 Yaqiong Feng1
Yaqiong Feng1 Zhiyuan Yin
Zhiyuan Yin Yuxing Wu
Yuxing Wu Lili Huang
Lili Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 05 November 2020
Sec. Fungi and Their Interactions
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.581203
Apple Valsa canker is a destructive disease caused by the ascomycete Valsa mali and poses a serious threat to apple production. Toxins synthesized by secondary metabolite biosynthetic gene clusters (SMBGCs) have been proven to be crucial for pathogen virulence. A previous study showed that V. mali genome contains remarkably expanded SMBGCs and some of their genes were significantly upregulated during infection. In this study, we focus on LaeA, a known regulator of secondary metabolism, for its role in SMBGC regulation, toxin production, and virulence of V. mali. Deletion of VmLaeA led to greatly reduced virulence with lesion length reduced by 48% on apple twigs. Toxicity tests proved that toxicity of secondary metabolites (SMs) produced by VmLaeA deletion mutant (ΔVmlaeA) was markedly decreased in comparison with wild-type (WT). Transcriptomic and proteomic analyses of WT and ΔVmlaeA indicated that a portion of transporters and about half (31/60) SMBGCs are regulated by VmLaeA. Function analysis of eight gene clusters including PKS7, PKS11, NRPS14, PKS16, PKS23, PKS31, NRPS/PKS33, and PKS39 that were differentially expressed at both transcriptional and translational levels showed that four of them (i.e., PKS11, PKS16, PKS23, and PKS31) were involved in pigment production and NRPS14 contributed to virulence. Our findings will provide new insights and gene resources for understanding the role of pathogenicity-related toxins in V. mali.
Apple Valsa canker, a destructive disease in apple trees which leads to severe yield losses, is caused by the ascomycete Valsa mali (Lee et al., 2006; Li et al., 2013; Wang X. et al., 2014). It was first identified in Japan and is now widespread in eastern Asia. Elucidation of its pathogenesis mechanisms is vital to develop rational, novel control strategies. Pathogenic fungi usually secrete copious amounts of toxins to rapidly kill host cells and establish colonization at the early stage of infection (Doehlemann et al., 2017). Electron microscopy observation of apple bark tissues infected with V. mali hyphae found severe tissue maceration and cell necrosis which suggests that toxins may involve in the early stage of infection in V. mali (Ke et al., 2013). Toxins, such as the trichothecenes of Fusarium graminearum (Kim et al., 2013), T-toxin of Cochliobolus heterostrophus (Wu et al., 2012), and tentoxin produced by Alternaria spp. (Li et al., 2016), all of which stem from secondary metabolism, are important virulence factors in phytopathogenic fungi. For the causal agent of southern corn leaf blight C. heterostrophus, its highly aggressive nature is mostly attributable to its ability to produce the host selective toxin (HST) T-toxin, which causes severe ultrastructural damage to mitochondria in nothing flat (Gregory et al., 1980; Baker et al., 2006). The non-host-specific toxin botrydial, produced by Botrytis cinerea, could induce chlorosis and collapse of French bean tissues, and deletion of the botrydial biosynthetic genes would lead to reduced virulence (Deighton et al., 2001; Siewers et al., 2005). Another well-known toxin fumonisin B1 produced by several Fusarium spp. can induce host cell death and is necessary for the development of disease symptoms on maize seedlings (Myung et al., 2012). These toxins are all crucial weapons for phytopathogenic fungi and are generally synthesized by secondary metabolite biosynthetic gene clusters (SMBGCs).
Previously, several toxins were isolated from culture filtrates of V. mali. They were identified as degradation products of phlorizin, including p-hydroxybenzoic acid, p-hydroxyacetophenone, phloroglucinol, 3-(p-hydroxyphenyl) propionic acid, and protocatechuic acid, and are responsible for the symptoms of Valsa canker (Koganezawa and Sakuma, 1982; Natsume et al., 1982; Wang C. et al., 2014). Five isocoumarins produced by V. mali were also shown to promote the infection of V. mali on apple trees (Okuno et al., 1986). Remarkably, secondary metabolism-related genes are remarkably expanded in V. mali and are significantly upregulated during infection (Ke et al., 2014; Yin et al., 2015). These findings prompt us to speculate that SMBGCs probably play essential roles in V. mali infection. However, which SMBGCs are involved in virulence or toxin production is still undefined.
LaeA, a global regulator for secondary metabolism, was first identified in Aspergillus nidulans and is now well characterized in many filamentous fungi (Bok and Keller, 2004). LaeA regulates secondary metabolism by forming velvet complexes to induce chromatin modification, which is important for modulation of fungal secondary metabolism (Bayram et al., 2008; Brakhage, 2013). In maize pathogen C. heterostrophus, ChLae1 (an ortholog of A. nidulans LaeA) positively regulates Tox1 expression and T-toxin production (Wu et al., 2012). In Penicillium expansum, LaeA regulates the patulin gene cluster and concomitant patulin synthesis in vitro, other SMBGCs, and secondary metabolism-modulated virulence (Kumar et al., 2017). By performing proteomic analysis of wild-type (WT) and ΔlaeA mutants in A. flavus, enzymes participating in aflatoxin, cyclopiazonic acid, and ustiloxin B biosynthesis were found to be dramatically decreased in the mutant (Lv et al., 2018). Furthermore, LaeA is capable of regulating some other infection-related functions in phytopathogenic fungi. For instance, in lae1 (the deletion mutant of LaeA in B. cinerea), a set of infection-related proteins was strongly underexpressed, including secreted proteins, carbohydrate-active enzymes (CAZymes), proteases, and transporters except for secondary metabolism-related enzymes (Müller et al., 2018). To date, LaeA is regarded as a regulator of the most common mycotoxins by controlling the expression of secondary metabolism-related genes, including sterigmatocystin (Bok and Keller, 2004), aflatoxin (Yao et al., 2018), fumonisin (Butchko et al., 2012), cyclopiazonic acid (Georgianna et al., 2010), T-toxin (Wu et al., 2012), patulin (Kumar et al., 2017), and ochratoxin (Crespo-Sempere et al., 2013).
Traditionally, toxins are isolated from culture filtrates of pathogenic fungi or diseased tissues. However, the amount of toxins is usually too low to perform further research such as structural identification. Moreover, many SMBGCs remain silent under standard fermentation conditions (Bergmann et al., 2007). Considering these limitations in toxin isolation and SMBGC identification, LaeA can be exploited as a tool to identify SMBGCs that might have an impact on virulence, allowing the correlation of specific SMBGCs with toxins and virulence (Perrin et al., 2007; Brakhage, 2013). Furthermore, genetic engineering, including overexpression of pathway-specific transcription factors (TFs), the use of strong inducible promoters, and manipulation of global regulators, could promote the identification of toxins produced by specific SMBGCs (Brakhage, 2013; Keller, 2019).
In this study, we showed that VmLaeA markedly affects virulence and toxic secondary metabolites (SMs) in V. mali. Transcriptomic and proteomic analyses found that VmLaeA regulates a large amount of transports and about half of the SMBGCs identified in V. mali. A non-ribosomal peptide synthesis (NRPS) gene cluster (NRPS14) was identified to be negatively regulated by VmLaeA and contribute to virulence. VmLaeA also seems to be involved in the regulation of pigment production by controlling specific polyketide synthesis (PKS) gene clusters. Our findings provide new insights on role of pathogenicity-related toxins in V. mali.
LaeA was reported to influence fungal virulence and secondary metabolism in many filamentous fungi. A. nidulans LaeA homologous was identified in V. mali by BLAST and termed VmLaeA. To determine the potential roles of VmLaeA, we generated a deletion mutant of VmLaeA (ΔVmlaeA) and a complemented strain (ΔVmlaeA-C). Phenotypic analysis showed that ΔVmlaeA exhibited no apparent difference on vegetative growth after cultivation on potato dextrose agar (PDA) medium for 48 h (Figure 1A). However, under light conditions, ΔVmlaeA produced remarkably decreased amount of pycnidia compared with WT on PDA after 30 days. Under dark conditions, both strains produced very few pycnidia (Supplementary Figure 1). Intriguingly, the fermentation broth and mycelium pellets of WT turned yellow after 7 days of culture in a synthetic liquid medium, while those of ΔVmlaeA remained colorless (Figure 1B). These results suggest that the conidiation and pigmentation is affected by VmLaeA.
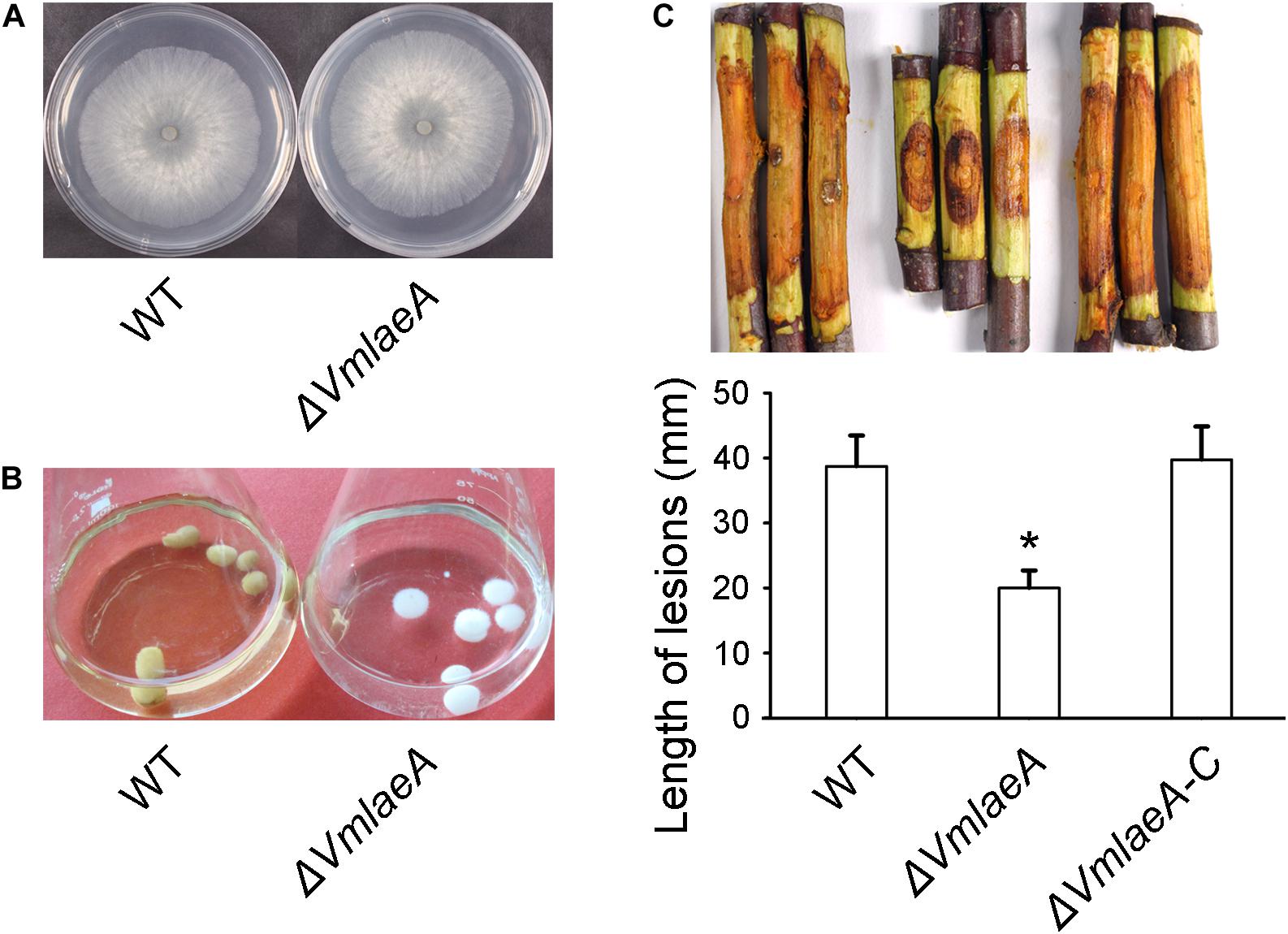
Figure 1. Growth and infection phenotype of Valsa mali WT and ΔVmlaeA. (A) Vegetative growth of V. mali WT and ΔVmlaeA mutant on potato dextrose agar (PDA) medium at 48 h. (B) Colony color of WT and ΔVmlaeA cultured in a synthetic liquid medium for 7 days. (C) Pathogenicity test of ΔVmlaeA on detached apple twigs. WT, ΔVmlaeA, and ΔVmlaeA-C were inoculated onto detached twigs of Malus domestica Borkh. cv. “Fuji” and incubated for 4 days. Bar graphs show the statistical analysis of at least three biological replicates, means ± SD are shown. Asterisk indicates a significant difference at P value <0.05.
To investigate whether VmLaeA contributes to virulence in V. mali, WT, ΔVmlaeA, and ΔVmlaeA-C strains were inoculated onto detached apple twigs. As was shown, lesion length of ΔVmlaeA was reduced by approximately 48% after 4 days of cultivation in comparison with WT (Figure 1C). By contrast, the complemented strain ΔVmlaeA-C displayed a comparable lesion size as the WT (Figure 1C). These results indicate that VmLaeA acts as a crucial virulence factor.
Considering the regulatory role of LaeA in toxin production in many phytopathogens, toxicity tests were conducted to investigate whether the production of SMs is affected by VmLaeA. We produced culture filtrates of WT, ΔVmlaeA, and uninoculated medium to obtain crude extracts of SMs. Toxicity of these crude SMs was tested on apple leaves as well as on tobacco leaves. Crude SMs of WT and ΔVmlaeA were able to induce necrotic symptom at 24 h after treatment. However, crude SMs from WT were more effective than those from ΔVmlaeA at the same concentration (Figure 2). On apple leaves, the areas of necrotic lesions caused by crude SMs of ΔVmlaeA amounted to just 59% of necrotic lesions caused by crude SMs of WT (Figure 2A). Similarly on tobacco leaves, necrotic lesions caused by crude SMs of ΔVmlaeA equated to only 45% of that cause by crude SMs of WT (Figure 2B). By contrast, the solvent and uninoculated medium control did not produce any noticeable lesions in the same time frame. These findings indicate that deletion of VmLaeA may lead to phyletic changes or reduction in toxic SMs.
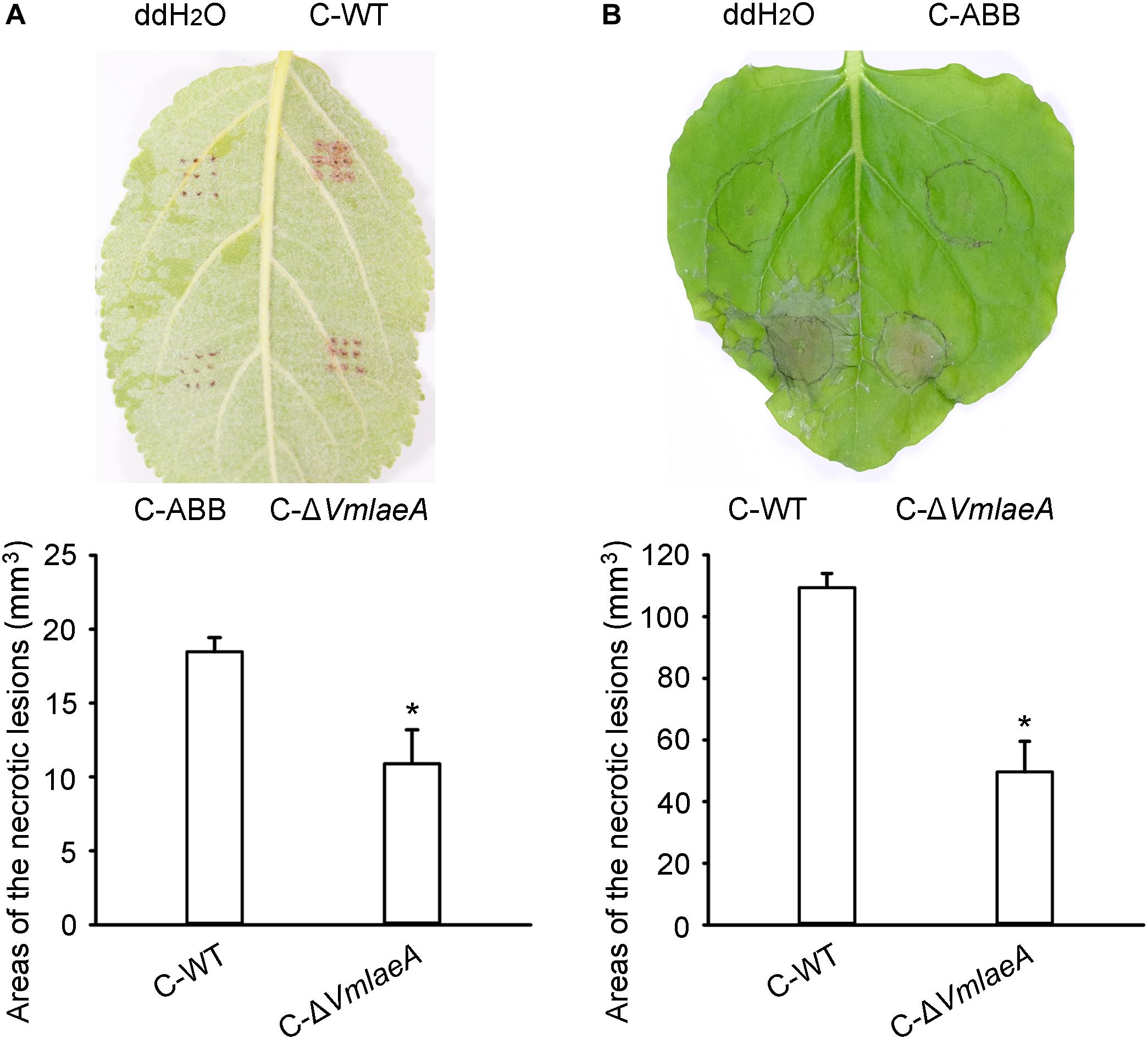
Figure 2. Toxicity of crude secondary metabolites (SMs) produced by Valsa mali WT and ΔVmlaeA. (A) Toxicity of crude SMs produced by WT and ΔVmlaeA on apple leaves. (B) Toxicity of crude SMs produced by WT and ΔVmlaeA on tobacco leaves. C-WT: crude SMs of WT; C-ABB: crude extracts of ABB medium; C-ΔVmlaeA: crude SMs of ΔVmlaeA. WT and ΔVmlaeA strains were grown in apple bark broth medium (ABB) for 10 days. Crude SMs of WT, ΔVmlaeA, and uninoculated medium (ABB) were obtained from culture filtrate by extraction with ethyl acetate and the dried solids were dissolved in ddH2O. Toxicity tests were carried out on apple leaves with a concentration of 50 mg/ml and on tobacco leaves with a concentration of 5 mg/ml. ddH2O and crude extracts of ABB were used as controls and pictures were taken at 24 h after treatment. Significant differences are indicated with asterisks (P value <0.05).
To explore how VmLaeA affects secondary metabolism and virulence in V. mali, RNA sequencing-based transcriptomic analysis and TMT-based proteomic analysis of both WT and ΔVmlaeA were performed. Totally, 1,095 of 9,945 genes identified from transcriptomes showed differential expression with fold changes >2 and P values <0.05 in ΔVmlaeA. Of these differentially expressed genes (DEGs), 514 were downregulated and 581 were upregulated (Figure 3A). Quantitative reverse transcription-PCR (qRT-PCR) analysis of randomly selected 10 downregulated DEGs and 10 upregulated DEGs displayed that these genes showed consistent up- or downregulation with their expression levels in transcriptomic analysis (Supplementary Figure 2). By comparing the proteomes of WT and ΔVmlaeA, 227 differentially expressed proteins (DEPs) of a total of 4,299 proteins identified (FDR < 0.01) were found, with fold changes >1.5 and P values <0.05. Among the 227 DEPs, 107 were decreased in abundance and 120 were increased in ΔVmlaeA (Figure 3B). The Venn diagram shows that transcriptomes and proteomes of WT and ΔVmlaeA share 131 DEGs (57.7% of all DEPs) in which 55 genes were downregulated and 75 genes were upregulated (Figure 3C). The Pearson correlation coefficient of the two omics is 0.7538 (P value = 0) which shows a significant positive correlation between the transcriptomic DEGs and proteomic DEPs (Figure 3D).
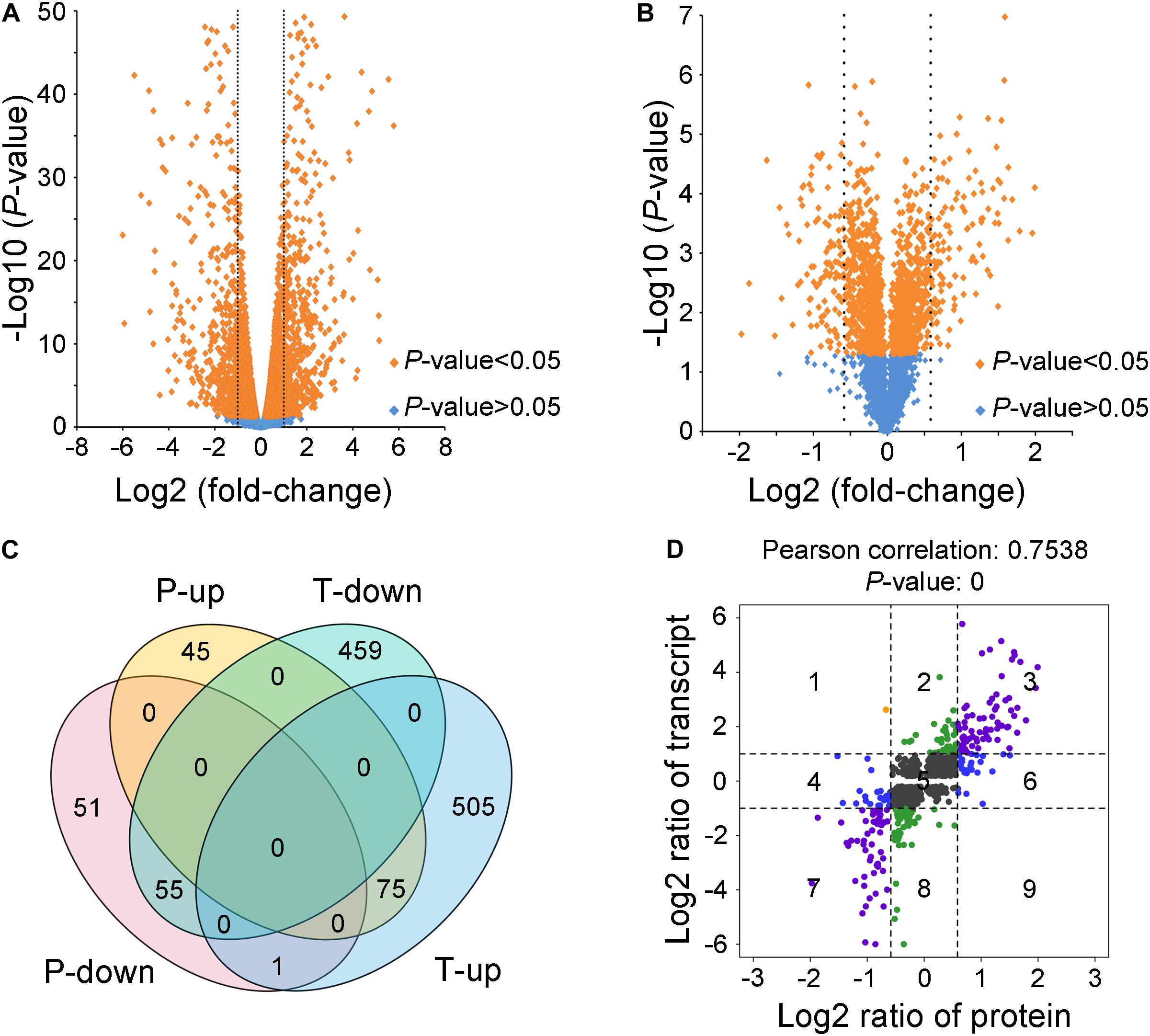
Figure 3. Differentially expressed genes (DEGs) and proteins (DEPs) in ΔVmlaeA. (A) Volcano plot of transcriptome data. The dotted lines represent fold change = 2/0.5, orange spots represent significant DEGs in ΔVmlaeA. (B) Volcano plot of proteome data. The dotted lines represent fold change = 1.5/0.67, orange spots represent significant DEPs in ΔVmlaeA. (C) Venn diagram of DEGs and DEPs in ΔVmlaeA. P-down: downregulated proteins in the proteome of ΔVmlaeA; P-up: upregulated proteins in the proteome of ΔVmlaeA; T-down: downregulated genes in the transcriptome of ΔVmlaeA; T-up: upregulated genes in the transcriptome of ΔVmlaeA. (D) Nine-quadrant diagram of DEGs and DEPs in ΔVmlaeA. The closer the Pearson correlation coefficient is to one, the higher the correlation between the two omics. P value <0.05 represents a significant correlation.
To determine potential functions of the DEGs and DEPs identified, annotation, functional classification, and enrichment analysis were performed. Gene ontology (GO) classification at level 2 GO terms was performed on transcriptomic DEGs and proteomic DEPs. The transcriptomic DEGs and proteomic DEPs showed very similar arrangement of GO classification (Supplementary Figure 3). The results of GO enrichment analysis showed that both transcriptomic DEGs and proteomic DEPs were mainly involved in transport and secondary metabolism-related biological processes (Figure 4 and Supplementary Tables 1 and 2). Transport-related biological processes that transcriptomic DEGs were involved in are consistent with the proteomic DEPs, with 43.49 and 31.33%, respectively, including transmembrane transport, carbohydrate transport, and single-organism transport (Figure 4). Secondary metabolism-related biological processes that transcriptomic DEGs were involved in include SM biosynthetic process, secondary metabolic process, polyketide biosynthetic process, tetracenomycin C metabolic process, and tetracenomycin C biosynthetic process, and the percentage of these DEGs is 14.13% (Figure 4A), whereas that of proteomic DEPs include alditol metabolic process, polyol metabolic process, glycerol metabolic process, organic hydroxy compound metabolic process, phenol-containing compound metabolic process, secondary metabolic process, and SM biosynthetic process, and the percentage of these DEPs is 12.67% (Figure 4B). The results of GO enrichment analysis demonstrate that VmLaeA conducts nearly uniform effects of transport and secondary metabolism in transcriptional and translational regulation.
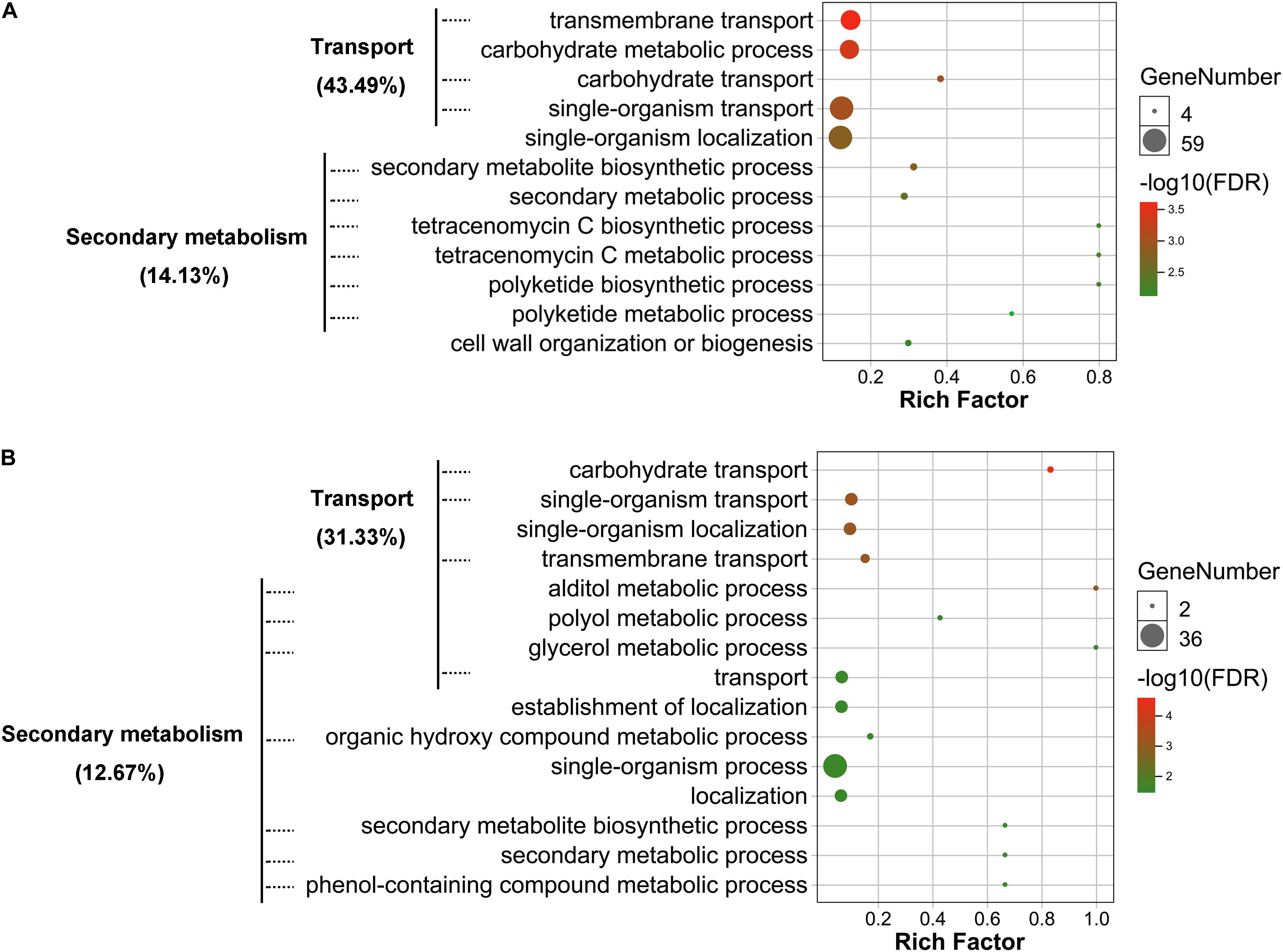
Figure 4. Gene ontology (GO) enrichment of biological process that differentially expressed genes (DEGs) and proteins (DEPs) were involved in. (A) Enriched biological process that transcriptomic DEGs were involved in. (B) Enriched biological process that proteomic DEPs were involved in. Processes with FDR <0.05 indicate that they were significantly enriched and are shown in the figures. Rich factor = DEGs (DEPs) number/total gene (protein) number identified from transcriptome or proteome of a certain process.
In addition, a portion of CAZymes and proteases were regulated by VmLaeA. In total, 91 CAZyme genes were under the control of VmLaeA, including 40 upregulated and 51 downregulated (Supplementary Table 3). Also, 26 protease genes were regulated by VmLaeA, including 14 upregulated and 12 downregulated (Supplementary Table 4).
Fungal membrane transporters play an important role in counteracting the physiological impact of exogenetic antifungal compounds from host plant, other microbes, or synthesized by self-defense system and transport nutrients. Out of the 746 genomic membrane transporter genes of V. mali, 166 were differentially expressed in ΔVmlaeA, including 84 upregulated and 82 downregulated (Supplementary Table 5). The multidrug-resistant transporter (MFS superfamily) genes were markedly regulated by VmLaeA, including drug: H+ antiporter-1 family (DHA1 family), drug: H+ antiporter-2 family (DHA2 family), sugar porter family (SP family), anion: cation symporter family (ACS family), and monocarboxylate porter family (MCP family; Figure 5A). Especially, the DHA1 family is involved in multidrug resistance and infection. Of the 17 DHA1 family genes regulated by VmLaeA, 12 were under the positive regulation of VmLaeA and five of them (VM1G_01696, VM1G_03590, VM1G_05084, VM1G_08059, and VM1G_08300) had been proved significantly upregulated during infection. However, most genes of DHA2 family and SP family that were regulated by VmLaeA were under the negative regulation of VmLaeA, as 12 of 18 DHA2 family genes and 19 of 23 SP family genes were upregulated in ΔVmlaeA. The voltage-gated K+ channel β-subunit family (Kvβ family) that mediates signal transduction and iron/lead transporter family (ILT family) that is implicated in heavy metal binding all belong to ion channels. Of the 10 Kvβ family genes regulated by VmLaeA, seven were under the positive regulation of VmLaeA. Three ILT family genes accounted for 30% of V. mali ILT family and were under the negative regulation of VmLaeA. These results suggest that VmLaeA possibly affects virulence by specifically regulating multidrug-resistant transporters and signaling through specific ion channel transporters.
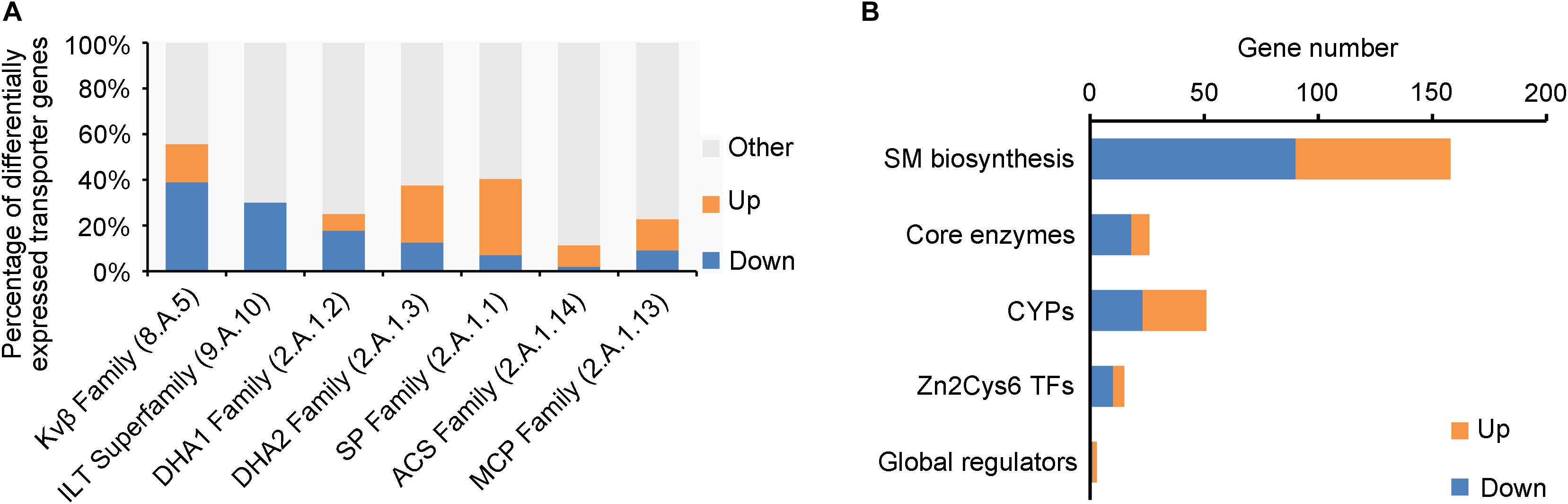
Figure 5. Enriched membrane transporter families and secondary metabolism-related genes from differentially expressed genes (DEGs) or proteins (DEPs). (A) Enriched membrane transporter families from DEGs or DEPs. Kvβ Family: the voltage-gated K+ channel β-subunit family; ILT Family: the iron/lead transporter family; DHA1 Family: the drug: H+ antiporter-1 (12 spanner) family; DHA2 Family: the drug: H+ antiporter-2 (14 spanner) family; SP Family: the sugar porter family; ACS Family: the anion: cation symporter family; MCP Family: the monocarboxylate porter family. (B) Enriched secondary metabolism-related genes from differentially expressed genes or proteins. CYPs: cytochrome p450s; Zn2Cys6 TFs: Zn2Cys6 transcription factors. Up: upregulated genes or proteins in ΔVmlaeA from transcriptomic and proteomic analyses; Down: downregulated genes or proteins in ΔVmlaeA from transcriptomic and proteomic analyses.
Genes involved in secondary metabolism consist of SM biosynthetic genes, core enzyme genes, cytochrome P450 (CYP) genes, Zn2Cys6 TF genes, and secondary metabolism-related global regulator genes (Figure 5B). Twenty-six core enzyme genes were found to be differentially expressed in ΔVmlaeA, including 8 upregulated and 18 downregulated (Figure 5B and Supplementary Table 6). Particularly, four of them that were positively regulated by VmLaeA were previously reported to be involved in infection, and one of them (VM1G_04745) has been proved to contribute to virulence. Fifty-one CYPs out of the 156 identified in V. mali were differentially expressed in ΔVmlaeA, including 28 upregulated and 23 downregulated (Figure 5B and Supplementary Table 7). Of them, five CYPs (three upregulated and two downregulated) belong to SMBGCs and one downregulated CYP gene (VM1G_03094) has been demonstrated previously to contribute to virulence. Furthermore, 15 genes for Zn2Cys6 TFs (5 upregulated and 10 downregulated) and three genes for global regulators were found to be differentially expressed in ΔVmlaeA (Figure 5B and Supplementary Tables 8 and 9). Of the 15 Zn2Cys6 TFs, five (two upregulated and three downregulated) belong to specific SMBGCs. In total, 158 SM biosynthetic genes (68 upregulated and 90 downregulated) were differentially expressed in ΔVmlaeA, including above 26 core enzyme genes, 5 CYP genes, 5 Zn2Cys6 TF genes, and 12 transporter genes. These results indicate that secondary metabolism-related genes are significantly regulated by VmLaeA.
To better understand the SM biosynthetic pathways regulated by VmLaeA, SMBGCs were re-predicted by antiSMASH (Medema et al., 2011) based on genome, transcriptome, and proteome data, and a total of 60 SMBGCs were identified in V. mali genome (Supplementary Table 10). All DEGs/DEPs involved in secondary metabolism were first matched with SMBGCs and clusters that contain two or more DEGs/DEPs were selected as differentially expressed SMBGCs (Supplementary Table 10). By analyzing differential expression of secondary metabolism-related genes in ΔVmlaeA, 31 differentially expressed SMBGCs were identified. Among these 31 SMBGCs, 17 gene clusters were downregulated, including 13 PKS gene clusters, 3 NRPS gene clusters, and 1 terpene synthesis gene cluster (Table 1). In addition, 14 upregulated gene clusters include seven PKS gene clusters, five NRPS gene clusters, and two NRPS/PKS hybrid gene clusters (Table 1). Especially, eight gene clusters showed completely consistent expression change at transcriptional and translational levels in ΔVmlaeA. Of the eight differentially expressed SMBGCs, five PKS gene clusters, including PKS7, PKS11, PKS16, PKS23, and PKS31, were downregulated, and NRPS14, PKS39, and hybrid gene cluster NRPS/PKS33 were upregulated (Table 1). Over half of the SMBGCs are under the command of VmLaeA, and all these findings explain that VmLaeA is the crucial regulator of secondary metabolism in V. mali.
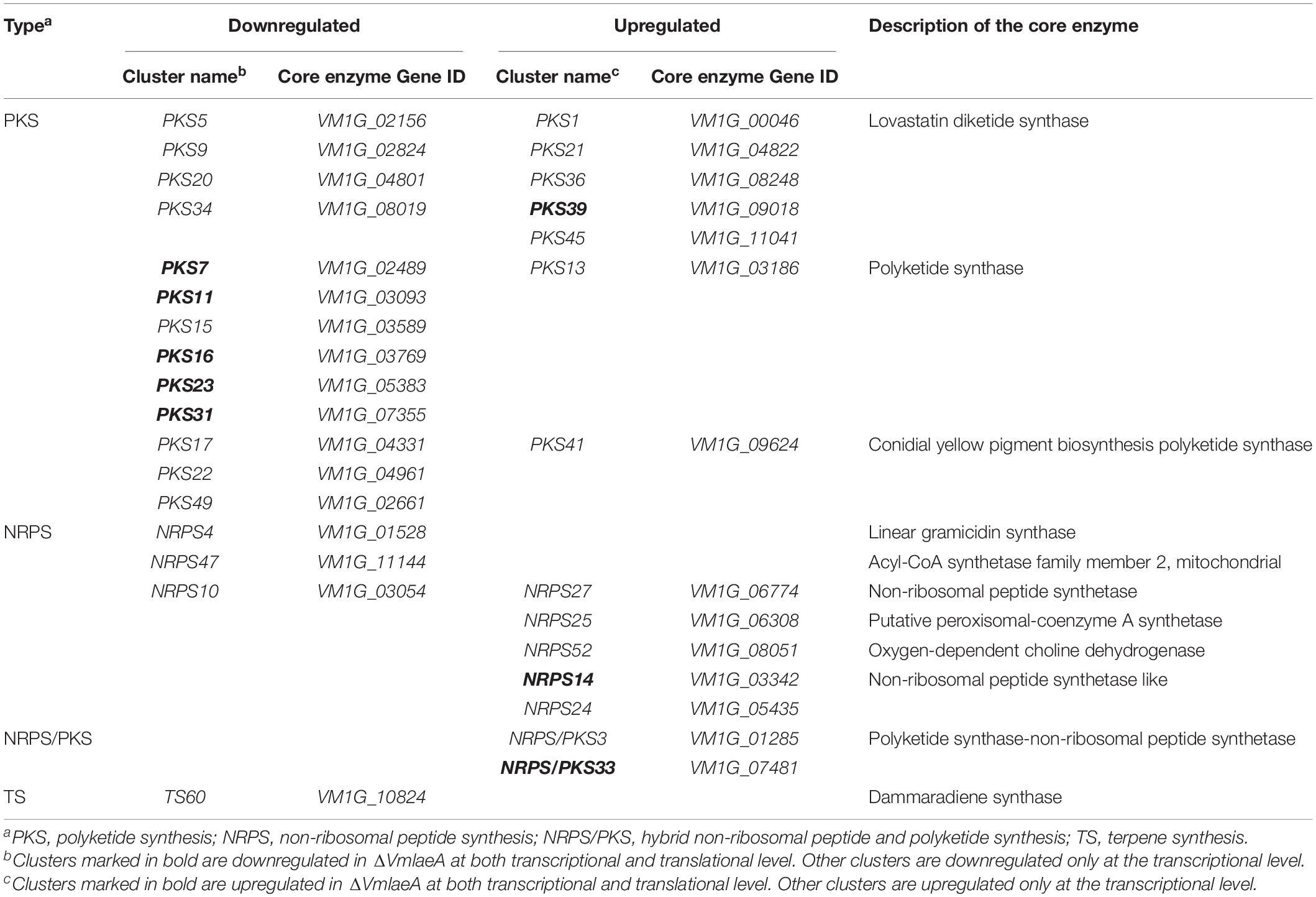
Table 1. Downregulated and upregulated secondary metabolite biosynthetic gene clusters (SMBGCs) in the ΔVmlaeA mutant.
To further determine the functions of SMBGCs regulated by VmLaeA, all eight gene clusters regulated by VmLaeA at both transcriptional and translational levels were chosen for further functional validation experiments using gene knockout and overexpression analyses. We were able to generate deletion mutants of six of the eight gene clusters. These are ΔVmpks11, ΔVmpks16, ΔVmpks23, ΔVmpks31, ΔVmpks39, and ΔVmnrps14. Deletion mutants of PKS7 and NRPS/PKS33 could not be obtained. All deletion mutants were confirmed by Southern hybridization (Supplementary Figure 4A). Moreover, overexpression mutants of the six gene clusters (ΔVmpks11OE, ΔVmpks16OE, ΔVmpks23OE, ΔVmpks31OE, ΔVmpks39OE, ΔVmnrps14OE-6, and ΔVmnrps14OE-13) were conducted through overexpressing the TF gene of corresponding clusters. ΔVmnrps14OE-6 and ΔVmnrps14OE-13 are the two overexpression mutants of cluster NRPS14. Overexpression mutants were verified by qRT-PCR and generally upregulated about threefolds (Supplementary Figure 4B).
To evaluate the roles of the six gene clusters on vegetative growth, all deletion and overexpression mutants along with the WT were cultured on PDA for 48 h to measure mycelia growth. All deletion and overexpression mutants showed no significant difference on vegetative growth (Supplementary Figure 5). However, ΔVmpks11OE, ΔVmpks16OE, ΔVmpks23OE, and ΔVmpks31OE exhibited obvious accumulation of pigments after cultivation on PDA medium for 5 days (Supplementary Figure 6). To determine whether PKS11, NRPS14, PKS16, PKS23, PKS31, or PKS39 are involved in pathogenesis, we next determined the virulence phenotype of their mutants. The results showed that all deletion mutants of the six gene clusters had no significant influence on V. mali virulence (Figure 6 and Supplementary Figure 7). Consistently, overexpression of PKS11, PKS16, PKS23, PKS31, and PKS39 also showed no significant influence on fungal virulence (Supplementary Figure 7). However, when NRPS14 was overexpressed, the lesion diameters were increased about 19.35% on detached apple leaves and 26.15% on detached apple twigs (Figure 6). To investigate whether the overexpression of NRPS14 affects the toxicity of SMs produced by V. mali, the crude SMs of WT and two overexpression mutants of NRPS14 (ΔVmnrps14OE-6 and ΔVmnrps14OE-13) were produced to conduct toxicity tests. The results showed that toxicity of the crude SMs of ΔVmnrps14OE-6 and ΔVmnrps14OE-13 increased 40 and 44%, respectively, in comparison with that of WT on apple leaves (Figure 7A). However, on tobacco leaves, the necrotic lesions caused by the crude SMs of WT and two overexpression mutants of NRPS14 are of uniform size at the same concentration (Figure 7B). The NRPS14 cluster contains 20 genes predicted by antiSMASH and six of these genes are significantly upregulated in ΔVmlaeA. Especially, VM1G_03342, the core enzyme gene of NRPS14, is upregulated by 8.17-fold at the transcriptional level in ΔVmlaeA (Supplementary Table 10). These results indicate that NRPS14 is negatively regulated by VmLaeA and contributes to virulence. Toxicity tests showed that overexpression of NRPS14 affected the production of toxic SMs of V. mali.
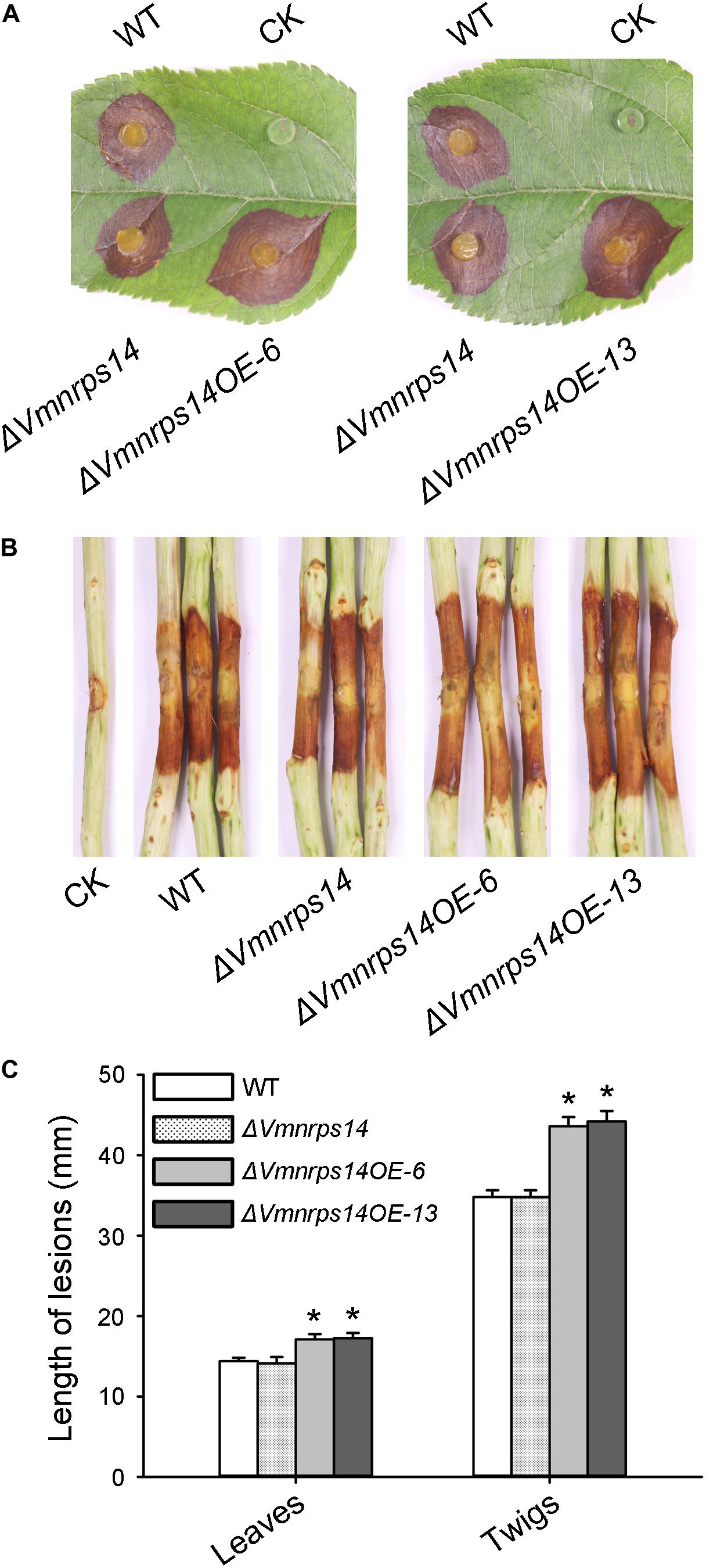
Figure 6. Infection assays of WT, ΔVmNRPS14, and ΔVmnrps14-OE. (A) Pathogenicity test of ΔVmNRPS14 and ΔVmnrps14-OE on detached apple leaves. WT, ΔVmNRPS14, and ΔVmnrps14-OE were inoculated on detached leaves of Malus domestica Borkh. cv. “Fuji” and were cultured for 3 days. (B) Pathogenicity test of ΔVmNRPS14 and ΔVmnrps14-OE on detached apple twigs. WT, ΔVmNRPS14, and ΔVmnrps14-OE were inoculated on detached twigs of Malus domestica Borkh. cv. “Fuji” and were cultured for 4 days. (C) Bar graph of lesions length cause by WT, ΔVmNRPS14, and ΔVmnrps14-OE. In bar graphs, means ± SD of at least three biological replicates are shown. Significant differences are indicated with asterisks (P value <0.05).
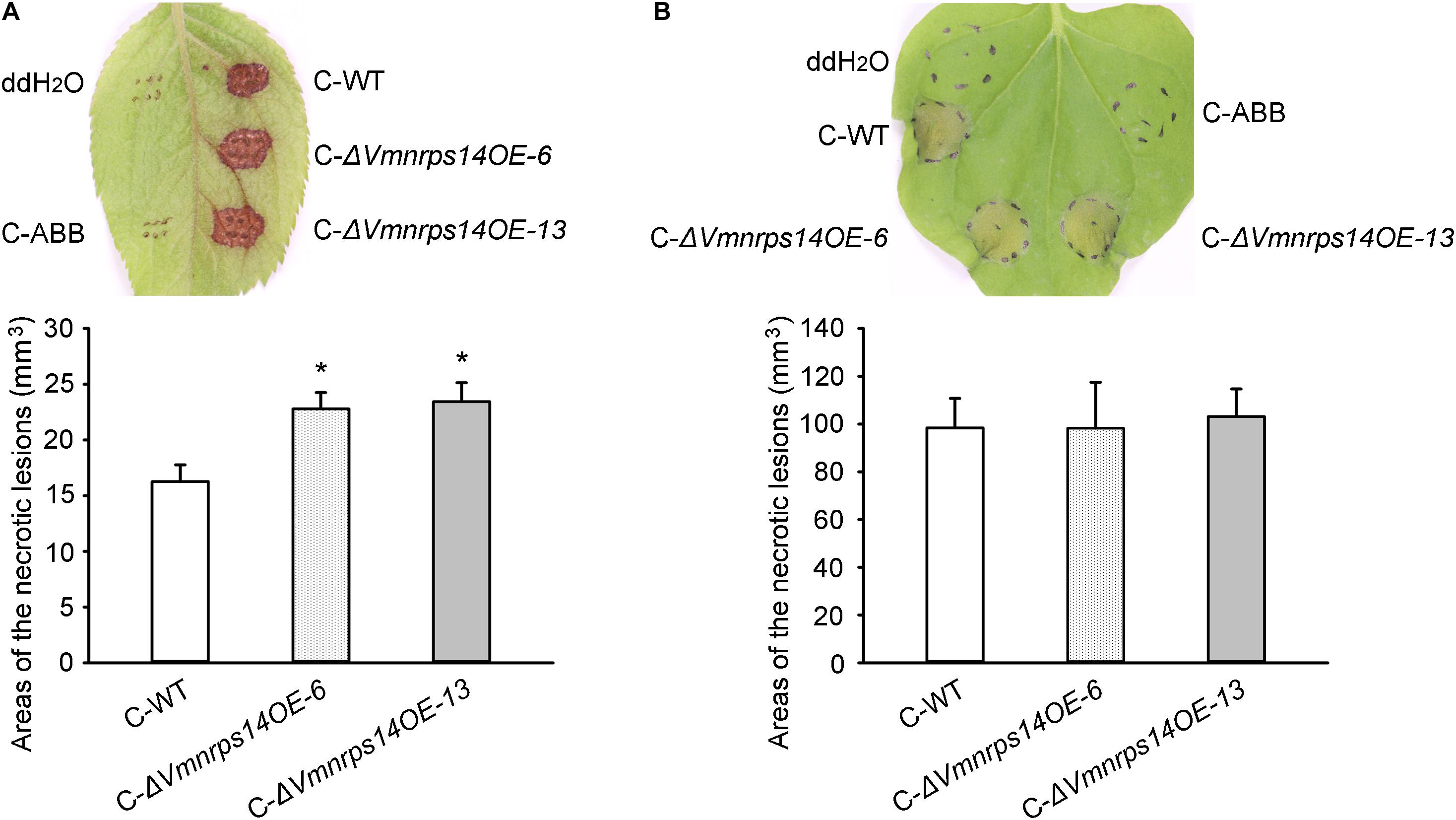
Figure 7. Toxicity of crude secondary metabolites (SMs) produced by Valsa mali WT and the overexpression mutants of NRPS14. (A) Toxicity of crude SMs produced by WT and the overexpression mutants of NRPS14 on apple leaves. (B) Toxicity of crude SMs produced by WT and the overexpression mutants of NRPS14 on tobacco leaves. C-WT: crude SMs of WT; C-ABB: crude extracts of ABB medium; C-ΔVmnrps14OE-6: crude SMs of ΔVmnrps14OE-6; C-ΔVmnrps14OE-13: crude SMs of ΔVmnrps14OE-13. WT and the overexpression mutants of NRPS14 (ΔVmnrps14OE-6 and ΔVmnrps14OE-13) were grown in apple bark broth medium (ABB) for 10 days. Crude SMs were obtained from culture filtrate by extraction with ethyl acetate and the dried solids were dissolving in ddH2O. Toxicity tests were carried out on apple leaves with a concentration of 50 mg/ml and on tobacco leaves with a concentration of 5 mg/ml. ddH2O and crude extracts of ABB were used as controls and pictures were taken at 24 h after treatment. Significant differences are indicated with asterisks (P value <0.05).
Toxins produced by plant pathogenic fungi are often crucial determinants of plant disease, as toxins can reproduce at least partial disease symptoms and non-toxic mutants are non-pathogenic or show reduced virulence (Richard, 2007). Previous research on the V. mali genome and transcriptome suggested that biosynthetic pathways of toxin are probably involved in virulence. Therefore, exploration of V. mali SMBGCs would facilitate revealing the pathogenesis mechanisms of V. mali. In this study, we investigated the function of VmLaeA and profiled transcriptomes and proteomes of V. mali WT and ΔVmlaeA. Absence of VmLaeA caused markedly compromised virulence of V. mali. Analysis of the two omics showed VmLaeA powerfully regulates secondary metabolism-related genes and SMBGCs.
LaeA has a minor impact on fungal vegetative growth but participates in light-dependent development through the interaction with the regulator of sporogenesis VosA (Bayram et al., 2008). Despite light triggering asexual development, deficiency of VmLaeA cut off the interaction between LaeA and VosA, corresponding to the decrease of pycnidia in ΔVmlaeA whether in dark or light. Deletion of laeA caused a loss of mycelial pigmentation in Aspergillus spp. as it was first identified as a global regulator (Bok and Keller, 2004). Similarly, deletion of LaeA in Pestalotiopsis microspore led to the retardation of pigmentation (Akhberdi et al., 2018). Consistent with these findings, the fermentation broth and mycelium pellets of ΔVmlaeA showed reduced pigmentation compared with WT in a synthetic liquid medium. However, LaeA is not always affecting pigmentation. In Aspergillus fumigatus, deletion of laeA did not affect pigmentation in strains B-5233 and AF293 (Sugui et al., 2007). More notably, overexpression of four PKS clusters (PKS11, PKS16, PKS23, and PKS31) that were positively regulated by VmLaeA led to obvious accumulation of pigments and the core enzymes of these four clusters were all identified as conidial yellow pigment biosynthesis polyketide synthases. Conidial pigmentation provides protection against desiccation, ultraviolet light, ionizing radiation, and oxidative stress and contributes to survival of fungi (Belozerskaya et al., 2017; Chang et al., 2020). Controlling conidial pigment biosynthesis could be a way that VmLaeA help V. mali cope with harsh environments.
With the identification of LaeA in plenty of pathogenic fungi, LaeA was regarded as a general virulence factor. Deletion of LaeA weakened the infection ability of A. ochraceus on pear (Wang et al., 2019). In maize pathogen C. heterostrophus, both T-toxin–mediated super virulence and basic pathogenic ability were affected when ChLae1 was deleted or overexpressed (Wu et al., 2012). LaeA mutants of two P. expansum isolates (Pe-21 from Israel and Pe-T01 from China) showed differential reduction in disease severity (Kumar et al., 2017). Deletion of BcLAE1 in B. cinerea resulted in the mutant losing the ability to colonize the host tissue and to produce oxalic acid (Schumacher et al., 2015). Similar in V. mali, deletion of VmLaeA led to greatly reduced virulence on apple twigs. As a conserved and crucial virulence factor, further exploration of the regulatory mechanism by VmLaeA is of great importance to reveal the pathogenic mechanism of V. mali.
Transcriptomic and proteomic analyses indicated plenty of genes that are controlled by VmLaeA seem to be involved in infection-related processes, including membrane transporter genes, CAZyme genes, and protease genes. Membrane transporters could counteract the physiological impact of host antifungal defense compounds and transport nutrients. Our research found DHA1 transporters, a member of MFS superfamily, were positively regulated by VmLaeA. DHA1 transporters were significantly upregulated during V. mali infection and involved in multidrug resistance (Meredith and Christian, 2008; Sá-Correia et al., 2009; Yin et al., 2015). In addition, 91 CAZyme genes and 26 protease genes were found to be under the control of VmLaeA. It was reported that CAZymes, in particular cell wall–degrading enzymes (CWDEs) and some proteases, target structural materials of plant cell wall and degrade cell walls to promote infection and colonization (Dow et al., 1998; Dunaevsky et al., 2007; Mohnen, 2008). These results indicate VmLaeA probably regulates virulence by controlling multiple infection-related processes including counteracting host antifungal compounds, cell wall degradation, killing host cells, and nutrient uptake from host cell and transport.
Toxicity and pathogenicity tests indicate that deletion of VmLaeA leads to reduced toxic SMs and decreased virulence in V. mali. In phytopathogenic fungi, LaeA modulates virulence by regulating the expression of many toxin biosynthetic gene clusters and the production of corresponding toxins. In P. expansum, LaeA affects patulin production and patulin-modulated virulence by positively regulating the expression of the patulin biosynthetic gene cluster (Kumar et al., 2017). Analysis of V. mali secondary metabolism-related genes revealed that more than half of the SMBGCs are under the control of VmLaeA. Of these SMBGCs, NRPS14, negatively regulated by VmLaeA, was shown to affect virulence. It was reported that many specialized SMBGCs keep silent and are activated only under some conditions by specific induction (Rutledge and Challis, 2015). For instance, the silent trichosetin gene cluster in F. fujikuroi is activated by the overexpression of the cluster-specific TF gene TF22 and then produces trichosetin (Janevska et al., 2017). This may explain why cluster NRPS14 influences virulence only when it is overexpressed but not deleted. The phenomenon that crude SMs of ΔVmnrps14OE-6 and ΔVmnrps14OE-13 exhibited increased toxicity on apple leaves instead on tobacco leaves suggested that overexpression of NRPS14 may lead to phyletic change or counts increase in toxic SMs, and the changed SMs could be host specific, like T-toxin in C. heterostrophus (Wu et al., 2012). Although the overexpression of NRPS14 was only verified by traditional PCR and RT-qPCR, current results including virulence and toxicity tests also proved that the gene was overexpressed. It seems there is still a contradiction as deletion of VmLaeA increased the expression of virulence-related NRPS14 but showed decreased virulence. As a matter of fact, reduction in virulence of ΔVmlaeA is most likely due to the combined effects of multiple virulence factors regulated by VmLaeA. For instance, one NRPS gene VM1G_04745 that was positively regulated by VmLaeA had been shown to contribute to virulence (Ma et al., 2016). VmLaeA also positively regulated two genes (VM1G_01526 and VM1G_01527) that belong to a virulence-related gene cluster NRPS4 (data not shown). CYPs can catalyze the conversion of hydrophobic intermediates of secondary metabolic pathways. One CYP gene VM1G_03094 that was positively regulated by VmLaeA had been proved to contribute to virulence (Gao et al., 2016). In addition, reduction in toxicity of the crude SMs of ΔVmlaeA also implied other virulence-related SMBGCs were involved. These SMBGCs might contribute to virulence by producing toxins. However, further research of toxins produced by these SMBGCs is still needed.
The phenomenon that deletion or silencing of some specific SMBGCs or regulatory genes may improve the biosynthesis of other SMs is known as cross-regulation (cross-talk) of SM biosynthetic pathways (Bergmann et al., 2010; Brakhage, 2013; Liu et al., 2013). P. chrysogenum mutants lacking the penicillin gene cluster produce increasing amounts of PR-toxin, and mutants of P. roqueforti silenced in the PR-toxin genes produce large amounts of mycophenolic acid (Martín, 2017). The LaeA-velvet complex could mediate pathway cross-talk of secondary metabolism. In ΔVmlaeA, 17 SMBGCs were downregulated and 14 were upregulated. Cross-talk may implicate in regulation of V. mali secondary metabolism and increased the complicacy of SMBGC expression. Apart from toxins and pigments, some SMBGCs may also be involved in the synthesis of SMs which are responsible for signal transduction, such as kojic acid that acts as a macrophage activator in Aspergillus spp. (Dufour and Rao, 2011; Rodrigues et al., 2011). These may be part of the reasons why some SMBGCs regulated by LaeA showed no effect on virulence after deletion and overexpression in our study.
In summary, VmLaeA is a crucial virulence factor of V. mali. SMBGCs are largely regulated by VmLaeA and contribute to virulence. SMBGCs that are involved in virulence provide genetic resources for elucidating pathogenicity-related toxins in V. mali.
The WT strain of V. mali was obtained from the stock culture of the Laboratory of Pathogen Biology and Integrated Control of Fruit Tree Diseases, College of Plant Protection, Northwest A&F University, Yangling, Shaanxi, China. Strains used in this study were cultured on PDA medium (20% potato extract, 2% glucose, and 1.5% agar) at 25°C. Potato dextrose broth medium (PDB; 20% potato extract, 2% dextrose) was used for cultivating mycelium used for RNA-sequencing and proteomic analysis. A synthetic liquid medium (10 g of sucrose, 2 g of L-asparagine monohydrate, 1 g of potassium dihydrogen phosphate, 0.5 g of magnesium sulfate heptahydrate, 0.88 g of zinc sulfate heptahydrate, 1.5 mg of ferric nitrate monohydrate, 0.44 mg of manganese sulfate pentahydrate, 5 μg of biotin, 100 μg of thiamine in 1 L) was used for fermentation to observe the color change of the mycelium. Apple bark broth (ABB) medium (30% apple branch extract, 0.2% yeast extract powder, 0.5% sucrose) was used for fermentation to extract crude SMs.
Three biological replicates of WT and ΔVmlaeA were cultivated in PDB at 25°C and 100 rpm for 48 h. Mycelium was collected and frozen in liquid nitrogen. Total RNA was extracted using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer’s protocol. RNA integrity number (RIN) of all RNA samples was assessed using a Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States) and samples of which the RIN are greater than six were considered to be of sufficient quality.
The cDNA libraries were prepared using the TruSeq Stranded mRNA Library Preparation kit (Illumina, San Diego, CA, United States). The cDNA was sequenced on a HiSeq2000 platform (Illumina). After filtering out low-quality reads (Phred ≤ 20) and adaptors, clean reads were mapped against predicted transcripts of the V. mali genome using Tophat v2.0.4 (Trapnell et al., 2009).
Extraction and purification of protein from mycelium was carried out according to the trichloroacetic acid (TCA)/acetone-sodium dodecyl sulfate (SDS)/phenol extraction method (Wang et al., 2003) with little modification. Mycelium dried at low temperature was ground into powder in liquid nitrogen. The ground powder was suspended in acetone containing 10% TCA and 0.2% dithiothreitol and allowed to stand for 2 h. After purification with methanol, the precipitate was extracted using SDS/phenol extraction buffer for 5 min on ice. The upper phase was transferred and precipitated by acetone containing 100 mmol ammonium acetate per liter. Proteins were obtained by centrifugation. SDS-PAGE was employed to control the quality of the extracted proteins.
Protein samples were digested using sequencing-grade trypsin for 24 h at 37°C. Trypsin-digested peptides were labeled with TMT 10-plex reagents, using 126-tag, 127N-tag, 127C-tag for the three biological replicates of the WT, and 129C-tag, 130N-tag, 130C-tag for the three biological replicates of ΔVmlaeA, respectively. The tag-labeled samples were then mixed followed by the first dimensional high pH reverse-phase (hpRP) separation of tryptic peptide mixtures using an UltiMate 3000 MDLC platform (Thermo-Dionex, Sunnyvale, CA, United States). A total of 12 fractions were collected and dried for further nanoLC-MS/MS analysis. The nanoLC-MS/MS analysis was performed by an Orbitrap Fusion (Thermo-Fisher Scientific, San Jose, CA, United States) mass spectrometer equipped with nano ion source using high-energy collision dissociation coupled with auto-sampler injection and NanoLC RP (NanoLC, 75 μm ID column). A 120-min NanoLC gradient on one LC fraction was used for MS/MS analysis as described previously (Qin et al., 2019). All MS and MS/MS raw spectra were processed and searched using Sequest HT software within the Proteome Discoverer 1.4 (PD1.4; Thermo). The V. mali protein sequence database containing 11,218 entries downloaded on NCBI1 were used for database searches. The default search settings used for 10-plex TMT quantitative processing and protein identification in PD1.4 searching software were two mis-cleavages for full trypsin with fixed carbamidomethyl modification of cysteine, fixed 10-plex TMT modifications on lysine and N-terminal amines, and variable modifications of methionine oxidation and deamidation on asparagines/glutamine residues. The peptide mass tolerance and fragment mass tolerance values were 10 ppm and 50 mDa, respectively. Identified peptides were filtered for maximum 1% FDR using the Percolator algorithm in PD 1.4 along with additional peptide confidence set to high. The TMT10plex quantification method within Proteome Discoverer 1.4 software was used to calculate the reporter ratios with mass tolerance ±10 ppm without applying the isotopic correction factors. Only peptide spectra containing all reporter ions were designated as “quantifiable spectra” and used for peptide/protein quantitation. A protein ratio was expressed as a median value of the ratios for all quantifiable spectra of the unique peptides pertaining to that protein. A precursor co-isolation filter of 50% was also applied for minimizing ratio compression caused by co-isolation of precursor ions. For each relative ratio group, normalization on protein median was applied. The comparison between groups was undertaken with Microsoft Excel software.
Transcriptomic DEGs of ΔVmlaeA were analyzed using the DESeq package (Anders and Huber, 2010) with fold change >2 (beta value >1 or <−1) and FDR ≤ 0.05 as thresholds. The database search of proteomic data was performed by Proteome Discoverer 2.1 (PD 2.1) with the Sequest HT search engine for both protein identification and quantification against the protein database of V. mali which consists of 11,218 sequence entries. Peptide identification was filtered by FDR less than 1% and at least two unique peptides were required. Differentially expressed proteins were determined by a 1.5-fold change cut-off. Blast2go (Bioinformatics Department, Valencia, Spain) and OmicShare2 were used for the GO and KEGG annotations and functional classification of identified genes. SMBGCs were predicted by antiSMASH.
VmLaeA and the core enzyme genes of cluster PKS7, PKS11, NRPS14, PKS16, PKS23, PKS31, NRPS/PKS33, and PKS39 were selected to construct deletion mutants. The gene knockout cassettes were assembled by double-joint PCR with upstream and downstream flanking sequences of the target gene and neo gene as selective marker (Yu et al., 2004; Supplementary Figure 8). The gene knockout cassettes were then transformed into protoplasts of V. mali mediated by polyethylene glycol (Gao et al., 2011). Putative deletion mutants filtered by selection with geneticin were confirmed by PCR and further confirmed by Southern blot hybridization using the DIG DNA Labeling and Detection Kit II (Roche, Mannheim, Germany). Gene complementation was conducted by cloning the target genes into plasmid PDL2 by the yeast gap repair approach and then transforming the recombined plasmids into the respective target gene deletion mutant (Bruno et al., 2004; Zhou et al., 2012). The complemented mutants were finally confirmed by PCR. All primers used in gene deletion and complementation are listed in Supplementary Table 11.
The TF genes of cluster PKS11, NRPS14, PKS16, PKS23, PKS31, and PKS39 were predicted by antiSMASH (Medema et al., 2011) and cloned into plasmid PDL2. The recombinant plasmids were then transformed into protoplasts of V. mali after sequencing. Transformants were firstly filtered by selection with hygromycin B and PCR was conducted to select overexpression mutants for each TF gene. Afterward, RNA of WT and the overexpression mutants were extracted and used for qRT-PCR analysis to determine the overexpression of the TF genes. G6PDH was used as reference gene (Yin et al., 2013) and relative expression levels were calculated with the 2–ΔΔCT method (Livak and Schmittgen, 2001).
WT, deletion, complemented, and overexpression mutants were inoculated onto PDA plates and the growth, color, and conidia formation were observed at 48 h, 5, and 30 days. Pathogenicity assays were conducted by inoculating WT and mutants on detached leaves or twigs of Malus domestica Borkh. cv. “Fuji” and cultivation for 3 days on leaves or 4 days on twigs at 25°C followed by lesion measuring and data analysis (Wei et al., 2010). Each experiment was replicated at least three times.
WT and ΔVmlaeA were cultivated in ABB medium at 25°C and 100 rpm for 10 days. Culture filtrates of WT, ΔVmlaeA, and ABB medium were dried at low temperature and extracted with ethyl acetate three times. The organic phase was concentrated to obtain crude extracts of SMs. All crude extracts were dissolved to 50 mg/ml in ddH2O and tested on apple leaves with the leaf puncture method and tested on tobacco leaves by infiltration with the concentration of 5 mg/ml (Sugawara et al., 1987; Wang C. et al., 2014). ABB crude extract–treated and ddH2O-treated samples were kept as control and symptoms were carefully examined 48 h every 12 h. The pictures were taken at 24 h after treatment with a ruler included. ImageJ software was used to calculate the areas of necrotic lesions. At least three biological repeats of necrotic lesions were used in the statistical analysis.
The transcriptomic data has been uploaded to SRA database (accession: PRJNA666017). The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021783.
YF, YW, and HD performed the experiment. YF, ZY, and LX analyzed the results. YF, ZY, YW, LX, NW, and LH composed the article. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (U1903206) and the Natural Science Basic Research Program of Shaanxi (2018JM3012).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Sheng Zhang at Cornell University for helping with proteomics and NIH SIG grant 1S10 OD017992-01 grant support for the Orbitrap Fusion mass spectrometer. We are grateful to Ralf T. Voegele at Universität Hohenheim for revising this article.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.581203/full#supplementary-material
Akhberdi, O., Zhang, Q., Wang, D., Wang, H., Hao, X., Liu, Y., et al. (2018). Distinct roles of velvet complex in the development, stress tolerance, and secondary metabolism in Pestalotiopsis microspora, a taxol producer. Genes 9:164. doi: 10.3390/genes9030164
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Baker, S. E., Kroken, S., Inderbitzin, P., Asvarak, T., Li, B., Shi, L., et al. (2006). Two polyketide synthase-encoding genes are required for biosynthesis of the polyketide virulence factor, T-toxin, by Cochliobolus heterostrophus. Mol. Plant Microbe Interact. 19, 139–149. doi: 10.1094/MPMI-19-0139
Bayram, Ö, Krappmann, S., Ni, M., Bok, J. W., Helmstaedt, K., Valerius, O., et al. (2008). VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320, 1504–1506. doi: 10.1126/science.1155888
Belozerskaya, T. A., Gessler, N. N., and Averyanov, A. A. (2017). “Melanin pigments of fungi,” in Fungal Metabolites, eds J. M. Mérillon and K. Ramawat (Cham: Springer), 263–291. doi: 10.1007/978-3-319-25001-4_29
Bergmann, S., Funk, A. N., Scherlach, K., Schroeckh, V., Shelest, E., Horn, U., et al. (2010). Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster. Appl. Environ. Microbiol. 76, 8143–8149. doi: 10.1128/AEM.00683-10
Bergmann, S., Schumann, J., Scherlach, K., Lange, C., Brakhage, A. A., and Hertweck, C. (2007). Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol. 3, 213–217. doi: 10.1038/nchembio869
Bok, J. W., and Keller, N. P. (2004). LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3, 527–535. doi: 10.1128/EC.3.2.527-535.2004
Brakhage, A. A. (2013). Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32. doi: 10.1038/nrmicro2916
Bruno, K. S., Tenjo, F., Li, L., Hamer, J. E., and Xu, J. R. (2004). Cellular localization and role of kinase activity of PMK1 in Magnaporthe grisea. Eukaryot. Cell 3, 1525–1532. doi: 10.1128/EC.3.6.1525-1532.2004
Butchko, R. A. E., Brown, D. W., Busman, M., Tudzynski, B., and Wiemann, P. (2012). Lae1 regulates expression of multiple secondary metabolite gene clusters in Fusarium verticillioides. Fungal Genet. Biol. 49, 602–612. doi: 10.1016/j.fgb.2012.06.003
Chang, P. K., Cary, J. W., and Lebar, M. D. (2020). Biosynthesis of conidial and sclerotial pigments in Aspergillus species. Fungal Biol. 104, 2277–2286. doi: 10.1007/s00253-020-10347-y
Crespo-Sempere, A., Marin, S., Sanchis, V., and Ramos, A. J. (2013). VeA and LaeA transcriptional factors regulate ochratoxin A biosynthesis in Aspergillus carbonarius. Int. J. Food Microbiol. 166, 479–486. doi: 10.1016/j.ijfoodmicro.2013.07.027
Deighton, N., Muckenschnabel, I., Colmenares, A. J., Collado, I. G., and Williamson, B. (2001). Botrydial is produced in plant tissues infected by Botrytis cinerea. Phytochemistry 57, 689–692. doi: 10.1016/S0031-9422(01)00088-7
Doehlemann, G., Ökmen, B., Zhu, W., and Sharon, A. (2017). Plant pathogenic fungi. Microbiol. Spectrum 5, 701–726. doi: 10.1128/microbiolspec
Dow, J. M., Davies, H. A., and Daniels, M. J. (1998). A metalloprotease from Xanthomonas campestris that specifically degrades proline/hydroxyprolinerich glycoproteins of the plant extracellular matrix. Mol. Plant. Microbe. Interact. 11, 1085–1093. doi: 10.1094/mpmi.1998.11.11.1085
Dufour, N., and Rao, R. P. (2011). Secondary metabolites and other small molecules as intercellular pathogenic signals. FEMS Microbiol. Lett. 314, 10–17. doi: 10.1111/j.1574-6968.2010.02154.x
Dunaevsky, Y. E., Matveeva, A. R., Beliakova, G. A., Domash, V. I., and Belozersky, M. A. (2007). Extracellular alkaline proteinase of Colletotrichum gloeosporioides. Biochemistry 72, 345–350. doi: 10.1134/S0006297907030145
Gao, J., Li, Y., Ke, X., Kang, Z., and Huang, L. (2011). Development of genetic transformation system of Valsa mali of apple mediated by PEG. Acta Microbiol. Sin. 51, 1194–1199.
Gao, M., Wu, Y., Zhu, B., Gao, X., Feng, H., and Huang, L. (2016). Characterization of cytochrome P450 gene Vmcyp5 in Valsa mali. Acta Microbiol. Sin. 58, 274–283.
Georgianna, D. R., Fedorova, N. D., Burroughs, J. L., Dolezal, A. L., Bok, J., Horowitz-Brown, S., et al. (2010). Beyond aflatoxin: four distinct expression patterns and functional roles associated with Aspergillus flavus secondary metabolism gene clusters. Mol. Plant Pathol. 11, 213–226. doi: 10.1111/j.1364-3703.2009.00594.x
Gregory, P., Earle, E. D., and Gracen, V. E. (1980). Effects of purified Helminthosporium maydis race T Toxin on the structure and function of corn mitochondria and protoplasts. Plant Physiol. 66, 477–481. doi: 10.1104/pp.66.3.477
Janevska, S., Arndt, B., Baumann, L., Apken, L. H., Marques, L. M. M., Humpf, H. U., et al. (2017). Establishment of the inducible Tet-on system for the activation of the silent trichosetin gene cluster in Fusarium fujikuroi. Toxins 9:126. doi: 10.3390/toxins9040126
Ke, X., Huang, L., Han, Q., Gao, X., and Kang, Z. (2013). Histological and cytological investigations of the infection and colonization of apple bark by Valsa mali var. mali. Austral. Plant Pathol. 42, 85–93. doi: 10.1007/s13313-012-0158-y
Ke, X., Yin, Z., Song, N., Dai, Q., Voegele, R. T., Liu, Y., et al. (2014). Transcriptome profiling to identify genes involved in pathogenicity of Valsa mali on apple tree. Fungal Genet. Biol. 68, 31–38. doi: 10.1016/j.fgb.2014.04.004
Keller, N. P. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180. doi: 10.1038/s41579-018-0121-1
Kim, H. K., Lee, S., Jo, S. M., McCormick, S. P., Butchko, R. A. E., Proctor, R. H., et al. (2013). Functional roles of FgLaeA in controlling secondary metabolism, sexual development, and virulence in Fusarium graminearum. PLoS One 8:e68441. doi: 10.1371/journal.pone.0068441
Koganezawa, H., and Sakuma, T. (1982). Possible role of breakdown products of phloridzin in symptom development by Valsa ceratosperma. Ann. Phytopath. Soc. Jpn. 48, 521–528. doi: 10.3186/jjphytopath.48.521
Kumar, D., Barad, S., Chen, Y., Luo, X., Tannous, J., Dubey, A., et al. (2017). LaeA regulation of secondary metabolism modulates virulence in Penicillium expansum and is mediated by sucrose. Mol. Plant Pathol. 18, 1150–1163. doi: 10.1111/mpp.12469
Lee, D. H., Lee, S. W., Choi, K. H., Kim, D. A., and Uhm, J. Y. (2006). Survey on the occurrence of apple diseases in Korea from 1992 to 2000. Plant Pathol. J. 22, 375–380. doi: 10.5423/ppj.2006.22.4.375
Li, Y., Han, W., Gui, X., Wei, T., Tang, S., and Jin, J. (2016). Putative nonribosomal peptide synthetase and cytochrome p450 genes responsible for tentoxin biosynthesis in Alternaria alternata ZJ33. Toxins 8:234. doi: 10.3390/toxins8080234
Li, Z., Gao, X., Du, Z., Hu, Y., Kang, Z., and Huang, L. (2013). Survey of apple Valsa canker in Weibei area of Shaanxi province. Boreali Occident. Sin. 22, 174–178.
Liu, G., Chater, K. F., Chandra, G., Niu, G., and Tan, H. (2013). Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143. doi: 10.1128/MMBR.00054-12
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lv, Y., Lv, A., Zhai, H., Zhang, S., Li, L., Cai, J., et al. (2018). Insight into the global regulation of laeA in Aspergillus flavus based on proteomic profiling. Int. J. Food Microbiol. 284, 11–21. doi: 10.1016/j.ijfoodmicro.2018.06.024
Ma, C., Li, Z., Dai, Q., Han, Q., and Huang, L. (2016). Function of nonribosomal peptide synthetase gene VmNRPS12 of Valsa mali. Acta Microbiol. Sin. 56, 1273–1281. doi: 10.13343/j.cnki.wsxb.20150504
Martín, J. F. (2017). Key role of LaeA and velvet complex proteins on expression of β-lactam and PR-toxin genes in Penicillium chrysogenum: cross-talk regulation of secondary metabolite pathways. J. Ind. Microbiol. Biotechnol. 44, 525–535. doi: 10.1007/s10295-016-1830-y
Medema, M. H., Blin, K., Cimermancic, P., Jager, V. D., Zakrzewski, P., Fischbach, M. A., et al. (2011). AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, 339–346. doi: 10.1093/nar/gkr466
Meredith, D., and Christian, H. C. (2008). The SLC16 monocaboxylate transporter family. Xenobiotica 38, 1072–1106. doi: 10.1080/00498250802010868
Mohnen, D. (2008). Pectin structure and biosynthesis. Curr. Opin. Plant Biol. 11, 266–277. doi: 10.1016/j.pbi.2008.03.006
Müller, N., Leroch, M., Schumacher, J., Zimmer, D., Konnel, A., Klug, K., et al. (2018). Investigations on VELVET regulatory mutants confirm the role of host tissue acidification and secretion of proteins in the pathogenesis of Botrytis cinerea. New Phytol. 219, 1062–1074. doi: 10.1111/nph.15221
Myung, K., Zitomer, N. C., Duvall, M., Glenn, A. E., Riley, R. T., and Calvo, A. M. (2012). The conserved global regulator VeA is necessary for symptom production and mycotoxin synthesis in maize seedlings by Fusarium verticillioides. Plant Pathol. 61, 152–160. doi: 10.1111/j.1365-3059.2011.02504.x
Natsume, H., Seto, H., and Otake, N. (1982). Studies on apple canker disease. The necrotic toxins produced by Valsa ceratosperma. Agric. Biol. Chem. 46, 2101–2106. doi: 10.1271/bbb1961.46.2101
Okuno, T., Oikawa, S., Goto, T., Sawai, K., Shirahama, H., and Matsumoto, T. (1986). Structures and phytotoxicity of metabolites from Valsa ceratosperma. Agric.Biol. Chem. 50, 997–1001. doi: 10.1271/bbb1961.50.997
Perrin, R. M., Fedorova, N. D., Jin, W. B., Cramer, R. A., Wortman, J. R., Kim, H. S., et al. (2007). Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 3:e0030050. doi: 10.1371/journal.ppat.0030050
Qin, L., Walk, T. C., Han, P., Chen, L., Zhang, S., Li, Y., et al. (2019). Adaption of roots to nitrogen deficiency revealed by 3D quantification and proteomic analysis. Plant Physiol. 179, 329–347. doi: 10.1104/pp.18.00716
Richard, J. L. (2007). Some major mycotoxins and their mycotoxicoses-an overview. Int. J. Food Microbiol. 119, 3–10. doi: 10.1016/j.ijfoodmicro
Rodrigues, A. P. D., Carvalho, A. S. C., Santos, A. S., Alves, C. N., Nascimento, J. L. M., and do Silva, E. O. (2011). Kojic acid, a secondary metabolite from Aspergillus sp., acts as an inducer of macrophage activation. Cell Biol. Int. 35, 335–343. doi: 10.1042/CBI20100083
Rutledge, P. J., and Challis, G. L. (2015). Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 13, 509–523. doi: 10.1038/nrmicro3496
Sá-Correia, I., dos Santos, S. C., Teixeira, M. C., Cabrito, T. R., and Mira, N. P. (2009). Drug: H+ antiporters in chemical stress response in yeast. Trends Microbiol. 17, 22–31. doi: 10.1016/j.tim.2008.09.007
Schumacher, J., Simon, A., Cohrs, K. C., Traeger, S., Porquier, A., Dalmais, B., et al. (2015). The VELVET complex in the gray mold fungus Botrytis cinerea: impact of BcLAE1 on differentiation, secondary metabolism, and virulence. Mol. Plant. Microbe. Interact. 28, 659–674. doi: 10.1094/MPMI-12-14-0411-R
Siewers, V., Viaud, M., Jimenez-Teja, D., Collado, I. G., Gronover, C. S., Pradier, J. M., et al. (2005). Functional analysis of the cytochrome P450 monooxygenase gene bcbot1 of Botrytis cinerea indicates that botrydial is a strain-specific virulence factor. Mol. Plant Microbe Interact. 18, 602–612. doi: 10.1094/MPMI-18-0602
Sugawara, F., Strobel, G., and Strange, R. N. (1987). Phytotoxins from the pathogenic fungi Drechslera maydis and Drechslera sorghicola. Proc. Natl. Acad. Sci. U.S.A. 84, 3081–3085. doi: 10.1073/pnas.84.10.3081
Sugui, J. A., Pardo, J., Chang, Y. C., Mullbacher, A., Zarember, K. A., Galvez, E. M., et al. (2007). Role of laeA in the regulation of alb1, gliP, conidial morphology, and virulence in Aspergillus fumigatus. Eukaryot. Cell 6, 1552–1561. doi: 10.1128/EC.00140-07
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Wang, C., Wang, G., Zhang, Q., Dong, X., Li, B., Li, C., et al. (2014). Toxins produced by Valsa mali var. mali and their relationship with pathogenicity. Toxins 6, 1139–1154. doi: 10.3390/toxins6031139
Wang, G., Zhang, H., Wang, Y., Liu, F., Li, E., Ma, J., et al. (2019). Requirement of LaeA, VeA, and VelB on asexual development, ochratoxin A biosynthesis, and fungal virulence in Aspergillus ochraceus. Front. Microbiol. 10:2759. doi: 10.3389/fmicb.2019.02759
Wang, W., Scali, M., Vignani, R., Spadafora, A., Sensi, E., Mazzuca, S., et al. (2003). Protein extraction for two-dimensional electrophoresis from olive leaf, a plant tissue containing high levels of interfering compounds. Electrophoresis 24, 2369–2375. doi: 10.1002/elps.200305500
Wang, X., Zang, R., Yin, Z., Kang, Z., and Huang, L. (2014). Delimiting cryptic pathogen species causing apple Valsa canker with multilocus data. Ecol. Evol. 4, 1369–1380. doi: 10.1002/ece3.1030
Wei, J., Huang, L., Gao, Z., Ke, X., and Kang, Z. (2010). Laboratory evaluation methods of apple Valsa canker disease caused by Valsa ceratosperma sensu Kobayashi. Acta Phytopathol. Sin. 40, 14–20.
Wu, D., Oide, S., Zhang, N., Choi, M. Y., and Turgeon, B. G. (2012). ChLae1 and ChVel1 regulate T-toxin production, virulence, oxidative stress response, and development of the maize pathogen Cochliobolus heterostrophus. PLoS Pathog. 8:e1002542. doi: 10.1371/journal.ppat.1002542
Yao, G., Yue, Y., Fu, Y., Fang, Z., Xu, Z., Ma, G., et al. (2018). Exploration of the regulatory mechanism of secondary metabolism by comparative transcriptomics in Aspergillus flavus. Front. Microbiol. 9:1568. doi: 10.3389/fmicb.2018.01568
Yin, Z., Ke, X., Huang, D., Gao, X., Voegele, R. T., Kang, Z., et al. (2013). Validation of reference genes for gene expression analysis in Valsa mali var. mali using real-time quantitative PCR. World J. Microbiol. Biotechnol. 29, 1563–1571. doi: 10.1007/s11274-013-1320-6
Yin, Z., Liu, H., Li, Z., Ke, X., Dou, D., Gao, X., et al. (2015). Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 208, 1202–1216. doi: 10.1111/nph.13544
Yu, J. H., Hamari, Z., Han, K. H., Seo, J. A., Reyes-Domínguez, Y., and Scazzocchio, C. (2004). Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 41, 973–981. doi: 10.1016/j.fgb.2004.08.001
Keywords: Valsa mali, LaeA, virulence, secondary metabolism, transcriptome, proteome, NRPS
Citation: Feng Y, Yin Z, Wu Y, Xu L, Du H, Wang N and Huang L (2020) LaeA Controls Virulence and Secondary Metabolism in Apple Canker Pathogen Valsa mali. Front. Microbiol. 11:581203. doi: 10.3389/fmicb.2020.581203
Received: 08 July 2020; Accepted: 07 October 2020;
Published: 05 November 2020.
Edited by:
Yuxian Xia, Chongqing University, ChinaReviewed by:
Caixia Wang, Qingdao Agricultural University, ChinaCopyright © 2020 Feng, Yin, Wu, Xu, Du, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Huang, aHVhbmdsaWxpQG53c3VhZi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.