
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 December 2020
Sec. Microbial Physiology and Metabolism
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.593217
 Jin Lü1
Jin Lü1 Qingshan Long1
Qingshan Long1 Zhilong Zhao2Lu Chen1Weijun He1Jiali Hong1Kai Liu1Yemin Wang1
Zhilong Zhao2Lu Chen1Weijun He1Jiali Hong1Kai Liu1Yemin Wang1 Xiuhua Pang2Zixin Deng1
Xiuhua Pang2Zixin Deng1 Meifeng Tao1*
Meifeng Tao1*Bacteria of the genus Saccharopolyspora produce important polyketide antibiotics, including erythromycin A (Sac. erythraea) and spinosad (Sac. spinosa). We herein report the development of an industrial erythromycin-producing strain, Sac. erythraea HOE107, into a host for the heterologous expression of polyketide biosynthetic gene clusters (BGCs) from other Saccharopolyspora species and related actinomycetes. To facilitate the integration of natural product BGCs and auxiliary genes beneficial for the production of natural products, the erythromycin polyketide synthase (ery) genes were replaced with two bacterial attB genomic integration sites associated with bacteriophages ϕC31 and ϕBT1. We also established a highly efficient conjugation protocol for the introduction of large bacterial artificial chromosome (BAC) clones into Sac. erythraea strains. Based on this optimized protocol, an arrayed BAC library was effectively transferred into Sac. erythraea. The large spinosad gene cluster from Sac. spinosa and the actinorhodin gene cluster from Streptomyces coelicolor were successfully expressed in the ery deletion mutant. Deletion of the endogenous giant polyketide synthase genes pkeA1-pkeA4, the product of which is not known, and the flaviolin gene cluster (rpp) from the bacterium increased the heterologous production of spinosad and actinorhodin. Furthermore, integration of pJTU6728 carrying additional beneficial genes dramatically improved the yield of actinorhodin in the engineered Sac. erythraea strains. Our study demonstrated that the engineered Sac. erythraea strains SLQ185, LJ161, and LJ162 are good hosts for the expression of heterologous antibiotics and should aid in expression-based genome-mining approaches for the discovery of new and cryptic antibiotics from Streptomyces and rare actinomycetes.
Bioactive natural products isolated through actinomycete fermentation processes are important sources of therapeutics and agrochemicals, including antibacterials (e.g., erythromycin A, tylosin, and vancomycin); antifungals (e.g., amphotericin B); immunosuppressants (e.g., FK-506 and rapamycin); anticancer agents (e.g., doxorubicin and epoxomicin); anthelmintics (e.g., avermectin); and insecticides (e.g., spinosad) (Challis, 2014; Pham et al., 2019). Most of these compounds are isolated from the most dominant actinomycete genus, Streptomyces. However, several important compounds come from non-Streptomyces actinomycetes, known as rare actinomycetes, such as vancomycin (Amycolatopsis orientalis), erythromycin (Saccharopolyspora erythraea), and spinosad (Saccharopolyspora spinosa) (Pham et al., 2019). Indeed, rare actinomycetes have been regarded as a storehouse of novel antibiotics (Tiwari and Gupta, 2012), and novel natural products are increasingly discovered from among this group (Nett et al., 2009; Choi et al., 2015).
Genome sequencing has revealed that each actinomycete genome usually harbors more than ten biosynthetic gene clusters (BGCs) for different secondary metabolites, and a majority of these BGCs are silent or cryptic under normal laboratory cultivation conditions (Nett et al., 2009; Choi et al., 2015). Heterologous biosynthesis has emerged as a viable route to access the beneficial properties of cryptic natural products. Intact BGC of interest is cloned into a suitable vector and delivered into a heterologous host for optimal production of the unknown compound, using the host’s gene expression machinery, precursor substances, and cofactors. Bioinformatics tools, such as antiSMASH, NP.searcher, and ClustScan, are available for the prediction of BGCs having the potential to synthesize novel compounds (Starcevic et al., 2008; Li et al., 2009; Blin et al., 2019). Various techniques, for example, the transformation-associated recombination system (Kouprina and Larionov, 2016), integrase-mediated recombination system (Olorunniji et al., 2019), and bacterial artificial chromosome (BAC) system (Sosio et al., 2000), have been adapted for cloning some large, intact BGCs. However, the current heterologous hosts of actinomycetes mostly belong to the Streptomyces genus, including Streptomyces coelicolor (Gomez-Escribano and Bibb, 2011); Streptomyces lividans (Xu et al., 2016, 2020; Zhao et al., 2016; Gao et al., 2017; Chen et al., 2018; Peng et al., 2018); Streptomyces avermitilis (Komatsu et al., 2013); and Streptomyces albus (Chater and Wilde, 1976; Myronovskyi et al., 2018). Few heterologous hosts are derived from rare actinomycetes, mainly due to the lack of efficient genetic manipulation systems that would enable the substantial strain engineering required for removing internal competitive biosynthetic pathways, such as polyketide BGCs (Pfeifer and Khosla, 2001).
Site-specific recombination systems enable one to construct recombinant plasmids in an experimentally tractable host such as Escherichia coli and then transfer the plasmids into recipients by conjugation (Flett et al., 2006). The well-characterized ϕC31 and ϕBT1 attachment/integration (att/int) systems have been engineered into many integrative Streptomyces plasmids, allowing the efficient integration of large plasmids into the highly conserved and relatively neutral attB sites in Streptomyces genomes (Zhang et al., 2016). Many actinomycetes, such as Sac. erythraea, lack a typical attB site, which has largely impeded efforts for the delivery of exogenous DNA. Indeed, previous studies have introduced an attBϕC31 site into the genome of Sac. erythraea for the purpose of improving production of erythromycin by transferring exogenous or endogenous genes (Rodriguez et al., 2003; Wu et al., 2011). In another approach to strain engineering, the entire spinosad BGC was assembled using a multi-step homologous recombination procedure and was used to replace the native erythromycin BGC in the wild-type strain Sac. erythraea ATCC 40137 (Huang et al., 2016); through substantial genetic engineering and mutagenesis, this heterologous expression system allowed spinosad production to reached a titer of 800 mg/L in the resulting Sac. erythraea strain, which is hundreds of times higher than those in Streptomyces hosts, including S. albus, S. coelicolor, and S. lividans (Huang et al., 2016; Tan et al., 2017; Zhao et al., 2017). These studies confirmed the suitability of Saccharopolyspora spp. for expression of heterologous BGCs from related actinobacterial species, and also suggested the necessity of non-Streptomyces actinomycete hosts for heterologous expression-based genome mining.
In this study, we engineered an erythromycin-producing strain of Saccharopolyspora into a suitable heterologous host for the expression of large size polyketide BGCs, by optimizing the genetic manipulation system, deleting background polyketide biosynthetic pathways, and other modifications of the host genome.
Strains used in this study are listed in Table 1. Sac. erythraea HOE107 and its derivatives were grown on ESM medium at 34°C for spore preparation as previously described (Li et al., 2013). E. coli strains, Staphylococcus aureus, and Bacillus mycoides were cultured in Luria-Bertani (LB) medium at 37°C. The following antibiotics were added to the medium when required: apramycin (50 μg/mL), hygromycin (100 μg/mL), spectinomycin (50 μg/mL), ampicillin (100 μg/mL), kanamycin (50 μg/mL), chloramphenicol (25 μg/mL). Sta. aureus and B. mycoides were used as indicator strains in the bioassay experiments. E. coli XL1-Blue (Stratagene) was used as the host for cosmid library construction, and E. coli DH10B (Invitrogen) was used for general cloning, plasmid maintenance, and as host for the BAC library. E. coli ET12567/pUB307 was used as a helper strain to mobilize oriTRK2-plasmids from E. coli into Sac. Erythraea by tri-parental conjugation (Flett et al., 2006).

Table 1. Bacterial strains used in this study.
pIJ4642 (Kieser et al., 1992) was used as template for PCR amplification of the aadA resistance marker. SuperCos 1 (Stratagene) was used to construct the genomic cosmid library of Sac. erythraea HOE107. pOJ260 (Bierman et al., 1992) was used as a non-replicating suicide vector for gene replacement in Sac. erythraea. pWHU2653, bearing an engineered clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 combined with the counterselection system CodA(sm) (Zeng et al., 2015), was used as gene deletion vector in Sac. erythraea. pHL931 (Zhao et al., 2016) was the vector for the genomic BAC library of Sac. spinosa NRRL18395. pJTU6728 is a derivative of the integrative vector pMS82 and carries the integration site attP and int loci of the Streptomyces temperate phage ϕBT1 (Gregory et al., 2003) and contains the transcription factor gene nusG, global regulator genes afsRScla, and the two drug efflux pump genes mdfAco and lrmAco (Peng et al., 2018). pJTU6728 was used to improve the production of heterologous antibiotics in Sac. erythraea.
Isolation of DNA and all subsequent manipulations were performed according to standard protocols (Kieser et al., 2000). Primers used in this study are listed in Table 2. BAC and plasmid constructs are listed in Table 3.

Table 2. Primers used in this study.
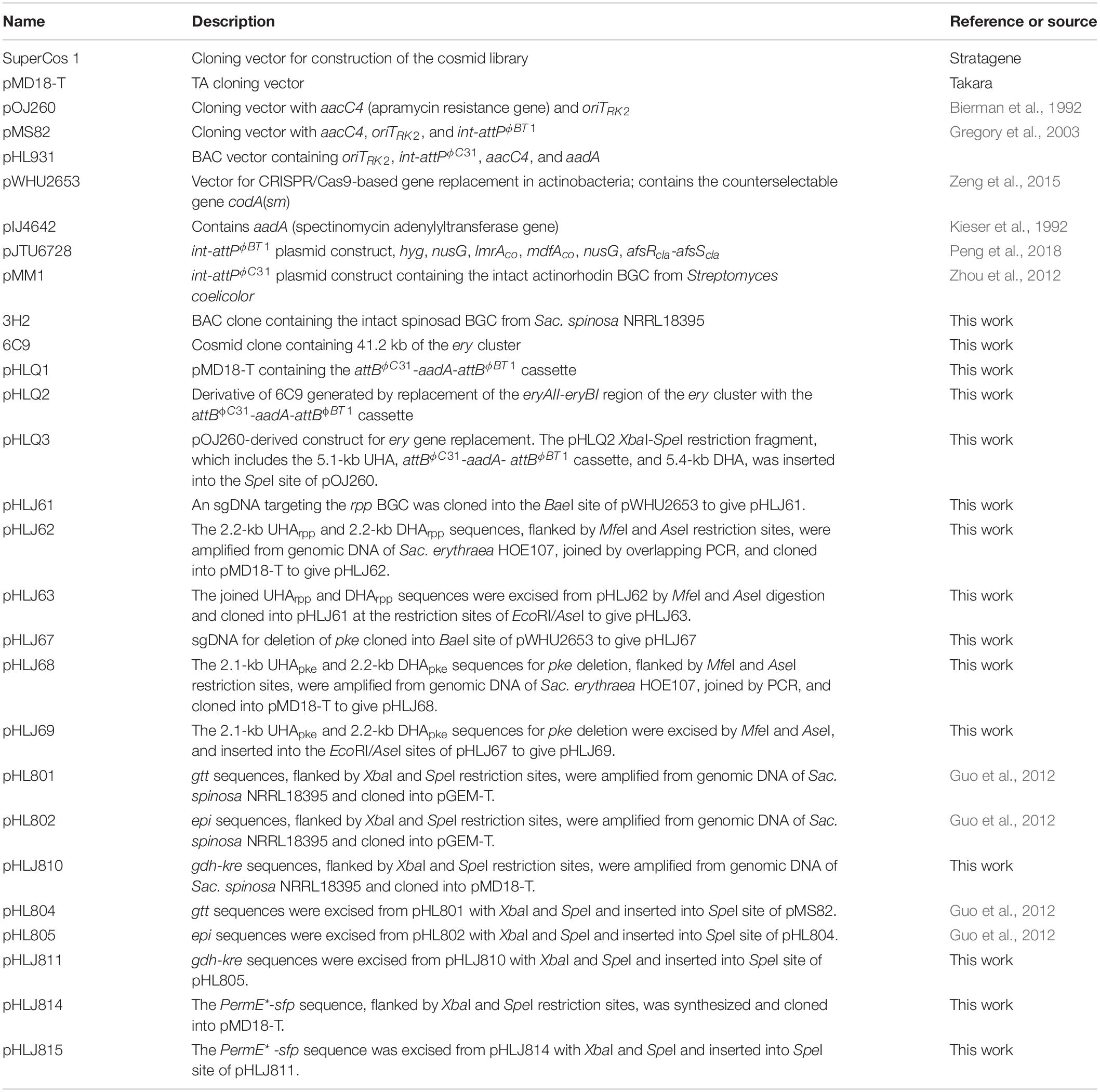
Table 3. BAC and plasmid constructs used in this study.
The Sac. spinosa NRRL18395 genomic BAC library was constructed according to a standard protocol (Wang et al., 2005; Zhao et al., 2016). An overnight culture of Sac. spinosa NRRL18395 was harvested, embedded in agarose plugs, and the cells in the plugs were digested with lysozyme and proteinase K and then partially digested with Sau3AI. The ca. 150 kb genomic DNA fragments were fractionated by pulsed-field gel electrophoresis (PFGE), recovered, and ligated with BamHI-linearized and dephosphorylated BAC vector pHL931. The ligation mixture was electroporated into E. coli DH10B competent cells and plated onto LB agar plates with apramycin (50 μg/mL) for an overnight incubation at 37°C. The resulting transformants were picked and stored in 96-well microtiter plates.
The genomic cosmid library of Sac. erythraea HOE107 was constructed as described previously (Gould et al., 1998). The genomic DNA of the bacterium was extracted and partially digested with MboI. The 35–45 kb fragments were isolated by PFGE, dephosphorylated, and ligated into the SuperCos 1 vector. The resulting ligation mixture was packaged into the λ phage, followed by phage transfection into the E. coli XL1-Blue MR strain. A cosmid, 6C9, was identified by PCR and end-sequencing as carrying a 40.9 kb eryCI-eryCVI region of the ery gene cluster.
The mobilization of the BAC clones from the E. coli host to Sac. erythraea was accomplished using a triparental conjugation approach, including E. coli ET12567/pUB307 (helper), E. coli DH10B/BACs (donors), and Sac. erythraea SLQ185 (recipient). The Sac. erythraea spores (108 cfu) were washed and resuspended in 250 μL 2 × YT broth (Kieser et al., 2000) at a concentration of 108 per mL before incubation at 50°C for 10 min. E. coli DH10B containing conjugative BAC clones and E. coli ET12567/pUB307 were grown in LB separately to an OD600 of 0.6 at 37°C. These cells were washed twice with LB and resuspended together in 250 μL 2 × YT. Subsequently, the E. coli donor-helper mixtures were added to the spores, mixed thoroughly by pipetting, and spread on the solid conjugation media ESM; 2CM (Li and Piel, 2002); IWL4 (Min et al., 2003); or ISP4 (Choi et al., 2004). After an incubation of 16–20 h, each plate was flooded with trimethoprim (50 μg/mL) and apramycin (50 μg/mL) and incubated for 5 days at 34°C. Transfer frequency was then calculated as the ratio between the number of exconjugants on an antibiotic-selective plate and the number of recipient cells. The average frequency of three independent experiments was calculated.
The Sac. erythraea spores (recipient) and E. coli ET12567/pUB307 (helper) were prepared as described above and resuspended together in 500 μL 2 × YT. Then, the mixtures of recipient and helper cells were spread on ISP4 plates, followed by air-drying. The BAC library (conjugation donor) was inoculated into 96-well plates with antibiotic-selective LB (130 μl per well) at 37°C overnight, and then transferred to antibiotic-free LB and cultured for 4-6 h until the OD600 reached 0.4 to 0.6. The E. coli DH10B/BACs were then transferred with a 48-pin replicator onto the helper-recipient pre-coated plates and incubated at 34°C for 20 h. Then, the conjugation plates were flooded with apramycin and trimethoprim to a final concentration of 50 μg/ml and incubated at 34°C for another 5 days.
The ery gene cluster was replaced with an attBϕC31-aadA-attBϕBT1 cassette via homologous recombination. Firstly, the aadA (aminoglycoside resistance gene) was amplified from pIJ4642 using primers aadA-F and aadA-R, which have, respectively, an attBϕC31 and attBϕBT1 sequence at the 5′-end. The resulting PCR product, attBϕC31-aadA-attBϕBT1, was cloned into pMD18-T vector (Takara) to generate pHLQ1. The attBϕC31-aadA-attBϕBT1 cassette on pHLQ1 was amplified by PCR with primer pairs eryT-F/eryT-R, which harbor overhang regions complementary to the boundaries of the eryAI-eryBIII genomic region. Genes eryAI-eryBIII in cosmid 6C9 were then replaced with the attBϕC31-aadA-attBϕBT1 cassette, using a λ-Red recombination protocol (Gust et al., 2003), to generate pHLQ2, which contains an ery gene cluster disruption cassette with an attBϕC31-aadA-attBϕBT1 fragment flanked with a 5.1-kb upper homologous arm (UHA) and a 5.4-kb downstream homologous arm (DHA) homologous to the ery gene cluster. The ery disruption cassette was excised from pHLQ2 with XbaI and SpeI and inserted into the SpeI site of pOJ260 (Bierman et al., 1992) to generate the ery gene replacement construct pHLQ3. pHLQ3, which does not contain an autonomous replication region or integration locus that function in actinobacteria, was introduced into Sac. erythraea HOE107 by the triparental conjugation approach. Spectinomycin-resistant and apramycin-sensitive mutant strains were verified by PCR and sequence analysis, using primer pairs eryV1-F/eryV1-R, eryV2-F/eryV2-R, and eryV3-F/eryV3-R. The confirmed mutant strains were renamed Sac. erythraea SLQ185.
For deletion of the pke gene cluster, we selected pkeA1-pkeA4 between ORFs SACE_RS20070 and SACE_RS20095 (RefSeq NC_009142.1) as the knock-out target, using a CRISPR/Cas9-CodA(sm)-based approach (Zeng et al., 2015). A double-stranded DNA fragment encoding a small guide RNA (sgRNA) scaffold was obtained by annealing the 5′-phosphorylated oligonucleotides pkeG-F and pkeG-R. The DNA fragment was cloned into vector pWHU2653 (Zeng et al., 2015) at the BaeI restriction site to generate pHLJ67. Next, 2.1-kb UHA and 2.2-kb DHA sequences were amplified from Sac. erythraea HOE107 genomic DNA using the primer pairs pkeU-F/pkeU-R and pkeD-F/pkeD-R, respectively, and the two DNA fragments were joined together by overlapping PCR using the primer pair pkeU-F/pkeD-R. The PCR product was cloned into the pMD18-T TA cloning vector to generate pHLJ68, and then the UHA-DHApke region from pHLJ68 was cloned into pHLJ67 at the EcoRI-AseI sites to generate the pke gene cluster replacement construct pHLJ69.
For deletion of the rpp gene cluster, we selected the orfA-orfF region (including rppAB) between SACE_RS06025 and SACE_RS06085 (RefSeq NC_009142.1) as the knock-out target, using the CRISPR-Cas9-based approach (Zeng et al., 2015). A double-stranded DNA encoding an sgRNA scaffold was obtained by annealing the 5′-phosphorylated oligonucleotides rppG-F and rppG-R. The DNA fragment was cloned into vector pWHU2653 via the BaeI restriction site to generate pHLJ61. The 2.2-kb UHA and 2.2-kb DHA sequences were amplified from Sac. erythraea HOE107 genomic DNA using the primer pairs rppU-F/rppU-R and rppD-F/rppD-R, respectively, and the two DNA fragments were joined together by overlapping PCR using the primer pair rppU-F/rppD-R. The PCR product was cloned into pMD18-T to generate pHLJ62, and then the UHA-DHArpp region from pHLJ62 was cloned into pHLJ61 at the EcoRI-AseI sites to generate the rpp gene cluster replacement construct pHLJ63.
pHLJ69 and pHLJ63 were separately transformed into Sac. erythraea SLQ185 by E. coli-Sac. erythraea triparental conjugation. Independent apramycin-resistant exconjugants were streaked onto solid ESM medium, containing 800 μg/mL 5-fluorocytosine (5FC, for selection against the vector backbone) and 25 μg/mL nalidixic acid (for selection against E. coli), and then grown in the dark for 5 days. The 5FC-resistant colonies were replicated onto plates with or without apramycin to confirm plasmid loss. Genomic DNA of single apramycin-sensitive colonies was extracted and used to screen for gene replacement mutants by PCR. The pkeA1–pkeA4 deletion mutant, named Sac. erythraea LJ161, was confirmed by PCR with primer pairs pkeV4-F/pkeV4-R and pkeV5-F/pkeV5-R and sequence analysis. The rpp deletion mutant, named Sac. erythraea LJ162, was confirmed by PCR with primer pairs rppV6-F/rppV6-R and rppV7-F/rppV7-R and sequence analysis.
The gtt, epi, and gdh-kre gene sequences were amplified from Sac. spinosa NRRL18395 genomic DNA using the primer pairs gtt-F1/gtt-R1, epi-F1/epi-R1, and GK-F1/GK-R1 respectively. The three PCR products were cloned into pMD18-T to generate pHL801, pHL802, and pHLJ810. The 1299-bp SpeI/XbaI fragment containing gtt from pHL801 was cloned into the SpeI site of pMS82 to give pHL804. The 1193-bp SpeI/XbaI fragment containing epi from pHL802 was cloned into the SpeI site of pHL804 to give pHL805. The 2459-bp SpeI/XbaI fragment containing gdh-kre from pHLJ810 was cloned into the SpeI site of pHL805 to give pHLJ811. The codon-optimized gene sfp was synthesized based on the protein sequence of Sfp of Bacillus subtilis (AEK64474.1), using the web server for codon optimization1. The strong, constitutive promoter PermE∗ (Bibb et al., 1985) was placed upstream of sfp to control sfp expression, and the PermE∗-sfp DNA fragment with flanking XbaI and SpeI sites was cloned into pMD18-T to generate pHLJ814. The 804-bp SpeI/XbaI restriction fragment containing PermE∗-sfp was excised from pHL814 and ligated with the SpeI-linearized pHLJ811 to give pHLJ815.
Fresh spores of Sac. erythraea HOE107 and SLQ185 were spread onto EFM supplemented with 3 mM Fe3+. After culturing at 28°C for 7 days, agar plugs (8-mm diameter) were taken and put onto the surface of LB agar plates previously inoculated with an overnight culture (1:100) of either Micrococcus luteus or B. subtilis, followed by incubation overnight at 37°C.
For erythromycin production, three pieces of culture lawn (ca. 1.5 cm2) were cut from the sporulating plates and inoculated into 25 mL of seed medium (0.5% glucose, 2.5% corn starch, 1% yeast extract, 1% whole-milk powder, 0.2% MgSO4⋅7H2O, pH 7.2) in a 250 mL flask and incubated at 28°C for 72 h on a rotary shaker at 250 rpm as described previously (Huang et al., 2016). Then, 2 mL of seed culture was inoculated in 30 mL of the fermentation medium EFM [4% cornstarch, 3% soybean flour, 3% dextrin, 0.2% (NH4)2SO4, 1% soybean oil, 6% CaCO3, pH7.2] (Li et al., 2013) in a 250 mL flask and incubated under the same conditions for 7 days. Erythromycin was extracted from the fermentation culture following the methods described previously (Le et al., 2001; Kirm et al., 2013). Briefly, the broth was adjusted to pH 10 and mixed with an equal volume of acetonitrile for 40 min. Then, 2 g NaCl was added per 10 mL broth, left to dissolve, and the acetonitrile phase was then separated by centrifugation.
For spinosad production, strains were cultured in seed medium as described above, and then 2 mL of cultured seed was inoculated into 30 mL of the fermentation media EFM or HJFM (9% glucose, 2% whole-milk powder, 2.5% cottonseed cake powder, 0.2% yeast powder, 0.1% lactic acid, 0.4% trisodium citrate, 0.2% K2HPO4, pH 7.2) (Huang et al., 2016) in a 250 mL flask under the same conditions for 10 days. After fermentation, 1 mL of each culture was extracted with 4 mL of methanol in an ultrasonic bath for 30 min, centrifuged, and the supernatant was analyzed by high-performance liquid chromatography (HPLC) to detect the spinosad production. At the meantime, each culture was sampled for measurement of the biomass dry weight: 1 mL culture was centrifuged at 9,000 rpm for 10 min to collect the pellet, which was hold in the desiccator at 75°C for 3 days to obtain a constant weight.
For actinorhodin production, strains were cultured in R3 agar medium (Shima et al., 1996) at 28°C for 7 days, and then 500 mg of each culture containing both bacteria and agar was taken from the plates and put into 1.5 mL Eppendorf tubes, followed by the addition of 500 μl of KOH or methanol to each tube. The tube contents were dispersed in a homogenizer with glass beads (0.1 mm in diameter) and centrifuged at 13,000 rpm for 10 min to remove the particulate matter and collect the liquid crude extract for the measurement of actinorhodin by optical absorbance.
The metabolite sample of erythromycin was applied to a Zorbax SB-C18 column (5 μm particle size, 4.6 × 250 mm, Agilent, Germany) installed on the Agilent 1260 Infinity II LC system. Isocratic elution was applied with the mobile phase consisting of 40% 50 mM K2HPO4 (pH adjusted to 9 with diluted phosphoric acid) and 60% acetonitrile. After injection of 10 μl of the sample solution, the HPLC system was operated at a flow rate of 0.6 mL/min, with a total run time of 50 min. The column temperature was set at 60°C and the detection wavelength at 206 nm as described previously (Kirm et al., 2013). Reference substance erythromycin A (purity > 98%) was obtained from Aladdin Bio-Chem Technology Co. (Shanghai, China). A standard solution was prepared by dissolving 5 mg of erythromycin in 5 mL of ethanol.
The metabolite sample of spinosad was analyzed by liquid chromatography/mass spectrometry (LC/MS) using the Agilent 1260 Infinity II LC system coupled to Agilent 6470 triple quadrupole mass spectrometry (MS) instruments. The separation was performed on a Zorbax SB-C18 column (5 μm particle size, 4.6 × 250 mm, Agilent) and elution was performed with an isocratic mobile phase consisting of methyl-acetonitrile-0.05% sodium acetate with the volume ratio of 45:45:10, at a flow rate of 0.35 mL/min, and detected at 250 nm as described previously (Huang et al., 2016). The MS analysis was conducted in the positive ion mode with capillary voltage and nozzle voltage set at 3,500 and 500 V, respectively. The gas temperature was set to 300°C at a flow rate of 5 L/min. Sheath gas temperature was set to 250°C at a flow rate of 11 L/min. Spinosad was monitored using multiple reaction monitoring transitions at 732.5 to 142.1 and 98.1 m/z for spinosyn A, and 746.5 to 142.1 and 98.1 m/z for spinosyn D with positive electrospray ionization. The obtained data were evaluated by Agilent MassHunter workstation software (Agilent Technologies). Data are representative of three independent experiments. Reference substance spinosad (66% spinosyn A and 28% spinosyn D) was obtained from Dr. Ehrenstorfer GmbH (Augsburg, Germany). A standard solution was prepared by dissolving 5 mg of spinosad in 5 mL of methanol.
For the metabolite sample of actinorhodin, the absorbance at 640 nm was read using a microplate reader (Bioteke Corporation), and the background absorbance of the culture extraction in the absence of bacteria was subtracted. Data are representative of three independent experiments.
Saccharopolyspora erythraea HOE107 has been used for the industrial production of erythromycin. It is an erythromycin-overproducing descendent of Sac. erythraea NRRL 23338 obtained by mutagenesis and selection. Genome sequencing of Sac. erythraea HOE107 revealed multiple point mutations, including a nonsense mutation in a polyketide biosynthetic gene cluster pks3 as reported in Sac. erythraea Px (Peano et al., 2012; Li et al., 2013). To re-engineer this strain for the heterologous production of other polyketides, we replaced a 30.7-kb fragment of the erythromycin BGC in Sac. erythraea HOE107 by an attBϕC31-aadA-attBϕBT1 cassette via double crossover between a gene replacement construct pHLQ3 and the Sac. erythraea HOE107 chromosome (Figure 1A). Intergenus conjugation between E. coli DH10B/pHLQ3 and Sac. erythraea HOE107 and subsequent screening of the resulting exconjugants gave rise to 20 spectinomycin-resistant and apramycin-sensitive colonies. The genotype of SLQ185 was verified by PCR, using primer pairs eryV1-F/R to confirm that the spectinomycin resistance gene aadA was successfully inserted at gene cluster ery (Figures 1A,B); using primer pairs eryV2-F/R to confirm deletion of the partial gene cluster ery; and using primer pairs eryV3-F/R to confirm the loss of the apramycin resistance gene (aacC4), indicating that the plasmid pHLQ3 was eliminated (Figure 1B). However, PCR indicated that only three of the 20 SpcRAprS colonies were gene replacement mutants, in which nine erythromycin biosynthetic genes, i.e., eryAII-eryBIII, and part of eryAI were deleted. One of these mutants was named SLQ185.
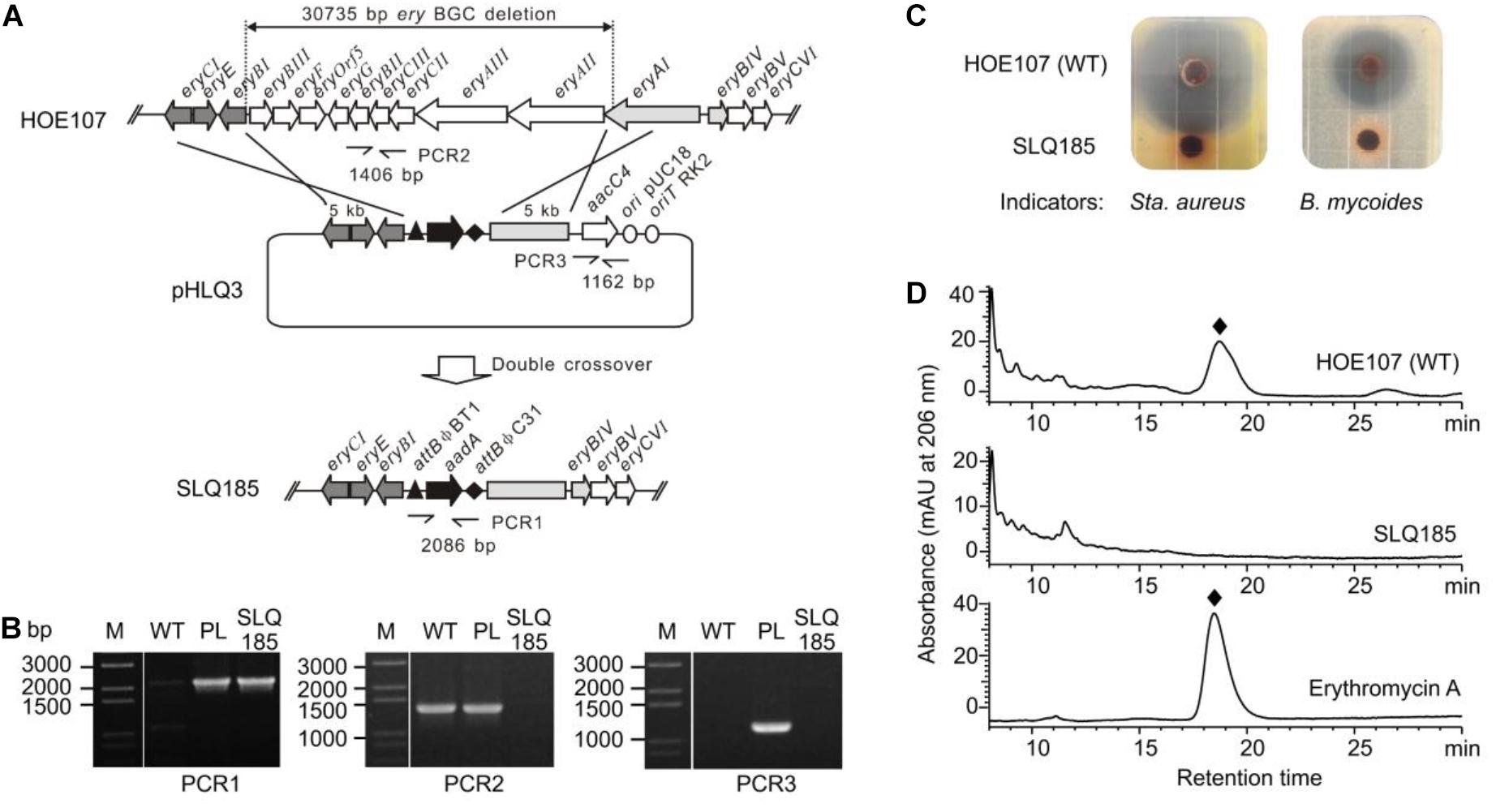
Figure 1. Replacement of the erythromycin BGC on the Sac. erythraea chromosome with the attBϕC31-aadA-attBϕBT1 cassette. (A) Schematic representation of the homologous recombination (double crossover) between the wild-type ery locus and the gene replacement vector pHLQ3 to give the gene replacement mutant SLQ185. PCR1, PCR2, and PCR3 are PCR reactions designed for the verification of the mutant. (B) Verification of the genotype of the mutant SLQ185 by PCR. M, molecular marker; WT, Sac. erythraea HOE107; PL, plasmid pHLQ3; SLQ185, Sac. erythraea SLQ185. (C) Analysis of the antibacterial activity of the Sac. erythraea strains by growth inhibition of Sta. aureus (left) and B. mycoides (right). (D) HPLC analysis of the production of erythromycin A in Sac. erythraea HOE107 and SLQ185. The bottom profile shows the erythromycin A standard.
Since erythromycin produced by Sac. erythraea displays broad-spectrum activity against Gram-positive organisms at very low concentrations, we tested the antibacterial phenotype of the gene replacement mutant strain Sac. erythraea SLQ185. The wild-type strain and SLQ185 were cultivated on EFM agar medium, which was supplemented with 3 mM Fe3+ to suppress the possible production of erythrochelin, a 2,5-diketopiperazine siderophore that shows weak antibacterial activity against Gram-positive organisms (Lazos et al., 2010). As shown in Figure 1C, the plugs of cultivated Sac. erythraea HOE107 produced large zones of inhibition against Sta. aureus or B. subtilis whereas the plugs of SLQ185 did not produce any inhibition zone, suggesting the loss of erythromycin production. HPLC analysis confirmed that the erythromycin biosynthesis was completely abolished in the gene replacement mutant SLQ185 (Figure 1D).
As one study indicated that the type I PKS BGC pke and the type III PKS BGC rpp were actively expressed in Sac. erythraea and competed with erythromycin production (Li et al., 2013), we decided to delete these two PKS BGCs from the SLQ185 genome to make a clean metabolic background and to avoid potential substrate competition between endogenous and heterologous biosynthetic pathways. To construct the deletion mutants in an efficient way, we chose the CRISPR/Cas9-CodA(sm) gene knockout system, which worked well in Streptomyces spp. (Zeng et al., 2015). The CRISPR/Cas9-based RNA-guided DNA digestion was utilized to stimulate homologous recombination between the target chromosome locus and the homologous repair template pairs provided by a gene-targeting construct. The counterselection marker codA(sm) on the plasmid, which confers 5-fluorocytosine (5FC) sensitivity to the host cell, was used for the selection of a recombinant that had lost the target genes and the plasmid backbone (Zeng et al., 2015).
To delete the PKS genes pkeA1–pkeA4 from the pke BGC, the pHLJ69 was constructed based on the CRISPR/Cas9-CodA vector pWHU2653 and introduced into SLQ185. pHLJ69 contains the codon-optimized CRISPR-Cas9 gene scas9; a sgRNA gene targeting the pke BGC; the codA(sm) gene; and two DNA fragments of 2.1 and 2.2 kb, which were homologous to the flanking regions of the pke BGC and were used as repair templates (Figure 2A). After conjugal transfer of pHLJ69 into SLQ185 and subsequent screening, 20 resulting 5FC-resistant and apramycin-sensitive colonies were randomly picked and tested by PCR. The PCR results confirmed that all colonies were pkeA1-pkeA4 deletion mutants, one of which is named Sac. erythraea LJ161 and shown in Figure 2B.
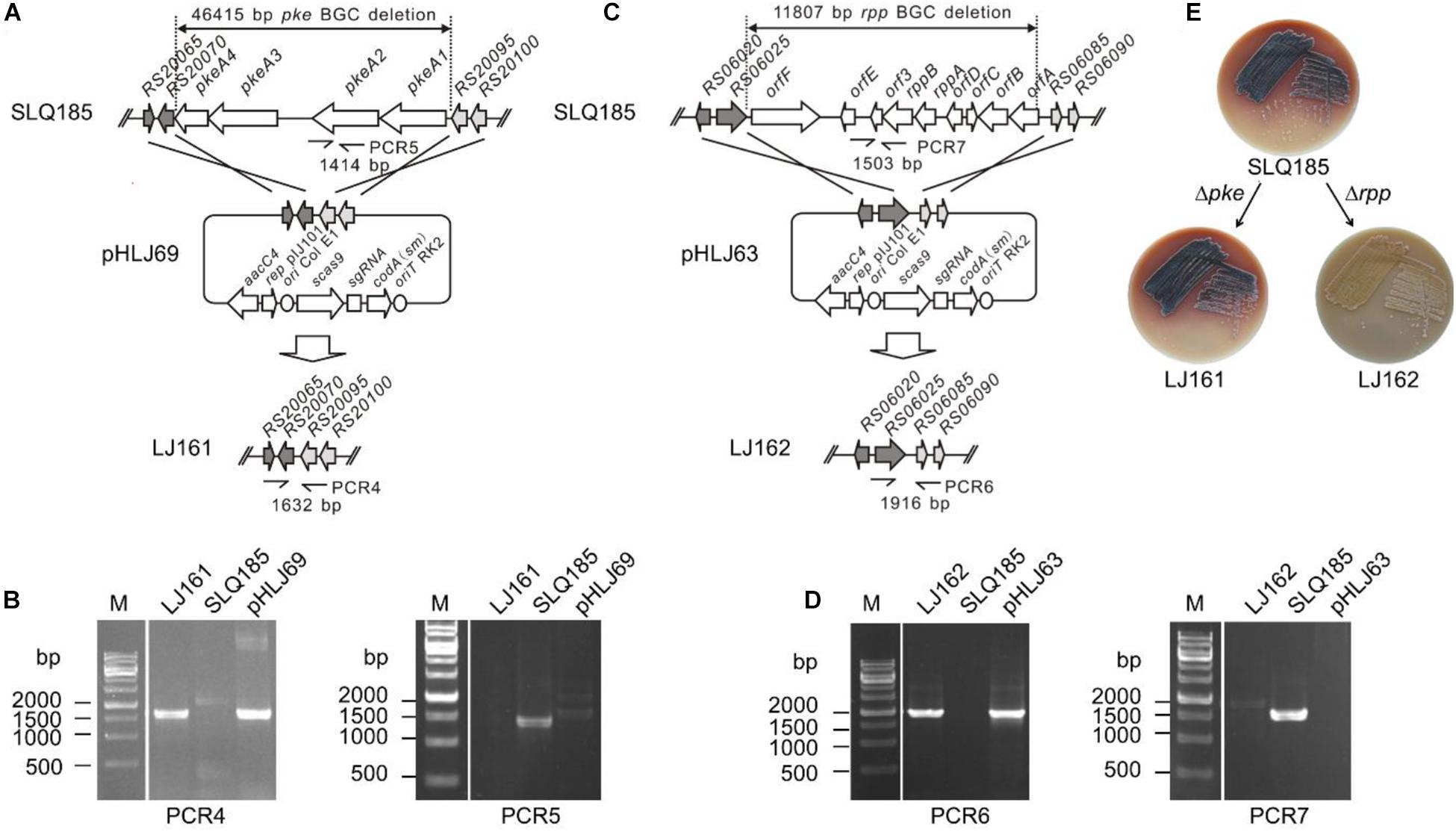
Figure 2. CRISPR/Cas9-CodA(sm)-mediated deletion of the pke and rpp PKS biosynthetic gene clusters. (A) Schematic representation of the homologous recombination between the pke locus and the gene deletion construct pHLJ69 generating the gene deletion mutant LJ161. PCR4 and PCR5 are PCR reactions designed for the verification of the mutant. (B) Verification of the genotype of LJ161 by PCR. M, molecular marker. (C) Schematic representation of the homologous recombination between the rpp locus and the gene deletion construct pHLJ63 generating the gene deletion mutant LJ162. PCR6 and PCR7 are PCR reactions designed for the verification of the mutant. (D) Verification of the genotype of LJ162 by PCR. (E) Photographs showing the surface growth and pigmentation production of the parental strain and deletion mutants. Photos were taken after 7 days of cultivation on ESM medium at 34°C.
To delete the rpp BGC, the rpp-targeting plasmid pHLJ63, containing two 2.2-kb homologous repair arms, was constructed and introduced into Sac. erythraea SLQ185 by intergenus conjugation (Figure 2C). Twenty 5FC-resistant and apramycin-sensitive colonies were randomly selected and all were confirmed by PCR to be rpp deletion mutants, one of which is named Sac. erythraea LJ162 and shown in Figure 2D. In contrast to the pkeA1-pkeA4-deletion strain Sac. erythraea LJ161, which showed no obvious difference in growth or colony morphology compared with the parental strain Sac. erythraea SLQ185, the rpp mutant strain exhibited an albino phenotype and failed to produce the diffusible brown pigment characteristic of Sac. Erythraea when grown on solid ESM medium (Figure 2E).
To test whether BAC clones harboring large size inserts could be efficiently transferred into the engineered Sac. erythraea strains from E. coli, we first constructed a BAC library using large fragments of genomic DNA from Sac. spinosa NRRL18395, the native producer of the macrolide polyketide insecticide spinosad. An integrative BAC vector pHL931 (Zhao et al., 2016), which contains an attP-int locus of phage ϕC31, was used as the cloning vector. All BAC clones would harbor the attPϕC31-int locus for mediating their own integration into actinomycete chromosomes at the attBϕC31 site via site-specific recombination. The resultant BAC library had 960 clones and an average insert size of 110-kb (Figure 3A).
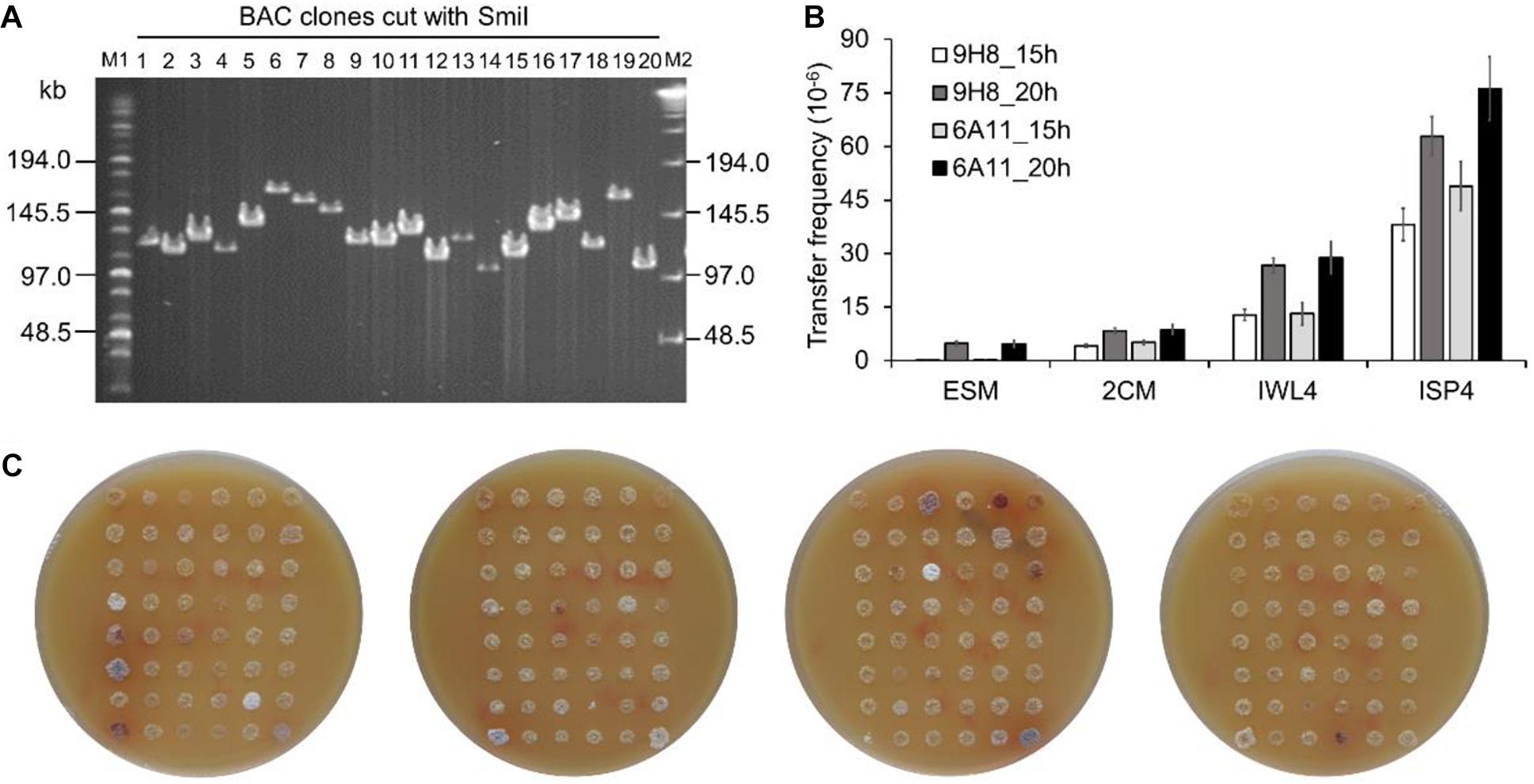
Figure 3. Transfer of large size BAC clones into Sac. erythraea. (A) Analysis of the insert size of Sac. spinosa NRRL18395 BAC clones by PFGE. M1, middle range PFG marker (NEB); M2, lambda ladder PFG marker (NEB); lanes 1–20, recombinant DNA digested with SmiI and obtained from alkaline lysis minipreps of randomly picked colonies. (B) Conjugation transfer of BAC clones 9H8 and 6A11 into Sac. erythraea SLQ185 evaluated on four types of agar media. The conjugation plates were incubated for 15 or 20 h before overlaying with antibiotics. Data are from three biological replicates. (C) High-throughput conjugation transfer of the BAC library into Sac. erythraea SLQ185 on the ISP4 agar plates. Photos of the conjugation plates were taken after 5 days of prolonged cultivation at 34°C after flooded with antibiotics for the selection of exconjugants. Spots indicate the exconjugants of corresponding clones of the Sac. spinosa genomic BAC library.
Two BAC clones, E. coli DH10B/9H8 and DH10B/6A11, which have sizes of 140 kb and 124 kb respectively, were randomly selected as the donors to explore a triparental conjugation protocol with E. coli ET12567/pUB307 as the helper and Sac. erythraea SLQ185 as the recipient. The donor, helper, and recipient cells were mixed and spread on four types of agar media (2CM, ISP4, IWL4 and ESM) separately and incubated for 15 or 20 h before overlaying with antibiotics for the selection of exconjugants. Two crucial parameters of conjugal transfer, i.e., the conjugation media and the time point for antibiotic overlay, were evaluated in these experiments. Of the media tested, ISP4 agar produced about ten times more exconjugants than 2CM and ESM did. A longer pre-incubation time (20 h) before antibiotic overlaying also led to slightly more exconjugants (p = 0.0037). The optimal conditions resulted in a high frequency of conjugal transfer of BAC clones from E. coli to Sac. erythraea SLQ185 (6.3 × 10–5 and 7.6 × 10–5 exconjugants/recipient) (Figure 3B). Under the same conditions, the smaller integrative vectors pSET152 (5.7 kb) and pHL931 (16.7 kb) gave two orders of magnitude more exconjugants than did these two BAC clones. When the SLQ185-derived strain LJ161 was used as the recipient, similar conjugation frequencies were observed. Since no spores were formed on the aerial hyphae of rpp-deletion strain Sac. erythraea LJ162 (Figure 2E), we had to use mycelium without heat-shock treatment instead of heat-shocked spores as the recipient during conjugation transfer, which reduced the frequency of conjugation slightly. We then tested the engineered strain Sac. erythraea SLQ185 as the recipient in high-throughput triparental conjugation with the BAC library. As shown in Figure 3C, the arrayed BAC genomic library of Sac. spinosa NRRL18395 was effectively transferred into SLQ185, in that all BAC clones gave growth spots consisting of exconjugants.
Spinosad is a mixture of spinosyns A and D produced by a type I PKS gene cluster (spn) from Sac. spinosa NRRL 18395. Most of the genes involved in spinosad biosynthesis are located in this cluster, which spans a 80-kb region, except for the four rhamnose biosynthetic genes gtt, epi, gdh, and kre, which are dispersed in the genome (Waldron et al., 2001). The sfp gene, which codes for a 4′-phosphopantetheinyl transferase, also contributes to spinosad synthesis, as introduction of this gene increased the heterologous production of spinosad in Sac. erythraea (Huang et al., 2016).
To express the spinosad biosynthetic pathway in our engineered Sac. erythraea strains, we screened the BAC library and isolated 3H2, a BAC clone harboring a 128-kb genomic insert covering the entire 80-kb spinosad BGC. We also constructed an attPϕBT1-based integrative plasmid, pHLJ815, carrying four rhamnose biosynthetic genes and a synthetic sfp gene. The attPϕC31-based BAC clone 3H2 and the attPϕBT1-based plasmid pHLJ815 were transferred into Sac. erythraea strains SLQ185, LJ161, and LJ162 for the heterologous expression of spinosad.
Firstly, we tested the effect of fermentation media on the heterologous production of spinosad. SLQ185/3H2/pHLJ815 was fermented in the erythromycin industrial fermentation medium (EFM) or HJFM, a medium previously used for the heterologous expression of spinosad (Huang et al., 2016). LC/MS analysis of the extracts of the fermented cultures showed that spinosad was produced in both media, although the yield of spinosad (i.e., sum of spinosyns A and D) with EFM was 102% higher than with HJFM (p = 0.00011) (Figure 4A). The increase of spinosad yield was partially due to the increase in dry weight, with EFM producing 69% (p = 0.0012) more biomass when compared with HJFM (Figure 4B).
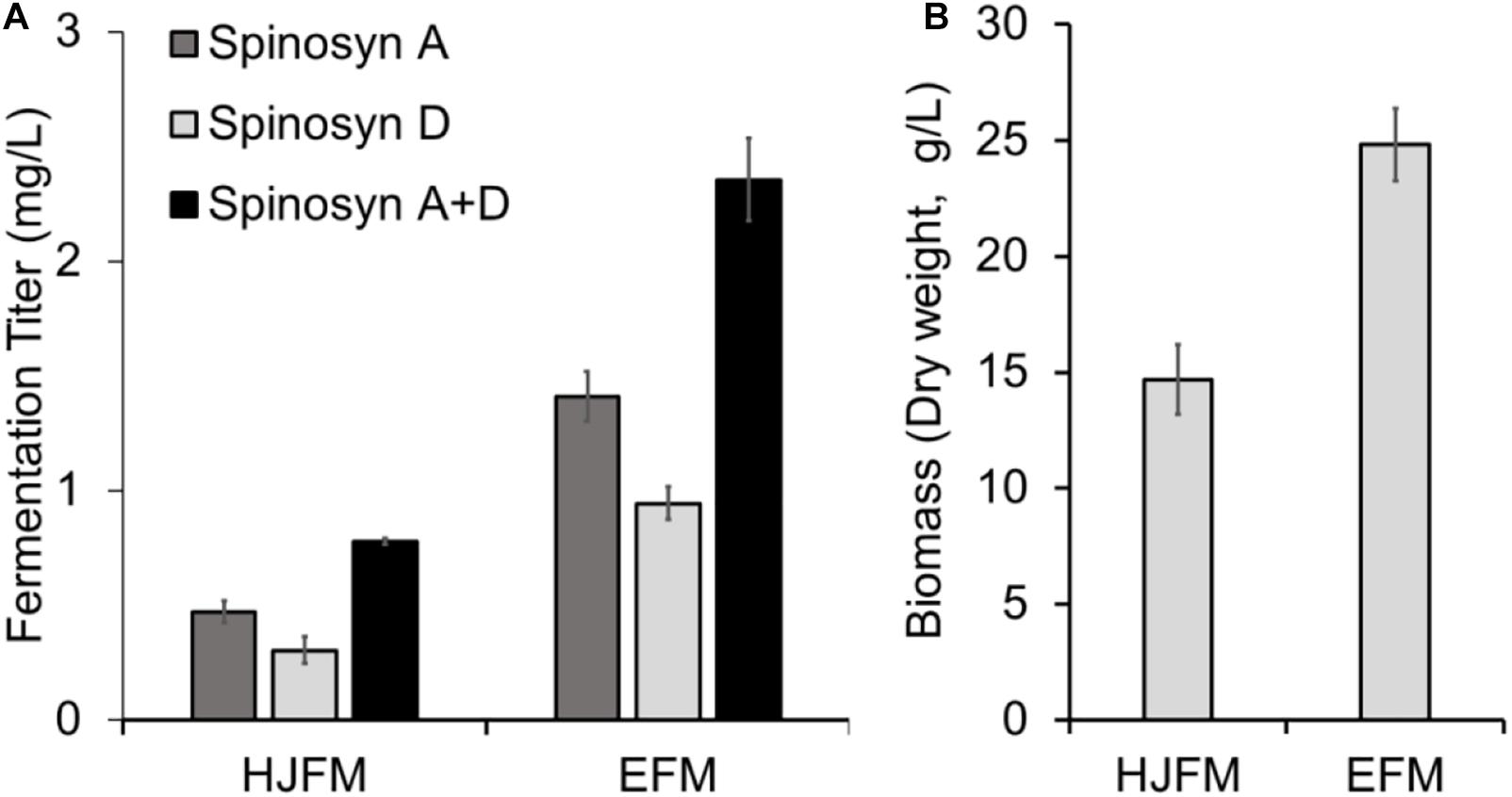
Figure 4. Heterologous production of spinosad by Sac. erythraea SLQ185/3H2/pHLJ815 in two types of fermentation media. (A) Production of spinosad in HJFM and EFM media. (B) Bacterial biomass. The dry weight of SLQ185/3H2/pHLJ815 was measured following growth in the two types of media. Data are from three biological replicates.
To evaluate the impact of the pke or rpp deletion on the production of spinosad, BAC clone 3H2 and pHLJ815 were transferred into the engineered strains Sac. erythraea strains LJ161 and LJ162, generating LJ161/3H2/pHLJ815 and LJ162/3H2/pHLJ815, respectively. We found that the yield of spinosad in LJ161/3H2/pHLJ815 and LJ162/3H2/pHLJ815 improved slightly, by 58% (p = 0.0070) and 34% (p = 0.011), respectively, when compared to SLQ185/3H2/pHLJ815. The highest yield of spinosad was produced by LJ161/3H2/pHLJ815, reaching a level of 3.73 mg/L (Figure 5A). In addition, the dry weight of LJ162/3H2/pHLJ815 was improved by 45% (p = 0.0078) in comparison with that of SLQ185/3H2/pHLJ815 (Figure 5B).
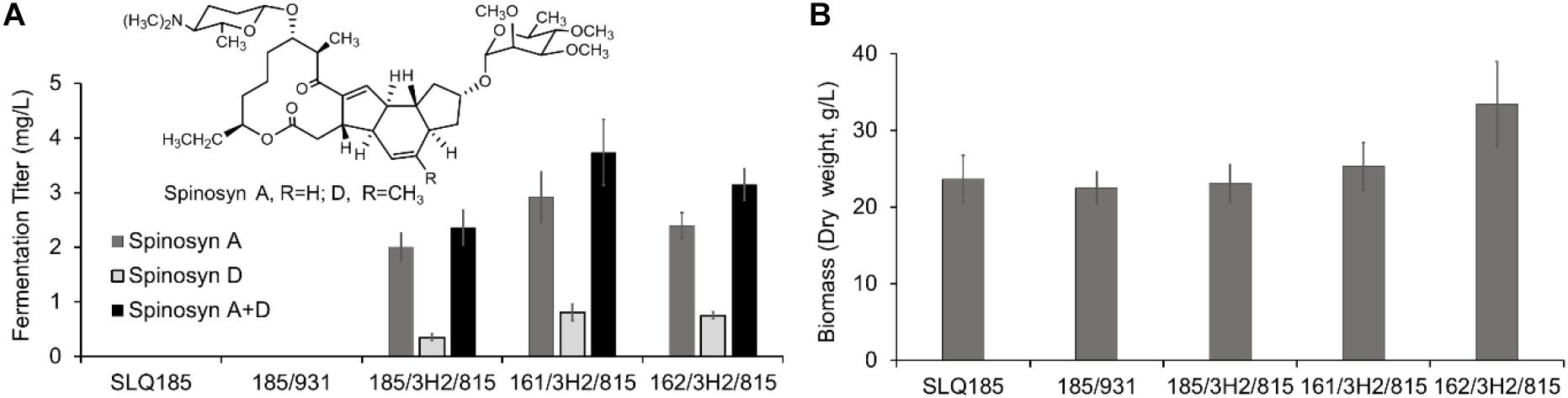
Figure 5. Heterologous production of spinosad by Sac. erythraea strains SLQ185, LJ161, and LJ162 carrying 3H2 and pHLJ815. 3H2 contains the 80-kb spn BGC, and pHLJ815 contains the rhamnose biosynthetic genes (gtt, epi, gdh, kre) and sfp. (A) Fermentation titer of spinosyns A and D. (B) Bacterial biomass. The dry weight of Sac. erythraea strains was measured following growth in the fermentation medium EFM for 10 days. Data are from three biological replicates.
Actinorhodin is synthesized from acetyl-CoA and malonyl-CoA by a type II PKS encoded by the act BGC in S. coelicolor A3(2) (Bystrykh et al., 1996). To assess the effects of the deletion of PKS BGCs (pkeA1-pkeA4 and rpp) on the heterologous biosynthesis of actinorhodin, the previously described attPϕC31-based integrative plasmid pMM1 (Zhou et al., 2012), which contains the entire actinorhodin BGC, was introduced into Sac. erythraea SLQ185, LJ161, and LJ162 to generate SLQ185/pMM1, LJ161/pMM1, and LJ162/pMM1, respectively. When the exconjugants were cultivated on solid R3 medium, the heterologous expression of the actinorhodin BGC resulted in the observable production of the blue-pigmented actinorhodin (Figure 6A). To compare actinorhodin production quantitatively, the cultures were extracted with either 1 M KOH as described previously (Bystrykh et al., 1996) or methanol, and the extracts were monitored by UV absorbance at 640 nm. We found that, from the same culture, extraction with methanol produced 1–2 times more blue pigment than did alkaline extraction (p < 0.001) (Figures 6B,C), suggesting that the methanol extraction data were more representative of the actual yields in the cells. Additionally, in the methanol extraction data, the yield of blue pigment from LJ161/pMM1 and LJ162/pMM1 was improved slightly (52%, p = 0.059; and 43%, p = 0.025, respectively) in comparison with that of SLQ185/pMM1 (Figure 6B).
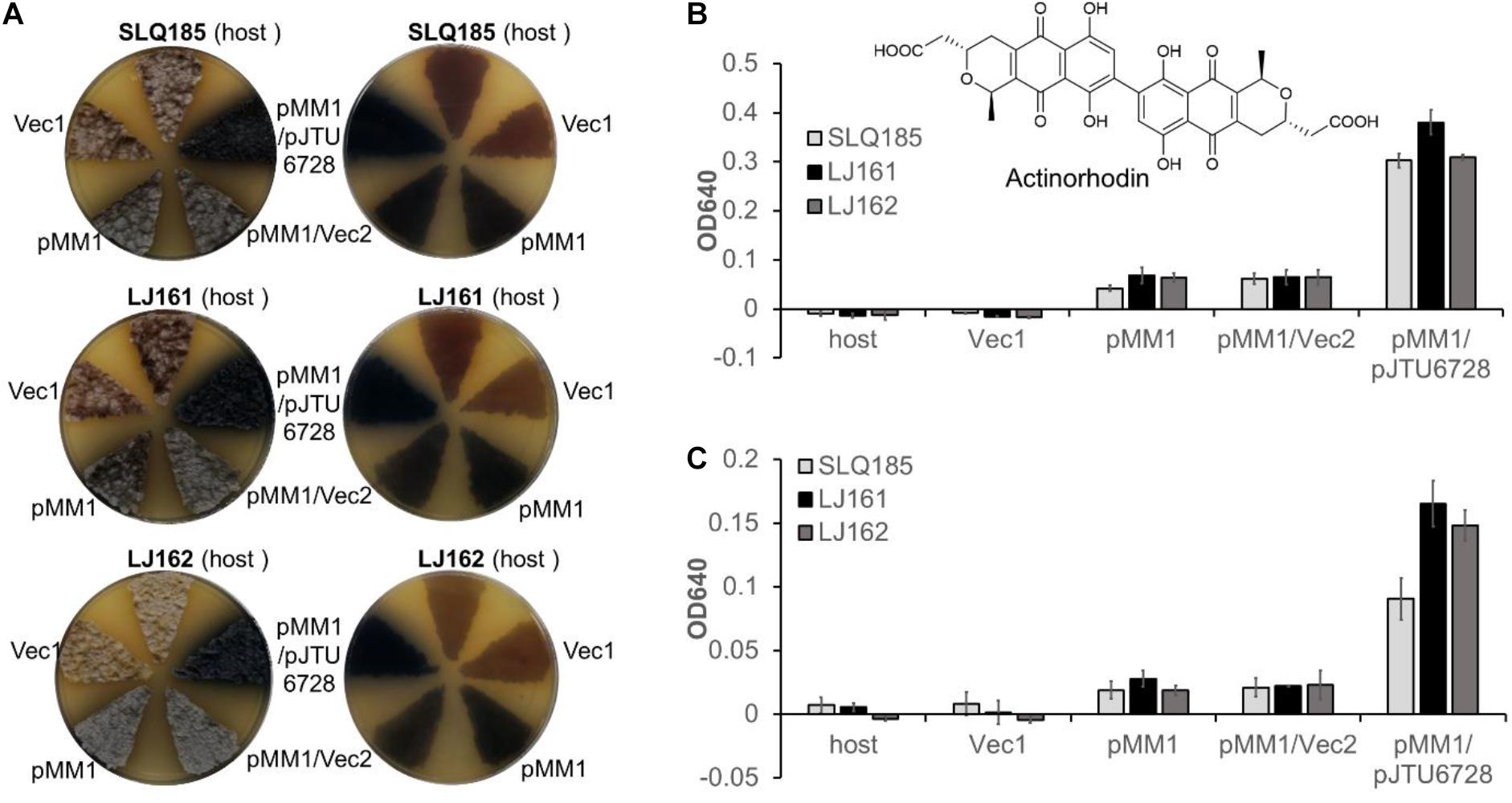
Figure 6. Heterologous production of actinorhodin by Sac. erythraea strains SLQ185, LJ161, and LJ162 carrying pMM1and pJTU6728. (A) Heterologous production of the blue pigment actinorhodin on R3 agar plates. The surfaces (left) and the backs (right) of the culture plates are shown. Vec1, the vector pMM1; Vec2, the vector pMS82. (B,C) Quantification of actinorhodin production in R3 medium. The cultures were extracted by either methanol (B) or 1 M KOH (C). The optical density (OD) was determined at 640 nm. Data are from three biological replicates.
We have previously demonstrated that pJTU6728, an attPϕBT1-based integrative plasmid carrying the transcription factor gene nusG, the global regulator gene afsScla, and the two drug-efflux pump genes mdfAco and lrmAco, increased the heterologous production of secondary metabolites in an S. lividans host (Peng et al., 2018). To assess the effects of pJTU6728 on the heterologous production of actinorhodin in Sac. erythraea, pJTU6728 and the empty vector pMS82 were individually introduced into SLQ185/pMM1, LJ161/pMM1, and LJ162/pMM1 and integrated into the host chromosome via the phage ϕBT1 att/int system. The yield of actinorhodin in the pJTU6728-containing strains SLQ185/pMM1/pJTU6728, LJ161/pMM1/pJTU6728, and LJ162/pMM1/pJTU6728 was 4.9, 5.9, and 4.8 times higher than in the corresponding vector control strains SLQ185/pMM1/pMS82, LJ161/pMM1/pMS82, and LJ162/pMM1/pMS82 (p < 0.0001) (Figure 6B). In addition, compared with the yield in SLQ185/pMM1/pJTU6728, the yield of blue pigment in the pke deletion mutant LJ161/pMM1/pJTU6728 was improved by 25% (p = 0.0089) in the methanol extraction and 82% (p = 0.0060) in the KOH extraction (Figures 6B,C).
Heterologous-expression hosts derived from rare actinomycetes are valuable for genome mining of bioactive natural products. Here, we described the optimization and application of the erythromycin-overproducing bacterium Sac. erythraea HOE107 as a host for the heterologous expression of polyketide BGCs. In our study, we disrupted the erythromycin biosynthetic PKS gene cluster and replaced it with two phage integration (attB) sites from the actinomycete phages ϕC31 and ϕBT1, yielding the gene replacement strain Sac. erythraea SLQ185, which harbors an attBϕC31-aadA-attBϕBT1 in place of the ery BGC. Two different attB sites were considered a useful modification since one site (attBϕC31) served as the integration site for the heterologous BGC, and the other (attBϕBT1) provided an integration site for beneficial supplemental factors, such as pJTU6728 and pHLJ815, to increase the production of actinorhodin and spinosad, respectively. We also applied a reported CRISPR/Cas9-CodA(sm) combined recombination system to delete the pke BGC, encoding a type I PKS, or the rpp BGC, encoding a type III PKS, from Sac. erythraea SLQ185, producing strains Sac. erythraea LJ161 and LJ162, respectively. Our results demonstrated that the CRISPR/Cas9-CodA(sm) combined homologous recombination system substantially improves the efficiency of gene replacement in Sac. erythraea.
Nine erythromycin biosynthetic genes, including eryAII, eryAIII (with a type I thioesterase domain), and eryORF5 (coding for a type II thioesterase, Hu et al., 2003), were deleted from the erythromycin overproducing strain. The 1.4-kb 3′-terminal part of eryAI was also deleted. The remaining parts of EryAI protein expressed in the strains would not load and release polyketide building blocks due to the lack of both type I and II thioesterases. Therefore, the deletion of ery BGC in this study would save biosynthetic substates for the heterologous expression of polyketide BGCs. Although the pke BGC was actively expressed in an erythromycin-producing strain of Sac. erythraea (Li et al., 2013), extensive searches using 50 different types of solid and liquid media have not detected the products of the pke BGC (Boakes et al., 2004; Oliynyk et al., 2007). In this study, the deletion of the pke BGC slightly increased the production of both spinosad and actinorhodin, indicating that the multifunctional PKS machinery encoded by the pke BGC was functional in these conditions and that it competed with the introduced heterologous PKS pathways for precursors such as malonyl-CoA. The rpp BGC is another actively expressed PKS gene cluster in Sac. erythraea (Cortés et al., 2002). Deletion of the rpp BGC abolished the production of the brown pigment associated with this strain, which also alleviated substrate competition for polyketide production.
We also established a highly efficient conjugation protocol for transferring large-sized BAC clones into Sac. erythraea strains. Based on the optimized method, the arrayed BAC library was effectively transferred into Sac. erythraea SLQ185 using the massive triparental conjugation approach. We successfully expressed the spinosad BGC from Sac. spinosa and the actinorhodin BGC from Streptomyces, as indicated by the substantial production of the blue pigment in strains containing pJTU6728; these findings suggest that the engineered Sac. erythraea strains can serve as heterologous hosts in function-driven, genome-mining approaches, e.g., LEXAS (Xu et al., 2016), for the discovery of cryptic and new antibiotics from Streptomyces and rare actinomycetes.
In summary, we modified the erythromycin-producing strain Sac. erythraea into a heterologous host with a cleaner, less competitive metabolic background and an amendable genetic manipulation system, and demonstrated its utilization for the heterologous expression of polyketide BGCs from Streptomyces and Sacchropolyspora.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
MT, JL, and ZD were responsible for the original concept and designed the experiments. MT, JL, XP, and YW analyzed the data. JL, QL, ZZ, LC, WH, JH, and KL performed the experimental work. JL and MT wrote the manuscript. All the authors read and approved the final manuscript.
This work was supported by the National Key Research and Development Program of China (grant no. 2018YFA0901901), the National Natural Science Foundation of China (No. 31770036), and the Science and Technology Commission of Shanghai Municipality (19430750600).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Yuhui Sun for the gift of pWHU2653.
Bibb, M. J., Janssen, G. R., and Ward, J. M. (1985). Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene 38, 215–226. doi: 10.1016/0378-1119(85)90220-3
Bierman, M., Logan, R., Obrien, K., Seno, E. T., Rao, R. N., and Schoner, B. E. (1992). Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49.
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Boakes, S., Oliynyk, M., Cortés, J., Böhm, I., Rudd, B. A. M., Revill, W. P., et al. (2004). A new modular polyketide synthase in the erythromycin producer Saccharopolyspora erythraea. J. Mol. Microbiol. Biotechnol. 8, 73–80. doi: 10.1159/000084562
Bystrykh, L. V., Fernández-Moreno, M. A., Herrema, J. K., Malpartida, F., Hopwood, D. A., and Dijkhuizen, L. (1996). Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J. Bacteriol. 178, 2238–2244. doi: 10.1128/jb.178.8.2238-2244.1996
Challis, G. L. (2014). Exploitation of the Streptomyces coelicolor A3(2) genome sequence for discovery of new natural products and biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 41, 219–232. doi: 10.1007/s10295-013-1383-2
Chater, K. F., and Wilde, L. C. (1976). Restriction of a bacteriophage of Streptomyces albus G involving endonuclease SalI. J. Bacteriol. 128, 644–650. doi: 10.1128/JB.128.2.644-650.1976
Chen, X., Xu, M., Lü, J., Xu, J., Wang, Y., Lin, S., et al. (2018). Biosynthesis of tropolones in Streptomyces spp.: interweaving biosynthesis and degradation of phenylacetic acid and hydroxylations on the tropone ring. Appl. Environ. Microbiol. 84:e00349-18. doi: 10.1128/AEM.00349-18
Choi, S.-S., Kim, H.-J., Lee, H.-S., Kim, P., and Kim, E.-S. (2015). Genome mining of rare actinomycetes and cryptic pathway awakening. Process Biochem. 50, 1184–1193. doi: 10.1016/j.procbio.2015.04.008
Choi, S.-U., Lee, C.-K., Hwang, Y.-I., Kinoshita, H., and Nihira, T. (2004). Intergeneric conjugal transfer of plasmid DNA from Escherichia coli to Kitasatospora setae, a bafilomycin B1 producer. Arch. Microbiol. 181, 294–298. doi: 10.1007/s00203-004-0654-8
Cortés, J., Velasco, J., Foster, G., Blackaby, A. P., Rudd, B. A. M., and Wilkinson, B. (2002). Identification and cloning of a type III polyketide synthase required for diffusible pigment biosynthesis in Saccharopolyspora erythraea. Mol. Microbiol. 44, 1213–1224. doi: 10.1046/j.1365-2958.2002.02975.x
Flett, F., Mersinias, V., and Smith, C. P. (2006). High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol. Lett. 155, 223–229. doi: 10.1111/j.1574-6968.1997.tb13882.x
Gao, G., Liu, X., Xu, M., Wang, Y., Zhang, F., Xu, L., et al. (2017). Formation of an angular aromatic polyketide from a linear anthrene precursor via oxidative rearrangement. Cell Chem. Biol. 24, 881.e4–891.e4. doi: 10.1016/j.chembiol.2017.06.008
Gomez-Escribano, J. P., and Bibb, M. J. (2011). Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 4, 207–215. doi: 10.1111/j.1751-7915.2010.00219.x
Gould, S. J., Hong, S. T., and Carney, J. R. (1998). Cloning and heterologous expression of genes from the kinamycin biosynthetic pathway of Streptomyces murayamaensis. J. Antibiot. 51, 50–57. doi: 10.7164/antibiotics.51.50
Gregory, M. A., Till, R., and Smith, M. C. M. (2003). Integration site for Streptomyces phage BT1 and development of site-specific integrating vectors. J. Bacteriol. 185, 5320–5323. doi: 10.1128/JB.185.17.5320-5323.2003
Guo, H., Bai, T., and Tao, M. (2012). Cloning and reconstitution of rhamose and forosamine biosynthetic gene of Saccharopolyspora spinosa NRRL18395. J. Huazhong Agric. Univ. 31, 298–302.
Gust, B., Challis, G. L., Fowler, K., Kieser, T., and Chater, K. F. (2003). PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 100, 1541–1546. doi: 10.1073/pnas.0337542100
Hu, Z., Pfeifer, B. A., Chao, E., Murli, S., Kealey, J., Carney, J. R., et al. (2003). A specific role of the Saccharopolyspora erythraea thioesterase II gene in the function of modular polyketide synthases. Microbiology 149, 2213–2225. doi: 10.1099/mic.0.26015-0
Huang, J., Yu, Z., Li, M.-H., Wang, J.-D., Bai, H., Zhou, J., et al. (2016). High level of spinosad production in the heterologous host Saccharopolyspora erythraea. Appl. Environ. Microbiol. 82, 5603–5611. doi: 10.1128/AEM.00618-16
Kieser, H. M., Kieser, T., and Hopwood, D. A. (1992). A combined genetic and physical map of the Streptomyces coelicolor A3(2) chromosome. J. Bacteriol. 174, 5496–5507. doi: 10.1128/jb.174.17.5496-5507.1992
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F., Hopwood, D. A., Charter, K., et al. (2000). Practical Streptomyces Genetics. Norwich: John Innes Foundation.
Kirm, B., Magdevska, V., Tome, M., Horvat, M., Karnièar, K., Petek, M., et al. (2013). SACE_5599, a putative regulatory protein, is involved in morphological differentiation and erythromycin production in Saccharopolyspora erythraea. Microb. Cell Fact. 12:126. doi: 10.1186/1475-2859-12-126
Komatsu, M., Komatsu, K., Koiwai, H., Yamada, Y., Kozone, I., Izumikawa, M., et al. (2013). Engineered Streptomyces avermitilis host for heterologous expression of biosynthetic gene cluster for secondary metabolites. ACS Synth. Biol. 2, 384–396. doi: 10.1021/sb3001003
Kouprina, N., and Larionov, V. (2016). Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 125, 621–632. doi: 10.1007/s00412-016-0588-3
Lazos, O., Tosin, M., Slusarczyk, A. L., Boakes, S., Cortés, J., Sidebottom, P. J., et al. (2010). Biosynthesis of the putative siderophore erythrochelin requires unprecedented crosstalk between separate nonribosomal peptide gene clusters. Chem. Biol. 17, 160–173. doi: 10.1016/j.chembiol.2010.01.011
Le, Q., Shong, L., and Shi, Y. (2001). Extraction of erythromycin from fermentation broth using salt-induced phase separation processes. Sep. Purif. Technol. 24, 85–91. doi: 10.1016/S1383-5866(00)00217-3
Li, A., and Piel, J. (2002). A gene cluster from a marine Streptomyces encoding the biosynthesis of the aromatic spiroketal polyketide griseorhodin A. Chem. Biol. 9, 1017–1026. doi: 10.1016/S1074-5521(02)00223-5
Li, M. H. T., Ung, P. M. U., Zajkowski, J., Garneau-Tsodikova, S., and Sherman, D. H. (2009). Automated genome mining for natural products. BMC Bioinformatics 10:185. doi: 10.1186/1471-2105-10-185
Li, Y.-Y., Chang, X., Yu, W.-B., Li, H., Ye, Z.-Q., Yu, H., et al. (2013). Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genomics 14:523. doi: 10.1186/1471-2164-14-523
Min, L., Wen, L., and Yuan, L. (2003). Cloning, expression and characterization of gene sgcD involved in the biosynthesis of novel antitumor lidamycin. Sci. China 46, 310–319. doi: 10.1360/03yc9033
Myronovskyi, M., Rosenkränzer, B., Nadmid, S., Pujic, P., Normand, P., and Luzhetskyy, A. (2018). Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 49, 316–324. doi: 10.1016/j.ymben.2018.09.004
Nett, M., Ikeda, H., and Moore, B. S. (2009). Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26:1362. doi: 10.1039/b817069j
Oliynyk, M., Samborskyy, M., Lester, J., Mironenko, T., Scott, N., Dickens, S., et al. (2007). Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol. 25, 447–453. doi: 10.1038/nbt1297
Olorunniji, F. J., Lawson-Williams, M., McPherson, A. L., Paget, J. E., Stark, W. M., and Rosser, S. J. (2019). Control of ϕC31 integrase-mediated site-specific recombination by protein trans-splicing. Nucleic Acids Res. 47, 11452–11460. doi: 10.1093/nar/gkz936
Peano, C., Tala, A., Corti, G., Pasanisi, D., Durante, M., Mita, G., et al. (2012). Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb. Cell Fact. 11:32. doi: 10.1186/1475-2859-11-32
Peng, Q., Gao, G., Lü, J., Long, Q., Chen, X., Zhang, F., et al. (2018). Engineered Streptomyces lividans strains for optimal identification and expression of cryptic biosynthetic gene clusters. Front. Microbiol. 9:3042. doi: 10.3389/fmicb.2018.03042
Pfeifer, B. A., and Khosla, C. (2001). Biosynthesis of polyketides in heterologous hosts. Microbiol. Mol. Biol. Rev. 65, 106–118.
Pham, J. V., Yilma, M. A., Feliz, A., Majid, M. T., Maffetone, N., Walker, J. R., et al. (2019). A review of the microbial production of bioactive natural products and biologics. Front. Microbiol. 10:1404. doi: 10.3389/fmicb.2019.01404
Rodriguez, E., Hu, Z., Ou, S., Volchegursky, Y., Hutchinson, C. R., and McDaniel, R. (2003). Rapid engineering of polyketide overproduction by gene transfer to industrially optimized strains. J. Ind. Microbiol. Biotechnol. 30, 480–488. doi: 10.1007/s10295-003-0045-1
Shima, J., Hesketh, A., Okamoto, S., Kawamoto, S., and Ochi, K. (1996). Induction of actinorhodin production by rpsL (encoding ribosomal protein S12) mutations that confer streptomycin resistance in Streptomyces lividans and Streptomyces coelicolor A3(2). J. Bacteriol. 178, 7276–7284. doi: 10.1128/jb.178.24.7276-7284.1996
Sosio, M., Giusino, F., Cappellano, C., Bossi, E., and Donadio, S. (2000). Artificial chromosomes for antibiotic-producing actinomycetes. Nat. Biotechnol. 18, 343–345. doi: 10.1038/73810
Starcevic, A., Zucko, J., Simunkovic, J., Long, P. F., Cullum, J., and Hranueli, D. (2008). ClustScan: an integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 36, 6882–6892. doi: 10.1093/nar/gkn685
Tan, G.-Y., Deng, K., Liu, X., Tao, H., Chang, Y., Chen, J., et al. (2017). An omics-guided large polyketide synthase gene cluster reconstitution in Streptomyces. ACS Synth. Biol. 6, 995–1005. doi: 10.1021/acssynbio.6b00330
Tiwari, K., and Gupta, R. K. (2012). Rare actinomycetes: a potential storehouse for novel antibiotics. Crit. Rev. Biotechnol. 32, 108–132. doi: 10.3109/07388551.2011.562482
Waldron, C., Matsushima, P., Rosteck, P. R., Broughton, M. C., Turner, J. R., Madduri, K., et al. (2001). Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa. Chem. Biol. 8, 487–499. doi: 10.1016/S1074-5521(01)00029-1
Wang, W., Tanurdzic, M., Luo, M., Sisneros, N., Kim, H. R., Weng, J.-K., et al. (2005). Construction of a bacterial artificial chromosome library from the spikemoss Selaginella moellendorffii: a new resource for plant comparative genomics. BMC Plant Biol. 5:10. doi: 10.1186/1471-2229-5-10
Wu, J., Zhang, Q., Deng, W., Qian, J., Zhang, S., and Liu, W. (2011). Toward improvement of erythromycin A production in an industrial Saccharopolyspora erythraea strain via facilitation of genetic manipulation with an artificial attB site for specific recombination. Appl. Environ. Microbiol. 77, 7508–7516. doi: 10.1128/AEM.06034-11
Xu, M., Wang, Y., Zhao, Z., Gao, G., Huang, S.-X., Kang, Q., et al. (2016). Functional genome mining for metabolites encoded by large gene clusters through heterologous expression of a whole-genome bacterial artificial chromosome library in Streptomyces spp. Appl. Environ. Microbiol. 82, 5795–5805. doi: 10.1128/AEM.01383-16
Xu, M., Zhang, F., Cheng, Z., Bashiri, G., Wang, J., Hong, J., et al. (2020). Functional genome mining reveals a novel class V lanthipeptide containing a D-amino acid introduced by an F420H2-dependent reductase. Angew. Chem. [Epub ahead of print]. doi: 10.1002/ange.202008035
Zeng, H., Wen, S., Xu, W., He, Z., Zhai, G., Liu, Y., et al. (2015). Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Appl. Microbiol. Biotechnol. 99, 10575–10585. doi: 10.1007/s00253-015-6931-4
Zhang, M. M., Wang, Y., Ang, E. L., and Zhao, H. (2016). Engineering microbial hosts for production of bacterial natural products. Nat. Prod. Rep. 33, 963–987. doi: 10.1039/C6NP00017G
Zhao, C., Huang, Y., Guo, C., Yang, B., Zhang, Y., Lan, Z., et al. (2017). Heterologous expression of spinosyn biosynthetic gene cluster in Streptomyces species is dependent on the expression of rhamnose biosynthesis genes. J. Mol. Microbiol. Biotechnol. 27, 190–198. doi: 10.1159/000477543
Zhao, Z., Shi, T., Xu, M., Brock, N. L., Zhao, Y.-L., Wang, Y., et al. (2016). Hybrubins: bipyrrole tetramic acids obtained by crosstalk between a truncated undecylprodigiosin pathway and heterologous tetramic acid biosynthetic genes. Org. Lett. 18, 572–575. doi: 10.1021/acs.orglett.5b03609
Keywords: Saccharopolyspora erythraea, CRISPR/Cas9-CodA(sm), heterologous expression, polyketides, antibiotic production
Citation: Lü J, Long Q, Zhao Z, Chen L, He W, Hong J, Liu K, Wang Y, Pang X, Deng Z and Tao M (2020) Engineering the Erythromycin-Producing Strain Saccharopolyspora erythraea HOE107 for the Heterologous Production of Polyketide Antibiotics. Front. Microbiol. 11:593217. doi: 10.3389/fmicb.2020.593217
Received: 10 August 2020; Accepted: 10 November 2020;
Published: 08 December 2020.
Edited by:
Christian Sohlenkamp, National Autonomous University of Mexico, MexicoReviewed by:
Pablo Cruz-Morales, Lawrence Berkeley National Laboratory, United StatesCopyright © 2020 Lü, Long, Zhao, Chen, He, Hong, Liu, Wang, Pang, Deng and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meifeng Tao, dGFvX21laWZlbmdAc2p0dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.