
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 January 2021
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.563885
 Manuel II García-Ulloa1
Manuel II García-Ulloa1 Ana Elena Escalante2
Ana Elena Escalante2 Alejandra Moreno-Letelier3
Alejandra Moreno-Letelier3 Luis E. Eguiarte1
Luis E. Eguiarte1 Valeria Souza1*
Valeria Souza1*Anthropogenic perturbations introduce novel selective pressures to natural environments, impacting the genomic variability of organisms and thus altering the evolutionary trajectory of populations. Water overexploitation for agricultural purposes and defective policies in Cuatro Cienegas, Coahuila, Mexico, have strongly impacted its water reservoir, pushing entire hydrological systems to the brink of extinction along with their native populations. Here, we studied the effects of continuous water overexploitation on an environmental aquatic lineage of Pseudomonas otitidis over a 13-year period which encompasses three desiccation events. By comparing the genomes of a population sample from 2003 (original state) and 2015 (perturbed state), we analyzed the demographic history and evolutionary response to perturbation of this lineage. Through coalescent simulations, we obtained a demographic model of contraction-expansion-contraction which points to the occurrence of an evolutionary rescue event. Loss of genomic and nucleotide variation alongside an increment in mean and variance of Tajima’s D, characteristic of sudden population expansions, support this observation. In addition, a significant increase in recombination rate (R/θ) was observed, pointing to horizontal gene transfer playing a role in population recovery. Furthermore, the gain of phosphorylation, DNA recombination, small-molecule metabolism and transport and loss of biosynthetic and regulatory genes suggest a functional shift in response to the environmental perturbation. Despite subsequent sampling events in the studied site, no pseudomonad was found until the lagoon completely dried in 2017. We speculate about the causes of P. otitidis final decline or possible extinction. Overall our results are evidence of adaptive responses at the genomic level of bacterial populations in a heavily exploited aquifer.
Anthropogenic perturbations alter evolutionary trajectories of bacterial populations by introducing novel and often strong selective pressures to biological systems (Davison, 1999; Krause et al., 2012; Tello et al., 2012; Evans and Wallenstein, 2014; Romero et al., 2019), in some cases leading to the breakdown of community dynamics and subsequent ecosystem collapse (Grant et al., 2017). The evolutionary response of species to these pressures depends on the standing genetic variation within populations, which allows for adaptation to the new environmental conditions (Holt, 1990).
Periodic selection is the main model that explains bacterial evolution (Cohan, 2001, 2005) and has been observed in multiple laboratory experiments on Escherichia coli (Atwood et al., 1951; Papadopoulos et al., 1999; Notley-McRobb and Ferenci, 2000), Pseudomonas fluorescens and P. aeruginosa in lung infections (Barrett et al., 2005; Caballero et al., 2015) and through time-series metagenomics in environmental aquatic Chlorobium and Bacteroidales (Bendall et al., 2016). Periodic selection consists on the succession or diversification of a bacterial population when a new advantageous variant appears, as it can either provide a fitness benefit within its current niche, out-competing its ancestors to extinction, thus sweeping through the population, or allow for the occupation of a new niche, generating a new ecotype (Cohan, 2001). This process applies well to mainly asexual bacterial populations in constant environments, where it has been mostly studied; evolution via periodic selection assumes that horizontal gene transfer (HGT) is extremely rare or completely absent (Cohan, 2005). However, this tends to not be the case in the face of a new environmental stress (Foster, 2005; Aminov, 2011).
In contrast, evolutionary rescue has been proposed as a process on which populations that suffer a sharp demographic decline as a consequence of environmental shock, escape from extinction by adaptive mechanisms (Orr and Unckless, 2008; Bell, 2013, 2017; Carlson et al., 2014). In evolutionary rescue, fast adaptive response can happen along with HGT, which accelerates genome evolution by the introduction of new advantageous genetic variation to populations (Jain et al., 2003; Pál et al., 2005; Baharoglu and Mazel, 2011; Baskett and Gomulkiewicz, 2011; Uecker and Hermisson, 2016). This creates a hallmark U-shaped demographic curve depicting the population decline and recovery, first predicted by Gomulkiewicz and Holt (1995) and later observed experimentally by others (Bell and Gonzalez, 2009; Agashe et al., 2011; Ramsayer et al., 2013).
Given the difficulties of in situ studies (Bell, 2017), evolutionary rescue in bacteria has only been studied in silico through mathematical models and simulations (Gomulkiewicz and Holt, 1995; Orr and Unckless, 2008, 2014; Uecker and Hermisson, 2016; Vogwill et al., 2016; Van Den Elzen et al., 2017; Anciaux et al., 2018, 2019; Carja and Plotkin, 2019) and in few laboratory experiments (Bell and Gonzalez, 2009; Agashe et al., 2011; Ramsayer et al., 2013). Long-term consequences of evolutionary rescue are is still under debate; for instance, does the resulting erosion in genetic diversity leaves populations vulnerable to extinction after evolutionary rescue (Carlson et al., 2014; Bell, 2017)?
Anthropogenic perturbation of natural environments are evolutionary experiments to look at the role of recombination and to evaluate periodic selection versus evolutionary rescue in bacterial populations. One worldwide example of an anthropogenic environmental perturbation is water overexploitation (Konikow and Kendy, 2005; Wada et al., 2010), which particularly affects arid ecosystems (Cui and Shao, 2005; McLendon et al., 2008). A dramatic example of this global trend is Cuatro Cienegas Basin (CCB), an oasis in the Chihuahua desert in northern Mexico. CCB is composed of various hydrological systems that used to include numerous pools, lagoons and rivers fed by underground springs (Minckley and Cole, 1968). Nowadays, there is only 5% or less of the original wetland left, as the overexploitation of groundwater through canals that have drained the wetlands since the 70’s has had a strong impact on CCB hydrological systems. Paradigm of this tragedy is the desiccation of the larger lagoon of our long-term study site, Churince (Souza et al., 2007, 2018; De Anda et al., 2018). This is particularly poignant, since Churince was described as a “lost world” (Souza et al., 2018), where unusually unbalanced stoichiometry (PO4 as low as 0.1 μM and often below detection and N:P > 200:1 for total nutrients) (Elser et al., 2005, 2018; Escalante et al., 2008; Lee et al., 2015, 2017; Ponce-Soto et al., 2015) had isolated this wetland from modern microbial communities, preserving a hyper diverse indigenous microbiota (Desnues et al., 2008; Moreno-Letelier et al., 2012; Gómez-Lunar et al., 2016; Souza et al., 2018; Taboada et al., 2018) composed of unique bacterial lineages of which a large fraction descended from marine ancestors (Souza et al., 2006; Alcaraz et al., 2008).
Previous studies of microbial populations of the Churince system have shown that the genus Pseudomonas have been impacted by past and recent environmental shocks of the Cuatro Cienegas aquatic system (Avitia et al., 2014; Ponce-Soto et al., 2015; García-Ulloa et al., 2019). Biology and ecology of pseudomonads from Churince has been described in different studies (Rodríguez-Verdugo et al., 2012; Avitia et al., 2014; Ponce-Soto et al., 2015) and a new lineage, P. cuatrocienegasensis, was previously described (Escalante et al., 2009). More recently two new strains of the P. aeruginosa PA14 clade native to Laguna Intermedia were sampled for the first time during the first desiccation event in 2011 (García-Ulloa et al., 2019). Pseudomonas otitidis isolated from CCB also represented a separate native lineage that was present, but rare, at Laguna Intermedia (Rodríguez-Verdugo et al., 2012). The availability of P. otitidis isolates from 2003 (normal state) and its subsequent isolation in 2015 (perturbed state) made it possible to observe the genomic evolution of this lineage in response to the desiccation events.
In this study we took advantage of a unique opportunity to study the evolution of natural populations of bacteria in the face of environmental perturbation. We studied the response of P. otitidis from Laguna Intermedia to a new selective pressure in the form of desiccation events. We used comparative genomics and population genetics techniques that allowed us to reconstruct the evolutionary response and demographic history of P. otitidis at Laguna Intermedia in a period of 12 years, from the first sampling (2003) to the last time it was found (2015), encompassing 3 desiccation events in 2011, 2012 and during the last sampling in 2015.
Laguna Intermedia from the Churince system in Cuatro Ciénegas, Coahuila, Mexico (26.848572 N, 102.141783 W) suffered four recent desiccation events: October 2011, November 2012, October 2015 and October 2016 before completely drying up in 2017 (Souza et al., 2018; García-Ulloa et al., 2019). Churince was a long-term study site, and water physico-chemical conditions were recorded periodically since 2002 with the multiparameter water quality sonde Hydrolab MS5. In Laguna Intermedia, salinity, specific conductivity and pH were particularly altered with each desiccation event (Supplementary Figure 1).
On October 2015, surface water samples were taken from multiple points of Laguna Intermedia using sterile BD Falcon vials (BD Biosciences, San Jose, CA, United States). Isolates were obtained by plating 200 μl of each sample on Pseudomonas Isolation Agar (PIA) plates, which were further incubated at 32°C for 24 h. Individual colonies were transferred to new PIA plates and grown at 32°C for 1 day. Each colony was grown in LB medium at 32°C for 1 day and then stored in 50% glycerol solution at −80°C. From October 2015 until 2017, when Laguna Intermedia became completely dry, no other pseudomonad was found, even if they were intensively looked after in different subsequent samplings and cultures through the implementation of this exact same protocol and sampling effort.
DNA extraction was done with Qiagen DNeasy Blood & Tissue kit. 16S rRNA gene was amplified with universal primers 27F and 1492R and sequenced by the Sanger method. 138 colonies from the October 2015 sampling were screened and 7 of them were identified as P. otitidis through a blast search against the RefSeq database. Additionally, all 5 existing isolates obtained from water in August 2003 previously identified as P. otitidis by Avitia et al. (2014) were used for the analysis as representatives of the undisturbed state of the lagoon.
Sequencing was carried out using the Illumina MiSeq platform in its PE 2x300 modality, obtaining a coverage larger than 30× for all genomes (Supplementary Table 1). Genomes were assembled de novo with MaSuRCA 3.2.4 using default settings (Zimin et al., 2013), followed by SPAdes 3.10.0 using the prior assembly with the –trusted_contigs option for refinement (Bankevich et al., 2012). Contigs less than 600 bp long and 10× of k-meric coverage were discarded. The quality and integrity evaluation of the assemblies was done with Quast v4 (Gurevich et al., 2013) and BUSCO v2 (Simão et al., 2015; Supplementary Table 1). All genomes were deposited on NCBI with the accession numbers JAANPJ000000000 (41M), JAANPK000000000 (39M), JAANPL000000000 (38M), JAAN PM000000000 (36M), JAANPN000000000 (35M), JAANPO00 0000000 (34M), JAANPP000000000 (32M), JAANPQ000000000 (15M), JAANR000000000 (13M), JAANPS000000000 (12M), JA ANPT000000000 (10M), JAANPU000000000 (9M). A database with all the complete annotated genomes of Pseudomonas to date was built as reference for annotation with Prokka 1.11 (Seemann, 2014).
Phylogenomic relationships between all lineages were assessed through a core genome genealogy. A genome alignment was done with progressiveMauve using default settings and the core genome was extracted with the stripSubsetsLCBs script (Darling et al., 2010). Core genome genealogy was performed with FastTree2.1.10 (Price et al., 2010) using the gamma and general time-reversible model parameters. NCBI accession numbers for the reference genomes used in the genealogy are: AE008922.1 (Xanthomonas campestris), NC_012660.1 (P. fluorescens SBW25), NC_002947.4 (P. putida KT2240), NZ_PXJI00000000.1 (P. otitidis PAM-1), NC_002516.2 (P. aeruginosa PAO1) and NZ_FOFP00000000.1 (P. cuatrocienegasensis).
The raw genomic reads were filtered by a quality of 20 with Sickle v1.33 (Joshi and Fass, 2011) and mapped against the genome of P. otitidis PAM-1 with the Burrows-Wheeler aligner (Li and Durbin, 2009). SNP calling was done using Genome Analysis Toolkit (Van der Auwera et al., 2013), SAMtools (Li et al., 2009) and VarScan2.3.9 (Koboldt et al., 2012) programs. The folded frequency spectrum was calculated with easySFS1.
Coalescence simulations were performed with fastsimcoal2 (Excoffier et al., 2013); 500 repetitions of 500,000 simulations were carried out with 40 optimization cycles and 1,000 parametric bootstraps of the best model to obtain 95% confidence intervals. Demographic models allowed the possibility of positive (expansion: E) and negative (contraction: K) population changes as well as absence of changes (constant population size: C) in three time intervals based on the desiccation events (August 2003 to October 2011; October 2011 to November 2012; November 2012 to October 2015) based on the dates of the desiccation events, resulting in 27 different models. The initial parameters used in the models were: generational time of P. otitidis sister clade, P. aeruginosa, in low phosphorus medium = 0.04 h-1 (Buch et al., 2008), range of generations from 2003 to 2015 = 6,300 to 2,100 (this study), mutation rate of non-mutator P. aeruginosa = 2.5 e-8 (Oliver et al., 2000), recombination rate calculated from R/θ = 5 e-9 (this study, using the mentioned mutation rate), effective population size range = 500,000 to 8,000,000 calculated using a coalescent-based approach on 4 concatenated housekeeping genes (acnB, gyrB, recA and rpoD) (Avitia et al., 2014). The evaluation of the models was done by calculating and comparing their weighted AIC values (wAIC) which serve as a measure of the strength of each model in comparison to the others given their likelihoods, from 0 to 100%.
Global core, cores by sampling, exclusive accessory and shared accessory genomes were obtained with the consensus of the three clustering algorithms (BDBH, OMCL and COG) of GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013) and aligned with ClustalW using default settings (Thompson et al., 1994). The orthologous genes of the global core genome were used to obtain the mean nucleotide diversity obtained from all possible pairwise comparisons within each cluster of orthologous group (π) and mean Tajima’s D with DnaSP v5 (Librado and Rozas, 2009). Tajima’s D is often used in population genetics studies to assess selection and demographic changes in populations comparing the pairwise differences with the number of segregating sites. From a demographic perspective, a neutral value of Tajima’s D signals constant population size, while a positive and negative values indicate a recent population contraction or expansion, respectively.
Functional annotation of the genes of the exclusive accessory genomes was done with Blast2GO (Götz et al., 2008), using Biological Process gene ontology category for the functional analysis. Furthermore, a protein blast against the non-redundant protein sequences database was performed on the translated genes from the exclusive accessory genomes.
Rarefaction curves of the unique gene clusters were done with the R-package micropan v 2.0.1 (Snipen and Liland, 2015) using its dclust algorithm on hmmer domains of the gene products of the accessory genomes of each sampling separately. 1,000 permutations were performed.
To assess the changes in the importance of HGT, recombination rate to mutation ratio (R/θ), insert size (δ), number of substitutions introduced per insert (δν) and the effect of recombination to mutation on genome level variation (r/m) were calculated with ClonalFrameML using default settings (Didelot and Wilson, 2015). 1,000 pseudoreplicates were performed to obtain 95% confidence intervals.
The genealogy of core genomes shows phylogenetic correspondence between isolates from 2003 and 2015, confirming their close relatedness which makes comparison between them valid (Figure 1). The lack of divergence regardless of the sampling date, as seen on the lack of separation within their phylogenetic group, points to all isolates forming part of the same population at different time points. In addition, the genealogy confirms the proximity of all isolates with P. otitidis PAM-1.
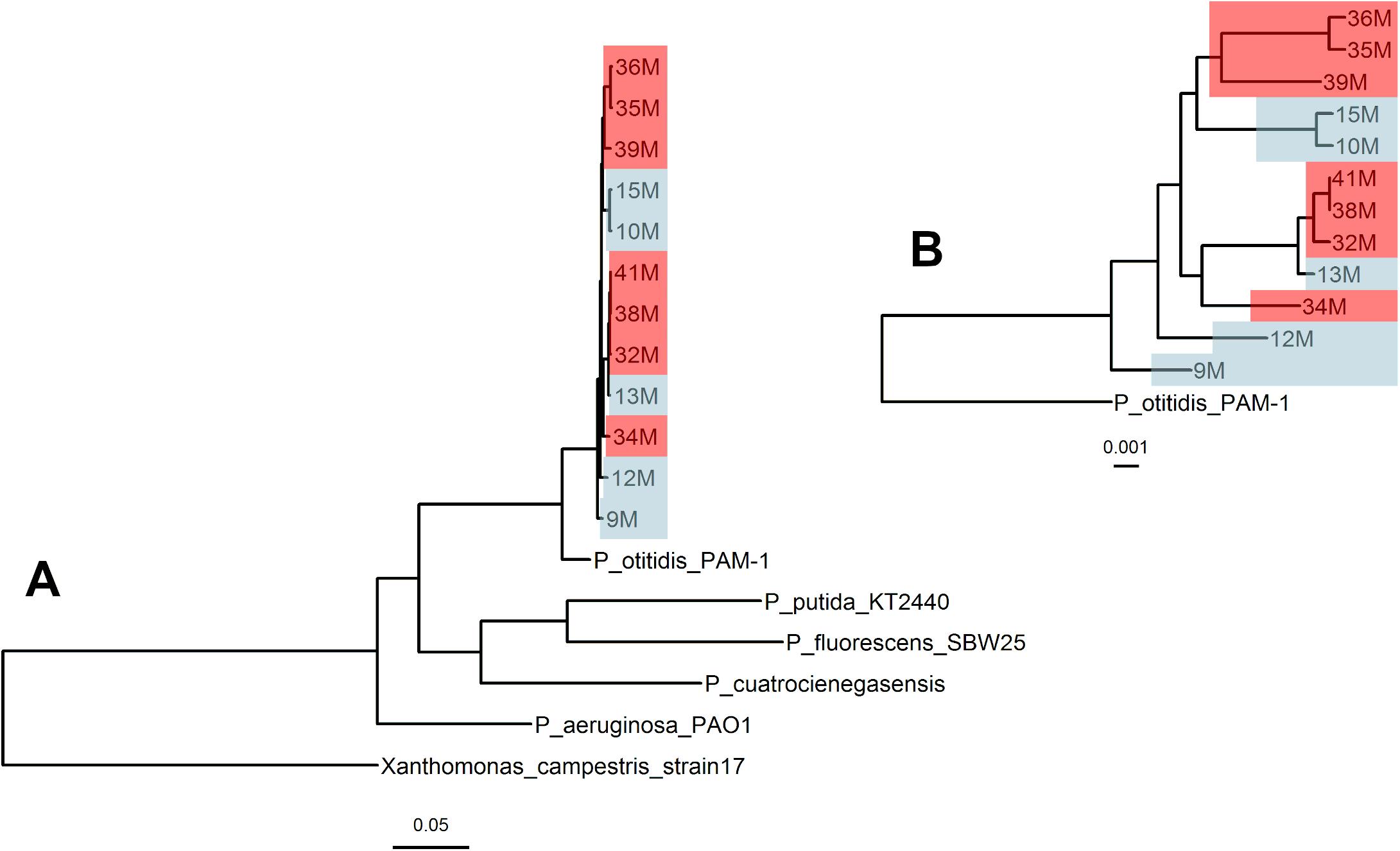
Figure 1. Genealogy of core genomes of Pseudomonas otitidis collected from Laguna Intermedia in the Churince system. Genealogy of the Pseudomonas genus including multiple outgroups (A) and zoomed at the clade that comprises the Churince isolates from this study (B). The isolates from August 2003 (original state) are in blue and from October 2015 (perturbed state) in red. The bar below the trees represents branch lengths. Both phylogenies were calculated by FastTree2.0. All genomes were aligned with progressiveMauve and the core genome was extracted with the stripSubsetsLCBS script (Darling et al., 2010).
The two best coalescent models according to their wAIC values are contraction-expansion-contraction (KEK, wAIC = 73.15%) and contraction-expansion-constant (KEC, wAIC = 18.43%), both coinciding in an initial population contraction from 2003 to the first desiccation event in 2011 of more than 99%, followed by a drastic population increase until the second desiccation event in 2012 (Figure 2 and Supplementary Table 2). Both models also reach a similar final population size in 2015, which is approximately 1,000 times larger than its initial size (Supplementary Table 2).
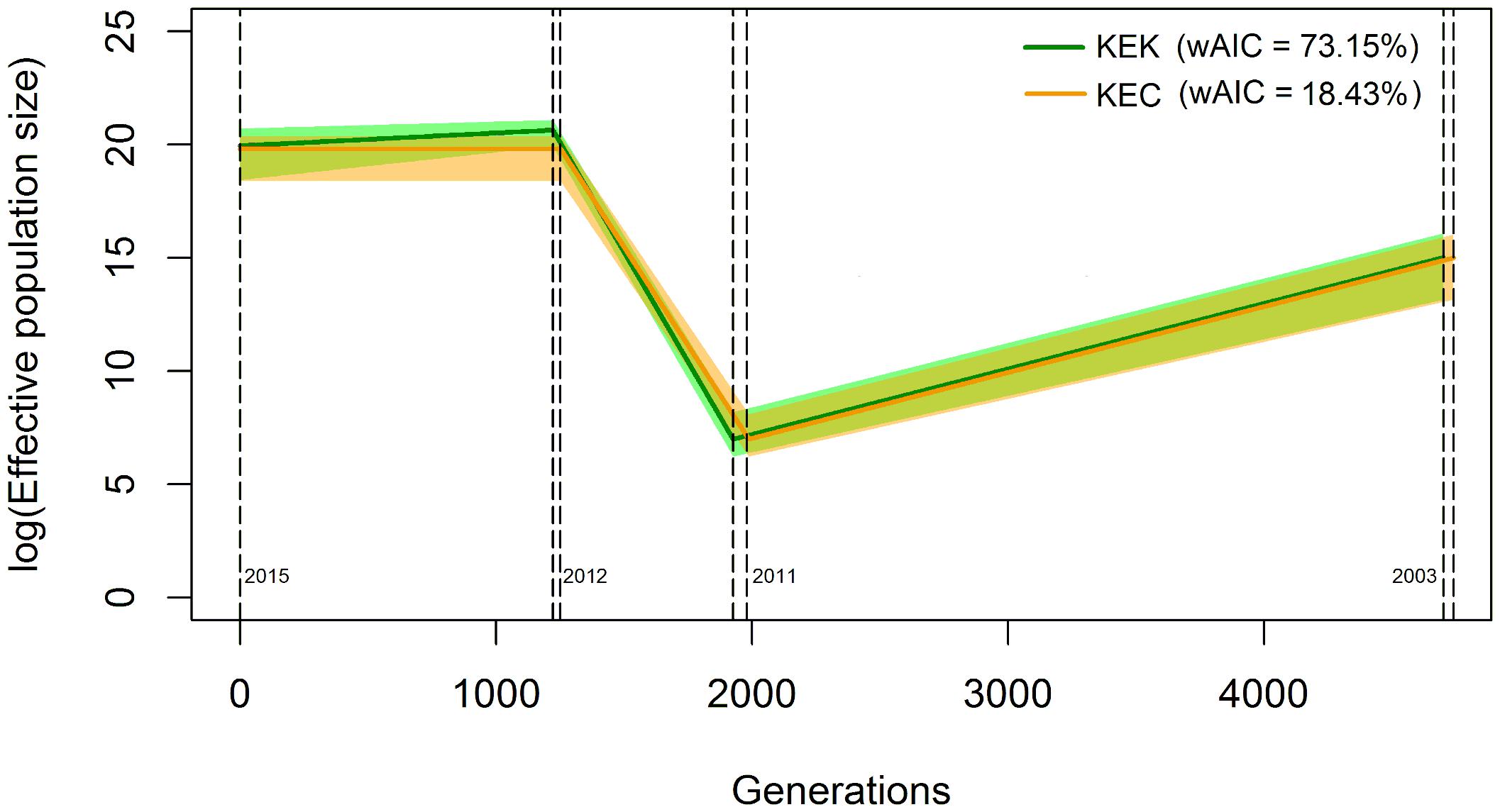
Figure 2. Graphical representation of the best two demographic models according to their weighted AIC value based on the results obtained from fastsimcoal2. Coalescent simulations were performed using the observed allele frequency spectrum of the Pseudomonas otitidis population from 2015, taking into account an initial population range of 500,000 to 8,000,000 estimated by Avitia et al. (2014), mutation rate of P. otitidis sister clade P. aeruginosa (wild tyfpe) of 5e-9 calculated by Oliver et al. (2000), generational time of P. aeruginosa in low phosphorus medium of 0.04 h-1 calculated by Buch et al. (2008), and range of generations from 2003 to 2015 of 6,300 to 2,100 and recombination rate R = 2.5 e-8 calculated from R/θ in this study. The solid colored lines are the average values and the shaded areas represent the 95% confidence intervals. Contraction-Expansion-Contraction (KEK) is the best model with a wAIC value of 73.15% (green) and Contraction-Expansion-Constant (KEC) follows with 18.45% (orange). Dotted vertical lines mark the desiccation events according to the generations calculated by fastsimcoal2. The population contraction from 2003 to 2011 for both models was approximately 99%. For detailed values see Supplementary Table 2.
There was a significant increase in mean Tajima’s D, which went from a marginally negative, almost neutral value (D = −0.03) in 2003 to a positive value (D = 0.26) in 2015 (paired two-sided Wilcoxon test: V = 2.07e6, p < 0.001). Variance in Tajima’s D increased from 0.50 to 0.61, and the change was also significant (Lavene’s test: F(1,7004) = 23.8, p < 0.001) (Figure 3).
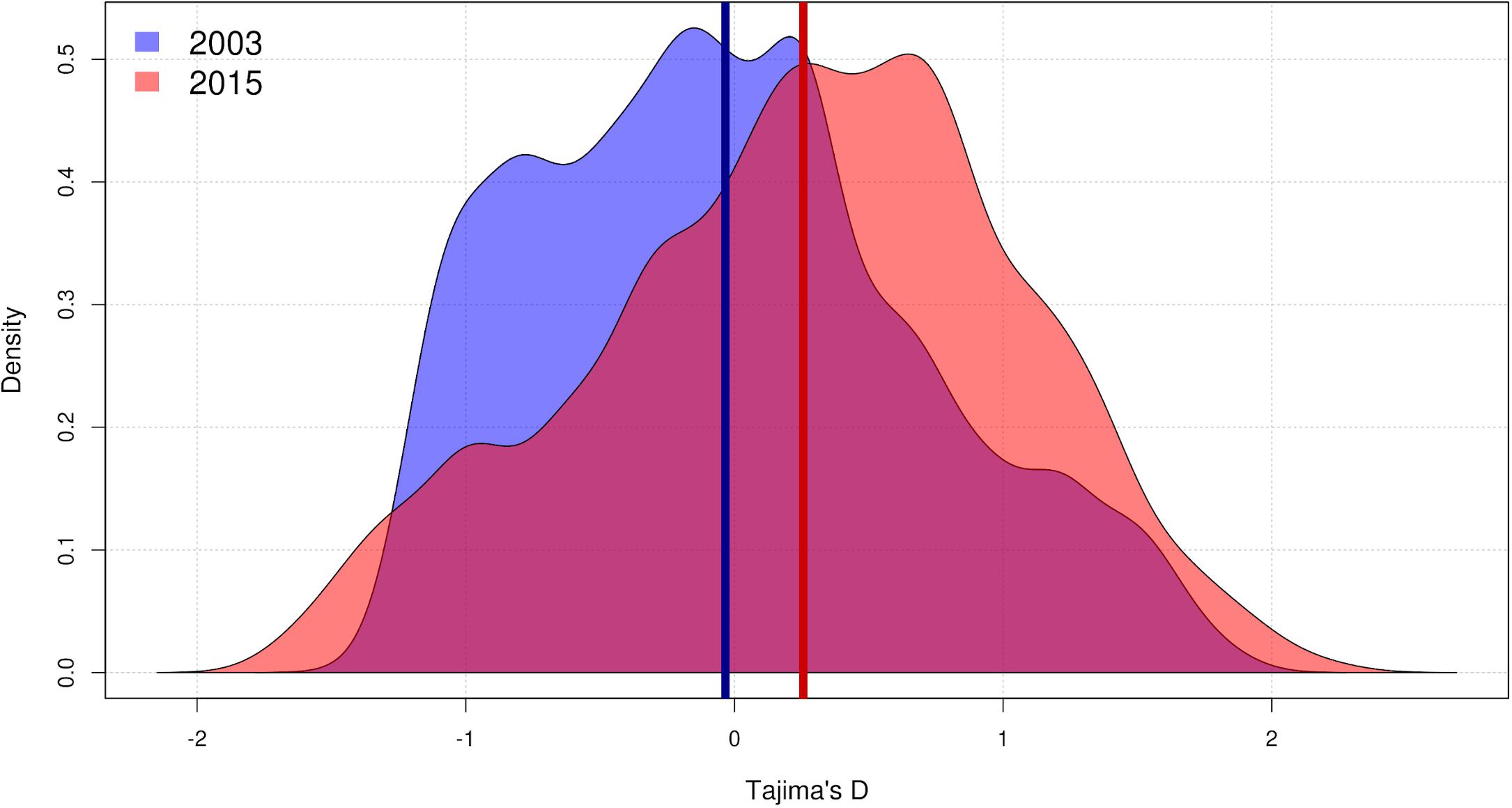
Figure 3. Density plot of the distribution of Pseudomonas otitidis Tajima’s D at core genome level. Samples of P. otitidis from 2003 and 2015 consist of 5 and 7 individuals, respectively. 3503 genes were used for the analysis due to the rest of the 3682 global core lacking any variation to measure. Kernel smoothing was used to plot the values. Vertical bars represent the means: –0.03 in 2003 (blue, left-skewed) and 0.26 in 2015 (red, right-skewed). The increase in mean value and the increase in variance are significant (paired two-sided Wilcoxon test: V = 2.07e6, p < 0.001; Lavene’s test: F(1,7004) = 23.8, p < 0.001).
Pseudomonas otitidis isolated from Laguna Intermedia suffered a significant decrease both in its genomic and mean nucleotide diversity (π). The core genome increased by 17.3% (559 genes), while the accessory decreased by 32.8% (486 genes). Similarly, mean π of the global core genome (3,682 genes) decreased from 0.0075 to 0.0058, which represents a significant loss of 22% of its mean π (paired two-sided Wilcoxon test: V = 4.42e6, p < 0.001), as well as a significant decrease in its variance from 1.39e-4 to 6.4e-5 (Brown-Forsythe test: F(1, 7362) = 27.6, p < 0.001) (Figure 4).
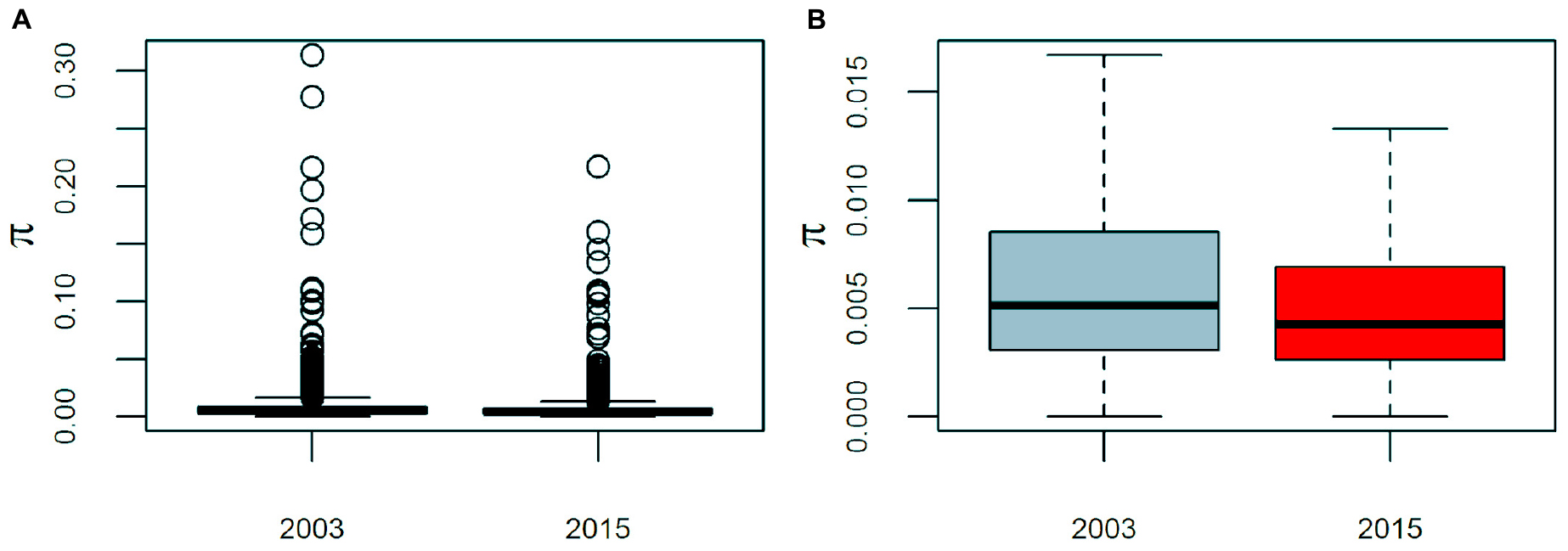
Figure 4. Boxplots of the average nucleotide diversity (π) of Pseudmonas otitidis at the global core genome level. The global core genome consists of 3,682 genes. Barplots are presented with (A) and with outliers (values outside 1.5 times the interquartile range) (B). The change of mean and variance of π are significant (paired two-sided Wilcoxon test: V = 4.42e6, p < 0.001; Brown-Forsythe test: F(1, 7362) = 27.6, p < 0.001).
The exclusive accessory genome comprises the unique genes of each sampling, those that were lost (only found in 2003) and gained (only found in 2015), with a net decrease of 41.7% (156 genes). Exclusive accessory genomes of both samplings (Figure 5, purple fraction) were functionally annotated (Supplementary Tables 3, 4), with 27.8% of genes (104 genes) for the population of 2003, and 36.7% (80 genes) for the population of 2015 being functionally identifiable under the Biological Process gene ontology category.
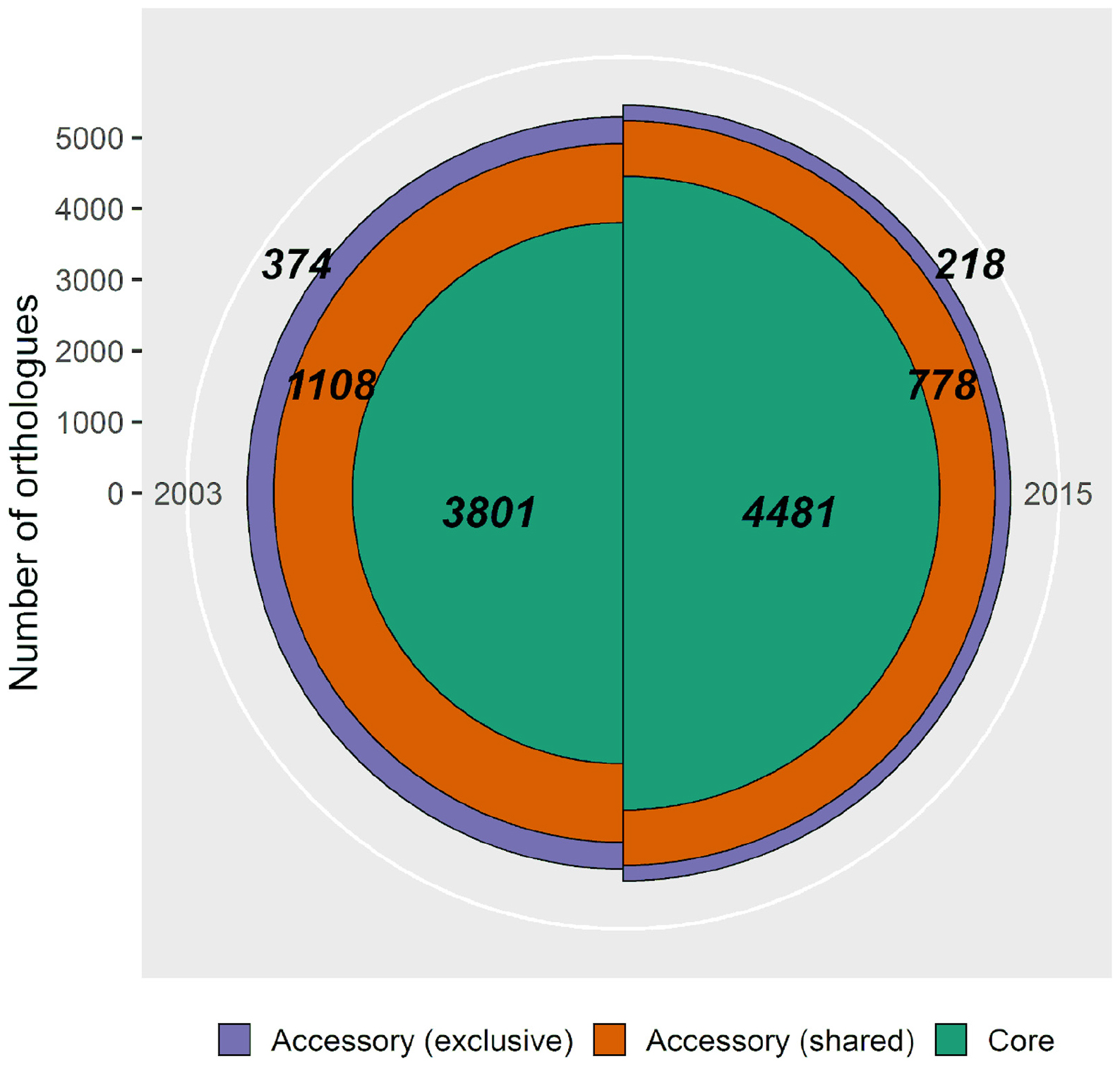
Figure 5. Pan-genomic comparison of Pseudomonas otitidis from 2003 and 2015 from Laguna Intermedia in the Churince system. The population from 2003 consists of 5 individuals and has a pan-genome of 5,283 genes and the one from 2015 consists of 7 individuals with a pan-genome of 5,477 genes. The green (innermost) fraction represents the core genome, the orange (middle) one the shared accessory genome and the purple one (outermost) the exclusive accessory genome of each sampling. Bold numbers indicate the quantity of genes in each category.
According to a multi-level functional analysis performed by Blast2GO, gained genes included functions related to metabolism of organic nitrogen compounds and small molecules, modification of macromolecules, phosphorylation and recombination. In contrast, genes involved in DNA and cellular metabolic process, biosynthesis of cellular macromolecules and nucleobase-containing compounds and regulation of cellular process were lost (Figure 6). Even though all other functional categories decreased in their number of genes, genes with transport functions, particularly of organic substrates, increased by 42.8% (Figure 6).
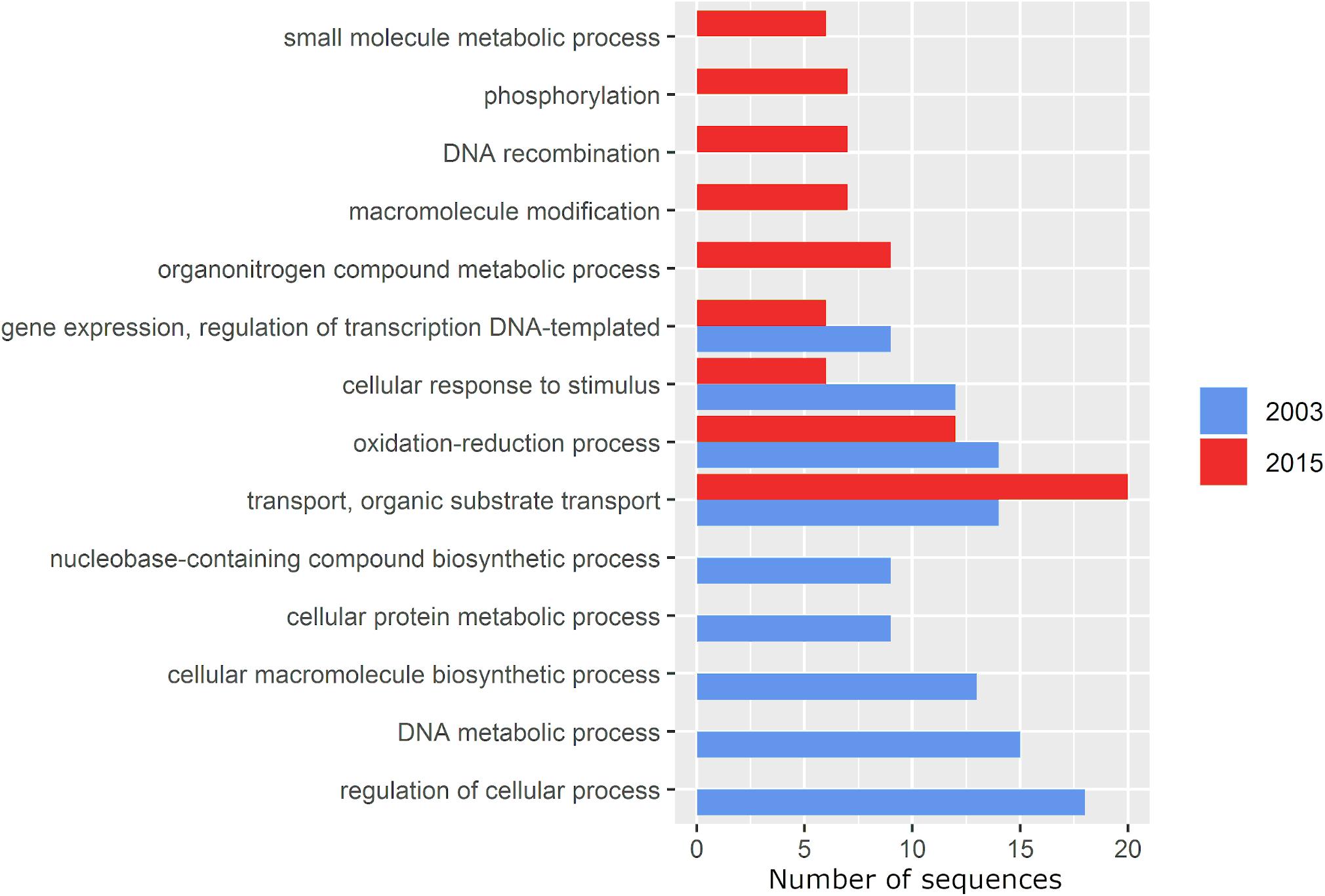
Figure 6. Multi-level gene ontology graph of the exclusive accessory genome of P. otitidis under the Biological Process category calculated by Blast2GO. The gene ontology terms (functions) are on the Y axis and the X axis represents the number of gene sequences assigned to each one. The 2003 and 2015 samplings are marked in blue and red, respectively. Bars alone represent functions lost (2003, blue) and gained (2015, red). Specific functions and annotation details are in Supplementary Tables 3, 4.
Blast searches showed an increase of accessory proteins of 3.4% from multiple closely related P. otitidis strains, in contrast with a decrease of 3.5% in proteins from other Pseudomonas species (2003: P. aeruginosa, P. syringae, P. stutzeri, P. oleovorans, P. citronellolis, P. asplenii, P. alcaligenes and P. corrugata; 2015: P. aeruginosa, P. panipatensis, P. xanthomarina, P. alcaligenes, P. syringae, P. compostae and P. pseudoalcaligenes). Proteins from other genera were less than 1% on either sampling, although with a slight increase of 0.1% in 2015. Best hits of proteins acquired from other genera point to them originally belonging to Sphaerotilus natans subsp. natans DSM 6575, Burkholderia gladioli and B. pseudomallei MSHR3709 in the 2003 sampling and Magnetospirillum marisnigri and Acinetobacter junii in 2015. Only the A. junii protein has a known function as a fatty acid desaturase.
Significant decreases in average size of recombinant fragments and substitutions per fragment reflect the loss in genetic diversity as seen in the decrease of δ by 23.6% (110 pb, CI 95% = 0.085 to 84 pb, CI 95% = 0.063; p > 0.001) and δν by 19.2% (5.5, CI 95% = 0.0043 to 4.5, CI 95% = 0.0035; p > 0.001). Moreover, an increase in recombination rate is depicted by a slight but significant increase in R/θ (0.19, CI 95% = 0.00017 to 0.20, CI 95% = 0.00018; p > 0.001). As a result, r/m also decreased significantly by 12.4% (1.05, CI 95% = 0.0011 to 0.92, CI 95% = 0.0009; p > 0.001) as the population became less genetically diverse and recombination occurred more often between more genetically similar individuals, thus, minimizing the role of recombination on the introduction of novel genetic variation.
According to the two best demographic models calculated by fastsimcoal2 (Excoffier et al., 2013), a population bottleneck drove P. otitidis near to extinction in 2011, followed by a drastic population expansion that peaked in 2012 (Figure 2). Although they differ on their last time interval (2012 to 2015; contraction vs. constant) the final population size in 2015 was about 1000 times greater than its initial size in 2003 in both models, with the 95% CI overlapping at all times (Figure 2 and Supplementary Table 2). An increase in mean Tajima’s D and its variance were also observed, supporting the observation of a population (Maruyama and Fuerst, 1985; Schmidt and Pool, 2002).
Another observable effect of evolutionary rescue is the loss of variation, since often a very small portion of the population survives and expands (Carlson et al., 2014; Bell, 2017). Accordingly, pan-genome analysis showed a decrease in both genomic variation (Figure 5) and mean π and its variance (Figure 4).
Rarefaction curves of the pan-genome unique gene clusters (Supplementary Figure 2; Snipen and Liland, 2015; Adamek et al., 2018) show that the 2015 population sample nearly reached its asymptote, while the 2003 sample did not. This observation points to a larger accessory genome in 2003 than the one we found (Figure 5) and, although its size is unknown, it is safe to assume it was larger than the one of 2015 (Supplementary Figure 2). As the 2003 sample may be incomplete in terms of genetic variation, conclusions regarding this matter must be taken with caution, since there are actually more lost genes than the ones found and there is a possibility that some of those genes are the ones we catalog as “gained” in 2015 but were not found in 2003 because of the small sample size (n = 5). Nonetheless, this result supports the observed decrease in diversity discussed before and, even though the 2003 population sample included the whole exclusive accessory genome from the 2015 sample, that would mean the latter is a subset of the former, which is consistent with an evolutionary rescue event.
The role of adaptive gene loss in bacteria has been previously studied (Hottes et al., 2013). Here, genes involved in regulation of transcription and cellular processes, response to stimulus, oxidation-reduction, biosynthesis of macromolecules and nucleobase-containing compounds and metabolism of DNA and proteins were lost (Figure 6). Loss of biosynthetic genes in bacteria is a general and well documented phenomenon by which lineages become auxotrophic in new environmental conditions due to the cost of maintaining genes that are no longer needed, thus becoming unable to produce some of the metabolites they needed to grow in their former environment (D’Souza et al., 2014). This can also be an adaptive response when the corresponding metabolite is sufficiently present in the environment (Morris et al., 2012). We believe the disruption of the original community interactions and their microbial “market” (Werner et al., 2014), may have rendered biosynthesis of certain metabolites an unnecessary burden, thus being lost as an adaptive response (Giovannoni et al., 2005). Albalat and Cañestro (2016) pointed out the importance of loss of regulatory functions in the adaptation of bacterial populations to new environments that require alternative or new metabolic pathways, which could be the case at Laguna Intermedia, given the increasing severity of the perturbations.
Functions gained include DNA recombination and phosphorylation, both of which have been observed to play a role in environmental stress response (Stock et al., 1989; Sutton et al., 2000; Lipa and Janczarek, 2020). Although a correlation cannot be drawn from our results, the gain of genes involved in DNA recombination coincides with a slight but significant increase in recombination rate (R/θ, 0.19 ± 0.00017 to 0.20 ± 0.00018, p < 0.001), suggesting a mechanism of evolutionary rescue assisted by HGT. Bacteria such as Escherichia coli (Little and Mount, 1982), Vibrio (Baharoglu and Mazel, 2011), Staphylococcus (Anderson et al., 2006) and Pseudomonas (Penterman et al., 2014) also possess a stress response known as SOS that consists of repairing DNA damage caused by multiple sources of stress, such as UV light, fungal metabolites, reactive oxygen species (Sutton et al., 2000), exposure to antibiotics and phages (Torres-Barceló and Hochberg, 2016). The SOS response could facilitate evolutionary rescue by integrating exogenous DNA into the genome. Contrastingly, r/m decreased significantly (12.4% decrease, p < 0.001), suggesting that although recombination increased, it occurred between a less diverse gene pool. This observation coincides with the general loss of genetic diversity, the less taxonomically diverse sources of the exclusive accessory genome in 2015, and that closer bacterial lineages tend to recombine more frequently (Nandi et al., 2015; Vazquez-Rosas-Landa et al., 2020).
Phosphorylation is a key mechanism involved in signal transduction as a response to environmental stimuli such as nitrogen deprivation, phosphorous availability, osmolarity and chemotaxis molecules (Stock et al., 1989). Experimental, experimental studies in rhizobia indicate that phosphorylation plays an essential role in many physiological processes in adaptation to various environmental factors (e.g., dicarboxylate transport, phosphate utilization and adaptation to pH stress and microaerobic conditions) (Liu et al., 2015) through multiple regulatory mechanisms (Lipa and Janczarek; 2020). As Laguna Intermedia became perturbed, the gain of phosphorylation-related genes may have helped P. otitidis to withstand the new environmental conditions and adapt to them. Additionaly, the increase in small-molecule metabolism genes correlates with an increase in metabolic versatility required under conditions of high competition (Torsvik et al., 2002), and a large variety of nutrients (Cases et al., 2003). Although no measurements of nutrients were taken in 2015, studies have found that cycles of hydration-desiccation in soil provide extra nutrients to surviving bacterial populations through bacterial remnants and osmolites (Bottner, 1985; Kieft et al., 1987; Fierer and Schimel, 2003; Št′ovíček et al., 2017), which may have been the case in Laguna Intermedia. We think that this change in nutrients translated in the transport of organic substrates functions being the only shared category of the exclusive accessory genome between samplings that increased (42.8%) in 2015 as an adaptation to a less oligotrophic Laguna Intermedia.
Together with the severity of the perturbation and population size (Gomulkiewicz and Holt, 1995), initial genetic diversity plays an important role in the survival of a population through evolutionary rescue (Ramsayer et al., 2013). Culturable bacterial lineages from the Churince system have been found to be highly clonal (Cerritos et al., 2011) and P. otitidis from Laguna Intermedia is no exception, with consistently low values of π and R/θ. In fact, clinical P. aeruginosa is considered clonal even at R/θ = 0.54 (Castañeda-Montes et al., 2018), which is almost 3 times the R/θ of P. otitidis of Laguna Intermedia.
Although the initial variation was enough to withstand the perturbation and enter evolutionary rescue, P. otitidis was not found after the 2015 desiccation event, even though subsequent attempts with the same sampling effort and protocol were done in March and October (same time of the year as the 2015 sampling) 2016. Bell (2017) has speculated that the loss of genetic variation after an evolutionary rescue event could render populations less likely to survive a subsequent (new) stress in fluctuating environments, as Laguna Intermedia became since 2011. However, P. otitidis only went through one bottleneck in the first desiccation event in 2011, not in the one of 2012 nor 2015. We propose two possible (non-mutually exclusive) explanations:
(1) Although population size remained high at the 2nd and 3rd desiccation events, we don’t know if genomic and/or nucleotide variation was being lost. One possible scenario is that the variation of P. otitidis progressively decreased to the point of it being incapable of responding to any new environmental pressure, as Bell (2017) proposed, and after going through three consecutive desiccation events, the fourth one in 2016 (Supplementary Figure 1) was the final nail in the coffin.
(2) The volume of water of Laguna Intermedia decreased with each desiccation event, which is reflected in the progressive increase in salinity, specific conductivity and pH shown in Supplementary Figure 1. In this scenario, the successive desiccation events could have progressively degraded the lagoon to the point of it becoming uninhabitable by P. otitidis, or harsh enough (on its environmental conditions, community dynamics or both) to keep P. otitidis at such low abundance that it could not be sampled again. Interestingly, in October 2016, using the same Pseudomonas isolation agar and the same sampling protocol with the same sampling effort, we only recovered multiple strains of environmental Vibrio cholerae for the first time ever in Laguna Intermedia. This serves as evidence that there was at least an important change in the community, supporting this scenario. Also, additional evidence of community disruption is provided by the finding of taxonomically very different non-pseudomonad genes on the exclusive accessory genomes, with 2 genes from Burkholderia and 1 from Sphaerotilus in 2003, and 1 from Magnetospirillum and 1 from Acinetobacter in 2015. Medium to long-term evolutionary rescue studies comparing constant and fluctuating environments with more samplings in time are needed in order to answer this question.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MG-U, AE, AM-L, LE, and VS conceived and designed the study. MG-U and AE performed the fieldwork. MG-U performed the experimental procedures, analyzed all the data and wrote the original draft of the manuscript. VS and LE contributed with reagents, materials, computational resources, and acquired the funding for the project. AE, AM-L, LE, and VS supervised the experimental work and reviewed and edited the manuscript. All authors approved the final manuscript.
This work was supported by World Wildlife Fund, Carlos Slim Foundation, DGAPA/UNAM-PAPIIT (grant number IG200319) and CONACyT (grant number 000000000220708).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MG-U is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and has received CONACyT fellowship 596613. We greatly acknowledge V.M.D Gabriel Manuel Rosas Barrera and Dr. Gabriel Yaxal Ponce-Soto for the help during the sample collection and lab assistance. We also thank Dr. Erika Aguirre Planter and Dr. Laura Espinosa-Asuar for the technical support. This manuscript was released as a pre-print at bioRxiv (García-Ulloa et al., 2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.563885/full#supplementary-material
Adamek, M., Alanjary, M., Sales-Ortells, H., Goodfellow, M., Bull, A. T., Winkler, A., et al. (2018). Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genomics 19:426. doi: 10.1186/s12864-018-4809-4
Agashe, D., Falk, J. J., and Bolnick, D. I. (2011). Effects of founding genetic variation on adaptation to a novel resource. Evolution 65, 2481–2491. doi: 10.1111/j.1558-5646.2011.01307.x
Albalat, R., and Cañestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Alcaraz, L. D., Olmedo, G., Bonilla, G., Cerritos, R., Hernández, G., Cruz, A., et al. (2008). The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment. Proc. Natl. Acad. Sci. U.S.A. 105, 5803–5808. doi: 10.1073/pnas.0800981105
Aminov, R. I. (2011). Horizontal gene exchange in environmental microbiota. Front. Microbiol. 2:158. doi: 10.3389/fmicb.2011.00158
Anciaux, Y., Chevin, L. M., Ronce, O., and Martin, G. (2018). Evolutionary rescue over a fitness landscape. Genetics 209, 265–279. doi: 10.1534/genetics.118.300908
Anciaux, Y., Lambert, A., Ronce, O., Roques, L., and Martin, G. (2019). Population persistence under high mutation rate: from evolutionary rescue to lethal mutagenesis. Evolution 73, 1517–1153. doi: 10.1111/evo.13771
Anderson, K. L., Roberts, C., Disz, T., Vonstein, V., Hwang, K., Overbeek, R., et al. (2006). Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 188, 6739–6756. doi: 10.1128/JB.00609-06
Atwood, K. C., Schneider, L. K., and Ryan, F. J. (1951). Periodic selection in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 37, 146–155. doi: 10.1073/pnas.37.3.146
Avitia, M., Escalante, A. E., Rebollar, E. A., Moreno-Letelier, A., Eguiarte, L. E., and Souza, V. (2014). Population expansions shared among coexisting bacterial lineages are revealed by genetic evidence. PeerJ 2:e696. doi: 10.7717/peerj.696
Baharoglu, Z., and Mazel, D. (2011). Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob. Agents Chemother. 55, 2438–2441. doi: 10.1128/AAC.01549-10
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barrett, R. D., MacLean, R. C., and Bell, G. (2005). Experimental evolution of Pseudomonas fluorescens in simple and complex environments. Am. Nat. 166, 470–480. doi: 10.1086/444440
Baskett, M. L., and Gomulkiewicz, R. (2011). Introgressive hybridization as a mechanism for species rescue. Theor. Ecol. 4, 223–239. doi: 10.1007/s12080-011-0118-0
Bell, G. (2013). Evolutionary rescue and the limits of adaptation. Philos. Trans. R. Soc. B Biol. Sci. 368:20120080. doi: 10.1098/rstb.2012.0080
Bell, G. (2017). Evolutionary rescue. Annu. Rev. Ecol. Evol. Syst. 48, 605–627. doi: 10.1146/annurev-ecolsys-110316-023011
Bell, G., and Gonzalez, A. (2009). Evolutionary rescue can prevent extinction following environmental change. Ecol. Lett. 12, 942–948. doi: 10.1111/j.1461-0248.2009.01350.x
Bendall, M. L., Stevens, S. L., Chan, L. K., Malfatti, S., Schwientek, P., Tremblay, J., et al. (2016). Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J. 10, 1589–1601. doi: 10.1038/ismej.2015.241
Bottner, P. (1985). Response of microbial biomass to alternate moist and dry conditions in a soil incubated with 14C-and 15N-labelled plant material. Soil Biol. Biochem. 17, 329–337. doi: 10.1016/0038-0717(85)90070-7
Buch, A., Archana, G., and Kumar, G. N. (2008). Metabolic channeling of glucose towards gluconate in phosphate-solubilizing Pseudomonas aeruginosa P4 under phosphorus deficiency. Res. Microbiol. 159, 635–642. doi: 10.1016/j.resmic.2008.09.012
Caballero, J. D., Clark, S. T., Coburn, B., Zhang, Y., Wang, P. W., Donaldson, S. L., et al. (2015). Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6:e00981-15. doi: 10.1128/mBio.00981-15
Carja, O., and Plotkin, J. B. (2019). Evolutionary rescue through partly heritable phenotypic variability. Genetics 211, 977–988. doi: 10.1534/genetics.118.301758
Carlson, S. M., Cunningham, C. J., and Westley, P. A. (2014). Evolutionary rescue in a changing world. Trends Ecol. Evol. 29, 521–530. doi: 10.1016/j.tree.2014.06.005
Cases, I., De Lorenzo, V., and Ouzounis, C. A. (2003). Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 11, 248–253. doi: 10.1016/S0966-842X(03)00103-3
Castañeda-Montes, F. J., Avitia, M., Sepúlveda-Robles, O., Cruz-Sánchez, V., Kameyama, L., Guarneros, G., et al. (2018). Population structure of Pseudomonas aeruginosa through a MLST approach and antibiotic resistance profiling of a Mexican clinical collection. Infect. Genet. Evol. 65, 43–54. doi: 10.1016/j.meegid.2018.06.009
Cerritos, R., Eguiarte, L. E., Avitia, M., Siefert, J., Travisano, M., Rodríguez-Verdugo, A., et al. (2011). Diversity of culturable thermo-resistant aquatic bacteria along an environmental gradient in Cuatro Ciénegas, Coahuila, México. Antonie Van Leeuwenhoek 99, 303–318. doi: 10.1007/s10482-010-9490-9
Cohan, F. M. (2001). Bacterial species and speciation. Syst. Biol. 50, 513–524. doi: 10.1080/10635150118398
Cohan, F. M. (2005). “Periodic selection and ecological diversity in bacteria,” in Selective sweep, ed D. Nurminsky, (Boston, MA: Springer), 78–93.
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cui, Y., and Shao, J. (2005). The role of ground water in arid/semiarid ecosystems, Northwest China. Groundwater 43, 471–477. doi: 10.1111/j.1745-6584.2005.0063.x
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Davison, J. (1999). Genetic exchange between bacteria in the environment. Plasmid 42, 73–91. doi: 10.1006/plas.1999.1421
De Anda, V., Zapata-Peñasco, I., Blaz, J., Poot-Hernández, A. C., Contreras-Moreira, B., González-Laffitte, M., et al. (2018). Understanding the mechanisms behind the response to environmental perturbation in microbial mats: a metagenomic-network based approach. Front. Microbiol. 9:2606. doi: 10.3389/fmicb.2018.02606
Desnues, C., Rodriguez-Brito, B., Rayhawk, S., Kelley, S., Tran, T., Haynes, M., et al. (2008). Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature 452, 340–343. doi: 10.1038/nature06735
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
D’Souza, G., Waschina, S., Pande, S., Bohl, K., Kaleta, C., and Kost, C. (2014). Less is more: selective advantages can explain the prevalent loss of biosynthetic genes in bacteria. Evolution 68, 2559–2570. doi: 10.1111/evo.12468
Elser, J., Okie, J., Lee, Z., and Souza, V. (2018). “The effect of nutrients and N: P ratio on microbial communities: testing the growth rate hypothesis and its extensions in Lagunita Pond (Churince),” in Ecosystem Ecology and Geochemistry of Cuatro Ciénegas, ed. F. Gracia-Oliva (Cham: Springer), 31–41.
Elser, J. J., Schampel, J. H., García-Pichel, F., Wade, B. D., Souza, V., Eguiarte, L., et al. (2005). Effects of phosphorus enrichment and grazing snails on modern stromatolitic microbial communities. Freshw. Biol. 50, 1808–1825. doi: 10.1111/j.1365-2427.2005.01451.x
Escalante, A. E., Caballero-Mellado, J., Martínez-Aguilar, L., Rodríguez-Verdugo, A., Gonzalez-Gonzalez, A., Toribio-Jimenez, J., et al. (2009). Pseudomonas cuatrocienegasensis sp. nov., isolated from an evaporating lagoon in the Cuatro Cienegas valley in Coahuila, Mexico. Int. J. Syst. Evol. Microbiol. 59, 1416–1420. doi: 10.1099/ijs.0.006189-0
Escalante, A. E., Eguiarte, L. E., Espinosa-Asuar, L., Forney, L. J., Noguez, A. M., and Souza Saldivar, V. (2008). Diversity of aquatic prokaryotic communities in the Cuatro Cienegas basin. FEMS Microbiol. Ecol. 65, 50–60. doi: 10.1111/j.1574-6941.2008.00496.x
Evans, S. E., and Wallenstein, M. D. (2014). Climate change alters ecological strategies of soil bacteria. Ecol. Lett. 17, 155–164. doi: 10.1111/ele.12206
Excoffier, L., Dupanloup, I., Huerta-Sánchez, E., Sousa, V. C., and Foll, M. (2013). Robust demographic inference from genomic and SNP data. PLoS Genet. 9:e1003905. doi: 10.1371/journal.pgen.1003905
Fierer, N., and Schimel, J. P. (2003). A proposed mechanism for the pulse in carbon dioxide production commonly observed following the rapid rewetting of a dry soil. Soil Sci. Soc. Am. J. 67, 798–805. doi: 10.2136/sssaj2003.7980
García-Ulloa, M., Escalante, A. E., Letelier, A. M., Eguiarte, L. E., and Souza, V. (2020). Fast eco-evolutionary changes in bacterial genomes after anthropogenic perturbation. bioRxiv doi: 10.1101/2020.03.13.990432
García-Ulloa, M., Ponce-Soto, G. Y., González-Valdez, A., González-Pedrajo, B., Díaz-Guerrero, M., Souza, V., et al. (2019). Two Pseudomonas aeruginosa clonal groups belonging to the PA14 clade are indigenous to the Churince system in Cuatro Ciénegas Coahuila, México. Environ. Microbiol. 21, 2964–2976. doi: 10.1111/1462-2920.14692
Giovannoni, S. J., Tripp, H. J., Givan, S., Podar, M., Vergin, K. L., Baptista, D., et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245. doi: 10.1126/science.1114057
Gómez-Lunar, Z., Hernández-González, I., Rodríguez-Torres, M. D., Souza, V., and Olmedo-Álvarez, G. (2016). Microevolution analysis of Bacillus coahuilensis unveils differences in phosphorus acquisition strategies and their regulation. Front. Microbiol. 7:58. doi: 10.3389/fmicb.2016.00058
Gomulkiewicz, R., and Holt, R. D. (1995). When does evolution by natural selection prevent extinction? Evolution 49, 201–207. doi: 10.1111/j.1558-5646.1995.tb05971.x
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grant, P. R., Grant, B. R., Huey, R. B., Johnson, M. T., Knoll, A. H., and Schmitt, J. (2017). Evolution caused by extreme events. Philos. Trans. R. Soc. B Biol. Sci. 372:20160146. doi: 10.1098/rstb.2016.0146
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Holt, R. D. (1990). The microevolutionary consequences of climate change. Trends Ecol. Evol. 5, 311–315. doi: 10.1016/0169-5347(90)90088-U
Hottes, A. K., Freddolino, P. L., Khare, A., Donnell, Z. N., Liu, J. C., and Tavazoie, S. (2013). Bacterial adaptation through loss of function. PLoS Genet. 9:e1003617. doi: 10.1371/journal.pgen.1003617
Jain, R., Rivera, M. C., Moore, J. E., and Lake, J. A. (2003). Horizontal gene transfer accelerates genome innovation and evolution. Mol. Biol. Evol. 20, 1598–1602. doi: 10.1093/molbev/msg154
Joshi, N. A., and Fass, J. N. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33)[Software].
Kieft, T. L., Soroker, E., and Firestone, M. K. (1987). Microbial biomass response to a rapid increase in water potential when dry soil is wetted. Soil Biol. Biochem. 19, 119–126. doi: 10.1016/0038-0717(87)90070-8
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Konikow, L. F., and Kendy, E. (2005). Groundwater depletion: a global problem. Hydrogeol. J. 13, 317–320. doi: 10.1007/s10040-004-0411-8
Krause, E., Wichels, A., Giménez, L., Lunau, M., Schilhabel, M. B., and Gerdts, G. (2012). Small changes in pH have direct effects on marine bacterial community composition: a microcosm approach. PLoS One 7:e47035. doi: 10.1371/journal.pone.0047035
Lee, Z. M., Steger, L., Corman, J. R., Neveu, M., Poret-Peterson, A. T., Souza, V., et al. (2015). Response of a stoichiometrically imbalanced ecosystem to manipulation of nutrient supplies and ratios. PLoS One 10:e0123949. doi: 10.1371/journal.pone.0123949
Lee, Z. M. P., Poret-Peterson, A. T., Siefert, J. L., Kaul, D., Moustafa, A., Allen, A. E., et al. (2017). Nutrient stoichiometry shapes microbial community structure in an evaporitic shallow pond. Front. Microbiol. 8:949. doi: 10.3389/fmicb.2017.00949
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lipa, P., and Janczarek, M. (2020). Phosphorylation systems in symbiotic nitrogen-fixing bacteria and their role in bacterial adaptation to various environmental stresses. PeerJ 8:e8466. doi: 10.7717/peerj.8466
Little, J. W., and Mount, D. W. (1982). The SOS regulatory system of Escherichia coli. Cell 29, 11–22. doi: 10.1016/0092-8674(82)90085-X
Liu, T., Tian, C. F., and Chen, W. X. (2015). Site-specific Ser/Thr/Tyr phosphoproteome of Sinorhizobium meliloti at stationary phase. PLoS One 10:e0139143. doi: 10.1371/journal.pone.0139143
Maruyama, T., and Fuerst, P. A. (1985). Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 111, 675–689.
McLendon, T., Hubbard, P. J., and Martin, D. W. (2008). Partitioning the use of precipitation-and groundwater-derived moisture by vegetation in an arid ecosystem in California. J. Arid Environ. 72, 986–1001. doi: 10.1016/j.jaridenv.2007.11.019
Minckley, W. L., and Cole, G. A. (1968). Preliminary limnologic information on waters of the Cuatro Cienegas Basin, Coahuila, Mexico. Southwest. Natural. 13, 421–431. doi: 10.2307/3668909
Moreno-Letelier, A., Olmedo-Alvarez, G., Eguiarte, L. E., and Souza, V. (2012). Divergence and phylogeny of Firmicutes from the Cuatro Ciénegas Basin, Mexico: a window to an ancient ocean. Astrobiology 12, 674–684. doi: 10.1089/ast.2011.0685
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. mBio 3:e00036-12. doi: 10.1128/mBio.00036-12
Nandi, T., Holden, M. T., Didelot, X., Mehershahi, K., Boddey, J. A., Beacham, I., et al. (2015). Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles. Genome Res. 25, 129–141. doi: 10.1101/gr.177543.114
Notley-McRobb, L., and Ferenci, T. (2000). Experimental analysis of molecular events during mutational periodic selections in bacterial evolution. Genetics 156, 1493–1501.
Oliver, A., Cantón, R., Campo, P., Baquero, F., and Blázquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253. doi: 10.1126/science.288.5469.1251
Orr, H. A., and Unckless, R. L. (2008). Population extinction and the genetics of adaptation. Am. Nat. 172, 160–169. doi: 10.1086/589460
Orr, H. A., and Unckless, R. L. (2014). The population genetics of evolutionary rescue. PLoS Genet. 10:e1004551. doi: 10.1371/journal.pgen.1004551
Pál, C., Papp, B., and Lercher, M. J. (2005). Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 37, 1372–1375. doi: 10.1038/ng1686
Papadopoulos, D., Schneider, D., Meier-Eiss, J., Arber, W., Lenski, R. E., and Blot, M. (1999). Genomic evolution during a 10,000-generation experiment with bacteria. Proc. Natl. Acad. Sci. U.S.A. 96, 3807–3812. doi: 10.1073/pnas.96.7.3807
Penterman, J., Singh, P. K., and Walker, G. C. (2014). Biological cost of pyocin production during the SOS response in Pseudomonas aeruginosa. J. Bacteriol. 196, 3351–3359. doi: 10.1128/JB.01889-14
Ponce-Soto, G. Y., Aguirre-von-Wobeser, E., Eguiarte, L. E., Elser, J. J., Lee, Z. M. P., and Souza, V. (2015). Enrichment experiment changes microbial interactions in an ultra-oligotrophic environment. Front. Microbiol. 6:246. doi: 10.3389/fmicb.2015.00246
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Ramsayer, J., Kaltz, O., and Hochberg, M. E. (2013). Evolutionary rescue in populations of Pseudomonas fluorescens across an antibiotic gradient. Evol. Appl. 6, 608–616. doi: 10.1111/eva.12046
Rodríguez-Verdugo, A., Souza, V., Eguiarte, L. E., and Escalante, A. E. (2012). Diversity across seasons of culturable Pseudomonas from a desiccation lagoon in Cuatro Cienegas, Mexico. Int. J. Microbiol. 2012:201389. doi: 10.1155/2012/201389
Romero, F., Sabater, S., Font, C., Balcázar, J. L., and Acuña, V. (2019). Desiccation events change the microbial response to gradients of wastewater effluent pollution. Water Res. 151, 371–380. doi: 10.1016/j.watres.2018.12.028
Schmidt, D., and Pool, J. (2002). The effect of population history on the distribution of the Tajima’s D statistic. J. Endocrinol. 33, 211–222.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Snipen, L., and Liland, K. H. (2015). micropan: an R-package for microbial pan-genomics. BMC Bioinform. 16:79. doi: 10.1186/s12859-015-0517-0
Souza, V., Espinosa-Asuar, L., Escalante, A. E., Eguiarte, L. E., Farmer, J., Forney, L., et al. (2006). An endangered oasis of aquatic microbial biodiversity in the Chihuahuan desert. Proc. Natl. Acad. Sci. U.S.A. 103, 6565–6570. doi: 10.1073/pnas.0601434103
Souza, V., Falcón, L. I., Elser, J. J., and Eguiarte, L. E. (2007). Protecting a window into the ancient Earth: towards a Precambrian park at Cuatro Cienegas, Mexico. Evol. Ecol. Res.
Souza, V., Moreno-Letelier, A., Travisano, M., Alcaraz, L. D., Olmedo, G., and Eguiarte, L. E. (2018). The lost world of Cuatro Cienegas Basin, a relictual bacterial niche in a desert oasis. eLife 7:e38278. doi: 10.7554/eLife.38278
Stock, J. B., Ninfa, A. J., and Stock, A. (1989). Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol. Mol. Biol. Rev. 53, 450–490.
Št′ovíček, A., Kim, M., Or, D., and Gillor, O. (2017). Microbial community response to hydration-desiccation cycles in desert soil. Sci. Rep. 7:45735. doi: 10.1038/srep45735
Sutton, M. D., Smith, B. T., Godoy, V. G., and Walker, G. C. (2000). The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34, 479–497. doi: 10.1146/annurev.genet.34.1.479
Taboada, B., Isa, P., Gutiérrez-Escolano, A. L., Del, Ángel, R. M., Ludert, J. E., et al. (2018). The geographic structure of viruses in the Cuatro Ciénegas Basin, a unique oasis in northern Mexico, reveals a highly diverse population on a small geographic scale. Appl. Environ. Microbiol. 84:e00465-18. doi: 10.1128/AEM.00465-18
Tello, A., Austin, B., and Telfer, T. C. (2012). Selective pressure of antibiotic pollution on bacteria of importance to public health. Environ. Health Perspect. 120, 1100–1106. doi: 10.1289/ehp.1104650
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Torres-Barceló, C., and Hochberg, M. E. (2016). Evolutionary rationale for phages as complements of antibiotics. Trends Microbiol. 24, 249–256. doi: 10.1016/j.tim.2015.12.011
Torsvik, V., Øvreås, L., and Thingstad, T. F. (2002). Prokaryotic diversity–magnitude, dynamics, and controlling factors. Science 296, 1064–1066. doi: 10.1126/science.1071698
Uecker, H., and Hermisson, J. (2016). The role of recombination in evolutionary rescue. Genetics 202, 721–732. doi: 10.1534/genetics.115.180299
Van Den Elzen, C. L., Kleynhans, E. J., and Otto, S. P. (2017). Asymmetric competition impacts evolutionary rescue in a changing environment. Proc. R. Soc. B Biol. Sci. 284:20170374. doi: 10.1098/rspb.2017.0374
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 1–10. doi: 10.1002/0471250953.bi1110s43
Vazquez-Rosas-Landa, M., Ponce-Soto, G. Y., Aguirre-Liguori, J. A., Thakur, S., Scheinvar, E., Barrera-Redondo, J., et al. (2020). Population genomics of Vibrionaceae isolated from an endangered oasis reveals local adaptation after an environmental perturbation. BMC Genomics 21:418. doi: 10.1186/s12864-020-06829-y
Vogwill, T., Phillips, R. L., Gifford, D. R., and MacLean, R. C. (2016). Divergent evolution peaks under intermediate population bottlenecks during bacterial experimental evolution. Proc. R. Soc. B Biol. Sci. 283:20160749. doi: 10.1098/rspb.2016.0749
Wada, Y., Van Beek, L. P., Van Kempen, C. M., Reckman, J. W., Vasak, S., and Bierkens, M. F. (2010). Global depletion of groundwater resources. Geophys. Res. Lett. 37:L20402. doi: 10.1029/2010GL044571
Werner, G. D., Strassmann, J. E., Ivens, A. B., Engelmoer, D. J., Verbruggen, E., Queller, D. C., et al. (2014). Evolution of microbial markets. Proc. Natl. Acad. Sci. U.S.A. 111, 1237–1244. doi: 10.1073/pnas.1315980111
Keywords: coalescence, Cuatro Cienegas, genomics, horizontal gene transfer, microbial ecology, population genetics, recombination, selection
Citation: García-Ulloa M II, Escalante AE, Moreno-Letelier A, Eguiarte LE and Souza V (2021) Evolutionary Rescue of an Environmental Pseudomonas otitidis in Response to Anthropogenic Perturbation. Front. Microbiol. 11:563885. doi: 10.3389/fmicb.2020.563885
Received: 20 May 2020; Accepted: 15 December 2020;
Published: 18 January 2021.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Heather Lyn Hendrickson, Massey University, New ZealandCopyright © 2021 García-Ulloa, Escalante, Moreno-Letelier, Eguiarte and Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Souza, c291emFAdW5hbS5teA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.