNAs of Closteroviridae. Front. Microbiol. 4:132. doi: 10.3389/fmicb.2013.00001
Bertazzon, N., Borgo, M., Vanin, S., and Angelini, E. (2010). Genetic variability and pathological properties of grapevine leafroll-associated virus 2 isolates. Eur. J. Plant Pathol. 127, 185–197. doi: 10.1007/s10658-010-9583-3
Blouin, A. G., Biccheri, R., Khalifa, M. E., Pearson, M. N., Poggi Pollini, C., Hamiaux, C., et al. (2018). Characterization of Actinidia virus 1, a new member of the family Closteroviridae encoding a thaumatin-like protein. Arch. Virol. 163, 229–234. doi: 10.1007/s00705-017-3610-z
Blouin, A. G., Chavan, R. R., Pearson, M. N., MacDiarmid, R. M., and Cohen, D. (2012). Detection and characterisation of two novel vitiviruses infecting Actinidia. Arch. Virol. 157, 713–722. doi: 10.1007/s00705-011-1219-1
Blouin, A. G., Pearson, M. N., Chavan, R. R., Woo, E. N. Y., Lebas, B. S. M., Veerakone, S., et al. (2013). Viruses of kiwifruit (Actinidia species). J. Plant Pathol. 95, 221–235. doi: 10.4454/JPP.V95I2.013
Boulila, M. (2010). Selective pressure, putative recombination events and evolutionary relationships among members of the family Closteroviridae: a proposal for a new classification. Biochem. Syst. Ecol. 38, 1185–1192. doi: 10.1016/j.bse.2010.12.002
Bratlie, M. S., and Drablos, F. (2005). Bioinformatic mapping of AlkB homology domains in viruses. BMC Genomics 6:1. doi: 10.1186/1471-2164-6-1
Chare, E. R., and Holmes, E. C. (2006). A phylogenetic survey of recombination frequency in plant RNA viruses. Arch. Virol. 151, 933–946. doi: 10.1007/s00705-005-0675-x
Chavan, R. R., Blouin, A. G., Cohen, D., and Pearson, M. N. (2013). Characterization of the complete genome of a novel citrivirus infecting Actinidia chinensis. Arch. Virol. 158, 1679–1686. doi: 10.1007/s00705-013-1654-2
Chavan, R. R., Cohen, D., Blouin, A. G., and Pearson, M. N. (2012). Characterization of the complete genome of ribgrass mosaic virus isolated from Plantago major L. from New Zealand and Actinidia spp.from China. Arch. Virol. 157, 1253–1260. doi: 10.1007/s00705-012-1292-0
Clover, G. R. G., Pearson, M. N., Elliott, D. R., Tang, Z., Smales, T. E., and Alexander, B. J. R. (2003). Characterization of a strain of apple stem grooving virus in Actinidia chinesis from China. Plant Pathol. 52, 371–378. doi: 10.1046/j.1365-3059.2003.00857.x
Cretazzo, E., and Velasco, L. (2017). High-throughput sequencing allowed the completion of the genome of grapevine red globe virus and revealed recurring co-infection with other tymoviruses in grapevine. Plant Pathol. 66, 1202–1213. doi: 10.1111/ppa.12669
Cuellar, W. J., Tairo, F., Kreuze, J. F., and Valkonen, J. P. T. (2008). Analysis of gene content in sweet potato chlorotic stunt virus RNA1 reveals the presence of the p22 RNA silencing suppressor in only a few isolates: implications for viral evolution and synergism. J. Gen. Virol. 89, 573–582. doi: 10.1099/vir.0
Dolja, V. V., Kreuze, J. F., and Valkonen, J. (2006). Comparative and functional genomics of closteroviruses. Virus Res. 117, 38–51. doi: 10.1016/j.virusres.2006.02.002
Donda, B. P., Jarugula, S., and Naidu, R. A. (2017). An analysis of the complete genome sequence and subgenomic RNAs reveals unique features of the Ampelovirus, grapevine leafroll-associated virus 1. Phytopathology 107, 1069–1079. doi: 10.1094/PHYTO-02-17-0061-R
Erwin, V. D. B., Omelchenko, M. V., Anders, B., Vibeke, L., Koonin, E. V., Dolja, V. V., et al. (2008). Viral AlkB proteins repair RNA damage by oxidative demethylation. Nucleic Acids Res. 17, 5451–5461. doi: 10.1093/nar/gkn519
Esteves, F., Teixeira Santos, M., EirasDias, J. E., and Fonseca, F. (2012). Occurrence of Grapevine leafroll-associated virus 5 in Portugal: genetic variability and population structure in field-grown grapevines. Arch. Virol. 157, 1747–1765. doi: 10.1007/s00705-012-1371-2
Fan, X., Hong, N., Dong, Y., Ma, Y., Zhang, Z., Ren, F., et al. (2015). Genetic diversity and recombination analysis of grapevine leafroll-associated virus 1 from China. Arch. Virol. 160, 1669–1678. doi: 10.1007/s00705-015-2437-8
Farooq, A. B. U., Ma, Y., Wang, Z., Zhuo, N., Xu, W., Wang, G., et al. (2012). Genetic diversity analyses reveal novel recombination events in grapevine leafroll-associated virus 3 in China. Virus Res. 171, 15–21. doi: 10.1016/j.virusres.2012.10.014
Folimonova, S. Y. (2020). Citrus tristeza virus: A large RNA virus with complex biology turned into a valuable tool for crop protection. Plos Pathog. 16:e1008416. doi: 10.1371/journal.ppat.1008416
García-Arenal, F., and Fraile, A. (2011). “Population dynamics and genetics of plant infection by viruses,” in Recent Advances in Plant Virology, eds C. Caranta, M. A. Aranda, M. Tepfer, and J. J. Lopez-Moya (Norfolk: Caister Academic Press), 263–281.
Ghanem-Sabanadzovic, N. A., Sabanadzovic, S., Gugerli, P., and Rowhani, A. (2012). Genome organization, serology and phylogeny of grapevine leafroll-associated viruses 4 and 6: taxonomic implications. Virus Res. 163, 120–128. doi: 10.1016/j.virusres.2011.09.001
Gowda, S., Tatineni, S., Ayllon, M. A., Moreno, P., Flores, R., and Dawson, W. O. (2003). The conserved structures of the 5’ nontranslated region of citrus tristeza virus are involved in replication and virion assembly. Virology 317, 50–64. doi: 10.1016/j.virol.2003.08.018
Hadidi, A., Flores, R., Candresse, T., and Barba, M. (2016). Next-generation sequencing and genome editing in plant virology. Front. Microbiol. 7:1325. doi: 10.3389/fmicb.2016.01325
Harper, S. J., Chooi, K. M., and Pearson, M. N. (2008). First report of citrus leaf blotch virus in New Zealand. Plant Dis. 92, 1470–1470. doi: 10.1094/PDIS-92-10-1470C
Holland, J., Spindler, K., Horodyski, F., Grabau, E., Nichol, S., and VandePol, S. (1982). Rapid evolution of RNA genomes. Science 215, 1577–1588. doi: 10.1126/science.7041255
Iglesias, N. G., Gago-Zachert, S. P., Robledo, G., Costa, N., Plata, M. I., Vera, O., et al. (2008). Population structure of citrus tristeza virus from field Argentinean isolates. Virus Genes 36, 199–207. doi: 10.1007/s11262-007-0169-x
Ito, T., Sato, A., and Suzaki, K. (2015). An assemblage of divergent variants of a novel putative closterovirus from American persimmo. Virus Genes 51, 105–111. doi: 10.1007/s11262-015-1202-0
James, D. (1999). A simple and reliable protocol for the detection of apple stem grooving virus by RT-PCR and in a multiplex PCR assay. J. Virol. Methods 83, 1–9. doi: 10.1016/S0166-0934(99)00078-6
James, D., and Phelan, J. (2016). Complete genome sequence of a strain of Actinidia virus X detected in Ribes nigrum cv. Baldwin showing unusual symptoms. Arch. Virol. 161, 507–511. doi: 10.1007/s00705-015-2678-6
Jarugula, S., Gowda, S., Dawson, W. O., and Naidu, R. A. (2010). 3’-Coterminal subgenomic RNAs and putative cis-acting elements of grapevine leafroll-associated virus 3 reveals ‘unique’ features of gene expression strategy in the genus Ampelovirus. Virol. J. 7:180. doi: 10.1186/1743-422X-7-180
Jarugula, S., Gowda, S., Dawson, W. O., and Naidu, R. A. (2018). Development of infectious cDNA clones of grapevine leafroll-associated virus 3 and analyses of the 5′ non-translated region for replication and virion formation. Virology 523, 89–99. doi: 10.1016/j.virol.2018.07.023
Kang, S., Atallah, O. O., Sun, Y., and Folimonova, S. Y. (2018). Functional diversification upon leader protease domain duplication in the citrus tristeza virus genome: role of RNA sequences and the encoded proteins. Virology 514, 192–202. doi: 10.1016/j.virol.2017.11.014
Karasev, A. V. (2000). Genetic diversity and evolution of closteroviruses. Annu. Rev. Phytopathol. 38, 293–324. doi: 10.1146/annurev.phyto.38.1.293
Katsiani, A. T., Maliogka, V. I., Amoutzias, G. D., Efthimiou, K. E., and Katis, N. I. (2015). Insights into the genetic diversity and evolution of little cherry virus 1. Plant Pathol. 64, 817–824. doi: 10.1111/ppa.12309
Kreuze, J. F., Perez, A., Untiveros, M., Quispe, D., Fuentes, S., Barker, I., et al. (2009). Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388, 1–7. doi: 10.1016/j.virol.2009.03.024
Kreuze, J. F., Savenkov, E. I, and Valkonen, J. P. T. (2002). Complete genome sequence and analyses of the subgenomic RNAs of sweet potato chlorotic stunt virus reveal several newfeatures for the genus Crinivirus. J. Virol. 76, 9260–9270. doi: 10.1128/JVI.76.18.9260-9270.2002
Liu, H., Wang, G., Yang, Z., Wang, Y., Zhang, Z., Li, L., et al. (2020). Identification and characterization of a pear chlorotic leaf spot-associated virus, a novel emaravirus associated with a severe disease of pear trees in China. Plant Dis. 104, 2786–2798. doi: 10.1094/PDIS-01-20-0040-RE
Liu, Y., Peremyslov, V. V., Vicente Medina, V., and Dolja, V. V. (2009). Tandem leader proteases of grapevine leafroll-associated virus 2: host-specific functions in the infection cycle. Virology 383, 291–299. doi: 10.1016/j.virol.2008.09.035
Liveratos, I. C., Eliasco, E., Muller, G., Olsthoorn, R. C. L., Salazar, L. F., Pleij, C. W. A., et al. (2004). Analysis of the RNA of potato yellow vein virus: evidence for a tripartite genome and conserved 3′-terminal structures among members of the genus Crinivirus. J. Gen. Virol. 85, 2065–2075. doi: 10.1099/vir.0.79910-0
Lopez, C., Ayllon, M. A., Navas-Castillo, J., Guerri, J., Moreno, P., and Flores, R. (1998). Molecular variability of the 5′- and 3′-terminal regions of citrus tristeza virus RNA. Phytopathology 88, 685–691. doi: 10.1094/PHYTO.1998.88.7.685
Maree, H. J., Freeborough, M. J., and Burger, J. T. (2008). Complete nucleotide sequence of a South African isolate of grapevine leafroll-associated virus 3 reveals a 5′ UTR of 737 nucleotides. Arch. Virol. 153, 755–757. doi: 10.1007/s00705-008-0040-y
Martelli, G. P., Agranovsky, A. A., Bar-Joseph, M., Boscia, D., Candresse, T., Coutts, R. H. A., et al. (2012). “Family Cloteroviridae,” in Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, eds A. M. Q. King, M. J. Adams, E. B. Carstens, and E. J. Lefkowitz (London: Elsevier Academic Press), 987–1001.
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Mawassi, M., Mietkiewska, E., Gofman, R., Yang, G., and Bar-Joseph, M. (1996). Unusual sequence relationships between two isolates of citrus tristeza virus. J. Gen. Virol. 77, 2359–2364. doi: 10.1099/0022-1317-77-9-2359
Meng, B., Li, C., Goszczynski, D. E., and Gonsalves, D. (2005). Genome sequences and structures of two biologically distinct strains of grapevine leafroll-associated virus 2 and sequence analysis. Virus Genes 31, 31–41. doi: 10.1007/s11262-004-2197-0
Mongkolsiriwattana, C., Zhou, J., and Ng, J. C. K. (2016). A 3′ end structure in RNA2 of a crinivirus is essential for viral RNA synthesis and contributes to replication-associated translation activity. Sci. Rep. 6:34482. doi: 10.1038/srep34482
Pearson, M. N., Cohen, D., Chavan, R., and Blouin, A. G. (2011). Actinidia is a natural host to a wide range of plant viruses. Acta Hort. 913, 467–472. doi: 10.17660/ActaHortic.2011.913.62
Peng, C., Peremyslov, V. V., Mushegian, A. R., Dawson, W. O., and Dolja, V. V. (2001). Functional specialization and evolution of leader proteinases in the family Closteroviridae. J. Virol. 75, 12153–12160. doi: 10.1128/JVI.75.24.12153-12160.2001
Peng, Q., Lv, R., Ning, J., Yang, T., Lin, H., Xi, D., et al. (2018). First report of Actinidia virus 1 infecting Actinidia chinensis in China. Plant Dis. 103, 782–782. doi: 10.1094/PDIS-08-18-1334-PDN
Qu, L., Cui, H., Wu, G., Zhou, J., Su, J., Wang, G., et al. (2014). Genetic diversity and molecular evolution of Plum bark necrosis stem pitting-associated virus from China. PLoS One 9:e105443. doi: 10.1371/journal.pone.0105443
Roossinck, M. J. (2011). The big unknown: plant virus biodiversity. Curr. Opin. Virol. 1, 63–67. doi: 10.1016/j.coviro.2011.05.022
Roossinck, M. J., Martin, D. P., and Roumagnac, P. (2015). Plant virus metagenomics: advances in virus discovery. Phytopathology 105, 716–727. doi: 10.1094/PHYTO-12-14-0356-RVW
Roy, A., and Brlansky, R. (2009). Population dynamics of a Florida citrus tristeza virus isolate and aphid-transmitted subisolates: identification of three genotypic groups and recombinants after aphid transmission. Phytopathology 99, 1297–1306. doi: 10.1094/PHYTO-99-11-1297
Roy, A., Manjunath, K., and Brlansky, R. (2005). Assessment of sequence diversity in the 5′ terminal region of citrus tristeza virus from India. Virus Res. 113, 132–142. doi: 10.1016/j.virusres.2005.04.023
Rubio, L., Ayllo, M. A., Kong, P., Fernandez, A., Polek, M., Guerri, J., et al. (2001). Genetic variation of citrus tristeza virus isolates from California and Spain: evidence for mixed infections and recombination. J. Virol. 75, 8054–8062. doi: 10.1128/JVI.75.17.8054-8062.2001
Rubio, L., Guerri, J., and Moreno, P. (2013). Genetic variability and evolutionary dynamics of viruses of the family Closteroviridae. Front. Microbiol. 4:151. doi: 10.3389/fmicb.2013.00151
Ruiz, L., Simon, A., Garcıa, C., Velasco, L., and Janssen, D. (2018). First natural crossover recombination between two distinct species of the family Closteroviridae leads to the emergence of a new disease. PLoS One 13:e0198228. doi: 10.1371/journal.pone.0198228
Sztuba-Solińska, J., Urbanowicz, A., Figlerowicz, M., and Bujarski, J. J. (2011). RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 49, 415–443. doi: 10.1146/annurev-phyto-072910-095351
Tatineni, S., Gowda, S., Ayllon, M. A., Albiach-Marti, M. R., and Dawson, W. O. (2002). Mutational analysis of the replication signals in the 3′-nontranslated region of citrus tristeza virus. Virology 300, 140–152. doi: 10.1006/viro.2002.1550
Thompson, J. R., Fuchs, M., and Perry, K. L. (2012). Genomic analysis of grapevine leafroll-associated virus 5 and related viruses. Virus Res. 163, 19–27. doi: 10.1016/j.virusres.2011.08.006
Turturo, C., Saldarelli, P., Dong, Y., Digiaro, M., Minafra, A., Savino, V., et al. (2005). Genetic variability and population structure of grapevine leafroll-associated virus 3 isolates. J. Gen. Virol. 86, 217–224. doi: 10.1099/vir.0.80395-0
Veerakone, S., Liefting, L. W., Tang, J., and Ward, L. I. (2018). The complete nucleotide sequence and genome organisation of a novel member of the family Betaflexiviridae from Actinidia chinensis. Arch. Virol. 163, 1367–1370. doi: 10.1007/s00705-017-3701-x
Villamor, D. E. V., Ho, T., Rwahnih, M. A., Martin, R. R., and Tzanetakis, I. E. (2019). High throughput sequencing for plant virus detection and discovery. Phytopathology 109, 716–725. doi: 10.1094/PHYTO-07-18-0257-RVW
Wang, D., Liu, X., Li, T., Li, Q., Liu, Y., Gong, G., et al. (2018). First report of cucumber mosaic virus infection in kiwifruit (Actinidia chinensis) in China. Plant Dis. 102, 1180–1180. doi: 10.1094/PDIS-06-17-0824-PDN
Wang, Y., Wang, G., Yang, Z., Wang, L., Li, L., and Hong, N. (2016). First report of the tospovirus tomato necrotic spot associated virus infecting kiwifruit (Actinidia sp.). Plant Dis. 100, 2539–2539. doi: 10.1094/PDIS-05-16-0629-PDN
Wang, Y., Zhai, L., Wen, S., Yang, Z., Wang, G., and Hong, N. (2020). Molecular characterization of a novel emaravrius infecting Actinidia spp. in China. Virus Res. 275:197736. doi: 10.1016/j.virusres.2019.197736
Wang, Y., Zhuang, H., Yang, Z., Wen, L., Wang, G., and Hong, N. (2018). Molecular characterization of an apple stem grooving virus isolate from kiwifruit (Actinidia chinensis) in China. Can. J. Plant Pathol. 40, 76–83. doi: 10.1094/PDIS-06-17-0824-PDN
Wen, S., Zhu, C., Yang, Z., Wang, Y., Wang, G., Wang, L., et al. (2018). First report of Actinidia virus 1 infecting kiwifruit in China. Plant Dis. 103:780. doi: 10.1094/PDIS-07-18-1123-PDN
Wu, G., Tang, M., Wang, G., Jin, F., Yang, Z., Cheng, L., et al. (2015). Genetic diversity and evolution of two capsid protein genes of citrus tristeza virus isolates from China. Arch. Virol. 160, 787–794. doi: 10.1007/s00705-014-2281-2
Wu, Q., Ding, S., Zhang, Y., and Zhu, S. (2015). Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Ann. Rev. Phytopathol. 53, 425–444. doi: 10.1146/annurev-phyto-080614-120030
Zhang, S., Yang, L., Ma, L., Tian, X., Li, R., Zhou, C., et al. (2020). Virome of Camellia japonica: discovery and molecular characterization of new viruses of different taxa in Camellias. Front. Microbiol. 11:945. doi: 10.3389/fmicb.2020.00945
Zhao, L., Cao, M., Huang, Q., Jing, M., Bao, W., Zhang, Y., et al. (2020). Occurrence and molecular characterization of Actinidia virus (AcVC), a novel vitivirus infecting kiwifruit (Actinidia spp.) in China. Plant Pathol. 69, 775–782. doi: 10.1111/ppa.13171
Zhao, L., Yang, W., Zhang, Y., Wu, Z., Wang, Q., and Wu, Y. (2019). Occurrence and molecular variability of kiwifruit viruses in Actinidia deliciosa “Xuxiang” in the Shaanxi province of China. Plant Dis. 103, 1309–1318. doi: 10.1094/PDIS-09-18-1570-RE
Zheng, Y., Navarro, B., Wang, G., Wang, Y., Yang, Z., Xu, W., et al. (2017). Actinidia chlorotic ringspot-associated virus: a novel emaravirus infecting kiwifruit plants. Mol. Plant Pathol. 18, 569–581. doi: 10.1111/mpp.12421
Keywords: next-generation sequencing, Actinidia spp., Actinidia virus 1, molecular diversity, recombination, RT-PCR
Citation: Wen S, Wang G, Yang Z, Wang Y, Rao M, Lu Q and Hong N (2020) Next-Generation Sequencing Combined With Conventional Sanger Sequencing Reveals High Molecular Diversity in Actinidia Virus 1 Populations From Kiwifruit Grown in China. Front. Microbiol. 11:602039. doi: 10.3389/fmicb.2020.602039
Received: 02 September 2020; Accepted: 11 November 2020;
Published: 16 December 2020.
Edited by:
John Wesley Randles, University of Adelaide, AustraliaReviewed by:
Michael Norman Pearson, The University of Auckland, New ZealandXuefeng Wang, Chinese Academy of Agricultural Sciences, China
Copyright © 2020 Wen, Wang, Yang, Wang, Rao, Lu and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ni Hong, d2huaUBtYWlsLmh6YXUuZWR1LmNu