
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 August 2020
Sec. Microbial Physiology and Metabolism
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.02000
 Po-Chuen Hsu1†
Po-Chuen Hsu1† Chien-Sheng Chen2†Shuying Wang3,4,5,6†
Chien-Sheng Chen2†Shuying Wang3,4,5,6† Masayuki Hashimoto1,4,5
Masayuki Hashimoto1,4,5 Wen-Chun Huang1,4
Wen-Chun Huang1,4 Ching-Hao Teng1,4,5*
Ching-Hao Teng1,4,5*Microbial proteases play pivotal roles in many aspects of bacterial physiological processes. Because a protease exerts its biological function by proteolytically regulating its substrates, the identification and characterization of the physiological substrates of a protease advance our understanding of the biological roles of the protease. Prc (also named Tsp) is an Escherichia coli periplasmic protease thought to be indispensable for E. coli to survive under low osmolality at 42°C. The accumulation of the Prc substrate MepS due to Prc deficiency contributes to the conditional growth defect. Because preventing MepS accumulation only partially restored the growth of Prc-deficient E. coli, we hypothesized that other unidentified Prc substrates intracellularly accumulate due to Prc deficiency and contribute to the conditional growth defect. To identify previously undiscovered substrates, 85 E. coli proteins able to physically interact with Prc were identified using E. coli proteome arrays. Ten proteins were shown to be cleavable by Prc in vitro. Among these candidates, MltG was able to interact with Prc in E. coli. Prc regulated the intracellular level of MltG, indicating that MltG is a physiological substrate of Prc. Prc deficiency induced the accumulation of MltG in the bacteria. Blocking MltG accumulation by deleting mltG partially restored the growth of Prc-deficient E. coli. In addition, Prc-deficient E. coli with blocked MltG and MepS expression exhibited higher growth levels than those with only the MltG or MepS expression blocked under low osmolality at 42°C, suggesting that these accumulated substrates additively contributed to the conditional growth defect. MltG is a lytic transglycosylase involved in the biogenesis of peptidoglycan (PG). In addition to MltG, the previously identified physiological Prc substrates MepS and PBP3 are involved in PG biogenesis, suggesting a potential role of Prc in regulating PG biogenesis.
Microbial intracellular proteases govern vital physiological processes through diverse actions, such as protein quality control and the promotion of turnover, maturation and modification, which are crucial for proper function or localization of bacterial proteins (Weichart et al., 2003; Clausen et al., 2011; Dalbey et al., 2012). A protease usually exerts its physiological function through the proteolytic regulation of its substrates in an organism. Thus, identifying and characterizing substrates of a protease will provide a fundamental understanding of the biological roles of the protease. Prc, an Escherichia coli periplasmic protease (also known as Tsp), is physiologically important in E. coli as indicated by Prc deficiency completely inhibiting bacterial growth at 42°C in a low-salt medium which is commonly referred to as a condition of combined high temperature and low osmolality (Hara et al., 1991, 1996; Bass et al., 1996; Tadokoro et al., 2004). To date, whether there are other unidentified Prc substrates that are dysregulated in Prc-deficient E. coli contributing to the conditional growth defect has not been fully elucidated.
A 76-kDa protein, Prc is composed of an N-terminal domain, a C-terminal catalytic domain, and a PDZ domain (Beebe et al., 2000). The active site residues of Prc, Ser-430, Asp-441 and Lys-455, are located near the C-terminus and execute the proteolytic activity of Prc. The N-terminal domain of Prc is required for its ancillary function in directly or indirectly facilitating catalytic activity (Keiler and Sauer, 1995; Beebe et al., 2000). The PDZ domain is essential for substrate recognition by recognizing and binding to the nonpolar or hydrophobic C-terminal residues of substrate proteins (Beebe et al., 2000). It has been shown that a number of natural or recombinant proteins can be cleaved by Prc in vitro and in vivo. Prc can degrade mammalian β-casein and a recombinant variant of the N-terminal λ-repressor (residues 1-102), with its C-terminal 5 residues replaced with nonpolar amino acids (Silber et al., 1992; Spiers et al., 2002). In addition, Prc can recognize and degrade proteins tagged by the small stable RNA A (ssrA)-encoded peptide in vitro, suggesting that this protease may be involved in protein quality control in E. coli (Karzai et al., 2000). Moreover, penicillin-binding protein 3 (PBP 3) and MepS (also named Spr) have been identified as physiological substrates of Prc in E. coli (Hara et al., 1989, 1991; Nagasawa et al., 1989; Singh et al., 2015; Chueh et al., 2019). PBP3 is a peptidoglycan (PG) transpeptidase involved in septal PG synthesis during cell division (Hara et al., 1991), while MepS is a PG endopeptidase involved in breaking the peptide linkage between the glycan strands of a PG (Singh et al., 2012). However, the fates of these natural substrates are different; MepS is fully degraded after being proteolyzed by Prc, while only 11 residues in the C-terminal of PBP3 are cleaved (Hara et al., 1989; Nagasawa et al., 1989).
The intracellular accumulation of MepS has been shown to contribute to the conditional growth defect of Prc-deficient E. coli (Singh et al., 2015). However, in this study, we found that blocking MepS accumulation only partially restored the growth of Prc-deficient E. coli under low osmolality at 42°C, suggesting that previously unidentified Prc substrates, in addition to MepS, may play roles in the growth defect of prc mutants.
To identify new Prc substrates involved in the conditional growth defect of prc mutants, an E. coli proteome microarray (Chen et al., 2008) was employed to screen for proteins able to physically interact with Prc. The E. coli proteome array is based on the K12 open reading frame (ORF) plasmid library constructed by Kitagawa et al. (2005), which enabled us to efficiently screen approximately 99% of the E. coli K12 proteins (Chen et al., 2008). The candidate proteins selected based on the screening were subjected to in vitro Prc proteolytic assays, gene mutation analyses, and in vivo functional assays. Finally, we identified a new Prc natural substrate, MltG, whose intracellular accumulation in Prc-deficient E. coli contributed to the conditional growth defect.
Previous studies have indicated that the growth of Prc-deficient E. coli strains is completely inhibited at 42°C in salt-free 1/2 LB medium (see Materials and Methods), referred to as low osmolality medium (Hara et al., 1991, 1996; Bass et al., 1996; Tadokoro et al., 2004). However, no direct evidence showed that the growth defect was due to the low osmolality of the medium. To confirm that low osmolality contributed to the growth defect of Prc-deficient E. coli at 42°C, we measured the growth of the wild-type E. coli K12 strain BW25113 (WT-BW25113) and its prc mutant (Δprc-BW25113) in salt-free 1/2 LB medium with the osmolality adjusted to the level of the regular LB medium by using different solutes. The osmolality of the salt-free 1/2 LB and normal LB medium was approximately 46 mOsm/kg and 390 mOsm/kg, respectively. The strains were cultured in normal LB medium and salt-free 1/2 LB medium in which the osmolality was adjusted to approximately 390 mOsm/kg using NaCl (172.5 mM), KCl (172.5 mM), MgCl2 (114.1 mM), LiCl (172.7 mM), sorbitol (300 mM), and glucose (344 mM). While Δprc-BW25113 did not show significant growth in salt-free 1/2 LB medium without osmolality adjustment at 42°C (Figure 1A), increased osmolality in salt-free 1/2 LB medium enabled the growth of Δprc-BW25113 at high temperature (Figures 1B–G). These findings support the commonly accepted notion that osmolality is a major factor contributing to the nongrowing phenotype of Prc-deficient E. coli at 42°C in the salt-free 1/2 LB.
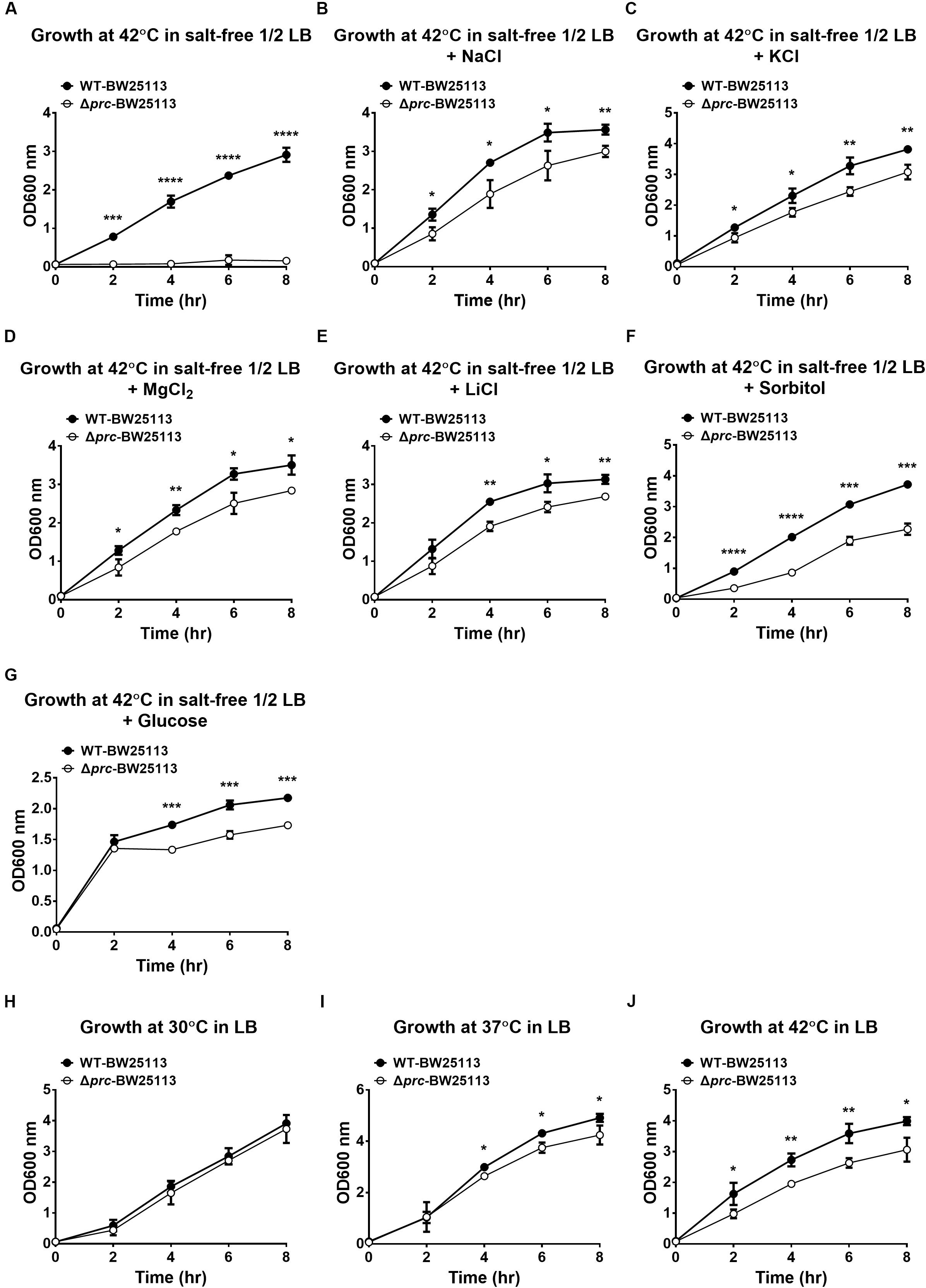
Figure 1. The growth of WT-BW25113 and Δprc-BW25113 in media with or without osmolality adjustment. (A–G) The growth of the bacteria at 42°C in salt-free 1/2 LB medium without osmolality adjustment (A), and with osmolality adjustment using NaCl (B), KCl (C), MgCl2 (D), LiCl (E), sorbitol (F), and glucose (G), respectively. (H–I) The growth of the bacteria in normal LB medium at 30°C (H), 37°C (I), and 42°C (J). The growth of the strains was determined by measuring the values of O.D.600 at the indicated time points using a spectrophotometer. Data are shown as the mean ± SD of three independent experiments. Asterisks denote significant differences between the O.D.600 values of WT-BW25113 and Δprc-BW25113 at the indicated time points (*, P-values of ≤ 0.05; **, P-values of ≤ 0.01; ***, P-values of ≤ 0.001; ****, P-values of ≤ 0.0001).
In addition, Δprc-BW25113 and WT-BW25113 exhibited similar growth in LB medium at 30°C (Figure 1H), suggesting Prc-deficiency does not interfere with E. coli growth in LB medium at 30°C, which is consistent with the finding of a previous study (Hara et al., 1991). However, the prc mutant showed a significant lower level of growth than the wild-type strain in LB medium at 37°C and 42°C (Figures 1I,J). These results consistently demonstrated that temperature is also a factor that interferes with the growth of Prc-deficient E. coli.
Although the growth defect of E. coli prc mutants under combined conditions of high temperature (42°C) and low osmolality can be alleviated by blocking intracellular MepS accumulation (Hara et al., 1996; Singh et al., 2015), it remains unclear whether MepS accumulation is the only factor contributing to the defect. To clarify this issue, we measured and compared the growth of the prc single mutant (Δprc-BW25113), the mepS and prc double mutant (ΔmepSΔprc-BW25113), and WT-BW25113. As shown in Figure 2A, the three strains showed similar growth in LB medium (390 mOsm/kg) at 30°C, which was the condition in which Prc deficiency did not affect bacterial growth (Figure 1H). When cultured in salt-free 1/2 LB medium (46 mOsm/kg) at 42°C, Δprc-BW25113 showed no growth. The ΔmepSΔprc-BW25113 strain, in which MepS accumulation due to Prc deficiency is blocked, exhibited a growth phenotype, as described previously (Hara et al., 1996; Singh et al., 2015). However, the growth of ΔmepSΔprc-BW25113 was significantly lower than that of WT-BW25113 (Figure 2B). These results suggest that relief of MepS accumulation may not fully rescue the conditional growth defect caused by prc deletion, i.e., the growth was not recovered to the level of the wild-type strain. It is likely that the conditional growth defect caused by the prc deletion was the result of the dysregulation of more than one Prc substrate in the prc mutant. Thus, relieving the accumulation of a single substrate, MepS, can only recover the growth of the Prc-deficient strain partially but not restore the growth to the level of the WT strain. Accordingly, we hypothesized that there are possibly other unidentified Prc substrate(s) whose intracellular accumulation in the prc mutant also contributes to the above stated growth defect. This hypothesis led us to search for a novel Prc substrate(s) contributing to the conditional growth defect of the prc mutant.
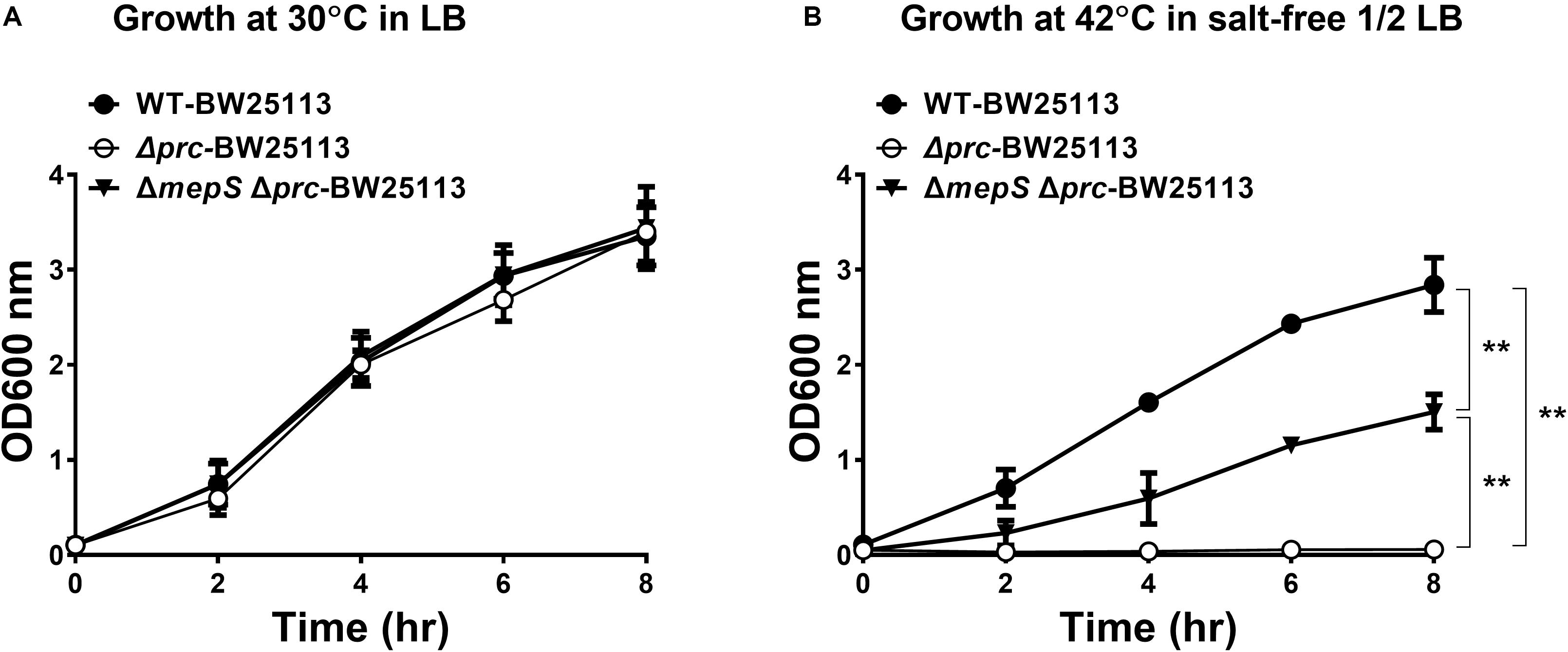
Figure 2. The growth of WT-BW25113, Δprc-BW25113, and ΔmepSΔprc-BW25113. (A) Growth in LB medium at 30°C. (B) Growth in salt-free 1/2 LB medium at 42°C. Data are shown as the mean ± SD of three independent experiments. Asterisks denote significant differences between the O.D.600 values of the indicated strains after 8 h of incubation (**, P-values of ≤ 0.01).
Physical interaction between a protease and its substrates is necessary for the protease to exert its proteolytic function on the substrate proteins. Thus, to identify novel substrates of Prc, we first screened for E. coli proteins that can physically interact with the protease. The availability of the E. coli K12 proteome arrays enabled us to efficiently screen 4,256 proteins to find those that interact with Prc (Chen et al., 2008). In the proteome array-based screen, fluorescently labeled Prc variants served as probes of the interactions with the proteins on the array. A potential challenge for such screening is that the substrate proteins physically associated with Prc may be degraded by the protease, leading to the loss of the fluorescence signals that would have reflected the protease-substrate interaction on the array. Thus, we generated a catalytically inactive Prc mutant (K455A-Prc) for the screening assay. The K455A-Prc protein has no catalytic activity because of the alanine substitution at Lys455 but still possesses a structure and substrate-binding ability similar to the wild-type protein (Keiler and Sauer, 1995). In addition, we generated the ΔPDZ-K455A-Prc recombinant protein, which is K455A-Prc with the deletion of the PDZ domain leading to its loss of substrate-binding ability. Fluorescently labeled Prc, K455A-Prc, and ΔPDZ-K455A-Prc were incubated with the proteome arrays. The substrates of Prc were expected to interact directly with Prc or K455A-Prc but not ΔPDZ-K455A-Prc. Using this approach, we screened for proteins that exhibited substantial signals of interaction with Prc or K455A-Prc, selecting those with signals that were significantly stronger than those with ΔPDZ-K455A-Prc (see section “MATERIALS AND METHODS”). Based on the criteria, 85 E. coli proteins were selected (Supplementary Table S1). Representative chip assay images of the proteins able to interact with Prc and/or K455A-Prc are shown in Figure 3. (Note: representative images of the proteins were selected and shown here because the proteins were shown to be cleavable in vitro in further Prc proteolytic assays).
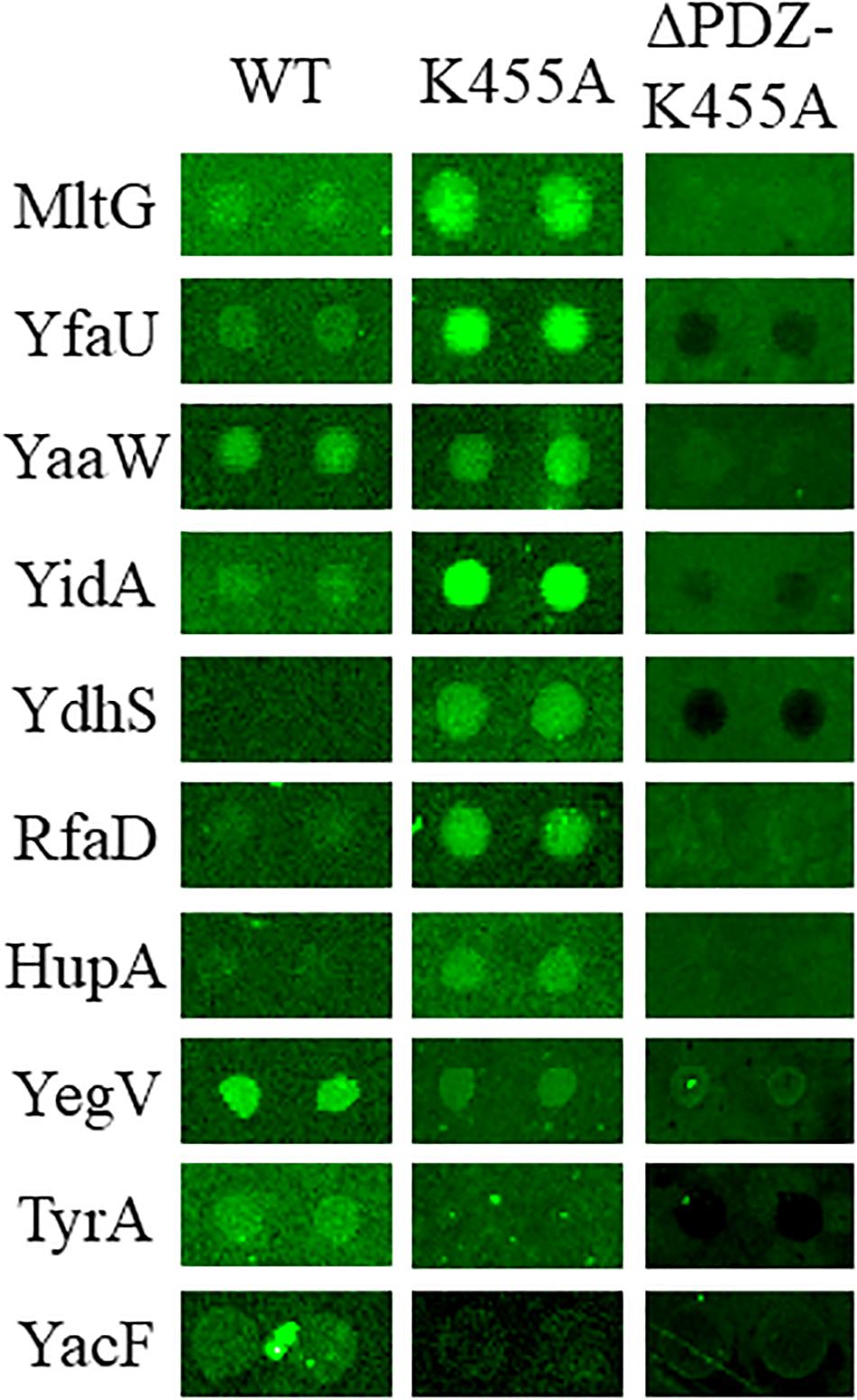
Figure 3. Representative images of the E. coli proteome array assays with the Dylight™ 549 fluorescence-labeled Prc protein variants Prc (WT), K455A-Prc (K455A), and ΔPDZ-K455A-Prc (ΔPDZ-K455A). Among these representative proteins, MltG, YfaU, YaaW, and YidA showed stronger interaction signals with Prc and K455A-Prc than with ΔPDZ-K455A-Prc. YdhS, RfaD, and HupA showed stronger interaction signals with Prc than with ΔPDZ-K455A-Prc. YegV, TyrA, and YacF showed stronger interaction signals with K455A-Prc than with ΔPDZ-K455A-Prc. The array analyses were performed in triplicate. The images of these representative protein spots were chosen from one of the triplicate experiments.
To further scrutinize whether the 85 E. coli proteins could be cleaved by Prc, we performed in vitro Prc proteolytic assays (Wang et al., 2012). The 85 N-terminally 6xHis-tagged recombinant protein candidates were overexpressed from the corresponding ORF clones in the ASKA library (Kitagawa et al., 2005). They were purified by the 96-well plate-based method developed previously (Chen et al., 2008), which is a high-throughput protocol allowing the simultaneous purification of 85 proteins in small amounts. Then, these recombinant proteins were examined by Prc proteolytic assays, with β-casein, a known Prc-cleavable protein, as a proteolytic positive control. Among these candidates, 10 of the E. coli proteins, MltG, YfaU, YaaW, YdhS, YidA, RfaD, HupA, YegV, TyrA, and YacF (Table 1), were shown to be fully or partially cleaved by the Prc protease in vitro (Figure 4), while the others were not affected by the protease. These in vitro cleavable proteins were potential Prc substrates in E. coli cells.
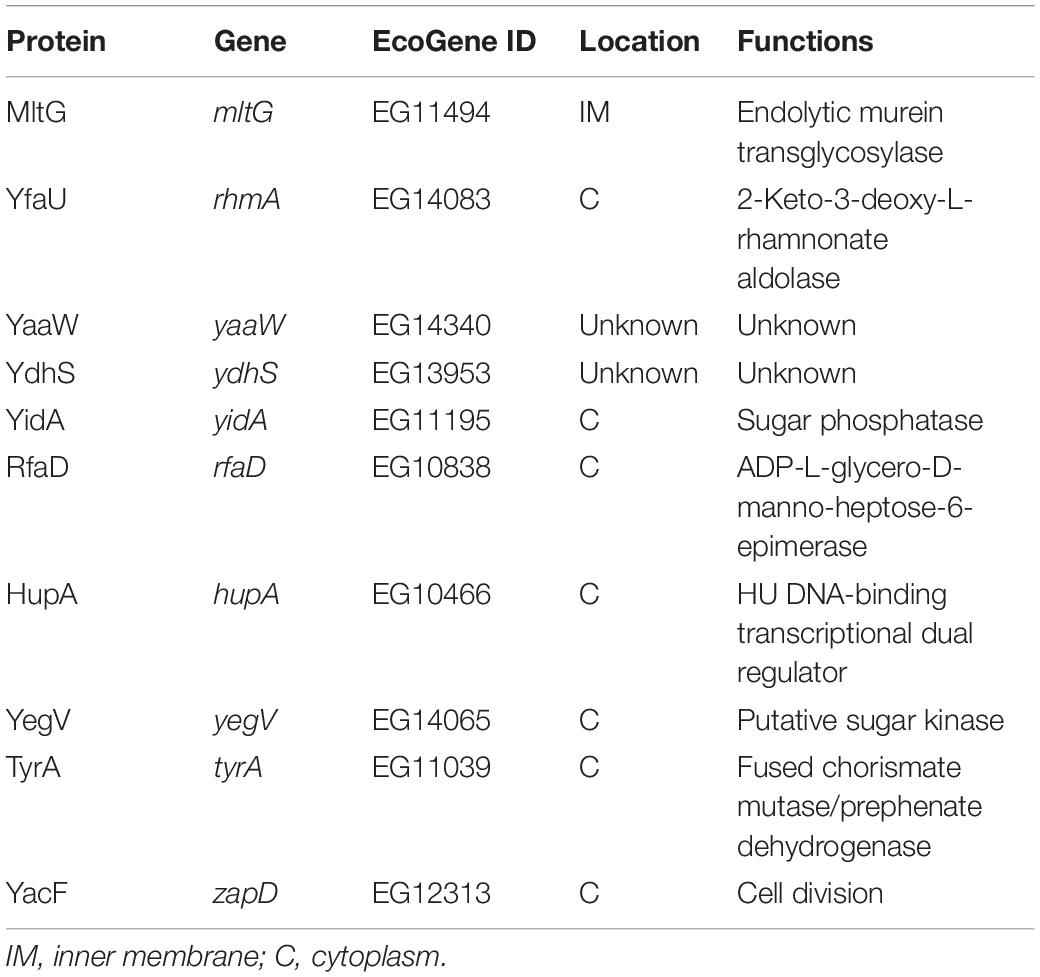
Table 1. The Prc-cleavable proteins.
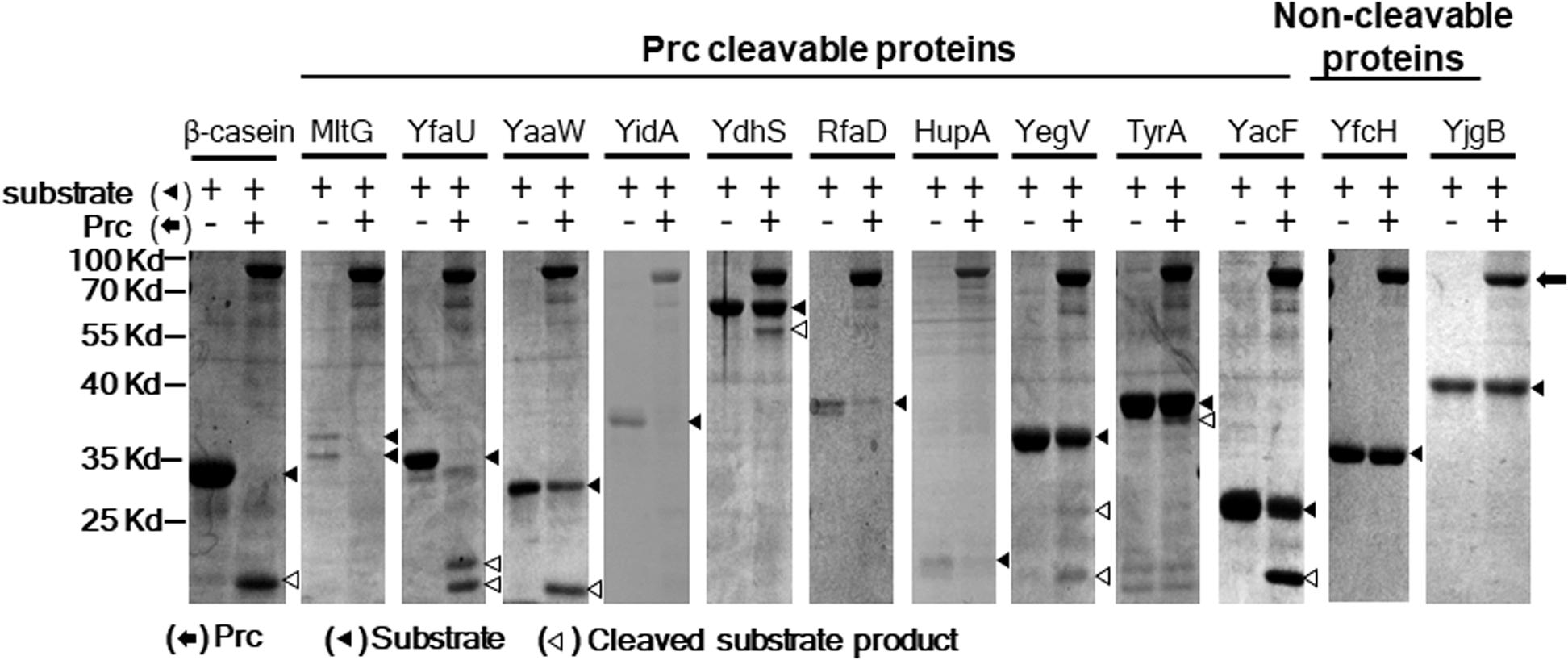
Figure 4. Prc proteolytic assays with E. coli proteins screened in the proteome assays. After incubation with or without Prc for 2.5 h, these proteins were separated by SDS–PAGE, and the protein bands on the gels were visualized by Coomassie blue staining. Among these proteins, 10 (MltG, YfaU, YaaW, YidA, YdhS, RfaD, HupA, YegV, TyrA, and YacF) were fully or partially cleaved by Prc, while the other proteins were not cleaved. YfcH and YjgB were randomly selected as representatives of the proteins that were not cleavable by Prc. β-casein, a protein known to be cleavable by Prc, served as the proteolytic positive control. Representative results of at least two independent experiments using at least two independent protein preparations are presented.
The 10 proteins shown to be cleavable by Prc (Figure 4) were further investigated for their involvement in the conditional growth defect of the E. coli prc mutant. Based on our hypothesis that the intracellular accumulation of Prc substrates, in addition to MepS, may also contribute to the defective growth of the prc mutant, deletion of the genes encoding the corresponding substrates may block their intracellular accumulation in the prc mutant and thus suppress the defective phenotype. Accordingly, the deletion mutations of the genes encoding these Prc-cleavable proteins were introduced into Δprc-BW25113. Then, the double mutants were subjected to screening for those able to grow in salt-free 1/2 LB medium at 42°C (Figures 5A–C). Among these mutants, only the ΔmltGΔprc double mutant (ΔmltGΔprc-BW25113) showed the ability to grow under this condition, while the other double mutants, similar to Δprc-BW25113, showed no growth. Consistent with these findings, trans-complementation with a copy of mltG in the lacZ chromosomal locus of ΔmltGΔprc-BW25113 (the resulting strain was lacZ::mltG-ΔmltGΔprc-BW25113) resulted in the same growth defect as that observed for Δprc-BW25113. However, the mltG single mutant (ΔmltG-BW25113) showed no significant growth defect (Figure 5D). In addition, WT-BW25113, Δprc-BW25113, ΔmltG-BW25113, ΔmltGΔprc-BW25113, and lacZ::mltG-ΔmltGΔprc-BW2511 showed similar growth in normal LB medium at 30°C (Figure 5E). These results indicated that blocking MltG expression suppresses the conditional growth defect caused by the prc deletion and suggest that MltG was a potential substrate responsible for the conditional growth defect of the prc mutant. Thus, we then focused on MltG in the following investigations.
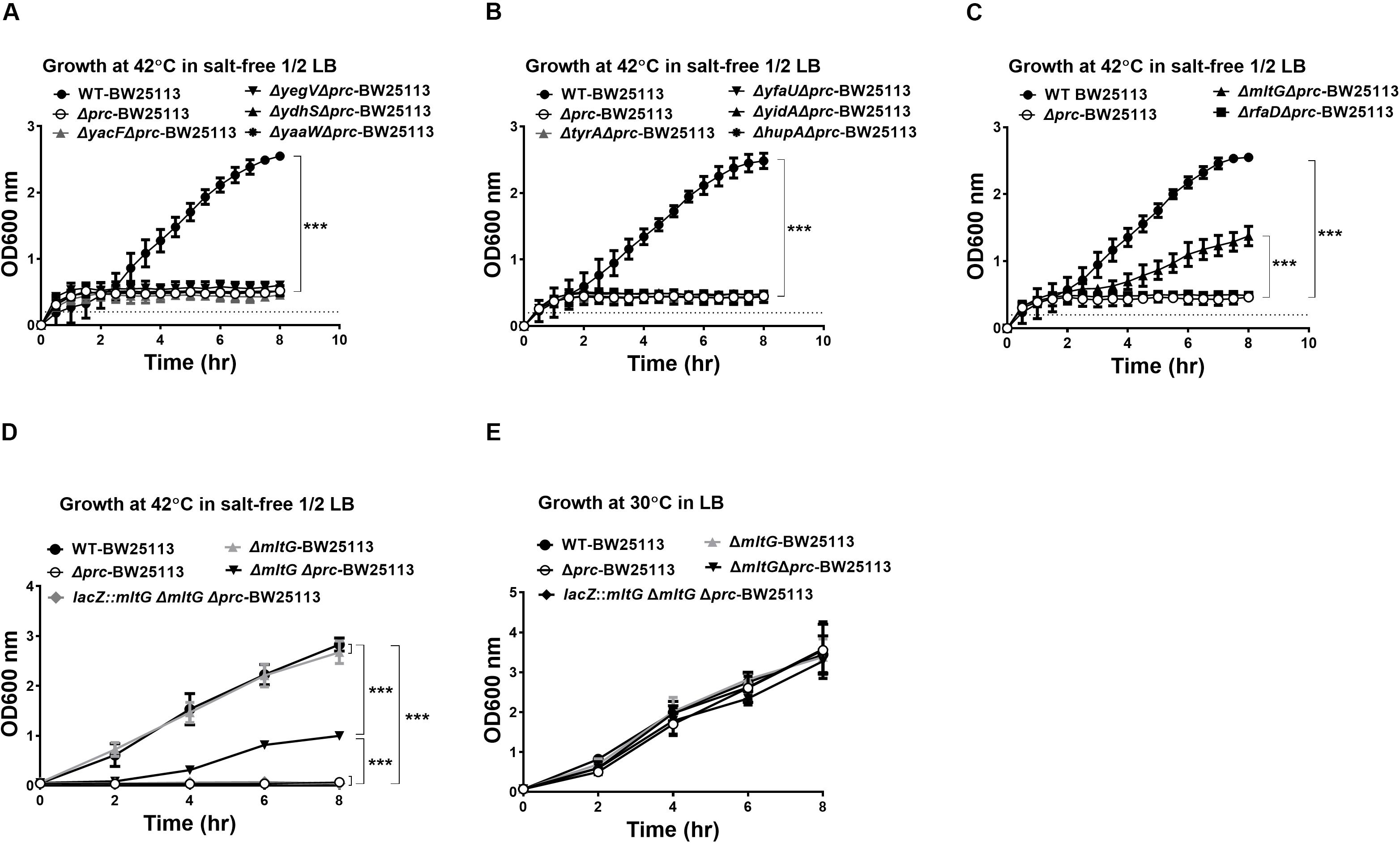
Figure 5. Screening for potential Prc substrates involved in the growth defect of Prc-deficient E. coli under low osmolality at high temperature. (A–C) The growth of the double mutants of the potential substrates and Prc in salt-free 1/2 LB medium at 42°C. WT-BW25113 and Δprc-BW25113 served as the growth positive and negative controls, respectively. The growth of the strains was measured by a noninvasive turbidity meter, which can automatically assess multiple samples simultaneously. (D,E) The growth of WT-BW25113, Δprc-BW25113, ΔmltG-BW25113, ΔmltGΔprc-BW25113, and lacZ::mltG-ΔmltGΔprc-BW25113 in salt-free 1/2 LB medium at 42°C (D) and in normal LB medium at 30°C (E). The growth of the strains was determined by measuring the values of O.D.600 at the indicated time points using a spectrophotometer. Data are shown as the mean ± SD of three independent experiments. Asterisks denote significant differences between the O.D.600 values of the indicated strains after 8 h of incubation (***, P-values of ≤ 0.001).
MltG is a protein associated with the inner membrane. The mltG gene encodes the pro-MltG protein, which contains an N-terminal signal peptide (Yunck et al., 2016). The signal peptide is removed after the MltG protein is translocated to the inner membrane. However, the MltG recombinant protein used in the Prc proteolytic assays (Figure 4) still contained the signal peptide. It cannot be excluded that the presence of the signal peptide makes the recombinant protein cleavable by Prc in vitro. Additionally, because the recombinant protein was purified with a high-throughput method, the protein preparation may have contained other bacterial factors. To further confirm whether the pure MltG protein can be cleaved by Prc in vitro, we cloned and purified N-terminal signal sequence-truncated MltG recombinant proteins with a C-terminal or N-terminal 6xHis tag. To obtain pure recombinant proteins, we used Ni-affinity and subsequent size exclusion chromatography. As shown in Figures 6A,B, the recombinant MltG proteins could be cleaved by Prc regardless of the location of the 6xHis tag but not by K455A-Prc. This result demonstrates that MltG can be cleaved by Prc in vitro although approximately 12 h were required for the protease to cleave one-half of the purified MltG proteins. In addition, the column-purified E. coli proteins, YfcH and YjgB, whose high throughput preparations were shown to be noncleavable by Prc (Figure 4), showed no degradation after incubation with Prc for up to 24 h (data not shown), suggesting that Prc degrades MltG specifically under this in vitro condition.
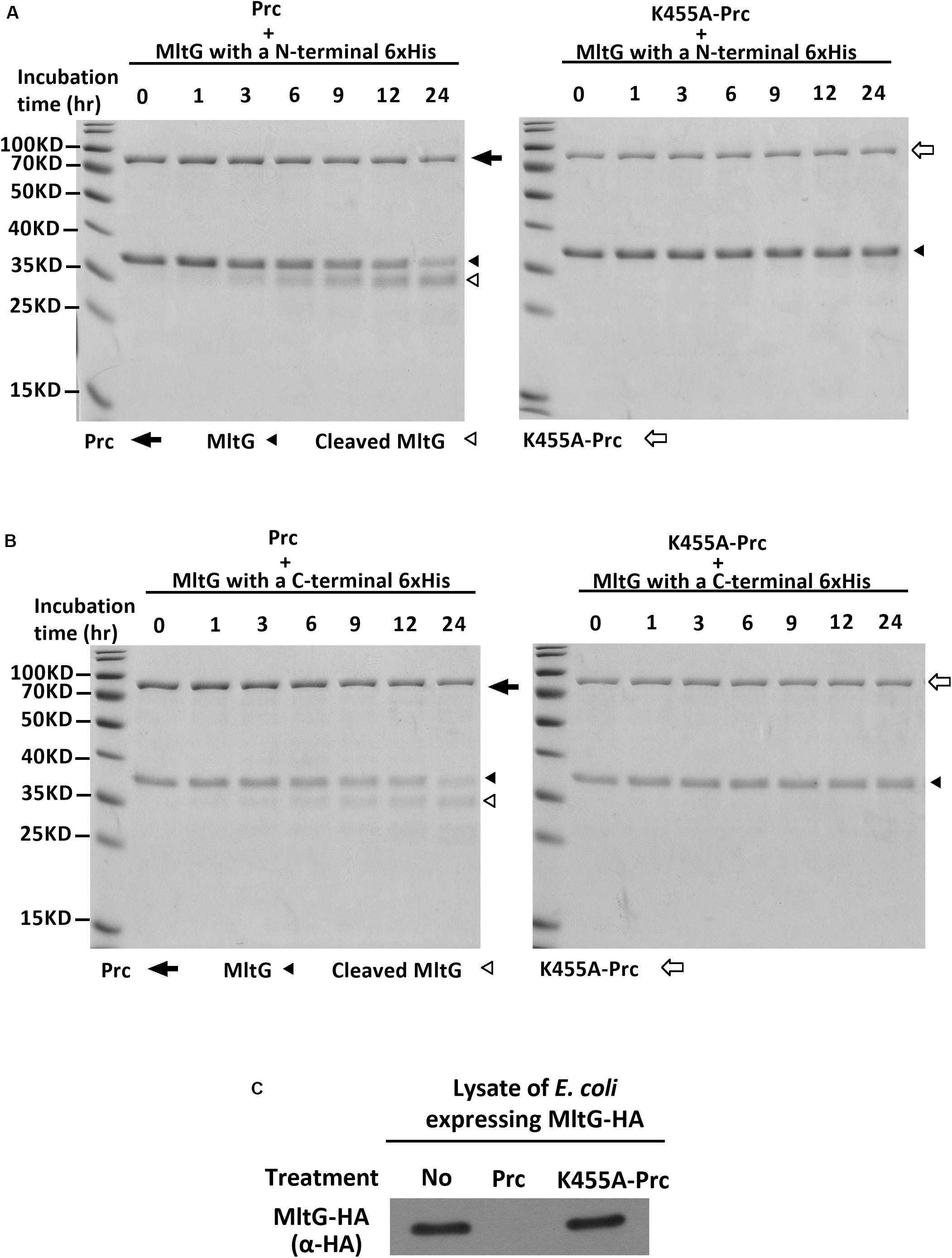
Figure 6. In vitro Prc-mediated proteolysis of MltG. (A) Proteolytic assays of N-terminally 6xHis–tagged MltG with Prc or K455A-Prc. (B) Proteolytic assays of C-terminally 6xHis–tagged MltG with Prc or K455A-Prc. For panels A and B, the Prc-MltG mixtures at the indicated time points post-incubation were subjected to SDS–PAGE and Coomassie blue staining. (C) The lysates of ΔmltGΔprc-BW25113/pBAD24-MltG-HA (Table 2), which expressed C-terminally HA-tagged MltG but not Prc, were subjected to proteolytic analyses with purified Prc or K455A-Prc. The levels of MltG in the lysates at 3 h post-incubation were determined by Western blotting with anti-HA antibodies. For (A–C), representative results of two independent experiments using two independently purified proteins are presented.
We investigated whether MltG could be more efficiently cleaved by Prc in the presence of other bacterial factors. Lysates of the E. coli strain expressing MltG but not Prc (ΔmltGΔprc-BW25113/pBAD24-MltG-HA) were subjected to proteolytic analysis with purified Prc or K455A-Prc. As shown in Figure 6C, MltG was fully degraded by Prc but not K455A-Prc after 3 h of incubation. This result may suggest presence of factors in vivo increases the efficiency of MltG degradation by Prc, which may mimic the intracellular condition of E. coli cells.
We further investigated whether MltG can be cleaved by Prc within bacterial cells. First, to examine whether Prc interacts with MltG in vivo, we performed coimmunoprecipitation assays. An MltG protein with a C-terminal HA tag was coexpressed with K455A-Prc, which contained a C-terminal 6xHis-tag (Spiers et al., 2002), in ΔmltG-BW25113. To precipitate K455A-Prc and the HA-tagged MltG, the lysate of the bacteria was incubated with anti-His and anti-HA antibodies, respectively, followed by antibody-antigen complex precipitation using protein G-coated magnetic beads. Immunoblot analysis of the K455A-Prc and MltG fractions pulled down with anti-HA and anti-His antibodies showed that MltG and K455A-Prc coprecipitated, respectively (Figures 7A,B). These results suggest that MltG interacts with the Prc protease in E. coli cells.
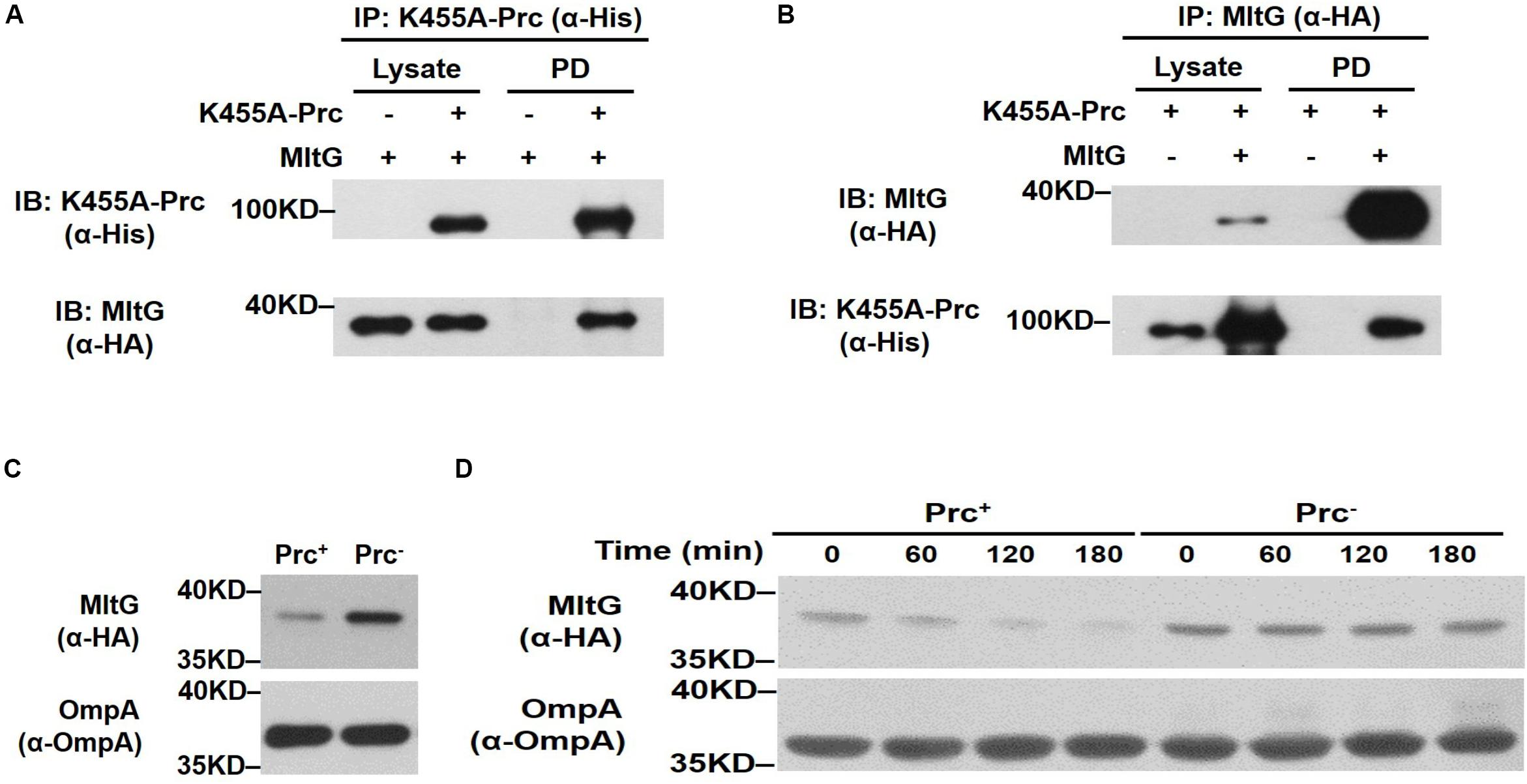
Figure 7. Effect of Prc on the intracellular level of MltG in E. coli. (A,B) Validation of the in vivo interaction between Prc and MltG. ΔmltG-BW25113 harboring the plasmids pTR163 (Spiers et al., 2002) and pACYC184-MltG-HA were used to coexpress K455A-Prc and HA-tagged MltG. K455A-Prc was immunoprecipitated (IP) with anti-His antibodies (A), while the MltG protein was precipitated with anti-HA antibodies (B). The pull-down fractions (PD) were analyzed by immunoblotting (IB) with anti-His or anti-HA antibodies. (C) The levels of MltG in E. coli strains with and without Prc. The levels of MltG in BW25113-MltG-HA (Prc+) (Table 2) and Δprc-BW25113-MltG-HA (Prc–) were determined by Western blot analysis of the log-phase bacterial cultures with anti-HA antibodies. (D) In vivo degradation assays of MltG in the bacterial strains with or without Prc. The MltG levels in BW25113-MltG-HA (Prc+) and Δprc-BW25113-MltG-HA (Prc–) were determined at the indicated time points after spectinomycin treatment. For (A,B), representative results of three independent experiments are presented. For panels (C,D), representative results of two independent experiments are presented. The outer membrane protein OmpA was used as a protein-loading control.
Second, to determine whether the presence of Prc affects the intracellular level of MltG in E. coli, the E. coli strains BW25113-MltG-HA and Δprc-BW25113-MltG-HA (Table 2) were constructed by fusing the HA tag sequences to the 3′ ends of the chromosomal mltG genes in WT-BW25113 and Δprc-BW25113, allowing the expression of HA-tagged MltG. The MltG levels in the E. coli strains in log phase with and without the prc gene were determined by Western blot analysis with anti-HA antibodies. As shown in Figure 7C, the MltG level in Δprc-BW25113-MltG-HA was significantly higher than that in BW25113-MltG-HA, suggesting that Prc regulates the intracellular level of MltG and that inactivation of prc causes the accumulation of MltG.

Table 2. Bacterial strains used in this study.
Finally, to further investigate the basis of the Prc-mediated regulation of MltG in E. coli, a pulse–chase experiment (in vivo degradation assay) was performed after the inhibition of protein synthesis by treatment with the ribosome inhibitor spectinomycin. As shown in Figure 7D, after blocking protein synthesis, the MltG level decreased significantly in BW25113-MltG-HA within 60 min, while this protein was maintained at a similar level in Δprc-BW25113-MltG-HA. This result demonstrated that the increased MltG level in the Prc-deficient strain (Figure 7C) is a consequence of enhanced posttranslational stability, suggesting that Prc regulates the intracellular level of MltG through its proteolytic function.
Taken together, these findings, in conjunction with the observation that the deletion of mltG can relieve the growth defect of the E. coli prc mutant at high temperature and low osmolality (Figures 5C,D), strongly suggest that the accumulation of MltG contributes to the growth defect in Prc-deficient E. coli.
We further investigated whether increasing the intracellular level of MltG without the loss of Prc function can interfere with E. coli growth in salt-free 1/2 LB medium at 42°C. To raise the intracellular MltG level, the plasmid pACYC184-MltG-HA was introduced into BW25113-MltG-HA (BW25113-MltG-HA/pACYC184-MltG-HA) to allow overexpression of MltG. BW25 113-MltG-HA/pACYC184-MltG-HA exhibited a significantly low level of grow than did the BW25113-MltG-HA strain harboring the empty plasmid vector (BW25113-MltG-HA/pACYC184) at 42°C in salt-free 1/2 LB medium, while the strains exhibited a similar level of growth at 30°C in normal LB medium (Figures 8A,B). Additionally, as expected, BW25113-MltG-HA/pACYC184-MltG-HA showed higher levels of MltG than BW25113-MltG-HA/pACYC184 in both 42°C/salt-free 1/2 LB medium and 30°C/LB medium conditions (Figure 8C). This result suggests that increasing the intracellular level of MltG alone can interfere with bacterial growth under low osmolality at high temperature, further supporting the notion that the increased MltG level in Prc-deficient E. coli contributes to the conditional growth defect of the bacteria.
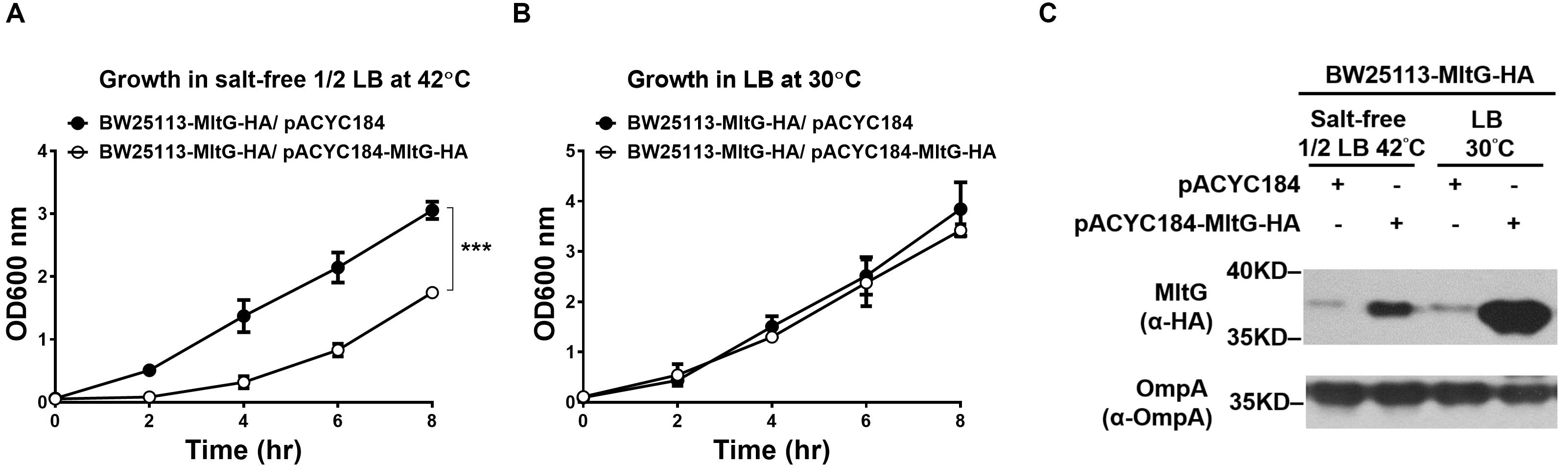
Figure 8. The effect of the intracellular accumulation of MltG on the growth of E. coli. (A) The growth of BW25113-MltG-HA harboring pACYC184-MltG-HA or the empty vector pACYC184 in salt-free 1/2 LB medium at 42°C. (B) The growth of the strains in normal LB medium at 30°C. For (A) and (B), the data are shown as the mean ± SD of three independent experiments. Asterisks denote significant differences between the O.D.600 values of the indicated strains after 8 h of incubation (***, P-values of ≤ 0.001). (C) The MltG levels in BW25113-MltG-HA with or without MltG overexpression under 42°C/salt-free 1/2 LB medium and 30°C/LB medium conditions. The levels of MltG were determined by Western blot analysis of the log-phase bacteria (2 h of incubation of 100-fold-diluted overnight culture) with anti-HA antibodies. The outer membrane protein OmpA was used as a protein-loading control. Representative results of two independent experiments are presented.
To clarify whether the accumulation of MltG affects the accumulation of MepS in the prc mutant, we compared the MepS levels in E. coli strains without mltG to those in the strains with mltG, in the backgrounds with and without prc. As shown in Figure 9, deletion of mltG did not aggravate or alleviate the levels of MepS in either the strains with Prc+ or Prc– backgrounds. Additionally, consistent with a previous study (Singh et al., 2015), the MepS levels in the strains without prc were significantly higher than those in the strains with prc. These results demonstrate that relieving MltG accumulation through the deletion of mltG does not affect the MepS level in the prc mutant. Thus, it is reasonable to conclude that the contribution of MltG accumulation to the conditional growth defect of the prc mutant does not work through affecting the MepS level.
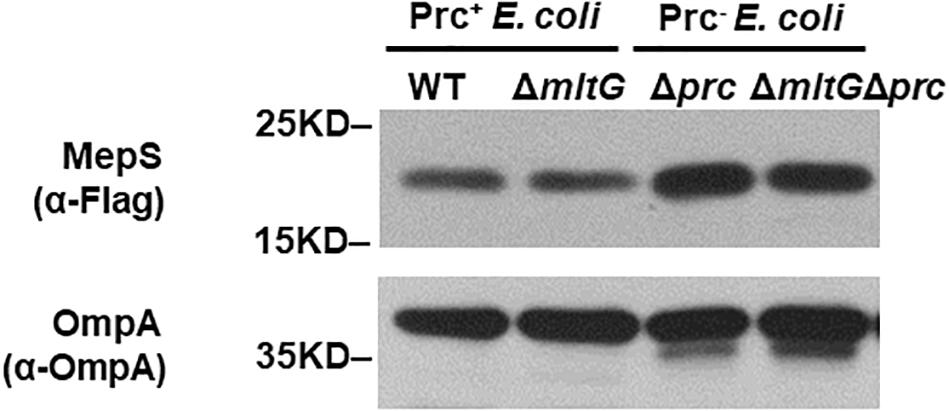
Figure 9. Effect of mltG deletion on the intracellular level of MepS. The levels of MepS in the E. coli strains were measured by Western blot analysis using a C-terminally 3xFLAG-tagged MepS encoded at the native chromosomal locus. A log-phase culture of each strain was collected and subjected to Western blot analysis. The outer membrane protein OmpA was used as a protein-loading control. Representative results of two independent experiments are presented. WT: BW25113-MepS-3xFlag; ΔmltG: ΔmltG-BW25113-MepS-3xFlag; Δprc: Δprc-BW25113-MepS-3xFlag; and ΔmltGΔprc: ΔmltGΔprc-BW25113-MepS-3xFlag (Table 2).
The interaction between the Prc protease and the MepS substrate requires the outer membrane-anchored protein NlpI as an adaptor (Singh et al., 2015). Deletion of nlpI also results in the intracellular accumulation of MepS in E. coli because the intracellular interaction of Prc and MepS is abolished. We further investigated whether NlpI is also involved in the Prc-mediated proteolytic regulation of MltG. We measured and compared the levels of MltG in the BW25113 strains with or without nlpI deleted (BW25113-MltG-HA-MepS-3xFlag and ΔnlpI-BW25113-MltG-HA-MepS-3xFlag; Table 2). As shown in Figure 10, deletion of nlpI did not increase the level of MltG in E. coli. However, the nlpI deletion caused MepS accumulation, which is consistent with the finding from a previous study (Singh et al., 2015). This result suggests that, in contrast to the MepS-Prc interaction, the interaction between MltG and Prc does not require NlpI as an adaptor (Figure 10).
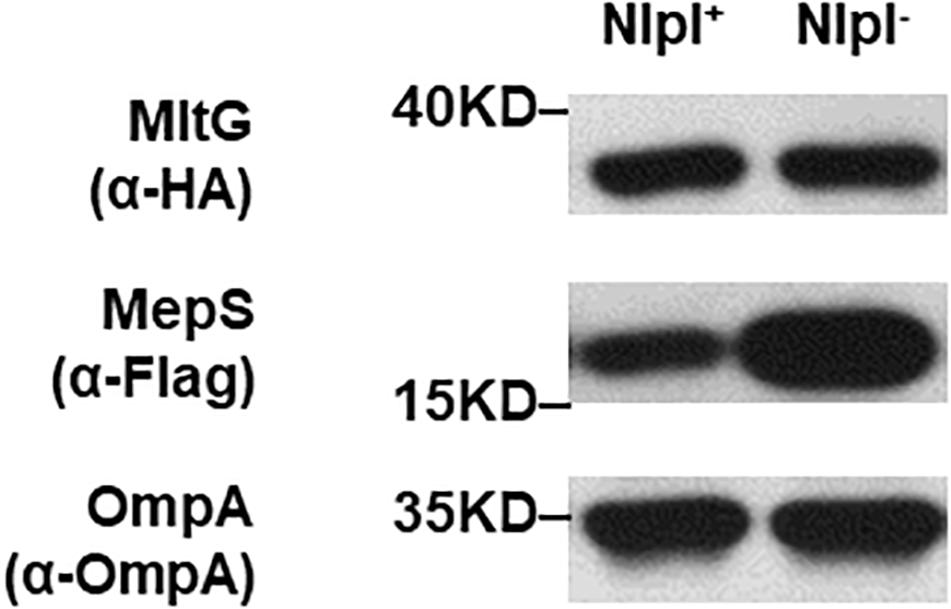
Figure 10. MltG levels in nlpI mutant strains. The levels of MltG in BW25113-MltG-HA-MepS-3xFlag and ΔnlpI-BW25113-MltG-HA- MepS-3xFlag (Table 2) were measured by Western blotting with anti-HA antibodies. The levels of MepS were measured with anti-Flag antibodies. The outer membrane protein OmpA was used as a protein-loading control. Representative results of two independent experiments are presented. NlpI+: BW25113-MltG-HA-MepS-3xFlag; NlpI–: ΔnlpI-BW25113-MltG-HA- MepS-3xFlag.
To investigate whether MltG regulation by Prc is growth phase-dependent, we measured the MitG levels in E. coli strains with or without Prc after different growth periods. As shown in Figure 11, the level of MltG in the Prc-expressing strain BW25113-MltG-HA was lower than that in the Prc-deficient strain Δprc-BW25113-MltG-HA (Table 2) in each of the indicated cultivation time of 2 h to 8 h (from the log phase to the stationary phase). These results suggest that Prc regulates the level of MltG in all bacterial growth stages.
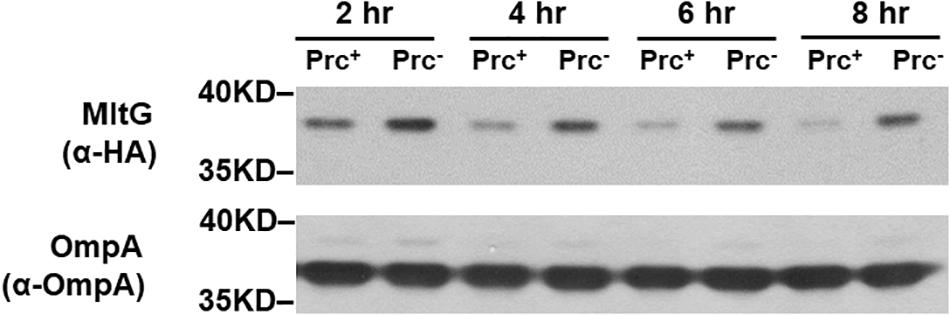
Figure 11. Levels of MltG in E. coli strains with or without Prc in cultures of different growth stages. Western blot analysis of HA-tagged MltG in BW25113-MltG-HA and Δprc-BW25113-MitG-HA (Table 2). Prc+, BW25113-MltG-HA; Prc+, Δprc-BW25113-MitG-HA. The outer membrane protein OmpA was used as a protein-loading control. Representative results of two independent experiments are presented.
To investigate whether blocking the accumulation of MepS and MltG restores the growth of Prc-deficient E. coli, the growth of ΔmepSΔmltGΔprc-BW25113, ΔmltGΔprc-BW25113, ΔmepSΔprc-BW25113, Δprc-BW25113, and WT-BW25113 in salt-free 1/2 LB medium at 42°C was measured. As shown in Figure 12, ΔmepSΔmltGΔprc-BW25113 exhibited a significantly higher level of growth than did ΔmltGΔprc-BW25113 or ΔmepSΔprc-BW25113 but did not reach the same growth level as WT-BW25113. These findings demonstrate that simultaneously blocking MltG and MepS accumulation enhances the growth of Prc-deficient E. coli to a greater extent than did blocking the accumulation of either of these proteins, thus suggesting that the accumulation of MltG and MepS addictively contributes to the conditional growth defect caused by Prc deficiency.
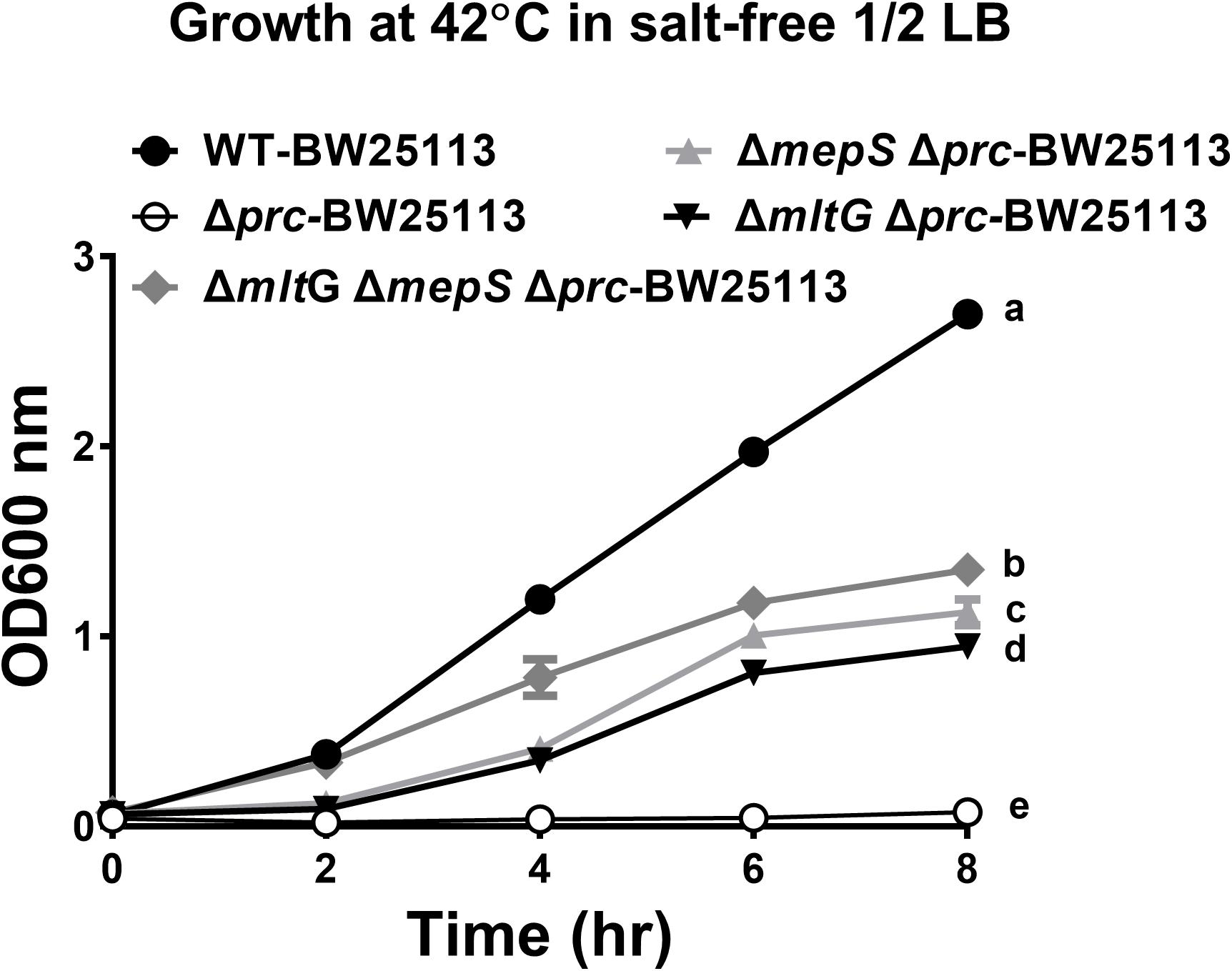
Figure 12. The growth of WT-BW25113, ΔmepSΔmltGΔprc-BW25113, ΔmltGΔprc-BW25113, ΔmepSΔprc-BW25113, and Δprc-BW25113 in salt-free 1/2 LB medium at 42°C. The growth curves of the strains were determined by measuring the values of O.D.600 at the indicated time points using a spectrophotometer. Data are shown as the mean ± SD of three independent experiments. Different letters (a,b,c,d, and e) indicate significances (P ≤ 0.05) between groups after 8 h of incubation. The data were analyzed by One-way ANOVA (Tukey’s multiple comparisons test).
In the present study, we identified a new Prc physiological substrate, MltG, whose intracellular level was increased by Prc deficiency. This dysregulated level was responsible for the growth defect of Prc-deficient E. coli under low osmolality at high temperature. The outcome of MltG dysregulation due to Prc deficiency was similar to that of the dysregulation of MepS, a previously identified substrate. The intracellular accumulation of MltG and MepS additively contributed to the conditional growth defect. MltG is a lytic transglycosylase (LT) involved in PG synthesis (Yunck et al., 2016). In addition to MltG, MepS and PBP3 (another previously identified substrate) are also involved in PG synthesis (Sauvage et al., 2008; Singh et al., 2012). Taking into account the functions of these substrates, Prc may play a potential role in regulating PG synthesis.
MltG is anchored to the inner membrane, with its functional domain exposed to the periplasm (Yunck et al., 2016). The LT activity of MltG is able to cleave nascent glycan strands of the PG meshwork and therefore terminate strand elongation during the course of PG polymerization (Yunck et al., 2016). It has been shown that a specific intracellular level of MltG is required for maintaining the integrity of the PG meshwork of bacteria. Deficiency of MltG alters the glycan strand length of PG in E. coli. On the other hand, a high level of MltG overexpression induces morphological defects and cell lysis, suggesting that a high level of MltG may damage the PG meshwork (Yunck et al., 2016).
The unfettered LT activity of MltG may be responsible for the conditional growth defect of Prc-deficient E. coli. PG is an essential structure that fortifies the cytoplasmic membrane against osmotic pressure. Thus, the cell wall is a key structure to withstand outwardly directed osmotic pressure (turgor pressure), thereby preventing cell lysis. Turgor pressure is determined by the difference in the osmolality of the cytoplasm and that of the external medium and can be calculated with the Morse equation, p = RT (Cin – Cout), where Cin is the osmolality of the cytoplasm, Cout is the osmolality of the external medium, R is the gas constant, and T is the temperature. When bacteria are transferred to a growth medium of lower osmolality, the turgor pressure increases. Then, the bacterial cells actively reduce cytoplasmic osmolality to downregulate the raised turgor pressure to adapt to the new environment (Record et al., 1998; Cayley et al., 2000). The PG damage caused by MltG accumulation may cripple the ability of the cell wall to withstand the increased turgor pressure in Prc-deficient E. coli before the active cytoplasmic osmolality modification takes place, thus resulting in the bacterial growth defect. In addition, it has been shown that in phosphate minimal medium the active reduction of cytoplasmic osmolality does not fully eliminate the turgor pressure rise caused by an external osmolality decrease (Record et al., 1998; Cayley et al., 2000). It is also likely that when E. coli are transferred from normal LB medium to salt-free 1/2 LB medium, the bacterial turgor pressure still rises in spite of the active modification of cytoplasmic osmolality, and the pressure increase is high enough to impair the growth of the Prc-deficient bacteria. Furthermore, based on the Morse equation, the turgor pressure increases as the temperature increases. Therefore, impaired growth of Prc-deficient E. coli in LB medium was observed at 42°C and 37°C but not at 30°C (Figure 1H–J).
Prc regulates the intracellular levels of MltG and MepS in different ways. The Prc-mediated degradation of MepS requires the adaptor protein NlpI to facilitate the protease-substrate interaction in vivo (Singh et al., 2015; Su et al., 2017). NlpI, however, is not involved in Prc-mediated MltG degradation (Figure 10). There may be other accessory bacterial factors facilitating the Prc-MltG interaction in E. coli. This prospect was indicated by Prc-mediated MltG degradation being more efficient in conditions with various bacterial factors, such as in E. coli cells (Figure 7D) and in E. coli lysates (Figure 6C), than in the condition using only the column-purified MltG and Prc without other bacterial factors (Figures 6A,B). In addition, the MltG protein purified with the high-throughput method was found to be efficiently degraded by Prc in vitro (Figure 4). There may be some residual accessory factors in the protein sample that facilitated the MltG degradation by Prc.
In addition to MltG, nine other E. coli proteins were shown to be able to interact with Prc or/and K455A-Prc (Figure 3) and were cleavable by Prc in vitro (Figure 4 and Table 1). Most of these proteins were likely not physiological substrates of Prc, as indicated by their cellular localization. As Prc is a periplasmic protease, its physiological substrates are expected to be located or at least partially exposed to the periplasmic space. However, of the nine proteins, seven, YfaU, YidA, RfaD, HupA, YegV, TyrA, and YacF, are cytoplasmic proteins (Lopez-Campistrous et al., 2005; Ishihama et al., 2008; Rea et al., 2008; Diaz-Mejia et al., 2009), suggesting that they may not be able to interact with Prc in vivo. Although the localization of two proteins, YaaW and YdhS, remains unknown, PSORTb software scoring showed that they are likely to be cytoplasmic proteins.
Even if all of the nine proteins are not physiological substrates, further understanding of their interaction with Prc would provide insights into how Prc recognizes and exerts its proteolytic function on substrates. However, it is worth noting that the interactions of the nine proteins with Prc were demonstrated by the high-throughput method, which may have retained some concomitant bacterial factors, as described above (Figures 3, 4). The possible involvement of certain unknown residual factors in the interaction with Prc cannot be excluded.
It has been reported that Prc preferentially recognizes and thus degrades proteins bearing a nonpolar C-terminal tail (Silber et al., 1992; Spiers et al., 2002). However, the C-termini of MltG, MepS, and PBP3 do not appear to bear this feature and, in fact, contain a substantial portion of polar residues (Supplementary Table S2). This finding suggests that a nonpolar C-terminal region may not be necessary for substrate recognition by Prc. Prc may recognize substrates without a nonpolar C-terminus, probably through the assistance of other bacterial factors, such as the adaptor NlpI for MepS and potential accessory bacterial factors for MltG. Alternatively, Prc may directly recognize regions of substrates not in the C-terminus. Further investigation will be required to understand how Prc recognizes its substrates.
Prc deficiency decreases the abilities of extraintestinal pathogenic E. coli (ExPEC) to cause bacteremia and urinary tract infections (UTIs) (Wang et al., 2012; Huang et al., 2020). The accumulation of MepS (Spr) contributes to the impaired ability of Prc-deficient ExPEC to colonize urinary tracts. It is worth further investigation to determine whether the dysregulation of MltG due to Prc deficiency is involved in the impaired ExPEC virulence in bacteremia and UTIs.
In summary, we identified MltG as a new physiological substrate of the periplasmic protease Prc. Prc deficiency induced MltG intracellular accumulation, which was responsible for the growth defect of Prc-deficient E. coli under low osmolality and high temperature. MltG is an LT involved in PG biogenesis. Increased MltG LT activity may impair the PG structure and attenuate the growth of Prc-deficient E. coli in a hypotonic medium at high temperature. Since the biological function of a protease is defined by its substrates, in addition to MltG, two other Prc substrates, MepS and PBP3, are also involved in PG synthesis (Sauvage et al., 2008; Singh et al., 2012). The functions of these substrates collectively suggest a potential role of Prc in regulating PG biogenesis.
The bacterial strains and plasmids used in this study are listed in Tables 2, 3. All the strains were derivatives of BW25113 (Baba et al., 2006) unless otherwise indicated. The bacteria were grown in 5 ml of Luria-Bertani broth (LB) (1% tryptone, 0.5% yeast extract, and 1% NaCl) in test tubes at 37°C with shaking at 200 rpm unless otherwise indicated. To investigate bacterial growth in 30°C/LB medium and 42°C/salt-free 1/2 LB medium conditions with or without osmolality adjustment, Overnight cultures of bacteria were adjusted to an optical density (O.D.600) of 4, transferred to fresh LB or salt-free 1/2 LB medium (0.5% tryptone and 0.25% yeast extract) (Hara et al., 1991) at a ratio of 1:100, and cultured at the indicated temperature.

Table 3. Plasmids used in this study.
Escherichia coli mutants were constructed by the λ-red-recombinase-based method or P1 phage transduction as described previously (Datsenko and Wanner, 2000; Thomason et al., 2007; Wang et al., 2012). The description of the construction of the mutants and plasmids used in this study are described in Supplementary Text S1. The primers used to construct the mutants and plasmids are listed in Supplementary Table S3.
The construction of the E. coli proteome chip was based on procedures described previously (Chen et al., 2008). Briefly, the E. coli K12 open reading frame (ORF) clone library (ASKA library), which was constructed by Dr. Mori and his colleagues and harbors 4,256 E. coli genes (Kitagawa et al., 2005), was used to overexpress E. coli proteins. Because the N-termini of the proteins were fused with a 6xHis tag, they were purified with Ni-NTA resin (Qiagen). The purified proteins were spotted in duplicate on Full Moon slides (Full Moon BioSystems) by using a ChipWriter Pro (Bio-Rad) with 48 pins. The printed microarrays were stored at −80°C until further use.
To fluorescently label the protein probes (Prc, K455A-Prc, and ΔPDZ-K455A-Prc), an amine-reactive fluorescence dye, Dylight™ 549 NHS Ester (Thermo Scientific), which contains N-hydroxysuccinimide (NHS) esters able to covalently bind to amines of proteins, was utilized to label the protein probes following the manufacturer’s instructions. The maximum excitation/emission wavelength of Dylight™ 549 is 562 nm/576 nm, which is spectrally similar to Cy3.
The arrays were blocked with 1% bovine serum albumin (BSA) (Sigma-Aldrich Co.) in Tris-buffered saline (TBS; 137 mM NaCl, 2.7 mM KCl, 24.8 mM Tris-base, pH 7.5) for 5 min at room temperature (RT) to reduce nonspecific binding. The chips were probed with 80 μ of 1 μM Dylight™ 549-labeled Prc, K455A-Prc, or ΔPDZ-K455A-Prc in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) containing 1% BSA in a hybridization chamber with shaking at RT for 1 h. After incubation, the chips were washed 3 times with Tris-buffered saline-Tween 20 (TBS-T; TBS containing 0.05% Tween 20, pH 7.5). After the final wash, the chips were dried by centrifugation at 201 × g and then scanned by a microarray scanner (GenePix® 4000B, Axon Instruments). Proteins binding to the fluorescently labeled Prc variants were detected at an excitation wavelength of 532 nm and emission wavelength of 570 nm.
The protein microarray experiments were done in triplicate. The results of the triplicate experiments were combined for bioinformatics analyses. Since there are duplicate spots for each protein on one array, there were 6 fluorescence readouts for each proteins. To analyze the images of the chip assay results, GenePix Pro 6.0 was used to align each protein spot and export all the imaged signals to text files. To adjust for probing and scanning procedure variations, inter-array signal intensities were quantile normalized (Bolstad et al., 2003). Then, ProCAT (Zhu et al., 2006) was applied to normalize (intra-array) the signals. The positive hits for Prc and K455A-Prc binding were selected based on a local cutoff, which was arbitrary defined as two standard deviation above the signal mean of for each spot. From the high hit candidates, we further selected for proteins whose interaction with Prc or K455A-Prc was significantly higher than ΔPDZ-K455A-Prc. Thus the proteins whose corresponding spots showed significantly higher levels of signals of interaction with Prc or K455A-Prc than those with ΔPDZ-K455A-Prc (p < 0.01, Student’s t-test) were selected (Supplementary Table S4).
In the systematic Prc proteolytic assay with the proteins selected from the E. coli proteome array assay, the proteins were expressed from the ASKA library and purified using the 96-well-based high-throughput method developed by Chen et al. (2008). Finally, the proteins purified from 800 μl of IPTG-induced E. coli cultures were eluted from Ni-NTA resins (Qiagen) in 50 μl of elution buffer (50 mM NaH2PO4, 150 mM NaCl, 25% glycerol, 250 mM imidazole and 0.01% Triton X-100, pH 7.5) (Chen et al., 2008).
Prc, K455A-Prc, and ΔPDZ-K455A-Prc were purified from DH5α strains harboring pTR147, pTR163, and pTR163-ΔPDZ, respectively. Expression of these 6xHis-tagged proteins was induced by adding 0.1 mM IPTG to the culture when the cells reached an O.D.600 of 0.6, and the culture was incubated at 30°C for another 5 h. The cells were centrifuged at 6000 × g for 20 min, resuspended in buffer A (20 mM Tris–HCl and 200 mM NaCl, pH 7.5) and disrupted by French press as described previously (Teng et al., 2010). The supernatant was loaded onto a Ni-NTA column (GE Healthcare), and the unbound proteins were washed away with 60 mM imidazole in buffer A. The proteins were eluted with 300 mM imidazole in buffer A. Fractions containing the proteins were pooled and further concentrated using a 50-kDa centrifugal membrane filter (Millipore). In this process, the solutions were dialyzed with buffer A to remove imidazole.
The C-terminally and N-terminally 6xHis-tagged MltG proteins were purified from DH5α strains harboring pBAD24-MltG-His and pBAD24-His-MltG, respectively. The expression of these MltG proteins was induced by adding 0.1 mM arabinose to the culture after the cells had reached an O.D.600 of 0.6, and the culture was further incubated at 30°C for 5 h. Then, the cells were subjected to the protein purification process using a Ni-NTA column, as described above. The fractions containing the proteins were pooled and further purified by size-exclusion chromatography on a Superdex™ 75 column (GE Healthcare). Subsequently, the proteins were dialyzed in buffer B (20 mM Tris–HCl, 150 mM NaCl, 0.5 mM EDTA, 5% glycerol, and 3 mM DTT, pH 7.5). The purified proteins were stored at 4°C until further use.
The assays were performed as described previously with some modification (Wang et al., 2012). For the assays with the proteins purified with the 96-well plate-based method (Figure 4), 10 μl of the purified proteins (see section “Protein Preparation”) were incubated with 2 μg of Prc, which was dissolved in 5 μl of buffer A, at 37°C for 2.5 h, while 6 μg of β-casein was utilized as a degradation-positive control. After the proteolytic assays, the protein mixtures were subjected to SDS–PAGE analyses with 12% polyacrylamide gels, and the proteins were visualized by staining with Coomassie brilliant blue.
For the assays with the recombinant MltG proteins purified by Ni affinity and size exclusion chromatography (Figures 6A,B), 3 μg of the recombinant MltG proteins were incubated with 2 μg of Prc or K455A-Prc in 35 μl of buffer A at 37°C for the indicated time period. Then, the protein mixtures were subjected to SDS–PAGE analyses with 15% polyacrylamide gels. The protein bands on the gels were visualized by staining with Coomassie Brilliant Blue.
For the assays with E. coli lysates (Figure 6C), 20 ml of the of Δprc-BW25113/pBAD24-MltG-HA that had been cultured overnight in LB medium with 0.2% L-arabinose was harvested by centrifugation, resuspended in 2 ml of buffer A, and subjected to French press disruption (Teng et al., 2010). The resulting bacterial lysate was diluted 10-fold with buffer A, and 5 μl of the diluted lysate was incubated with 2 μg of Prc or K455A-Prc in 15 μl of buffer A at 37°C for 3 h. Then, the samples were subjected to Western blotting with an anti-HA antibody (Sigma-Aldrich) to detect HA-tagged MltG.
The BW25113 strain harboring both pTR163 and pACYC184- MltG-HA or one of the plasmids was cultured overnight and then was transferred to fresh LB at a ratio of 1:100 and cultured for 2.5 h at 37°C. Then, to induce the expression of the 6xHis-tagged K455A-Prc, IPTG was added to the culture to a final concentration of 0.1 mM. After 5 h of incubation at 30°C, the bacteria in 50 ml of this culture were harvested by centrifugation and resuspended in 5 ml of buffer A (20 mM Tris–HCl and 200 mM NaCl, pH 7.5) containing 2% (v/v) Triton X-100 and 250 μg/ml lysozyme. The bacterial suspension was incubated on ice for 30 min, and the bacteria were disrupted by sonication. Unbroken cells and cell debris were removed by centrifugation at 20,000 × g for 25 min at 4°C. The protein concentrations of the resulting supernatants were determined using a Pierce™ BCA protein assay kit (Thermo Fisher Scientific, Carlsbad, CA, United States) and adjusted to 1 mg/ml using buffer A with 2% Triton X-100. For the pull-down assays, 1000 μl of the resulting protein solution was incubated with 1 μg (0.5 μl) of anti-His antibody (Sigma-Aldrich) or 3 μg (0.5 μl) of anti-HA antibody (Sigma-Aldrich) for 2 h at 4°C with gentle rocking in an Eppendorf tube. To precipitate the antibody-bound proteins, 5 μl of magnetic Dynabeads™ protein G (Thermo Fisher Scientific) was added to the solution. The solution was incubated for an additional 1 h at 4°C with gentle rocking, and the beads were precipitated by placing the tube on a magnet for 1 min. The beads were washed three times with 1 ml of buffer A containing 2% Triton X-100 and resuspended in 20 μl of SDS–PAGE sample buffer, followed by 10 min of incubation at 100°C. After precipitating the magnetic beads, the supernatant was subjected to Western blot analysis with anti-His and anti-HA antibodies to detect the presence of K455A-Prc and MltG-HA.
To measure whether Prc degrades MltG in E. coli cells, an in vivo assay of MltG degradation was performed as previously described with some modification (Singh et al., 2015). The overnight culture (200 μl) consisting of MltG-HA-BW25113 or MltG-HA-Δprc-BW25113 was inoculated into 20 ml of fresh LB and incubated for 4 h at 37°C, and then, spectinomycin was added at a concentration of 300 μg/mL to block translation in the cells. Then, 2-ml aliquots of the culture were incubated at 37°C. At the indicated time points, one aliquot of the sample was subjected to Western blot analysis with anti-HA antibodies to determine the amount of MltG in the bacteria. The outer membrane protein OmpA was used as a protein-loading control, which was measure with a mouse anti-OmpA serum described previously (Huang et al., 2020).
The osmolality of media was measured with the Micro-Digital i-Osmometer Type 7iM (Vogel Loser, Germany) by using freezing point depression (Pena-Verdeal et al., 2015). The osmolality was expressed as milliosmoles per liter per kilogram (mOsm/kg).
Student’s t-test was used to analyze the results of bacterial growth experiments, except that of the growth experiment of ΔmepSΔmltGΔprc-BW25113, ΔmltGΔprc-BW25113, ΔmepSΔprc-BW25113, Δprc-BW25113, and WT-BW25113 in salt-free 1/2LB medium at 42°C was measured, which was analyzed by One-way ANOVA (Tukey’s multiple comparisons test).
All datasets generated for this study are included in the article/Supplementary Material.
P-CH carried out the experiments in this study. SW, C-SC, and C-HT contributed to the study conception, planning experiments, and data analysis and interpretation. MH and W-CH participated in the result discussion and technical support. All authors contributed to the article and approved the submitted version.
This work was supported by the Ministry of Science and Technology of Taiwan, R.O.C. (Grant nos. NSC 102-2628-B-006-004-MY3 and MOST 105-2320-B-006-023) and the National Cheng Kung University Hospital, Tainan, Taiwan (Grant no. NCKUH-10509002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.02000/full#supplementary-material
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006–2008.
Bass, S., Gu, Q., and Christen, A. (1996). Multicopy suppressors of prc mutant Escherichia coli include two HtrA (DegP) protease homologs (HhoAB), DksA, and a truncated R1pA. J. Bacteriol. 178, 1154–1161. doi: 10.1128/jb.178.4.1154-1161.1996
Beebe, K. D., Shin, J., Peng, J., Chaudhury, C., Khera, J., and Pei, D. (2000). Substrate recognition through a PDZ domain in tail-specific protease. Biochemistry 39, 3149–3155. doi: 10.1021/bi992709s
Bolstad, B. M., Irizarry, R. A., Astrand, M., and Speed, T. P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. doi: 10.1093/bioinformatics/19.2.185
Cayley, D. S., Guttman, H. J., and Record, M. T. Jr. (2000). Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress. Biophys. J. 78, 1748–1764. doi: 10.1016/s0006-3495(00)76726-9
Chang, A. C., and Cohen, S. N. (1978). Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134, 1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978
Chen, C. S., Korobkova, E., Chen, H., Zhu, J., Jian, X., Tao, S. C., et al. (2008). A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat. Methods 5, 69–74. doi: 10.1038/nmeth1148
Chueh, C. K., Som, N., Ke, L. C., Ho, M. R., Reddy, M., and Chang, C. I. (2019). Structural basis for the differential regulatory roles of the pdz domain in c-terminal processing proteases. mBio 10:e01129-19.
Clausen, T., Kaiser, M., Huber, R., and Ehrmann, M. (2011). HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell. Biol. 12, 152–162. doi: 10.1038/nrm3065
Dalbey, R. E., Wang, P., and Van Dijl, J. M. (2012). Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol. Mol. Biol. Rev. 76, 311–330. doi: 10.1128/mmbr.05019-11
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Diaz-Mejia, J. J., Babu, M., and Emili, A. (2009). Computational and experimental approaches to chart the Escherichia coli cell-envelope-associated proteome and interactome. FEMS Microbiol. Rev. 33, 66–97. doi: 10.1111/j.1574-6976.2008.00141.x
Guzman, L. M., Belin, D., Carson, M. J., and Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995
Hara, H., Abe, N., Nakakouji, M., Nishimura, Y., and Horiuchi, K. (1996). Overproduction of penicillin-binding protein 7 suppresses thermosensitive growth defect at low osmolarity due to an spr mutation of Escherichia coli. Microb. Drug Resist. 2, 63–72. doi: 10.1089/mdr.1996.2.63
Hara, H., Nishimura, Y., Kato, J., Suzuki, H., Nagasawa, H., Suzuki, A., et al. (1989). Genetic analyses of processing involving C-terminal cleavage in penicillin-binding protein 3 of Escherichia coli. J. Bacteriol. 171, 5882–5889. doi: 10.1128/jb.171.11.5882-5889.1989
Hara, H., Yamamoto, Y., Higashitani, A., Suzuki, H., and Nishimura, Y. (1991). Cloning, mapping, and characterization of the Escherichia coli prc gene, which is involved in C-terminal processing of penicillin-binding protein 3. J. Bacteriol. 173, 4799–4813. doi: 10.1128/jb.173.15.4799-4813.1991
Huang, W. C., Lin, C. Y., Hashimoto, M., Wu, J. J., Wang, M. C., Lin, W. H., et al. (2020). The role of the bacterial protease Prc in the uropathogenesis of extraintestinal pathogenic Escherichia coli. J. Biomed. Sci. 27:14.
Ishihama, Y., Schmidt, T., Rappsilber, J., Mann, M., Hartl, F. U., Kerner, M. J., et al. (2008). Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9:102. doi: 10.1186/1471-2164-9-102
Karzai, A. W., Roche, E. D., and Sauer, R. T. (2000). The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Biol. 7, 449–455.
Keiler, K. C., and Sauer, R. T. (1995). Identification of active site residues of the Tsp protease. J. Biol. Chem. 270, 28864–28868. doi: 10.1074/jbc.270.48.28864
Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., et al. (2005). Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299. doi: 10.1093/dnares/dsi012
Lopez-Campistrous, A., Semchuk, P., Burke, L., Palmer-Stone, T., Brokx, S. J., Broderick, G., et al. (2005). Localization, annotation, and comparison of the Escherichia coli K-12 proteome under two states of growth. Mol. Cell. Proteomics 4, 1205–1209. doi: 10.1074/mcp.d500006-mcp200
Nagasawa, H., Sakagami, Y., Suzuki, A., Suzuki, H., Hara, H., and Hirota, Y. (1989). Determination of the cleavage site involved in C-terminal processing of penicillin-binding protein 3 of Escherichia coli. J. Bacteriol. 171, 5890–5893. doi: 10.1128/jb.171.11.5890-5893.1989
Pena-Verdeal, H., Garcia-Resua, C., Minones, M., Giraldez, M. J., and Yebra-Pimentel, E. (2015). Accuracy of a freezing point depression technique osmometer. Optom Vis. Sci. 92, e273–e283. doi: 10.1097/opx.0000000000000669
Rea, D., Hovington, R., Rakus, J. F., Gerlt, J. A., Fulop, V., Bugg, T. D., et al. (2008). Crystal structure and functional assignment of YfaU, a metal ion dependent class II aldolase from Escherichia coli K12. Biochemistry 47, 9955–9965. doi: 10.1021/bi800943g
Record, M. T. Jr., Courtenay, E. S., Cayley, D. S., and Guttman, H. J. (1998). Responses of E. coli to osmotic stress: large changes in amounts of cytoplasmic solutes and water. Trends Biochem. Sci. 23, 143–148. doi: 10.1016/s0968-0004(98)01196-7
Sauvage, E., Kerff, F., Terrak, M., Ayala, J. A., and Charlier, P. (2008). The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258. doi: 10.1111/j.1574-6976.2008.00105.x
Silber, K. R., Keiler, K. C., and Sauer, R. T. (1992). Tsp: a tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc. Natl. Acad. Sci. U.S.A. 89, 295–299. doi: 10.1073/pnas.89.1.295
Singh, S. K., Parveen, S., Saisree, L., and Reddy, M. (2015). Regulated proteolysis of a cross-link-specific peptidoglycan hydrolase contributes to bacterial morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 112, 10956–10961. doi: 10.1073/pnas.1507760112
Singh, S. K., Saisree, L., Amrutha, R. N., and Reddy, M. (2012). Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Mol. Microbiol. 86, 1036–1051. doi: 10.1111/mmi.12058
Spiers, A., Lamb, H. K., Cocklin, S., Wheeler, K. A., Budworth, J., Dodds, A. L., et al. (2002). PDZ domains facilitate binding of high temperature requirement protease A (HtrA) and tail-specific protease (Tsp) to heterologous substrates through recognition of the small stable RNA A (ssrA)-encoded peptide. J. Biol. Chem. 277, 39443–39449. doi: 10.1074/jbc.m202790200
Su, M. Y., Som, N., Wu, C. Y., Su, S. C., Kuo, Y. T., Ke, L. C., et al. (2017). Structural basis of adaptor-mediated protein degradation by the tail-specific PDZ-protease Prc. Nat. Commun. 8:1516.
Tadokoro, A., Hayashi, H., Kishimoto, T., Makino, Y., Fujisaki, S., and Nishimura, Y. (2004). Interaction of the Escherichia coli lipoprotein NlpI with periplasmic Prc (Tsp) protease. J. Biochem. 135, 185–191. doi: 10.1093/jb/mvh022
Teng, C. H., Tseng, Y. T., Maruvada, R., Pearce, D., Xie, Y., Paul-Satyaseela, M., et al. (2010). NlpI contributes to Escherichia coli K1 strain RS218 interaction with human brain microvascular endothelial cells. Infect. Immun. 78, 3090–3096. doi: 10.1128/iai.00034-10
Thomason, L. C., Costantino, N., and Court, D. L. (2007). E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. 79, 1.17.1–1.17.8. doi: 10.1002/0471142727.mb0117s79
Wang, C. Y., Wang, S. W., Huang, W. C., Kim, K. S., Chang, N. S., Wang, Y. H., et al. (2012). Prc contributes to Escherichia coli evasion of classical complement-mediated serum killing. Infect. Immun. 80, 3399–3409. doi: 10.1128/iai.00321-12
Weichart, D., Querfurth, N., Dreger, M., and Hengge-Aronis, R. (2003). Global role for ClpP-containing proteases in stationary-phase adaptation of Escherichia coli. J. Bacteriol. 185, 115–125. doi: 10.1128/jb.185.1.115-125.2003
Yamamoto, H., Hashimoto, M., Higashitsuji, Y., Harada, H., Hariyama, N., Takahashi, L., et al. (2008). Post-translational control of vegetative cell separation enzymes through a direct interaction with specific inhibitor IseA in Bacillus subtilis. Mol. Microbiol. 70, 168–182. doi: 10.1111/j.1365-2958.2008.06398.x
Yunck, R., Cho, H., and Bernhardt, T. G. (2016). Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol. Microbiol. 99, 700–718. doi: 10.1111/mmi.13258
Keywords: Prc, Tsp, MltG, YceG, MepS, Spr, protease, peptidoglycan
Citation: Hsu P-C, Chen C-S, Wang S, Hashimoto M, Huang W-C and Teng C-H (2020) Identification of MltG as a Prc Protease Substrate Whose Dysregulation Contributes to the Conditional Growth Defect of Prc-Deficient Escherichia coli. Front. Microbiol. 11:2000. doi: 10.3389/fmicb.2020.02000
Received: 24 October 2019; Accepted: 28 July 2020;
Published: 27 August 2020.
Edited by:
Kenneth C. Keiler, The Pennsylvania State University (PSU), United StatesReviewed by:
Manjula Reddy, Centre for Cellular & Molecular Biology (CCMB), IndiaCopyright © 2020 Hsu, Chen, Wang, Hashimoto, Huang and Teng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ching-Hao Teng, Y2h0ZW5nQG1haWwubmNrdS5lZHUudHc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.