 Yuying Fu1,2
Yuying Fu1,2 Lishan Zhang1,2
Lishan Zhang1,2 Guibin Wang1,2,3
Guibin Wang1,2,3 Yuexu Lin1,2
Yuexu Lin1,2 Srinivasan Ramanathan1,2Guidi Yang1,2Wenxiong Lin1,2*
Srinivasan Ramanathan1,2Guidi Yang1,2Wenxiong Lin1,2* Xiangmin Lin1,2,4*
Xiangmin Lin1,2,4*- 1Fujian Provincial Key Laboratory of Agroecological Processing and Safety Monitoring, School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, China
- 2Key Laboratory of Crop Ecology and Molecular Physiology (Fujian Agriculture and Forestry University), Fujian Province University, Fuzhou, China
- 3State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, Beijing, China
- 4Key Laboratory of Marine Biotechnology of Fujian Province, Institute of Oceanology, Fujian Agriculture and Forestry University, Fuzhou, China
Aeromonas hydrophila is an aquatic pathogen of freshwater fish. The emergence of widespread antimicrobial-resistance strains of this pathogen has caused increasing rates of fish infections. Our previous research reported that A. hydrophila yeeY, a LysR-type transcriptional regulator (LTTR), negatively regulated furazolidone (FZ) resistance. Although, it’s intrinsic regulatory mechanism is still unclear. In this study, a data-independent acquisition (DIA) quantitative proteomics method was used to compare the differentially expressed proteins (DEPs) between the ΔyeeY and wild-type strain under FZ treatment. When compared to the control, a total of 594 DEPs were identified in ΔyeeY. Among which, 293 and 301 proteins were substantially increased and decreased in abundance, respectively. Bioinformatics analysis showed that several biological pathways such as the secretion system and protein transport were mainly involved in FZ resistance. Subsequently, the antibiotics susceptibility assays of several gene deletion strains identified from the proteomics results showed that YeeY may regulate some important genes such as cysD, AHA_2766, AHA_3195, and AHA_4275, which affects the FZ resistance in A. hydrophila. Furthermore, 34 antimicrobial resistance genes (ARGs) from the bacterial drug resistance gene database (CARD) were found to be directly or indirectly regulated by YeeY. A subsequent assay of several ARGs mutants showed that ΔAHA_3222 increased the susceptibility of A. hydrophila to FZ, while ΔcysN and ΔAHA_3753 decreased the susceptibility rate. Finally, the chromatin immunoprecipitation (ChIP) PCR and an electrophoretic mobility shift assay (EMSA) have revealed that the genes such as AHA_3222 and AHA_4275 were directly and transcriptionally regulated by YeeY. Taken together, our findings demonstrated that YeeY may participate in antimicrobial resistance of A. hydrophila to FZ, which provides a new target for the development of novel antimicrobial agents in the future.
Introduction
Aeromonas hydrophila is a well-known pathogen of freshwater fish, which is widely distributed in aquatic habitats including surface water, oceans, sewage and even chlorinated water (Liu et al., 2019). The number of reported cases caused by A. hydrophila has been grown worldwide. Therefore, this pathogen has received an increasing attention to combat their infections. Further, a several earlier studies have reported that A. hydrophila is a causative agent of hemorrhagic septicemia and motile aeromonad septicemia (MAS) in fish (Zahran et al., 2018; Abdelhamed et al., 2019). Outbreaks of A. hydrophila in humans have been recorded since 1992, which thought to be related to diarrhea, septicemia and soft-tissue wound infections in humans (de la Morena et al., 1993; Soltan Dallal et al., 2016). Further, a tons of antibiotics are used in aquaculture each year for controlling the infections in aquaculture worldwide, which causes a significant bacterial antibiotic resistance (Dahanayake et al., 2019). Therefore, the fish vaccines are used to the control the spread of diseases connected with A. hydrophila. However, only a few aquatic vaccines have obtained the national veterinary drug certifications in China, meaning that control measures are far from meeting the needs of the status quo (Wang Q. et al., 2020). Thus, it is necessary to understand the mechanism of antibiotic resistance in this pathogen to develop new and novel therapeutic agents.
It is generally agreed that the production of enzymes that inactivate antibiotics, changes in the target of antibiotics, increases in the permeability of cell membrane pore proteins and activation of efflux pumps are classic mechanisms of bacterial resistance (Das et al., 2020). Moreover, the previous studies have documented the outer membrane proteins (e.g., LamB and OmpA), fatty acid biosynthesis and the central metabolism of A. hydrophila also play an important roles in antibiotic resistance (Lin et al., 2014; Li et al., 2016; Yao et al., 2016). Nevertheless, specific mechanisms of antibiotic resistance are still largely elusive; although the proteins involved in antibiotic resistance yet need to be discovered.
LysR-type transcriptional regulators (LTTRs) are belonging to the transcription regulation families, which are widespread in prokaryotes. They are generally reported to be involved in bacterial antibiotic resistance. For example, the LTTR protein CidR was reported to positively regulate the Staphylococcus aureus cidABC operon, which enhances murein hydrolase activity and affects antibiotic tolerance against penicillin, vancomycin and rifampicin (Yang et al., 2005). A another LTTR protein OxyR regulates the cell division superfamily (RND) efflux pump gene acrB in Klebsiella pneumoniae and thereby confers resistance to chloramphenicol, erythromycin, nalidixic acid and trimethoprim (Rice et al., 2005). Since, LTTRs control diverse multi-functional genes and proteins as a well-known transcriptional regulator family, the antibiotic resistance regulatory functions of LTTRs are needed to be further investigated.
YeeY in A. hydrophila ATCC 7966 belongs to the LTTRs family that encodes a putative HTH-type transcriptional regulator. A multiple sequence alignment analysis showed that it’s DNA binding domain with an HTH motif at the N terminal and a regulatory domain at the C terminal is highly conserved with those of other LTTRs (Kim et al., 2018). Interestingly, the deletion of yeeY (AHA_3980 as well) has displayed multidrug characteristics, which significantly increased resistance to chloramphenicol, chlortetracycline, ciprofloxacin, furazolidone and balofloxacin. So, it clearly suggests that the YeeY act as a negative transcriptional regulator during antibiotic stresses (Fu et al., 2019). However, the intrinsic antibiotic resistance mechanism regulated by A. hydrophila YeeY remains unclear.
In this study, to further understand the role of YeeY in antibiotic resistance, we compared the differentially expressed proteins (DEPs) between ΔyeeY and wild-type strains under the stress of FZ using data-independent acquisition (DIA) quantitative proteomics technology. The bioinformatics analysis showed that the deletion of yeeY affected several key biological processes and metabolic pathways. Several DEPs were subsequently validated by Western blotting and several related genes were deleted and their antimicrobial susceptibilities were evaluated. Furthermore, the outcome of chromatin immunoprecipitation (ChIP) PCR and electrophoretic mobility shift assay (EMSA) were suggested that some antimicrobial resistance related genes were directly and transcriptionally upregulated by YeeY. Overall, the obtained results of this study provide a novel understanding on the regulatory role of LTTRs YeeY in antibiotic resistance of A. hydrophila against FZ.
Materials and Methods
Bacterial Strain and Sample Preparation
A. hydrophila ATCC 7966 (WT), yeeY deleted strain (ΔyeeY), the rescue strain (ΔyeeY + pACYC184-yeeY), a negative control strain (ΔyeeY + pACYC184) and other gene deletion strains were constructed and stored in our laboratory as previously described (Pang et al., 2018). Bacterial strains were cultured overnight in Luria-Bertani (LB) medium at 30°C on a shaker at 200 rpm and then transferred to 100 mL LB medium at a ratio of 1:100 with a final concentration of 1.5 μg/mL of FZ treatment. The cells were collected by centrifugation at 8000 g for 15 min at 4°C when the OD of the bacteria culture reached 1.0 at 600 nm. After washed three times with pre-cooled PBS buffer, the cell pellets were resuspended in lysis buffer (6 M urea, 2 M thiourea, 100 mM Tris–HCl pH 8.5, protease inhibitor) and then ultrasonically disrupted by sonication on ice for 15 min at 30% power with intervals of 9 s. Then, the whole proteins were isolated by centrifuging at 12000 × g for 30 min at 4°C and the concentrations were determined by the Bradford method.
Trypsin Digestion
A total of 50 μg protein of each sample was reduced with dithiothreitol (DTT), alkylated by iodoacetamide (IAA) and then digested with trypsin at a ratio of 1:50 for 16–18 h at 37°C. Subsequently, the digested peptides were desalted using a Sep-Pak Vac C18 Column (Waters Inc., Milford, MA) and dried using a CentriVap Concentrator (Labconco, Inc., Kansas City, MO) (Sun et al., 2019; Wisniewski, 2019; Yao et al., 2019).
DIA Based LC-MS/MS Analysis
The digested peptides were resuspended in 25 μL of 0.1% formic acid (FA) containing iRT standard peptide (Biognosys) and then submitted to analysis on an Q Exactive HF mass spectrometer (Thermo Scientific, United States) with an EASY-nano-LC1200 chromatographic system (Thermo Scientific, United States) as previously described (Doerr, 2014). Briefly, peptides were loaded onto a C18 Trap column EASY-nano-LC at a rate of 4.5 μL/min with 12 μL phase A (2% acetonitrile and 0.1% FA) at a maximum pressure of 280 bars and then eluted using a C18 column with a runtime of 120 min at a rate of 600 nL/min using phase A (2% acetonitrile and 0.1% FA) and phase B (98% acetonitrile and 0.1% FA) with the following gradient elution program: 0–18 min, 6–12% B; 18–77 min, 12–20% B; 77–109 min, 20–32% B; 109–110 min, 32–90% B; 111–120 min, 90% B hold. The data were collected in the nano-spray ion source with an ion-spray voltage of 2.1 kV and an ion source temperature of 320°C. The scan range was 350 to 1400 m/z at a resolution of 60000 FWHM (at m/z 200) with automatic gain control (AGC) set to 3 E6 (maximum injection time of 20 ms). Further, the MS/MS was scanned at a resolution of 15000 FWHM (at m/z 200) with an isolation window of 1.6 Da with AGC set to 5 E4 (maximum injection time of 45 ms). DIA was scanned at a resolution of 30000 FWHM (at m/z 200) with a set of 45 variable overlapping windows covering the precursor mass range of 350–1400 m/z.
A confidential spectral library generated as previously study was used for spectral library (Wang G. B. et al., 2020). The DIA-generated data were imported into Spectronaut Pulsar X for protein qualitative and quantitative analyses using the following parameters: the iTR curve preceded local (non-linear) regression and protein identification was performed with Precursor Q value Cutoff 0.01 software. The identified proteins with at least two peptides matching and the peptide and protein FDR (false discovery rate) less than 0.01 were further analyzed.
Bioinformatics Analysis
The Gene Ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of DEPs were analyzed with DAVID online software1 (Li H. L. et al., 2020; Zhang et al., 2020). The GO and KEGG results were displayed in a diagram using GOCircle and GOChord plot functions of the GOplot R package. The predicted antibiotic resistance of DEPs was analyzed with the Comprehensive Antibiotic Research Database (CARD2) and visualized with loop heat map using the TBtool software (Chen et al., 2019, 2020).
Western Blotting Validation
To validate the proteomics results, several specific antibodies were developed in house and Western blotting was performed as described previously (Li L. et al., 2020). Protein samples from each group were run on SDS-PAGE gels and transferred to PVDF membranes (Millipore, Billerica, MA, United States) for 15 min at 25 V using a Trans-Blot SD Cell (Bio-Rad, Hercules, CA, United States). After that, the PVDF membranes were washed three times with PBST and blocked in 5% skim milk with PBS buffer containing 0.05% Tween 20 (PBST) for 1 h and then probed with primary antibody before being incubated for 1 h at room temperature. Then, the membranes were washed with PBST five times and incubated in HRP goat anti-mouse or anti-rabbit IgG for 1 h at room temperature. Finally, the PVDF membranes were washed five times with PBST, exposed using the ECL system and visualized with a ChemiDocTM MP imager (Bio-Rad, Hercules, CA, United States). The band signals were quantified with Image J software.
Antibiotics Susceptibility Assay
The antibiotics susceptibility assay was performed as described previously (Yao et al., 2018; Zhang et al., 2019). Briefly, the mutant and WT strains were incubated at 30°C for overnight and then diluted to ratio of 1:100 with fresh LB medium. After being treated with a series of concentrations of 0.25, 0.5, 1.0, and 2.0 μg/mL of FZ, the samples were divided into a HONEYCOMB® Sterile 100 well plate and the OD600 value was determined at 16 h using a Bioscreen C instrument (Oy Growth Curves AB Ltd., Helsinki, Finland).
ChIP-PCR
To further investigate the regulations between transcriptional regulator YeeY and candidate genes, ChIP-PCR was performed as previously described with slight modifications (Shen et al., 2020). Briefly, ΔyeeY containing pACYC184-His-yeeY or pACYC184-His were incubated at 30°C for overnight and then collected by centrifugation at 6000 g for 10 min at 4°C. After being washed three times with pre-cooled PBS buffer, the pellets were resuspended in 40 μL of 1% formaldehyde containing PBS buffer and placed on ice for 5 min; then, 3 M glycine solution was added for 15 min to terminate the crosslinking reaction. After crosslinking, the chromatin was collected and resuspended in 40 μL PBS buffer. After that, the chromatin was fragmented by sonication on ice for 20 min at 30% power with intervals of 9 s on ice to obtain an average length of 300 to 500 bps. Then, the nickel beaver beads were incubated with the crosslinking products for 2 h at 4°C and then the immunoprotein was eluted using 400 μL of 500 mM imidazole. After immunoprecipitation, 10% SDS and 2 μL of 100 mg/mL proteinase K was added to the precipitate and the samples were stored at 37°C. Next, the crosslinking was reversed and the DNA was extracted using phenol, chloroform and isoamyl alcohol at a ratio of 25:24:1 and then dissolved in 100 μL sterile water. Finally, the purified DNA was analyzed by ChIP-PCR using the primers listed in Table 1.
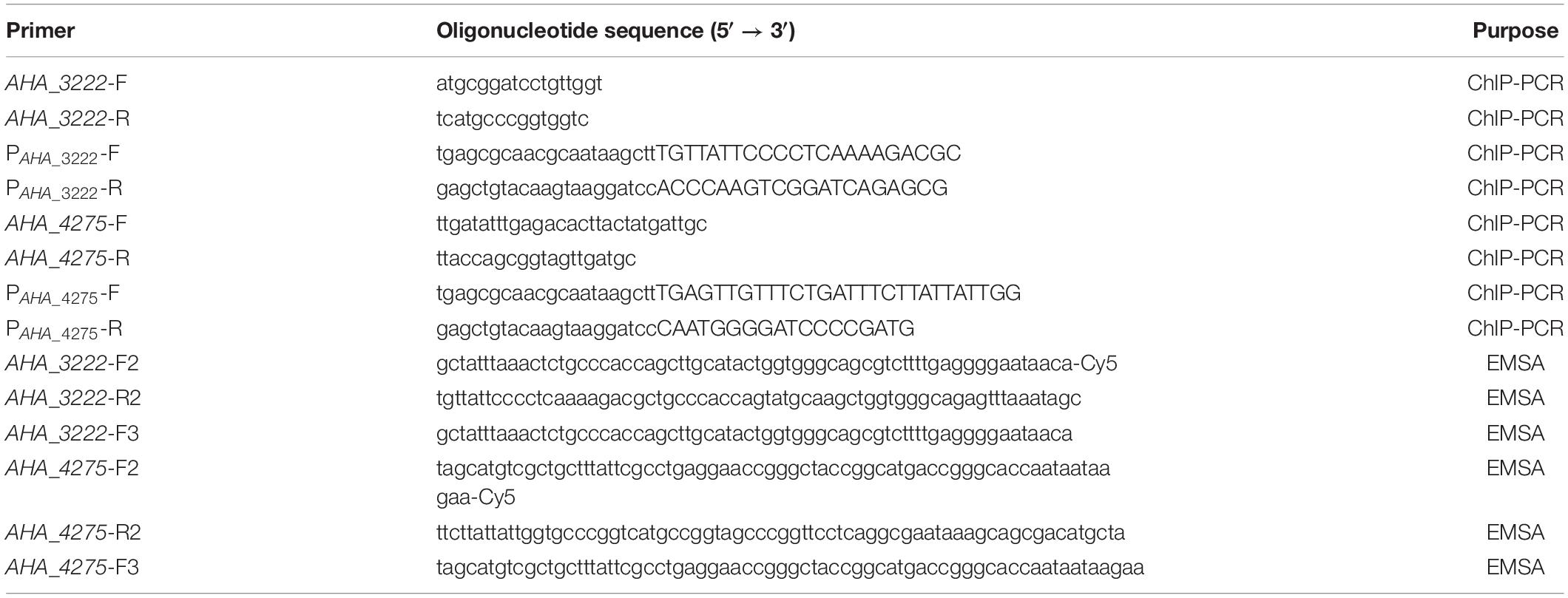
Table 1. Primer sequences used for ChIP-PCR and EMSA in this study.
EMSA
To investigate the binding of YeeY to the target genes’ promoters, the EMSA was performed as described previously (Heravi and Altenbuchner, 2014; Cheng et al., 2019). First, the recombinant proteins of pET-32a-YeeY were obtained via a prokaryotic expression system. Briefly, the pET-32a plasmid containing yeeY was transformed into E. coli BL21 (DE3). The recombinant proteins were induced with IPTG (isopropyl β-D-1-thiogalactopyranoside, 0.05 mM) at 30°C for 6 h and then purified using Ni-TNA column as described in previous study (Li et al., 2017). Second, two DNA probes were synthesized by PCR using two pairs of single-strand primers (Table 1) in which the upstream primer was labeled by Cy5-labeled oligonucleotides at the 5′ end, whereas two cold probes as a competitor were synthesized with the primers without the Cy5-labeled at the upstream primer 5′ end (Table 1). To ensure the specificity of the probes, we randomly selected pET-32a-Hcp, which was kept in our laboratory as a negative control and then the recombinant proteins pET-32a-YeeY and pET-32a-Hcp were incubated with probe and binding buffer (1 M pH 7.5 Tris–HCl, 5 M NaCl, 1 M KCl, 1 M MgCl2, 0.5 M pH 8.0 EDTA, 10 mg/mL BSA), respectively for 30 min at 4°C in the dark. Finally, the mixture was separated by PAGE gels and then scanned using Odyssey@ CLX (LI-COR, United States).
Results
The Deletion of A. hydrophila yeeY Increases the Antibiotics Resistance to FZ
The antimicrobial susceptibility rate of deletion and rescued strains were assessed when challenged with a series of FZ concentrations by a percentage of survival (Figure 1). At lower FZ concentrations (0.25 and 0.5 μg/mL), no significant differences were observed in the susceptibility rate of ΔyeeY or ΔyeeY + pACYC184 when compared to the wild-type and ΔyeeY + pACYC184-yeeY strains, respectively. However, at the higher doses of FZ (1 to 2 μg/mL), the ΔyeeY strain showed significantly increased susceptibility rate, whereas the rescued strain showed decreased susceptibility rate when compared to the controls.
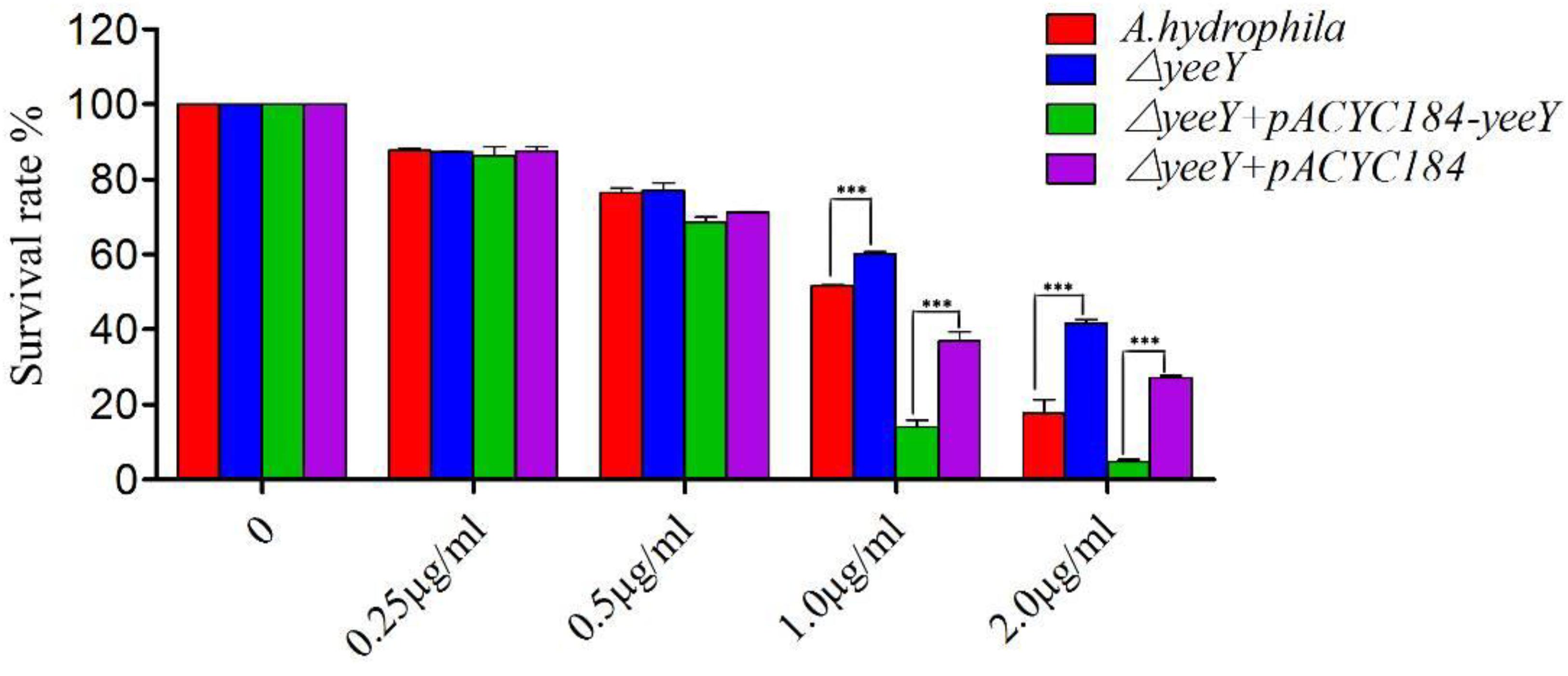
Figure 1. The antibiotics susceptibility of ΔyeeY derivatives to FZ. The survival rates of ΔyeeY, wild-type, ΔyeeY carrying pACYC184-yeeY (rescued strain) and ΔyeeY carrying an empty vector when exposed to various FZ concentrations.
Quantitative Proteomics of DEPs Between WT and △yeeY in Response to FZ
To further understand the effects of yeeY deletion on protein expression in A. hydrophila, whole protein samples between WT and ΔyeeY under 1.5 μg/mL of FZ antibiotic stress were extracted and then digested to peptides by trypsin to quantify in the protein levels by using DIA-based proteomics method. Each sample was independently repeated three times as biological replicates. Consequently, a total of 2066 proteins were identified with a considerable conservative threshold (protein and peptide false discovery rate <1%; listed in Supplementary Table S1) by LC-MS/MS. The protein MS intensities of WT and ΔyeeY were highly correlated among biological replicates (Pearson correction factor > 0.9) suggesting that the quantification analysis had high reliability (Figure 2A). Besides, as shown in the volcano plot, 593 DEPs were identified, including 293 increased and 300 decreased in abundance with fold change (FC) > 2 and p-value < 0.05 (Figure 2B).
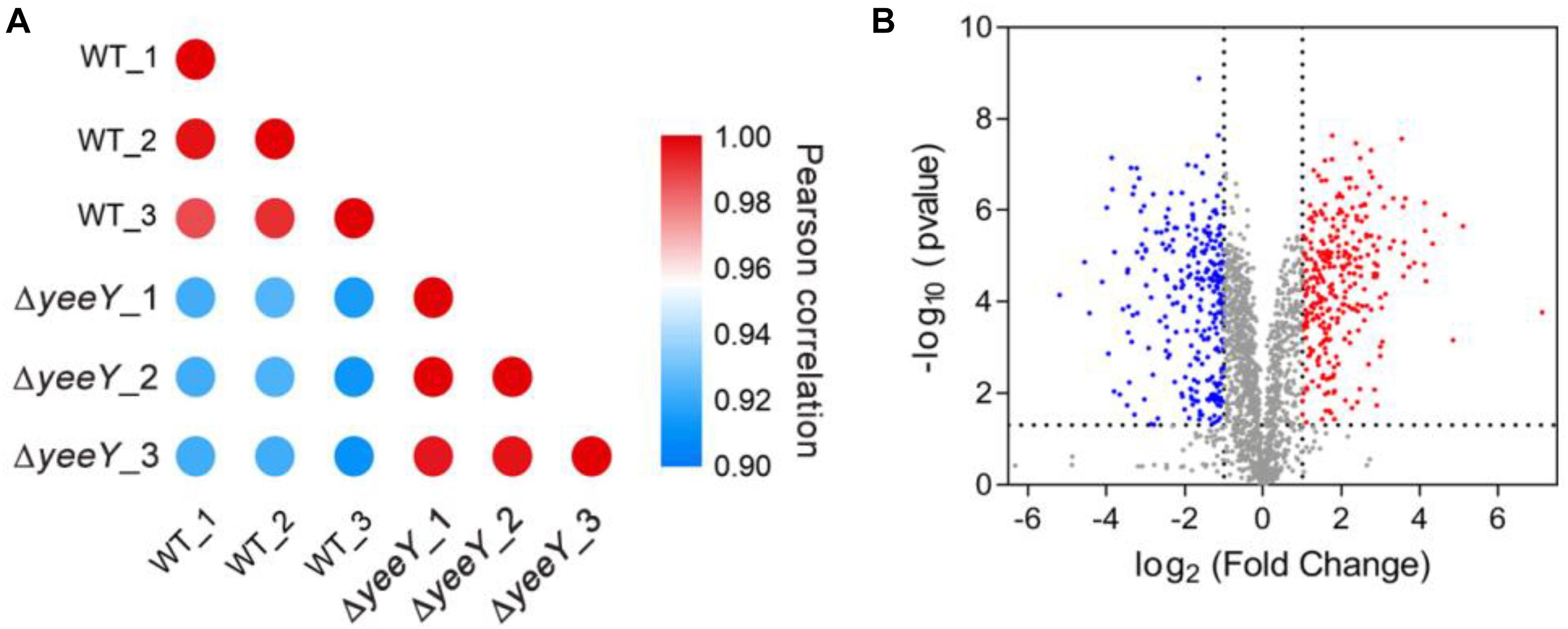
Figure 2. DIA based quantitative proteomics data analysis. (A) Correlation coefficients were used to analyze the associations of protein strength in three biological replications; (B) Volcano map comparing the abundance ratios of proteins with significant differences in expression (Fold change > 2; p < 0.05). The blue dots in the figure represent differentially down-regulated proteins; the red dots represent differentially increasing abundance proteins and the gray color represents non-differentially expressed proteins.
Go Enrichment of DEPs Under FZ Stress
GO analysis of DEPs between WT and ΔyeeY strains under FZ stress was performed with DAVID and visualized using the GOplot package in the R software. In the biological process (BP) classification clusters, the significantly enriched GO terms were oxidation-reduction process (5.6%, 25 increased and 8 decreased in abundance), aerobic respiration (2.5%, 11 increased and 4 decreased in abundance), energy derivation by oxidation of organic compounds (3.7%, 17 increased and 5 decreased in abundance), tricarboxylic acid cycle (2.2%, 9 increased and 4 decreased in abundance), cellular respiration (3.2%, 15 increased and 4 decreased in abundance) and cellular catabolic process (5.4%, 9 increased and 23 decreased in abundance) (Figure 3A). In the molecular functioning (MF) classification clusters, the significantly enriched GO terms were oxidoreductase activity, electron carrier activity, metal ion binding, tetrapyrrole binding and cation binding. Of these clusters, many proteins were increased in abundance in tetrapyrrole binding, electron carrier activity and heme-binding (Figure 3B). In the cell component (CC) classification clusters, the most significantly enriched GO terms were external encapsulating structure, cell envelope, cell outer membrane, external encapsulating structure part and outer membrane and envelope. Further, the protein expression showed that lamB had the highest fold change, while the hgpB exhibited the lowest expression in the CC enrichment (Figure 3C).
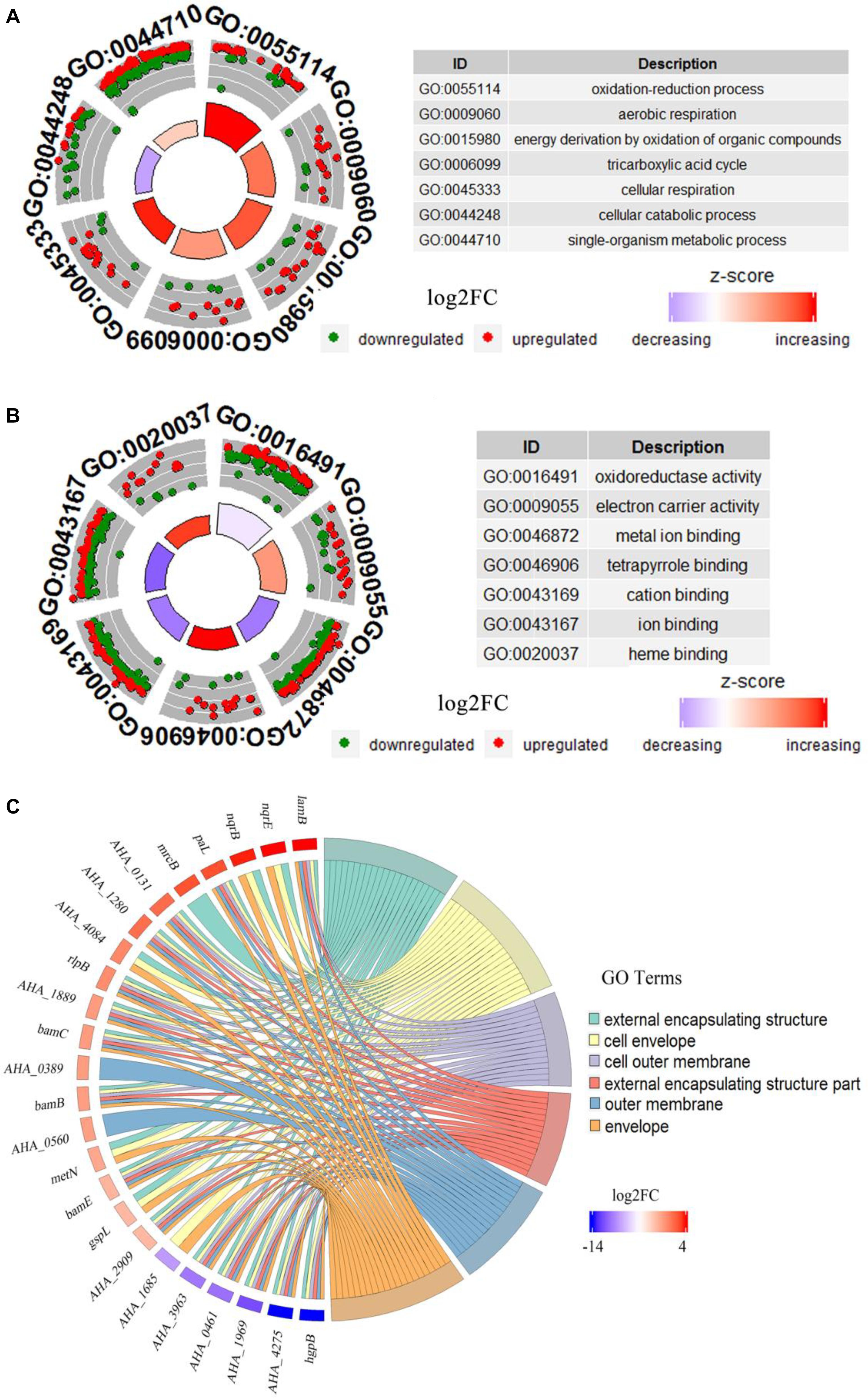
Figure 3. Bioinformatics analysis of DEPs between WT and ΔyeeY under FZ stress. (A) The cluster analysis of GO annotation in biological processes; (B) The cluster analysis of GO annotation in molecular functions; (C) The chord chart analysis of GO annotation in cellular components.
KEGG Enrichment of DEPs in FZ Stress
KEGG pathways enrichment analysis was further performed for the DEPs between the WT and △yeeY under FZ stress. The results showed that the most representative pathways were carbon metabolism (19 proteins increased and 23 proteins decreased in abundance), microbial metabolism in diverse environments (29 proteins increased and 29 proteins decreased in abundance), biosynthesis of antibiotics (33 proteins increased and 21 proteins decreased in abundance), TCA cycle (11 proteins increased and 3 proteins decreased in abundance), bacterial secretion system, protein export and pyruvate metabolism (Figure 4). Meanwhile, many pyruvate metabolism-related proteins were decreased in abundance, whereas bacterial secretion system and protein export-related proteins were found to be largely enriched with increased abundance. In general, most of these proteins are involved in key metabolic functions and protein export and these processes may contribute to FZ resistance.
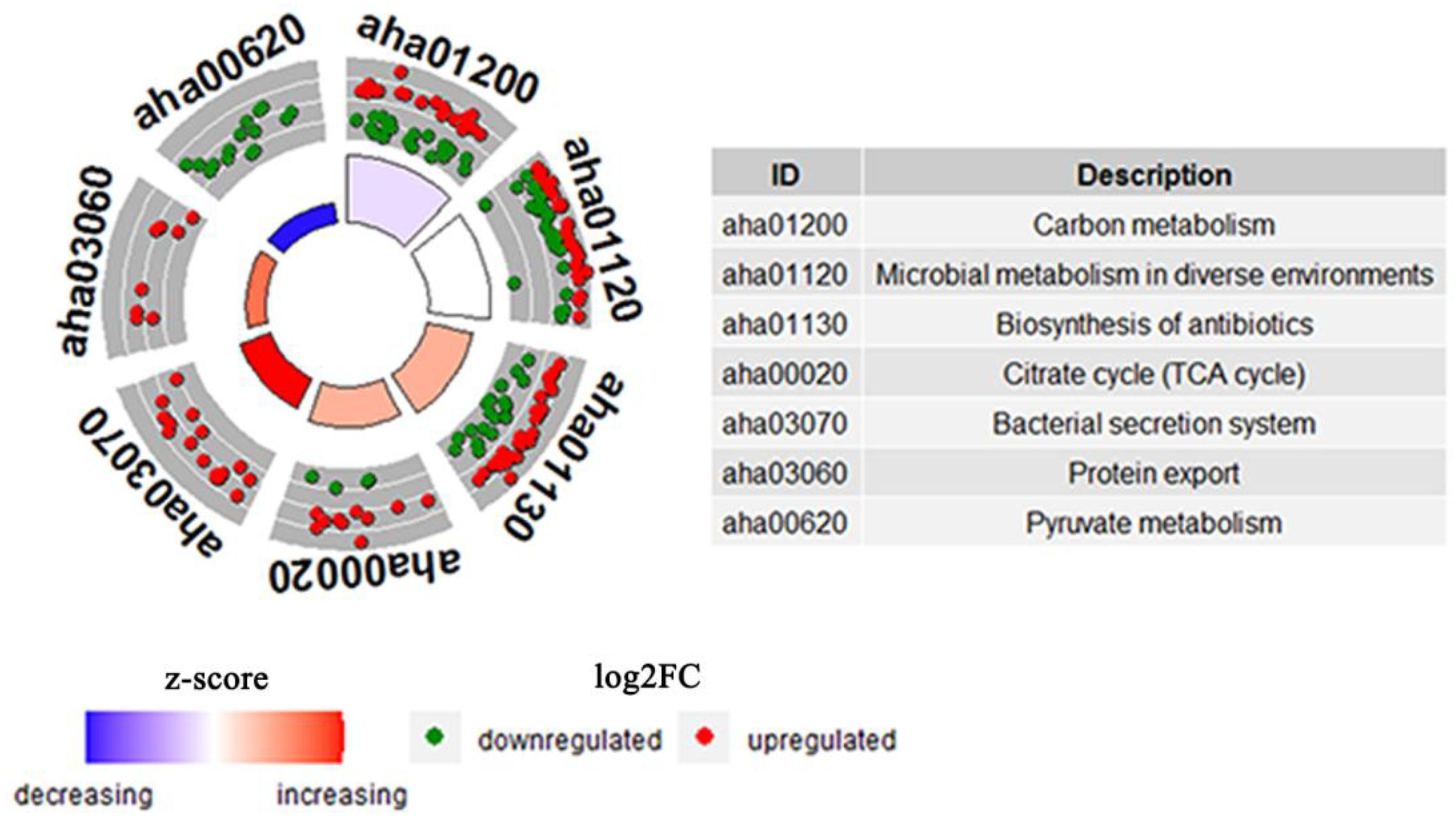
Figure 4. The cluster enrichment analysis of KEGG metabolic pathway of DEPs between WT and ΔyeeY under FZ stress. The three circles from outside to inside are indicated GO terms; a scatter plot of the expression levels (log2FC) for the genes in each, where red dots represent increasing abundance while green dots represent down-regulated; and a bar plot where the bar height indicates the significance of the KEGG pathway (log10pvalue) and the color indicates the z-score gradient.
Western Blotting Validation of Proteomics Results
To validate the proteomics results, two increased abundance proteins (AhyI and Hcp) and three decreased abundance proteins (A0KGN7, A0KLX0 and A0KFG8) were selected and analyzed by Western blotting using the primary antibodies that were previously developed by our group. The Western blotting results showed that Acyl-homoserine lactone synthase (AhyI) and hemolysin co-regulated protein (Hcp) were increased in abundance in ΔyeeY, while oligopeptide ABC transporter (A0KFG8), phosphate acetyltransferase (A0KGN7) and cytochrome c553 (A0KLX0) were down-regulated in ΔyeeY, compared to the WT strain under FZ stress (Figure 5A). All experiments were repeated at least three times, then the target bands intensity were quantified using Image J software and visualized with histogram (Figure 5B). Our data showed that the Western blotting results were relatively consistent with the mass spectrometry data, which indicates that our proteomics results were reliable.
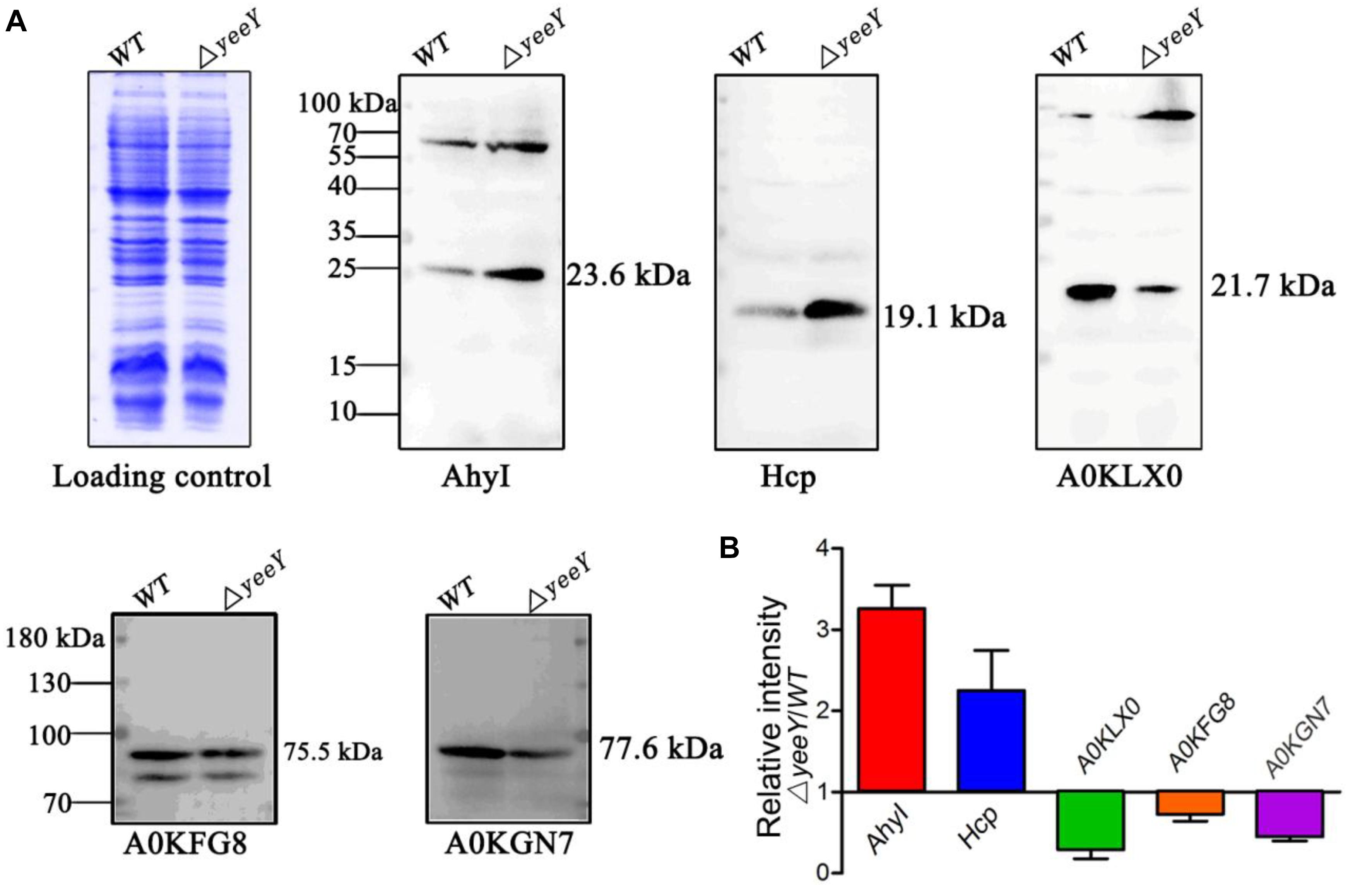
Figure 5. Western blotting analysis of the selected DEPs between WT and ΔyeeY under FZ stress. (A) The CBB-stained SDS-PAGE map of A. hydrophila WT and ΔyeeY under 1.0 μg/ml FZ treatment on the left acted as a loading control; (B) Gray level analysis of western blotting band signals by image J software.
Survival Capability Assay of Related Mutants in Response to FZ Stress
To further understand the roles of YeeY on the altered proteins under FZ resistance, the antimicrobial susceptibilities of seven deletion mutants (ΔahyI, ΔcysD, ΔmrcA, ΔAHA_0389, ΔAHA_2766, ΔAHA_2831 and ΔAHA_4275), which were stored in our laboratory, were evaluated based on survival rate in a series of FZ concentrations treatment. Among these, cysD, mrcA and AHA_4275 genes were involved in metabolic pathways and microbial metabolism in diverse environments. Further, the cysD gene was also involved in the biosynthesis of antibiotics and AHA_2766 was involved in the part of metabolic pathways. The results showed that ΔAHA_2766 and ΔAHA_4275 had significantly increased survival rate, while ΔcysD and ΔmrcA had decreased survival rate when compared to the survival rate of the wild-type strain. Although ahyI and AHA_0389 were showed increasing abundance in the proteomics results, whereas these two deleted mutants were showed no significant differences in survival rates, compared to the WT strain. Interestingly, ΔAHA_2831 was showed increased survival rate in lower concentrations of antibiotics (0.25 to 0.5 μg/mL), while it was showed sharply decreased survival rate in 1.0 μg/mL of FZ. In general, our results indicated that AHA_2766, AHA_4275, mrcA and cysD may be regulated by YeeY and may be involved in FZ resistance (Figure 6).
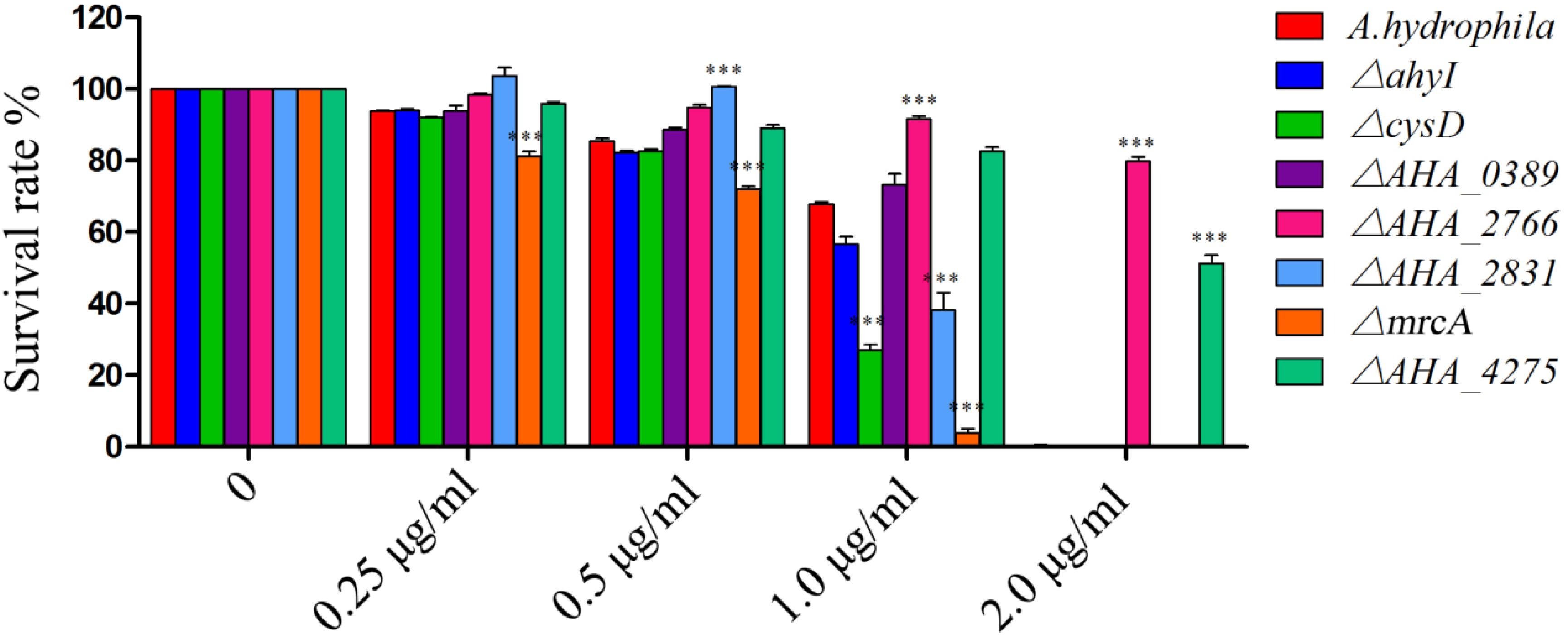
Figure 6. Survival capability assay of selected gene deleted strains under different doses of furazolidone treatment. ∗∗∗means p < 0.001.
The Roles of CARD Drug-Resistant Genes on FZ Resistance
To further understanding the effect of A. hydrophila YeeY on antimicrobial resistance genes (ARGs), all DEPs from proteomics analysis were submitted to ARGs detection by searching the CARD that provides well-known ARGs from diverse bacterial species. A total 30 resistance genes were found to be directly or indirectly regulated by YeeY under FZ stress, of which 22 and 12 proteins were increased and decreased in abundance, respectively (Figure 7A, Table 2). Subsequently, the antimicrobial susceptibilities of four resistance deletion mutants (ΔcysN, ΔsecD, ΔAHA_3222 and ΔAHA_3753), which were kept in our laboratory, were evaluated based on survival rate when treated with a series of FZ concentrations (Figure 7B). The results showed that the survival rates of ΔAHA_3222 and ΔsecD were increased while those of ΔcysN and ΔAHA_3753 were significantly decreased under the stress of FZ. Therefore, our results indicated that the genes cysN, AHA_3222 and AHA_3753 were regulated by YeeY, which may participate in the resistance process of A. hydrophila to FZ.
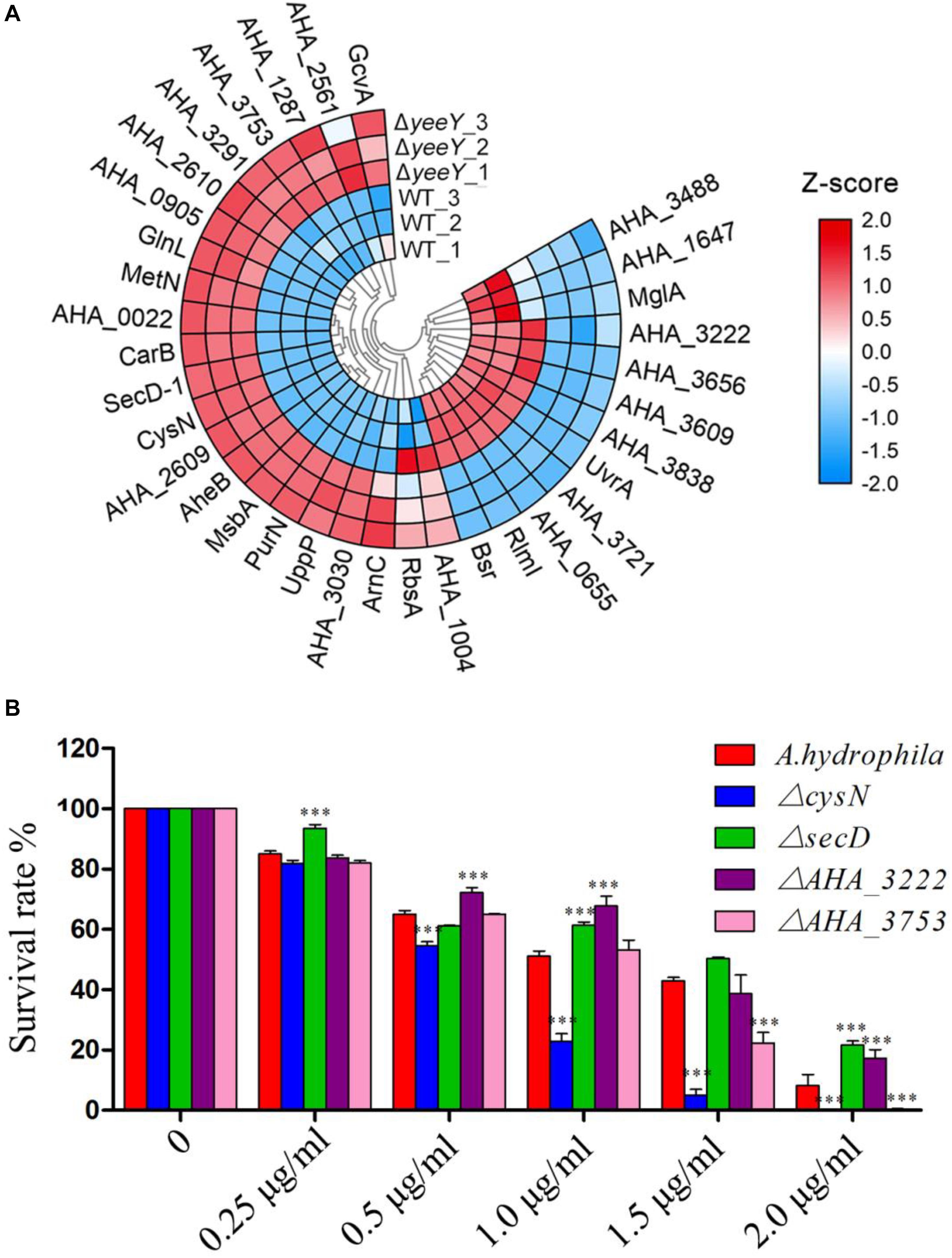
Figure 7. CARD ARGs analysis of DEPs between WT and ΔyeeY under FZ stress. (A) Loop heat map analysis of ARGs in ΔyeeY differentially expressed genes. The colors of squares indicate the z-scores, red represents increased and blue represents decreased. The outer cycle of the heat map represents the gene names of DEPs in ΔyeeY; (B) survival capability assay of four ARG deleted strains under different doses of furazolidone treatment.
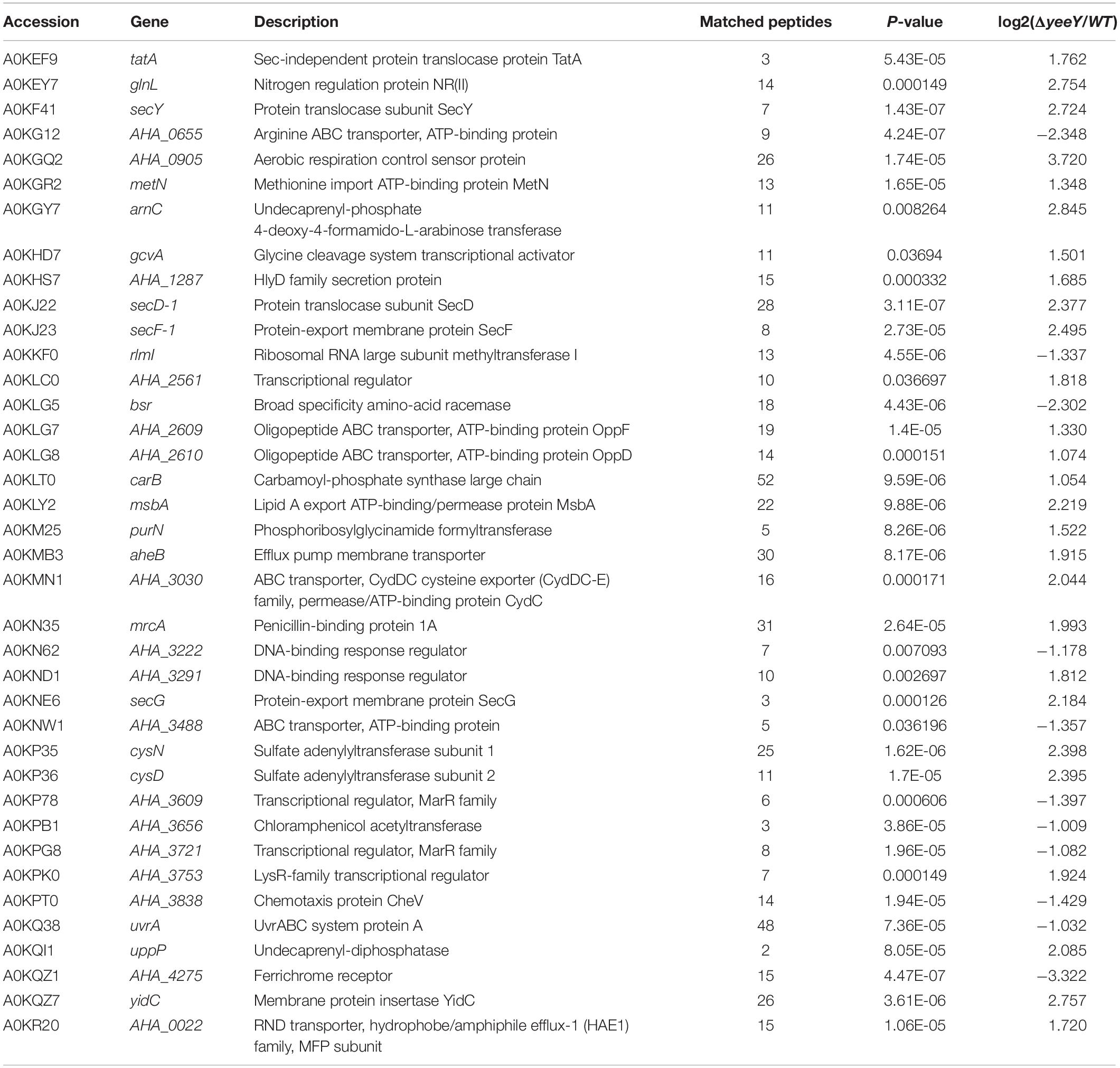
Table 2. DEPs related to ARGs between ΔyeeY and WT A. hydrophila under FZ stress by DIA-LC-MS/MS.
AHA_3222 and AHA_4275 Were Directly Regulated by YeeY in FZ Resistance
The data of this study indicates that YeeY may affect the FZ resistance of A. hydrophila via regulating the expression of some proteins, such as cysN, mrcA, AHA_3222 and AHA_4275. Therefore, we have validated this possibility using ChIP-PCR technique. The A. hydrophila chromatin was isolated and immunoprecipitation with anti-His antibody and the acquired recovered DNA was used as a template for PCR with the target gene and its predicted promoter region primer pairs. The results showed that only the predicted promoter regions of AHA_3222 and AHA_4275 produced product in the ΔyeeY: pACYC184-His-yeeY IP sample, whereas AHA_3222 and AHA_4275 could not be amplified (Figures 8A,B). So, it is suggesting that AHA_3222 and AHA_4275 may be directly targeted by YeeY. To further confirm these results, EMSA was performed to investigate the binding of YeeY to AHA_3222 and AHA_4275 promoters in vitro. The results showed that promoter fragments of both genes were bound by YeeY (Figures 8C–E). Therefore, the outcome of EMSA clearly suggested that AHA_3222 and AHA_4275 can be directly regulated by YeeY.
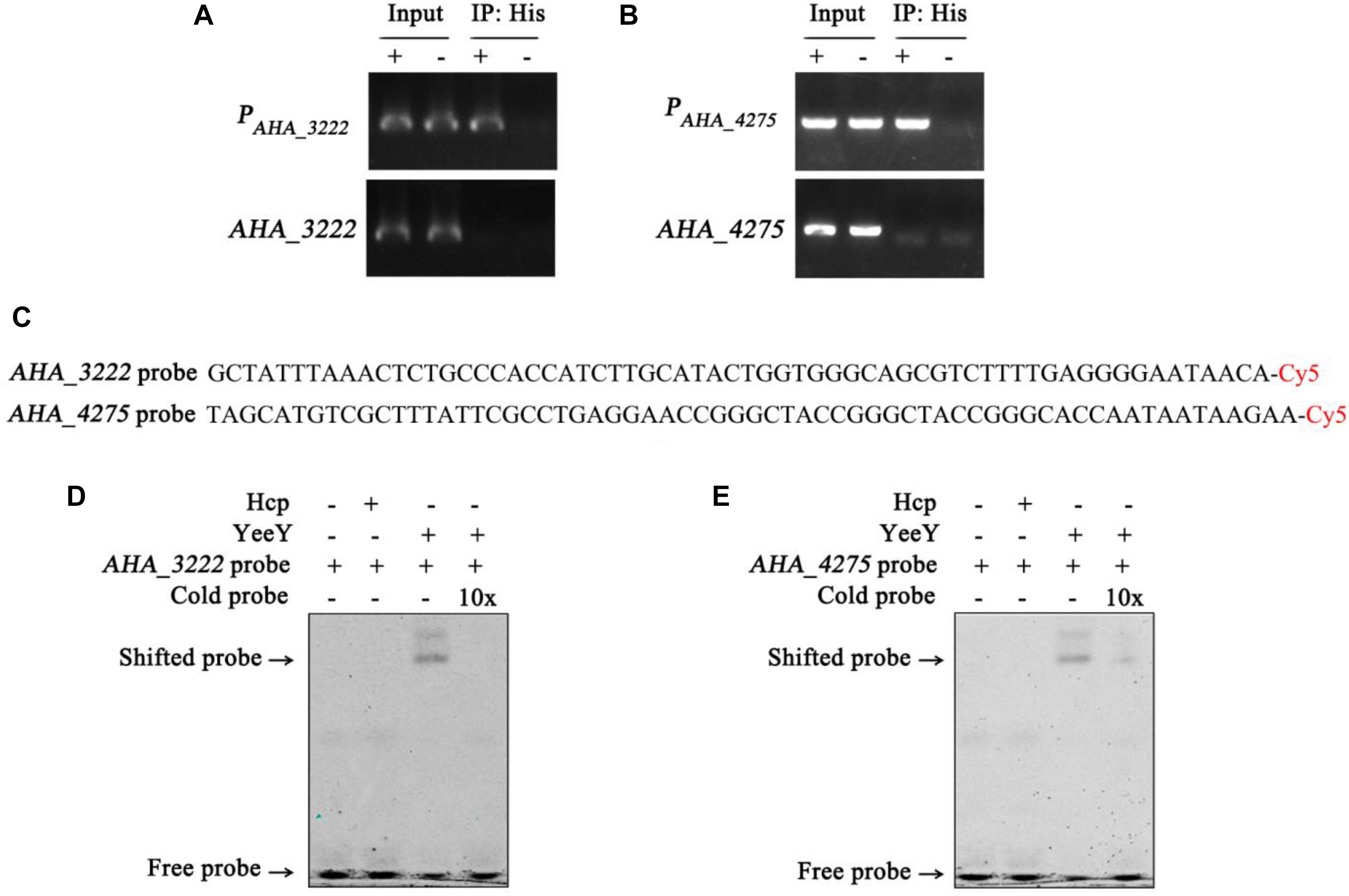
Figure 8. The binding of YeeY with the promoters of AHA_3222 and AHA_4275. (A,B) ChIP-PCR analysis of the binding of YeeY and the promoter fragments of AHA_3222 and AHA_4275. The promoter fragments of both genes are marked as PAHA_3222 and PAHA_4275, respectively; (C) the nucleotide sequences of AHA_3222 and AHA_4275 DNA probes; (D,E) EMSA analysis of the binding of YeeY and the promoter fragments of AHA_3222 and AHA_4275, respectively; The Cold probes competition were performed with 10 × unlabeled probes.
Discussion
A. hydrophila is an important aquatic bacterial pathogen in freshwater aquaculture and it has been reported that the threat of infection caused by this pathogen in fish and even in humans is on the rise worldwide (Awan et al., 2018). Although, the antibiotic treatment is an effective tactic to control the bacterial diseases in aquaculture, side however the effects such as antibiotic residue and antibiotic resistance cannot be ignored. Further, more than two million people who are infected with antibiotics resistant bacterial pathogens each year in the United States on average 23000 people die, meaning that antibiotic resistance has become one of the greatest threats faced by modern medicine (Laxminarayan et al., 2013). Therefore, elucidating the antibiotic resistance mechanism of A. hydrophila is of great significance for the development of new antibiotic therapeutic strategies in the future. YeeY, an LTTR, was reported to be associated with the rapid emergence of persistence in E. coli (Girgis et al., 2012). However, the biological function of this protein is still largely unknown, especially concerning antibiotic resistance. Although furazolidone has been prohibited in many parts of the world due to its toxic and carcinogenic side effects, we found that the minimal inhibitory concentration (MIC) of FZ was decreased when the YeeY was overexpressed and the absence of yeeY led to an increase the FZ resistance in our previous work (Fu et al., 2019). To further understand the biological behavior of YeeY on antibiotic resistance, differentially expressed proteins between ΔyeeY and WT strains were compared under FZ stress by DIA-quantitative proteomics in this study. A total of 594 DEPs were identified in ΔyeeY, with 293 proteins that were increased in abundance and 301 were decreased in abundance.
Of these DEPs, 15 outer membrane proteins were affected by the loss of yeeY under the FZ stress when compared to the wild-type strain. Among them, the proteins such as LamB, OprM, and OmpA were increased in abundance, whereas A0KQZ1 (AHA_4275) and A0KN35 (AHA_2766) were decreased in abundance. The OmpA has been reported to play an important role in the transport of antibiotics. For example, the MICs of chloramphenicol, aztreonam and nalidixic acid were decreased when the ompA was disrupted in A. baumannii (Smani et al., 2014). Further, in our previous study, we have observed that the MBCs (minimum bactericidal concentrations) of ceftriaxone sodium, apramycin, neomycin sulfate and gentamicin in ΔAHA_2766 were increased more than 4-fold, whereas the MBCs of norfloxacin and chloramphenicol in ΔAHA_4275 were decreased at least 2-fold (Li et al., 2019). Therefore, we have evaluated the antibiotic susceptibility of these both mutants to FZ antibiotic in this study. The survival test showed that the survival rates of ΔAHA_2766 and ΔAHA_4275 were significantly higher, compared to the wild strains under various doses of FZ. It suggests that A. hydrophila YeeY may regulate several antibiotic resistance-related outer membrane proteins during FZ stress.
MrcA (penicillin-binding protein 1a, PBP1a) and MrcB (penicillin-binding protein 1b, PBP1b) were both increased in abundance in ΔyeeY strain under FZ stress. Both proteins are essential for cell wall peptidoglycan biosynthesis and maintenance of cell growth. More both of these proteins are considered as antibiotic targets by beta-lactams (Kumar et al., 2012; King et al., 2017). Apart from their involvement in beta-lactams resistance, these both proteins were increased in abundance within the chlortetracycline (CTC) resistant strain in our previous study, whereas their actual functions in CTC resistance are still unknown (Li et al., 2018). In this study, the survival rate of ΔmrcA was decreased under high doses of FZ, which suggesting that the MrcA may be involved in FZ resistance as well. This finding indicates that YeeY may negatively regulate penicillin-binding protein against FZ stress.
The KEGG analysis showed that the bacterial secretion system was enriched in increased abundance proteins, including general secretion pathway (gspC, gspG), type II secretion system T2SS (gspM, gspL), type VI secretion system T6SS (AHA_1826, AHA_1827, AHA_1840, AHA_1841, and AHA_1845), Sec secretion system (secD, yidC, secE, secF, secG, secY, and lepB) and Tat-dependent system (tatA). Some studies have documented that the bacterial secretion system related proteins are involved in antibiotics resistance. For example, T6SS functioned not only as a virulence system, which also contributed to antimicrobial resistance in A. baumannii ATCC 19606, it was proved that upon the T6SS core component vgrG was deleted, the antimicrobial resistance to ampicillin, chloramphenicol and β-lactam antibiotics was altered (Wang et al., 2018). It indicated that T6SS may be closely related to furazolidone resistance in A. hydrophila since AHA_1827 was highly homolog to A. baumannii ATCC 19606’s gene vgrG by comparative amino acid sequence analysis. Besides, beta-lactamase, a target of beta-lactam antibiotics, was transported through the Sec and Tat-dependent secretion systems (Pradel et al., 2009). Thus, protein transport may be an adaptive strategy in the FZ resistance in A. hydrophila.
The DEPs were also involved in complex cellular metabolic processes, especially the TCA cycle and sulfur metabolism. It was reported that the down-regulation of bacterial energy generation systems such as the TCA cycle may be a potential antibiotic resistance tactic in many bacterial species (Rosato et al., 2014; Peng et al., 2015; Su et al., 2018). However, many proteins associated with the TCA cycle, such as SucA, SucB, SucC, SucD, GltA, IcD, and SdhA were increased in abundance in this study, indicating that the role of the TCA cycle in FZ resistance in A. hydrophila may be alleviated by YeeY regulation. Sulfur, an essential element, exists in hundreds of metabolites with various oxidation states that are not only related to the virulence and antioxidant stress of Mycobacterium tuberculosis but also play important roles in rifampicin resistance. In the current study, four sulfur metabolism-related proteins (CysD, CysI, CysH, and CysN) were increased in abundance in ΔyeeY under FZ stress. The survival assay showed that the survival rate of ΔcysD and ΔcysN mutant strains were decreased when compared to the wild-type strain, which suggesting that sulfur metabolism is also a part of the mechanism of bacterial resistance by YeeY regulation. Taken together, these findings suggest that bacterial intracellular metabolism may be an important bacterial resistance strategy.
Also, we estimated the FZ susceptibilities of four well-known ARGs homologs (AHA_3222, AHA_3753, cysN, and secD) from the CARD by measuring the survival rates of their gene deletion strains under different concentrations of FZ. According to the annotation of CARD, cysN is related to tetracycline resistance; AHA_3222 homolog is related to fluoroquinolone and acridine dye resistance; AHA_3753 homolog is related to cephalosporin and secD is related to cephalosporin. There is considerable research documenting the antibiotic resistance properties of these ARGs or their homologs. For example, both SecD and SecF belong to the resistance-nodulation-cell division (RND) family of multidrug exporters and they are involved in the export of antimicrobial resistance proteins in Staphylococcus aureus (Quiblier et al., 2011). AHA_3222 is a DNA-binding response regulator and its homologous protein ArlR (98% identity) in S. aureus was reported to positively regulate the expression of the efflux pump gene NorA and to be involved in bacterial multidrug resistance (Fournier et al., 2000); In addition to these, Mycobacterium tuberculosis sulfur metabolism genes including CysN were found to participate in certain antibiotics resistance as well (Hatzios and Bertozzi, 2011). In the current proteomics results, the protein level of AHA_3222 (A0KN62) was decreased, while CysN and AHA_3753 (A0KPK0) were increased in the ΔyeeY strain under FZ stress. Moreover, the survival of ΔAHA_3222 was increased, while those of ΔcysN and ΔAHA_3753 were decreased. It indicates that YeeY may positively regulate AHA_3222 and negatively regulate cysN and AHA_3753 by direct or indirect pathways contributing to survival under FZ stress. In addition, we found SecD was increased in the proteomics analysis, whereas the deletion of secD showed increased survival rate under FZ stress. This apparent inconsistency could be the result of a tradeoff between adaption and survival. As an important component of the Sec system for protein secretion, the deletion of secD could sharply reduce the membrane permeability and thereby prevent antibiotics entry. However, the loss of some important proteins such as outer membrane proteins will be disadvantageous for survival in the long term. Thus, the regulation of secD should be complex or there may be other transcriptional regulators involved.
Additionally, ChIP-PCR and EMSA were performed to explore the relationships of the LTTR protein YeeY with those genes. Interestingly, we found that YeeY can directly regulate AHA_3222, which may regulate the expression of efflux pump-related genes involved in bacterial multidrug resistance. Further, it also can directly regulate the outer membrane related gene (AHA_4275) during FZ stress. In general, our results provide evidence that the YeeY protein can bind directly with certain ARGs’ promoters that contribute to FZ resistance in A. hydrophila.
Conclusion
In this study, we found that A. hydrophila YeeY could directly and positively regulate the ARG such as AHA_3222 and AHA_4275 and could indirectly or directly regulate several drug resistance-related genes as well as genes involved in key energy biosynthetic pathways such as metabolism and the bacterial secretion system. Overall, the outcomes of this study gave a view to understand the complicated regulatory mechanisms of transcription factors on bacterial physiological functions. More, it provided a new target for the treatment of pathogenic bacteria and the development of new antimicrobial agents.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
XL and WL conceived and supervised the project. YF constructed strains, performed the experimental work, and drafted the manuscript. LZ and GW contributed to quantitative proteomics and data analysis. YL, SR, and GY improved the manuscript. All authors reviewed and approved the final manuscript.
Funding
This work was sponsored by grants from NSFC projects (Nos. 31670129, 31470238, and 31802343) and China Postdoctoral Science Foundation (No. 2019M662214).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the support of the program for Innovative Research Team in Fujian Agricultural and Forestry University (No. 712018009), Key Laboratory of Marine Biotechnology of Fujian Province (2020MB04), and the Fujian-Taiwan Joint Innovative Center for Germplasm Resources and Cultivation of Crop (FJ 2011 Program, No. 2015-75, China). We also thank Mr. Sheng Yang for providing technical guidance on ChIP-PCR and EMSA.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.577376/full#supplementary-material
TABLE S1 | Identification and quantification results between WT and ΔyeeY using DIA method in 1.5 g/mL FZ stress.
Footnotes
References
Abdelhamed, H., Banes, M., Karsi, A., and Lawrence, M. L. (2019). Recombinant ATPase of virulent Aeromonas hydrophila protects channel catfish against motile Aeromonas septicemia. Front. Immunol. 10:1641. doi: 10.3389/fimmu.2019.01641
Awan, F., Dong, Y., Wang, N., Liu, J., Ma, K., and Liu, Y. (2018). The fight for invincibility: environmental stress response mechanisms and Aeromonas hydrophila. Microb. Pathog. 116, 135–145. doi: 10.1016/j.micpath.2018.01.023
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv [Preprint]. doi: 10.1101/289660
Chen, C. Y., Clark, C. G., Langner, S., Boyd, D. A., Bharat, A., McCorrister, S. J., et al. (2019). Detection of antimicrobial resistance using proteomics and the comprehensive antibiotic resistance database: a case study. Proteomics Clin. Appl. 14:e1800182. doi: 10.1002/prca.201800182
Cheng, Z. X., Guo, C., Chen, Z. G., Yang, T. C., Zhang, J. Y., Wang, J., et al. (2019). Glycine, serine and threonine metabolism confounds efficacy of complement-mediated killing. Nat. Commun. 10:3325. doi: 10.1038/s41467-019-11129-5
Dahanayake, P. S., Hossain, S., Wickramanayake, M., and Heo, G. J. (2019). Antibiotic and heavy metal resistance genes in Aeromonas spp. isolated from marketed Manila Clam (Ruditapes philippinarum) in Korea. J. Appl. Microbiol. 127, 941–952. doi: 10.1111/jam.14355
Das, B., Verma, J., Kumar, P., Ghosh, A., and Ramamurthy, T. (2020). Antibiotic resistance in Vibrio cholerae: understanding the ecology of resistance genes and mechanisms. Vaccine 38(Suppl. 1), A83–A92. doi: 10.1016/j.vaccine.2019.06.031
de la Morena, M. L., Van, R., Singh, K., Brian, M., Murray, M. E., and Pickering, L. K. (1993). Diarrhea associated with Aeromonas species in children in day care centers. J. Infect. Dis. 168, 215–218. doi: 10.1093/infdis/168.1.215
Fournier, B., Aras, R., and Hooper, D. C. (2000). Expression of the multidrug resistance transporter NorA from Staphylococcus aureus is modified by a two-component regulatory system. J. Bacteriol. 182, 664–671. doi: 10.1128/jb.182.3.664-671.2000
Fu, Y. Y., Cai, Q. L., Wang, Y. Q., Li, W. X., Yu, J., Yang, G. D., et al. (2019). Four LysR-type transcriptional regulator family proteins (LTTRs) involved in antibiotic resistance in Aeromonas hydrophila. World J. Microbiol. Biotechnol. 35:127. doi: 10.1007/s11274-019-2700-3
Girgis, H. S., Harris, K., and Tavazoie, S. (2012). Large mutational target size for rapid emergence of bacterial persistence. PNAS 109, 12740–12745. doi: 10.1073/pnas.1205124109
Hatzios, S. K., and Bertozzi, C. R. (2011). The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 7:e1002036. doi: 10.1371/journal.ppat.1002036
Heravi, K. M., and Altenbuchner, J. (2014). Regulation of the Bacillus subtilis mannitol utilization genes: promoter structure and transcriptional activation by the wild-type regulator (MtlR) and its mutants. Microbiology 160, 91–101. doi: 10.1099/mic.0.071233-0
Kim, Y., Chhor, G., Tsai, C. S., Winans, J. B., Jedrzejczak, R., Joachimiak, A., et al. (2018). Crystal structure of the ligand-binding domain of a LysR-type transcriptional regulator: transcriptional activation via a rotary switch. Mol. Microbiol. 110, 550–561. doi: 10.1111/mmi.14115
King, D. T., Wasney, G. A., Nosella, M., Fong, A., and Strynadka, N. C. (2017). Structural insights into inhibition of Escherichia coli penicillin-binding protein 1b. J. Biol. Chem. 292, 979–993. doi: 10.1074/jbc.M116.718403
Kumar, A., Sarkar, S. K., Ghosh, D., and Ghosh, A. S. (2012). Deletion of penicillin-binding protein 1b impairs biofilm formation and motility in Escherichia coli. Res. Microbiol. 163, 254–257. doi: 10.1016/j.resmic.2012.01.006
Laxminarayan, R., Duse, A., Wattal, C., Zaidi, A. K., Wertheim, H. F., Sumpradit, N., et al. (2013). Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 13, 1057–1098. doi: 10.1016/S1473-3099(13)70318-9
Li, H., Zhu, Q. F., Peng, X. X., and Peng, B. (2017). Interactome of E. piscicida and grouper liver proteins reveals strategies of bacterial infection and host immune response. Sci. Rep. 7:39824. doi: 10.1038/srep39824
Li, H. L., Li, Y. J., Sun, T. S., Du, W., Zhang, Z. J., Li, D. C., et al. (2020). Integrative proteome and acetylome analyses of murine responses to Cryptococcus neoformans infection. Front. Microbiol. 11:575. doi: 10.3389/fmicb.2020.00575
Li, L., Su, Y. B., Peng, B., Peng, X. X., and Li, H. (2020). Metabolic mechanism of colistin resistance and its reverting in Vibrio alginolyticus. Environ. Microbiol. doi: 10.1111/1462-2920.15021
Li, W. X., Ali, F., Cai, Q. L., Yao, Z. J., Sun, L. N., Lin, W. X., et al. (2018). Quantitative proteomic analysis reveals that chemotaxis is involved in chlortetracycline resistance of Aeromonas hydrophila. J. Proteomics 172, 143–151. doi: 10.1016/j.jprot.2017.09.011
Li, W. X., Yao, Z. J., Sun, L. N., Hu, W. J., Cao, J. J., Lin, W. X., et al. (2016). Proteomics analysis reveals a potential antibiotic cocktail therapy strategy for Aeromonas hydrophila infection in biofilm. J. Proteome Res. 15, 1810–1820. doi: 10.1021/acs.jproteome.5b01127
Li, Z. Q., Wang, Y. Q., Li, X. Y., Lin, Z. P., Lin, Y. X., Srinivasan, R., et al. (2019). The characteristics of antibiotic resistance and phenotypes in 29 outer-membrane protein mutant strains in Aeromonas hydrophila. Environ. Microbiol. 21, 4614–4628. doi: 10.1111/1462-2920.14761
Lin, X. M., Kang, L. Q., Li, H., and Peng, X. X. (2014). Fluctuation of multiple metabolic pathways is required for Escherichia coli in response to chlortetracycline stress. Mol. Biosyst. 10, 901–908. doi: 10.1039/c3mb70522f
Liu, J. Q., Xie, L. F., Zhao, D., Yang, T. T., Hu, Y. F., Sun, Z. L., et al. (2019). A fatal diarrhoea outbreak in farm-raised Deinagkistrodon acutus in China is newly linked to potentially zoonotic Aeromonas hydrophila. Transbound Emerg. Dis. 66, 287–298. doi: 10.1111/tbed.13020
Pang, H. Y., Qiu, M. S., Zhao, J. M., Hoare, R., Monaghan, S. J., Song, D. W., et al. (2018). Construction of a Vibrio alginolyticus hopPmaJ (hop) mutant and evaluation of its potential as a live attenuated vaccine in orange-spotted grouper (Epinephelus coioides). Fish Shellfish Immunol. 76, 93–100. doi: 10.1016/j.fsi.2018.02.012
Peng, B., Su, Y. B., Li, H., Han, Y., Guo, C., Tian, Y. M., et al. (2015). Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell. Metab. 21, 249–262. doi: 10.1016/j.cmet.2015.01.008
Pradel, N., Delmas, J., Wu, L. F., Santini, C. L., and Bonnet, R. (2009). Sec- and Tat-dependent translocation of beta-lactamases across the Escherichia coli inner membrane. Antimicrob. Agents Chemother. 53, 242–248. doi: 10.1128/AAC.00642-08
Quiblier, C., Zinkernagel, A. S., Schuepbach, R. A., Berger-Bachi, B., and Senn, M. M. (2011). Contribution of SecDF to Staphylococcus aureus resistance and expression of virulence factors. BMC Microbiol. 11:72. doi: 10.1186/1471-2180-11-72
Rice, K. C., Nelson, J. B., Patton, T. G., Yang, S. J., and Bayles, K. W. (2005). Acetic acid induces expression of the Staphylococcus aureus cidABC and lrgAB murein hydrolase regulator operons. J. Bacteriol. 187, 813–821. doi: 10.1128/JB.187.3.813-821.2005
Rosato, R. R., Fernandez, R., Paz, L. I., Singh, C. R., and Rosato, A. E. (2014). TCA cycle-mediated generation of ROS is a key mediator for HeR-MRSA survival under beta-lactam antibiotic exposure. PLoS One 9:e99605. doi: 10.1371/journal.pone.0099605
Shen, L., Yang, S., Yang, F., Guan, D., and He, S. L. (2020). CaCBL1 acts as a positive regulator in pepper response to Ralstonia solanacearum. Mol. Plant Microbe Interact. 33, 945–957. doi: 10.1094/MPMI-08-19-0241-R
Smani, Y., Fabrega, A., Roca, I., Sanchez-Encinales, V., Vila, J., and Pachon, J. (2014). Role of OmpA in the multidrug resistance phenotype of Acinetobacter baumannii. Antimicrob. Agents Chemother. 58, 1806–1808. doi: 10.1128/AAC.02101-13
Soltan Dallal, M. M., Mazaheri Nezhad Fard, R., Kavan Talkhabi, M., Aghaiyan, L., and Salehipour, Z. (2016). Prevalence, virulence and antimicrobial resistance patterns of Aeromonas spp. isolated from children with diarrhea. Germs 6, 91–96. doi: 10.11599/germs.2016.1094
Su, Y. B., Peng, B., Li, H., Cheng, Z. X., Zhang, T. T., Zhu, J. X., et al. (2018). Pyruvate cycle increases aminoglycoside efficacy and provides respiratory energy in bacteria. PNAS 115, E1578–E1587. doi: 10.1073/pnas.1714645115
Sun, L. N., Yao, Z. J., Guo, Z., Zhang, L. S., Wang, Y. Q., Mao, R. R., et al. (2019). Comprehensive analysis of the lysine acetylome in Aeromonas hydrophila reveals cross-talk between lysine acetylation and succinylation in LuxS. Emerg. Microbes Infect. 8, 1229–1239. doi: 10.1080/22221751.2019.1656549
Wang, G. B., Wang, Y. Q., Zhang, L. S., Cai, Q. L., Lin, Y. X., Lin, L., et al. (2020). Proteomics analysis reveals the effect of Aeromonas hydrophila sirtuin CobB on biological functions. J. Proteomics 225:103848. doi: 10.1016/j.jprot.2020.103848
Wang, J. F., Zhou, Z. H., He, F., Ruan, Z., Jiang, Y., Hua, X. T., et al. (2018). The role of the type VI secretion system vgrG gene in the virulence and antimicrobial resistance of Acinetobacter baumannii ATCC 19606. PLoS One 13:e0192288. doi: 10.1371/journal.pone.0192288
Wang, Q., Ji, W., and Xu, Z. (2020). Current use and development of fish vaccines in China. Fish Shellfish Immunol. 96, 223–234. doi: 10.1016/j.fsi.2019.12.010
Wisniewski, J. R. (2019). Filter aided sample preparation-A tutorial. Anal. Chim. Acta 1090, 23–30. doi: 10.1016/j.aca.2019.08.032
Yang, S. J., Rice, K. C., Brown, R. J., Patton, T. G., Liou, L. E., Park, Y. H., et al. (2005). A LysR-type regulator, CidR, is required for induction of the Staphylococcus aureus cidABC operon. J. Bacteriol. 187, 5893–5900. doi: 10.1128/JB.187.17.5893-5900.2005
Yao, Z. J., Guo, Z., Wang, Y. Q., Li, W. X., Fu, Y. Y., Lin, Y. X., et al. (2019). Integrated succinylome and metabolome profiling reveals crucial role of s-ribosylhomocysteine lyase in quorum sensing and metabolism of Aeromonas hydrophila. Mol. Cell. Proteomics 18, 200–215. doi: 10.1074/mcp.RA118.001035
Yao, Z. J., Li, W. X., Lin, Y. X., Wu, Q., Yu, F. F., Lin, W. X., et al. (2016). Proteomic analysis reveals that metabolic flows affect the susceptibility of Aeromonas hydrophila to antibiotics. Sci. Rep. 6:39413. doi: 10.1038/srep39413
Yao, Z. J., Sun, L. N., Wang, Y. Q., Lin, L., Guo, Z., Li, D., et al. (2018). Quantitative proteomics reveals antibiotics resistance function of outer membrane proteins in Aeromonas hydrophila. Front. Cell. Infect. Mi 8:390. doi: 10.3389/fcimb.2018.00390
Zahran, E., Abd El-Gawad, E. A., and Risha, E. (2018). Dietary Withania sominefera root confers protective and immunotherapeutic effects against Aeromonas hydrophila infection in Nile tilapia (Oreochromis niloticus). Fish Shellfish Immunol. 80, 641–650. doi: 10.1016/j.fsi.2018.06.009
Zhang, L. S., Li, W. X., Sun, L. N., Wang, Y. Q., Lin, Y. X., and Lin, X. M. (2020). Quantitative proteomics reveals the molecular mechanism of Aeromonas hydrophila in enoxacin stress. J. Proteomics 211:103561. doi: 10.1016/j.jprot.2019.103561
Keywords: Aeromonas hydrophila, DIA based proteomics, YeeY, furazolidone, antimicrobial resistance
Citation: Fu Y, Zhang L, Wang G, Lin Y, Ramanathan S, Yang G, Lin W and Lin X (2020) The LysR-Type Transcriptional Regulator YeeY Plays Important Roles in the Regulatory of Furazolidone Resistance in Aeromonas hydrophila. Front. Microbiol. 11:577376. doi: 10.3389/fmicb.2020.577376
Received: 29 June 2020; Accepted: 19 August 2020;
Published: 09 September 2020.
Edited by:
Bo Peng, Sun Yat-sen University, ChinaReviewed by:
Shanmuganathan – Balakrishnan, Upstate Medical University, United StatesSuganthy Natarajan, Alagappa University, India
Copyright © 2020 Fu, Zhang, Wang, Lin, Ramanathan, Yang, Lin and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenxiong Lin, bHd4QGZhZnUuZWR1LmNu; Xiangmin Lin, eGlhbmdtaW5AZmFmdS5lZHUuY24=