 Hui-Ying Ko
Hui-Ying Ko Gielenny M. Salem1
Gielenny M. Salem1 Day-Yu Chao
Day-Yu Chao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 19 June 2020
Sec. Virology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01371
Dengue viral (DENV) infection results in a wide spectrum of clinical manifestations from asymptomatic, mild fever to severe hemorrhage diseases upon infection. Severe dengue is the leading cause of pediatric deaths and/or hospitalizations, which are a major public health burden in dengue-endemic or hyperendemic countries. Like other RNA viruses, DENV continues to evolve. Adaptive mutations are obscured by the major consensus sequence (so-called wild-type sequences) and can only be identified once they become the dominant viruses in the virus population, a process that can take months or years. Traditional surveillance systems still rely on Sanger consensus sequencing. However, with the recent advancement of high-throughput next-generation sequencing (NGS) technologies, the genome-wide investigation of virus population within-host and between-hosts becomes achievable. Thus, viral population sequencing by NGS can increase our understanding of the changing epidemiology and evolution of viral genomics at the molecular level. This review focuses on the studies within the recent decade utilizing NGS in different experimental and epidemiological settings to understand how the adaptive evolution of dengue variants shapes the dengue epidemic and disease severity through its transmission. We propose three types of studies that can be pursued in the future to enhance our surveillance for epidemic prediction and better medical management.
Dengue Virus (DENV), a member of the genus Flavivirus in the family of Flaviviridae, is a single-strand, positive-sense RNA virus (King et al., 2011). Dengue viruses can be classified into four serotypes, DENV-1 to DENV-4. Humans infected with DENV develop only serotype-specific but life-long protective immunity. Antigenic differences between different serotypes limit the cross-protection from other uninfected serotypes. Therefore, secondary or heterologous sequential DENV infections are commonly observed (Guzmán et al., 2002a, b; Yacoub et al., 2013). Humans infected by dengue virus show a wide range of clinical manifestations from asymptomatic, mild fever to severe hemorrhagic diseases. Most DENV infections were asymptomatic or self-limited dengue fever (DF) characterized by fever, joint pain, muscles sore, and severe headache; a small portion of infections manifest the severe form of the disease resulting in dengue hemorrhagic fever (DHF), which is characterized by bleeding, plasma leakage, thrombocytopenia, and increased vascular permeability. Additionally, DHF can lead to fatal complications known as dengue shock syndrome (DSS). The severe dengue is the leading cause of pediatric hospitalizations and death, and a major public health burden in dengue-endemic or hyperendemic countries (World Health Organization [WHO], 2009).
The genome of DENV contains one single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTR). The ORF is translated as a single, long polypeptide, followed by post-translational process by host and virus-encoded proteases to generate three structural proteins, capsid (C), pre-membrane (prM) and envelope (E), and seven non-structural (NS) proteins, NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5 (Chambers et al., 1990). Due to a lack of proofreading function in the RNA-dependent RNA polymerase (RdRp), in each virus replication cycle RdRp will induce random mutations in the genome. Therefore, RNA viruses usually have an exceptionally high mutation rate resulting in the diversification of genomic sequences within an infected individual, so-called intra-host genetic variants or quasispecies. The quasispecies theory, proposed by Manfred Eigen, first illustrates the evolution of self-replicating RNA molecules (Eigen et al., 1989). Detailed implications on virus evolution are reviewed by Domingo and colleagues (Domingo et al., 2012). Like other RNA viruses, DENV continues to evolve and the evolutionary forces acting on the viral population include genetic mechanisms [i.e., mutations and recombination (Holmes et al., 1999)], demographic processes (i.e., genetic drift; Lequime et al., 2016) and natural selection (Bennett et al., 2003; Holmes and Twiddy, 2003). Paradoxically, unlike other single-stranded RNA viruses (such as poliomyelitis virus), DENV undergoes substantially slower rates of evolution. Comparing the non-synonymous variation in mutations between hosts (within the same host) and inter-host (among different hosts) obtained from traditional Sanger consensus sequencing, non-synonymous mutations occurred in viral genome were found more frequently within hosts rather than inter-host implying the strong negative/purified selection of DENVs (Holmes, 2003). The “trade-off” hypothesis (Figure 1) has been postulated to explain this disparity (Vasilakis et al., 2009; Deardorff et al., 2011; Sim et al., 2015; Hanley et al., 2019). Alternating replication between vertebrate and arthropod hosts constrains arbovirus evolution due to the different fitness landscapes shaped by apparently distinct physiological environments within the two different hosts.
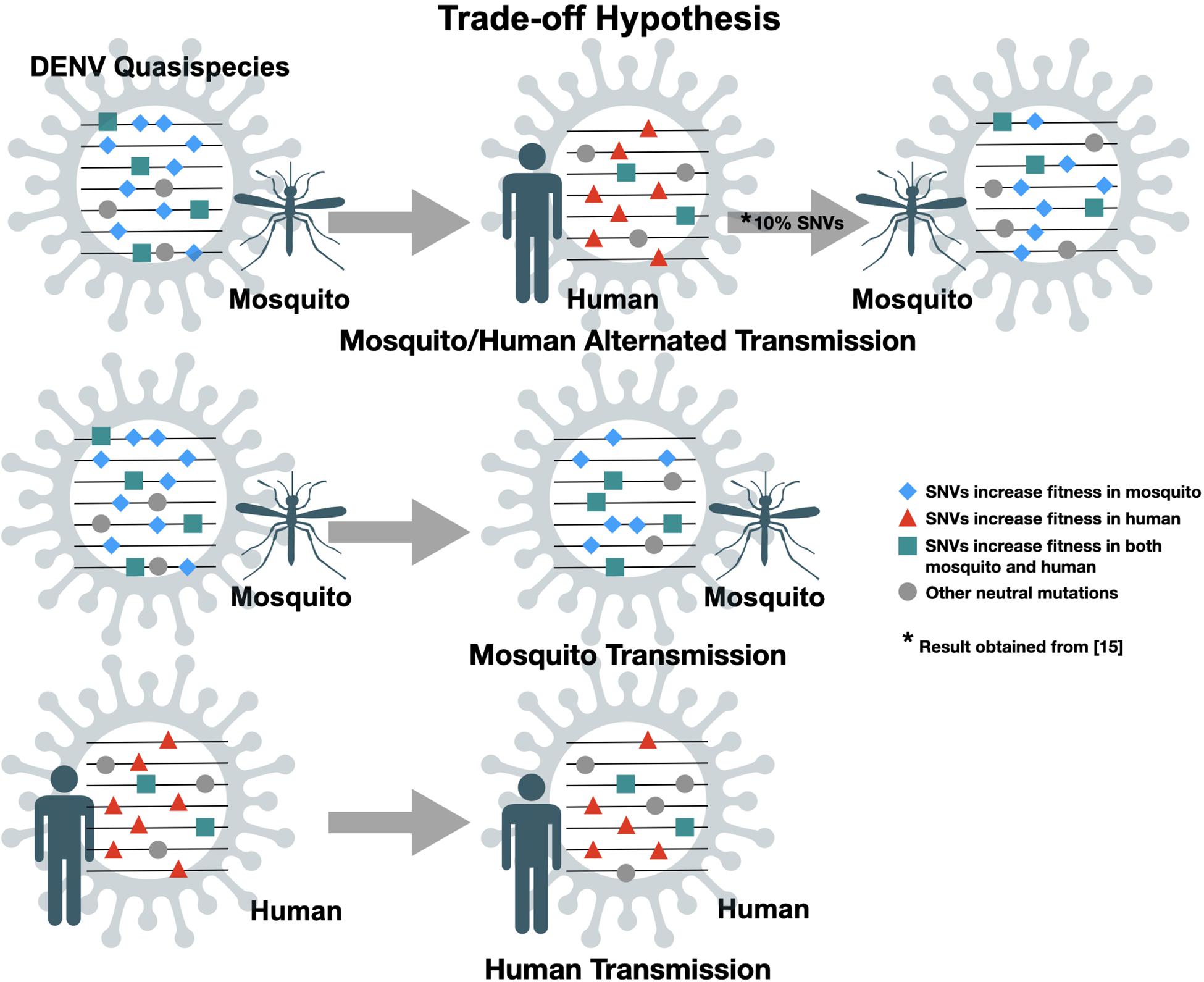
Figure 1. Trade-off hypotheses of dengue virus quasispecies transmission. Random mutations are introduced into virus genome during replication cycles. Transmission involves transferring minor population with variants of genome, practically assayed by single nucleotide variants (SNVs). Only SNVs with increased fitness in both mosquito and human will be kept after mosquito/human alternated transmission.
The four serotypes of DENVs, which circulated between humans and peridomestic Aedes mosquitoes, emerged in four independent events from sylvatic progenitors. The sylvatic strains were maintained in non-human primates and jungle mosquito species within sylvatic cycles (Wang et al., 2000). Although dengue viruses can be transmitted between humans by various jungle mosquito species, such as Ae. furcifer, Ae. Luteocephalus, and Ae. taylori, the urban cycle is maintained by urban mosquito species, mainly by Ae. aegypti and Ae. albopictus (Chen and Vasilakis, 2011). The urban DENV lineages are ecologically and evolutionarily independent from the ancestral sylvatic viruses, but might undergo selection to increase its virulence in humans. Epidemiological studies showed that globally severe dengue, including DHF and DSS, increased its frequency since its first documentation in the 1950s (Gubler, 2002). Phylogenetic evidence indicates that some DENV strains or genotypes, such as Southeast Asian (SEA) DENV-2 genotype, have the tendency to cause severe diseases. This DENV-2 SEA genotype tends to cause DHF/DSS than the original American genotype after its introduction to the Americas in middle twentieth-century (Rico-Hesse et al., 1997). These traditional studies attempted to address viral genetic variation had to rely on using Sanger sequencing, which was developed by Frederick Sanger in 1977. The method was invented based on the selective incorporation of labeled dideoxynucleotides terminator during DNA replication (Sanger, 1988). Although this has been the most widely performed sequencing method with advantages of convenience, relative longer reads length and lower cost for each reaction, the proportion of genetic variants needs molecular cloning plus Sanger sequencing (Lin et al., 2004). The process is characterized as laborious (Chao et al., 2005; Gong et al., 2013), and biological properties of the genetic variant may directly bias the chance of recovery (Forns et al., 1997). Unfortunately, the current viral surveillance system remains mostly dependent on Sanger sequencing. Adaptive mutations within the minor population are obscured by wild-type sequences and only revealed once they become dominant in the virus population, a process that can take months or years.
With the advancement of high-throughput next-generation sequencing technologies (NGS), it is achievable to conduct the genome-wide interrogation of virus population within-host (human or mosquito) and transmission between hosts (human and mosquito). High-throughput NGS techniques, including next-generation and third-generation sequencing methods, have been developed with longer reads, greater amount of sequencing data, real-time basecalling with lower cost per reads, aiming to unbiasedly explore the massive information of sequence variations. However, detecting viral variants from NGS data is not straightforward. The process usually involves aligning reads (i.e., short sequence segments) to the reference sequence and compare a spectrum of nucleotides with the reference nucleotide at each position. Between-host variations are determined by constructing the nucleotide sequences with the highest frequency at each position. In contrast, within-host variation usually requires further tests on the distribution of nucleotide at each position. Specifically, statistical methods will be applied to determine if the occurrence of a nucleotide is generated by chance, considering error rate and quality of sequencing. Minor variants are thus defined as nucleotide sequences which are detected with significant frequency but different from the highest frequency at a specific position. Various computational tools have been developed to detect minor variants. Among these tools, LoFreq (Wilm et al., 2012), ViVAN (Isakov et al., 2015), DeepSNV (Gerstung et al., 2012), and Varscan (Koboldt et al., 2009) are widely used in viral genome. Thus far, accurately calling minor variants within viral genome remains a growing field of study.
This review will focus on the NGS studies in different experimental and epidemiological settings to understand how the adaptive evolution of dengue variants shapes the dengue epidemic and disease severity through its transmission. We will discuss the discrepancy of the results between studies and the potential future directions to dissolve the gap.
DENV faces different physiological conditions in mosquitoes and humans. Replication cycles between alternate mosquitoes and humans necessitate a fitness trade-off (Vasilakis et al., 2009). To understand how different forces play a role within different hosts requires dissection of the transmission process by either infecting mosquitoes with DENV isolated from humans or infecting humans with DENV isolates from mosquito. Due to ethical considerations, human challenge studies are rarely performed under this purpose (Lyons, 2014; Mammen et al., 2014; Kestelyn et al., 2019); therefore, within-human genetic variations of DENV can only be observed under epidemiological settings. In this section, we reviewed the studies applying NGS to understand the different evolutionary forces acting on mosquito vectors fed with blood meal mixed with dengue virus isolates, or viremic blood directly from dengue patients in the well-defined laboratory settings (Table 1).
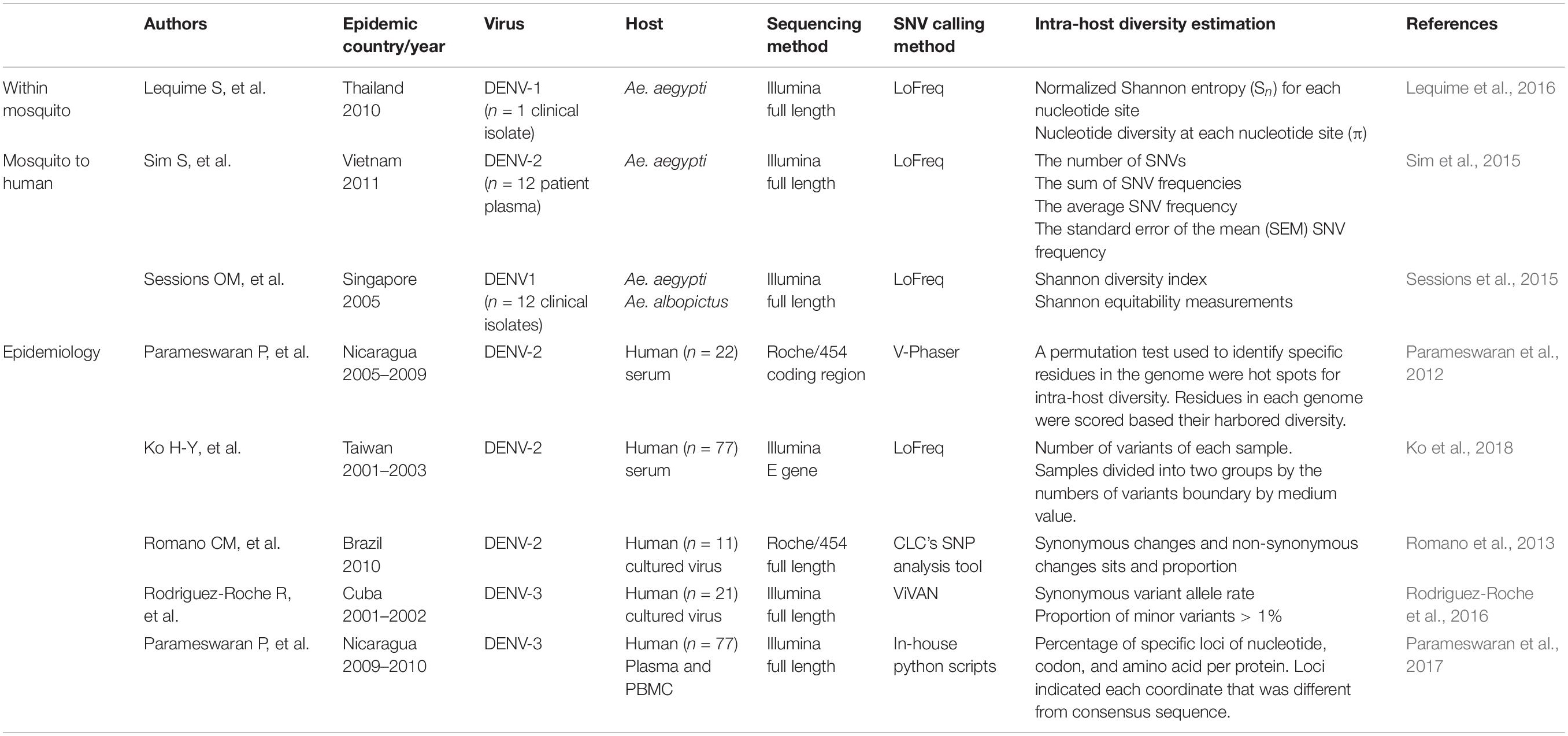
Table 1. Studies utilizing deep sequencing on DENV genetic variants.
Although studies by traditional Sanger sequencing have shown that DENV exists as heterogeneous populations in mosquito vectors (Craig et al., 2003; Lin et al., 2004), the population may differ while virus traverses through different organs of the mosquito (Lequime et al., 2016). Anatomical barriers can lead to population bottlenecks when viruses travel across different organs. Advances in deep sequencing have greatly expanded our ability to examine intra-host diversity by tracking different genetic variants passing through different anatomical barriers of the mosquito vectors. Lequime and colleagues monitored the intra-mosquito evolution of a wild-type dengue virus isolate by infecting mosquitoes with different genetic backgrounds (Lequime et al., 2016). Deep sequencing of full-length viral genomes from various mosquito organs harvested at different time points after infection confirmed that the overall genetic diversity of viral population during infection was predominantly under purifying selection. However, the genetic diversity differed significantly between mosquitoes of different genetic backgrounds despite similar initial bottleneck size. Following a stochastic reduction in genetic diversity, the population size and diversity of DENV rapidly recovered with minor genetic variants after traveling through different compartments and further expanding into the next saliva tissue. In the end, the loss of fitness by inoculating salivary gland-derived virus into the Vero mammalian cell culture confirmed the “trade-off” hypothesis. Since virus diversification could be enhanced by tissue-specific sequence-dependent small interfering RNA (siRNA) during the transversion from the midgut to the saliva of mosquitoes (Brackney et al., 2009), it remains unknown how other factors, such as viral genotypes/strains/serotypes (Parameswaran et al., 2012; Ko et al., 2018) and genetic background in mosquito (Sessions et al., 2015; Grubaugh et al., 2016), would interact with tissue-specific siRNA and impinge on the genetic breadth of virus population as well as its association with the fitness (Figure 2).
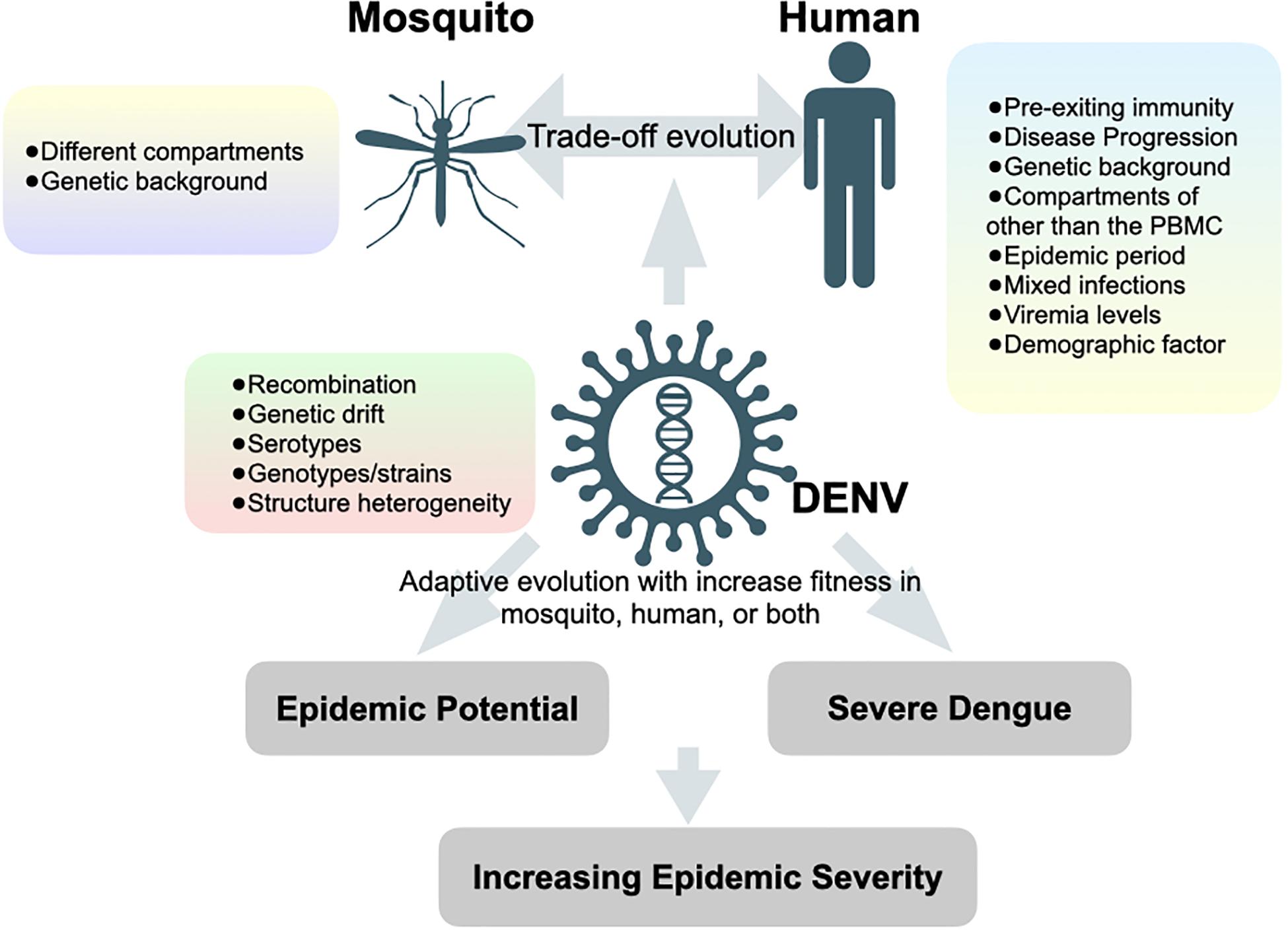
Figure 2. Hypotheses of factors that contributing to increasing epidemic severity. Forces and factors determine the evolution of dengue viruses and the potential outcomes of dengue epidemiology.
NGS has been utilized to link the transmission route and to track minor viral variants by single nucleotide variants (SNVs) of intra-host genetic variation of RNA viruses shared by different hosts or individuals (Forbi et al., 2014; Plummer et al., 2015; Nasheri et al., 2017). Sim and colleagues tracked genetic variants transmitted from human to mosquito in the laboratory setting by feeding the mosquitoes, Aedes aegypti, with viremic blood from twelve Vietnamese DENV patients (Sim et al., 2015). Viral RNA from both patients’ blood and mosquitoes were analyzed using whole-genome deep sequencing technology. The results showed that the frequency of genetic diversity of DENV, evaluated by different methods based on the SNVs, was similar between human and mosquito. However only approximately one-tenth of SNVs (in a total of 267 SNVs) found in the human was repeatedly shown in the mosquito. This result suggested that most mutations within viremic patients are either deleterious or with poor fitness once transmitted to mosquitoes. Comparing the genetic diversity between two hosts, the genetic mutations of intra-human derived viral variants were mainly located in prM, E and NS1; on the contrary, the genetic mutations of mosquito-derived variants were mainly located in NS3 and 3′ UTR. Similarly, the study by Sessions et al. (2015) using DENV-1 isolated from human viremic sera or two mosquito species after the inoculated intrathoracically found that two residues in the NS5 protein were exclusively acquired from infected Ae. aegypti but not from Ae. albopictus. The authors also identified a region within the NS3 protein that is refractory to mutations from infected humans and mosquitoes. In summary, although the sequential bottleneck transmission eventually leads to the extinction of many variants, rapid emergence and maintenance of SNVs during DENV transmission from viremic human to mosquitoes were observed, which might lead to the emergence of a new clade or lineage. In the following section, we will review the extent to which the newly evolved clade is associated with disease manifestation during major DENV epidemics.
Studying intra-host variations under an epidemiological setting aims to provide insight into inter-host evolution through human-mosquito transmission cycles and to give implications of viral fitness, which may lead to increased epidemic potential and/or severe clinical outcomes. Increased epidemic potential and disease severity are primarily influenced by two viral factors: transmissibility and virulence. A deep-sequencing tool was recently applied to study genetic variation and evolutionary dynamics of DENV under different epidemiological settings, which differed in the scale of transmissibility (numbers of infection in the human population) and virulence (numbers of severe dengue cases during the epidemics) (Figure 2). Here, we categorize three major epidemiological phenomena related to viral transmissibility and virulence, commonly observed during DENV outbreaks. We summarize those studies in Table 1 and review below:
The epidemic potential is the ability of pathogens to rapidly spread and infect many people in the population. Strong purifying selection during alternating transmission between human and mosquito hosts increases the possibility of extinction due to repeated bottleneck transmissions imposed by mosquitoes. Regular lineage extinction and replacement by the variants with better fitness for supporting viral replication in both hosts is one of its dynamic features characterizing the transmissibility of DENV with epidemic potential.
Ko et al. (2018) analyzed viral sequences from the sera of the representative dengue patients during two consecutive DENV-2 outbreaks from 2001 to 2003 in Taiwan. The study identified three consensus genetic variants: groups Ia, Ib, and II, with different spatio-temporal population dynamics. Deep sequencing of the viral envelope gene further confirmed the emergence of those three variants from patient sera. Signature nucleotide changes carried by Ib and II were detected within the quasispecies of Ia virus. The finding indicated the initial Ia variants with low-frequency were gradually replaced by variants of Ib and II with high-frequency or consensus sequence. Combined with the surveillance data and phylogenetic analyses, it showed that no DENV-confirmed cases detected but with group Ib variants continuously circulating during two consecutive years. The observations indicated the potential of group Ib as the “sheltered overwintering” lineage maintained in an undefined ecological niche. On the other hand, group II, which presented higher epidemic potential, was responsible for the larger scale of epidemics during 2002/03. Similar findings were also observed by other group in Singapore studied in between-host DENV-1 diversity during the epidemic in 2013–2014. With more than 40,000 dengue-confirmed cases recorded by the end of 2014, the unprecedented increase in dengue incidence was associated with the switch from DENV-2 to DENV-1 in early 2014. This epidemic strain of DENV-1 initially emerged from an mixed viral population and went through drastically bottleneck transmission with decreased between-host genetic diversity, which led to clade replacement by the emergence of a new lineage of genotype III of DENV-1 (Hapuarachchi et al., 2016). The spatial and temporal dynamics of the inter-human genetic variations of DENV-1 identified by NGS suggests the direct relationship of the numbers of viral variants and the transmission intensity, as reflected by confirmed dengue case-load and transmission hot-spot.
The results from both studies are consistent with the findings by mosquito inoculations as reviewed in section “Track Genetic Variant From Human to Mosquito.” Intensified transmission of DENV may accumulate the viral variants and increase the opportunity of selecting genetic variant(s) with better fitness for replicating both in mosquito and human and lead to the increase of epidemic potential (Romano et al., 2013; Rodriguez-Roche et al., 2016; Ko et al., 2018). The close interaction between viral evolution and transmissibility indicated that tracking genetic diversity during an outbreak is a potential tool to infer DENV transmission dynamics and thereby, to assess the epidemic risk and public health control measures.
The importance of viral genetics in determining the outcome of DENV infection has been suggested in the context of primary versus secondary infections (Vaughn et al., 2000), extent of primary epidemics in naïve populations (Barnes and Rosen, 1974; Gubler et al., 1978), viral variants with different virulence (Rico-Hesse, 2010), sequential order of infecting serotypes (Halstead, 2003), and the influence of pre-existing, heterotypical immunity (OhAinle et al., 2011; OhAinle and Harris, 2014). However, the large body of work has been mainly conducted by the consensus Sanger sequencing. Thus far, five studies have performed to measure genetic diversity within human hosts by deep sequencing and only three studies have considered the epidemiological conditions: clinical features (DF versus DHF) (Parameswaran et al., 2012, 2017) and immune status (primary versus secondary infection) (Rodriguez-Roche et al., 2016; Parameswaran et al., 2017; Ko et al., 2018).
Two studies evaluated the genetic mutation of DENV-3 from patients’ plasma samples and compared the intra-host diversity between DF and DHF cases in Nicaragua within two cohorts (Parameswaran et al., 2012, 2017). The first study on DENV-2 found that the clade replacement of DENV-2, NI-1 clade by a novel NI-2B clade was associated with increased virus replication in native mosquitoes (viral fitness) and clinical severity (Quiner et al., 2014). Further investigation on the patterns of intra-host diversity of the two clades found that direct correlations between viral diversity and clinical outcome were not significant, although NI-2B viruses had lower intra-host diversity than NI-1 viruses (Parameswaran et al., 2012). Consistently, Parameswaran et al. evaluated non-synonymous variants with DENV-3 patient plasma and PBMCs from the cohort during 2009–2010 in Nicaragua, and the results also failed to connect intra-host diversity to disease severity (Parameswaran et al., 2017).
During two separate epidemics caused by DENV-3 and DENV-2 in Cuba and Taiwan, higher genetic variation, defined by arbitrary thresholds of minor variants, was observed in patients with secondary infection than those with primary infection (Rodriguez-Roche et al., 2016; Ko et al., 2018). These studies proposed that the immune status of infected humans is the driving force contributing to the increase in genetic variants and the derivation of antibody-escape mutant(s) as previously suggested by Guzman and colleagues (Guzman et al., 2000), although no experimental data has been provided to verify the results. On the contrary, the study in Nicaragua by Parameswaran et al. found significantly fewer unique loci both in peripheral blood mononuclear cells (PBMC) and plasma obtained from DENV patients with secondary infection than those with primary infection (Parameswaran et al., 2017). It needs to be noted that the strength of positive selection differed in the complete genomic RNAs between PBMC and plasma samples from the same paired DENV patients. Significantly higher abundances of non-synonymous variants were presented in plasma than in PBMCs for prM/M and NS3 regions. Since the prM/M and NS3 regions were suggested to be the major targets of the human B-cell and T-cell immune responses, Parameswaran et al. also concluded that the immune-driven selection pressures could shape the differences in the DENV population released into plasma from PBMC or site(s) supporting viral replication.
In summary, applying NGS technology under epidemiological settings has enhanced our current knowledge on the dynamics of dengue viral population shaped by replication site(s) and serotype-specific immunity, and, in turn, can influence the disease severity during an epidemic. However, the current knowledge gap regarding the association between disease severity and sequence diversity is impeded by different methods and definition in estimating intra-host diversity and the un-controlled confounding factors, such as sampling days, age, immune status with different disease severity, from the epidemiology studies.
An increase in the proportion of DHF/DSS cases during the DENV epidemic was first documented in the South Pacific islands in the 1970s (Gubler et al., 1978). Similar epidemiological feature was also reported in several outbreaks in Taiwan (1998, 2002) (Chao et al., 2004; Chen et al., 2008), Cuba (1987, 1997, 2001–2002) (Valdés et al., 1999; Guzmán et al., 2000, 2002b; Peláez et al., 2004; Rodriguez-Roche et al., 2016), Brazil (1981–2002) (Nogueira et al., 2005; Siqueira et al., 2005) and Nicaragua (2004–2008) (OhAinle et al., 2011; Quiner et al., 2014). Deep sequencing technology was applied to explore the dynamic process of viral evolution and its association with increasing clinical severity during the DENV-3 outbreak in Cuba, 2001 (Rodriguez-Roche et al., 2016). This epidemic resulted in 12,889 confirmed cases, including 78 cases of DHF/DSS. A significant monthly increase in the proportion of DHF/DSS cases was observed during this epidemic. A mixed population of DENV-3 within genotype III was identified during the epidemic and the significant number of minor variants were selected and become major variants toward the end of the epidemic. In nostructural protein, the study conducted in Cuba also identified an increasing trend of synonymous variant toward the end of the epidemic, particularly among dengue patients with secondary infections (Rodriguez-Roche et al., 2016). However, the association between specific SNVs or the variability of quasispecies and disease severity was not evident, which was consistent with the other studies (Parameswaran et al., 2012, 2017).
Consistent with the findings of NGS studies from sections “Epidemic Potential” and “Severe Dengue” sections of this review, increasing both the numbers of infected patients and the proportion of DHF/DSS cases usually accompanies each other. This suggests that the gain of fitness by selecting viral variant in favor of replicating in both humans and mosquitoes is accompanied by serial bottleneck transmission during the large DENV outbreak. Prior immunity further contributed to the genetic diversity of DENV due to secondary infection. Selection of variants with growth advantage resulted in a higher probability to cause severe disease during the late phase of the outbreak (Quiner et al., 2014).
Like other viruses, the clinical outcomes of dengue infections were determined by complex interactions between the hosts’ immune status and viral genetics factors (Quinlivan et al., 2007; Tanaka and Mizokami, 2007; OhAinle et al., 2011). Host immunity is expected to exert selective pressure on viral replication. Insect hosts carry several arms of antiviral immunity including RNAi, apoptosis, ubiquitin-proteasome pathway, autophagy, and the heat-shock response (Yadav et al., 2005; Marques and Imler, 2016; Troupin et al., 2016). Rare viral variants having replicative advantages could be selected and become the dominant viral population if their sequence is sufficiently divergent to escape the antiviral immunity (Brackney et al., 2009; Lambrechts and Lequime, 2016).
In addition to innate immunity, antiviral adaptive immunity (in T- and B-cells) in humans can also shape the landscape of viral genomic population. The study in Puerto Rico showed that Clade PR-2B, which emerged during the 1994 DENV-2 epidemic and replaced an endemic PR-1 clade, generated increased levels of subgenomic flavivirus RNA (sfRNA). This sequence-dependent sfRNA is capable of binding to tripartite motif 25 (TRIM25) protein and prevents TRIM25 deubiquitylation, which therefore decreases type I interferon expression through the retinoic acid-inducible gene 1 (RIG-I) pathway (Manokaran et al., 2015).
Intensified transmission of DENV could generate defective interfering (DI) viral particles. Such DI particles have a replication advantage over the full-length parental virus, although they only replicate by complementation in the presence of co-infecting functional viruses. The emergence of defective viruses, including nonsense mutations in the envelope gene, was detected in both humans and mosquitoes over a period of 18 months in Myanmar (Aaskov et al., 2006). Combining dynamic modeling and phylogenetic analysis of viral populations for both intra-host and inter-host dynamics, the results indicated that co-transmission of defective and functional DENV can result in increased transmission and epidemic potential (Ke et al., 2013). These findings provide the underlying mechanism of how the interaction of viral genetic variants and host protein can evade the innate immune response and increases the fitness of the viral population during an epidemic, which leads to an increased epidemic severity.
Efforts on studies of phylogenetic and molecular evolution on viral genomic sequences have provided us valuable insights of DENV evolution and epidemiology within and among different hosts (Bennett et al., 2009; Weaver and Vasilakis, 2009). Although a stochastic process may play a major role in shaping viral genetic diversity, examples of lineage replacement and its association with an increased incidence of DENV infection during outbreaks have been reported (Ko et al., 2018). Furthermore, such genetic variants may also affect the vaccine efficacy, which significantly decreased when the amino acid distance from the DENV-4 vaccine increased and significantly elevated when eight signature positions matched against DENV-4 vaccine strain (Juraska et al., 2018). Unfortunately, such variants with epidemic potential can only be identified retrospectively after the epidemic has occurred due to the wide-scale monitoring of circulating strains is time-consuming and labor-intensive. Here we propose three future directions aiming to establish a link between varying viral diversity and the increased risk of epidemics or severe dengue diseases.
Predicting its evolution toward epidemic potential is complicated by ecological (mosquito) and epidemiological (human) factors. The extrinsic incubation period (EIP), defined as the time interval between pathogen acquisition and pathogen transmission by the vector, is one of the most influential parameters determining the epidemic potential. A study by Fontaine and colleagues found significant variations in EIP in Aedes aegypti mosquitoes among eight different isolates of DENV-1, representing the worldwide diversity of recently circulating viruses (Fontaine et al., 2018). Combining in silico outbreak simulation, it is predicted that the observed EIP variation in systemic mosquito infection may drive significant differences in the probability of dengue outbreak and the number of human infections. In principle, the shorter the EIP, the higher is the epidemic risk of the virus, potentially resulting in the higher magnitude of dengue outbreaks. If mosquito infection study is not possible for the lab, examining the phylogenetic tree to check any new clade formation may also give a hint of the emergence of potential epidemic strain.
Another attempt to follow evolutionary trajectories during virus transmission between humans and mosquito recapitulating the emergence of genetic variants with epidemic potential can be found for the Chikungunya virus (CHIKV). CHIKV, normally circulating in Aedes aegypti mosquitoes, occurred in the Indian Ocean islands before the 2005–2006 epidemic. It was found that an alanine-to-valine mutation at residue 226 of the E1 glycoprotein (A226V) enhanced the infectivity of another mosquito vector, Aedes albopictus, and expanded the affected geographical area (Tsetsarkin et al., 2007). Stapleford and colleagues studied the spatial and temporal evolution of CHIKV during natural transmission between mosquitoes and mice. Their experiments recapitulated the emergence of V80I: A129V from currently circulating A226V variants in the saliva of multiple mosquito strains (Stapleford et al., 2014). More adaptive mutations can also be found while further alternating the virus transmission between two hosts. This mosquito-mouse transmission model could be a useful tool to predict evolutionary trajectories and epidemic potentials of viral variants.
Replication of DENV within human host generates genetic variants of the viral population (intra-host diversity), which are thought to serve as a template on which evolutionary mechanisms, such as recombination, genetic drift, bottlenecking, or positive/negative selective pressure, acts to shape variation at the consensus level between hosts (inter-host diversity). The disease outcomes of viral evolutionary dynamics are determined by multiple factors interplayed between host and virus. However, direct comparisons between different epidemiological studies are difficult, mainly due to the following reasons: (1) the confounding factors from each epidemiological study, (2) different SNV basecalling method used, and (3) intra-host diversity estimation analyzed (Table 1). First, the studies in Cuba and Taiwan had more adult dengue cases, which were different from the study conducted in Nicaragua with mostly pediatric cases. A previous study has shown that asymptomatic or pre-symptomatic patients retain transmissibility to the mosquito, and at a given level of viremia, asymptomatic and pre-symptomatic people significantly and more readily transmit the virus to mosquitoes than symptomatic individuals (Duong et al., 2015). Given the possible high asymptomatic rate of dengue infections (Shepard et al., 2004), the viral intra-host variation within asymptomatic or pre-symptomatic humans is not only a missing part of studying viral micro-evolution but also an important question regarding virus pathogenesis. Second, the SNV calling methods and the statistical methods to infer the frequency of intra-host diversity might also drastically influence the results of epidemiological or clinical inferences. Despite variants calling pipelines such as LoFreq and DeepSNV have been commonly used, the accuracy in characterizing intra-host diversity has rarely been validated under the same sample condition (Barbezange et al., 2018) and it might be sensitive to the input virus titers and the setup of suitable quality thresholds (McCrone and Lauring, 2016). Third, studies in Cuba and Taiwan considered SNV in nucleotide level using the SNV counts or proportions to estimate intra-host diversity, and the study in Nicaragua considered the amino acid level by defining unique loci with non-synonymous minor variants to estimate the intra-host diversity. Currently, there’s no common rules or best approaches to estimate intra-host diversity, which make direct comparison between different epidemiological studies difficult and warrants further investigation.
Recently, more and more studies have applied deep sequencing technique to obtain consensus sequences in individual level. However, rich information embeded in output data have not been fully explored. Data deposited in Sequence Read Archive (SRA) under NCBI, for example, could therefore become a useful resource for researchers to adress clinical or epidmiological questions. Applying deep learning technology on big data analysis will be crucial to elucidate underlying mechanisms by integrating intra-host sequencing data with a systemic collection of epidemiological data acquired from DENV-infected individuals with or without symptoms.
H-YK and GS reviewed and drafted the manuscript. G-JC commented and edited the manuscript. D-YC coordinated the review, edited and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Ministry of Science and Technology Taiwan (MOST 107-2313-B-005-038-MY3).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aaskov, J., Buzacott, K., Thu, H. M., Lowry, K., and Holmes, E. C. (2006). Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 311, 236–238. doi: 10.1126/science.1115030
Barbezange, C., Jones, L., Blanc, H., Isakov, O., Celniker, G., Enouf, V., et al. (2018). Seasonal genetic drift of human influenza A virus quasispecies revealed by deep sequencing. Front. Microbiol. 9:2596. doi: 10.3389/fmicb.2018.02596
Barnes, W. J., and Rosen, L. (1974). Fatal hemorrhagic disease and shock associated with primary dengue infection on a Pacific island. Am. J. Trop. Med. Hyg. 23, 495–506. doi: 10.4269/ajtmh.1974.23.495
Bennett, S., Drummond, A., Kapan, D., Suchard, M., Munoz-Jordan, J., Pybus, O., et al. (2009). Epidemic dynamics revealed in dengue evolution. Mol. Biol. Evol. 27, 811–818. doi: 10.1093/molbev/msp285
Bennett, S. N., Holmes, E. C., Chirivella, M., Rodriguez, D. M., Beltran, M., Vorndam, V., et al. (2003). Selection-driven evolution of emergent dengue virus. Mol. Biol. Evol. 20, 1650–1658. doi: 10.1093/molbev/msg182
Brackney, D. E., Beane, J. E., and Ebel, G. D. (2009). RNAi targeting of West Nile virus in mosquito midguts promotes (virus). diversification. PLoS Pathog. 5:e1000502. doi: 10.1371/journal.ppat.1000502
Chambers, T. J., Hahn, C. S., Galler, R., and Rice, C. M. (1990). Flavivirus genome organization, expression, and replication. Ann. Rev. Microbiol. 44, 649–688. doi: 10.1146/annurev.mi.44.100190.003245
Chao, D.-Y., King, C.-C., Wang, W.-K., Chen, W.-J., Wu, H.-L., and Chang, G.-J. J. (2005). Strategically examining the full-genome of dengue virus type 3 in clinical isolates reveals its mutation spectra. Virol. J. 2:72.
Chao, D.-Y., Lin, T.-H., Hwang, K.-P., Huang, J.-H., Liu, C.-C., and King, C.-C. (2004). 1998 dengue hemorrhagic fever epidemic in Taiwan. Emerg. Infect. Dis. 10:552. doi: 10.3201/eid1003.020518
Chen, H.-L., Lin, S.-R., Liu, H.-F., King, C.-C., Hsieh, S.-C., and Wang, W.-K. (2008). Evolution of dengue virus type 2 during two consecutive outbreaks with an increase in severity in southern Taiwan in 2001–2002. Am. J. Trop. Med. Hyg. 79, 495–505. doi: 10.4269/ajtmh.2008.79.495
Chen, R., and Vasilakis, N. (2011). Dengue—quo tu et quo vadis? Viruses 3, 1562–1608. doi: 10.3390/v3091562
Craig, S., Thu, H. M., Lowry, K., Wang, X., Holmes, E. C., and Aaskov, J. (2003). Diverse dengue type 2 virus populations contain recombinant and both parental viruses in a single mosquito host. J. Virol. 77, 4463–4467. doi: 10.1128/jvi.77.7.4463-4467.2003
Deardorff, E. R., Fitzpatrick, K. A., Jerzak, G. V., Shi, P.-Y., Kramer, L. D., and Ebel, G. D. (2011). West Nile virus experimental evolution in vivo and the trade-off hypothesis. PLoS Pathog. 7:e1002335. doi: 10.1371/journal.ppat.1002335
Domingo, E., Sheldon, J., and Perales, C. (2012). Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76, 159–216. doi: 10.1128/mmbr.05023-11
Duong, V., Lambrechts, L., Paul, R. E., Ly, S., Lay, R. S., Long, K. C., et al. (2015). Asymptomatic humans transmit dengue virus to mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 112, 14688–14693. doi: 10.1073/pnas.1508114112
Eigen, M., McCaskill, J., and Schuster, P. (1989). The molecular quasi-species. Adv. Chem. Phys. 75, 149–263. doi: 10.1002/9780470141243.ch4
Fontaine, A., Lequime, S., Moltini-Conclois, I., Jiolle, D., Leparc-Goffart, I., Reiner, R. C., et al. (2018). Epidemiological significance of dengue virus genetic variation in mosquito infection dynamics. PLoS Pathog. 14:e1007187. doi: 10.1371/journal.ppat.1007187
Forbi, J. C., Campo, D. S., Purdy, M. A., Dimitrova, Z. E., Skums, P., Xia, G., et al. (2014). Intra-host diversity and evolution of hepatitis C virus endemic to Côte d’Ivoire. J. Med. Virol. 86, 765–771. doi: 10.1002/jmv.23897
Forns, X., Bukh, J., Purcell, R. H., and Emerson, S. U. (1997). How Escherichia coli can bias the results of molecular cloning: preferential selection of defective genomes of hepatitis C virus during the cloning procedure. Proc. Natl. Acad. Sci. U.S.A. 94, 13909–13914. doi: 10.1073/pnas.94.25.13909
Gerstung, M., Beisel, C., Rechsteiner, M., Wild, P., Schraml, P., Moch, H., et al. (2012). Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat. Commun. 3, 1–8.
Gong, L., Han, Y., Chen, L., Liu, F., Hao, P., Sheng, J., et al. (2013). Comparison of next-generation sequencing and clone-based sequencing in analysis of hepatitis B virus reverse transcriptase quasispecies heterogeneity. J. Clin. Microbiol. 51, 4087–4094. doi: 10.1128/jcm.01723-13
Grubaugh, N. D., Weger-Lucarelli, J., Murrieta, R. A., Fauver, J. R., Garcia-Luna, S. M., Prasad, A. N., et al. (2016). Genetic drift during systemic arbovirus infection of mosquito vectors leads to decreased relative fitness during host switching. Cell Host Microb. 19, 481–492. doi: 10.1016/j.chom.2016.03.002
Gubler, D. J. (2002). Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends microbiol. 10, 100–103. doi: 10.1016/s0966-842x(01)02288-0
Gubler, D. J., Reed, D., Rosen, L., and Hitchcock, J. C. Jr. (1978). Epidemiologic, clinical, and virologic observations on dengue in the Kingdom of Tonga. Am. J. Trop. Med. Hyg. 27, 581–589. doi: 10.4269/ajtmh.1978.27.581
Guzmán, M. G., Kouri, G., Bravo, J., Valdes, L., Susana, V., and Halstead, S. B. (2002a). Effect of age on outcome of secondary dengue 2 infections. Intern. J. Infect. Dis. 6, 118–124. doi: 10.1016/s1201-9712(02)90072-x
Guzmán, M. G., Kourí, G., Valdés, L., Bravo, J., Vázquez, S., and Halstead, S. B. (2002b). Enhanced severity of secondary dengue-2 infections: death rates in 1981 and 1997 Cuban outbreaks. Rev. Panam. Salud Públ. 11, 223–227. doi: 10.1590/s1020-49892002000400003
Guzman, M. G., Kouri, G., and Halstead, S. B. (2000). Do escape mutants explain rapid increases in dengue case-fatality rates within epidemics? Lancet 355, 1902–1903. doi: 10.1016/S0140-6736(00)02303-5
Guzmán, M. G., Kouri, G., Valdes, L., Bravo, J., Alvarez, M., Vazques, S., et al. (2000). Epidemiologic studies on Dengue in Santiago de Cuba, 1997. Am. J. Epidemiol. 152, 793–799. doi: 10.1093/aje/152.9.793
Halstead, S. B. (2003). Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 60, 421–467. doi: 10.1016/s0065-3527(03)60011-4
Hanley, K. A., Azar, S. R., Campos, R. K., Vasilakis, N., and Rossi, S. L. (2019). Support for the transmission-clearance trade-off hypothesis from a study of Zika virus delivered by mosquito bite to mice. Viruses 11:1072. doi: 10.3390/v11111072
Hapuarachchi, H. C., Koo, C., Kek, R., Xu, H., Lai, Y. L., Liu, L., et al. (2016). Intra-epidemic evolutionary dynamics of a Dengue virus type 1 population reveal mutant spectra that correlate with disease transmission. Sci. Rep. 6:22592.
Holmes, E. C. (2003). Patterns of intra-and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. J. Virol. 77, 11296–11298. doi: 10.1128/jvi.77.20.11296-11298.2003
Holmes, E. C., and Twiddy, S. S. (2003). The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evolut. 3, 19–28. doi: 10.1016/s1567-1348(03)00004-2
Holmes, E. C., Worobey, M., and Rambaut, A. (1999). Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 16, 405–409. doi: 10.1093/oxfordjournals.molbev.a026121
Isakov, O., Bordería, A. V., Golan, D., Hamenahem, A., Celniker, G., Yoffe, L., et al. (2015). Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics 31, 2141–2150. doi: 10.1093/bioinformatics/btv101
Juraska, M., Magaret, C. A., Shao, J., Carpp, L. N., Fiore-Gartland, A. J., Benkeser, D., et al. (2018). Viral genetic diversity and protective efficacy of a tetravalent dengue vaccine in two phase 3 trials. Proc. Natl. Acad. Sci. U.S.A. 115, E8378–E8387.
Ke, R., Aaskov, J., Holmes, E. C., and Lloyd-Smith, J. O. (2013). Phylodynamic analysis of the emergence and epidemiological impact of transmissible defective dengue viruses. PLoS Pathog. 9:e1003193. doi: 10.1371/journal.ppat.1003193
Kestelyn, E., Le Phuong, C., Van Nuil, J. I., Thi, H. T. D., Nguyen, N. M., The, T. D., et al. (2019). Expert voices and equal partnerships: establishing controlled human infection models (CHIMs) in Vietnam. Wellcome Open Res. 4:143. doi: 10.12688/wellcomeopenres.15337.1
King, A. M., Lefkowitz, E., Adams, M. J., and Carstens, E. B. (2011). Virus Taxonomy: Ninth Report Of The International Committee On Taxonomy Of Viruses. Amsterdam: Elsevier.
Ko, H.-Y., Li, Y.-T., Chao, D.-Y., Chang, Y.-C., Li, Z.-R. T., Wang, M., et al. (2018). Inter-and intra-host sequence diversity reveal the emergence of viral variants during an overwintering epidemic caused by dengue virus serotype 2 in southern Taiwan. PLoS Negl. Trop. Dis. 12:e0006827. doi: 10.1371/journal.ppat.1006827
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Lambrechts, L., and Lequime, S. (2016). Evolutionary dynamics of dengue virus populations within the mosquito vector. Curr. Opin. Virol. 21, 47–53. doi: 10.1016/j.coviro.2016.07.013
Lequime, S., Fontaine, A., Gouilh, M. A., Moltini-Conclois, I., and Lambrechts, L. (2016). Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. PLoS Genet. 12:e1006111. doi: 10.1371/journal.ppat.1006111
Lin, S.-R., Hsieh, S.-C., Yueh, Y.-Y., Lin, T.-H., Chao, D.-Y., Chen, W.-J., et al. (2004). Study of sequence variation of dengue type 3 virus in naturally infected mosquitoes and human hosts: implications for transmission and evolution. J. Virol. 78, 12717–12721. doi: 10.1128/jvi.78.22.12717-12721.2004
Lyons, A. G. (2014). The human dengue challenge experience at the walter reed army institute of research. J. Infect. Dis. 209(Suppl._2), S49–S55.
Mammen, M., Lyons, A., Innis, B., Sun, W., McKinney, D., Chung, R., et al. (2014). Evaluation of dengue virus strains for human challenge studies. Vaccine 32, 1488–1494. doi: 10.1016/j.vaccine.2013.12.040
Manokaran, G., Finol, E., Wang, C., Gunaratne, J., Bahl, J., Ong, E. Z., et al. (2015). Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221. doi: 10.1126/science.aab3369
Marques, J. T., and Imler, J.-L. (2016). The diversity of insect antiviral immunity: insights from viruses. Curr. Opin. Microbiol. 32, 71–76. doi: 10.1016/j.mib.2016.05.002
McCrone, J. T., and Lauring, A. S. (2016). Measurements of intrahost viral diversity are extremely sensitive to systematic errors in variant calling. J. Virol. 90, 6884–6895. doi: 10.1128/jvi.00667-16
Nasheri, N., Petronella, N., Ronholm, J., Bidawid, S., and Corneau, N. (2017). Characterization of the genomic diversity of norovirus in linked patients using a metagenomic deep sequencing approach. Front. Microbiol. 8:73. doi: 10.3389/fmicb.2018.00073
Nogueira, R. M. R., Schatzmayr, H. G., De Filippis, A. M. B., Dos Santos, F. B., Da Cunha, R. V., Coelho, J. O., et al. (2005). Dengue virus type 3, Brazil, 2002. Emerg. Infect. Dis. 11:1376.
OhAinle, M., Balmaseda, A., Macalalad, A. R., Tellez, Y., Zody, M. C., Saborío, S., et al. (2011). Dynamics of dengue disease severity determined by the interplay between viral genetics and serotype-specific immunity. Sci. Transl. Med. 3:ra28.
OhAinle, M., and Harris, E. (2014). 12 dengue pathogenesis: viral factors. Dengue Dengue Hemorrhagic Fever 2014:229. doi: 10.1079/9781845939649.0229
Parameswaran, P., Charlebois, P., Tellez, Y., Nunez, A., Ryan, E. M., Malboeuf, C. M., et al. (2012). Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity. J. Virol. 86, 8546–8558. doi: 10.1128/jvi.00736-12
Parameswaran, P., Wang, C., Trivedi, S. B., Eswarappa, M., Montoya, M., Balmaseda, A., et al. (2017). Intrahost selection pressures drive rapid dengue virus microevolution in acute human infections. Cell Host Microb. 22, 400–410.
Peláez, O., Guzmán, M. G., Kourí, G., Pérez, R., San Martín, J. L., Vázquez, S., et al. (2004). Dengue 3 epidemic, Havana, 2001. Emerg. Infect. Dis. 10:719.
Plummer, E., Buck, M. D., Sanchez, M., Greenbaum, J. A., Turner, J., Grewal, R., et al. (2015). Dengue virus evolution under a host-targeted antiviral. J. Virol. 89, 5592–5601. doi: 10.1128/jvi.00028-15
Quiner, C. A., Parameswaran, P., Ciota, A. T., Ehrbar, D. J., Dodson, B. L., Schlesinger, S., et al. (2014). Increased replicative fitness of a dengue virus 2 clade in native mosquitoes: potential contribution to a clade replacement event in Nicaragua. J. Virol. 88, 13125–13134.
Quinlivan, M. L., Gershon, A. A., Al Bassam, M. M., Steinberg, S. P., LaRussa, P., Nichols, R. A., et al. (2007). Natural selection for rash-forming genotypes of the varicella-zoster vaccine virus detected within immunized human hosts. Proc. Natl. Acad. Sci. U.S.A. 104, 208–212. doi: 10.1073/pnas.0605688104
Rico-Hesse, R., Harrison, L. M., Salas, R. A., Tovar, D., Nisalak, A., Ramos, C., et al. (1997). Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 230, 244–251. doi: 10.1006/viro.1997.8504
Rodriguez-Roche, R., Blanc, H., Bordería, A. V., Díaz, G., Henningsson, R., Gonzalez, D., et al. (2016). Increasing clinical severity during a dengue virus type 3 Cuban epidemic: deep sequencing of evolving viral populations. J. Virol. 90, 4320–4333. doi: 10.1128/jvi.02647-15
Romano, C. M., Lauck, M., Salvador, F. S., Lima, C. R., Villas-Boas, L. S., Araújo, E. S. A., et al. (2013). Inter-and intra-host viral diversity in a large seasonal DENV2 outbreak. PLoS One 8:e70318. doi: 10.1371/journal.ppat.070318
Sanger, F. (1988). Sequences, sequences, and sequences. Annu. Rev. Biochem. 57, 1–29. doi: 10.1146/annurev.bi.57.070188.000245
Sessions, O. M., Wilm, A., Kamaraj, U. S., Choy, M. M., Chow, A., Chong, Y., et al. (2015). Analysis of dengue virus genetic diversity during human and mosquito infection reveals genetic constraints. PLoS Negl. Trop. Dis. 9:e0004044. doi: 10.1371/journal.ppat.0004044
Shepard, D. S., Suaya, J. A., Halstead, S. B., Nathan, M. B., Gubler, D. J., Mahoney, R. T., et al. (2004). Cost-effectiveness of a pediatric dengue vaccine. Vaccine 22, 1275–1280. doi: 10.1016/j.vaccine.2003.09.019
Sim, S., Aw, P. P., Wilm, A., Teoh, G., Hue, K. D. T., Nguyen, N. M., et al. (2015). Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 9:e0004052. doi: 10.1371/journal.ppat.0004052
Siqueira, J. B. Jr., Martelli, C. M. T., Coelho, G. E., da Rocha Simplício, A. C., and Hatch, D. L. (2005). Dengue and dengue hemorrhagic fever, Brazil, 1981–2002. Emerg. Infect. Dis. 11:48.
Stapleford, K. A., Coffey, L. L., Lay, S., Bordería, A. V., Duong, V., Isakov, O., et al. (2014). Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microb. 15, 706–716. doi: 10.1016/j.chom.2014.05.008
Tanaka, Y., and Mizokami, M. (2007). Genetic diversity of hepatitis B virus as an important factor associated with differences in clinical outcomes. J. Infect. Dis. 195, 1–4. doi: 10.1086/509898
Troupin, A., Londono-Renteria, B., Conway, M. J., Cloherty, E., Jameson, S., Higgs, S., et al. (2016). A novel mosquito ubiquitin targets viral envelope protein for degradation and reduces virion production during dengue virus infection. Biochim. Biophys. Acta Gen. Subj. 1860, 1898–1909. doi: 10.1016/j.bbagen.2016.05.033
Tsetsarkin, K. A., Vanlandingham, D. L., McGee, C. E., and Higgs, S. (2007). A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3:e201. doi: 10.1371/journal.ppat.000201
Valdés, L., Guzmán, M., Kourí, G., Delgado, J., Carbonell, I., Cabrera, M., et al. (1999). Epidemiology of dengue and hemorrhagic dengue in Santiago, Cuba 1997. Rev. Panam. Salud Pub. 6, 16–25.
Vasilakis, N., Deardorff, E. R., Kenney, J. L., Rossi, S. L., Hanley, K. A., and Weaver, S. C. (2009). Mosquitoes put the brake on arbovirus evolution: experimental evolution reveals slower mutation accumulation in mosquito than vertebrate cells. PLoS Pathog. 5:e1000467. doi: 10.1371/journal.ppat.1000467
Vaughn, D. W., Green, S., Kalayanarooj, S., Innis, B. L., Nimmannitya, S., Suntayakorn, S., et al. (2000). Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181, 2–9. doi: 10.1086/315215
Wang, E., Ni, H., Xu, R., Barrett, A. D., Watowich, S. J., Gubler, D. J., et al. (2000). Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J. Virol. 74, 3227–3234. doi: 10.1128/jvi.74.7.3227-3234.2000
Weaver, S. C., and Vasilakis, N. (2009). Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 9, 523–540. doi: 10.1016/j.meegid.2009.02.003
Wilm, A., Aw, P. P. K., Bertrand, D., Yeo, G. H. T., Ong, S. H., Wong, C. H., et al. (2012). LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201. doi: 10.1093/nar/gks918
World Health Organization [WHO] (2009). Dengue: Guidelines For Diagnosis, Treatment, Prevention And Control. Geneva: World Health Organization.
Yacoub, S., Mongkolsapaya, J., and Screaton, G. (2013). The pathogenesis of dengue. Curr. Opin. Infect. Dis. 26, 284–289.
Keywords: dengue, evolution, next-generation sequencing, increasing severity epidemiology, genetic variants
Citation: Ko H-Y, Salem GM, Chang G-JJ and Chao D-Y (2020) Application of Next-Generation Sequencing to Reveal How Evolutionary Dynamics of Viral Population Shape Dengue Epidemiology. Front. Microbiol. 11:1371. doi: 10.3389/fmicb.2020.01371
Received: 16 March 2020; Accepted: 27 May 2020;
Published: 19 June 2020.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Anon Srikiatkhachorn, University of Rhode Island, United StatesCopyright © 2020 Ko, Salem, Chang and Chao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Day-Yu Chao, ZHljaGFvQG5jaHUuZWR1LnR3
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.