 Jianghua Yang
Jianghua Yang Mengzhi Liu2
Mengzhi Liu2 Zeliang Chen
Zeliang Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 15 December 2020
Sec. Microbial Physiology and Metabolism
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.598797
Brucellosis, an important bacterial zoonosis caused by Brucella species, has drawn increasing attention worldwide. As an intracellular pathogen, the ability of Brucella to deal with stress within the host cell is closely related to its virulence. Due to the similarity between the survival pressure on Brucella within host cells and that during the stationary phase, a label-free proteomics approach was used to study the adaptive response of Brucella abortus in the stationary stage to reveal the possible intracellular adaptation mechanism in this study. A total of 182 downregulated and 140 upregulated proteins were found in the stationary-phase B. abortus. B. abortus adapted to adverse environmental changes by regulating virulence, reproduction, transcription, translation, stress response, and energy production. In addition, both exponential- and stationary-phase B. abortus were treated with short-term starvation. The exponential B. abortus restricted cell reproduction and energy utilization and enhanced material transport in response to nutritional stress. Compared with the exponential phase, stationary Brucella adjusted their protein expression to a lesser extent under starvation. Therefore, B. abortus in the two growth stages significantly differed in the regulation of protein expression in response to the same stress. Overall, we outlined the adaptive mechanisms that B. abortus may employ during growth and compared the differences between exponential- and stationary-phase B. abortus in response to starvation.
Brucellosis, an important bacterial zoonosis, has drawn increasing attention worldwide. Each year, there are approximately more than 500,000 new cases of brucellosis worldwide, resulting in major losses in animal husbandry. Humans are mainly infected through direct contact with infected animals or the consumption of unpasteurized animal products, such as raw milk (Pappas et al., 2006a; McDermott et al., 2013). Aerosols are also an important transmission route, which makes Brucella spp. a potential biological weapon (Pappas et al., 2006b).
Several studies have focused on the virulence factors of Brucella, such as lipopolysaccharide (LPS) with atypical lipid A (Cardoso et al., 2006), type IV secretion system (T4SS) VirB (Ke et al., 2015), and VjbR (Ke et al., 2012). However, the pathogenic mechanisms of Brucella are not yet well understood. After invasion, Brucella sp. stealthily evade the host’s immune system, establish a replication niche inside the host cells, and eventually cause long-term infection (Ahmed et al., 2016). During invasion and the establishment of infection, Brucella must overcome a variety of environmental pressures, such as acidity, oxidative stress, and nutritional deficiency (Roop et al., 2009). Remarkably, the survival pressure on Brucella within a phagosome is similar to that during the stationary phase (Roop et al., 2003). In the early stage of intracellular infection, the levels of carbon center metabolism, material transport, protein synthesis, and transcription of Brucella were decreased at the proteomic level (Lamontagne et al., 2009); of note, similar changes were observed in gram-negative bacteria during the stationary phase (Navarro Llorens et al., 2010). Additionally, inhibition of growth, one of the characteristics of the stationary phase, was also observed early after intracellular infection in Brucella (Deghelt et al., 2014). The survival of Brucella in the stationary phase and within the host cells is regulated by the same virulence factor Hfq (Robertson and Roop, 1999). The absence of Hfq in Brucella leads to impaired stationary-phase physiology and intracellular growth. Therefore, the assessment of changes in protein expression patterns occurring during the switch from the exponential to the stationary phase may assist in the understanding of the pathogenesis of brucellosis. Proteomic techniques have been widely used to compare the protein expression patterns of pathogenic microorganisms under different conditions. Combined with bioinformatics technology, previous studies have annotated, classified, and enriched differentially expressed proteins (DEPs) to screen for potentially important proteins (Al Dahouk et al., 2009; Huang and Chiou, 2011; Al Dahouk et al., 2013; Albrethsen et al., 2013; Zai et al., 2017).
In this study, we investigated the adaptive regulation of Brucella during the stationary growth period using a label-free proteomics approach. In addition, Brucella melitensis 16 M in the exponential phase was more invasive to epithelial cells than bacteria in the stationary growth phase (Rossetti et al., 2009). Therefore, Brucella abortus harvested from exponential and stationary stages were treated with short-term nutritional stress, one of the most common stresses for Brucella within host cells. Then, the differences in the response of B. abortus to starvation in different growth phases were examined. We aimed to provide important information regarding the intracellular adaptation mechanism of Brucella and lay a foundation for further investigation.
Virulent B. abortus 2,308 was obtained from Tecon Biological Co., Ltd., (Urumqi, China) and cultured in tryptone soya agar (TSA) or tryptone soya broth (TSB). All experiments related to live B. abortus 2,308 were conducted in biosafety level 3 (BSL-3) laboratories. A single colony of Brucella was obtained using the streak plate method. The colonies were cultured in TSB medium in a shaking incubator set at 37°C. Bacteria were cultured to exponential or stationary growth stages and then harvested. The GEM medium was used for starvation treatment of B. abortus. The components of the GEM medium are as follows: MgSO47H2O (0.2 g/L), citric acidH2O (2.0 g/L), K2HPO4 (10.0 g/L), NaNH4HPO44H2O (3.5 g/L), and glucose (20 g/L; Wang et al., 2019). The cells harvested from the exponential or stationary growth stage were resuspended in the GEM medium and then incubated at 37°C by shaking the incubator for 1.5 h. The pH values of all media in this study were adjusted to 7.0. The exponential- and stationary-phase B. abortus cultured separately in TSB medium were called TSBL and TSBS, respectively. The exponential- and stationary-phase B. abortus cultures treated with starvation were called GEML and GEMS, respectively. The experimental design and sampling time are shown in Supplementary Figure S1.
Cells were harvested, centrifuged (7,000 × g at 4°C for 15 min), and then washed three times with sterile phosphate-buffered saline buffer. The bacteria were resuspended in lysis buffer, placed on ice, and lysed on ice through ultrasonic wave breaking. When the lysate became clear, it was centrifuged (40,000 × g at 4°C for 30 min). A protein quantification kit based on the bicinchoninic acid method (Thermo Fisher Scientific, Waltham, MA, United States) was used for concentration detection of protein in the supernatant. The cell protein extracts were reduced in 5 mM dithiothreitol at 56°C for 30 min. Then, the reduced cell protein extracts were added to iodoacetamide and incubated at room temperature away from light for 15 min. Finally, trypsin was added at a ratio of 1:50 (trypsin:protein, w/w), and the protein solution was digested overnight at 37°C. Trypsin was added again at a ratio of 1:100 (trypsin:protein, w/w) for a second 4-h digestion. For each condition, three biological replicates were sampled for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
The tryptic samples were fractionated into fractions by high-pH reverse HPLC with Agilent 300 Extend C18 (5-μm particles, 4.6-mm ID, and 250-mm length). The operation was as follows: peptides were separated with a gradient of 8–32% acetonitrile (pH = 9.0) into 60 fractions over 60 min; then, the peptides were combined into four fractions and dried by vacuum centrifuging for further operation.
The tryptic peptides were dissolved in 0.1% (v/v) formic acid (solvent A) and then separated using EASY-nLC 1,200 Ultra High Performance Liquid System. Solvent A contained 0.1% formic acid and 2% acetonitrile, while solvent B contained 0.1% formic acid and 90% acetonitrile. Gradient settings were as follows: 0–30 min, 8–16% solvent B; 30–55 min, 18–32% solvent B; 55–57 min, 32–80% solvent B; and 57–60 min, 80% solvent B. The flow rate was maintained at 400 nl/min.
The peptides were subjected to a nano-electrospray ionization (NSI) source and then analyzed by Orbitrap FusionTM Lumos mass spectrometry (MS). The electrospray voltage applied was 2.0 kV. The intact peptides were detected in the Orbitrap at a resolution of 60,000. The peptides were selected for tandem mass spectrometry (MS/MS) using normalized collision energy setting as 30, and ion fragments were detected in the Orbitrap at a resolution of 15,000. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 5E4 in the MS survey scan, with 30.0 s dynamic exclusion. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; the 5E4 ions were accumulated for the generation of MS/MS spectra. For MS scans, the m/z scan range was 350–1,550.
The resulting MS/MS data were processed using MaxQuant search engine (v.1.5.2.8). Tandem mass spectra were searched against the UniProt B. abortus (3,023 sequences) database concatenated with the reverse decoy database. Trypsin/P was specified as the cleavage enzyme allowing up to two missing cleavages. The mass tolerance for precursor ions of the first search and main search was set to 20 and 5 ppm, respectively. The mass tolerance for fragment ions was set as 0.02 Da. Carbamidomethyl on cysteine was specified as fixed modification, and oxidation on methionine, N-terminal acetylation of proteins, and deamidation of asparagine were specified as variable modifications. The false discovery rate (FDR) for protein identification and peptides and modification sites were set at 1%.
Gene Ontology (GO) annotation proteome was derived from the Genebank database1. Then, proteins were classified by GO annotation based on three categories: biological process, cellular component, and molecular function. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to annotate the pathways of the identified proteins. The Clusters of Orthologous Groups (COG2) system software program was used to determine the functional distribution of identified proteins.
The total RNA of B. abortus was extracted using TRIzol (Invitrogen), as recommended by the manufacturer. First, 1 μg of total RNA was reverse transcribed to cDNA using HiScript II Q RT SuperMix (Vazyme Biotech). Then, 1 μl of cDNA was amplified in 10 μl volumes using the ChamQ Universal SYBR qPCR Master Mix kit (Vazyme Biotech) and the following thermocycling program: (1) 95°C for 30 s; (2) 40 cycles at 95°C for 10 s and 60°C for 30 s; and (3) 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Real-time quantitative RT-PCR (qRT-PCR) analysis was performed in the Applied Biosystems QuantStudio 3 RT-PCR System. The transcript abundances for the target genes in each sample were assayed three times and normalized to that of 16S rRNA, which served as the internal control. Relative gene expression was calculated using the 2–Δ Δ Ct method, and the specific primers used in this study are listed in Supplementary Table S1.
Protein samples of Brucella obtained from different growth stages and treated with or without GEM medium were subjected to label-free proteomic analysis. A total of 1,290,057 secondary spectra were obtained using liquid chromatography with MS/MS analysis, and the available effective spectrum number was 605,083 (46.9%). A total of 32,477 peptide fragments were identified, of which the specific peptide fragments were 32,433. We identified 2,185 proteins, which accounted for about 72.3% of the predicted proteome, and 2,001 of these proteins were quantifiable (Supplementary Figure S2A and Supplementary Table S2). The MS/MS data were tested for quality control (Supplementary Figure S2). The molecular weight of the proteins was negatively correlated with their coverage (Supplementary Figure S2B) because proteins with higher molecular weights could produce more enzymatic peptide segments. To achieve the same coverage, larger proteins were needed to identify more peptide segments. The primary mass error of most spectrograms was less than 10 ppm, which is in accordance with the high-precision characteristics of the Orbitrap mass spectrometer, indicating that the mass accuracy of the mass spectrometer was normal (Supplementary Figure S2C). The length distribution of the peptides identified by MS/MS analysis met the quality control requirements. Most of the peptides were distributed in 7–20 amino acid fragments, which is in accordance with the general rules based on trypsin enzymatic hydrolysis and higher-energy collisional dissociation (Supplementary Figure S2D).
Brucella abortus grown in TSB medium were cultured to the exponential or stationary growth stage, harvested, and then starved for 1.5 h. Protein samples were subjected to label-free quantitative proteomic analysis. The proteins were divided into the following four comparison groups for analysis: TSBSL (TSBS vs. TSBL), GEMSL (GEMS vs. GEML), GTLL (GEML vs. TSBL), and GTSS (GEMS vs. TSBS). The TSBSL group was employed to analyze the adaptive regulation of B. abortus in the stationary phase, whereas the latter three groups were used to study the response of Brucella to nutritional stress in different growth stages.
Differentially expressed proteins in the four groups were selected using a cutoff of 1.5 fold change and a p value of less than 0.05 (Supplementary Table S3). The expression level was upregulated for 140 proteins and downregulated for 182 proteins in the TSBSL group. In the GEMSL group, 150 proteins were more abundant, whereas 235 proteins were less abundant. In the GTLL group, 63 and 29 proteins were upregulated and downregulated, respectively. Lastly, in the GTSS group, 14 proteins were upregulated and seven were downregulated (Figure 1A). Under the same nutritional stress, stationary-phase B. abortus induced fewer DEPs than the exponential phase, suggesting that there are differences in the protein regulation patterns in B. abortus at different growth stages. In addition, Venn diagrams were constructed to display the relationship between the four comparison groups (Figures 1B,C). In response to nutritional deficiency, one common downregulated protein was found in the two growth stages (Figure 1C) but was not upregulated (Figure 1B). This protein was lipoyl synthase (lipA), and lipoate synthase activity may have a significant role in the adaptive regulation of Brucella.
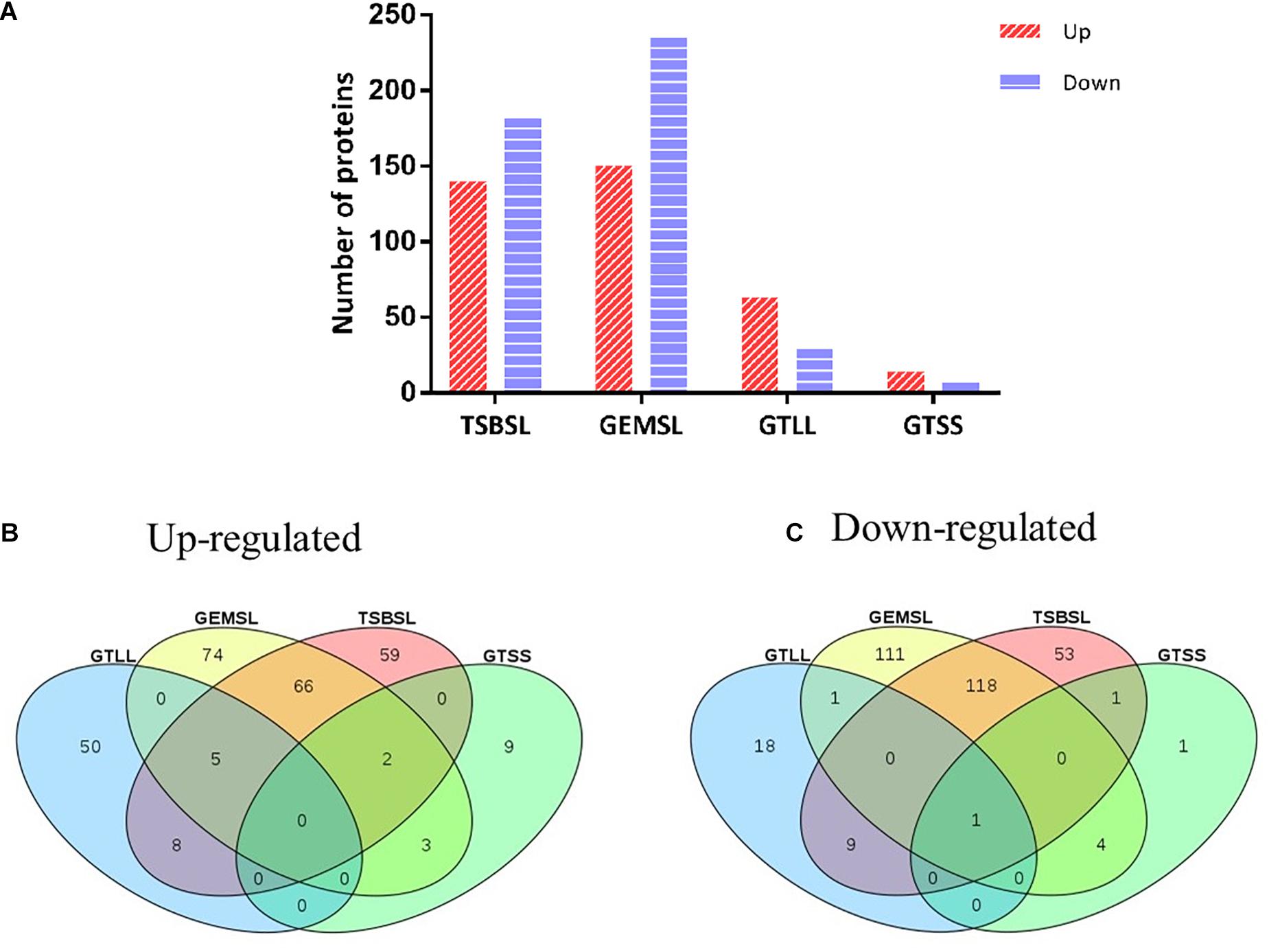
Figure 1. Statistical analysis of differentially expressed proteins (DEPs). (A) Number of up- or downregulated proteins in the groups of TSBSL, GEMSL, GTLL, and GTSS; (B) a Venn diagram of upregulated proteins in the groups of TSBSL, GEMSL, GTLL, and GTSS; and (C) a Venn diagram of downregulated proteins in the groups of TSBSL, GEMSL, GTLL, and GTSS. TSBSL group: stationary-phase Brucella abortus versus exponential-phase B. abortus cultured in the tryptone soya broth (TSB) medium; GEMSL group: stationary-phase B. abortus treated with starvation versus exponential-phase B. abortus treated with starvation; GTLL group: exponential-phase B. abortus treated with starvation versus untreated exponential-phase B. abortus; and GTSS group: stationary-phase B. abortus treated with starvation versus untreated stationary-phase B. abortus.
The functional distribution of the regulated proteins contributes to the exploration of the adaptation and response of B. abortus in the stationary growth phase and under nutritional deficiency. In the stationary phase (Figure 2A), we found that the majority of the downregulated proteins were clustered functionally in the categories of translation, ribosomal structure and biogenesis, transcription, transport, and metabolism of multiple substances as well as cell wall/membrane/envelope biogenesis. These results showed reduced metabolic activity in the stationary phase for B. abortus. Seven upregulated DEPs were predicted to be involved in intracellular trafficking, secretion, and vesicular transport, which might suggest that material transport of the stationary-phase B. abortus was vigorous. The functional distribution of GEMSL was similar to that of TSBSL (Figure 2B). Under short-term starvation stress, the COG analysis results of DEPs between exponential-phase (Figure 2C) and stationary-phase B. abortus (Figure 2D) were different. First, more DEPs were found in the exponential-phase B. abortus (Figure 2A). We found that the categories of amino acid as well as inorganic ion transport and metabolism contained 12 and 10 upregulated proteins, respectively, in the stationary-phase B. abortus (Figure 2C), whereas only one upregulated protein was found respectively in these categories in the exponential-phase B. abortus (Figure 2D). In addition, the transport and metabolism of carbohydrates were different. None of the upregulated proteins were observed in this category in the stationary-phase B. abortus, while nine upregulated proteins were found in the exponential-phase B. abortus. The COG functional classifications of the four groups are shown in Supplementary Table S4.
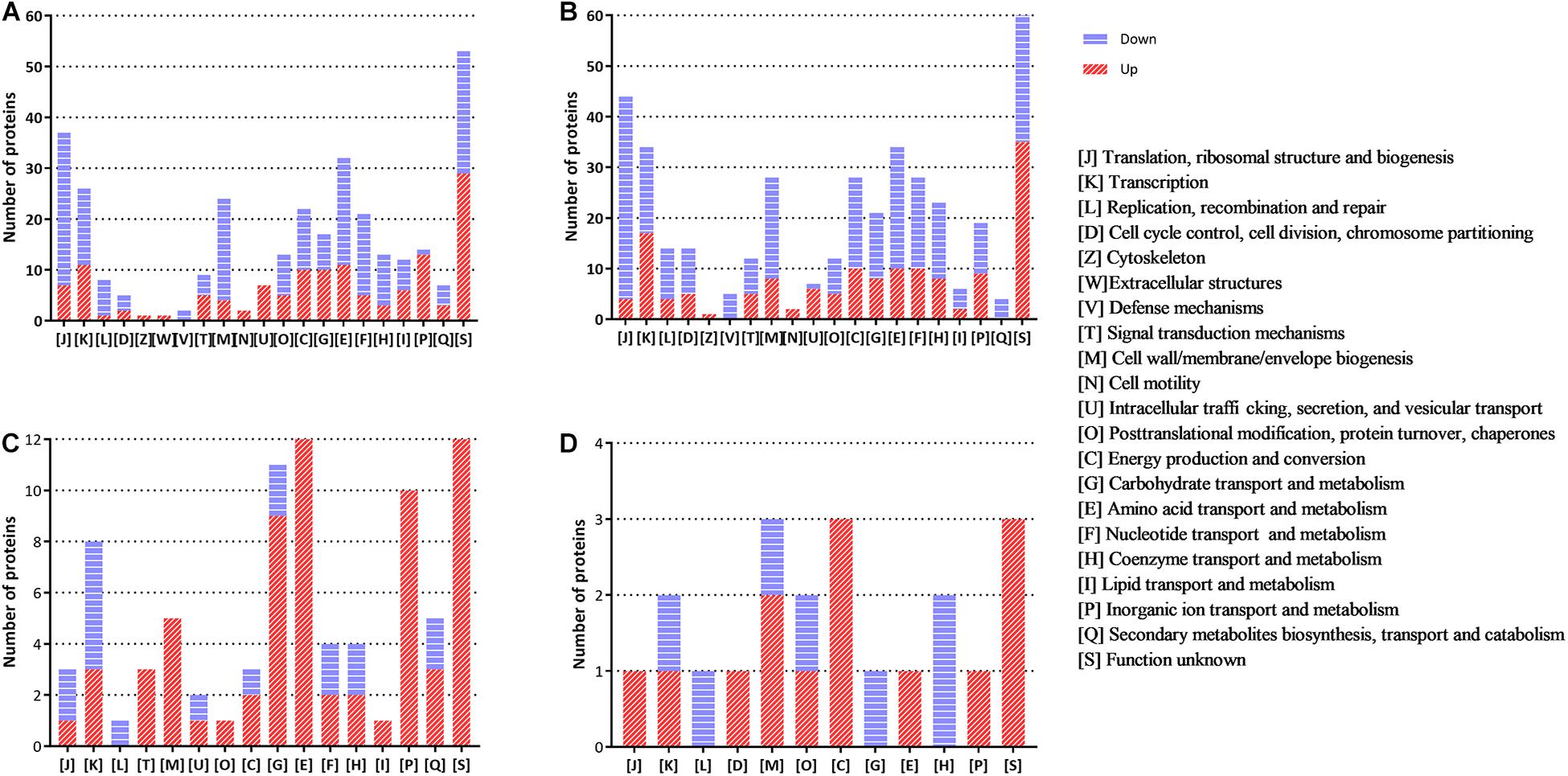
Figure 2. The clusters of orthologous groups (COG) analysis of the differentially expressed proteins for the groups of TSBSL, GEMSL, GTLL, and GTSS. (A) COG analysis of differentially expressed proteins (DEPs) for the group of TSBSL; (B) COG analysis of DEPs for the group of GEMSL; (C) COG analysis of DEPs for the group of GTLL group; and (D) COG analysis of DEPs for the group of GTSS group.
The DEPs were annotated using GO functional classification (Supplementary Figure S3). GO annotations can be divided into three major categories: biological process, cellular component, and molecular function. In the biological process category for the TSBSL, GEMSL, and GTLL groups, over 60% of the regulated proteins were related to metabolic and cellular processes. For the GTSS group, the metabolic process was detected in 28.57% of the cases when the cellular process was 25.00%. In terms of cellular component, the majority of the regulated proteins were located in the cell, followed by membrane, protein-containing complex, and organelle. Finally, in the molecular function category, over 80% of the regulated proteins in the four groups were related to catalytic activity and binding, followed by structural molecule activity and transporter activity. The GO functional classifications of the four groups are shown in Supplementary Table S5.
As shown in Table 1, the DEPs in the four groups of TSBSL, GEMSL, GTLL, and GTSS were further investigated using the KEGG database. In the TSBSL group, upregulated proteins were enriched in four pathways, namely ABC transporters, sulfur metabolism, thiamine metabolism, and tuberculosis. The downregulated proteins were enriched in the ribosome. In the GEMSL group, the upregulated proteins were enriched in the two-component system, histidine metabolism, thiamine metabolism, and tuberculosis, and downregulated proteins were enriched in amino sugar and nucleotide sugar metabolism; ribosome, starch, and sucrose metabolism; and galactose metabolism. In the GTLL group, we found two upregulated and downregulated pathways, respectively, including quorum sensing (QS), ABC transporters, histidine metabolism, and the biosynthesis of the siderophore group nonribosomal peptides. In the GTSS group, only one upregulated pathway was observed: glyoxylate and dicarboxylate metabolism.
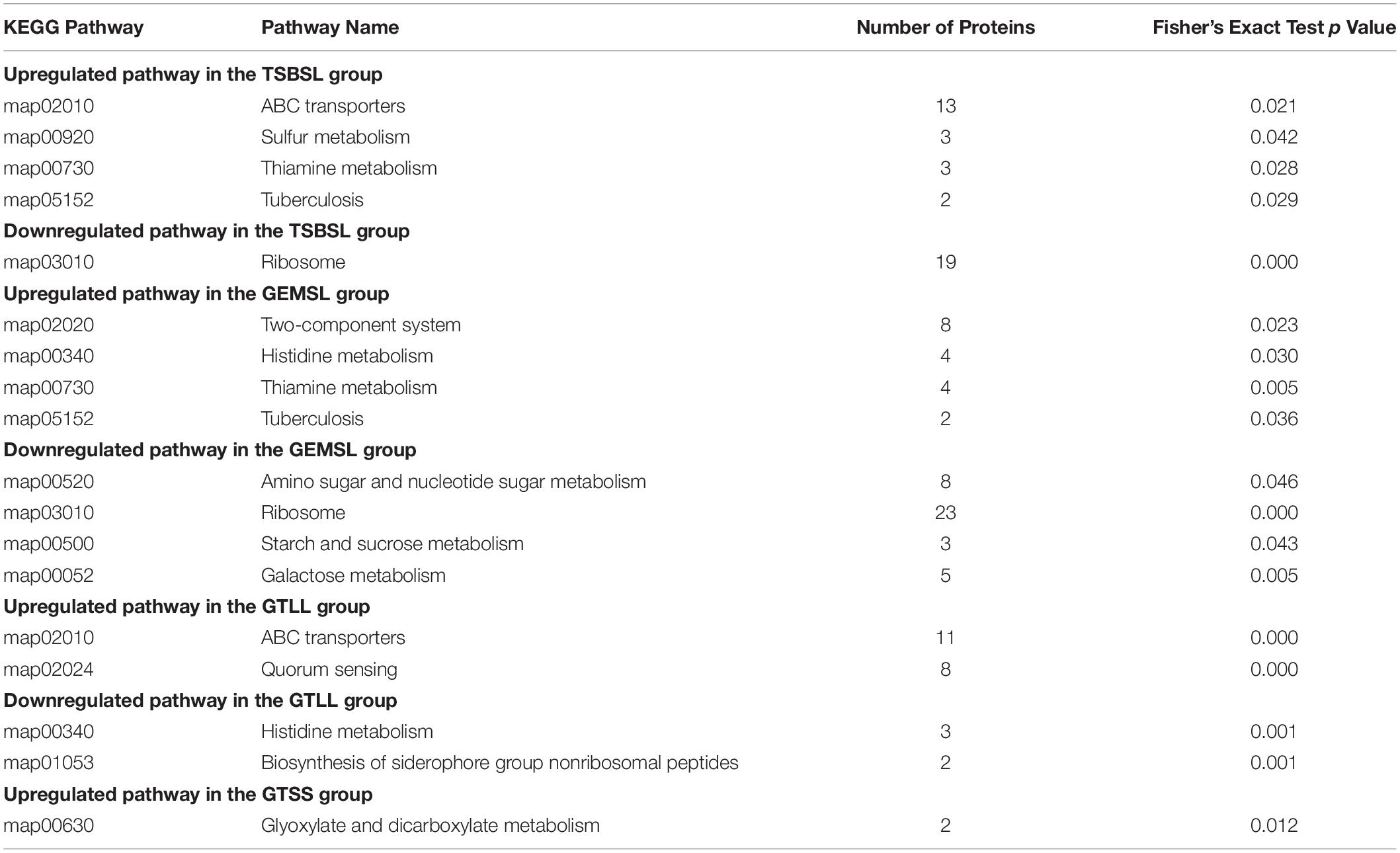
Table 1. Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis for DEPs (Fisher’s exact test, p < 0.05).
In this study, the expression levels of some virulence-associated factors were regulated to varying degrees (Table 2). Upregulated virulence factors found in the GEMSL and TSBSL groups were mainly T4SS proteins, transcriptional regulator VjbR (vjbR), copper-zinc superoxide dismutase (sodC), and integration host factor subunit α (ihfA). Decreased expression of the urease subunit α2 (ureC2) and accessory protein UreG2 (ureG2) was found in the GEMSL and TSBSL groups. Divalent metal cation transporter MntH (mntH) was upregulated in the GTLL group, while no regulated virulence factor was found in the GTSS group.
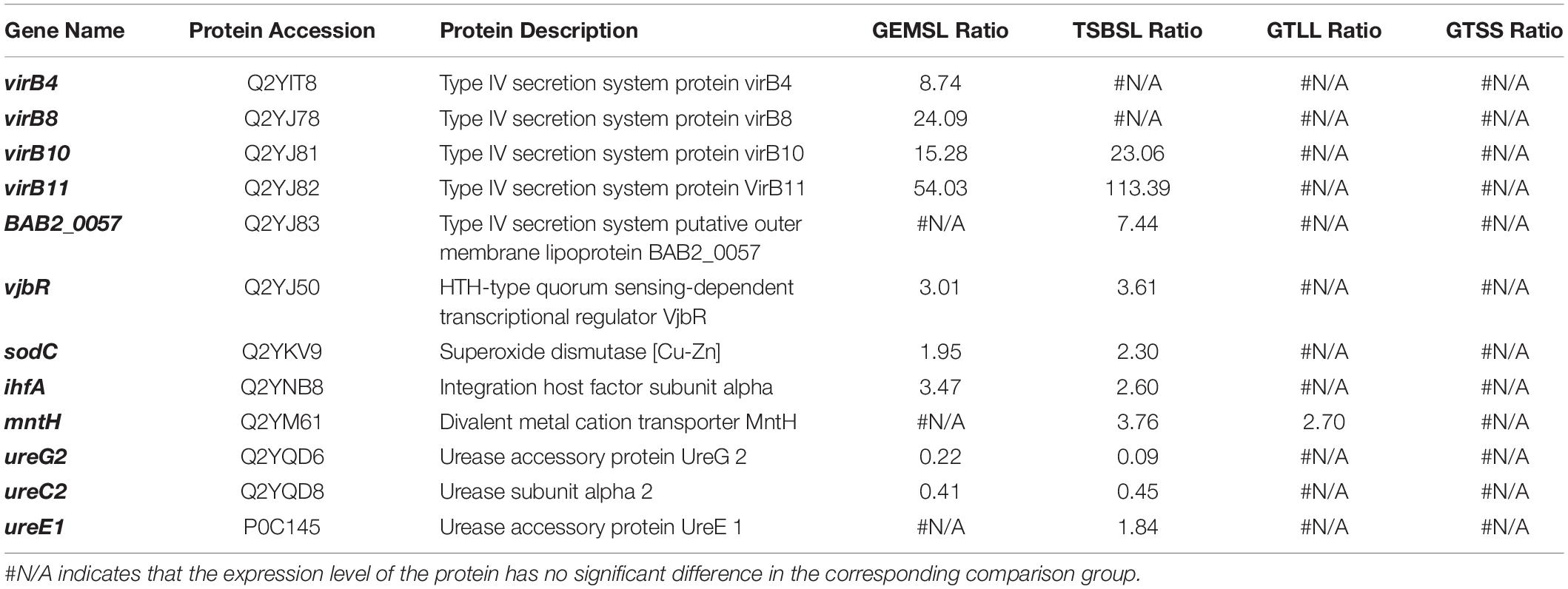
Table 2. Regulation of classical virulence factors.
mRNA levels are typically used to infer the corresponding protein abundances (Kuchta et al., 2018). Therefore, to verify the results of proteomics, we selected 10 DEPs and tested their expression at the mRNA level. These DEPs were mainly related to virulence, stress response, material metabolism, and cell division. Most of the results were consistent with those of the proteomic analysis (Table 3); qRT-PCR showed decreased expression of nrdE, ureG2, nuoD, minD, and recA in the TSBSL and GEMSL groups. The transcription level of lipA decreased in the four groups (data in the groups of GTLL and GTSS not shown), and clpA decreased in the TSBSL group. However, the changes in transcription of BAB2_0427 and vjbR were not consistent with those of protein expression. It is possible that there is a poor linear relationship between mRNA and protein levels.
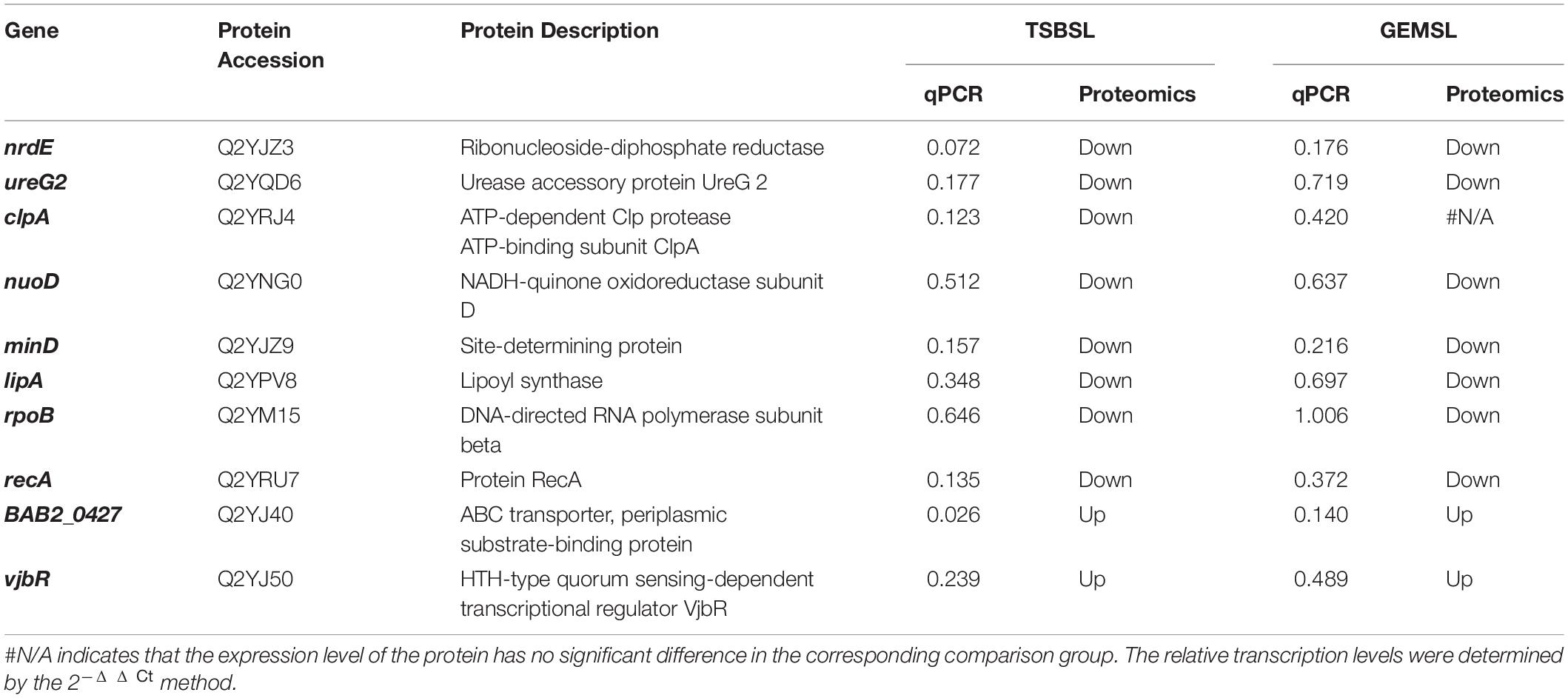
Table 3. Transcriptional level of 10 selected genes by RT-qPCR in the groups of TSBSL and GEMSL.
Brucella is an intracellular bacterial pathogen that encounters a broad range of stresses during its life cycle (Roop et al., 2009). The pressures Brucella faces within a phagosome are similar to those faced during the stationary phase (Roop et al., 2003). Therefore, studying the proteomic differences of B. abortus in the stationary phase may contribute toward a better understanding of its adaptive mechanism within the host cells. Therefore, we analyzed the main protein changes during the transition of B. abortus to the stationary phase at the proteomic level. Additionally, to compare the differences between the two growth stages in response to environmental stress, exponential- and stationary-phase B. abortus were treated with the same starvation stress and analyzed by the proteomic method. Based on the results of the COG, GO, and KEGG analyses, the DEPs were divided into the following different categories and discussed.
T4SS encoded by the virB operon (virB1-12) plays a crucial role in the establishment of chronic infection in B. abortus (Ke et al., 2015). For B. abortus, three T4SS proteins were identified as being induced markedly from exponential to stationary phase, especially virB11 (113.4-fold). The persistent establishment of Brucella infection depends on its ability to survive and proliferate within the host cells (Byndloss and Tsolis, 2016). After invasion, Brucella is contained within a vacuole (BCV), which can control its traffic within the host cell. After a brief interaction between BCV and lysosomes, the undigested Brucella reaches the endoplasmic reticulum and establishes the replicative niche (Celli, 2019). T4SS helps BCVs escape from lysosomal killing in host cells (Macedo et al., 2015). In addition, T4SS is required for the biogenesis of the endoplasmic reticulum-derived replication niche through the secretion and translocation of several effector proteins (Marchesini et al., 2011; Myeni et al., 2013). Several proteins participate in T4SS regulation, including LuxR-type regulator VjbR and integration host factor (Sieira et al., 2004; de Jong et al., 2008; Caswell et al., 2012), which showed increased abundance during the stationary phase in this study.
Copper-zinc superoxide dismutase (Cu/ZnSOD) was more abundant in the TSBSL and GEMSL groups. Cu/ZnSOD protects B. abortus from oxidative damage, thus supporting intracellular survival and replication (Jacobsen et al., 2005; Pratt et al., 2015). In addition, the divalent metal cation transporter MntH showed an increased expression level in the TSBSL group. MntH is responsible for manganese transport and plays a critical role in the virulence of B. abortus 2,308 (Anderson et al., 2009). Surprisingly, two urease-related proteins were repressed during the switch from the exponential to the stationary phase. Urease is a nickel-containing polysubunit enzyme that catalyzes the hydrolysis of urea, resulting in the production of two molecules of ammonia and one molecule of carbon dioxide (Mobley and Hausinger, 1989; Burne and Chen, 2000). Urease enhances the resistance of many pathogenic bacteria to an acidic environment, such as Helicobacter pylori (Debowski et al., 2017) and B. abortus (Sangari et al., 2007). Brucella sp. have two urease operons: ure1 and ure2. The ure1 operon encoded by ureDABCEFG genes is indispensable for urease activity, and ure2 is composed of ureABCEFGDT genes that encode the nickel and urea transport systems (Bandara et al., 2007; Sangari et al., 2007, 2010). The reduction of urease-related proteins could be due to a reduction in the demand for ammonia assimilation. Generally, during the stationary phase of B. abortus, the expression levels of some virulence-related factors were induced.
In stationary-phase B. abortus, the expression levels of some important proteins involved in cell division and DNA replication were downregulated, such as cell division topological specificity factor MinE (minE), site-determining protein MinD (minD), chromosomal replication initiator protein DnaA (dnaA), DNA-directed DNA polymerase (BAB1_0845), and replicative DNA helicase (BAB1_0475). During cell division, the FtsZ ring divides cells equally by forming a diaphragm in the middle of the cell. In gram-negative bacteria, three Min proteins (MinC, MinD, and MinE) prevent the placement of the FtsZ ring in the wrong region (Margolin, 2001; Weiss, 2004). In addition, the expression level of ribonucleoside-diphosphate reductase (nrdE) was 20.4-fold downregulated. The enzyme is responsible for catalyzing the biosynthesis of deoxyribonucleotides, which are essential precursors for DNA replication (Reichard, 1993). The decreased expression of these proteins indicates that cell replication of B. abortus is inhibited during the stationary stage. Invasive B. abortus is mainly in the Gap 1 phase of the cell cycle and maintains a non-proliferative state during the first stage of host cell infection (Deghelt et al., 2014). DNA replication occurring in harsh environments may be detrimental. For example, an acidic pH and reactive oxygen species can lead to DNA damage. Therefore, the inhibition of cell division and DNA replication may be a common strategy for Brucella to cope with environmental stress.
Under starvation treatment, DNA replication and the cell division of B. abortus in the two growth stages were not significantly affected because a few DEPs were observed in the above two categories. The duration of starvation treatment may be short for B. abortus, and the effects on cell division were not significant. Prolonging the starvation treatment time may yield different results.
Two components of RNA polymerase (RNAP) core enzymes, rpoB and rpoC, were repressed in the stationary phase. RNAP core enzymes participate in the synthesis of various RNAs and play an important role in the bacterial transcription process (Sutherland and Murakami, 2018). Three proteins, described as the cold shock DNA-binding domain, encoded by BAB1_1284 and BAB1_1512, respectively, showed reduced expression levels. Some cold shock proteins play a role in RNA chaperone activity and participate in the RNA folding process (Rennella et al., 2017). In addition, the expression levels of some bacterial regulatory proteins related to transcriptional regulation decreased. These findings suggest that B. abortus significantly reduced its own transcription level to adapt to environmental changes during the stationary growth period.
For stationary-phase B. abortus, two bacterial regulatory proteins were regulated under nutrient restriction. However, for exponential-phase B. abortus, more transcription-related DEPs were found according to the COG analysis, including three upregulated and five downregulated proteins. The downregulated proteins were four bacterial regulatory proteins, and one σ factor belonging to the σ70 family was involved in transcription initiation (Paget and Helmann, 2003). This result is easy to be accepted because reduction of transcription levels leads to energy saving, which may be crucial for B. abortus in response to starvation.
The expression of 18 ribosomal proteins was downregulated, representing approximately 10% of coverage of all downregulated proteins during the stationary stage. Ribosomes are molecular machines for protein synthesis and are responsible for transforming genetic codes into amino acid sequences (Opron and Burton, 2018). Reduced expression levels occurred for some enzymes involved in the synthesis of various amino acids, such as glutamate, tryptophan, and branched-chain amino acids, suggesting that B. abortus reduced their metabolic activity. It is not surprising that the level of protein synthesis showed a decline because the stationary phase does not require large amounts of proteins; these are usually needed for rapid cell division.
Under nutritional stress, the expression levels of one ribosomal protein increased in stationary-phase B. abortus. Interestingly, the translational regulation in the starved exponential-phase B. abortus was different, and methionine-tRNA ligase (metG) showed a decreased abundance, which is a critical enzyme for the translation initiation and elongation of protein synthesis by catalyzing the incorporation of methionine to its transfer RNA (tRNA; Wiltrout et al., 2012). The simultaneous repression of transcription- and translation-related processes may indicate that the exponential-phase B. abortus deals with starvation stress by reducing its metabolic activity.
This group of proteins is mainly related to DNA repair, protein folding, or oxidative stress. One reduced protein involved in the SOS response, protein RecA (recA), was observed during the transition from the exponential to stationary phase. The SOS response is an important regulatory system for bacteria to cope with DNA damage, which is regulated by the promoter protein RecA (Janion, 2008; Žgur-Bertok, 2013). In addition, the abundance of some proteins involved in protein folding also decreased. Chaperonin clpA (clpA) can promote specific proteins to unfold and degrade (Singh et al., 2001). The chaperone protein DnaJ (BAB1_2025) as co-chaperone of heat shock protein (Hsp) 70 assists in the folding of nascent proteins, refolding, and degradation of misfolded proteins (Qiu et al., 2006). The reduction of these proteins in the stationary phase indicates that DNA damage and protein misfolding occur more frequently during the exponential phase. In contrast, peroxiredoxin (BAB1_0504), glutaredoxin (grxC), and Cu/ZnSOD (sodC) were upregulated, and these are required for the detoxification of reactive oxygen species (Miao and St Clair, 2009; Rhee, 2016; Ma et al., 2018). The induced expression of stress-related proteins is not surprising because of the rapid consumption of nutrients and the accumulation of toxic metabolites in the stationary phase.
The overexpression of more stress-related proteins was found in the exponential-phase B. abortus in response to starvation. The protein-methionine-sulfoxide reductase catalytic subunit MsrP (msrP) and glutathione S-transferase (BAB2_0230) were 1.71- and 3.06-fold upregulated, respectively, and are responsible for maintaining the redox state of cells and protecting cells from oxidative damage (Allocati et al., 2009; Tarrago et al., 2018). For stationary-phase B. abortus, there was no upregulated protein closely related to the stress response. Therefore, B. abortus in the exponential phase shows a broader response and mobilizes antistress proteins in time to deal with adverse environmental changes compared with stationary-phase B. abortus.
During the transition from the exponential to stationary phase, metabolic downshift occurred in B. abortus. Two NADH-quinone oxidoreductase subunits encoded by nuoB and nuoD, respectively, showed decreased expression levels during the stationary growth period. NADH-quinone oxidoreductase plays a pivotal role in energy production (Novakovsky et al., 2016). The limited utilization of energy may be an important strategy for stationary-phase B. abortus to adapt to survival pressure. The abundance of holo-[acyl carrier protein] (ACP) synthase (a) related to the fatty acid biosynthetic process increased. ACP shuttles acyl intermediates to the fatty acid synthase system. ACP initially exists in the form of inactive apo-ACP and is then modified by holo-ACP synthase to become active (White et al., 2005; Chan and Vogel, 2010; Hiltunen et al., 2010). The increased synthesis of fatty acids is likely for energy storage, which might be conducive to the long-term survival of B. abortus under starvation.
Under starvation treatment, decreased expression levels of phosphogluconate dehydratase (edd) involved in the Entner–Doudoroff (ED) pathway were observed in the exponential-phase B. abortus. The ED pathway is one of the most common glycolytic routes in bacteria but has an inferior energy efficiency (Klingner et al., 2015). The above results show that the ED pathway might not be the optimum method of acquiring energy under starvation stress. Additionally, some enzymes related to the transport and metabolism of amino acids and carbohydrates were significantly upregulated. This suggests that the exponential-phase B. abortus carries out complex nutrient metabolism and transformation in response to starvation. For the stationary-phase B. abortus, glycine cleavage system (GCS) H-protein (gcvH) showed an increased abundance in the starved stationary-phase B. abortus. The GCS is an important pathway in glycine degradation and is widely distributed in animals, plants, and bacteria (Kikuchi et al., 2008). Increased levels of H-protein may suggest an increased utilization of glycine in the starved stationary-phase B. abortus. Of note, it is remarkable that lipA, which is responsible for lipoic acid biosynthesis, was downregulated in all groups (Figure 1C). Lipoic acid as a sulfur-containing cofactor is required for the lipoylation of pyruvate dehydrogenase or α-ketoglutarate dehydrogenase in the citric acid cycle. Therefore, this result suggests that energy production may be limited in B. abortus under growth phase transition or nutritional restriction.
In the stationary phase, one upregulated pathway was ABC transporters in Brucella. These transporters are located in the cytoplasmic membrane or periplasmic space and are responsible for the transport and metabolism of carbohydrate, amino acid, and ion (data not shown). Additionally, the upregulation of sulfur and thiamine metabolism was observed, indicating that the utilization of sulfur and thiamine in the stationary-phase Brucella increased. The ribosome related pathway was downregulated, consistent with the results concerning protein synthesis in the stationary phase.
For exponential B. abortus under starvation, the ABC transporter pathway was also upregulated, suggesting that there was an active substance transport in exponential-phase Brucella in response to starvation. Five of the transporters were also involved in the QS pathway, which allows bacteria to adapt to environmental changes as a collective entity (Waters and Bassler, 2005). Meanwhile, the histidine metabolism pathway was potentially downregulated, and the expression of HutI and Huth, two important enzymes in the histidine utilization (Hut) system (Bender, 2012), decreased synchronously. This may be due to the sudden decrease in histidine availability in the medium, which also indicates that histidine is an important carbon or nitrogen source for the growth and metabolism of Brucella. Interestingly, fewer pathway changes were observed in B. abortus under starvation in the stationary phase, compared with exponential B. abortus; only the glyoxylate and dicarboxylate metabolism pathway, crucial for the synthesis of carbohydrates via the conversion of acetyl CoA (Borjian et al., 2017), was upregulated.
Currently, the proteomic changes of intracellular Brucella have been comprehensively analyzed. Compared with the published results (Lamontagne et al., 2009), we found that the adaptation of Brucella in the stationary phase was similar to that in the early stage of intracellular infection. Most significantly, the expression of proteins (including RNA and protein synthesis-related ones) decreased in Brucella under both conditions. Remarkably, the decreased expression of NADH-1 subunits indicated that a low oxygen tension-based respiration was adopted in the stationary phase and early stage of intracellular infection. Moreover, in the early stage of infection, LPS (one of the cell wall components) synthesis-related protein expression decreased. Similarly, during the stationary phase, the expression of peptidoglycan synthesis-related enzymes, such as glycosyltransferase (BAB1_0932 and BAB1_0114), decreased. However, there were some differences with respect to cell division. The division of Brucella seems to be inactive in both the early stage of intracellular infection and the stationary phase. However, several proteins related to DNA replication and cell division were upregulated in the early stage of intracellular infection. Overall, our results suggest that in the context of proteomics, the physiological status of Brucella in the stationary phase in vitro and in the early stage of intracellular infection is indeed similar. Therefore, stationary-phase Brucella may be a good model for the study of intracellular adaptation.
Interestingly, B. abortus in the two growth stages showed different proteomic changes in response to starvation stress. Compared with the stationary phase, the exponential B. abortus induced more DEPs. Protein synthesis and transcription were significantly inhibited when increased expression of oxidative stress-associated proteins was observed in starved exponential-phase B. abortus. Additionally, more active substance material transport occurred in exponential-phase B. abortus. A previous published study indicated that B. melitensis at the exponential phase were more invasive to host cells than at the stationary growth phase (Rossetti et al., 2009). Our results suggest that exponential-phase B. abortus can make timely metabolic adjustments in response to the challenge of starvation, which contributes to the high invasiveness of exponential-phase Brucella.
To summarize, we explored the proteomic changes in exponential- and stationary-phase B. abortus, employing a label-free quantitative proteomics approach. B. abortus actively adapted to environmental changes in the stationary phase by regulating virulence, reproduction, transcription, translation, stress response, and energy production. Additionally, the protein expression profiles of exponential- and stationary-phase B. abortus, treated with nutrient restriction, were compared and analyzed. First, more DEPs were identified in exponential-phase B. abortus, including 63 upregulated and 29 downregulated proteins. Exponential-phase B. abortus attempt to restrict metabolic activity, resulting in energy savings. Overall, based on the proteomic data, several insights are provided for the intracellular adaptation mechanism of Brucella. In the future, efforts will focus on some of the important DEPs identified in this study.
The datasets generated in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/pride/archive/, PXD020869.
ZC designed the experiments. ML prepared protein samples for LC-MS/MS analysis. ZC, BL, CH, and JL carried out the revision of manuscript. JY analyzed the data and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program Projects of China (2017YFD0500305 and 2017YFD0500901), the National Key Program for Infectious Disease of China (2018ZX10101002-002), the State Key Program of National Natural Science of China (U1808202), the NSFC International (regional) cooperation and exchange program (31961143024), the Major science and technology projects of Inner Mongolia of China (2019ZD-006), and the National Science Foundation for Young Scientists of China (31702276).
ML was employed by the company of Tecon Biological Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank PTM Bio for the help in data analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.598797/full#supplementary-material
Supplementary Figure 1 | Experimental design of the quantitative proteomics. A label-free proteomics approach was used to study the stationary growth stage adaption and nutritional deficiency response of Brucella abortus. Protein samples were collected from the exponential (letter a) and stationary phases (letter b) and then treated with a short-term starvation stress. The identified proteins were analyzed using the GO, COG and KEGG databases.
Supplementary Figure 2 | Coverage of the Brucella abortus proteome and quality control testing of mass spectrometry.(A) Coverage of the B. abortus proteome. The coverage of proteins identified and quantitated in this work was 72.3% and 66.2%, respectively; (B) Protein mass and coverage distribution; (C) Peptide mass tolerance distribution; (D) Identified peptide length distribution.
Supplementary Figure 3 | GO analysis of DEPs for the groups of TSBSL, GEMSL, GTLL, and GTSS. (A) GO analysis of DEPs for the group of TSBSL; (B) GO analysis of DEPs for the group of GEMSL; (C) GO analysis of DEPs for the group of GTLL group; and (D) GO analysis of DEPs for the group of GTSS group.
Ahmed, W., Zheng, K., and Liu, Z. F. (2016). Establishment of chronic infection: Brucella’s stealth strategy. Front. Cell. Infect. Microbiol. 6:30. doi: 10.3389/Fcimb.2016.00030
Al Dahouk, S., Jubier-Maurin, V., Neubauer, H., and Köhler, S. (2013). Quantitative analysis of the Brucella suis proteome reveals metabolic adaptation to long-term nutrient starvation. BMC Microbiol. 13:199. doi: 10.1186/1471-2180-13-199
Al Dahouk, S., Loisel-Meyer, S., Scholz, H. C., Tomaso, H., Kersten, M., Harder, A., et al. (2009). Proteomic analysis of Brucella suis under oxygen deficiency reveals flexibility in adaptive expression of various pathways. Proteomics 9, 3011–3021. doi: 10.1002/Pmic.200800266
Albrethsen, J., Agner, J., Piersma, S. R., Højrup, P., Pham, T. V., Weldingh, K., et al. (2013). Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell Proteomics 12, 1180–1191. doi: 10.1074/Mcp.M112.018846
Allocati, N., Federici, L., Masulli, M., and Di Ilio, C. (2009). Glutathione transferases in bacteria. FEBS J. 276, 58–75. doi: 10.1111/J.1742-4658.2008.06743.X
Anderson, E. S., Paulley, J. T., Gaines, J. M., Valderas, M. W., Martin, D. W., Menscher, E., et al. (2009). The manganese transporter Mnth is a critical virulence determinant for Brucella abortus 2308 in experimentally infected mice. Infect. Immun. 77, 3466–3474. doi: 10.1128/Iai.00444-09
Bandara, A. B., Contreras, A., Contreras-Rodriguez, A., Martins, A. M., Dobrean, V., Poff-Reichow, S., et al. (2007). Brucella suis urease encoded by Ure1 but not Ure2 is necessary for intestinal infection of Balb/C mice. BMC Microbiol. 7:57. doi: 10.1186/1471-2180-7-57
Bender, R. A. (2012). Regulation of the histidine utilization (Hut) system in bacteria. Microbiol. Mol. Biol. Rev. 76, 565–584. doi: 10.1128/Mmbr.00014-12
Borjian, F., Johnsen, U., Schönheit, P., and Berg, I. A. (2017). Succinyl-coa:mesaconate coa-transferase and Mesaconyl-Coa hydratase, enzymes of the methylaspartate cycle in haloarcula hispanica. Front. Microbiol. 8:1683. doi: 10.3389/Fmicb.2017.01683
Burne, R. A., and Chen, Y. Y. (2000). Bacterial ureases in infectious diseases. Microbes Infect. 2, 533–542. doi: 10.1016/S1286-4579(00)00312-9
Byndloss, M. X., and Tsolis, R. M. (2016). Brucella Spp. virulence factors and immunity. Annu. Rev. Anim. Biosci. 4, 111–127. doi: 10.1146/Annurev-Animal-021815-111326
Cardoso, P. G., Macedo, G. C., Azevedo, V., and Oliveira, S. C. (2006). Brucella spp noncanonical lps: structure, biosynthesis, and interaction with host immune system. Microb. Cell Fact. 5:13. doi: 10.1186/1475-2859-5-13
Caswell, C. C., Gaines, J. M., and Roop, R. M. II (2012). The RNA chaperone Hfq independently coordinates expression of the virb type Iv secretion system and the Luxr-type regulator babr in Brucella abortus 2308. J. Bacteriol. 194, 3–14. doi: 10.1128/Jb.05623-11
Celli, J. (2019). The intracellular life cycle of Brucella Spp. Microbiol. Spectr. 7:6. doi: 10.1128/Microbiolspec.Bai-0006-2019
Chan, D. I., and Vogel, H. J. (2010). Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430, 1–19. doi: 10.1042/Bj20100462
de Jong, M. F., Sun, Y. H., Den Hartigh, A. B., Van Dijl, J. M., and Tsolis, R. M. (2008). Identification of Vcea and Vcec, two members of the Vjbr regulon that are translocated into macrophages by the Brucella type Iv secretion system. Mol. Microbiol. 70, 1378–1396. doi: 10.1111/J.1365-2958.2008.06487.X
Debowski, A. W., Walton, S. M., Chua, E. G., Tay, A. C., Liao, T., Lamichhane, B., et al. (2017). Helicobacter pylori gene silencing in vivo demonstrates urease is essential for chronic infection. PLoS Pathog. 13:E1006464. doi: 10.1371/Journal.Ppat.1006464
Deghelt, M., Mullier, C., Sternon, J. F., Francis, N., Laloux, G., Dotreppe, D., et al. (2014). G1-arrested newborn cells are the predominant infectious form of the pathogen Brucella abortus. Nat. Commun. 5:4366. doi: 10.1038/Ncomms5366
Hiltunen, J. K., Chen, Z., Haapalainen, A. M., Wierenga, R. K., and Kastaniotis, A. J. (2010). Mitochondrial fatty acid synthesis–an adopted set of enzymes making a pathway of major importance for the cellular metabolism. Prog. Lipid Res. 49, 27–45. doi: 10.1016/J.Plipres.2009.08.001
Huang, C. H., and Chiou, S. H. (2011). Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J. Med. Sci. 27, 544–553. doi: 10.1016/J.Kjms.2011.06.019
Jacobsen, I., Gerstenberger, J., Gruber, A. D., Bossé, J. T., Langford, P. R., Hennig-Pauka, I., et al. (2005). Deletion of the ferric uptake regulator fur impairs the in vitro growth and virulence of Actinobacillus pleuropneumoniae. Infect. Immun. 73, 3740–3744. doi: 10.1128/Iai.73.6.3740-3744.2005
Janion, C. (2008). Inducible sos response system of dna repair and mutagenesis in Escherichia Coli. Int. J. Biol. Sci. 4, 338–344. doi: 10.7150/Ijbs.4.338
Ke, Y., Wang, Y., Li, W., and Chen, Z. (2015). Type iv secretion system of Brucella Spp. and its effectors. Front. Cell. Infect. Microbiol. 5:72. doi: 10.3389/Fcimb.2015.00072
Ke, Y., Wang, Y., Yuan, X., Zhong, Z., Qu, Q., Zhou, D., et al. (2012). Altered transcriptome Of The B. melitensis vaccine candidate 16mδvjbr, implications for development of genetically marked live vaccine. Indian J. Microbiol. 52, 575–580. doi: 10.1007/S12088-012-0293-8
Kikuchi, G., Motokawa, Y., Yoshida, T., and Hiraga, K. (2008). Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 84, 246–263. doi: 10.2183/Pjab.84.246
Klingner, A., Bartsch, A., Dogs, M., Wagner-Döbler, I., Jahn, D., Simon, M., et al. (2015). Large-scale 13c flux profiling reveals conservation of the entner-doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl. Environ. Microbiol. 81, 2408–2422. doi: 10.1128/Aem.03157-14
Kuchta, K., Towpik, J., Biernacka, A., Kutner, J., Kudlicki, A., Ginalski, K., et al. (2018). Predicting proteome dynamics using gene expression data. Sci. Rep. 8:13866. doi: 10.1038/S41598-018-31752-4
Lamontagne, J., Forest, A., Marazzo, E., Denis, F., Butler, H., Michaud, J. F., et al. (2009). Intracellular adaptation of Brucella abortus. J. Proteome Res. 8, 1594–1609. doi: 10.1021/Pr800978p
Ma, H., Wang, M., Gai, Y., Fu, H., Zhang, B., Ruan, R., et al. (2018). Thioredoxin and glutaredoxin systems required for oxidative stress resistance, fungicide sensitivity, and virulence of alternaria alternata. Appl. Environ. Microbiol. 84:e00086-18. doi: 10.1128/Aem.00086-18
Macedo, A. A., Silva, A. P., Mol, J. P., Costa, L. F., Garcia, L. N., Araújo, M. S., et al. (2015). The abcedcba-encoded abc transporter and the virb operon-encoded type IV secretion system of Brucella ovis are critical for intracellular trafficking and survival in ovine monocyte-derived macrophages. PLoS One 10:E0138131. doi: 10.1371/Journal.Pone.0138131
Marchesini, M. I., Herrmann, C. K., Salcedo, S. P., Gorvel, J. P., and Comerci, D. J. (2011). In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through Virb system. Cell. Microbiol. 13, 1261–1274. doi: 10.1111/J.1462-5822.2011.01618.X
Margolin, W. (2001). Bacterial cell division: a moving mine sweeper boggles the mind. Curr. Biol. 11, R395–R398. doi: 10.1016/S0960-9822(01)00217-2
McDermott, J., Grace, D., and Zinsstag, J. (2013). Economics of Brucellosis impact and control in low-income countries. Rev. Off. Int. Epizoot. 32, 249–261. doi: 10.20506/Rst.32.1.2197
Miao, L., and St Clair, D. K. (2009). Regulation of superoxide dismutase genes: implications in disease. Free Radic. Biol. Med. 47, 344–356. doi: 10.1016/J.Freeradbiomed.2009.05.018
Mobley, H. L., and Hausinger, R. P. (1989). Microbial ureases: significance, regulation, and molecular characterization. Microbiol. Rev. 53, 85–108.
Myeni, S., Child, R., Ng, T. W., Kupko, J. J. III, Wehrly, T. D., Porcella, S. F., et al. (2013). Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathog. 9:E1003556. doi: 10.1371/Journal.Ppat.1003556
Navarro Llorens, J. M., Tormo, A., and Martínez-García, E. (2010). Stationary phase in gram-negative bacteria. FEMS Microbiol. Rev. 34, 476–495. doi: 10.1111/J.1574-6976.2010.00213.X
Novakovsky, G. E., Dibrova, D. V., and Mulkidjanian, A. Y. (2016). Phylogenomic analysis of type 1 nadh:quinone oxidoreductase. Biochem. Mosc. 81, 770–784. doi: 10.1134/S0006297916070142
Opron, K., and Burton, Z. F. (2018). Ribosome structure, function, and early evolution. Int. J. Mol. Sci. 20:40. doi: 10.3390/Ijms20010040
Paget, M. S., and Helmann, J. D. (2003). The Sigma70 family of sigma factors. Genome Biol. 4:203. doi: 10.1186/Gb-2003-4-1-203
Pappas, G., Papadimitriou, P., Akritidis, N., Christou, L., and Tsianos, E. V. (2006a). The new global map of human Brucellosis. Lancet Infect. Dis. 6, 91–99. doi: 10.1016/S1473-3099(06)70382-6
Pappas, G., Panagopoulou, P., Christou, L., and Akritidis, N. (2006b). Brucella as a biological weapon. Cell. Mol. Life Sci. 63, 2229–2236. doi: 10.1007/S00018-006-6311-4
Pratt, A. J., Didonato, M., Shin, D. S., Cabelli, D. E., Bruns, C. K., Belzer, C. A., et al. (2015). Structural, functional, and immunogenic insights on Cu, Zn superoxide dismutase pathogenic virulence factors from Neisseria meningitidis and Brucella abortus. J. Bacteriol. 197, 3834–3847. doi: 10.1128/Jb.00343-15
Qiu, X. B., Shao, Y. M., Miao, S., and Wang, L. (2006). The diversity of the Dnaj/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 63, 2560–2570. doi: 10.1007/S00018-006-6192-6
Reichard, P. (1993). From RNA To DNA, why so many ribonucleotide reductases. Science 260, 1773–1777. doi: 10.1126/Science.8511586
Rennella, E., Sára, T., Juen, M., Wunderlich, C., Imbert, L., Solyom, Z., et al. (2017). RNA binding and chaperone activity of the E. coli cold-shock protein CSPA. Nucleic Acids Res. 45, 4255–4268. doi: 10.1093/Nar/Gkx044
Robertson, G. T., and Roop, R. M. Jr. (1999). The Brucella abortus host factor I (Hf-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol. Microbiol. 34, 690–700. doi: 10.1046/J.1365-2958.1999.01629.X
Roop, R. M. II, Gaines, J. M., Anderson, E. S., Caswell, C. C., and Martin, D. W. (2009). Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Med. Microbiol. Immunol. 198, 221–238. doi: 10.1007/S00430-009-0123-8
Roop, R. M. II, Gee, J. M., Robertson, G. T., Richardson, J. M., Ng, W. L., and Winkler, M. E. (2003). Brucella stationary-phase gene expression and virulence. Annu. Rev. Microbiol. 57, 57–76. doi: 10.1146/Annurev.Micro.57.030502.090803
Rossetti, C. A., Galindo, C. L., Lawhon, S. D., Garner, H. R., and Adams, L. G. (2009). Brucella melitensis global gene expression study provides novel information on growth phase-specific gene regulation with potential insights for understanding Brucella:host initial interactions. BMC Microbiol. 9:81. doi: 10.1186/1471-2180-9-81
Sangari, F. J., Cayón, A. M., Seoane, A., and García-Lobo, J. M. (2010). Brucella abortus Ure2 region contains an acid-activated urea transporter and a nickel transport system. BMC Microbiol. 10:107. doi: 10.1186/1471-2180-10-107
Sangari, F. J., Seoane, A., Rodríguez, M. C., Agüero, J., and García Lobo, J. M. (2007). Characterization of the urease operon of Brucella abortus and assessment of its role in virulence of the bacterium. Infect. Immun. 75, 774–780. doi: 10.1128/Iai.01244-06
Sieira, R., Comerci, D. J., Pietrasanta, L. I., and Ugalde, R. A. (2004). Integration host factor is involved in transcriptional regulation of the Brucella abortus virb operon. Mol. Microbiol. 54, 808–822. doi: 10.1111/J.1365-2958.2004.04316.X
Singh, S. K., Rozycki, J., Ortega, J., Ishikawa, T., Lo, J., Steven, A. C., et al. (2001). Functional domains of the Clpa and Clpx molecular chaperones identified by limited proteolysis and deletion analysis. J. Biol. Chem. 276, 29420–29429. doi: 10.1074/Jbc.M103489200
Sutherland, C., and Murakami, K. S. (2018). An introduction to the structure and function of the catalytic core enzyme of Escherichia Coli RNA polymerase. Ecosal Plus 8:e0004-2018. doi: 10.1128/Ecosalplus.Esp-0004-2018
Tarrago, L., Grosse, S., Siponen, M. I., Lemaire, D., Alonso, B., Miotello, G., et al. (2018). Rhodobactersphaeroides methionine sulfoxide reductase P reduces R- and S-diastereomers of methionine sulfoxide from a broad-spectrum of protein substrates. Biochem. J. 475, 3779–3795. doi: 10.1042/Bcj20180706
Wang, Y., Ke, Y., Duan, C., Ma, X., Hao, Q., Song, L., et al. (2019). A small non-coding RNA facilitates Brucella melitensis intracellular survival by regulating the expression of virulence factor. Int. J. Med. Microbiol. 309, 225–231. doi: 10.1016/J.Ijmm.2019.04.002
Waters, C. M., and Bassler, B. L. (2005). Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21, 319–346. doi: 10.1146/Annurev.Cellbio.21.012704.131001
Weiss, D. S. (2004). Bacterial cell division and the septal ring. Mol. Microbiol. 54, 588–597. doi: 10.1111/J.1365-2958.2004.04283.X
White, S. W., Zheng, J., Zhang, Y. M., and Rock (2005). The structural biology of type ii fatty acid biosynthesis. Annu. Rev. Biochem. 74, 791–831. doi: 10.1146/Annurev.Biochem.74.082803.133524
Wiltrout, E., Goodenbour, J. M., Fréchin, M., and Pan, T. (2012). Misacylation Of TRNA with methionine in Saccharomyces cerevisiae. Nucleic Acids Res. 40, 10494–10506. doi: 10.1093/Nar/Gks805
Zai, X., Yang, Q., Yin, Y., Li, R., Qian, M., Zhao, T., et al. (2017). Relative quantitative proteomic analysis of Brucella abortus reveals metabolic adaptation to multiple environmental stresses. Front. Microbiol. 8:2347. doi: 10.3389/Fmicb.2017.02347
Keywords: Brucella abortus, proteomics, exponential phase, stationary phase, nutritional stress
Citation: Yang J, Liu M, Liu J, Liu B, He C and Chen Z (2020) Proteomic Analysis of Stationary Growth Stage Adaptation and Nutritional Deficiency Response of Brucella abortus. Front. Microbiol. 11:598797. doi: 10.3389/fmicb.2020.598797
Received: 25 August 2020; Accepted: 11 November 2020;
Published: 15 December 2020.
Edited by:
Harold J. Schreier, University of Maryland, College Park, United StatesReviewed by:
Diego J. Comerci, CONICET Institute of Biotechnological Research (IIB-INTECH), ArgentinaCopyright © 2020 Yang, Liu, Liu, Liu, He and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoshan Liu, bGJzbGd5QHN5YXUuZWR1LmNu; Chuanyu He, aGN5MDYxMTIwQDE2My5jb20=; Zeliang Chen, emVsaWFuZ2NoZW5AeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.