 Chih-Jung Chen
Chih-Jung Chen Yhu-Chering Huang1,2
Yhu-Chering Huang1,2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 July 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01414
The genomic evolution in vivo in persistent infection was critical information for understanding how methicillin-resistant Staphylococcus aureus (MRSA) was adapted to host environments with high antibiotic selective pressure. Thirty-two successive MRSA blood isolates with incremental non-susceptibility to vancomycin (VISA), daptomycin (DRSA), and/or linezolid (LRSA) were isolated from a patient failing multiple courses of antimicrobial therapy during 1,356 days of bacteremia. Whole genome sequencing (WGS) for all consecutive isolates were conducted to characterize the evolutionary pathways, resistance-associated mutations and their temporal relationship with antimicrobial treatment. The WGS-based phylogeny categorized the isogenic strains into three major clades, I (22 isolates), II (7 isolates), and III (3 isolates), respectively, harboring a median (range) of 7 (1–30), 62 (53–65), and 118 (100–130) non-synonymous mutations when compared to the very first isolate. Clade I strains were further grouped into early and late subclades, which, respectively, shared the most recent common ancestor with Clade III strains at day 393.7 and Clade II strain at day 662.5. Clade I and Clade III strains were characterized, respectively, with high rates of VISA (9/22, 40.9%) and VISA-and-DRSA phenotype (2/3, 66.7%). Linezolid-resistance including VISA-DRSA-and-LRSA phenotype was exclusively identified in Clade II strains after eight courses of linezolid treatment. The LRSA displayed a small colony variant phenotype and were associated with G2576T mutations in domain V region of 23S rRNA. Substantial loss of mobile elements or alleles mediating resistance or virulence were identified during the evolution of multi-resistance. However, the gene loss might not be correlated to the development of VISA, DRSA, or LRSA phenotype. In conclusion, MRSA in persistent bacteremia was adapted to harsh host environment through multiple pathways involving both resistance-associated mutations and extensive gene loss.
Methicillin-resistant Staphylococcus aureus (MRSA) bacteremia is a devastating disease associated with high rates of complications and mortality especially in those with underlying conditions (Charles et al., 2004; Tong et al., 2020). Treatment failures including persistent bacteremia were frequently identified in patients with medical implant or prothesis, with greater number of comorbidities and greater disease severity or infections with non-susceptible strains (Charles et al., 2004; Murray et al., 2013; Moise et al., 2016; Yang et al., 2018; Tong et al., 2020). Glycopeptide remains the drug of choice for treatment of MRSA bacteremia though accumulating evidence suggested daptomycin treatment may be associated with improved outcome, especially in patients whose diseases were caused by MRSA strains with reduced susceptibility to vancomycin (Murray et al., 2013; Claeys et al., 2016; Yang et al., 2018). Nevertheless, with the currently available anti-MRSA agents, the clinical and microbiological cure rates of MRSA bacteremia remain unsatisfactory.
Duration of bacteremia was an important factor predicting the outcome of patient with MRSA bloodstream infections (Hamdy et al., 2017). Development of drug resistance during treatment was a common observation in persistent MRSA bacteremia and may play an important role for the failure to eradicate the MRSA strains from bloodstreams (Friedman et al., 2006; Chen et al., 2014b, 2015). An understanding of the in vivo evolution of MRSA strains during the development of incremental resistance to the commonly used anti-MRSA agents including vancomycin, daptomycin, and linezolid may help deploy effective therapeutic strategy against the devastating infectious disease.
In the present study, we characterized the genomic evolution in a series of isogenic MRSA blood isolates exhibiting incremental non-susceptibilities to vancomycin, daptomycin, and linezolid. The isolates were belonged to sequence type (ST) 5 clone and consecutively isolated from a patient failing multiple courses of antimicrobial therapy against persistent bacteremia for 3 years before his death. By whole-genome sequencing (WGS), we were able to track all in vivo evolutionary clades, determined the phylogenetic relationship between isolates of different clades and identify the genetic alterations including mutations and recombinations associated with resistance to each of the three anti-MRSA agents. We also directly observed a correlation between the use of antimicrobial agents and the genetic changes in distinct evolutionary clades.
The study was approved by the by the institute review boards in Chang Gung Memorial Hospital (CMRPG3C1883), which allowed review of the medical data of the patient. A waiver of consent was granted given the retrospective nature of the project and anonymous analysis of the clinical information of the patient.
Approximately 1 month after an aortic repair of aneurysm and replacement with a vascular graft, a middle-aged patient developed MRSA bacteremia on January 10, 2006, for the first time. The bacteremia appeared to be temporarily controlled by administration of vancomycin followed by teicoplanin for 3 months. Unfortunately, the MRSA was cultivated from the bloodstream again in September 08, 2006 (the first available isolate, day 241 of bacteremia) and was afterward identified intermittently in blood cultures in the following 3 years until death on October 20, 2009 (the last available isolate, day 1,356). The anti-MRSA agents including glycopeptides, linezolid, daptomycin, tigecycline, and trimethoprim-sulfamethoxazole were used interchangeably to treat the refractory bacteremia, but the attempts were all unsuccessful. A total of 32 MRSA were isolated from blood cultures, and the isolates gradually developed non-susceptibility to the anti-MRSA agents (Table 1), with the first strain exhibiting vancomycin-intermediate S. aureus (VISA) phenotype at day 453 (LTF06, April 08, 2007), the first strain exhibiting both vancomycin-intermediate and daptomycin-resistant (VISA-DRSA) phenotype at day 936 (LTF15, August 03, 2008), and the first strain exhibiting resistance to vancomycin, daptomycin, and linezolid (VISA-DRSA-LRSA) at day 1196 (LTF22, April 20, 2009). Initial genotyping of the 32 strains disclosed that they were belonged to ST5 or its single locus variant, spa type t002 or t13754 and universally harboring SCCmec II (Supplementary Table S1).
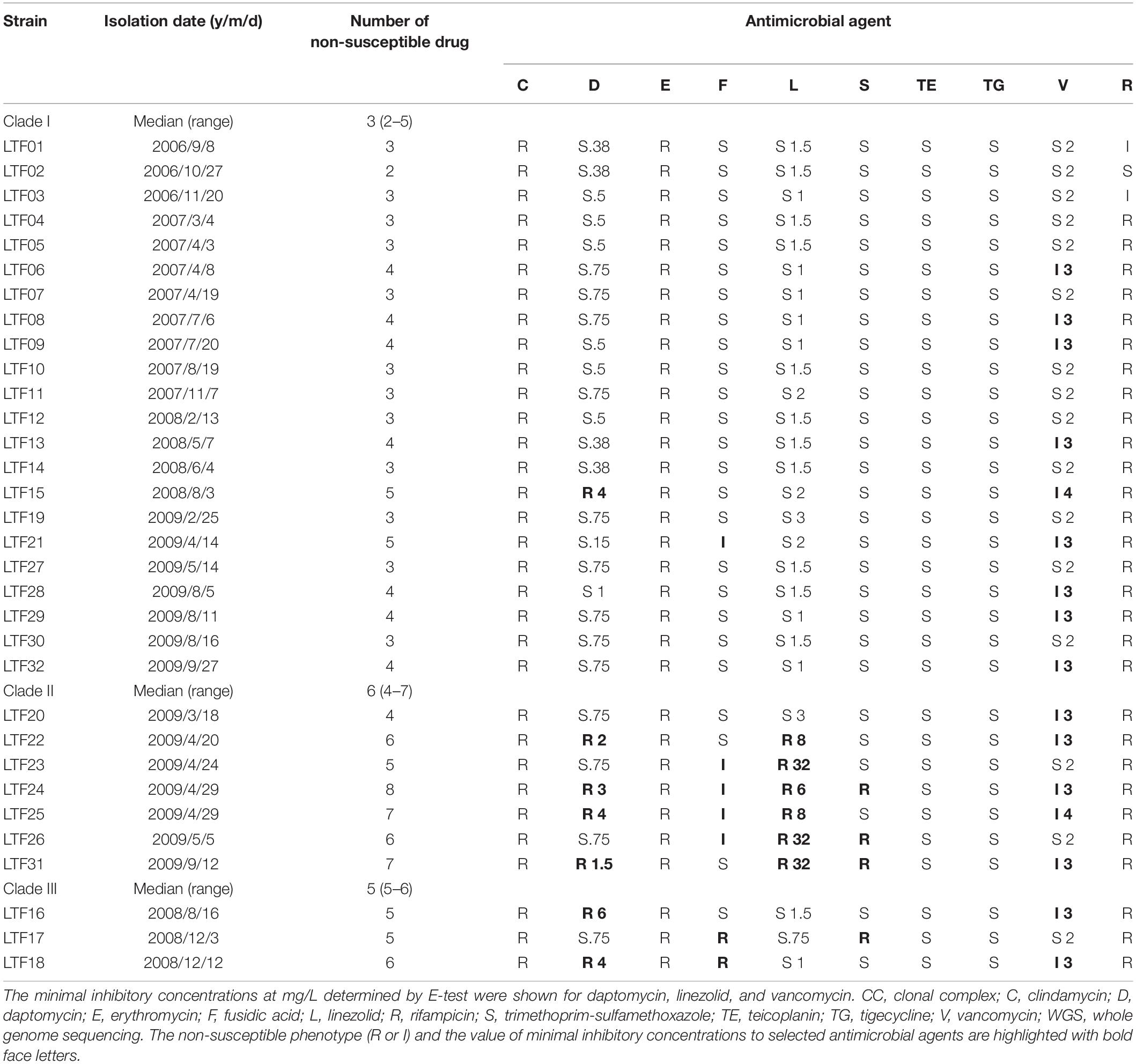
Table 1. Susceptibilities to 10 non-beta-lactam antimicrobial agents in 32 successive MRSA isolates belonging to CC5-SCCmecII and three WGS-defined clades in a patient with persistent bacteremia during 3 years.
A total of 32 MRSA strains (LTF01–LTF32, Table 1) were stored at −80°C after their first isolations from the patient. The strains used in this study were grown from their very first stocked cultures. Bacteria was grown in liquid or on solid basic medium (BM) (1% Pepton, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, 0.1% glucose) or tryptic soy broth (TSB) (Sigma, Munich, Germany) unless otherwise noted.
To clarify the phylogeny and the evolution rate of the 32 successive MRSA strains, another set of 71 non-duplicated clinical MRSA strains belonging to the same ST5 lineage were obtained from the strain bank. The strains were collected from the major hospitals in Taiwan during 1997 and 2014. The information of the strains including the molecular features had been characterized and presented elsewhere (Huang et al., 2006, 2008; Chen et al., 2010, 2014a).
The chromosomal DNA was extracted via QIAamp DNA Mini Kit (Qiagen, CA, United States). All isolates were detected for mecA gene and SCCmec types by multiplex PCR strategy as described previously (Kondo et al., 2007). The control strains for SCCmec types I, II, III, and IVa, kindly provided by Keiichi Hiramatsu, was as follows: type I, NCTC10442; type II, N315; type III, 85/2082; and type IVa, JCSC4744.
Susceptibilities to clindamycin, erythromycin, fusidic acid, trimethoprim-sulfamethoxazole, tigecycline, and rifampicin were determined using the standard disk diffusion method according to the CLSI guidelines (CLSI, 2017). Briefly, S. aureus isolates were grown in TSB at 37°C to a cell density equivalent to that of a 0.5 McFarland standard. The bacteria were then streaked onto Mueller–Hinton agar plates, and the plates were incubated at 37°C for 24 h before reading. The minimal inhibitory concentrations (MICs) of vancomycin, daptomycin, and linezolid were determined by Etest (bioMe’ rieux) according to the manufacturer’s instructions.
To observe the colony morphologies, the bacteria were refreshed from frozen stock on TSA with 5% sheep blood. A single colony was picked and streaked on BM and TSA with 5% sheep blood, respectively. Agar plates were incubated at 37°C with 5% CO2 for 24 h before reading.
The chromosomal DNA was extracted via QIAamp DNA Mini Kit (Qiagen, CA, United States). The WGS of the strains was performed using the next-generation sequencing technology, the Illumina MiSeq sequencer (Illumina, San Diego, CA, United States). The raw data of each strain was cleaned by adapter trimming and exclusion of reads in which greater than 45% of bases were of quality score < 20 (<Q20). A range of 199 million to 318 million bases was generated for each strain, with 71.1- to 113.6-fold base coverage across the genome. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under accession number PRJNA495118. The analysis strategy was as follows.
De novo assembly of each of the strains was conducted using SPAdes 3.11.1 (Bankevich et al., 2012). The default k-mer lengths of 21, 33, 55, 77, 99, and 127 were set and –careful option was used during assembly. The scaffolds outputs greater than 200 bp were used in the following analysis.
The multi-alignment of the core genomes of 32 successive MRSA strains was conducted with the parsnp program (Treangen et al., 2014). The ST5 S. aureus strain N315 was used as reference strain during the procedure. The multi-alignment file generated by the parsnp script was subsequently used for maximum likelihood phylogeny construction based on the single nucleotide polymorphisms (SNPs) and short insertions or deletions (INDELs) outside of the potential recombination regions with the Gubbins procedure (Croucher et al., 2015). The final phylogenetic tree was input into Interactive Tree of Life1 for further annotation.
In addition to the maximum likelihood phylogeny, the time-scaled phylogeny was constructed with the Bayesian analysis of the molecular sequences using Markov chain Monte Carlo. The BEAST program (v 1.10.4) was used to estimate the clock rates of evolution and the time to the most recent common ancestor (MRCA) (Drummond and Rambaut, 2007). An HKY model with estimated base frequencies, uncorrelated relaxed clock type, with lognormal distribution and constant size of tree prior were used. Three independent chains were run for 100 million generations and sampling every 1,000 generations. The effect sample size values were checked by tracer (v1.7.1) and were greater than 200. A burn-in of 10 million states was removed from each of the three independent runs before combining the results from those runs with the logcombiner program (v 1.10.4) from the BEAST package. The final tree was output and annotated with Figtree program2.
The annotation was conducted using the Rapid Annotation Server provided by the National Microbial Pathogen Data Resource3.
The cleaned raw data of WGS of 32 consecutive MRSA isolates were, respectively, aligned to the reference genome (N315, GenBank: BA000018.3) using bowtie2 and samtools (Li and Durbin, 2009; Johnson et al., 2014). The variants call was firstly conducted between N315 and 32 MRSA isolates using the bcftools with the command of call4. The variants call then was conducted between the first isolate (LTF01) and the sequential isolates (LTF02–LTF32) using bcftools with the command of isec. The identified variants in LTF02-LTF32 were verified by direct visualization and comparisons of the BAM files of LTF01 and those of the other isolates in the Intergrative Genomics Viewer (Robinson et al., 2011). The variants annotations and predicted effects of SNPs on amino acid alterations were conducted using the snpEFF (Cingolani et al., 2012). The variants appeared in the resistant strains but not in susceptible strains were considered to be associated with drug resistance. The position, effect, and affected alleles of the VISA or DRSA-associated mutations including SNPs and short INDELs were illustrated in the circus plot created with Circa5.
The carriage of plasmids, resistance, and virulence genes among the MRSA strains were detected by srst2 procedure using the PlasmidFinder, ARG-ANNOT and virulence factor of pathogenic bacteria database6, respectively. To visualize and compare the draft genomes of 32 stains, the Circular Genome Viewer (CGView) was used to create the map (Stothard and Wishart, 2005).
The descriptive statistics were conducted with Stata 13.0 (StataCorp, College Station, TX, United States). The comparisons of numbers of SNPs or INDELs between vancomycin-susceptible S. aureus (VSSA) and VISA strains and numbers of antimicrobial agents to which the strains of different clades were non-susceptible to were conducted with the Mann Whitney non-parametric test method. The comparisons of proportions of non-synonymous SNPs or INDELs and between strains of different genetic clades were conducted with Chi-square or Fisher’s exact test where appropriate. The data was analyzed and figures were output by using the GraphPad Prism 5 for Windows (GraphPad Software, San Diego, CA, United States). P-value < 0.05 was considered of statistical significance.
The long course of bacteremia across 3 years, the subtle change of ST and spa types, and variable antibiograms in the successive strains raised the first question whether all of the successive MRSA strains were derived from the same ancestor or the persistent bacteremia was actually resulted from repeated infections by new strains. To clarify this issue, we conducted the whole genome sequencing of the 32 strains along with a reference set of 71 non-duplicated clonal complex (CC) 5 MRSA clinical isolates collected nationwide from 1997 to 2014 (Supplementary Table S2). The core genome-based phylogeny of the 103 strains disclosed that the 32 successive bacteremic strains were clustered together, which could be clearly distinguished from the other contemporary clinical ST5 isolates (Figure 1). The finding confirmed that the strains causing persistent bacteremia were diverged from the same parental strain and were of isogenic nature.
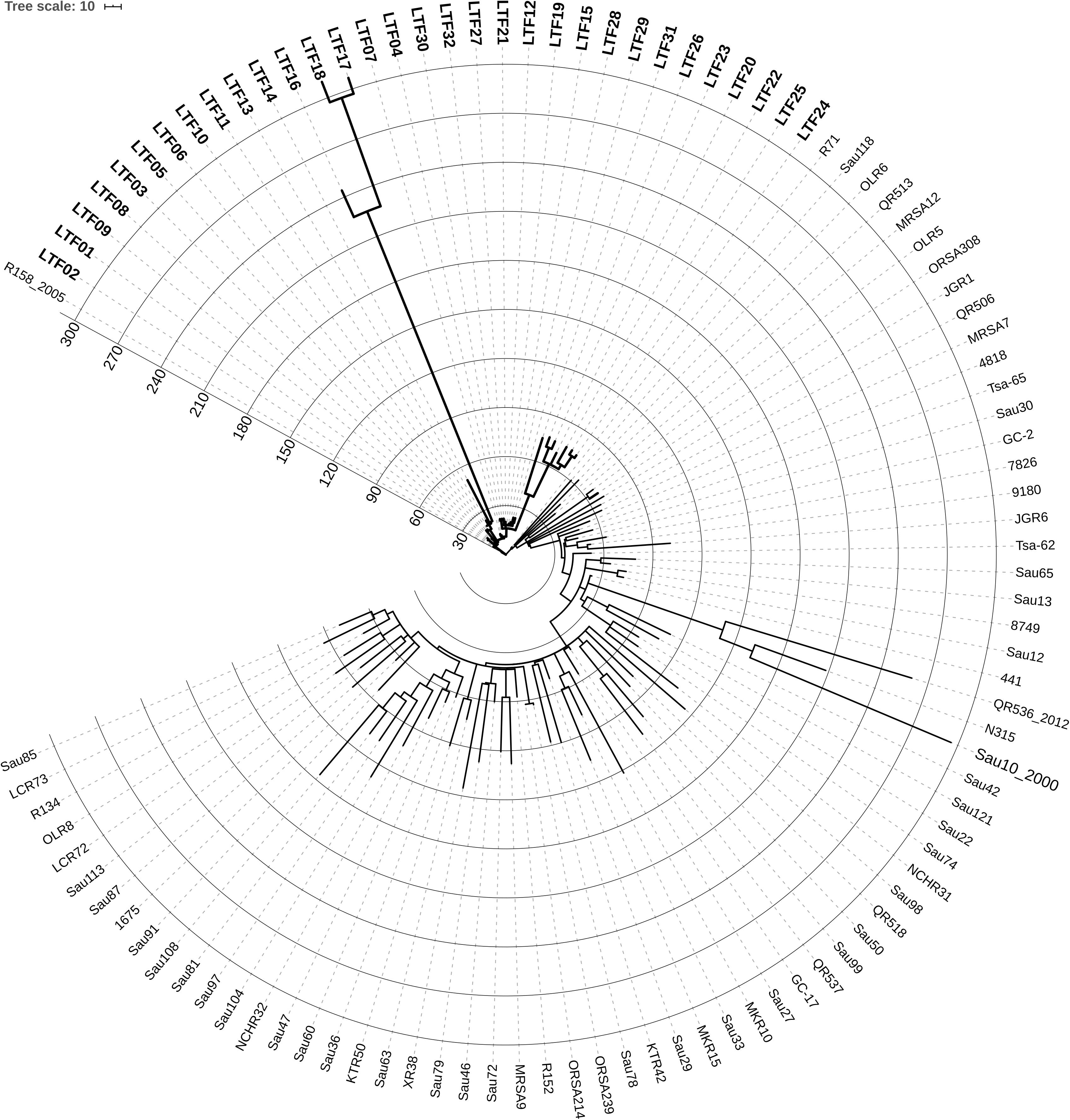
Figure 1. Circular display mode showing the core genome-based phylogeny of 32 successive ST5 MRSA strains (labeled with bold-face letters) from a patient with persistent bacteremia and 71 ST5 MRSA strains (Supplementary Table S2 for detailed information) collected nationwide in Taiwan from 1997 to 2014. The N315 strain belonging to ST5 was included for reference. The numbers at the starts of circles indicates the number of SNPs or short INDELs mutations in the strains, which was larger in certain strains of the persistent bacteremia than in the nationwide strains. The first available MRSA isolate (LTF01) from the patient with persistent bacteremia was closely related to a strain (R158), which was isolated from another patient in the same hospital few years earlier, suggesting the persistent bacteremia was initially caused by the endemic ST5 MRSA in this facility.
When compared to the first isolate (LTF01), a total of 709 SNPs or short INDELs including 320 (45.1%) non-synonymous mutations were identified in the core genomes of the 31 successive isolates (Table 2). The detailed information including the positions of mutations, altered nucleotides, effect of the mutations, and the affected alleles are displayed in the Supplementary Data Sheet S1. Multiple likelihood analysis of the 709 SNP and short INDELs mutations divided the isolates into three major clades (Clades I, II, and III, Figure 2). Clade I strains could be further divided into two subclades, Ia and Ib, respectively, appeared in early course (before day 876 when strain LTF14 was isolated) and late course (after day 764 when strain LTF12 was isolated). The phylogeny demonstrated that strains of Clade III were derived from the early Clade Ia, whereas strains of Clade II were genetically closer to the late Clade Ib.
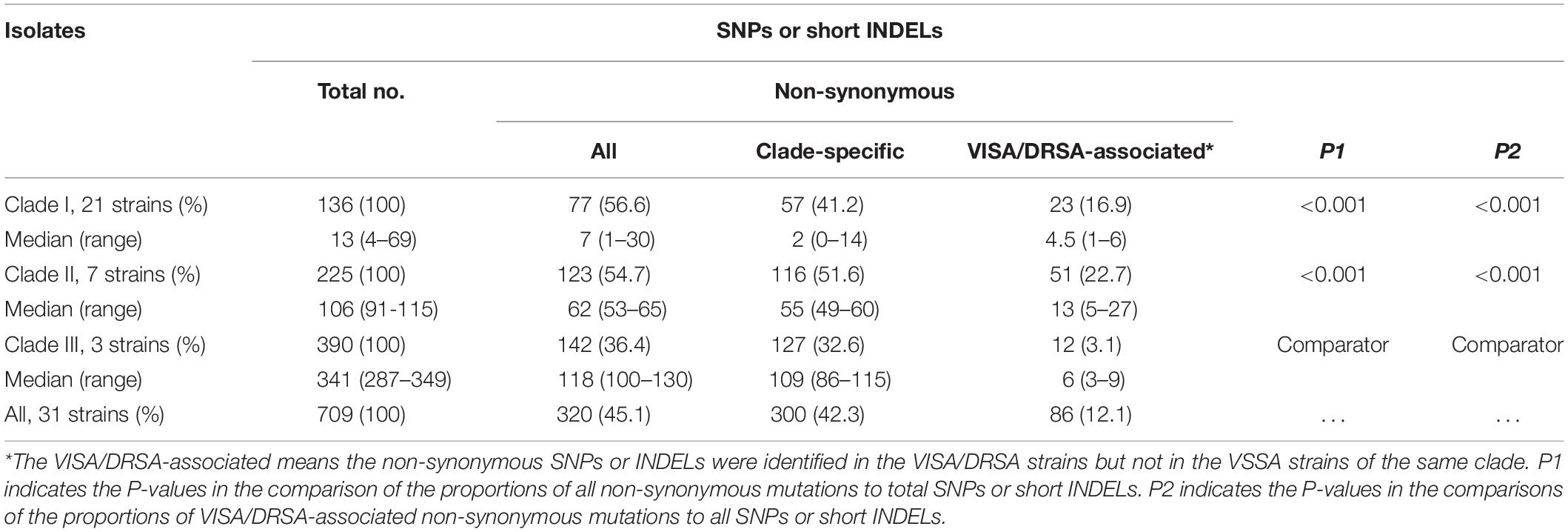
Table 2. Numbers of single nucleotide polymorphisms (SNPs) or short insertions/deletions (INDELs) among three clades of MRSA isolates causing persistent bacteremia during 3 years, comparison to the parenteral strain, LTF01.
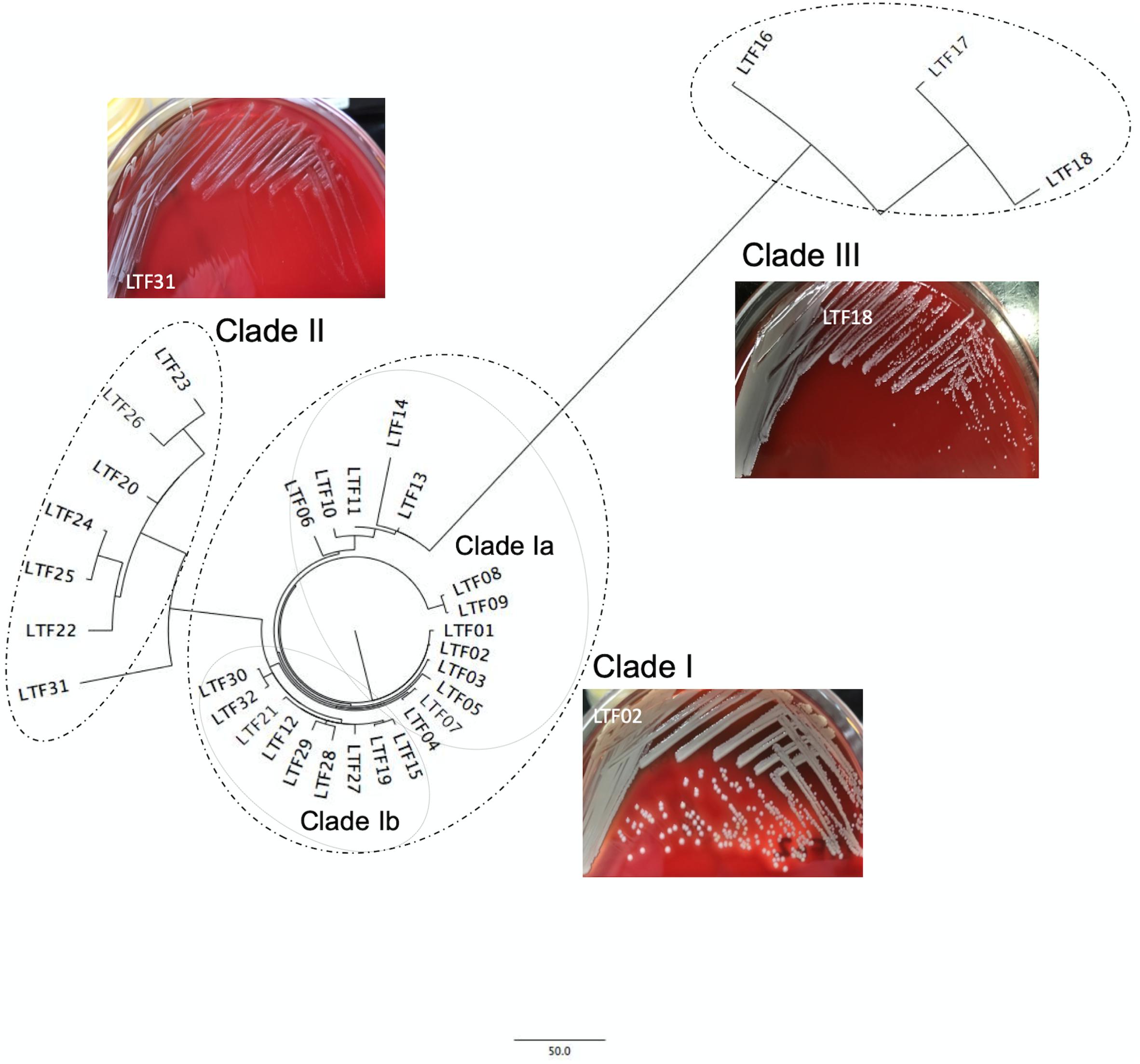
Figure 2. Core genome-based phylogenetic analysis of 32 ST5 MRSA strains from a patient with persistent bacteremia. The successive bacteremic strains were belonged to three major genetic clades with two subclades, Ia and Ib, in Clade I. The bacterial colonies of representative strains on blood agar plates were shown for each clade. The colonies of Clade II strains were at significantly smaller size and whiter color when compared to the strains of Clade I.
A significant morphology change of the bacterial colonies was identified for Clade II and Clade III strains. The colonies of the Clade II strains in agar plates were all at tiny sizes and in whiter color compared to the Clade I strains, indicating a small colony variant (SCV) phenotype (Figure 2 and Supplementary Presentation S1). The sizes of the Clade III strains appeared to be in-between of the Clade I and Clade II strains. Along with the morphological change, the Clade II strains also exhibited greater drug resistance when compared to the strains in Clade I. A range of 4–7 (median, 6) non-susceptible antimicrobial agents was identified in Clade II strains, which was significantly greater than the numbers of non-susceptible drugs in Clade I strains (2–5 drugs [median, 3], P < 0.001, Table 1). The LRSA strains including those of VISA-DRSA-LRSA phenotype were identified exclusively in strains of Clade II (LTF22, 24, 25, and 31).
The proportions of non-synonymous mutations were similar in strains of Clade I and Clade II (56.6% vs. 54.7%, P = 0.718, Table 2) but was significantly lower in strains of Clade III (36.4%, P < 0.001 for both comparisons to Clade I and Clade II, Table 2). A majority (300, 93.8%) of the 320 non-synonymous mutations occurred in only strains of certain clade and were considered clade-specific. The remaining 20 mutations harbored by strains of different clades are listed in Supplementary Table S3. The distributions of the 20 mutations among the strains further supported that the Clade II and Clade Ib strain shared the MRCA, whereas Clade III strains were more likely derived from Clade Ia.
To estimate the rate of evolution in the bacteremic strains of persistent infections, the BEAST program was applied using the reference set of 71 CC5 isolates (Supplementary Table S2) as the comparator. The mean clock rate for the 32 successive strains was 1.4518E-5 [95% highest posterior density (HPD) interval, 8.6295E-6, 2.0502E-5], which was approximately 6.4-fold faster than the clock rate of the nationwide collection isolates (mean clock rate, 2.2845E-6, 95% HPD interval, 2.0256E-6, 2.5491E-6). Further, the accumulation number of SNPs or short INDELs reached >300 in some of the successive bacteremic strains within 3 years, whereas a majority of the nationwide isolates in the reference set had less than 180 mutations during a time frame of 17 years (1997–2014) (Figure 1).
To understand the role of recombinations during the evolution of MRSA strains exhibiting increasing drugs resistance in this case, we compared 32 strains by screening the plasmid, resistance, and virulence genes with the srst2 procedure and viewing the graphic genome maps with the CGview program (Table 3 and Figures 3, 4). The srst2 procedure detected three plasmids, which were structurally similar to pSAS (BX571858), pWGB1773 (EF537646), and pPV141 (U82607) in the first three strains (LTF01, LTF02, and LTF03). The plasmids were gradually lost in the successive strains and complete missing in two strains of Clade Ib (LTF19 and LTF28) and all strains of Clade II. Several prophage-associated fragments were also missing in some of the successive strains. Of them, the beta-hemolysin converting prophage containing the immune evasion cluster (sak, scn, chp, and sep) was lost in some Clade I strains and all three strains of Clade III. The other lost resistance/virulence genes including fosB, mecA, bleO, ant(4′)-Ia, sdrD, and sdrE appeared not to be prophage or plasmid related. Together, a total of 11 recombination patterns involving three plasmids and four resistance/virulence genes-contained fragments were identified among the 32 successive strains (Table 3). The numbers of gene loss were significantly greater in the strains isolated in the later course of bacteremia (Clade Ib and Clade II strains, P = 0.0005 and P = 0.0003, respectively) compared to the Clade Ia strains being isolated at early course of bacteremia (Figure 5).
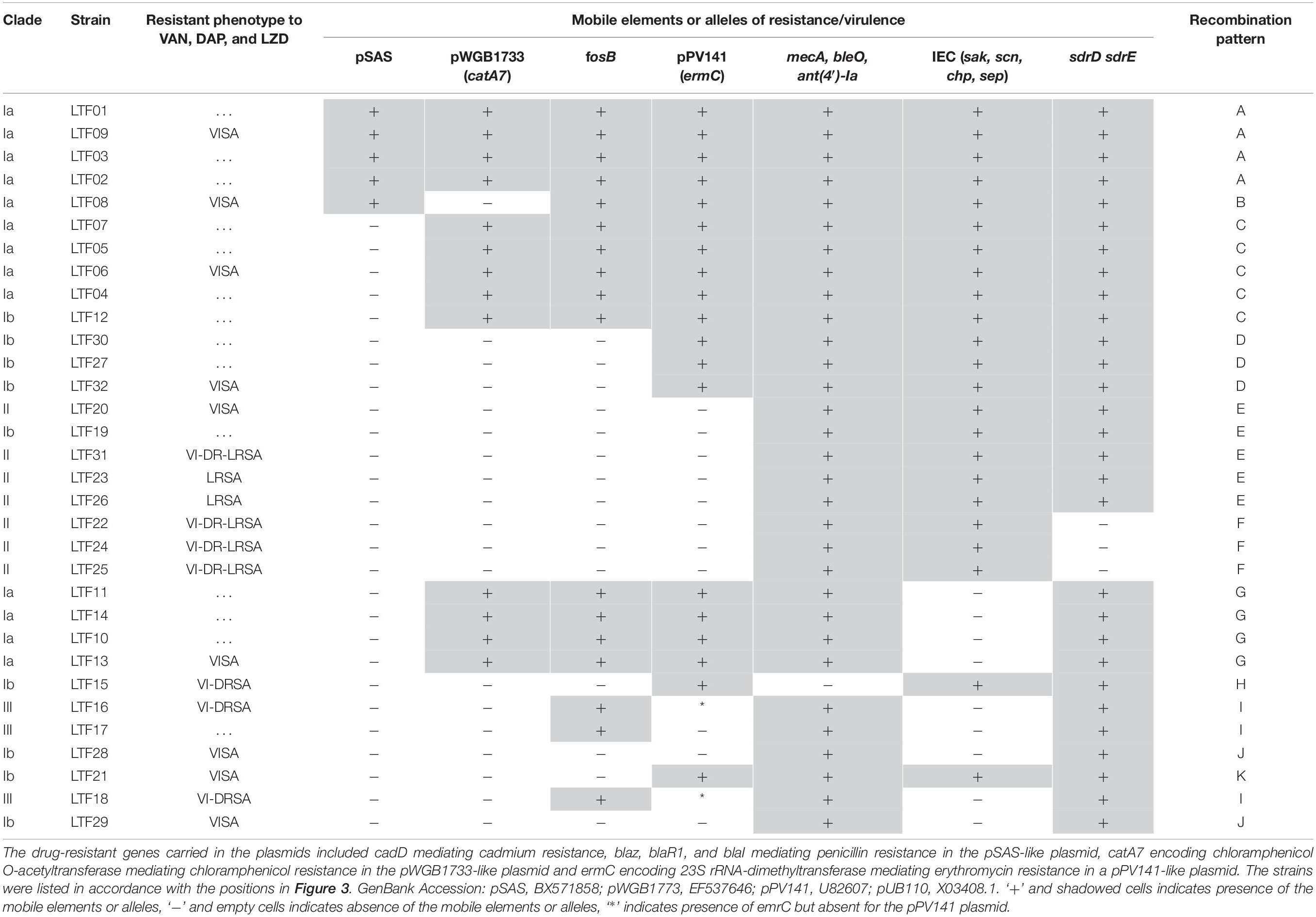
Table 3. Distributions of mobile elements and alleles encoding molecules of drug resistance or virulence in 32 successive MRSA strains isolated from a patient with persistent bacteremia in 3 years.
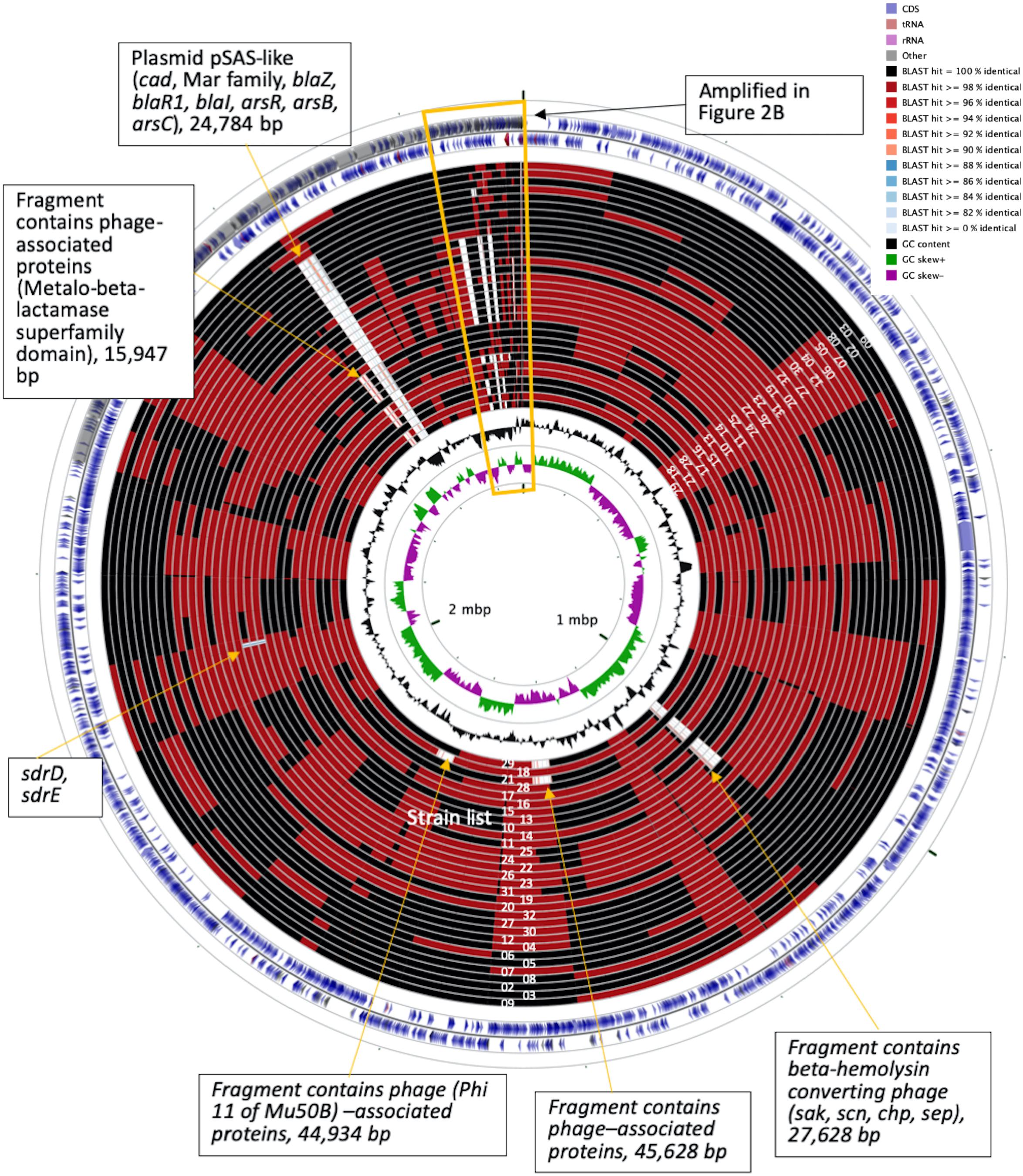
Figure 3. CGview of the genome map of 32 MRSA isolates from a patient with persistent bacteremia. The empty regions indicated missing of the plasmids or chromosomal fragments.
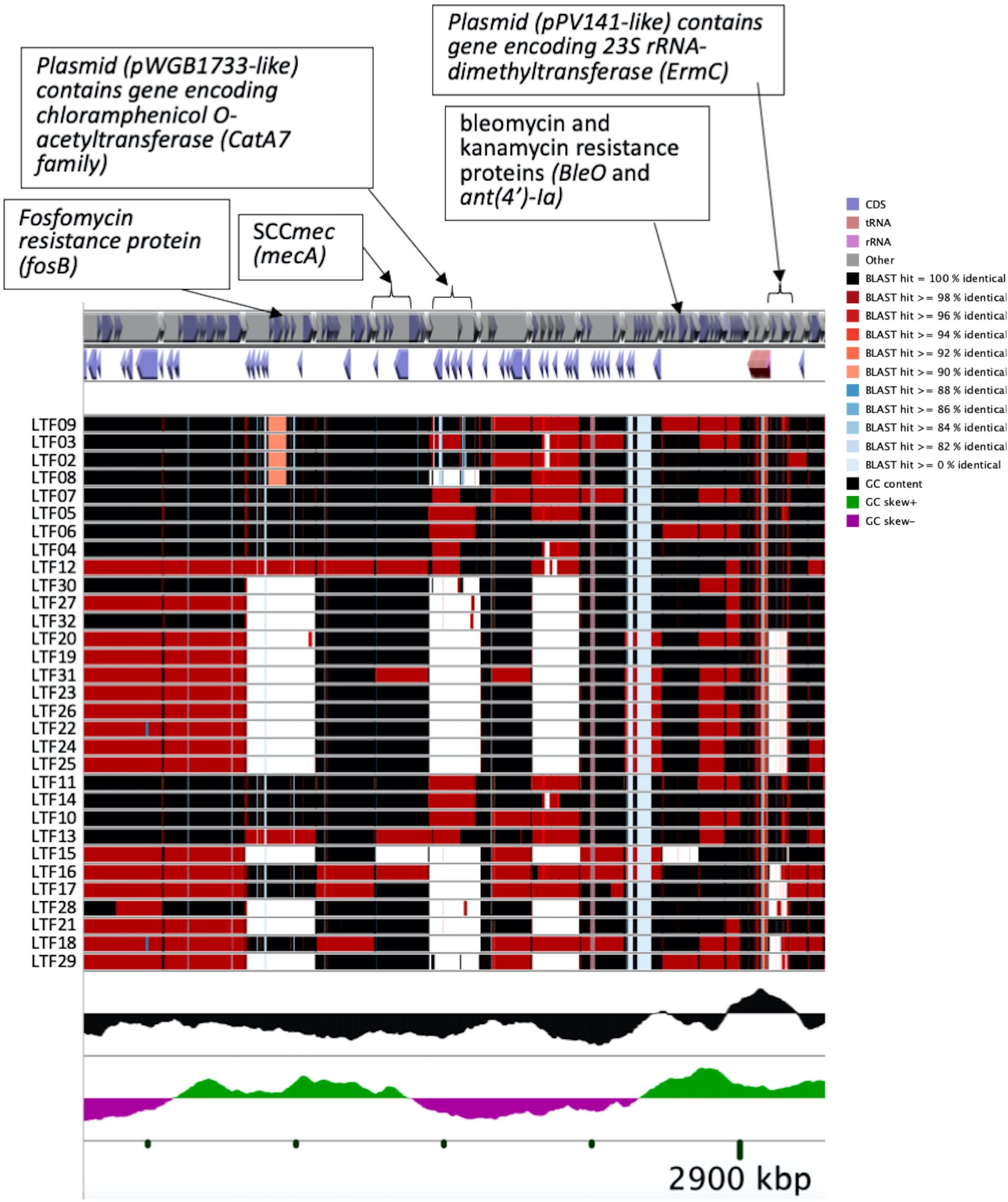
Figure 4. CGview of the genome map of 32 MRSA isolates from a patient with persistent bacteremia. Amplification of a region containing two plasmids (pWGB1733-like and pPV141-like) and drug-resistant genes in Figure 3.
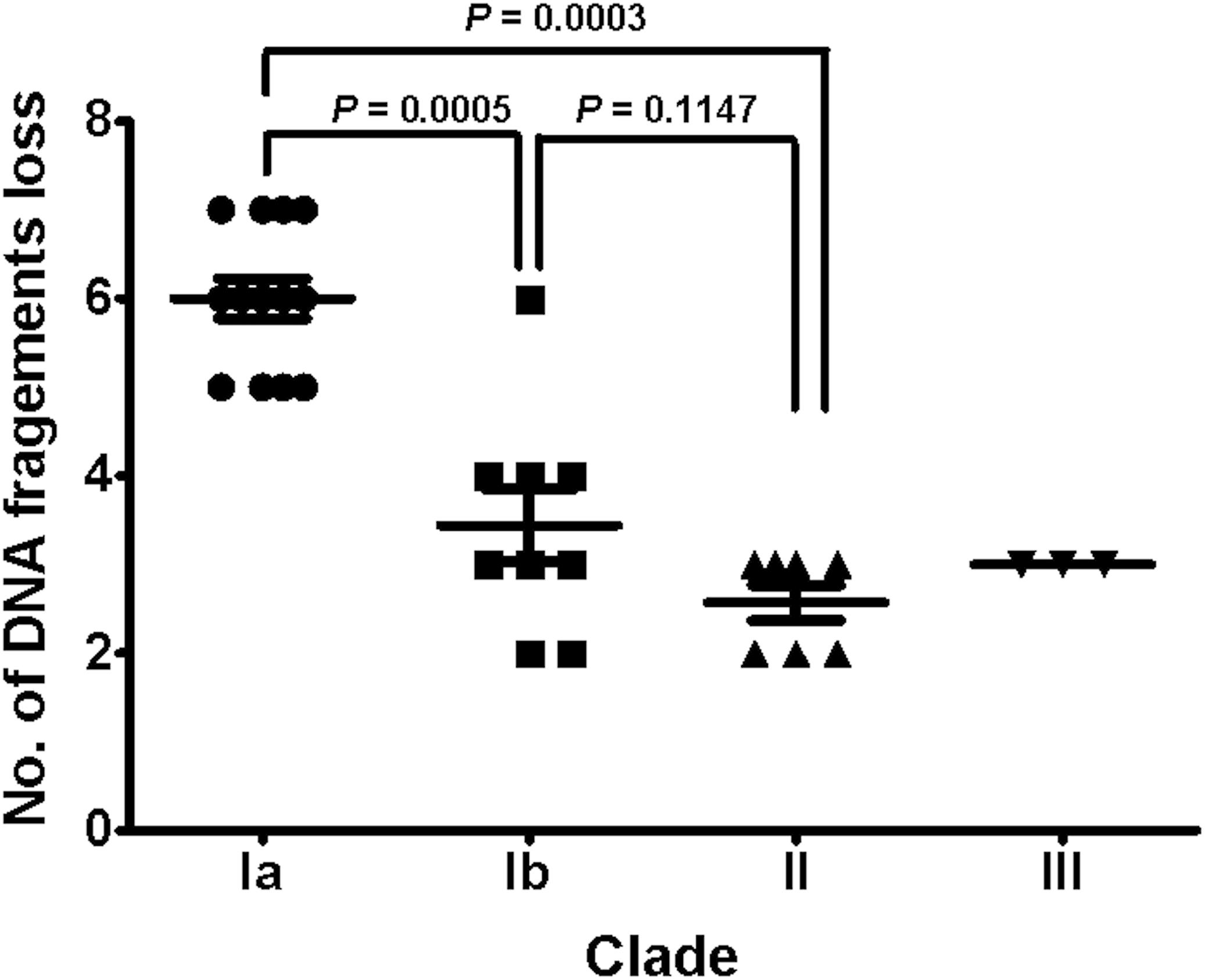
Figure 5. The distributions of numbers of gene loss in different clades of 32 successive MRSA isolates. The numbers of gene loss were significantly greater for strains (Clades Ib and II) isolated in later course of bacteremia than for strains (Clade Ia) in early course. Comparison to isolates in Clade III was not done due to the small number of isolates.
The VISA phenotype (vancomycin MIC of 3–4 mg/L) occurred in strains at an intermittent manner since the isolation of LTF06 and accounted for 40.9, 71.4, and 66.7% of strains of Clades I, II, and III, respectively. The DRSA phenotype was exclusively identified in strains with VISA phenotype irrespective of clades, suggesting a linked machinery of incremental non-susceptibility to vancomycin and daptomycin. A total of 87 mutations in 86 alleles occurring in VISA or VISA-DRSA strains but not in VSSA strains were considered to be associated with VISA/DRSA phenotypes and are displayed in Figure 6 and Supplementary Data Sheet S2. Of them, nine mutations in eight alleles associated with VISA/DRSA phenotype that had been reported in the literature are listed in Table 4. One isolate (LTF15) exhibited resistance to daptomycin among Clade I strains harbored a gain-in-function mutation in mprF (S295L) (Figure 6), which was not identified in the other VISA-DRSA strains in Clades II and III. When compared to the VSSA strains, the VISA or VISA-DRSA strains tended to harbor greater numbers of non-synonymous and clade-specific mutations (Supplementary Table S4).
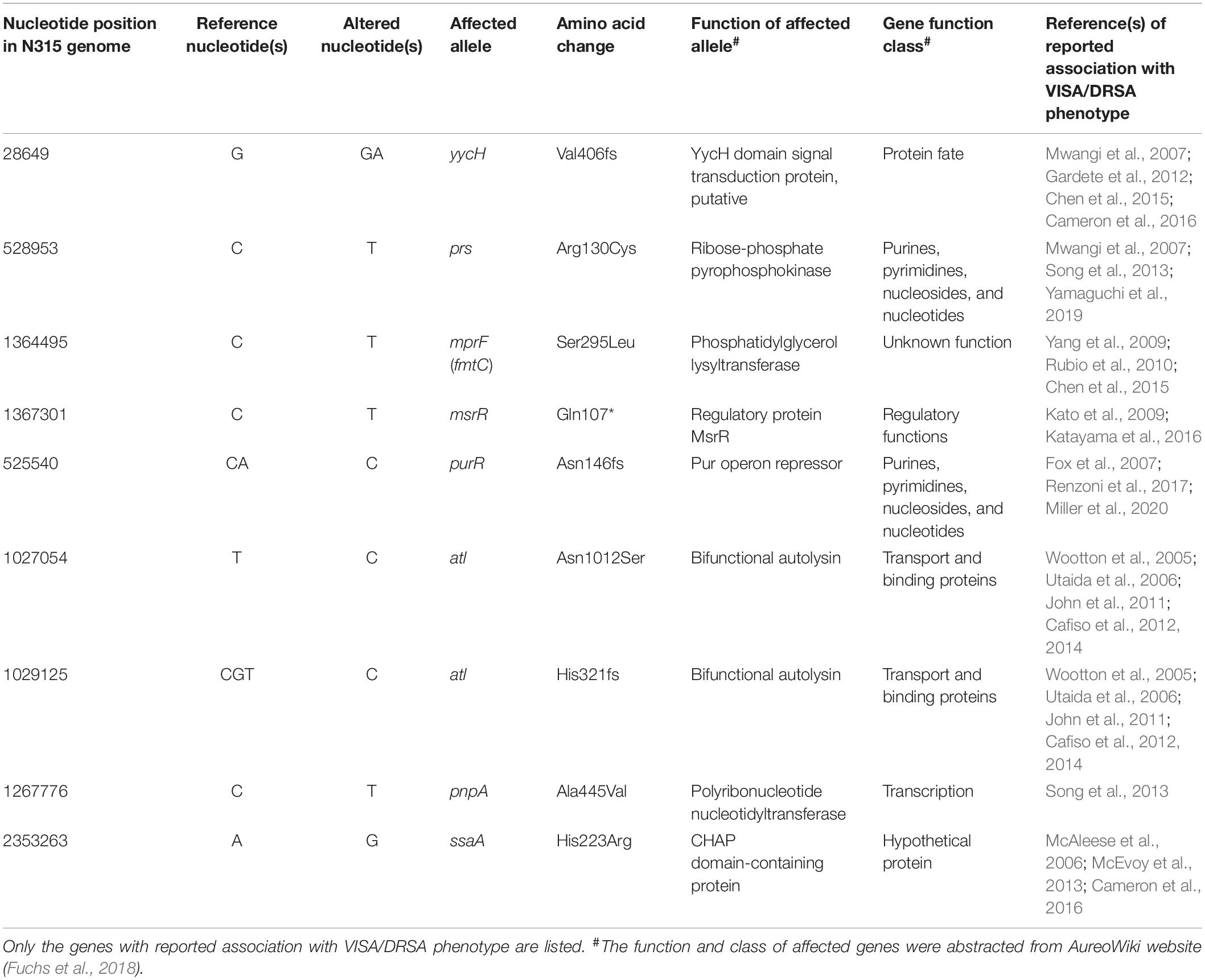
Table 4. Selected genetic alterations associated with vancomycin-intermediate S. aureus (VISA) and/or daptomycin-resistant S. aureus-associated (DRSA) phenotypes in successive ST5 MRSA strains causing persistent bacteremia.
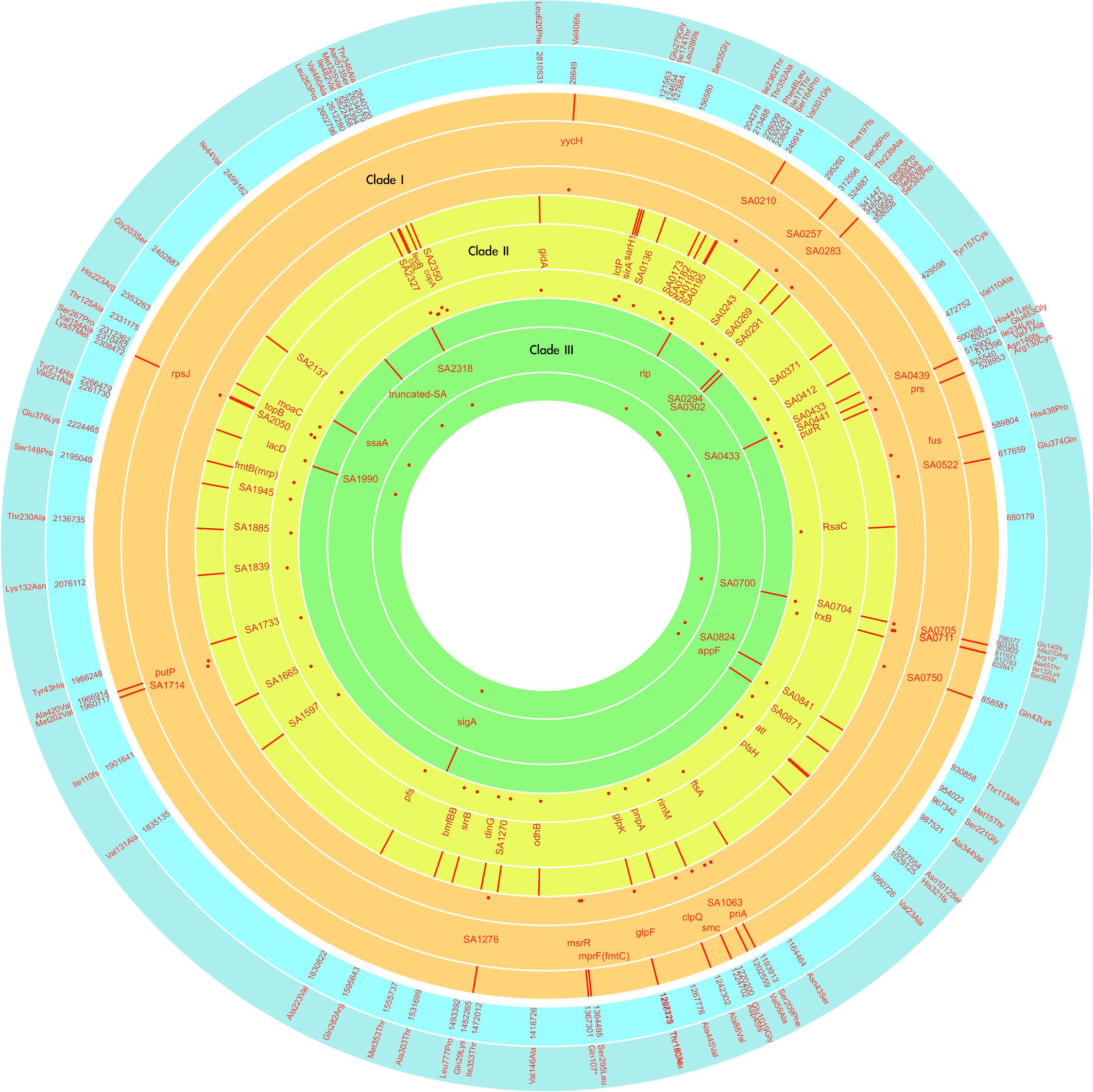
Figure 6. Map of the VISA/DRSA-associated genetic alteration among the successive MRSA strains of three clades marked with different colors, with the outer ring showing the position, middle ring showing the affected alleles, and inner ring showing the rates of affected strains in the indicated clade. Inner and outer ring in blue color, respectively, indicates the positions (in N315) and effects of mutations. The red dot indicates the carriage rate of the indicated mutation for strains in that clade. The plot was generated with the Circa program.
Resistance to rifampin was firstly identified in strains LTF04, which harbored a non-synonymous SNP (L527L) in rpoB gene. The other rifampin-resistant strains also had mutations in rpoB gene though with distinct SNPs (i.e., L482A, S529L, S717L, Supplementary Data Sheet S1). Another VISA strain (LTF21) exhibited resistance to fusidic acid, which was associated with the H438P mutation in fus gene (Figure 6). The resistance to linezolid was associated with mutations in 23s rRNA. All of the linezolid-resistant strains harbored two to three G2576T mutations in domain V regions of 23S rRNA (Table 5).
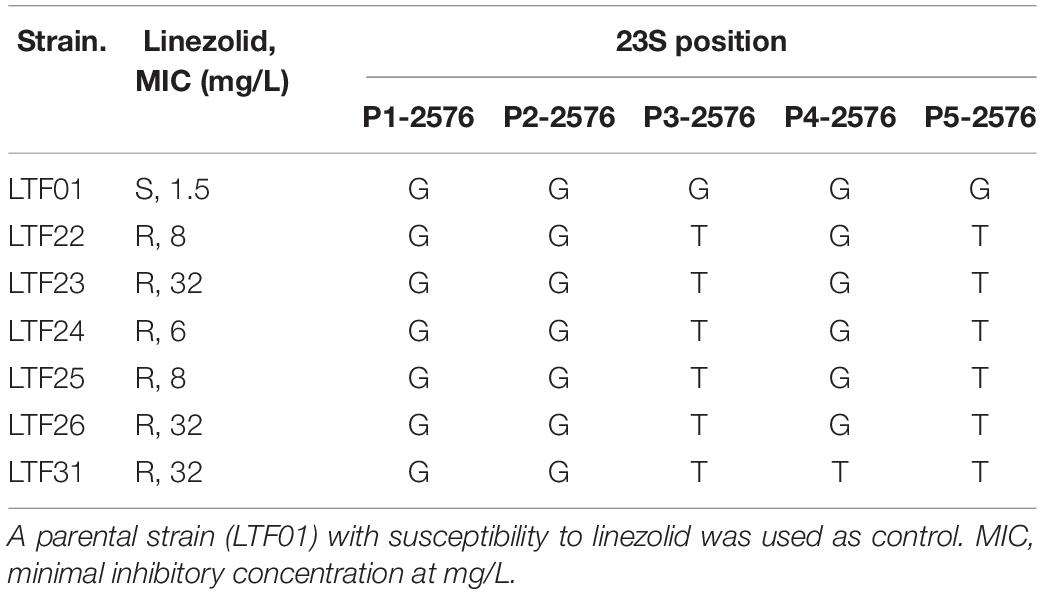
Table 5. Frequency of G2576T mutations in domain V regions of isolates with resistance to linezolid.
The time-scaled phylogeny of the 32 bacteremic strains and the use of antimicrobial agents in different time frames during the persistent bacteremia are shown in Figure 7. For most of the time, the patient received monotherapy for the bacteremia, with glycopeptide being mainly used between day 0 and 600. A majority (10 of 13 strains) of the Clade Ia strains were isolated in this period, with strains LTF11, LTF13, and LTF14 being the exceptions that shared the MRCA (day 393.7) with Clade III strains and survived beyond day 600. The MRCA of strains of Clade Ia (LTF04, LTF05, and LTF07), Clade Ib, and Clade II were also predicted to appear during this time frame at day 385.0.
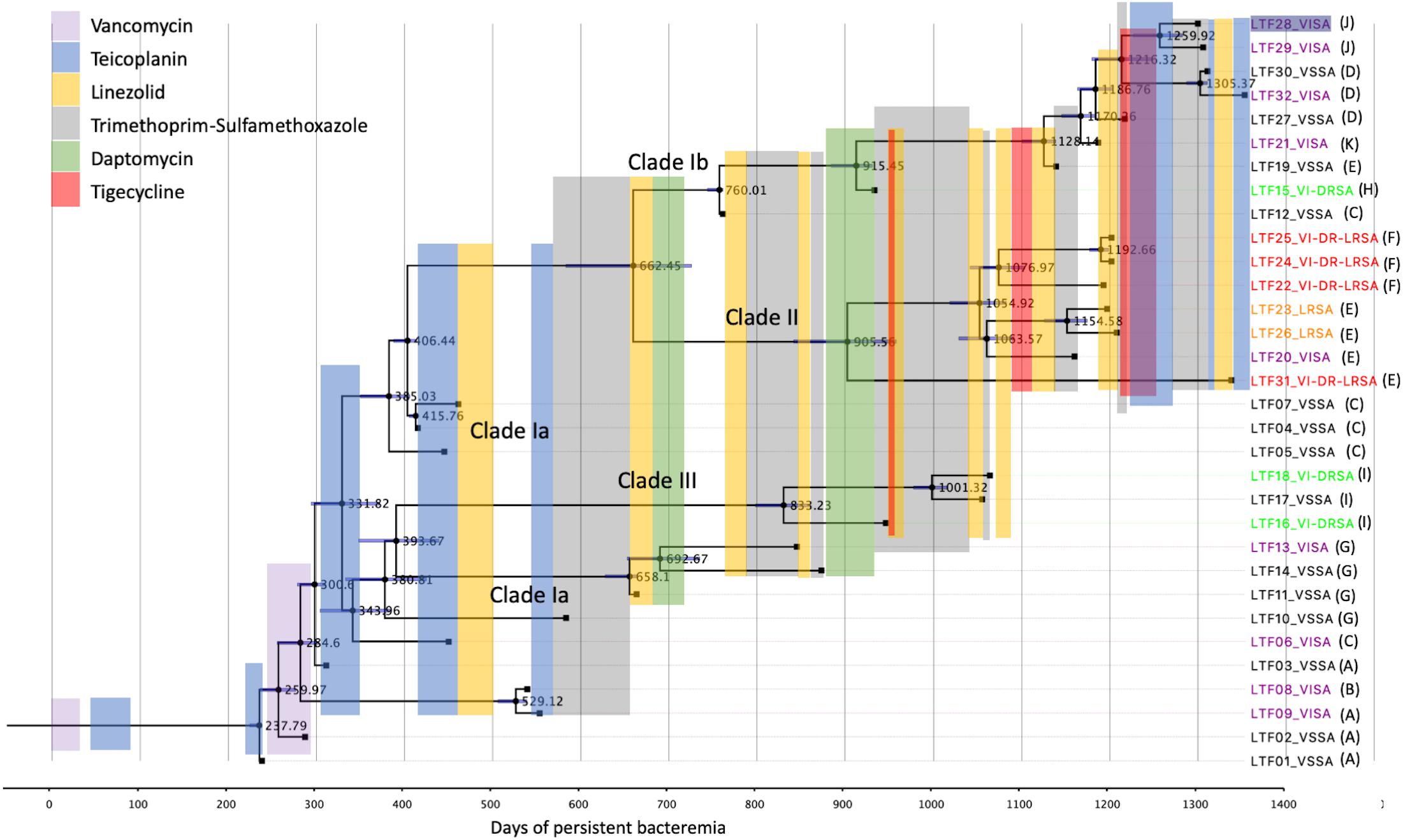
Figure 7. Antimicrobial treatment courses and the time-scaled phylogeny of 32 successive MRSA strains in a patient with persistent bacteremia by BEAST program. The node ages are shown as days after the onset of bacteremia, and the 95% highest poster density are displayed as blue bars. The tips are labeled with strain names followed by drug-resistant phenotypes and the patterns of recombinations indicated in Table 3. VISA, vancomycin-intermediate S. aureus; VSSA, vancomycin-susceptible S. aureus; VI-DRSA, vancomycin-intermediate and daptomycin-resistant S. aureus; LRSA, linezolid-resistant S. aureus; VI-DR-LRSA, vancomycin-intermediate, daptomycin-resistant, and linezolid-resistant S. aureus.
Between days 600 and 1,200, monotherapy with TMP-SXT, linezolid, daptomycin, and tigecycline was interchangeably used. Combination therapy with tigecycline followed by linezolid was only shortly used with TMP-SXT to treat the VISA-DRSA strains (LTF15 and LTF16) in this period. The use of a second course of daptomycin successfully eradicated the remaining Clade Ia strains LTF11, LTF13, and LTF14 but rapidly select the VISA-DRSA strains of Clade III (LTF16 and LTF18) and Clade Ib (LTF15). The VISA-DRSA-LRSA strains of Clade II started to emerge at the end of this time frame. After day 1,200, because of the continually isolated VISA-DRSA-LRSA strains, combination therapy with tigecycline and glycopeptide was launched. Unfortunately, the attempt was unsuccessful and one Clade II strain of VISA-DRSA-LRSA phenotype (LTF31) remained being isolated. In addition, the regimen was unable to stop the less-resistant strains, and three VISA strains and one VSSA strain of Clade Ib survived the intensive combination treatment.
Our study demonstrated that to adapt to the host environment with high antibiotic selective pressure, the MRSA ST5 isolates underwent rapid genetic evolution through three major pathways from strains of Clade Ia, respectively, to Clade Ib, Clade II, and Clade III. The genetic alterations during the evolution involved both mutations (SNP or short INDELs) and substantial genes loss likely by recombinations. It has been well documented that mutations in certain alleles in MRSA strains could lead to increased non-susceptibility to the anti-MRSA agents of last resort, including vancomycin, daptomycin, and linezolid (Howden et al., 2010; Gu et al., 2013; Yang et al., 2013; Chen et al., 2015). By comparing the mutations among VISA/DRSA strains to the VSSA strain in the same clade, we were able to identify dozens of mutations potentially associated with VISA/DRSA phenotypes (Figure 6 and Supplementary Data Sheet S2). Indeed, several alleles harboring mutations (i.e., rpoB, mrpF, and 23S rRNA) identified in the current study had been previously confirmed to be accountable for the VISA, DRSA, or LRSA phenotypes (Matsuo et al., 2011; Gu et al., 2013; Yang et al., 2013). Of note, the G2576T mutation in domain V regions of 23S rRNA has been shown to be one of the major machineries of linezolid resistance and was exclusively identified in all isolates resistant to linezolid in this study. Further, the only isolate (LTF15) exhibiting resistance to daptomycin in Clade I harbored a point mutation S295L in mprF gene. MprF is the synthase for positively charged lysylphosphatidyl-glycerol (L-PG), which is considered to be one of the major contributors to the positive charge of S. aureus bacterial surface. The MprFS295L has been demonstrated to be a gain-in-function mutation associated with enhancing the synthesis of L-PG and reducing the binding of daptomycin to S. aureus (Yang et al., 2013). Moreover, correlation of resistance to other anti-MRSA agents including rifampin and fusidic acid and the mutations in rpoB (Lys482Asn, Ile527Leu, Ser529Leu, and Ser717Leu) and fus (Val86Ala) genes, respectively, was also identified in this study (Table 1 and Supplementary Data Sheet S1). The data combined together strongly suggested the SNPs/INDELs mutations played an important role in MRSA exhibiting incremental drug resistance. The importance of mutations in resistance to vancomycin or daptomycin was further supported by the finding that the VISA/DRSA strains harboring a significantly greater number of SNPs and/or INDELs compared to the VSSA strains (Supplementary Table S4).
In addition to mprF, at least seven alleles with mutations identified in the current study had ever been reported to be associated with VISA/DRSA phenotypes (Table 4). Of them, the alleles including yycH, atl, and ssaA are involved in the WalKR cell wall regulatory operon controlling cell wall synthesis and autolysis (Dubrac et al., 2008). Of note, the frameshift mutation of yycH resulting in truncated YycH in VISA/DRSA strains was reported in previous studies by us and other groups (Mwangi et al., 2007; Gardete et al., 2012; Chen et al., 2015; Cameron et al., 2016). yycH together with yycI and yycJ is located directly downstream of the walKR two-component regulatory (TCR) system and belonged to the walKR/yycHIJ operon. It has been demonstrated that the YycH and YycI interact with the sensor kinase WalK and had positive regulatory role for walKR TCR (Szurmant et al., 2007). Mutations in yycH or yycI in VISA strains had a deleterious impact on the YycHI/WalKR complex through down-regulation of walKR regulon resulting in impaired autolysis. Allele atl together with another gene lytM are autolysins genes encoding the products majorly involved in the cell-wall expansion, remodeling, and daughter cell separation under the regulation of the WalKR system (Dubrac et al., 2008). Mutations or down-regulation of atl had been shown to be associated with VISA phenotype most likely by reducing autolysis and increased thickness of the cell wall (Howden et al., 2010). ssaA is also an autolysin gene encoding protein with CHAP amidase domain and regulated by WalKR system through directly binding by WalR to the promoter region (Dubrac et al., 2008; Howden et al., 2011). It has been demonstrated that the expression of ssaA was down-regulated in strains exhibiting VISA phenotype (McAleese et al., 2006; Cameron et al., 2016).
The VISA/DRSA-associated alleles including prs and purR were belonged to functional class of purines, pyrimidines, nucleosides, and nucleotides. Association of prs mutation and VISA or DRSA phenotype had been reported in at least three studies, but the role of this gene on drug resistance had not been directly addressed (Mwangi et al., 2007; Song et al., 2013; Yamaguchi et al., 2019). The prs gene encodes ribose-phosphate pyrophosphokinase, and mutation in prs may have a negative effect on the biosynthesis of purine and pyrimidine and further affect the growth rate of bacteria. However, the importance of prs mutation on drug resistance requires further study and should be carefully evaluated. Indeed, as shown in previous reports, mutation in another putative purine regulator, purR, has been observed in VISA/DRSA strains but a casual association was excluded by follow-up study investigating its biochemical and physiological characteristics (Fox et al., 2007). The other two VISA/DRSA-associated alleles (msrR and pnpA) are of regulatory or transcriptional function. Introduction of a mutation (E146K) in msrR had been reported to convert the VSSA strain of N315 background into heterogenous VISA phenotype (Katayama et al., 2016). msrR is a member of the LytR-CspA-Psr family of cell envelope-related transcriptional attenuators and was shown to be inducible by cell wall active agents, such as β-lactams, glycopeptides, and lysostaphin (Kato et al., 2009). It was suggested that the msrR mutation promotes the tethering of wall teichoic acid and the capsule to the cell wall, which then leads to decreased autolytic activity and the development of VISA phenotype. Allele pnpA is a gene-encoding polyribonucleotide nucleotidyltransferase, and mutation of this gene had ever been identified in a laboratory-derived DRSA strain (Song et al., 2013). PnpA is involved in the quality control of RNA precursors in Escherichia coli and appeared to have a role in the persistent infection of Salmonella enterica (Clements et al., 2002; De Lay and Gottesman, 2011). Its role in the resistance to vancomycin and/or daptomycin in S. aureus remained not determined.
In contrary to the SNPs or INDELs mutations, the role of recombination during the incremental drug resistance in persistent MRSA infections was largely unknown. In the current study, recombinations with the forms of loss of plasmids, loss of prophage with or without carrying virulence/resistance genes, and missing certain genetic fragments unrelated to plasmids or prophage (i.e., sdrD, sdrE, and fosB, Table 3) appeared to be common events during the evolution. It was intriguing to learn that the numbers of gene losses were greater in the strains being isolated at the later course of bacteremia (Clade Ib, Clade II, and Clade III strains) but appeared not necessarily related to the development of drug resistance. Of note, the susceptible strain (e.g., LTF19, VSSA strain in Clade Ib) had the same pattern of recombination as the strains in Clade II with multi-resistance to vancomycin, daptomycin, and linezolid (LTF31, VI-DR-LRSA, Table 3 and Figure 7). The observation suggested that the recombination leading to reduction in genome size may not be the critical step for the development of drug resistances but may be an important advantage for the fitness and persistence of MRSA strains in the extremely harsh condition (Albalat and Cañestro, 2016). Another intriguing observation was the loss of a fragment in SCCmec containing mecA and other two drug-resistant genes [bleO and ant(4′)-Ia] in the strain LTF15 exhibiting resistance to vancomycin and daptomycin. The strain was also of high-level resistance to beta-lactams including oxacillin and cefoxitin with minimal inhibitory concentrations of 32 mg/L and >256 mg/L, respectively. The mecA-negative MRSA has been reported elsewhere and the proposed machinery mediating beta-lactam resistance included mutation in other genes encoding penicillin-binding proteins (PBP) (i.e., PBP4) and through regulatory pathways (Banerjee et al., 2010; Roemer et al., 2013). A follow-up study is ongoing to address the mechanism of beta-lactams resistance in the MRSA strain absent for mecA gene.
The aggressive antimicrobial treatment in this patient was believed to be the major driving force of the evolution, which fostered a 6.4-fold faster clock rate of mutations in this particular case than that in other clinical isolates in the general health-care settings. The molecular evolution rate is generally determined by a range of factors including the background mutation rate, the strength of selection, generation time, and population size (Albalat and Cañestro, 2016). In this study, the “strength of selection” was majorly coming from aggressive antimicrobial therapy. To minimize the confounding effects of background mutation rate, generation time, and population size, we selected a group of MRSA strains with the same genetic background (CC 5) as the comparator. The comparator strains were non-duplicated clinical isolates collected from different patients nationwide during a time span of 17 years. They were not accountable for persistent infections or from longitudinal studies and should be suitable for estimating the evolution rate of contemporary MRSA in the healthcare facilities.
The initial glycopeptide treatment courses in this case was unsuccessful, which was not only failure to completely eradicate the Clade Ia strains but rapidly select a couple of VISA strains of the same clade. More importantly, the later isolated strains of VISA-DRSA and VISA-DRSA-LRSA phenotype appeared to be directly diverged from the Clade Ia strains in this critical period of time. The speculation was supported by the time-scaled phylogeny that the MRCA of the Clade II and III strains was closely related to a Clade Ia strain (LTF03) and predicted to appear during the fifth course of glycopeptide treatment (day 331.8, Figure 7). Two descendants of the MRCA appearing on day 393.7 and day 406.4 was, respectively, becoming the common ancestors of Clades Ia/III and Clades Ia/Ib/II. The glycopeptide monotherapy was not only ineffective in eliminating the bacteria but eventually driving the evolution of multi-resistance to other anti-MRSA agents through multiple genetic pathways. We believe that a more effective strategy with regimens that combine different classes of antimicrobial agents or early transition to an alternative treatment against MRSA will be required after the initial failed attempt to improve the outcome of patient with persistent bacteremia.
Clade II strains shared a MRCA with Clade Ib strains on day 662.5 though a majority of the strains were isolated on approximately day 1,200 of bacteremia. The Clade II strains were characterized with the colonies that are smaller, less hemolytic, and less pigmented (SCV phenotype) than the strain in other two clades and mostly exhibited resistance to linezolid in addition to vancomycin and daptomycin. These multiple phenotypic abnormalities suggest that the Clade II strains have alterations of global gene expression and/or cell signaling pathways. It has been demonstrated that the SCVs were heterogenous bacterial population and was usually unstable and dynamic during the chronic infections (Proctor et al., 2006; Garcia et al., 2013; Tuchscherr et al., 2016). Before identification of the first LRSA strains (LTF22), the patient had undergone eight courses of linezolid treatment for a total of 144 days during approximately 24 months of bacteremia. The LRSA phenotype was rarely identified in S. aureus, with an estimated rate of 0.05% for clinical isolates (Gu et al., 2013). Our observation was consistent with previous report that the patients were usually treated with linezolid prior to isolation of LRSA for a mean duration of 20 months (Gu et al., 2013). The multi-resistant strains appeared to be inhibited by administration of TMP-SXT, tigecycline, and glycopeptide in this study, but one VISA-DRSA-LRSA strain (LTF31) shared the MRCA with other Clade II strains on day 905.5 still escaped the intensive course of anti-MRSA treatment. The emergence of LRSA phenotype in MRSA strains remained a significant treatment challenge.
Clade III strains had 26.2- and 3.2-fold more mutations than that in the Clade I and Clade II strains, respectively. However, the colony morphology in Clade III strains was not significantly affected when compared to Clade I strains. A majority of the mutations in Clade III strains appeared not directly related to the drug resistance. Indeed, with a total of 390 SNPs or INDELs in Clade III strains, only 12 (3.1%) mutations was present in two VISA/DRSA strains but absent in the VSSA strain suggesting association with VISA/DRSA phenotype. The proportion of VISA/DRSA-associated mutations to all SNPs or INDELs was significantly greater for Clade II strains (51/225, 22.7%, Table 2). In addition to loss of all three plasmids, the Clade III strains were characterized with loss the beta-hemolysin prophage carrying the immune evasion cluster (IEC) including sak, scn, chp, and sep genes. Loss of the IEC in the multi-resistant strains suggested that the dispensability of the genes may confer adaptive advantage for MRSA in an environment with highly selective pressure.
We did have limitations in this study. First, the very first MRSA isolate in this case was not available and the evolution of the MRSA strains during the first 3 months of bacteremia were not captured in this study. However, the study was aimed to delineate the genomic evolution of MRSA during the transition from susceptible phenotypes to tripe-resistance to vancomycin, daptomycin, and linezolid. The first available isolate (LTF01) remained susceptible to the three antimicrobial agents, and the important genetic changes related to the incremental drug resistance should have been revealed in this study. Second, it has been reported that the evolutionary rate of bacteria might be negatively associated with the sampling time. The 6.4-fold faster clock rate for the MRSA strains in persistent bacteremia than that of the contemporary MRSA strains in the general health-care facilities might be offset to certain degree due to the shorter sampling time for the successive strains (approx. 3 years vs. 17 years).
Genomic evolution of a MRSA ST5 strain during long-term persistent bacteremia on aggressive antimicrobial therapy was approximately 6.4-fold faster than that of the contemporary ST5 clinical isolates. The development of incremental resistance to vancomycin, daptomycin, and linezolid was coupled with substantial morphological changes and proceeded through multiple pathways. Both mutations and recombinations were common events during the evolution. While the mutations were frequently associated with the development of drug resistance, the recombination resulting in the gene loss appeared to provide adaptive advantage in harsh host environment rather than mediating drug non-susceptibility.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA495118.
C-JC and Y-CH designed the study. C-JC and S-SS performed the samples and data collection and conducted the genome sequencing and data analysis. C-JC wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by grants from Ministry of Science and Technology in Taiwan (MOST 105-2314-B-182A-140-MY3 and MOST 108-2314-B-182A-055).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01414/full#supplementary-material
TABLE S1 | MLST, spa types and SCCmec types in 32 serial MRSA isolates from a patient with persistent bacteremia.
TABLE S2 | Information of 71 non-duplicated clinical MRSA strains belonging to ST5 or its single locus variants collected island wide from 1997 to 2014 in Taiwan.
TABLE S3 | Non-synonymous SNPs and short INDELs strains across different clades, comparison to the parenteral strain, LTF01.
TABLE S4 | The comparisons of numbers of SNPs/INDELs between the VISA strains and VSSA strains. The median and range are shown. Statistic was not performed for Clade II and Clade III strains due to small number of strains in the two clades: (A) comparisons of number of all SNPs/INDELs including synonymous and non-synonymous mutations; (B) only non-synonymous mutations were included for comparisons; and (C) only non-synonymous and clade-specific mutations were included for comparisons.
DATA SHEET 1 | The positions of mutations, altered nucleotides, effect of the mutations, and the affected alleles among the successive MRSA isolates from a patient with persistent bacteremia. The first available isolates LTF01 was used as the comparator.
DATA SHEET 2 | VISA/DRSA-associated genetic alteration among the successive MRSA strains. The positions of mutations, altered nucleotides, effect of the mutations, and function of the affected alleles are displayed.
PRESENTATION 1 | The colony morphologies on blood agar and BM agar for 32 successive MRSA blood isolates from a patient with persistent bacteremia.
Albalat, R., and Cañestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Banerjee, R., Gretes, M., Harlem, C., Basuino, L., and Chambers, H. F. (2010). A mecA-Negative Strain of Methicillin-Resistant Staphylococcusaureus with High-Level β-Lactam resistance contains mutations in three genes. Antimicrob. Agents Chemother. 54, 4900–4902. doi: 10.1128/AAC.00594-510
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Cafiso, V., Bertuccio, T., Purrello, S., Campanile, F., Mammina, C., Sartor, A., et al. (2014). dltA overexpression: A strain-independent keystone of daptomycin resistance in methicillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 43, 26–31. doi: 10.1016/j.ijantimicag.2013.10.001
Cafiso, V., Bertuccio, T., Spina, D., Purrello, S., Campanile, F., Di Pietro, C., et al. (2012). Modulating Activity of Vancomycin and Daptomycin on the Expression of Autolysis Cell-Wall Turnover and Membrane Charge Genes in hVISA and VISA Strains. PLoS One 7:e29573. doi: 10.1371/journal.pone.0029573
Cameron, D. R., Jiang, J.-H., Kostoulias, X., Foxwell, D. J., and Peleg, A. Y. (2016). Vancomycin susceptibility in methicillin-resistant Staphylococcus aureus is mediated by YycHI activation of the WalRK essential two-component regulatory system. Sci. Rep. 6:30823. doi: 10.1038/srep30823
Charles, P. G. P., Ward, P. B., Johnson, P. D. R., Howden, B. P., and Grayson, M. L. (2004). Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin. Infect. Dis. 38, 448–451. doi: 10.1086/381093
Chen, C.-J., Huang, Y.-C., and Chiu, C.-H. (2015). Multiple pathways of cross-resistance to glycopeptides and daptomycin in persistent MRSA bacteraemia. J. Antimicrob. Chemother. 70, 2965–2972. doi: 10.1093/jac/dkv225
Chen, C.-J., Huang, Y.-C., Su, L.-H., Wu, T.-L., Huang, S.-H., Chien, C.-C., et al. (2014a). Molecular epidemiology and antimicrobial resistance of methicillin-resistant Staphylococcus aureus bloodstream isolates in Taiwan, 2010. PLoS One 9:e101184. doi: 10.1371/journal.pone.0101184
Chen, C.-J., Lin, M.-H., Shu, J.-C., and Lu, J.-J. (2014b). Reduced susceptibility to vancomycin in isogenic Staphylococcus aureus strains of sequence type 59: tracking evolution and identifying mutations by whole-genome sequencing. J. Antimicrob. Chemother. 69, 349–354. doi: 10.1093/jac/dkt395
Chen, C.-J., Su, L.-H., Lin, T.-Y., and Huang, Y.-C. (2010). Molecular Analysis of Repeated Methicillin-Resistant Staphylococcus aureus Infections in Children. PLoS One 5:e14431. doi: 10.1371/journal.pone.0014431
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Claeys, K. C., Zasowski, E. J., Casapao, A. M., Lagnf, A. M., Nagel, J. L., Nguyen, C. T., et al. (2016). Daptomycin Improves Outcomes Regardless of Vancomycin MIC in a propensity-matched analysis of methicillin-resistant Staphylococcus aureus Bloodstream Infections. Antimicrob. Agents Chemother. 60, 5841–5848. doi: 10.1128/AAC.00227-216
Clements, M. O., Eriksson, S., Thompson, A., Lucchini, S., Hinton, J. C. D., Normark, S., et al. (2002). Polynucleotide phosphorylase is a global regulator of virulence and persistency in Salmonella enterica. Proc. Natl. Acad. Sci. 99, 8784–8789. doi: 10.1073/pnas.132047099
CLSI (2017). M100S24EM100-S24 Performance Standards for Antimicrobial Susceptibility Testing Twenty-Fourth Informational Supplement M100. Wayne, PA: Clinical and Laboratory Standard Institute.
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
De Lay, N., and Gottesman, S. (2011). Role of polynucleotide phosphorylase in sRNA function in Escherichia coli. RNA 17, 1172–1189. doi: 10.1261/rna.2531211
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Dubrac, S., Bisicchia, P., Devine, K. M., and Msadek, T. (2008). A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70, 1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x
Fox, P. M., Climo, M. W., and Archer, G. L. (2007). Lack of relationship between purine biosynthesis and vancomycin resistance in Staphylococcus aureus: a cautionary tale for microarray interpretation. Antimicrob. Agents Chemother. 51, 1274–1280. doi: 10.1128/AAC.01060-1066
Friedman, L., Alder, J. D., and Silverman, J. A. (2006). Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 50, 2137–2145. doi: 10.1128/AAC.00039-36
Fuchs, S., Mehlan, H., Bernhardt, J., Hennig, A., Michalik, S., Surmann, K., et al. (2018). AureoWiki - The repository of the Staphylococcus aureus research and annotation community. Int. J. Med. Microbiol. 308, 558–568. doi: 10.1016/j.ijmm.2017.11.011
Garcia, L. G., Lemaire, S., Kahl, B. C., Becker, K., Proctor, R. A., Denis, O., et al. (2013). Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J. Antimicrob. Chemother. 68, 1455–1464. doi: 10.1093/jac/dkt072
Gardete, S., Kim, C., Hartmann, B. M., Mwangi, M., Roux, C. M., Dunman, P. M., et al. (2012). Genetic pathway in acquisition and loss of vancomycin resistance in a methicillin resistant Staphylococcus aureus (MRSA) strain of clonal type USA300. PLoS Pathog. 8:e1002505. doi: 10.1371/journal.ppat.1002505.t003
Gu, B., Kelesidis, T., Tsiodras, S., Hindler, J., and Humphries, R. M. (2013). The emerging problem of linezolid-resistant Staphylococcus. J. Antimicrob. Chemother. 68, 4–11. doi: 10.1093/jac/dks354
Hamdy, R. F., Hsu, A. J., Stockmann, C., Olson, J. A., Bryan, M., Hersh, A. L., et al. (2017). Epidemiology of methicillin-resistant Staphylococcus aureus bacteremia in children. Pediatrics 139:e20170183. doi: 10.1542/peds.2017-2183
Howden, B. P., Allen, D. L., Chua, K., Gao, W., Harrison, P. F., Davies, J. K., et al. (2011). Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 7:e1002359. doi: 10.1371/journal.ppat.1002359
Howden, B. P., Davies, J. K., Johnson, P. D. R., Stinear, T. P., and Grayson, M. L. (2010). Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23:139. doi: 10.1128/CMR.00042-49
Huang, Y. C., Ho, C.-F., Chen, C.-J., Su, L.-H., and Lin, T. Y. (2008). Comparative molecular analysis of community-associated and healthcare-associated methicillin-resistant Staphylococcus aureus isolates from children in northern Taiwan. J. Clin. Microbiol. 14, 1167–1172. doi: 10.1111/j.1469-0691.2008.02115.x
Huang, Y.-C., Su, L.-H., Wu, T.-L., and Lin, T.-Y. (2006). Changing molecular epidemiology of methicillin-resistant Staphylococcus aureus bloodstream isolates from a teaching hospital in Northern Taiwan. J. Clin. Microbiol. 44, 2268–2270. doi: 10.1128/JCM.00776-776
John, A.-K., Schmaler, M., Khanna, N., and Landmann, R. (2011). Reversible Daptomycin Tolerance of Adherent Staphylococci in an Implant Infection Model. Antimicrob. Agents Chemother. 55, 3510–3516. doi: 10.1128/AAC.00172-111
Johnson, S., Trost, B., Long, J. R., Pittet, V., and Kusalik, A. (2014). A better sequence-read simulator program for metagenomics. BMC Bioinformatics 15:S14. doi: 10.1186/1471-2105-15-S9-S14
Katayama, Y., Sekine, M., Hishinuma, T., Aiba, Y., and Hiramatsu, K. (2016). Complete Reconstitution of the Vancomycin-Intermediate Staphylococcus aureus Phenotype of Strain Mu50 in Vancomycin-Susceptible S. aureus. Antimicrob. Agents Chemother. 60, 3730–3742. doi: 10.1128/AAC.00420-416
Kato, Y., Suzuki, T., Ida, T., and Maebashi, K. (2009). Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65, 37–45. doi: 10.1093/jac/dkp394
Kondo, Y., Ito, T., Ma, X. X., Watanabe, S., Kreiswirth, B. N., Etienne, J., et al. (2007). Combination of multiplex PCRs for staphylococcal cassette chromosome mec type assignment: rapid identification system for mec, ccr, and major differences in junkyard regions. Antimicrob. Agents Chemother. 51, 264–274. doi: 10.1128/AAC.00165-166
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Matsuo, M., Hishinuma, T., Katayama, Y., Cui, L., Kapi, M., and Hiramatsu, K. (2011). Mutation of RNA polymerase beta subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob. Agents Chemother. 55:4195. doi: 10.1128/AAC.00398-311
McAleese, F., Wu, S. W., Sieradzki, K., Dunman, P., Murphy, E., Projan, S., et al. (2006). Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate- S. aureus-type resistance to vancomycin. J. Bacteriol. 188, 1120–1133. doi: 10.1128/JB.188.3.1120-1133.2006
McEvoy, C. R. E., Tsuji, B., Gao, W., Seemann, T., Porter, J. L., Doig, K., et al. (2013). Decreased vancomycin susceptibility in Staphylococcus aureus caused by IS256 tempering of WalKR expression. Antimicrob. Agents Chemother. 57, 3240–3249. doi: 10.1128/AAC.00279-213
Miller, C. R., Dey, S., Smolenski, P. D., Kulkarni, P. S., Monk, J. M., Szubin, R., et al. (2020). Distinct Subpopulations of Intravalvular Methicillin-Resistant Staphylococcus aureuswith variable susceptibility to daptomycin in tricuspid valve endocarditis. Antimicrob. Agents Chemother. 64, 1455–1425. doi: 10.1128/AAC.01593-1519
Moise, P. A., Culshaw, D. L., Wong-Beringer, A., Bensman, J., Lamp, K. C., Smith, W. J., et al. (2016). Comparative Effectiveness of Vancomycin Versus Daptomycin for MRSA Bacteremia With Vancomycin MIC >1 mg/L: a multicenter evaluation. Clin. Ther. 38, 16–30. doi: 10.1016/j.clinthera.2015.09.017
Murray, K. P., Zhao, J. J., Davis, S. L., Kullar, R., Kaye, K. S., Lephart, P., et al. (2013). Early use of daptomycin versus vancomycin for methicillin-resistant Staphylococcus aureus bacteremia with vancomycin minimum inhibitory concentration >1 mg/L: a matched cohort study. Clin. Infect. Dis. 56, 1562–1569. doi: 10.1093/cid/cit112
Mwangi, M. M., Wu, S. W., Zhou, Y., Sieradzki, K., de Lencastre, H., Richardson, P., et al. (2007). Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 104, 9451–9456. doi: 10.1073/pnas.0609839104
Proctor, R. A., von Eiff, C., Kahl, B. C., Becker, K., McNamara, P., Herrmann, M., et al. (2006). Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305. doi: 10.1038/nrmicro1384
Renzoni, A., Von Dach, E., Landelle, C., Diene, S. M., Manzano, C., Gonzales, R., et al. (2017). Impact of Exposure of Methicillin-Resistant Staphylococcus aureus to Polyhexanide in vitro and in vivo. Antimicrob. Agents Chemother. 61, 2547–2549. doi: 10.1128/AAC.00272-17
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29:26. doi: 10.1038/nbt.1754
Roemer, T., Schneider, T., and Pinho, M. G. (2013). Auxiliary factors: a chink in the armor of MRSA resistance to β-lactam antibiotics. Curr. Opin. Microbiol. 16, 538–548. doi: 10.1016/j.mib.2013.06.012
Rubio, A., Conrad, M., Haselbeck, R. J., G. C. K., Brown-Driver, V., Finn, J., et al. (2010). Regulation of mprF by antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 55, 364–367. doi: 10.1128/AAC.00429-410
Song, Y., Rubio, A., Jayaswal, R. K., Silverman, J. A., and Wilkinson, B. J. (2013). Additional routes to Staphylococcus aureus daptomycin resistance as revealed by comparative genome sequencing, transcriptional profiling, and phenotypic studies. PLoS One 8:e58469. doi: 10.1371/journal.pone.0058469
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Szurmant, H., Mohan, M. A., Imus, P. M., and Hoch, J. A. (2007). YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 189, 3280–3289. doi: 10.1128/JB.01936-1936
Tong, S. Y. C., Lye, D. C., Yahav, D., Sud, A., Robinson, J. O., Nelson, J., et al. (2020). Effect of Vancomycin or Daptomycin With vs Without an Antistaphylococcal β-Lactam on Mortality, bacteremia, relapse, or treatment failure in patients with MRSA bacteremia: a randomized clinical trial. JAMA 323, 527–537. doi: 10.1001/jama.2020.0103
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/PREACCEPT-2573980311437212
Tuchscherr, L., Kreis, C. A., Hoerr, V., Flint, L., Hachmeister, M., Geraci, J., et al. (2016). Staphylococcus aureus develops increased resistance to antibiotics by forming dynamic small colony variants during chronic osteomyelitis. J. Antimicrob. Chemother. 71, 438–448. doi: 10.1093/jac/dkv371
Utaida, S., Pfeltz, R. F., Jayaswal, R. K., and Wilkinson, B. J. (2006). Autolytic properties of glycopeptide-intermediate Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 50, 1541–1545. doi: 10.1128/AAC.50.4.1541-1545.2006
Wootton, M., Bennett, P. M., MacGowan, A. P., and Walsh, T. R. (2005). Reduced expression of the atl autolysin gene and susceptibility to autolysis in clinical heterogeneous glycopeptide-intermediate Staphylococcus aureus (hGISA) and GISA strains. J. Antimicrob. Chemother. 56, 944–947. doi: 10.1093/jac/dki289
Yamaguchi, T., Ando, R., Matsumoto, T., Ishii, Y., and Tateda, K. (2019). Association between cell growth and vancomycin resistance in clinical community-associated methicillin-resistant Staphylococcus aureus. Infect. Drug Resist. 12, 2379–2390. doi: 10.2147/IDR.S209591
Yang, C.-C., Sy, C. L., Huang, Y.-C., Shie, S.-S., Shu, J.-C., Hsieh, P.-H., et al. (2018). Risk factors of treatment failure and 30-day mortality in patients with bacteremia due to MRSA with reduced vancomycin susceptibility. Sci. Rep. 8:7868. doi: 10.1038/s41598-018-26277-26279
Yang, S.-J., Mishra, N. N., Rubio, A., and Bayer, A. S. (2013). Causal role of single nucleotide polymorphisms within the mprF gene of Staphylococcus aureus in daptomycin resistance. Antimicrob. Agents Chemother. 57, 5658–5664. doi: 10.1128/AAC.01184-1113
Keywords: methicillin-resistant Staphylococcus aureus, persistent bacteremia, vancomycin-intermediate Staphylococcus aureus, daptomycin, linezolid, multi-resistance, genomic evolution
Citation: Chen C-J, Huang Y-C and Shie S-S (2020) Evolution of Multi-Resistance to Vancomycin, Daptomycin, and Linezolid in Methicillin-Resistant Staphylococcus aureus Causing Persistent Bacteremia. Front. Microbiol. 11:1414. doi: 10.3389/fmicb.2020.01414
Received: 07 April 2020; Accepted: 02 June 2020;
Published: 07 July 2020.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Dafne Bongiorno, University of Catania, ItalyCopyright © 2020 Chen, Huang and Shie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chih-Jung Chen, Y2hpbmp1bmdAY2dtaC5vcmcudHc=; amFtZXMucGVkQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.