
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 June 2020
Sec. Systems Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01310
 Zhenghui Li1
Zhenghui Li1 Kwok Lee1Urvi Rajyaguru1C. Hal Jones1
Kwok Lee1Urvi Rajyaguru1C. Hal Jones1 Sandra Janezic2,3
Sandra Janezic2,3 Maja Rupnik2,3
Maja Rupnik2,3 Annaliesa S. Anderson1
Annaliesa S. Anderson1 Paul Liberator1*
Paul Liberator1*Clostridioides (Clostridium) difficile is the most commonly recognized cause of infectious diarrhea in healthcare settings. Currently there is no vaccine to prevent initial or recurrent C. difficile infection (CDI). Two large clostridial toxins, TcdA and TcdB, are the primary virulence factors for CDI. Immunological approaches to prevent CDI include antibody-mediated neutralization of the cytotoxicity of these toxins. An understanding of the sequence diversity of the two toxins expressed by disease causing isolates is critical for the interpretation of the immune response to the toxins. In this study, we determined the whole genome sequence (WGS) of 478 C. difficile isolates collected in 12 countries between 2004 and 2018 to probe toxin variant diversity. A total of 44 unique TcdA variants and 37 unique TcdB variants were identified. The amino acid sequence conservation among the TcdA variants (≥98%) is considerably greater than among the TcdB variants (as low as 86.1%), suggesting that different selection pressures may have contributed to the evolution of the two toxins. Phylogenomic analysis of the WGS data demonstrate that isolates grouped together based on ribotype or MLST code for multiple different toxin variants. These findings illustrate the importance of determining not only the ribotype but also the toxin sequence when evaluating strain coverage using vaccine strategies that target these virulence factors. We recommend that toxin variant type and sequence type (ST), be used together with ribotype data to provide a more comprehensive strain classification scheme for C. difficile surveillance during vaccine development objectives.
Clostridioides (Clostridium) difficile, a Gram-positive, spore-forming, obligate anaerobe, is the main cause of nosocomial infectious diarrhea in industrialized countries (Kelly et al., 1994). The bacterium accounts for 20–30% of cases of antibiotic-associated diarrhea and is the most commonly recognized cause of infectious diarrhea in healthcare settings (Cohen et al., 2010). The main risk factors for an initial episode of C. difficile infection (CDI) are antibiotic therapy, hospitalization, and underlying comorbidities. Older adults (≥65 years of age) are at increased risk for CDI, particularly when exposed to health care settings (Asempa and Nicolau, 2017).
C. difficile can produce 3 toxins, toxin A (TcdA), toxin B (TcdB), and binary toxin (CDT) (Kuehne et al., 2014). TcdA and TcdB are large single subunit proteins (approximately 308 and 270 kDa, respectively), and are considered the principal virulence factors contributing to CDI (Voth and Ballard, 2005). These two toxins have similar structural features delineated by four functional domains but share just 50% overall amino acid sequence identity. The C-terminal domain of the toxin proteins, known as the combined repetitive oligopeptide (CROP) domain, facilitates toxin binding to the surface of intestinal epithelial cells. The toxins enter the cell by endocytosis where the reduced pH of endocytic vesicles triggers a conformational change in the cell entry domain of the toxin, resulting in pore formation and translocation of the glucosyltransferase domain (GTD) and autoprocessing domain (APD) to the cytosolic face of the membrane. Binding of the cytosolic cofactor InsP6 activates the APD, resulting in cleavage and release of the GTD. GTD-catalyzed transfer of glucose inactivates small cytoplasmic GTPases of the Rho family of proteins, leading to the disruption of the cytoskeleton. This manifests as a cytopathic rounding effect and cell death in the epithelium which results in diarrhea. CDT belongs to the family of binary ADP-ribosylating toxins consisting of two components: CDTa (ADP-ribosyltransferase) and CDTb (responsible for host cell binding and translocation of CDTa to the cytosol). As cdtA and cdtB are not detected in all toxigenic isolates, the significance of CDT as a virulence factor contributing to CDI remains in question (Gerding et al., 2014).
C. difficile infection often occurs when the integrity of the normal intestinal microbiota is disturbed. The spectrum of CDI presentation includes mild self-limiting to severe diarrhea which may progress to pseudomembranous colitis, toxic megacolon, intestinal perforation, and death (Bartlett, 2002). Although most patients experiencing a first episode of CDI respond well to standard antibiotic treatment (which can include metronidazole, vancomycin or fidaxomicin), approximately 15–35% of patients suffer from at least one recurrence (Louie et al., 2011). Immunoprophylactic approaches that target C. difficile toxins have been developed for the prevention of recurrent CDI (Sheldon et al., 2016). Vaccines have been successfully developed to prevent other toxin mediated diseases, such as tetanus and diphtheria, by inducing antibodies that neutralize the cytopathic effect of the toxin (Looney et al., 1956). The proposed mechanism of action of these approaches is through antibody mediated (both monoclonal antibody as well as vaccine-elicited polyclonal responses) neutralization of the cytotoxic activity of toxins produced by disease-causing C. difficile isolates.
Several molecular methods have been used to type C. difficile isolates for epidemiological studies. These include restriction endonuclease analysis (REA), pulse field gel electrophoresis (PFGE), and ribotyping, a PCR-based method that takes advantage of the size heterogeneity of the intergenic spacer region (ISR) between 16S and 23S rRNA genes (Loo et al., 2005; McDonald et al., 2005; Goorhuis et al., 2008). Isolates can also be classified by toxinotype, a restriction fragment length polymorphism (RFLP) method that is based on changes in the C. difficile pathogenicity locus (PaLoc) (Rupnik and Janezic, 2016). C. difficile may also be grouped into five main clades and at least three additional cryptic clades based on the clustering of the concatenate multiple locus sequence typing (MLST) alleles (Janezic et al., 2018). While each of these methods provides value for isolate characterization in epidemiological studies, none provides detailed insight to the diversity of full length TcdA and TcdB proteins. Reports on sequence-based variability within TcdA and TcdB toxins in large strain collections are uncommon. Sequence diversity within a 199 amino acid fragment of the CROP domain of TcdB (corresponding to the receptor binding domain of the toxin), has been used to differentiate toxin variant types (Dingle et al., 2011). An understanding of toxin diversity among contemporary disease-causing isolates is essential for assessment of the immune response to the toxins. In this manuscript we report on the deduced amino acid sequence of TcdA and TcdB toxin proteins determined from whole genome nucleotide sequence data of 478 C. difficile isolates. A total of 44 TcdA and 37 TcdB protein variants have been identified among these isolates and are presented together with detailed molecular analyses.
Initially, a total of 504 C. difficile isolates, collected from multiple sources across several geographic regions between the years 2004–2018 were included in this study (Table 1). The isolates do not represent a prevalence-based collection. Whole genome sequence (WGS) data was collected for all isolates and the PCR ribotype was determined for each of the isolates. Toxinotype assignments were available for a subset of the isolates.

Table 1. Clostridioides difficile isolates included in this study.
C. difficile isolates were either provided as glycerol stocks or were purified from stool specimens. All microbiology was conducted under anaerobic conditions. Glycerol stocks were plated onto Tryptone Soya Agar (TSA) + 5% sheep blood and incubated overnight at 37°C. On day 2, a single colony was selected and re-streaked onto a new TSA +5% sheep blood agar plate and again incubated overnight at 37°C. On day 3, colonies were transferred to a 96-well plate for lysis and genomic DNA was extracted using Beckman Coulter GenFind V2 kit (Indianapolis, IN, United States). For stool specimens, a 10 μl loop was used to transfer stool to 700 μL 95% ethanol; 20 μL of this suspension was then used to inoculate a cycloserine cefoxitin fructose agar plate, supplemented with horse blood and taurocholate (CCFA-HT) and incubated for 2 days at 37°C. On day 3, a single colony was picked from CCFA-HT plate and streaked onto a TSA blood agar plate followed by overnight incubation at 37°C. On day 4, the colonies were transferred to a 96-well plate for lysis and genomic DNA was extracted using the Beckman Coulter GenFind V2 kit.
For the majority of strains, PCR-ribotyping was performed using the protocol described in Svenungsson et al. (2003). For added discrimination, PCR products are analyzed using an Agilent 2100 Bioanalyzer. Ribotype and toxinotype assignments for the toxiontype diversity subset of strains were determined as described in Bidet et al. (1999) and Rupnik et al. (1998), respectively. The nucleotide sequence of adk, atpA, dxr, glyA, recA, sodA, and tpi genes was extracted from the whole genome sequence of each strain and used to assign a sequence type (ST) and clade at PubMLST1.
C. difficile sequencing libraries were prepared by using either the TruSeq DNA Sample Prep Kits (“TruSeq”) or the Nextera DNA Flex Library Prep Kit (“Flex”), both from Illumina (San Diego, CA, United States). When using the TruSeq kit, genomic DNAs are first mechanically sheared by a Covaris ME220 (Woburn, MA, United States) sonication instrument using the settings recommended by the manufacturer. After sonication, the TruSeq universal adapters are added to the ends of the genomic DNA fragments by ligation, followed by bead cleanup, size selection and quantification according to the manufacturer’s protocol. When using the Flex kit, genomic DNAs are first tagmented (transposon-mediated fragmentation) and universal adapters are added to the ends of the DNA fragments by PCR, according to the manufacturer’s protocol. DNA libraries (paired end, 2 × 300) were loaded on the Illumina Miseq for whole genome sequencing of the respective C. difficile genomes.
Primary nucleotide sequence data for the 478 C. difficile isolates from this study has been deposited to NCBI SRA (BioProject PRJNA600974). Primary C. difficile DNA sequence reads were run through the “Merge Overlapping Pairs” program followed by assembly into contigs using the “De Novo Assembly” program in the Qiagen CLC Genomics Workbench (Redwood City, CA, United States) using default parameters. The tcdA and tcdB allele sequences were inferred by aligning the assembled contigs to the tcdA and tcdB sequences of the CD630 isolate (GenBank accession AM180355) (Sebaihia et al., 2006). The deduced amino acid sequences of the genes coding for TcdA and TcdB in CD630 are labeled as TcdA001 and TcdB001, respectively.
A unique toxin variant is defined as a TcdA or TcdB open reading frame (ORF) that includes all four of the functional domains and whose amino acid sequence differs by at least one amino acid from any other entry. The most challenging region for collection of high-quality DNA sequence was in the C-terminal CROP domain, particularly in the TcdA ORF. In some instances, these challenges were addressed by repeat sequencing with fewer input strains to achieve greater depth and/or by performing the de novo assembly program a second time without merging overlapping pairs. If the toxin variant could not be determined by the de novo assembly program, primary sequence data was aligned to the CD630 reference genome to confirm the presence of the gene.
The nomenclature developed by den Dunnen et al. (2016) has been used to describe a series of truncations and in-frame deletions predicted from the tcdA nucleotide sequence of some isolates. In these instances, the TcdA001 sequence is used as the reference when describing position in the ORF. A unique TcdA variant number has been assigned if sequence corresponding to the CROP domain is included in the truncated ORF. However, if the deduced amino acid sequence is truncated prior to the CROP domain, a TcdA variant was not assigned and the isolate has been labeled as “truncated at TcdA.” In those isolates where the entire tcdA sequence is missing, the strain has been classified as “TcdA deletion.” Acceptance criteria for each novel TcdA or TcdB variant identified required a minimum 35X nucleotide sequence coverage across the respective ORFs with less than 10% sequence heterogeneity. If these acceptance criteria were not met, the isolate was not included in the phylogenomic analysis. The WGS from 26 of the 504 isolates in the collection did not meet these acceptance criteria and were not included in these analyses. TcdA and TcdB amino acid sequences were aligned using CLC Genomics Workbench and aligned sequences were used to construct the phylogenetic tree using the unweighted pair group method with arithmetic mean (UPGMA) with bootstrapping. The phylogenomic figure was created by aligning assembled C. difficile genomes using Parsnp (Treangen et al., 2014) with recombination filtration option enabled. The metadata (clade, ST, ribotype, TcdA variant type, and TcdB variant type) was plotted as concentric rings using a custom-built R program.
The Simpson’s Diversity Index (SDI) was used to quantitatively measure the diversity and distribution of toxin variants within phylogenetic clades of C. difficile. The higher the index obtained, the greater the number and distribution of the toxin variants observed within a clade. Likewise, the lower the index, the fewer number of variants and/or a more restricted distribution of the toxin variants within a clade.
Nucleotide sequence corresponding to tcdA and tcdB was extracted from the WGS data and used to generate the deduced amino acid sequence of TcdA and TcdB protein variants, respectively. To serve as a point of reference, the TcdA and TcdB proteins coded for by strain CD630 are variants TcdA001 and TcdB001. A total of 43 unique TcdA variants that differ from variant TcdA001 were identified. All TcdA variants were closely related, with pairwise amino acid sequence identity to TcdA001 that ranged from 98.0 to >99.9%. From the deduced amino acid sequence of TcdA variants, an unrooted phylogenetic tree was constructed using the UPGMA (Figure 1A). The toxin variant used as template to make the TxdA vaccine antigen (TcdA001) is grouped together with 21 other TcdA variants, with each variant sharing >99.7% amino acid sequence identity with TcdA001. Of the TcdA variants whose ORF is at least 2,710 amino acids in length, TcdA013 was the most diverse (98.0% sequence identity with TcdA001) and is grouped with three others. Variant TcdA007 and TcdA016 (98.2 and 98.4% sequence identity with TcdA001, respectively), are phylogenetically related to several other full-length variants. Finally, TcdA019 which shares 98.5% identity with TcdA001, is representative of a fourth group of variants on the TcdA phylogenetic tree. Most of the sequence variation among the 44 TcdA variants is due to single amino acid substitutions. The greatest amino acid sequence diversity among the TcdA variants is found within the CROP domain of the proteins (Table 2). The tcdA nucleotide sequence of 25 isolates predict ORFs that are shorter than TcdA001 (summarized in Supplementary Table 1). A termination codon in the TcdA sequence from 14 isolates occurs following amino acid residue 46 (p.Q47∗) (von Eichel-Streiber et al., 1999). While these isolates were collected in different geographic regions, including the United States and multiple European countries, each is genotyped as ST37/RT017. The tcdA sequence of three additional isolates also predicted termination codons within the GTD domain of the toxin (p.V57∗, p.D108∗, p.P196∗). A common termination codon (p.G699∗) in the APD domain is shared by three isolates. Since the predicted TcdA ORFs of these 20 isolates lack at least 3 of the functional toxin domains, TcdA variants were not assigned. Novel TcdA variants were assigned for 5 isolates whose tcdA sequence predict a deletion of a portion of the CROP domain of the toxin. In-frame deletions of 33 (TcdA050) and 175 amino acids (TcdA051) were detected in two isolates, while three isolates had termination codons in the TcdA CROP domain (TcdA052, TcdA053, TcdA054). Critical catalytic residues within the GTD (D285 and D287) and APD domains (C700) of the toxins are conserved among each of the 44 unique TcdA variants.
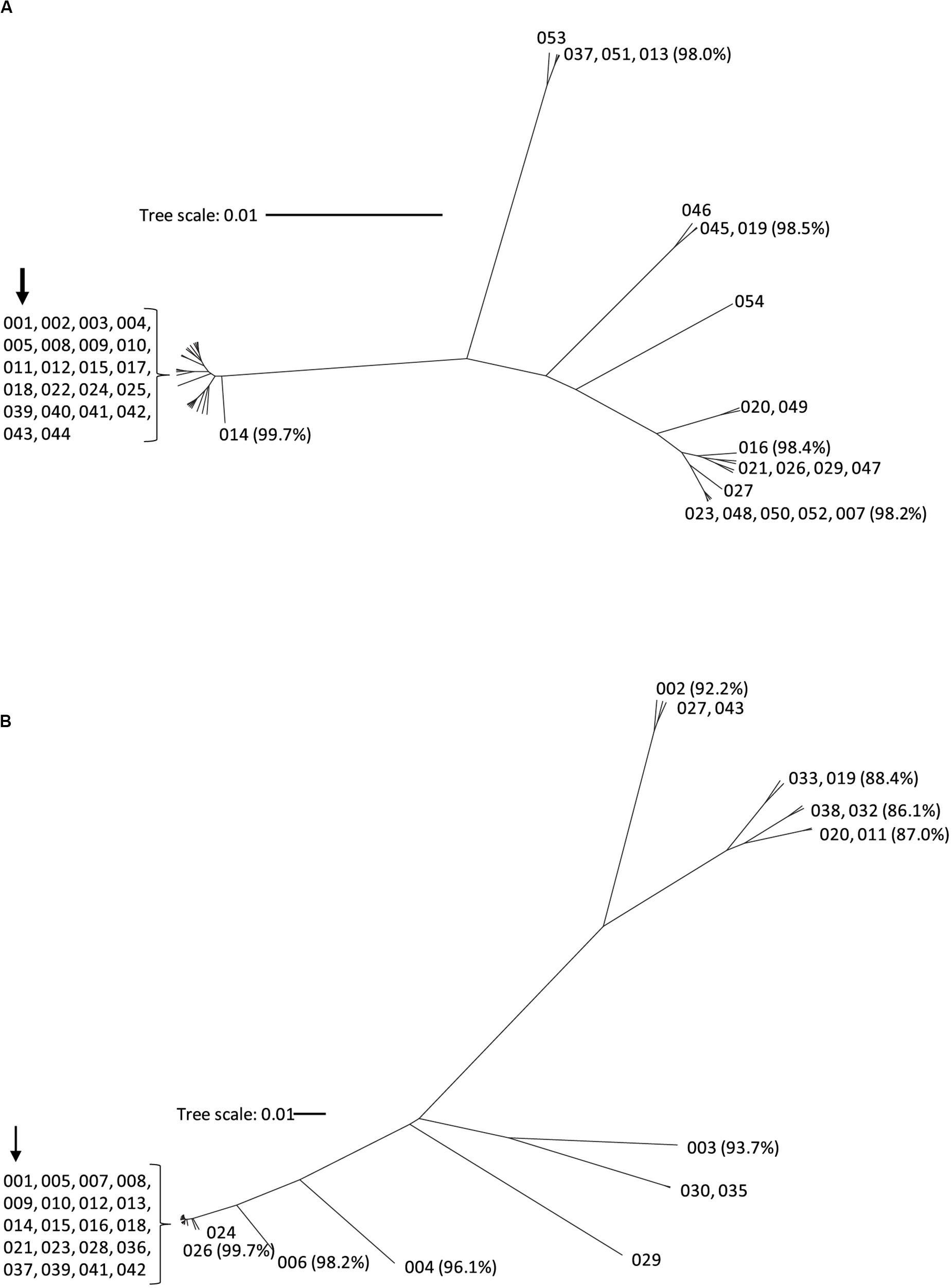
Figure 1. Phylogenetic tree of TcdA (A) and TcdB (B) toxin variants. The amino acid sequences of the 44 TcdA variants and 37 TcdB variants were used to build the phylogenetic trees using CLC Genomics workbench with the UPGMA method. Each branch represents a unique toxin variant. The length of each branch corresponds to the amino acid sequence diversity from the TcdA001 (A) or TcdB001 (B) variants. The numbers in parentheses represent the pairwise amino acid sequence identity of representative variants to TcdA001 or TcdB001. The position of the TcdA001 and TcdB001 variant sequences from the CD630 isolate is highlighted with an arrow.
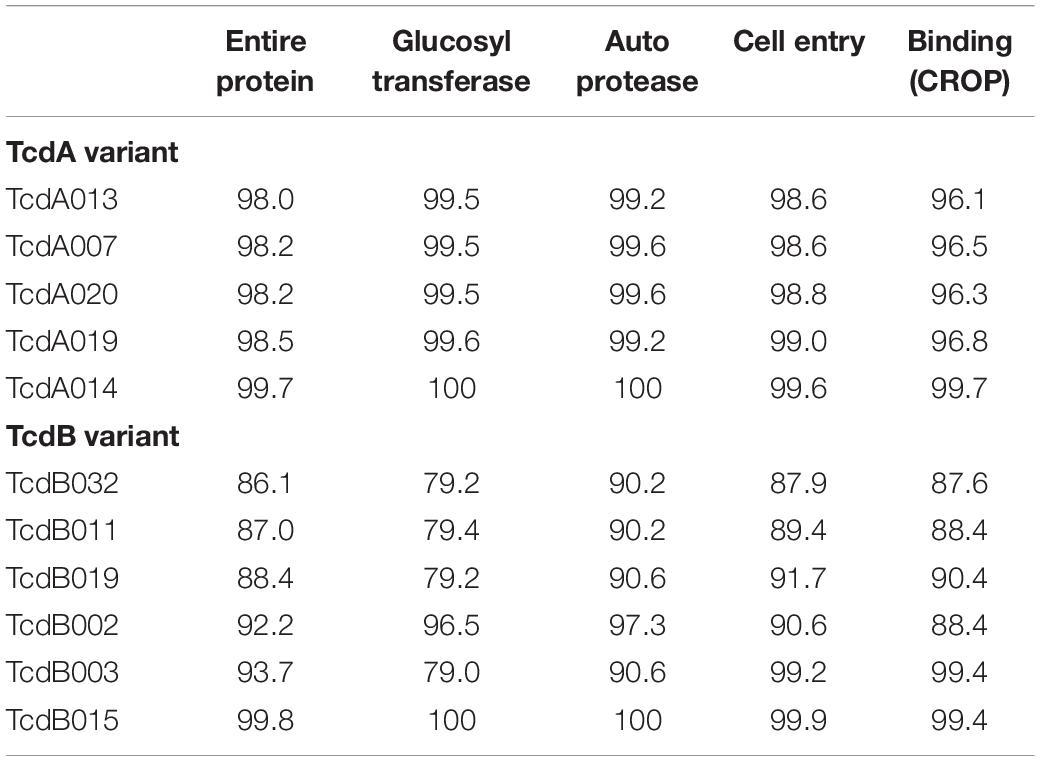
Table 2. Pairwise amino acid sequence identity (%) of a subset of diverse TcdA and TcdB variants to TcdA001 and TcdB001, respectively.
There were 31 non-toxigenic isolates, lacking any sequence corresponding to TcdA and TcdB (TcdA−/TcdB−). These were not restricted to a single ST or RT. A subset of 3 strains code for a full length TcdB variant, but without any tcdA sequence (TcdA−/TcdB+). These TcdA−/TcdB+ strains were also associated with multiple ribotypes. Precedent for both TcdA−/TcdB+ as well as non-toxigenic TcdA−/TcdB− C. difficile strains is well established in the literature (Rupnik, 2008).
The amino acid sequences of TcdB variants were more diverse than TcdA. Of the 36 variants that differed from TcdB001, pairwise amino acid sequence identity with TcdB001 ranged from 86.1 to >99.9%. This is illustrated in the phylogenetic tree of TcdB variants (Figure 1B). A total of 19 variants group together with TcdB001 (>99.8% pairwise sequence identity). Variants TcdB032 and TcdB038 are the most diverse, sharing 86.1 and 86.2% sequence identity with TcdB001, respectively. TcdB032 is detected in one isolate in the collection (typed as ST62 and RT591) and two isolates code for TcdB038 (ST567 and RT095). While sequence diversity is detected in each of the functional domains of the TcdB toxin, the greatest diversity is found within the GTD domain (Table 2). Unlike TcdB001, eleven of the TcdB variants have a lysine inserted in the GTD domain (p.Val307_Thr308insLys). Based on the structure of the TcdB GTD domain (PDB ID 2BVM2) (Reinert et al., 2005), this additional amino acid residue is not predicted to impact the conformation of the GTD domain. The eleven sequence variants that contain the additional amino acid cluster to two groups on the TcdB phylogenetic tree (Figure 1B). Critical catalytic residues of GTD (D286 and D288) and autoprotease (C698) domains are conserved among all TcdB variants. Although the TcdA and TcdB toxins share similar architectural homology with respect to the functional domains of the molecules, the pairwise amino acid sequence identity between TcdA001 and TcdB001 is only 42%.
More than half of the isolates (n = 292, 61%) in this study did not code for binary toxin (Supplementary Table 2). Each of the 31 non-toxigenic TcdA−/TcdB− isolates was binary toxin negative (genotyped as cdtA−/cdtB−). Considerable differences in TcdA and TcdB variant type and prevalence were also noted when comparing binary toxin positive and negative subsets of isolates. With very few exceptions, TcdA and TcdB variant types were selectively associated with either binary positive or binary negative isolates. Exceptions include variants TcdA007 and TcdB002. TcdA007 is coded for by just one of 292 binary negative isolates (0.3%) and 94/186 (50.5%) binary positive isolates. Similarly, the gene coding for TcdB002 is detected in just one binary negative strain but in 108/186 (58.1%) binary positive strains. This is particularly noteworthy as the C. difficile pathogenicity locus is not linked to the binary toxin alleles (Gerding et al., 2014).
The 478 C. difficile genomes were assembled to construct a phylogenomic tree using Parsnp (Figure 2). The nucleotide sequence of isolate CD630 was used as the reference genome. Each branch of the dendrogram is representative of a C. difficile isolate. Metadata descriptive of each isolate (Supplementary Table 2), including clade, ST, ribotype and toxin variant type, has been added as concentric rings to the circumference of the phylogenomic tree. Included among the 478 isolates are 61 ribotypes and 71 sequence types (STs). Comparative analysis afforded by the figure helps to illustrate that these epidemiological markers are not predictive of toxin variant type. Starting at 12 o’clock on the tree and traveling counterclockwise to 10 o’clock is a cluster of 107 isolates that are typed as ST1 and all code for TcdB variant TcdB002. Although most isolates in this cluster are ribotype 027, other ribotypes such as 081, 176, 027/198, and 198 are also identified. Continuing counter-clockwise on the tree is a cluster of 42 isolates that are typed as ST11 and code for TcdB variant TcdB004. Despite sharing considerable phylogenomic, ST and TcdB variant type similarities, the ribotype variability among these isolates is considerable, including ribotypes 045, 078, 126, 078/126, and 413.
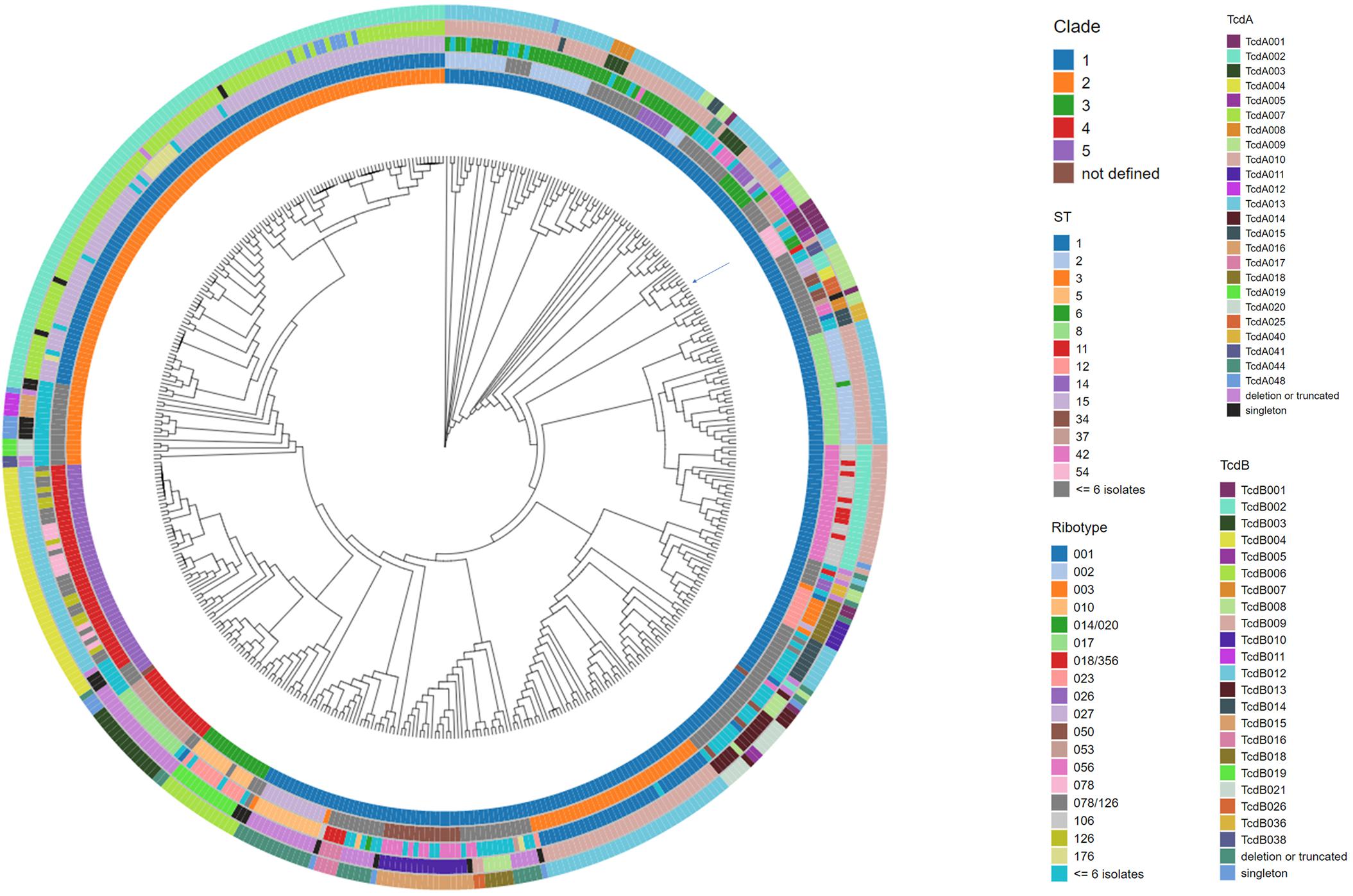
Figure 2. Phylogenomic tree of C. difficile isolates with accompanying meta data. The whole genome sequence of C. difficile isolates was used to construct a tree to illustrate the phylogenomic relationship among the isolates. C. difficile isolate CD630 (GenBank accession AM180355) was used as the reference genome during the alignment. The position of the CD630 genome is highlighted by an arrow on the tree. Each branch of the dendrogram tree corresponds to a C. difficile isolate in the collection. From the innermost circle to the outermost circle, the five color-coded concentric circles identify clade, ST, ribotype, TcdA, and TcdB variant type assignments for each isolate. For clarity, ST and ribotype assignments identified in less than or equal to 6 isolates were colored as dark gray and light blue, respectively. Clade assignment could not be made in two instances (entered as not defined). TcdA and TcdB variants detected just once in the collection (singletons) are colored as black and blue, respectively.
Among the numerous methods that have been developed for molecular typing of C. difficile isolates, ribotype is most commonly cited. Analysis of WGS data in this study illustrates that isolates grouped by ribotype code for sequence-diverse toxin variants (Tables 3, 4). Among the 21 isolates that are grouped as ribotype 078/126, genes coding for three TcdA variant types (TcdA013, TcdA015, TcdA046) were identified. The ribotype 078/126 isolates also code for three TcdB variants (TcdB004, TcdB006, TcdB012) whose amino acid sequence identity to TcdB001 ranges from 96.1 to 99.9%. Ribotype 014/020 isolates in the collection code for four unique TcdA and six TcdB variants. The pairwise identity of the six TcdB variants to TcdB001 ranges from 92.2 to 99.9%.
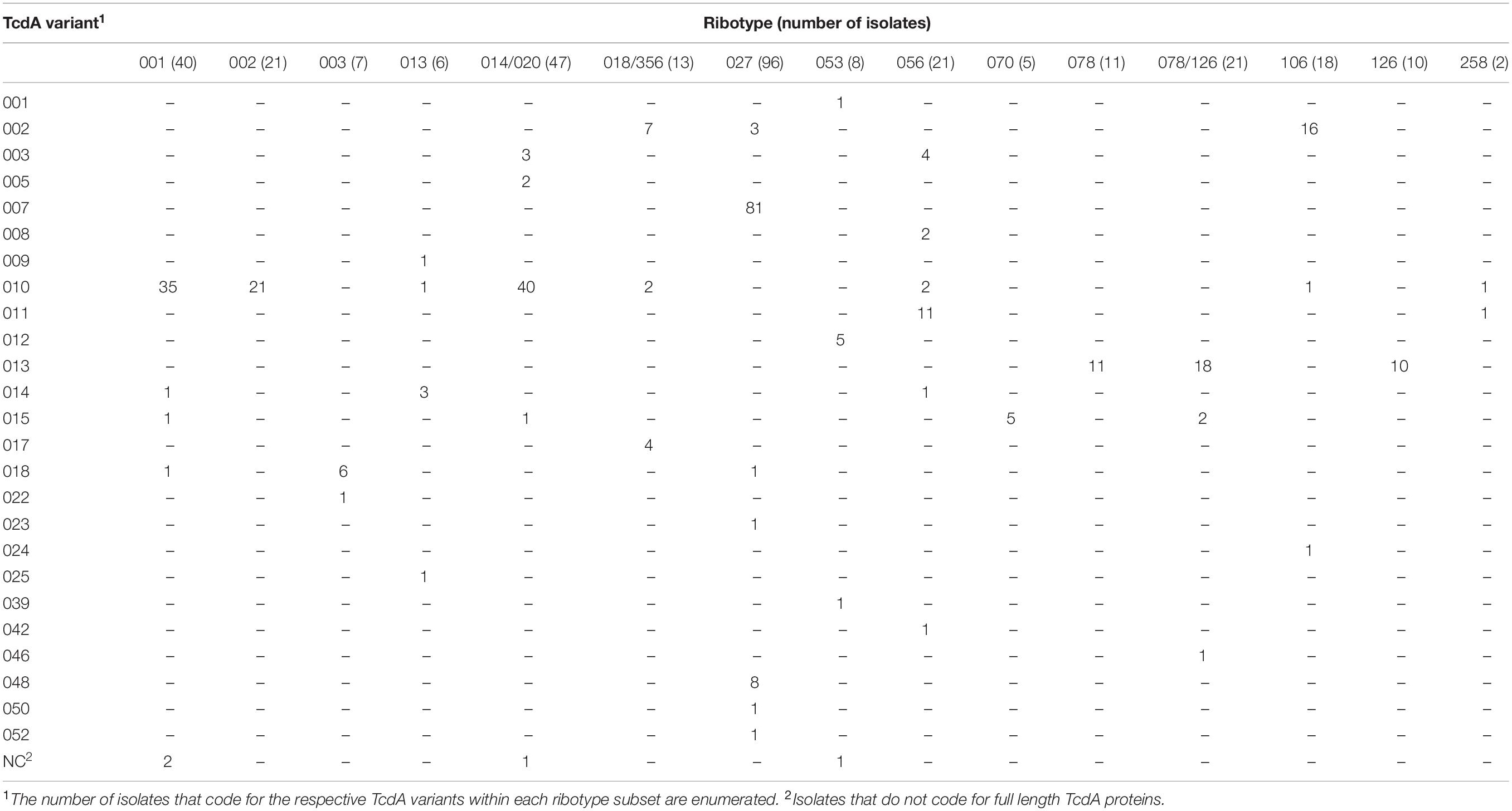
Table 3. C. difficile isolates grouped by ribotype can code for multiple and sequence diverse TcdA variants.
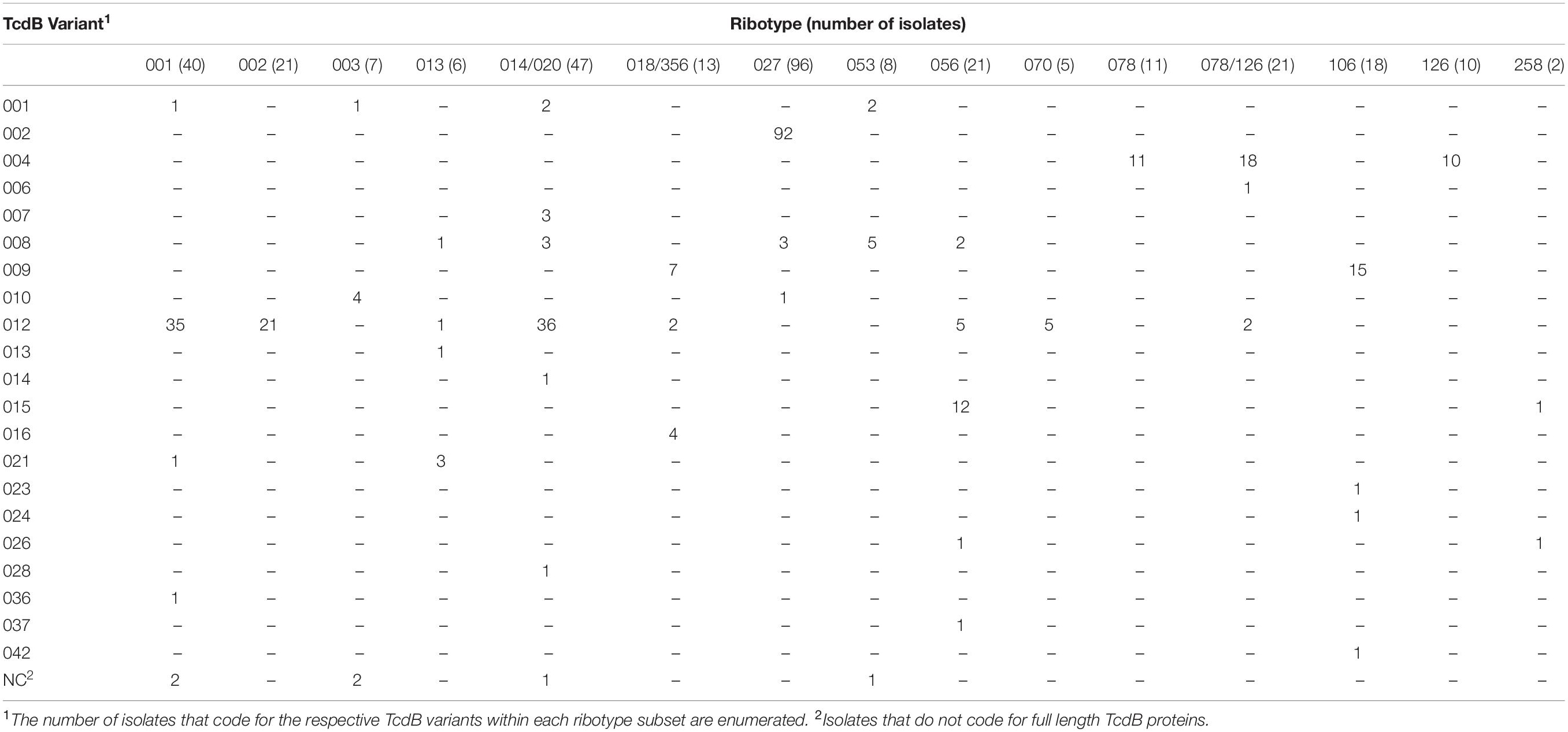
Table 4. C. difficile isolates grouped by ribotype can code for multiple and sequence diverse TcdB variants.
Using ST assignments, all but two of the isolates from this study could be grouped into one of five clades. With the exception of clade 4, the number and distribution of TcdA variants within each of the clades is more diverse than TcdB variants (Supplementary Figure 1).
C. difficile infection is a worldwide public health issue and is now the most common healthcare-associated bacterial pathogen. It was estimated that during 2011 there were nearly half a million cases of CDI in the United States, associated with approximately 29,000 deaths (Lessa et al., 2015). Several vaccine strategies for the prevention of CDI have been, or are currently, being evaluated in clinical trials and most of these have focused on the large single subunit glucosylating toxins, TcdA and TcdB as antigens (Bezay et al., 2016). Clinical proof of concept for the selection of these toxins as C. difficile vaccine antigens has come from studies that were used to support the approval of Bezlotoxumab (ZINPLAVATM, Merck) to reduce the recurrence of CDI in adult patients (Alonso and Mahoney, 2019). Bezlotoxumab is a human monoclonal antibody that binds to a discontinuous epitope located within the CROP domain of TcdB (Orth et al., 2014). Since the CROP domain accounts for approximately 20% of the entire protein, a prophylactic vaccine approach able to generate a functional polyclonal response to additional and multiple epitopes across the entire toxin has the potential to prevent primary disease. Three prophylactic vaccines composed of full-length toxin open reading frames, or portions thereof, have been evaluated in clinical studies. A recombinant fusion protein that includes a portion of the TcdA and TcdB CROP domain from a single strain of C. difficile (Bezay et al., 2016) has completed phase 2 testing (NCT02316470). The antigens included in the other investigational vaccines correspond to inactivated full length toxin proteins (i.e., toxoids TxdA, and TxdB) (Donald et al., 2013). Following a planned interim analysis in a phase 3 clinical study for one of the toxoid based vaccines (NCT01887912), the study was discontinued based on the low probability of meeting its primary objective (Sanofi, 2017). A bivalent toxoid-based vaccine composed of genetically and chemically inactivated toxoid antigens is in phase 3 clinical development (NCT03918629). Active immunization with these genetically and chemically modified full length versions of TcdA and TcdB has elicited a robust polyclonal antibody response in subjects 65–85 years of age (Sheldon et al., 2016).
Several molecular methods including WGS have been utilized to characterize C. difficile toxin genes for epidemiological analysis of CDI (Huber et al., 2013; Knetsch et al., 2013; Janezic and Rupnik, 2019). Two of these, toxinotype (Rupnik and Janezic, 2016) and the deduced amino acid sequence of the Receptor Binding Domain (RBD) of TcdB variants (Dingle et al., 2011), have focused on toxin genotype of C. difficile isolates. However, neither method provides detailed insight into the sequence diversity of the complete open reading frame of the two large toxin proteins. An understanding of the toxin variants expressed by CDI isolates is critically important for an assessment of the breadth of the functional antibody response elicited following immunization with a bivalent toxoid vaccine. Demonstration that the polyclonal immune sera can neutralize the cytotoxicity of sequence diverse toxins is required for an evaluation of vaccine efficacy. Prediction of the breadth of the functional immune response to a toxin-based vaccine antigen needs to be framed in the context of toxin variant type determined from WGS, because isolate classification by ribotype is not representative of the diversity of toxin protein sequences.
The availability of C. difficile WGS data with deep sequence coverage, across the PaLoc in particular, is limited. In a recent study, analysis of WGS data from 906 C. difficile strains focused on questions related to bacterial adaptation for healthcare-mediated transmission, but did not investigate the diversity of the tcdA and tcdB alleles (Kumar et al., 2019). The WGS of 478 C. difficile isolates have been determined in our study. Acceptance criteria for toxin variant assignment required greater than 35X coverage across both tcdA and tcdB with less than 10% sequence heterogeneity at any nucleotide position for at least one representative isolate. A total of 44 unique TcdA variants and 37 unique TcdB variants were identified, many of which had not previously been reported. The toxin variants with pairwise amino acid sequence identity furthest removed from TcdA001 and TcdB001 were TcdA013 (98% sequence identity) and TcdB032 (86.1% sequence identity), respectively. The ability of the immune response elicited by a toxoid antigen to neutralize the cytotoxicity of sequence diverse toxins remains to be experimentally tested. It should be noted that the origin of the C. difficile isolates evaluated in this study was largely restricted to North America and Europe. The TcdA and TcdB sequences of isolates from different geographic regions may uncover further toxin diversity. In addition, as the isolates collected in this study are not prevalence based the most frequent TcdA and TcdB variant types identified here may not be representative of prevalence among circulating CDI isolates.
Although both TcdA and TcdB play a role in CDI, several observations suggest that TcdB is the major virulence factor. In both murine and hamster models of C. difficile infection, TcdB was responsible for most of the intestinal damage whereas TcdA caused more superficial and localized damage (Carter et al., 2015). It is also worth noting that TcdA+/TcdB− C. difficile strains are extremely rare (Monot et al., 2015), whereas TcdA−/TcdB+ strains are not uncommon and have been associated with multiple disease outbreaks (Drudy et al., 2007; Rupnik, 2008). The greater amino acid sequence diversity noted among the 37 TcdB variants compared with the 44 TcdA variants identified in this study could in part be the consequence of host immune pressure applied to the more virulent TcdB toxins.
Several isolates with deduced amino acid sequences shorter than TcdA001 have been identified in this collection. The majority of these are the product of in-frame stop codons in the glucosyl transferase or auto protease domains and therefore are not likely to code for a functional toxin. TcdA variants with either a deletion or a termination codon within the CROP domain have also been identified. Cell-culture based cytotoxicity assays are required to demonstrate whether these TcdA variants are functional. Previous work by Rupnik and Janezic has indicated that toxinotype VI and VII strains, with deletions in the CROP domain, do produce functional TcdA (Rupnik and Janezic, 2016). It has also been reported that a TcdA mutant lacking the entire CROP domain is cytotoxic, able to enter human cells and cause cytotoxicity, albeit with reduced uptake (Gerhard et al., 2013). Among the isolates evaluated in this study, similar genetic changes in the sequences corresponding to the CROP domain of TcdB variants have not been identified. The larger number and length of repeat units in the TcdA CROP domain may render this locus more prone to genetic rearrangements than TcdB. Differences between the genes coding for the two toxins extend beyond the CROP domain. Examples of toxigenic isolates lacking any sequence coding for TcdA (TcdA−/TcdB+) are included in this collection and have been described in the literature (von Eichel-Streiber et al., 1999). As noted earlier and despite their close proximity in the PaLoc, examples of TcdA+/TcdB− isolates, lacking tcdB sequences are not common (Monot et al., 2015). Our current understanding of the molecular detail of epithelial cell binding and uptake also differentiates the toxins from one another (Chandrasekaran and Lacy, 2017). Taken together, these observations suggest that evolution of the two toxins has been and will continue to be subject to different selection pressures. Relative to the highly conserved TcdA variants, the greater sequence diversity among TcdB variants described in this manuscript is consistent with this idea.
The same acceptance criteria for deep sequence coverage were applied in the analysis of WGS data corresponding to the genes coding for both subunits of the C. difficile binary toxin. The majority of isolates (n = 292, 61%) in this study did not code for binary toxin (e.g., genotyped as cdtA−/cdtB−). Consistent with literature reports indicating that CDI isolates typed as TcdA−/TcdB−/CDT+ are rare (Geric et al., 2003; Gerding et al., 2014), there were no TcdA−/TcdB−/CDT+ isolates identified in this study. A study in a hamster model of CDI suggested that vaccination with bivalent TxdA and TxdB antigens did not fully protect from challenge with a TcdA+/TcdB+/CDT+ NAP1 strain and that the addition of binary toxin antigens to the vaccine greatly enhanced protection (Secore et al., 2017). Interestingly, human epidemic isolates 027/BI/NAP1 and 078/BK/NAP7 are both positive for binary toxin (Gerding et al., 2014), whereas the emerging RT106 as well as RT017 isolates common in Asia are negative for binary toxin (Liu et al., 2018; Imwattana et al., 2019). The contribution of binary toxin to clinical CDI remains controversial.
C. difficile isolates can be grouped into five different clades based on ST assignments (Griffiths et al., 2010). Dingle et al. showed that 17 alleles coding for 13 deduced peptide sequences corresponding to the TcdB RBD segregate in parallel with clade assignment (Dingle et al., 2011). That is, no single RBD allele was found in more than one clade. However, this study focused on just a small section of TcdB and did not capture the full sequence diversity of TcdB variants. In addition, the diversity of TcdA has not been taken into consideration. Using ST assignments from WGS data collected in our study, 476 of the 478 isolates can be placed into one of the 5 clades. Greater than half of the isolates (n = 276) are grouped into Clade 1 and these are genotypically diverse, including 49 different STs, 39 ribotypes, 24 TcdA variants and 22 TcdB variants. However, much like the clade-restricted observation for the RBD alleles, none of the 24 TcdA and 22 TcdB variants are coded for by strains other than those grouped to clade 1. The same conclusion can be drawn for strains associated with each clade. For example, while clade 5 strains are fewer in number (n = 45) and much less diverse (42 of these are ST11 and each code for TcdB004), all TcdA013 and TcdB004 variants are coded for by strains that are grouped to clade 5.
The Simpson’s Diversity Index (SDI) analysis indicated that the distribution of TcdA variants within the respective clades is generally more diverse than the variants of TcdB (Supplementary Figure 1). Although both TcdA and TcdB are co-localized to the PaLoc, the variation in SDI suggested that TcdA and TcdB might have evolved in response to different evolutionary pressures. One interpretation is that point mutations in TcdA are sporadic and equally represented across phylogenetic clades (excluding clade 4 in this collection of strains). The variability noted among TcdB protein sequences can be postulated as the sum of sporadic point mutations together with pressure exerted from additional mechanisms. It is interesting to note that while three distinct cell surface receptors for TcdB have been identified, [PVRL3 (or NECTIN3), CSPG4 and members of the Frizzled protein family] (LaFrance et al., 2015; Yuan et al., 2015; Tao et al., 2016), similar molecular binding detail has not been uncovered for TcdA. In fact, none of these receptors bind TcdA. In contrast to the CROP domain of TcdA, high-affinity glycan mediated binding for TcdB CROP at the cell surface has not been experimentally demonstrated (Gerhard, 2017). The molecular difference in host cell entry mechanisms utilized by TcdA and TcdB may contribute to the variation of SDI noted here.
To our knowledge, this is the first comprehensive WGS characterization of a large C. difficile isolate collection with a focused analysis of the sequence diversity among TcdA and TcdB toxins. From WGS data, we identified 44 TcdA variants and 37 TcdB variants, many of which were not previously documented. The relative lack of deep WGS data deposited in public databases could be due to technical challenges associated with the sequencing of C. difficile isolates using conventional methods, and in particular sequence determination across repetitive regions of the pathogenicity locus. We observed that neither ribotype nor ST assignment is predictive of the toxin genotype of C. difficile isolates. Specifically, isolates that are grouped together based on ribotype assignment can code for multiple sequence diverse TcdA and TcdB variants. We propose an amended nomenclature that describes both the ribotype and the TcdA and TcdB variant type for the purpose of C. difficile strain surveillance during vaccine efficacy trials. As the immune response to toxoid antigens will be polyclonal and not restricted to the RBD, it is essential that sequence diversity of the entire toxin proteins be considered in efforts to estimate vaccine efficacy. While challenges are associated with obtaining deep sequence coverage across the pathogenicity locus, WGS should be applied for C. difficile isolate typing together with ribotype determination to better guide C. difficile epidemiological studies and to help evaluate immunological approaches for the prevention of C. difficile disease.
The datasets generated for this study can be found in the NCBI BioProject ID PRJNA600974.
ZL and PL contributed substantially to conception and design and acquisition of the data, conducted the analysis and interpretation of the data, and drafted and revised the manuscript, and gave final approval of the version to be published, and agreed to act as guarantor of the work (ensuring that questions related to any part of the work are appropriately investigated and resolved). AA, MR, and SJ contributed substantially to conception and design, analysis and interpretation of the data, and revised the article critically for important intellectual content and gave final approval of the version to be published, and agreed to act as guarantor of the work (ensuring that questions related to any part of the work are appropriately investigated and resolved). KL, UR, and CJ contributed substantially to acquisition of the data and revised the manuscript critically for important intellectual content and gave final approval of the version to be published, and agreed to act as guarantor of the work (ensuring that questions related to any part of the work are appropriately investigated and resolved). All authors contributed to the article and approved the submitted version.
ZL, KL, UR, CJ, PL, and AA are current employees of the Pfizer and may hold stock options.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Pfizer Inc. The funder had the following involvement in the study: Pfizer paid for work required to prepare and transfer C. difficile isolates from collaborator.
We would like to thank Richard Goering (Creighton University School of Medicine) for ribotype analysis, Li Hao (Pfizer, Inc.), Jonathan Lee (Pfizer, Inc.), and Leonid Chindelevitch (Simon Fraser University) for review and comments, and Dawn DeThomas (Pfizer, Inc.) for graphics.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01310/full#supplementary-material
Alonso, C. D., and Mahoney, M. V. (2019). Bezlotoxumab for the prevention of Clostridium difficile infection: a review of current evidence and safety profile. Infect. Drug Resist. 12, 1–9. doi: 10.2147/idr.s159957
Asempa, T. E., and Nicolau, D. P. (2017). Clostridium difficile infection in the elderly: an update on management. Clin. Interv. Aging 12, 1799–1809. doi: 10.2147/cia.s149089
Bartlett, J. G. (2002). Clinical practice. Antibiotic-associated diarrhea. N. Engl. J. Med. 346, 334–339.
Bezay, N., Ayad, A., Dubischar, K., Firbas, C., Hochreiter, R., Kiermayr, S., et al. (2016). Safety, immunogenicity and dose response of VLA84, a new vaccine candidate against Clostridium difficile, in healthy volunteers. Vaccine 34, 2585–2592. doi: 10.1016/j.vaccine.2016.03.098
Bidet, P., Barbut, F., Lalande, V., Burghoffer, B., and Petit, J. C. (1999). Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS Microbiol. Lett. 175, 261–266. doi: 10.1111/j.1574-6968.1999.tb13629.x
Carter, G. P., Chakravorty, A., Pham Nguyen, T. A., Mileto, S., Schreiber, F., Li, L., et al. (2015). Defining the Roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during clostridium difficile infections. mBio 6:e00551.
Chandrasekaran, R., and Lacy, D. B. (2017). The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 41, 723–750. doi: 10.1093/femsre/fux048
Cohen, S. H., Gerding, D. N., Johnson, S., Kelly, C. P., Loo, V. G., Mcdonald, L. C., et al. (2010). Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31, 431–455. doi: 10.1086/651706
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., Mcgowan-Jordan, J., et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37, 564–569. doi: 10.1002/humu.22981
Dingle, K. E., Griffiths, D., Didelot, X., Evans, J., Vaughan, A., Kachrimanidou, M., et al. (2011). Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One 6:e19993. doi: 10.1371/journal.pone.0019993
Donald, R. G., Flint, M., Kalyan, N., Johnson, E., Witko, S. E., Kotash, C., et al. (2013). A novel approach to generate a recombinant toxoid vaccine against Clostridium difficile. Microbiology 159, 1254–1266. doi: 10.1099/mic.0.066712-0
Drudy, D., Fanning, S., and Kyne, L. (2007). Toxin A-negative, toxin B-positive Clostridium difficile. Int. J. Infect. Dis. 11, 5–10. doi: 10.1016/j.ijid.2006.04.003
Gerding, D. N., Johnson, S., Rupnik, M., and Aktories, K. (2014). Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5, 15–27. doi: 10.4161/gmic.26854
Gerhard, R. (2017). Receptors and Binding Structures for Clostridium difficile Toxins A and B. Curr. Top. Microbiol. Immunol. 406, 79–96. doi: 10.1007/82_2016_17
Gerhard, R., Frenzel, E., Goy, S., and Olling, A. (2013). Cellular uptake of Clostridium difficile TcdA and truncated TcdA lacking the receptor binding domain. J. Med. Microbiol. 62, 1414–1422. doi: 10.1099/jmm.0.057828-0
Geric, B., Johnson, S., Gerding, D. N., Grabnar, M., and Rupnik, M. (2003). Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J. Clin. Microbiol. 41, 5227–5232. doi: 10.1128/jcm.41.11.5227-5232.2003
Goorhuis, A., Bakker, D., Corver, J., Debast, S. B., Harmanus, C., Notermans, D. W., et al. (2008). Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin. Infect. Dis. 47, 1162–1170. doi: 10.1086/592257
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778.
Huber, C. A., Foster, N. F., Riley, T. V., and Paterson, D. L. (2013). Challenges for standardization of Clostridium difficile typing methods. J. Clin. Microbiol. 51, 2810–2814. doi: 10.1128/jcm.00143-13
Imwattana, K., Wangroongsarb, P., and Riley, T. V. (2019). High prevalence and diversity of tcdA-negative and tcdB-positive, and non-toxigenic. Clostridium difficile in Thailand. Anaerobe 57, 4–10. doi: 10.1016/j.anaerobe.2019.03.008
Janezic, S., Garneau, J. R., and Monot, M. (2018). Comparative Genomics of Clostridium difficile. Adv. Exp. Med. Biol. 1050, 59–75. doi: 10.1007/978-3-319-72799-8_5
Janezic, S., and Rupnik, M. (2019). Development and implementation of whole genome sequencing-based typing schemes for Clostridioides difficile. Front. Public Health 7:309. doi: 10.3389/fpubh.2019.00309
Kelly, C. P., Pothoulakis, C., and Lamont, J. T. (1994). Clostridium difficile colitis. N. Engl. J. Med. 330, 257–262.
Knetsch, C. W., Lawley, T. D., Hensgens, M. P., Corver, J., Wilcox, M. W., and Kuijper, E. J. (2013). Current application and future perspectives of molecular typing methods to study Clostridium difficile infections. Euro. Surveill. 18:20381.
Kuehne, S. A., Collery, M. M., Kelly, M. L., Cartman, S. T., Cockayne, A., and Minton, N. P. (2014). Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J. Infect. Dis. 209, 83–86. doi: 10.1093/infdis/jit426
Kumar, N., Browne, H. P., Viciani, E., Forster, S. C., Clare, S., Harcourt, K., et al. (2019). Adaptation of host transmission cycle during Clostridium difficile speciation. Nat Genet. 51, 1–6.
LaFrance, M. E., Farrow, M. A., Chandrasekaran, R., Sheng, J., Rubin, D. H., and Lacy, D. B. (2015). Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 7073–7078. doi: 10.1073/pnas.1500791112
Lessa, F. C., Mu, Y., Bamberg, W. M., Beldavs, Z. G., Dumyati, G. K., Dunn, J. R., et al. (2015). Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 372, 825–834.
Liu, X. S., Li, W. G., Zhang, W. Z., Wu, Y., and Lu, J. X. (2018). Molecular Characterization of Clostridium difficile Isolates in China from 2010 to 2015. Front Microbiol 9:845. doi: 10.3389/fmicb.2018.00845
Loo, V. G., Poirier, L., Miller, M. A., Oughton, M., Libman, M. D., Michaud, S., et al. (2005). A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 353, 2442–2449. doi: 10.1056/nejmoa051639
Looney, J. M., Edsall, G., Ipsen, J. Jr., and Chasen, W. H. (1956). Persistence of antitoxin levels after tetanus-toxoid inoculation in adults, and effect of a booster dose after various intervals. N. Engl. J. Med. 254, 6–12. doi: 10.1056/nejm195601052540102
Louie, T. J., Miller, M. A., Mullane, K. M., Weiss, K., Lentnek, A., Golan, Y., et al. (2011). Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 364, 422–431.
McDonald, L. C., Killgore, G. E., Thompson, A., Owens, R. C. Jr., Kazakova, S. V., et al. (2005). An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 353, 2433–2441.
Monot, M., Eckert, C., Lemire, A., Hamiot, A., Dubois, T., Tessier, C., et al. (2015). Clostridium difficile: new insights into the evolution of the pathogenicity locus. Sci. Rep. 5:15023.
Orth, P., Xiao, L., Hernandez, L. D., Reichert, P., Sheth, P. R., Beaumont, M., et al. (2014). Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 289, 18008–18021. doi: 10.1074/jbc.m114.560748
Reinert, D. J., Jank, T., Aktories, K., and Schulz, G. E. (2005). Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 351, 973–981. doi: 10.1016/j.jmb.2005.06.071
Rupnik, M. (2008). Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol. Rev. 32, 541–555. doi: 10.1111/j.1574-6976.2008.00110.x
Rupnik, M., Avesani, V., Janc, M., Von Eichel-Streiber, C., and Delmee, M. (1998). A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J. Clin. Microbiol. 36, 2240–2247. doi: 10.1128/jcm.36.8.2240-2247.1998
Rupnik, M., and Janezic, S. (2016). An upate on Clostridium difficile toxinotyping. J. Clin. Microbiol. 54, 13–18. doi: 10.1128/jcm.02083-15
Sebaihia, M., Wren, B. W., Mullany, P., Fairweather, N. F., Minton, N., Stabler, R., et al. (2006). The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38, 779–786. doi: 10.1038/ng1830
Secore, S., Wang, S., Doughtry, J., Xie, J., Miezeiewski, M., Rustandi, R. R., et al. (2017). Development of a Novel Vaccine Containing Binary Toxin for the Prevention of Clostridium difficile disease with enhanced efficacy against NAP1 strains. PLoS One 12:e0170640. doi: 10.1371/journal.pone.0170640
Sheldon, E., Kitchin, N., Peng, Y., Eiden, J., Gruber, W., Johnson, E., et al. (2016). A phase 1, placebo-controlled, randomized study of the safety, tolerability, and immunogenicity of a Clostridium difficile vaccine administered with or without aluminum hydroxide in healthy adults. Vaccine 34, 2082–2091. doi: 10.1016/j.vaccine.2016.03.010
Svenungsson, B., Burman, L. G., Jalakas-Pornull, K., Lagergren, A., Struwe, J., and Akerlund, T. (2003). Epidemiology and molecular characterization of Clostridium difficile strains from patients with diarrhea: low disease incidence and evidence of limited cross-infection in a Swedish teaching hospital. J. Clin. Microbiol. 41, 4031–4037. doi: 10.1128/jcm.41.9.4031-4037.2003
Tao, L., Zhang, J., Meraner, P., Tovaglieri, A., Wu, X., Gerhard, R., et al. (2016). Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355.
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15: 524.
von Eichel-Streiber, C., Zec-Pirnat, I., Grabnar, M., and Rupnik, M. (1999). A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol. Lett. 178, 163–168. doi: 10.1016/s0378-1097(99)00327-4
Voth, D. E., and Ballard, J. D. (2005). Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263. doi: 10.1128/cmr.18.2.247-263.2005
Keywords: Clostridioides difficile, Clostridium difficile, TcdA, TCDB, vaccines, whole genome sequencing
Citation: Li Z, Lee K, Rajyaguru U, Jones CH, Janezic S, Rupnik M, Anderson AS and Liberator P (2020) Ribotype Classification of Clostridioides difficile Isolates Is Not Predictive of the Amino Acid Sequence Diversity of the Toxin Virulence Factors TcdA and TcdB. Front. Microbiol. 11:1310. doi: 10.3389/fmicb.2020.01310
Received: 11 December 2019; Accepted: 25 May 2020;
Published: 19 June 2020.
Edited by:
Spyridon Ntougias, Democritus University of Thrace, GreeceReviewed by:
Felipe Luiz Pereira, Instituto Federal Catarinense, BrazilCopyright © 2020 Li, Lee, Rajyaguru, Jones, Janezic, Rupnik, Anderson and Liberator. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Liberator, UGF1bC5saWJlcmF0b3JAcGZpemVyLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.