
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 July 2020
Sec. Food Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01534
This article is part of the Research Topic Probiotic Trigger Molecules in Action View all 12 articles
 Panagiotis Manasian1,2
Panagiotis Manasian1,2 Atma-Sol Bustos2,3Björn Pålsson1Andreas Håkansson2
Atma-Sol Bustos2,3Björn Pålsson1Andreas Håkansson2 J. Mauricio Peñarrieta3Lars Nilsson2*
J. Mauricio Peñarrieta3Lars Nilsson2* Javier A. Linares-Pastén1*
Javier A. Linares-Pastén1*Lactobacillus rhamnosus GG (ATCC 53103) and Bifidobacterium longum NCC 2705 are among the most studied probiotics. However, the first evidence of acyl hydrolase/lipase of two annotated proteins, one in each genome of these strains, is reported in this work. Signal peptide analysis has predicted that these proteins are exported to the extracellular medium. Both proteins were produced in Escherichia coli, purified and characterized. Molecular masses (without signal peptides) were 27 and 52.3 kDa for the proteins of L. rhamnosus and B. longum, respectively. Asymmetrical flow field-flow fractionation analysis has shown that both proteins are present as monomers in their native forms at pH 7. Both have shown enzymatic activity on pNP-laurate at pH 7 and 37°C. The enzyme from L. rhamnosus was characterized deeper, showing preference on pNP-esters with short chain fatty acids. In addition, a computational model of the 3D structure has allowed the prediction of the catalytic amino acids. The enzymatic activities using synthetic substrates were very low for both enzymes. The investigation of natural substrates and biological functions of these enzymes is still open.
Probiotics are microorganisms that provide beneficial effects for health and are gaining great interest both in the prevention as well as in the treatment of different diseases. The WHO (FAO/WHO) defines probiotic as “live microorganisms that, when administered in adequate amounts, confer a health benefit on the host” (Hill et al., 2014). Lactobacillus rhamnosus GG (ATCC 53103) and Bifidobacterium longum NCC 2705 are among the most studied probiotics. Several evidences support the positive effect of these strains against obesity, diabetes, inflammatory bowel disease, cancer, mental disorders, infections, diarrhea, immune disorders, systemic inflammation and others (Brusaferro et al., 2018; Consortium, 2018; Capurso, 2019). Indeed, a recent search in Google Patents gave more than 1250 and 1050 patents related with B. longum NCC 2705 and L. rhamnosus GG, respectively. In addition, L. rhamnosus is one of the most used bacteria in the probiotic food industry, which includes the manufacture of milk-based products such as yogurt-type drinks, but also, probiotic fruit juices, berry soups and products based in fermented cereals and soy (Saxelin, 2008).
L. rhamnosus GG (ATCC 53103) is Gram positive anaerobic bacteria that inhabit in the gastrointestinal tract (GIT) but could also be recovered from oral cavity, tonsils, and vagina (Segers and Lebeer, 2014). L. rhamnosus GG (ATCC 53103) was isolated from the gastrointestinal tract in 1983 (Gorbach and Goldin, 1989) and its genome sequenced in 2009 (Kankainen et al., 2009). It is a circular chromosome of 300,5051 pb with 2834 predicted protein-coding genes. B. longum is a non-halophilic, anaerobic, Gram positive bacteria commonly present in the animal and human intestines (Consortium, 2018). It is associated with the gastrointestinal tract and promotes several health benefits, including reduction of irritable bowel syndrome (Brigidi et al., 2001), improves the immune response in infants (Young et al., 2004) and also helps to maintain homeostasis in the intestinal ecosystem promoting normal digestion (Consortium, 2018). B. longum NCC 2705 contains a 2,256,646 bp genome organized in a circular chromosome (Schell et al., 2002). Most of the studies at enzymatic level are focused on proteins involved in the metabolism of carbohydrates and amino acids, transport systems and defense mechanism. Indeed, the genomes of L. rhamnosus and B. longum have shown a relatively high number of these type of putative proteins comparing with other intestinal lactobacilli (Schell et al., 2002; Kankainen et al., 2009).
Despite the number of studies on L. rhamnosus and B. longum, to the best of our knowledge, there is lack of reports on lipolytic activity in these probiotic strains. Lipolytic enzymes include carboxylesterases and lipases, which are classified into 19 families based on phylogenetic criteria, conserved motifs and biological function (Kovacic et al., 2019). The metabolism of lipids, including esters of large and short chain fatty acids, in the gastrointestinal tract plays an important role in several cellular functions associated with disorders, such as obesity and pathological conditions. Thus, the study of lipolytic enzymes secreted by gastrointestinal microorganisms, including probiotics, could contribute to understand the potential effects of these strains in the metabolism of lipids in the intestinal tract and their health implications. The genome analysis of L. rhamnosus GG and B. longum NCC 2705 has shown two putative lipase/acyl-hydrolases containing signal peptides in the N-terminal, one in each strain, which indicates that these proteins can be secreted to the extracellular medium. Therefore, the purpose of this work was to investigate the activity of these proteins, named LrLyp and BlLyp (from L. rhamnosus and B. longum, respectively).
LrLyp and BlLyp were produced in Escherichia coli and their enzymatic and biophysical properties were studied. Substrate selectivity and activity were evaluated with series of synthetic para-nitrophenyl esters, while apparent kinetic constants were obtained at pH 7 and 37°C, using para-nitrophenyl laurate (pNP-laurate) as substrate. This synthetic ester has a 12-carbon atom chain, corresponding to a medium-size fatty acid, and it is widely used for characterizing lipase activity from different sources, including bacteria (Castro-Ochoa et al., 2005). The biophysical characterization was focused on the determination of: (1) optimal temperature; (2) protein stability, in terms of melting temperature determined by differential scanning fluorimetry (DSF) in a range of pH from 4 to 10; and (3) multimerization, studied by asymmetrical flow field-flow fractionation (AF4) coupled to multiangle light scattering (MALS). AF4 separates multimeric proteins and aggregates based on their diffusion coefficient and, compared to size exclusion chromatography (SEC), does not have a stationary phase which reduce the potential loss of analytes from adsorption or shear-induced degradation (Wahlund and Nilsson, 2012; Choi et al., 2018).
Finally, there are no structures with significant homology to LrLyp neither BlLyp in the protein data bank (PDB) (last search on 2019.10.27). This has limited the construction of a reliable computational 3D model for BlLyp. Nevertheless, a model for LrLyp was obtained using remote homology modeling techniques, allowing the prediction of the catalytic residues among other structural features.
The biological function and natural substrates of LrLyp and BlLyp remains unknown. However, this work contributes with the first evidences of acyl hydrolase/lipase activity for these enzymes encoded by the genomes of Lactobacillus rhamnosus GG (ATCC 53103) and Bifidobacterium longum NCC 2705, respectively.
Annotated esterases and lipases from the genomes of Lactobacillus rhamnosus GG (ATCC 53103) (GenBanck access code: NC_013198.1) and Bifidobacterium longum NCC 2705 (NC_004307) were subjected to signal peptide analysis using SignalP 4.0 Server (Petersen et al., 2011). Loci LGG_RS06710 (L. rhamnosus) and BL1109 (B. longum) were synthesized (GenScript, United States) with codon optimized for Escherichia coli, excluding the region encoding the signal peptides. These genes were cloned in a pET21b(+) vector between the NdeI and XhoI restriction sites. A 6-histidine tag encoded by the vector backbone was added in the C-terminal of each gene. The resulting plasmids were named pET21b:LGG_RS06710 and pET21b:BL1109.
Motifs around the catalytic serine of different families of lipolytic enzymes (Kovacic et al., 2019) were searched in the aminoacidic sequences of LrLyp and BlLyp in order to determine their classification. Other conserved motifs were determined by multiple sequence alignments with selected characterized enzymes. The alignments were performed using the Clustal Omega (1.2.4) server.1
Different strains of E. coli, including BL21(DE3), Origami2, and Artic Express, were transformed with the plasmids pET21b:LGG_RS06710 and pET21b:BL1109. Each plasmid was introduced into chemically competent cells (Novagen, United States) by thermic shock (4°C 10 min, 42°C 2 min, 4°C 10 min). All cells were grown in LB medium supplemented with 100 μg/mL of ampicillin, additional 20 μg/mL gentamicin was added in the cultivation media of E. coli Artic Express. All cultivations were incubated in shake flasks (with agitation) at 37°C until reach an ODλ = 600 nm of 0.6. Then, 1 mM IPTG was added and the incubation temperature for the recombinants E. coli BL21(DE3) and Origami2 was decreased to 30°C for 12 h, while for the recombinant E. coli Artic Express was decreased to 8°C for 24 h.
Cell pellets were harvested by centrifugation at 4500 g for 20 min and resuspended in 1/10 volume of binding buffer (100 mM tris, NaCl 500 mM adjusted to pH 7.4 with HCl). Thereafter, the cell suspensions were sonicated for 5 min and centrifuged at 14,000 g. The precipitates were discarded, and the supernatants were subjected to SDS-PAGE analysis in order to determine the level of expression of the recombinant proteins. Protein purification was performed by IMAC, each supernatant was injected in a 5-mL HisTrap column FF (GE Healthcare, Uppsala, Sweden) previously equilibrated with binding buffer. Next, the column was washed with 15 column volumes of binding buffer. The recombinant protein was eluted with elution buffer pH 7.4 (50 mM sodium phosphate, 0.5 M NaCl, and 0.5 M imidazole). Finally, imidazole was removed by dialysis. Protein purity was determined by SDS-PAGE. Second purifications, using a 5-mL HisTrap column HP (GE Healthcare, Uppsala, Sweden), were performed when needed. The concentration of pure proteins was quantified spectrophotometrically at 280 nm.
Thermal stability of the enzyme in different pH values was determined using the Prometheus NT 48 nanoDSF (NanoTemper Technologies, GmbH, Munich Germany) (Choi et al., 2018). Enzyme samples at 0.5 g/L were prepared in pH 4.3, 5.3, 6.2, 7, 8 (McIlvaine buffer system, 100 mM), 9 and 9.6 (glycin-NaOH buffer, 100 mM). Then, 10 μL of every sample was directly loaded in the instrument capillaries. Intrinsic fluorescence at emission wavelengths of 330 and 350 nm was monitored in a temperature gradient from 20 to 90°C with a temperature ramp of 1°C per min. All data analysis was performed using the PR control software (Version 2.0, Munich Germany).
Stock solutions of the following para-nitrophenyl esters: pNP-acetate (C2), pNP-butyrate (C4), pNP-caprylate (C8), pNP-laurate (C12), pNP-myristate (C14), pNP-palmitate (C16), and pNP-stearate (C18), were prepared in concentrations of 200 (C2,C4,C8), 100 (C12, C14), and 50 (C16, C18) mM. A mixture of acetonitrile and 2-propanol (1:1) was used as solvent. The mixtures were subjected to ultrasound bath until a clear solution was obtained. All substrates and solvents were purchased from Sigma Aldrich (Darmstadt, Germany). The reactions were prepared in volumes of 210 μL, containing 50 mM phosphate buffer pH 7.2, 1 mM substrate and 0.074 g/L of enzyme. The reactions started by adding the enzyme and monitored at 410 nm of wavelength in a microplate spectrophotometer (Thermo ScientificTM, MultiskanTM GO) during 30 min at 37°C.
The optimal temperature was determined for LrLyp in a range from 20 to 60°C. The pH was set to 7.2 with 20 mM McIlvain buffers and 5 mM of p-nitrophenyl laurate was used as substrate. The reactions were initiated by adding the enzyme (0.133 g/L) and stopped after 40 min by heating at 94°C for 10 min and cooling down to 4°C. All reactions were performed in triplicates and using controls, which consisted in same reactions mixtures, except enzyme. All reactions, including controls, were incubated in thermocycler (Biometra T-gradient from AnalyticJena, Germany). The formed product, p-nitrophenol, was quantified in a microplate spectrophotometer (Thermo ScientificTM, MultiskanTM GO) at 410 nm.
A stock solution of pNP-laurate, was prepared in a concentration of 10 mM, dissolving para-nitrophenyl laurate flakes in a mixture of water, DMSO and triton-X 100 (2:1:1, respectively) (modified from Mastihuba et al., 2002). The mixture was subjected to ultrasonic bath at 50°C for 10 min until an oily clear solution was obtained. Initial rates were determined in a range of 0.1–9 mM p-nitrophenyl laurate. Reactions were initiated by adding the respective enzyme at concentrations of 0.133 g/L of LrLyp and 0.358g/L of BlLyp. The pH 7 was set with 20 mM McIlvaine buffer. The reactions were monitored quantifying the nitrophenol formed along the time, using a microplate spectrophotometer (Thermo ScientificTM, MultiskanTM GO) at 410 nm. All reactions were performed by triplicates and using controls, which were prepared with the same composition of reaction mixtures, except enzyme. The correlation between initial rates and substrate concentration was plotted and adjusted to the Michaelis-Menten (Eq. 1) or substrate inhibition kinetics (Eq. 2) model, by non-linear regression methods by the generalized reduced gradient non-linear algorithm, used as implemented in the Solver of Microsoft Excel:
where v0 is the initial rate, . is the apparent maximum velocity, [S] is substrate concentration, . is the apparent Michaelis Menten constant and . is the apparent dissociation constant for binding.
The stability of the enzyme LrLyp, at 37°C and pH 7, was monitored for 35 days. Residual activities were determined every day the first 5 days, after, once every week. Reactions were performed in triplicates, using 5 mM p-nitrophenyl laurate as substrate and quantifying the production of p-nitrophenol spectrophotometrically, such as is described before.
A model of the three-dimensional structure of LrLyp was obtained from the amino acid sequence, excluding the N-terminal signal peptide. Remote homology recognition techniques were applied using the Phyre2 Protein Fold Recognition Server (Kelley et al., 2015). The refinement of the model was performed by molecular dynamic simulations (MD) of 500 ps (Krieger et al., 2004). This protocol generates snapshots every 25 ps (in format pdb) with a table of energy to identify the best snapshot. The force field used was YAMBER2, implemented in the program YASARA (Krieger and Vriend, 2014). The MD conditions were pH 7.4, temperature 280°K and density 0.997. Additional validation of the model was analyzed using ProSA-web (Wiederstein and Sippl, 2007).
The instrumentation consisted in an Eclipse 3 + Separation System coupled to a Dawn Heleos II multi-angle light scattering (MALS) detector (Wyatt Technology, Dernbach, Germany) with a wavelength of 663.8 nm and to a UV detector (Jasco UV-975 detector, Corp., Tokyo, Japan) with a wavelength of 280 nm. An Agilent 1100 series pump coupled to a vacuum degasser (Agilent Technologies, Waldbronn, Germany was used to deliver the carrier liquid. For the sample injection into the channel, the system was equipped with an auto sampler Agilent 1100 series (Agilent Technologies). The channel used was a trapezoidal long channel (Wyatt Technology) with length of 26.0 cm and outlet inlet widths of 0.6 cm and 2.15 cm, respectively and 350 μm of nominal thickness. A membrane of regenerated cellulose (RC), cut-off of 10 kDa, was used as the accumulation wall (Merck Millipore, Bedford, MA, United States).
The samples were produced and purified as described in previous sections, resulting in a final concentration of 0.88 mg/mL for BlLyp and 1.2 mg/mL for LrLyp. Before the injection, the samples were centrifuged at 11,000 g for 15 min at 4°C. Bovine serum albumin (BSA) at concentration of 2 mg/mL was used as standard to check the performance of the AF4 system and to normalize de MALS detector.
First, the channel was flushed in elution and focus mode, 1 min each. After that, 100 μL of sample was injected onto the channel at 0.2 mL/min for 2 min. Once the sample was injected (0.088 mg of BlLyp and 0.12 mg of LrLyp), 3 min of focus mode were applied before the elution step. A constant cross flow of 5 mL/min was used during the elution period for 15 min, followed by 15 min without cross flow to remove any remnant material. The detector flow was always 1 mL/min. The parameters previously described were used with 5 different carrier liquids: 20 mM of phosphate-citrate buffer at pH 7, 6, 5, 4, and 3 in both lipase samples. A new channel was assembled for every lipase.
Astra software 6.1 (Wyatt Technology Europe) was used for the data analysis. For the molecular weight (Mw) of BlLyp, the value was determined at pH 6 using 13 scattering angles, from 42.8° to 163.3°. The calculations were performed with 1st order Zimm model (Zimm, 1948). The dn/dc value used was 0.185 mL/mg and the UV extinction coefficient 0.863 mL/(mg cm). The second virial coefficient was considered negligible. For both lipases, the hydrodynamic radius (rh) was estimated by the Stokes-Einstein equation using FFhydRad 2.1-MATLAB Apps (Håkansson et al., 2012) for the calculations. The channel thickness (w) for LrLyp and for BlLyp was determined using BSA as a standard.
Putative extracellular lipases were searched in the genomes of Lactobacillus rhamnosus, strain GG (ATCC 53103) and Bifidobacterium longum NCC 2705. Only one locus of a putative annotated lipase/acyl-hydrolase was found in each genome, LGG_RS06710 in L. rhamnosus, and BL1109 in B. longum. LGG_RS06710 encodes a 281 amino acids protein of 30728.20 Da including a predicted N-terminal signal peptide of 34-amino acids. BL1109 encodes a 480 amino acids protein of 51163.93 Da with 34-amino acids in the N-terminal predicted as signal peptide. Therefore, it is predicted that both proteins are exported to the extracellular medium. The genes encoding these proteins, excluding signal peptides, were synthesized with codons optimized for Escherichia coli and successfully produced in strains BL21(DE3), ArticExpres and Origami2. However, the last gave the highest fraction of soluble recombinant protein, estimated in 80% according to the SDS-PAGE analysis. The theoretical molecular weights of these truncated forms (without signal peptide) are 27058.74 Da (LrLyp) and 52360.25 Da (BlLyp) from L. rhamnosus and B. longum, respectively; which is consistent with the molecular mass determined by SDS-PAGE (Figure 1).
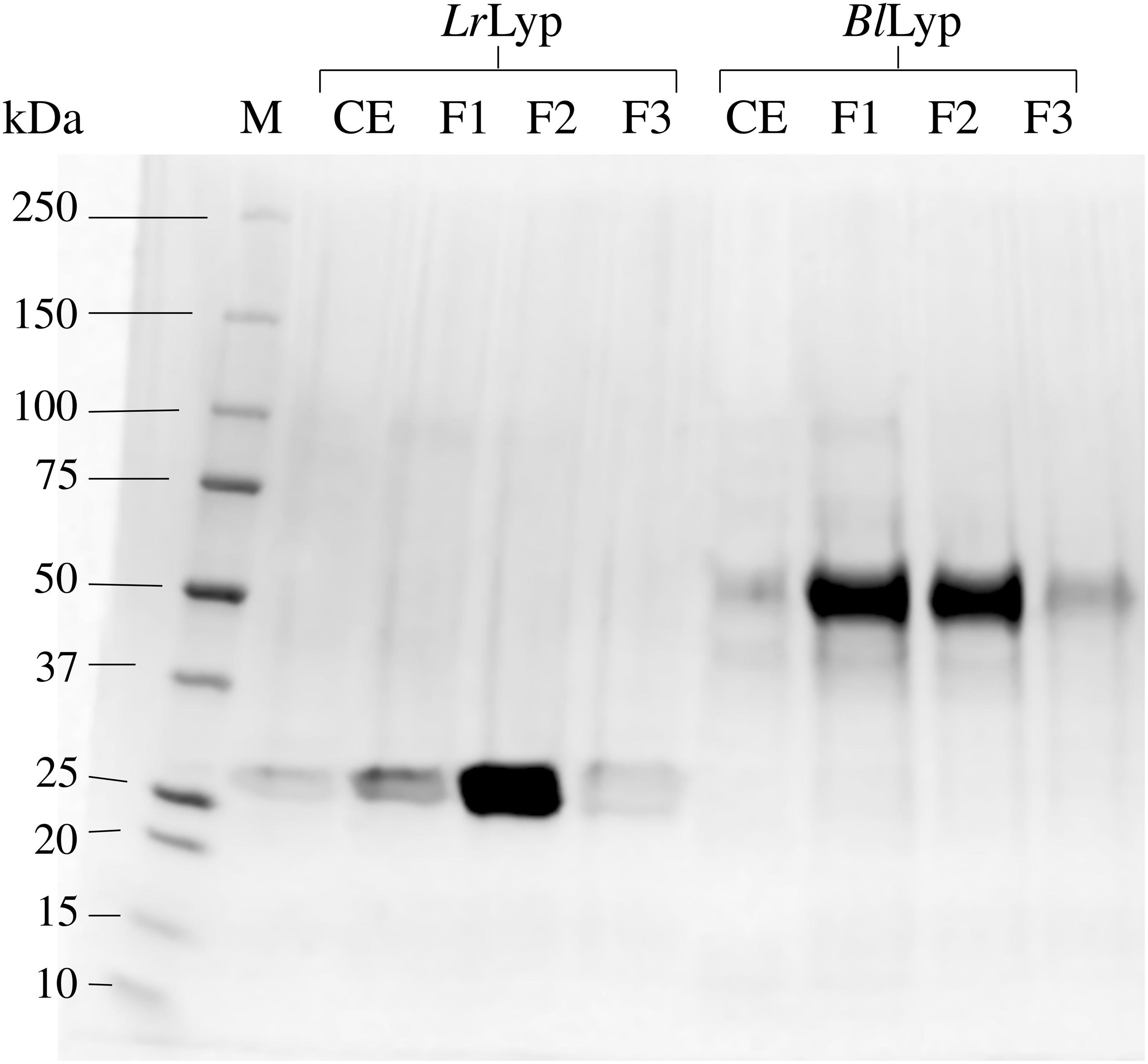
Figure 1. Production and purification of the recombinant LrLyp (locus LGG_RS06710) and BlLyp, without signal peptides. SDS-PAGE is showing the molecular weight marker M (Lane 1), the cell extracts (CE) and the different chromatographic fractions (F1–F3) corresponding to each purified protein: LrLyp (Lanes 3–5) and BlLyp (Lanes 7–9).
Amino acid sequence analysis of LrLyp and BlLyp has revealed that these enzymes contain conserved motif corresponding to lipolytic enzymes families II and IV respectively. LrLyp has four blocks: GDSLT (amino acids 53–56), GVSG (aa. 91–94), GGND (aa. 125–128), and HPN (aa. 256–258); as shown in the multiple alignment with other family II enzymes (Figure 2). Previously characterized bacterial GDSL enzymes, including Sc1 from Streptomyces coelicolor (Côté and Shareck, 2008), TesA and EstA from Pseudomonas aeruginosa, SrLip from Streptomyces rimosus, EstP from Pseudomonas putida (Leščić Ašler et al., 2010), XvEstE from Xanthomonas vesicatoriaand (Talker-Huiber et al., 2003), and EstHE1 obtained from a metagenome (Okamura et al., 2010), were selected for the multiple alignment (Supplementary Information S1).
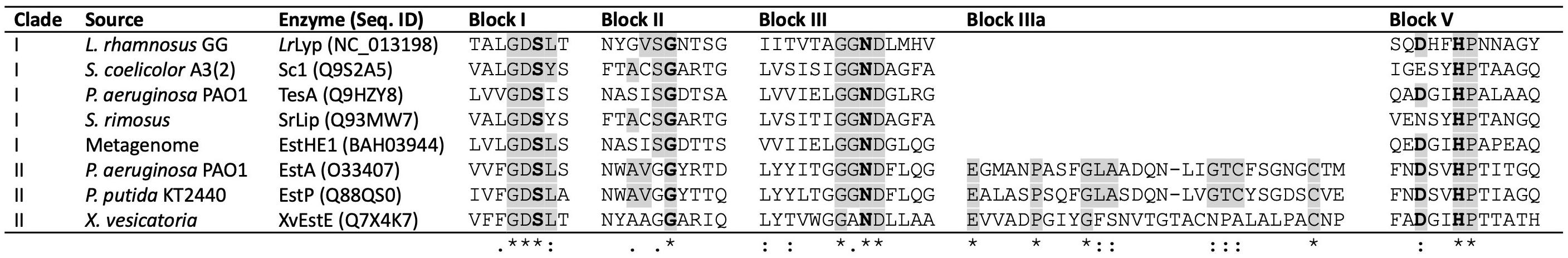
Figure 2. Multiple alignment of conserved blocks in lipolytic enzymes family II. Characterized enzymes were selected for the comparison with LrLyp from L. rhamnosus. Conserved catalytic amino acids are in bolds, S in the block I is the nucleophile, D and H in the block V are the donor proton and catalytic base respectively. G and N in the blocks II and III belong to the oxyanion hole. LrLyp belongs to clade I, since it lacks of block IIIa (Leščić Ašler et al., 2010). Where (*) denotes conserved or identical amino acid, (:) conservative or amino acid with similar biochemical property, (.) semiconservative and (_) non conservative amino acid.
BlLyp has five conserved motifs present in family IV as shown in Figure 3. The conserved sequences are in five blocks: HGGG (aa. 225–229), YRLA (aa. 257–260), GDSAGGNL (aa. 328–335), CPL (aa. 430–432), and HGI (aa. 460–462). The sequences selected for the multiple alignment correspond to previously characterized family IV lipolytic enzymes, including Est3k obtained from a metagenome (Kim et al., 2015), LipP from Pseudomonas sp. (Choo et al., 1998), Lip2 from Moraxella sp. (Feller et al., 1991), Est2 from Alicyclobacillus acidocaldarius (Mandrich et al., 2008), arylesterase from Saccharolobus solfataricus (PDB accession code 5L2P) and Aes from Saccharolobus shibatae (Ohara et al., 2014). It is remarkable the presence of cysteine in the motif CPL in BlLyp, instead of conserved aspartate (DPL) in the rest of the enzymes compared (Supplementary Information S2).
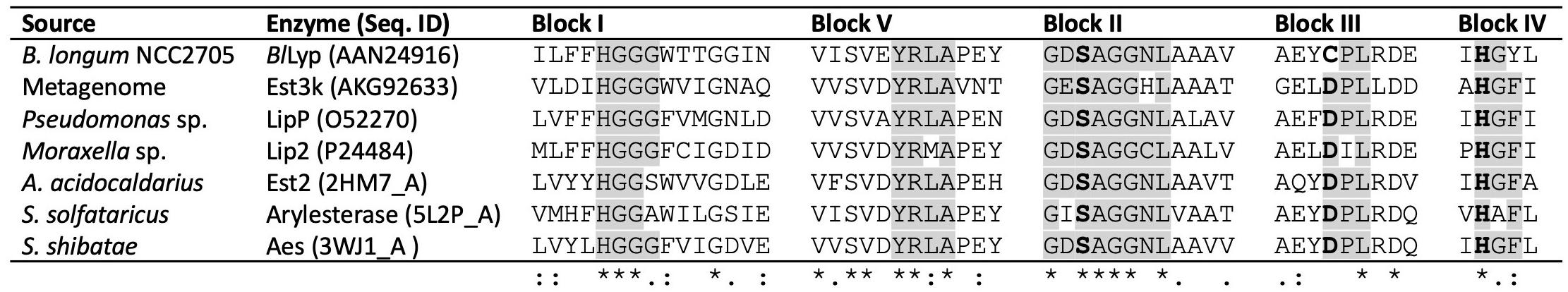
Figure 3. Multiple alignment of conserved blocks in lipolytic enzymes family IV. Characterized enzymes were selected for the comparison with BlLyp from B. longum. Conserved catalytic amino acids are in bolds, S in the block I is the nucleophile, H in block IV is the base, and the proton donor D located in bock III, interestingly is a C in BlLyp. The recently suggested block V (Kim et al., 2015) containing the consensus sequence YRLA is also present in BlLyp. Where (*) denotes conserved or identical amino acid, (:) conservative or amino acid with similar biochemical property, (.) semiconservative and (_) non conservative amino acid.
Both optimal temperature and pH were determined in terms of thermal stability in a range of pH from 4.3 to 9.9 by DSF. The protein has show the highest stability in a range of pH from 6 to 8 with the optimal melting point of 53.1°C at a pH close to 7 (Figure 4A). Indeed, the data adjusted to the quadratic model Tm = −1.22pH2 + 16.83pH. – 3.19, with a coefficient of determination, R2, of 98%, which first derivative equal to zero, gave a critical point corresponding to pH 6.88. Therefore, the optimal temperature in terms of relative activity was determined at pH 7, giving maximum activity (100%) in a range from 37 to 40°C (Figure 4B), which is consistent with the physiological temperature. The activity decreases to 90% in a range of 20–30°C, while decreases dramatically below 60% at 60°C.
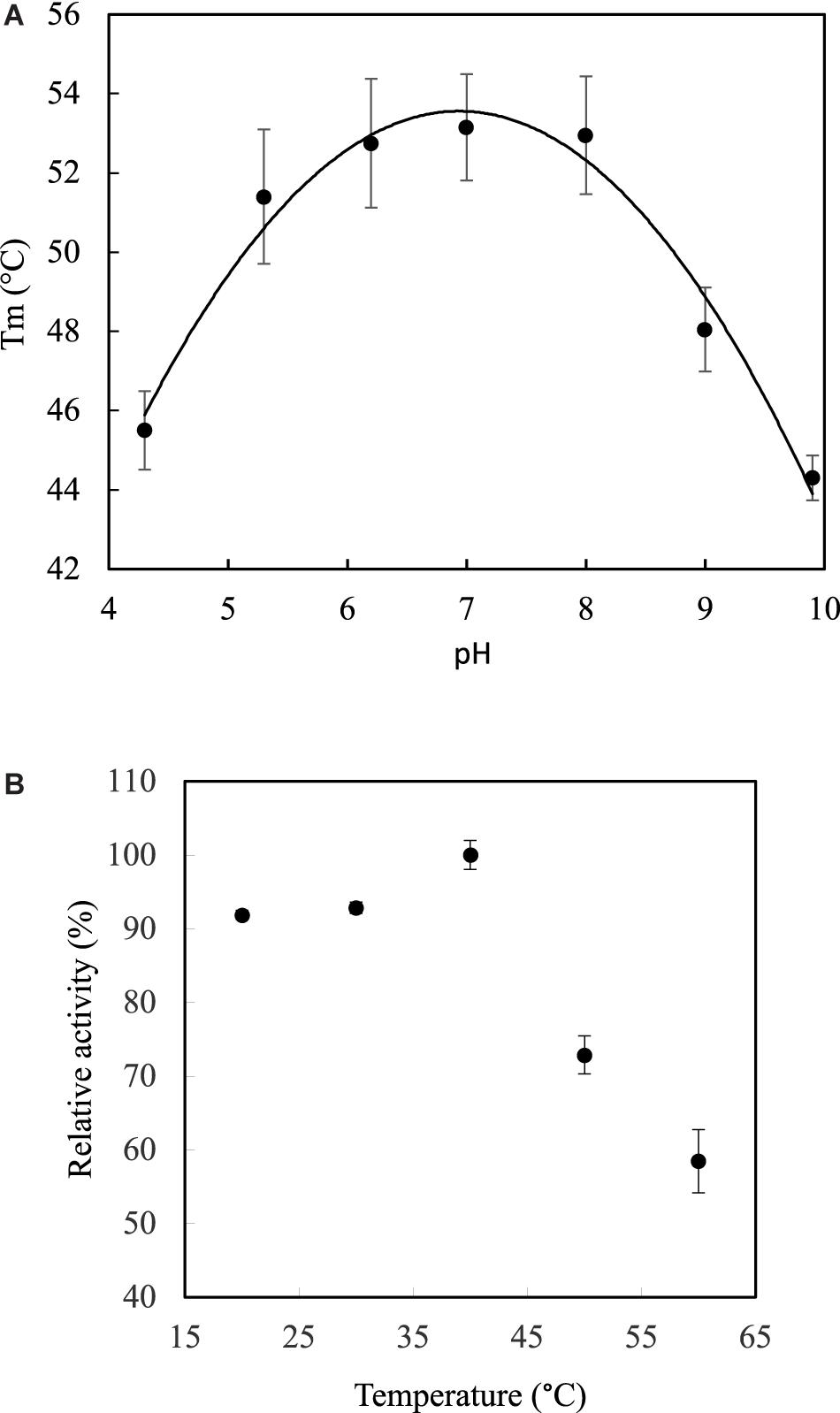
Figure 4. Optimum pH and temperature. (A) DSF analysis shows optimum pH between 6 and 8 with a highest transition temperature (melting point Tm) at 53.1°C. (B) Temperature curve shows maximum relative activity at 40°C. The pH was 7 and pNP-laurate was used as substrate. Error bars denote plus/minus one standard deviation.
The substrate selectivity was evaluated with seven synthetic para-nitrophenyl esters (pNP-esters) with linear hydrocarbon chains from C2 to C18. On the other hand, the biodegradable polyester-plastic polycaprolactonate (CAPA 6000) was tested as potential substrate and the phospholipase D activity was assessed as well. Activity was detected only on pNP-esters. The highest activity was on pNP-butyrate while the lowest was on pNP-sterate. Thus, these results show that LrLyp has preference for short hydrocarbon chain pNP-esters (Figure 5 and Table 1). No activity was detected on CAPA 6000 neither phospholipase D activity. The activity of BlLyp is dramatically lower than the LrLyp when pNP-laurate is used as substrate (Table 1).
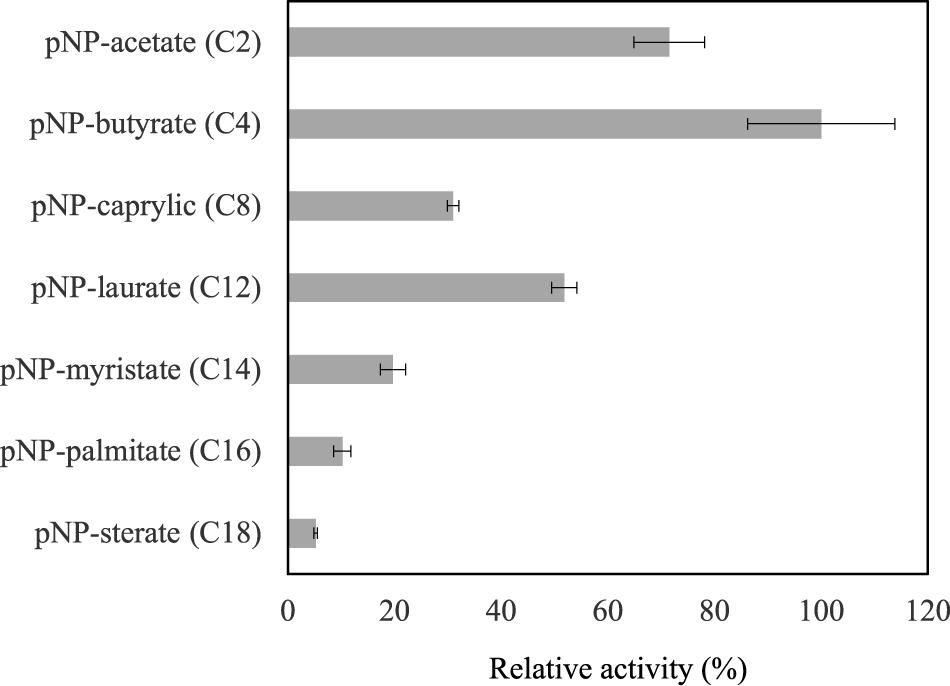
Figure 5. Substrate selectivity for LrLyp. The reactions were carryout using 1 mM of substrate at 37°C and pH 7.2. Error bars denote plus/minus one standard deviation.

Table 1. Enzymatic activity on pNP-esters. The reaction conditions were pH 7.2, 37°C, and 1 mM substrate.
Among the large chain esters, pNP-laurate (C12) has shown highest activity (52% comparing with the best substrate pNP-butyrate) and it was used for further studies of stability for LrLyp over time and determination of kinetic constants. Incubating LrLyp at 37°C, the activity dropped to an average of 5.3% after the 7th day and remained so until more than 35 days (Figure 6).
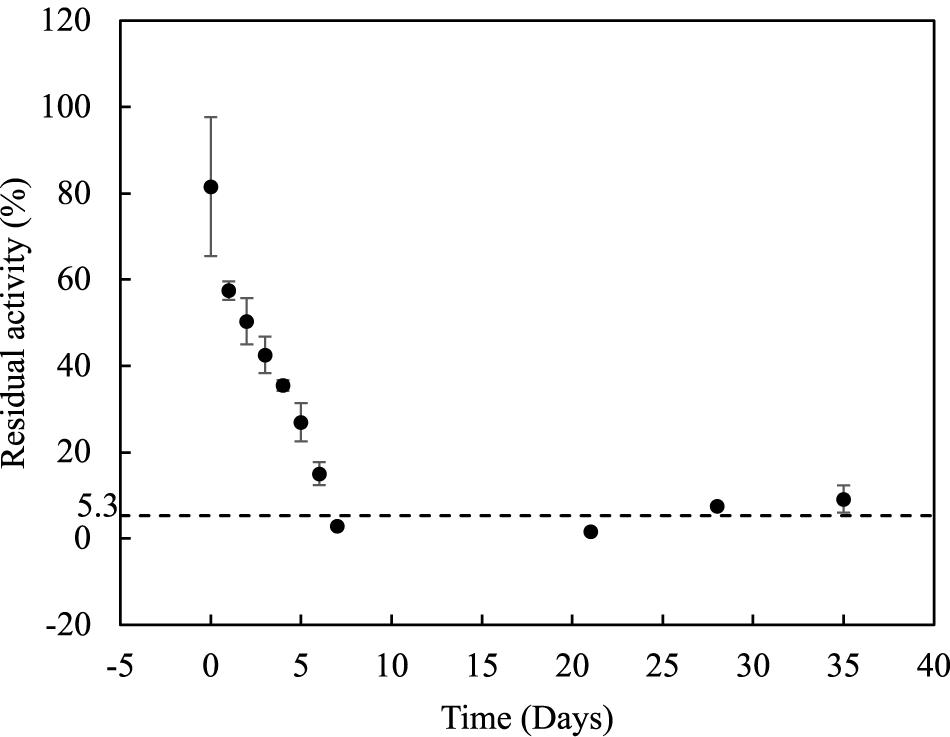
Figure 6. Stability of LrLyp along the time.
LrLyp kinetics does not follow a typical Michaelis-Menten model (Figure 7A). It seems that LrLyp reaches a saturation point at 5mM of substrate, and at higher substrate concentrations the reaction rate decreases. Which suggest that LrLyp suffer substrate inhibition at substrate concentrations higher than 5 mM. Therefore, the data were adjusted to the substrate inhibition model to obtain the kinetic constants (Table 2). The coefficient of determination, R2, of the model was 96%. On the other hand, for BlLyp Figure 7B shows a typical Michaelis-Menten behavior. The determined apparent kinetic constants are reported in Table 2, and the coefficient of determination was 99%.
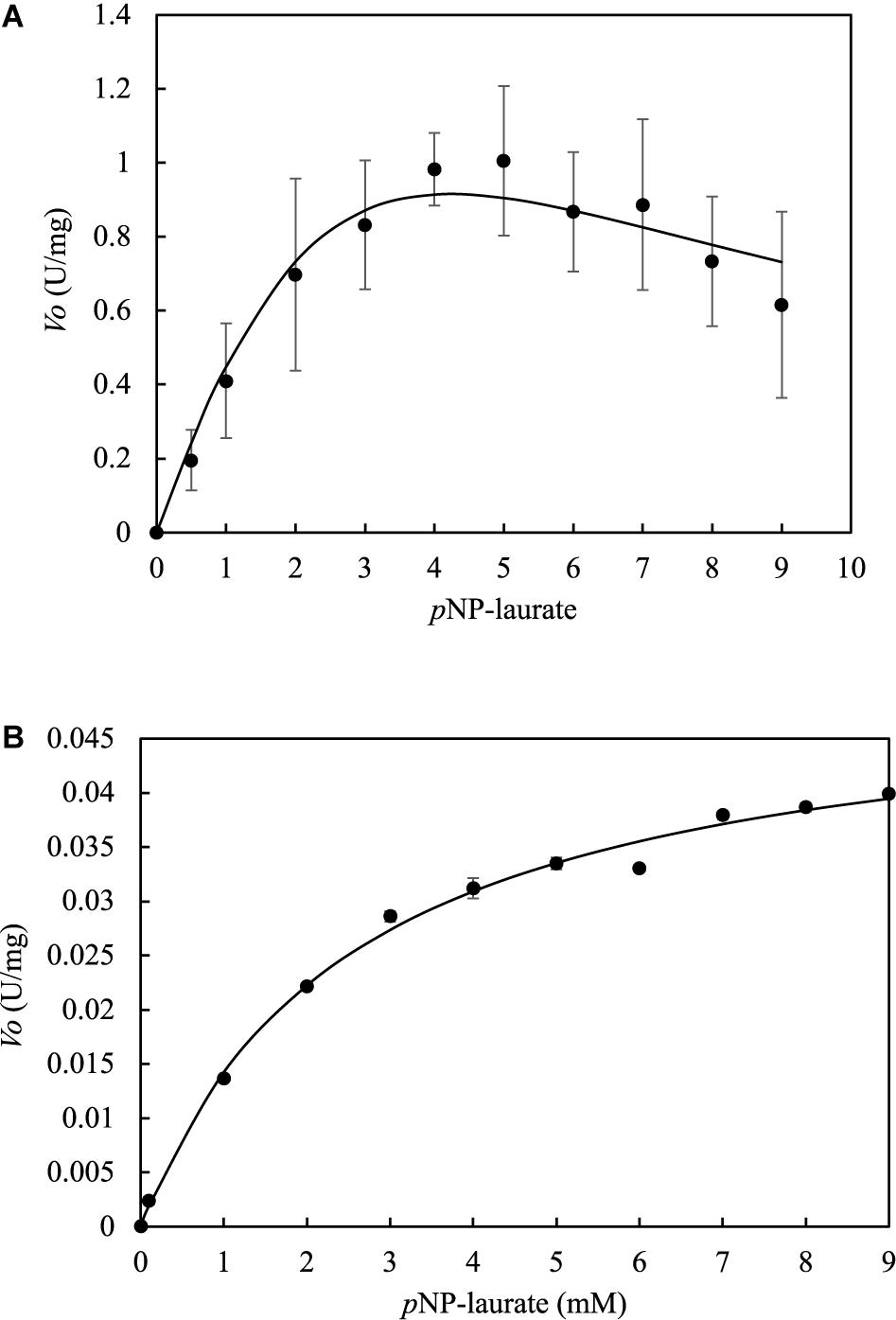
Figure 7. Determination of kinetic constants at pH 7 and 37°C for LrLyp and BlLyp by no-linear regression. Error bars represent the standard deviation. LrLyp (A) shows substrate inhibition kinetics, while BlLyp (B) shows a typical Michaelis Menten kinetics. Error bars denote plus/minus one standard deviation.

Table 2. Apparent kinetic constants.
Multimerization and aggregation studies of both LrLyp and BlLyp were performed by asymmetric flow field-flow fractionation (AF4). Figure 8A shows the UV – fractograms for BlLyp at different pHs where two main peaks were identified and characterized respect to their hydrodynamic rh and molar mass. For pH 7, peak 2 absorbs 10 times less than peak 1 (considering peak height). At pH 6 the concentration of peak 1 decrease and peak 2 absorb <10% than peak 1. In pH 5 only peak 2 is present. pH 4 does not show any notable peak, while at pH 3 peak 1 is missing and peak 2 present a tail of other compounds that were not separated. The UV – fractograms for LrLyp are plotted in Figure 8B, where the hydrodynamic rh for only the first peak was measured. At pH 7 we have the highest absorption for this peak, followed for other peaks that were not completely separated, for pH 6 this peak absorbs <10% compared with pH 7. This peak is prominent at pH 5 again and it disappears completely at pH 4.
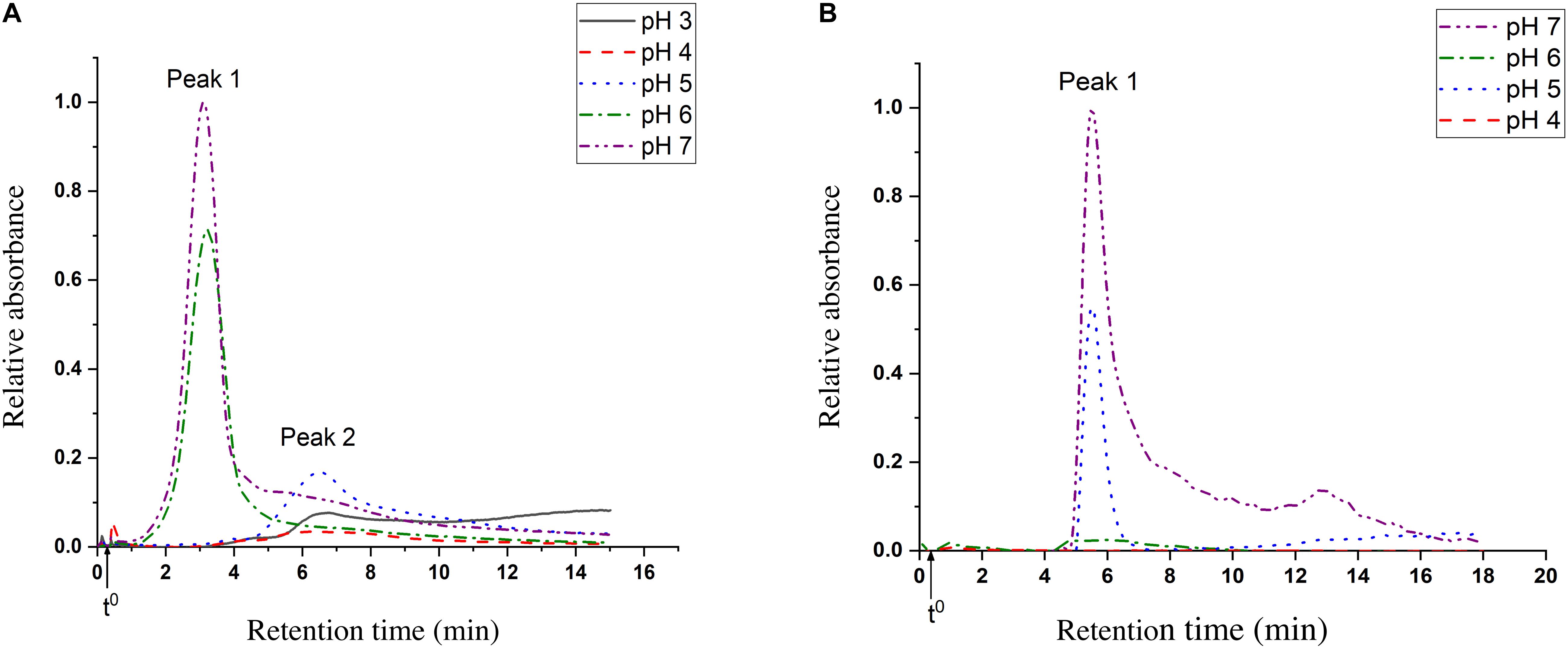
Figure 8. AF4 – UV fractograms of the proteins at different pH. (A) BlLyp, the injected mass is 0.088 mg and the void time t0 17 s. (B) LrLyp, the injected mass is 0.12 mg and the void time (t0) is 22 s.
For the molecular weight calculations, it is necessary to have both, MALS and UV fractograms. But, due to the MALS signal was not sufficient for LrLyp, the molecular weight was only calculated for BlLyp. A characteristic plot for this is shown in Figure 8, where the molecular weight is plotted in the y axis. The resulting molecular weight is the average of the top of the peak (from 2.6 to 3.7 min) and is reported in Table 3 together with the hydrodynamic radii for peak 1 and 2 in Figures 8A,B.
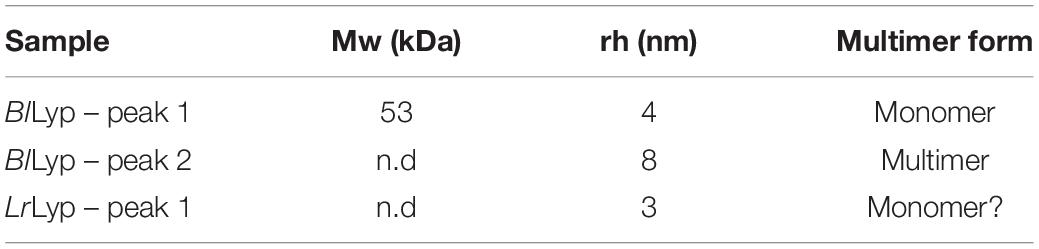
Table 3. Multimerization analysis.
The results from Table 3 indicate that the Mw obtained from peak 1 (BlLyp), 53 kDa, agrees with the theoretical Mw (52.36 kDa), therefore it corresponds to the monomeric form with a rh higher than BSA (3.3 nm). The Mw for peak 2 was not calculated because the LS signal-to-noise ratio was insufficient. Figure 8 suggests that BlLyp is predominantly monomeric at pHs 6 and 7, while at pH 5 it forms multimers. At lower pH no significant peaks were observed, suggesting that the enzyme precipitate or is degraded and lost from the analysis. On the other hand, the rh calculated for LrLyp suggest that the molecule appears somewhat smaller than BSA (Magnusson et al., 2012) (rh = 3.9 nm and Mw = 66.4 kDa), therefore it could correspond to the monomeric form. If that is the case, then at pHs 5 and 7 the main form would be monomeric, while at the other pH neither multimer nor monomer forms were identified.
The search of homologous crystalized proteins in the Protein Data Bank (PDB) via Blast, has given no significant homologous for both enzymes. However, a computational model for LrLyp was possible to obtain using remote homology techniques with Phyre2 (Kelley et al., 2015). This initial model was validated with ProSa-web (Wiederstein and Sippl, 2007) giving a Z-score of −3.56. This score was improved to −5.21 by refining the initial model by short molecular dynamic simulations (500 ps) with YASARA (Krieger and Vriend, 2014). The refinement has shown improvements in the whole structure, especially in the region of the C-terminal (Supplementary Information S3).
The molecular model of LrLyp resulted in a single domain protein with α/β-hydrolase fold, typical in lipases, esterases and other hydrolases (Figure 9A). The β-sheet is formed by five parallel β-strands and seven α-helixes, in addition, 210 helixes were predicted. In the active site region, long loops are predicted, Val98-Ala117, Gly147-Ser160, Ile183-Lys240 among the longest. These loops can be unmodeled regions due to the lack of crystallographic homologous available in the PDB. On the other hand these loops also can be flexible regions that affect the selectivity of substrates as is discussed in the next section. Despite of the difficulties to model the active site region, interestingly, superimposing the model with the no homologous crystallographic structure (PDB code: 1ESC) of a esterase from Streptomyces scabies (Figure 9B), it was found that the catalytic amino acids are well conserved. Thus, Ser21, His222, and Asn93 were predicted as potential catalytic amino acids in LrLyp (Figure 9C).
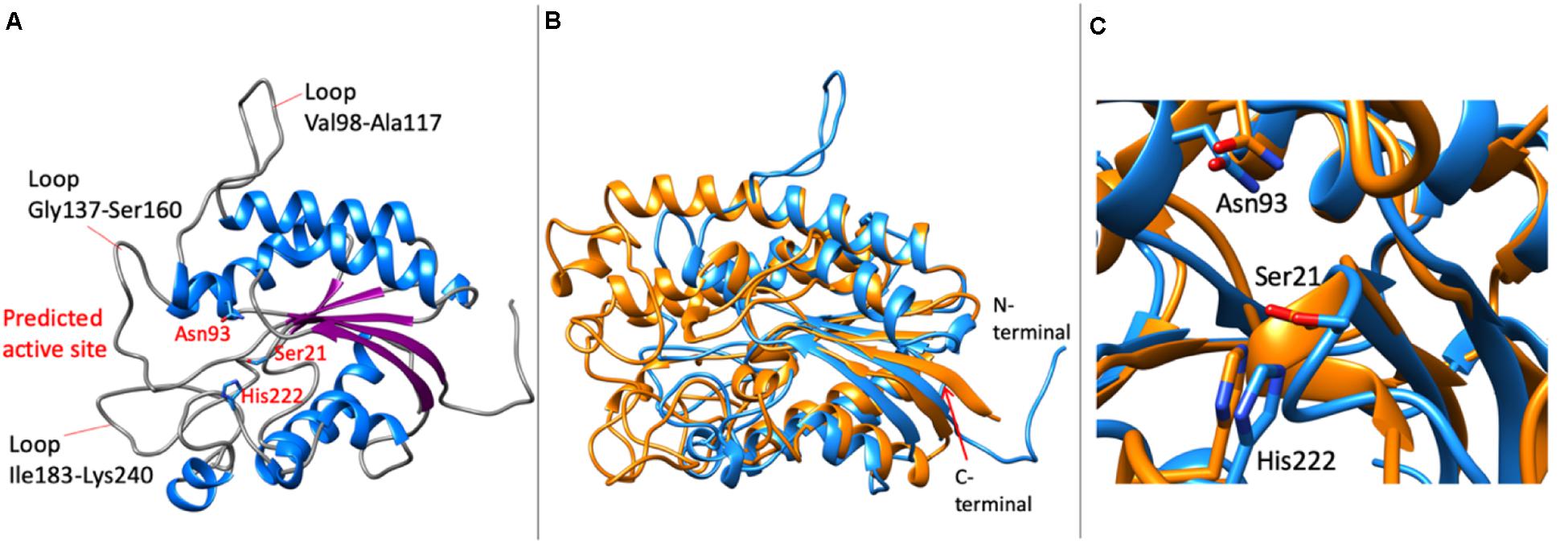
Figure 9. Molecular model of LrLyp. (A) Overall model showing the loops predicted surrounding the catalytic site and putative catalytic amino acids. (B) Superimposition of the molecular model (in blue) with the crystallographic structure (in orange) of an esterase from Streptomyces scabies (PDB code: 1ESC). (C) Detail of the conserved catalytic amino acids predicted by superimposition of the crystallographic structure of the esterase from S. scabies and the model of LrLyp. The catalytic amino acids in the structure 1ESC are Asn106, Ser14, and His283.
Both Lactobacillus rhamnosus GG and Bifidobacterium longum NCC 2705 are among of the most studied human probiotic strains. Despite that, the molecular bases of many mechanisms involved in the probiotic – host interactions are not clear yet. Probiotic proteins excreted to the extracellular medium could have effect in the interaction with the host. Thus, putative lipolytic enzymes (denoted as Lyp in this work) annotated in the genomes of L. rhamnosus GG and B. longum have been selected due to the presence of a signal peptide in the N-terminal, which indicates that these proteins are excreted and could have activity on host lipids, fat ingested by the host and even on other metabolites and drugs containing acyl ester bonds.
Based on amino acid sequences analysis, LrLyp and BlLyp belong to the lipolytic enzymes families II and IV, respectively. LrLyp has the GDSL motif, which define the family II. This family differs from the others because contain the amino acid sequence motif GDSL (block I) instead of the typical GXSXG, located in the active site, where S is the catalytic serine (Akoh et al., 2004). There are additional three conserved blocks that characterize this family. Blocks II and III contain conserved glycine (G) and asparagine (N), respectively; both serve as proton donors to the oxyanion hole (Kovacic et al., 2019). Block V contains the catalytic base, histidine (H), which deprotonates the hydroxyl group of the catalytic serine located in the block I, increasing its nucleophilicity (Figure 2). This family is subdivided in two clades, where clade II differs from I because it has an additional motif named block IIIa, no directly involved in catalysis (Leščić Ašler et al., 2010). Because LrLyp lacks block IIIa, it is subclassified in clade I (Figure 2). Enzymes of this family usually have a broad range of different substrates (Leščić Ašler et al., 2010).
BlLyp contains the characteristic family IV motif, GDSAGG. This family have four conserved blocks (Arpigny and Jaeger, 1999). The block I contains the consensus sequence HGGG, involved in stabilization of oxyanion hole through hydrogen bonds. Block II contains the catalytic serine (S), while the block IV the catalytic base, histidine (H). Interestingly, the proton donor predicted is a cysteine (C) in BlLyp, instead of the conserved aspartate (D) (Figure 3). The role of this unusual C will be clarified by mutational analysis in conjunction with structural studies. BlLyp also contains the sequence YRLA, conserved in the family IV and suggested as block V (Kim et al., 2015). Remarkably, this family groups enzymes with similarity to the mammalian hormone-sensitive lipases (HSL). Nevertheless, the substrate specificity in the bacterial HSL-family differs significantly. Human HSL can hydrolyze a broad range of substrates, both short esters as well as water insoluble substrates, however, bacterial HSL have shown activity on esters of short chain fatty acids (Chahinian et al., 2005).
Despite the classification based on amino acid sequences, these families are still poorly characterized. The experimental results have shown activity on a number of pNP-esters, with preference on esters of short chain fatty acids in the case of LrLyp (Figure 5), which is consistent with the gene annotation. The highest activity was on pNP-butyrate; indeed, butyrate and other fatty acids are commonly found in esterified form in plant oils and animal fat. For instance, bovine milk contains a broad variety of both short and large chain fatty acids esterified with glycerol in specific positions (Lindmark Månsson, 2008). The activities reported in this work are significantly lower comparing with the activities of other bacterial lipolytic enzymes. Therefore, it would be interesting to study the activity of LrLyp and BlLyp using natural substrates. In fact, the natural substrate and biological function of these novel enzymes remains unknown.
The multimerization study by AF4 indicated that LrLyp is monomer at pHs 5 and 7, while BlLyp is monomer at pHs 6 and 7, which suggests that these enzymes are active in monomeric form. Regarding to the kinetic studies, it was difficult to determine accurate kinetic constants for LrLyp due to the insolubility of the substrates in high concentrations, however the kinetic curve for the substrate pNP-laurate shows substrate inhibition when de concentration of the substrate is higher than 5 mM. In the case of BlLyp, the kinetic constant obeys a Michaelis-Menten behavior for pNP-laurate. Comparing the KM obtained from both enzymes, LrLyp shows the highest substrate affinity respect to pNP-laurate.
On the other hand, the molecular model of LrLyp obtained has allowed the prediction of catalytic amino acids. Interestingly, the superimposition of the molecular model with the crystallographic structure of a no homologous esterase from Streptomyces scabies (Wei et al., 1995) has shown highly conserved catalytic amino acids consistent with the catalytic mechanism suggested for this type of esterases. It is also suggested that the loops surrounding the active site can adopt open and close conformations in lipolytic enzymes, determining the substrate specificity. Thus, the loops predicted in the modeled LrLyp enzyme could be flexible structures that affect the interactions with the substrates. Crystallographic studies of LrLyp together with mutational analysis will support to understand the mechanism of reaction of this enzyme.
It was found that the putative extracellular acyl-hydrolase/lipase from the human probiotic bacteria Lactobacillus rhamnosus and Bifidobacterium longum have activity on pNP-esters. L. rhamnosus enzyme has shown preference for pNP-esters of short chain fatty acids, and its optimal activity is near to the pH 7 and 37°C. The bioinformatic analysis and computational model has allowed the prediction of catalytic amino acids for the Lactobacillus rhamnosus enzyme and suggests that large loops surrounding the active site could determine the substrate specificity. B. longum enzyme has been tested on pNP-laurate, showing activity at pH 7 and human physiological temperature. On the other hand, both enzymes are active in their monomeric form at pH 7.
All datasets generated for this study are included in the article/Supplementary Material.
JL-P designed the study, did the bioinformatic analysis and modeling, and wrote and corrected the manuscript. LN designed the study, analyzed the data, and corrected the manuscript. PM performed the experiments related to protein purification and kinetic, as well as wrote the first draft of the manuscript. A-SB performed the experiments about multimerization/aggregation analysis. BP produced and purified protein and did the DSF analysis. JP and AH participated in the revision of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Swedish International Development Agency (SIDA): a collaboration project between Universidad Mayor de San Andrés (Bolivia) and Lund University (Sweden).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Maria Gourdon and the Lund Protein Production Platform (PL3) staff for their assistance in using the DSF instrument.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01534/full#supplementary-material
Akoh, C. C., Lee, G.-C., Liaw, Y.-C., Huang, T.-H., and Shaw, J.-F. (2004). GDSL family of serine esterases/lipases. Prog. Lipid Res. 43, 534–552. doi: 10.1016/j.plipres.2004.09.002
Arpigny, J. L., and Jaeger, K.-E. (1999). Bacterial lipolytic enzymes: classification and properties. Biochem. J. 343, 177–183. doi: 10.1042/bj3430177
Brigidi, P., Vitali, B., Swennen, E., Bazzocchi, G., and Matteuzzi, D. (2001). Effects of probiotic administration upon the composition and enzymatic activity of human fecal microbiota in patients with irritable bowel syndrome or functional diarrhea. Res. Microbiol. 152, 735–741. doi: 10.1016/s0923-2508(01)01254-2
Brusaferro, A., Cozzali, R., Orabona, C., Biscarini, A., Farinelli, E., Cavalli, E., et al. (2018). Is it time to use probiotics to prevent or treat obesity? Nutrients 10:1613. doi: 10.3390/nu10111613
Capurso, L. (2019). Thirty years of Lactobacillus rhamnosus GG: a review. J. Clin. Gastroenterol. 53, S1–S41.
Castro-Ochoa, L. D., Rodríguez-Gómez, C., Valerio-Alfaro, G., and Ros, R. O. (2005). Screening, purification and characterization of the thermoalkalophilic lipase produced by Bacillus thermoleovorans CCR11. Enzyme Microb. Technol. 37, 648–654. doi: 10.1016/j.enzmictec.2005.06.003
Chahinian, H., Ali, Y. B., Abousalham, A., Petry, S., Mandrich, L., Manco, G., et al. (2005). Substrate specificity and kinetic properties of enzymes belonging to the hormone-sensitive lipase family: comparison with non-lipolytic and lipolytic carboxylesterases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1738, 29–36. doi: 10.1016/j.bbalip.2005.11.003
Choi, J., Lee, S., Linares-Pastén, J. A., and Nilsson, L. (2018). Study on oligomerization of glutamate decarboxylase from Lactobacillus brevis using asymmetrical flow field-flow fractionation (AF4) with light scattering techniques. Anal. Bioanal. Chem. 410, 451–458. doi: 10.1007/s00216-017-0735-6
Choo, D.-W., Kurihara, T., Suzuki, T., Soda, K., and Esaki, N. (1998). A cold-adapted lipase of an Alaskan psychrotroph, Pseudomonas sp. strain B11-1: gene cloning and enzyme purification and characterization. Appl. Environ. Microbiol. 64, 486–491. doi: 10.1128/aem.64.2.486-491.1998
Consortium, T. U. (2018). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515.
Côté, A., and Shareck, F. (2008). Cloning, purification and characterization of two lipases from Streptomyces coelicolor A3 (2). Enzyme Microb. Technol. 42, 381–388. doi: 10.1016/j.enzmictec.2008.01.009
Feller, G., Thiry, M., and Gerday, C. (1991). Nucleotide sequence of the lipase gene lip2 from the Antarctic psychrotroph Moraxella TA 144 and site-specific mutagenesis of the conserved serine and histidine residues. DNA Cell Biol. 10, 381–388. doi: 10.1089/dna.1991.10.381
Gorbach, S. L., and Goldin, B. R. (1989). Lactobacillus Strains and Methods of Selection. Google Patents US4839281A. Boston, MA: New England Medical Center Hospitals.
Håkansson, A., Magnusson, E., Bergenståhl, B., and Nilsson, L. (2012). Hydrodynamic radius determination with asymmetrical flow field-flow fractionation using decaying cross-flows. Part I. A theoretical approach. J. Chromatogr. A 1253, 120–126. doi: 10.1016/j.chroma.2012.07.029
Hill, C., Guarner, F., Reid, G., Gibson, G. R., Merenstein, D. J., Pot, B., et al. (2014). The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 11, 506–514. doi: 10.1038/nrgastro.2014.66
Kankainen, M., Paulin, L., Tynkkynen, S., Von Ossowski, I., Reunanen, J., Partanen, P., et al. (2009). Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human-mucus binding protein. Proc. Natl. Acad. Sci. 106, 17193–17198. doi: 10.1073/pnas.0908876106
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10:845. doi: 10.1038/nprot.2015.053
Kim, H. J., Jeong, Y. S., Jung, W. K., Kim, S. K., Lee, H. W., Kahng, H.-Y., et al. (2015). Characterization of novel family IV esterase and family I. 3 lipase from an oil-polluted mud flat metagenome. Mol. Biotechnol. 57, 781–792. doi: 10.1007/s12033-015-9871-4
Kovacic, F., Babic, N., Krauss, U., and Jaeger, K. (2019). “Classification of lipolytic enzymes from bacteria,” in Aerobic Utilization of Hydrocarbons, Oils and Lipids, ed. F. Rojo (Cham: Springer), 1–35. doi: 10.1007/978-3-319-39782-5_39-1
Krieger, E., Darden, T., Nabuurs, S. B., Finkelstein, A., and Vriend, G. (2004). Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins Struct. Funct. Bioinformatics 57, 678–683. doi: 10.1002/prot.20251
Krieger, E., and Vriend, G. (2014). YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30, 2981–2982. doi: 10.1093/bioinformatics/btu426
Leščić Ašler, I., Ivić, N., Kovačić, F., Schell, S., Knorr, J., Krauss, U., et al. (2010). Probing enzyme promiscuity of SGNH hydrolases. Chembiochem 11, 2158–2167. doi: 10.1002/cbic.201000398
Lindmark Månsson, H. (2008). Fatty acids in bovine milk fat. Food Nutr. Res. 52:1821. doi: 10.3402/fnr.v52i0.1821
Magnusson, E., Håkansson, A., Janiak, J., Bergenståhl, B., and Nilsson, L. (2012). Hydrodynamic radius determination with asymmetrical flow field-flow fractionation using decaying cross-flows. Part II. Experimental evaluation. J. Chromatogr. A 1253, 127–133. doi: 10.1016/j.chroma.2012.07.005
Mandrich, L., Menchise, V., Alterio, V., De Simone, G., Pedone, C., Rossi, M., et al. (2008). Functional and structural features of the oxyanion hole in a thermophilic esterase from Alicyclobacillus acidocaldarius. Proteins Struct. Funct. Bioinformatics 71, 1721–1731. doi: 10.1002/prot.21877
Mastihuba, V., Kremnický, L. R., Mastihubová, M., Willett, J., and Côté, G. L. (2002). A spectrophotometric assay for feruloyl esterases. Anal. Biochem. 309, 96–101. doi: 10.1016/s0003-2697(02)00241-5
Ohara, K., Unno, H., Oshima, Y., Hosoya, M., Fujino, N., Hirooka, K., et al. (2014). Structural insights into the low pH adaptation of a unique carboxylesterase from ferroplasma altering the pH optima of two carboxylesterases. J. Biol. Chem. 289, 24499–24510. doi: 10.1074/jbc.m113.521856
Okamura, Y., Kimura, T., Yokouchi, H., Meneses-Osorio, M., Katoh, M., Matsunaga, T., et al. (2010). Isolation and characterization of a GDSL esterase from the metagenome of a marine sponge-associated bacteria. Mar. Biotechnol. 12, 395–402. doi: 10.1007/s10126-009-9226-x
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785. doi: 10.1038/nmeth.1701
Saxelin, M. (2008). Probiotic formulations and applications, the current probiotics market, and changes in the marketplace: a European perspective. Clin. Infect. Dis. 46, S76–S79.
Schell, M. A., Karmirantzou, M., Snel, B., Vilanova, D., Berger, B., Pessi, G., et al. (2002). The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. U.S.A. 99, 14422–14427. doi: 10.1073/pnas.212527599
Segers, M. E., and Lebeer, S. (2014). Towards a better understanding of Lactobacillus rhamnosus GG-host interactions. Microb. Cell Fact. 13, (Suppl. 1):S7.
Talker-Huiber, D., Jose, J., Glieder, A., Pressnig, M., Stubenrauch, G., and Schwab, H. (2003). Esterase EstE from Xanthomonas vesicatoria (Xv_EstE) is an outer membrane protein capable of hydrolyzing long-chain polar esters. Appl. Microbiol. Biotechnol. 61, 479–487. doi: 10.1007/s00253-003-1227-5
Wahlund, K.-G., and Nilsson, L. (2012). “Flow FFF–basics and key applications,” in Field-Flow Fractionation in Biopolymer Analysis, eds S. Williams and K. Caldwell (Berlin: Springer), 1–21. doi: 10.1007/978-3-7091-0154-4_1
Wei, Y., Schottel, J. L., Derewenda, U., Swenson, L., Patkar, S., and Derewenda, Z. S. (1995). A novel variant of the catalytic triad in the Streptomyces scabies esterase. Nat. Struct. Biol. 2:218. doi: 10.1038/nsb0395-218
Wiederstein, M., and Sippl, M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410.
Young, S. L., Simon, M. A., Baird, M. A., Tannock, G. W., Bibiloni, R., Spencely, K., et al. (2004). Bifidobacterial species differentially affect expression of cell surface markers and cytokines of dendritic cells harvested from cord blood. Clin. Diagn. Lab. Immunol. 11, 686–690. doi: 10.1128/cdli.11.4.686-690.2004
Keywords: acyl-hydrolase/lipase, Lactobacillus rhamnosus GG, Biffidobacterium longum, asymmetrical flow field-flow fractionation, differential scanning fluorimetry
Citation: Manasian P, Bustos A-S, Pålsson B, Håkansson A, Peñarrieta JM, Nilsson L and Linares-Pastén JA (2020) First Evidence of Acyl-Hydrolase/Lipase Activity From Human Probiotic Bacteria: Lactobacillus rhamnosus GG and Bifidobacterium longum NCC 2705. Front. Microbiol. 11:1534. doi: 10.3389/fmicb.2020.01534
Received: 12 March 2020; Accepted: 12 June 2020;
Published: 24 July 2020.
Edited by:
Afef Najjari, Tunis El Manar University, TunisiaReviewed by:
Armen Trchounian, Yerevan State University, ArmeniaCopyright © 2020 Manasian, Bustos, Pålsson, Håkansson, Peñarrieta, Nilsson and Linares-Pastén. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Nilsson, bGFycy5uaWxzc29uQGZvb2QubHRoLnNl; Javier A. Linares-Pastén, amF2aWVyLmxpbmFyZXNfcGFzdGVuQGJpb3Rlay5sdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.