 Min Lin
Min Lin Hui Xiong
Hui Xiong Xuechuan Xiang
Xuechuan Xiang Zelin Zhou
Zelin Zhou Lifeng Liang
Lifeng Liang Zhinan Mei
Zhinan Mei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 June 2020
Sec. Plant Pathogen Interactions
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01257
Gymnadenia conopsea (L.) R. Br. is an important perennial terrestrial photosynthetic orchid species whose microbiomes are considered to play an important role in helping its germination and growth. However, the assemblage of G. conopsea root-associated microbial communities is poorly understood. The compositions of fungal and bacterial communities from the roots and corresponding soil samples in G. conopsea across distinct biogeographical regions from two significantly different altitudes were characterized at the vegetative and reproductive growth stages. The geographical location, developmental stage and compartment were factors contributing to microbiome variation in G. conopsea. Predominant fungal taxa include Ascomycota, Basidiomycota, Mortierellomycota and Chytridiomycota, whereas Proteobacteria, Bacteroidetes, Acidobacteria, Actinobacteria, Verrucomicrobia, Chloroflexi, TM7 and Planctomycetes were predominant bacterial taxa. Using G. conopsea as a model, the structural and functional composition in G. conopsea root-associated microbiomes were comprehensive analyzed. Contrary to previous studies, biogeography was the main factor influencing the microbial community in this study. Besides, compartment and developmental stage should also be considered to analyze the variation of microbiota composition. Although the microbial composition varied greatly by location, the symbiotic microorganisms of G. conopsea still have certain specificity. This study gives an abundant information of G. conopsea root-associated microbiomes and provides new clues to better understanding the factors affecting the composition and diversity of fungal/bacterial communities associated with orchids. Our results also laying a foundation for harnessing the microbiome for sustainable G. conopsea cultivation. Moreover, these results might be generally applicable to other orchidaceae plants.
Terrestrial plants harbor abundant and diverse microbes that affect plant distribution, growth and health by different ways. Previous research revealed that plants can recruit beneficial/specific communities to cope with pathogen encounters, modify the nutrient status as well as adapt to new or changing environmental conditions (Qin et al., 2016; Berendsen et al., 2018; Yuan et al., 2018). In addition, the spatial distribution of soil microorganisms, such as microbial mutualists and pathogens, probably acts as a driver of spatial patterns of palnt species and plant community diversity (Ettema and Wardle, 2002). These communities can be taken as plant host’s second genome, providing a wide diversity of promising functional capacities (Berendsen et al., 2012; Turner et al., 2013). To maintain plant healthy growth, it is valuable to investigate the community structure of plant-associated microorganisms and their functions. So far, most research work has made on certain model and crop plants, such as Arabidopsis (Lundberg et al., 2012; Bai et al., 2015), rice (Sessitsch et al., 2012), maize (Peiffer et al., 2013), millet (Jin et al., 2017), populus (Cregger et al., 2018), and citrus (Xu et al., 2018), the results revealed clear taxonomically structured microbiome and the factors that determine assembly of the microbiome. This work may enable new plant management approaches and provide novel tools to improve the robustness of crop plant performance.
Gymnadenia conopsea (L.) R. Br. (Orchidaceae) is a terrestrial photosynthetic orchid species that widely distributed in Eurasia, including England, Ireland, Russia, Nepal, China, Japan, and the Korean peninsula (Hultén and Fries, 1986). G. conopsea exist in a wide range of habitats with an altitude ranging from 200 to 4700 m in China. For many years, G. conopsea was taken as a primary component in many preparations that listed in the Pharmacopoeia of the People’s Republic of China, and used for improving the body in traditional medicines, such as tonify Qi of the kidney and nourish the lungs and treat various diseases, especially in Tibetan medicine and Mongolian medicine. Whereas G. conopsea has been investigated as an important fragrant orchid plant in some European countries. G. conopsea is declining very rapidly like most other Orchidaceae. Now, G. conopsea was list in Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES). Two factors resulted in the rapid decline in the number of this species. The external factors were due to the high market demand, over exploitation as well as the habitat destruction, the internal endangered factors were its weak fertility and low breeding efficiency.
It was known that orchidaceae seeds are remarkably small, extremely light, and lacking endosperm (food reserves). Known seed weights extend from 0.31 to 24 μg, the diameter generally range from 0.01 to 0.9 mm, with the difference between the ‘widest’ and ‘thinnest’ known seeds in the family being 90-fold (Arditti and Ghani, 2000). Because orchidaceae seeds lack of endosperm, germination under natural conditions depend on some fungi colonization supplying carbohydrates (Smith and Read, 2008). In addition, Orchidaceae invariably rely on mycorrhizal fungi in their seedling and adulthood as well (Waterman et al., 2011; McCormick and Jacquemyn, 2014; Rasmussen et al., 2015). Early studies on the relationship between Orchidaceae plants and “orchid mycorrhiza” were mainly via germination experiments or cultivation methods in the laboratory conditions and found a diversity of associated fungi (Curtis, 1939). Though it is well-known that fungi have crucial roles in Orchidaceae plants, the effect of root-associated fungi on plant growth and distribution is less well-understood. Meanwhile, fungi cultivation under laboratory conditions does not reflect the complexity of interactions under natural conditions (Masuhara and Katsuya, 1994; Perkins et al., 1995). Furthermore, on account of their hidden lifestyle, a great deal of fungi cannot be cultivated under normal laboratory conditions, and it is difficult to guarantee the exist and diversity of fungi without contamination, or the contaminants may affects these early experimental results.
Amplicon based community profiling methods that amplify a wide taxonomic spectrum of fungi largely get rid of these effects as mentioned above and enable a direct evaluation of the fungal diversity in the orchid roots (Kristiansen et al., 2001; Taylor and McCormick, 2008; Voyron et al., 2017; McCormick et al., 2018). Previous molecular studies of the fungal communities associated with G. conopsea have indicated that this species is a mycorrhizal generalist, in that it can interact with a wide variety of fungi species (Stark et al., 2009; Jacquemyn et al., 2012; Tesitelova et al., 2013; Waud et al., 2016; Chen Y. et al., 2019). It mainly associated with the Tulasnellaceae and Ceratobasidiaceae (Tesitelova et al., 2013; Waud et al., 2016; Xing et al., 2020) and ectomycorrhizal fungi from Thelephoraceae, Russulaceae, Inocybaceae, and Cortinariaceae (Stark et al., 2009; Xing et al., 2020). Though the relevance and detail function of these fungi remain unknown, the available knowledge suggests that their exist have an ecological functions at least in certain other photosynthetic plant species (Jacquemyn et al., 2017). Moreover, the fungal community of G. conopsea demonstrated a obvious spatial structure, the mycorrhizal communities found in the roots of adult plants differ clearly between plants growing in different geographically distinct regions (Stark et al., 2009; Xing et al., 2020). Recent research in the seed germination experiments have revealed that G. conopsea is regarded a mycorrhizal generalist, however, it needs specific fungi to help protocorm formation and seedling development (Gao et al., 2020), it indicated the fungi of G. conopsea may vary among different developmental stage. Besides, previously investigations showed that bacteria isolated from orchid also benefit for plant growth and/or resistance to pathogens (Tsavkelova et al., 2007; Faria et al., 2013). Nevertheless, the information of the actual distribution and diversity of fungi in natural populations of G. conopsea in China are limited and how these fungi influence spatial and temporal patterns of recruitment and establishment of G. conopsea still unclear. Meanwhile, the composition structure and potentially functional activity of the G. conopsea-related bacteria remains largely unexplored.
In our study, the root and soil samples of the wild-grown G. conopsea plants from two different growth locations (habitats) and two developmental stages were collected. The dynamics of fungal and bacterial communities in G. conopsea of two distinct compartments (root and corresponding soil) from two distinct growth locations at two developmental stages were examined by ITS2 and 16S rRNA gene amplicon sequencing. Multiple factors contributing to microbial community variation were evaluated, and fungal and bacterial functions were predicted according to their taxonomy. Using the dynamic studies of the microbiome composition in large datasets from the different conditions sampled in this study, it offers new insights into the process of microbiome assembly in G. conopsea, and lays a foundation for sustainable use of the microbial community in G. conopsea cultivation.
Representative G. conopsea root samples and corresponding soil samples were collected uniformly from the natural habitats at Linzhi (29°38′59′′ N/94°25′37′′ E, 3600 m asl, Tibet, China) and Greater Khingan Mountains (51°42′N/124°20′E, 496 m asl, Heilongjiang, China). Linzhi (LZ) has a plateau temperate semi-humid and humid monsoon climate with annual mean rainfall 650 mm and annual mean temperature 8.7°C. Whereas, Greater Khingan Mountains (DXAL) belongs to the cold-temperate humid continental climate zone with annual mean rainfall 746 mm and annual mean temperature −2.8°C. The soil with alkali-hydrolyzale N 256.30/265.57 mg/kg, available P 48.00/27.76 mg/kg, available K 290.07/293.97 mg/kg; organic matter 121.67/66.55 g/kg, total N 4.58/5.92 g/kg, and a pH of 5.62/5.94 in LZ and DXAL, respectively. To rule out the effect of age on developmental stages, the plants with the consistent growth vigor at two developmental stages (vegetative growth stage and reproductive growth stage) were selected. The root and corresponding soil were sampled two times in LZ and DXAL, corresponding to two growth stages of plant. Plants were dug out with their surrounding soil, rapidly transferred into sterile plastic sampling bags and transported to the laboratory on ice as quickly as possible. The adjacent soil layers (about 1 cm thick) in the roots’ surface were manually separated and collected as the corresponding soil compartment. To remove fine roots and large organic debris, the soil samples were sieved through 2 mm mesh and are defined as the soil compartment. A total of 14 individual plants were collected in LZ and DXAL. The corresponding bulk soil of each plant were sampled five biological replicates. The roots were manually separated and then the adhering soil particles were removed by gently shaking. Five-seven roots (replicates) were used for each plant. The roots were cut into small pieces and washed thoroughly using pre-cooled sterile distilled water to take away the soil particles, and then washed successively with 75% pre-cooled ethanol, 0.25% NaOCl to further clean the root’ surfaces, and finally washed several times by sterile water. These step roots were defined as microbially-enriched root compartment. These treated soil and root samples were transferred to 2 mL sterile centrifuge tubes. All the samples were stored immediately at −80°C until subsequent use. In total, 70 soil samples and 75 root samples were obtained from two representative locations and two developmental stages in China.
Total DNA was extracted from aforementioned samples using the NucleoSpin® Soil Kit (Macherey-Nagel GmbH & Co. KG, Germany) according to the manufacturer’s instructions. The DNA quality and quantity were evaluated using the NanoDrop device (Thermo Fisher Scientific, United States) and agarose gel electrophoresis (1%). All the qualified DNA (total DNA concentration more than 50 ng and no protein, RNA and salt ion contamination) were used to construct library. All samples were carried out by sequencing the V3–V4 region of 16S rRNA gene in bacteria (341F-806R) and the ITS2 region of fungal nrDNA. These primers were designed to amplify target sequence and they include dual index and adapters for annealing to the Illumina Miseq flow cell. In both cases, only the qualified library can be used for sequencing. The qualified library was defined based on the following criteria: the amplified fragment length in the library consistent with expectation, total amount of PCR products ≥ 3 μg and concentration ≥ 30 ng/μ8L and agarose gel electrophoresis showed clear bands without dispersion. After DNA libraries quality control (QC) and quantification, as well as normalization, 300-bp paired-end reads were obtained from MiSeq platform (BGI, China) for the amplicon (16S rRNA and ITS2) analyses, respectively (Fadrosh et al., 2014).
Fungal and bacterial community composition was detected by sequencing ITS2 and 16S rRNA gene amplicons from the G. conopsea root and corresponding soil samples. The high-quality illumina paired-end reads were merged using FLASH software with the default setting (Magoc and Salzberg, 2011). The operational taxonomic units (OTUs) were clustered using the UPARSE algorithm (Edgar, 2013) at 97% sequence identity, the chimeras were filtered by UCHIME (Edgar et al., 2011). To obtain the taxonomic information of the OTUs, the original reads were mapped back to their OTUs with the USEARCH (global alignment algorithm) (Edgar, 2010), the representative sequences in each OTU were obtained and mapped to Greengenes (v13_8 release/2013_5_99) and UNITE (2019_version8) reference databases by the Ribosomal Database Project classifier (Wang et al., 2007) for the bacterial and fungal communities, respectively. OTUs classified as mitochondria or chloroplast, or less than 5 sequences, were filtered from the datasets.
The alpha-diversity was computed for each sample by Chao and Shannon indices using mothur (Schloss et al., 2009) from the final OTU table. Boxplot were plotted using R software. The significant differences in alpha-diversity from different groups were determined by the Kruskal–Wallis and Dunn’s post hoc tests (P < 0.05). The OTU table was normalized by the cumulative sum scaling (CSS) method to calculate beta-diversity (Paulson et al., 2013). Unweighted UniFrac (UUF) and Weighted UniFrac (WUF) distances between samples were computed from the above-mentioned normalized OTU table. The taxonomic dissimilarity analyses between samples was performed based on principal coordinate analysis (PCoA) based on WUF and UUF. To assess the influence of the different factors (site, compartment, and stage) on the beta-diversity, permutational multivariate analysis of variance (PERMANOVA) analysis was carried out (adonis function in vegan R package).
Operational taxonomic units with frequency of occurrence above 90% from the root or soil samples in each group were defined as “core OTUs.” To identify significantly differential core OTUs between each pair of groups that need to compare, the Wilcoxon signed rank test was employed in each group, and with relative abundance (RA) considered separately in each sample. The resulting P-values were corrected for multiple testing via the Benjamini-Hochberg (BH) method. The core OTUs enriched in each group (P < 0.05) were then referred to as “differential core OTUs.” The comparison of core taxa of the root/soil microbiome between each pair of groups were based on differential core OTUs.
In order to confirm if fungal functional groups differed in relative abundance between host biogeography within compartment (root, soil) at two developmental stages, the FUNGuild (Nguyen et al., 2016) was used to classify each core OTU into an ecological guild. Core OTUs assigned to a guild with the confidence ranking of “probable” or “highly probable” were retained for further use, whereas those with “possible” were removed. Bacteria microbial function prediction was carried out by the PICRUSt software. Differentially enriched KO pathways or modules were identified on the basis of the reporter score from Z-scores of individual KOs. One tail Wilcoxon rank sum test was applied to all Kos, and corrected for multiple testing by the Benjamin-Hochberg method. The Z-score of each KO could then be computed. The detection threshold of significantly differentiating pathways was set as absolute value for reporter score ≥ 1.96 (95% confidence in either tail basing on normal distribution).
We analyzed fungal and bacterial microbiomes from two separate rhizocompartments (the root and corresponding soil), a total of 145 samples that included 69 samples from LZ and 76 samples from DXAL (Supplementary Table S1). The ITS2 and V3–V4 region of 16S rRNA gene were amplified by PCR and sequenced by Illumina MiSeq platform. Approximately 12.1 and 5.1 million high-quality sequences were obtained from the ITS2 and 16S rRNA gene sequencing data, respectively. 6,667,165 and 5,460,693 ITS2 tags and 3,883,175 and 1,206,448 16S rRNA tags were obtained from the corresponding soil and root sample, respectively (Supplementary Tables S2, S3). After discarding non-bacterial or non-fungi, mitochondrial, chloroplast and low-abundance OTUs, we obtained 5,193 fungi OTUs and 9,712 bacterial OTUs, respectively (Supplementary Tables S2, S3). For the microbial richness Chao index estimated based on the fungal dataset, it revealed that the microbial richness from soil to the root was decreased in both sites (Figure 1A, P < 0.05), except the soil sample in DXAL at reproductive growth stage. Significant differences were found in comparisons between two different developmental stages in root samples include LZ and DXAL, the richness value was lower in reproductive growth stage than the vegetative growth stage. Similar results were retrieved from microbial diversity shannon index values, with higher diversity in soil samples (Figure 1B, P < 0.05). For the bacterial microbiomes, measures of within-sample diversity (α-diversity indices) showed a decrease of microbial richness and diversity from soil samples to root samples based on the chao index and shannon index in both sites (Figures 1C,D, P < 0.05). In both bacterial microbial richness and diversity, higher richness and diversity were observed in vegetative growth stage than reproductive growth stage at two sides (P < 0.05).
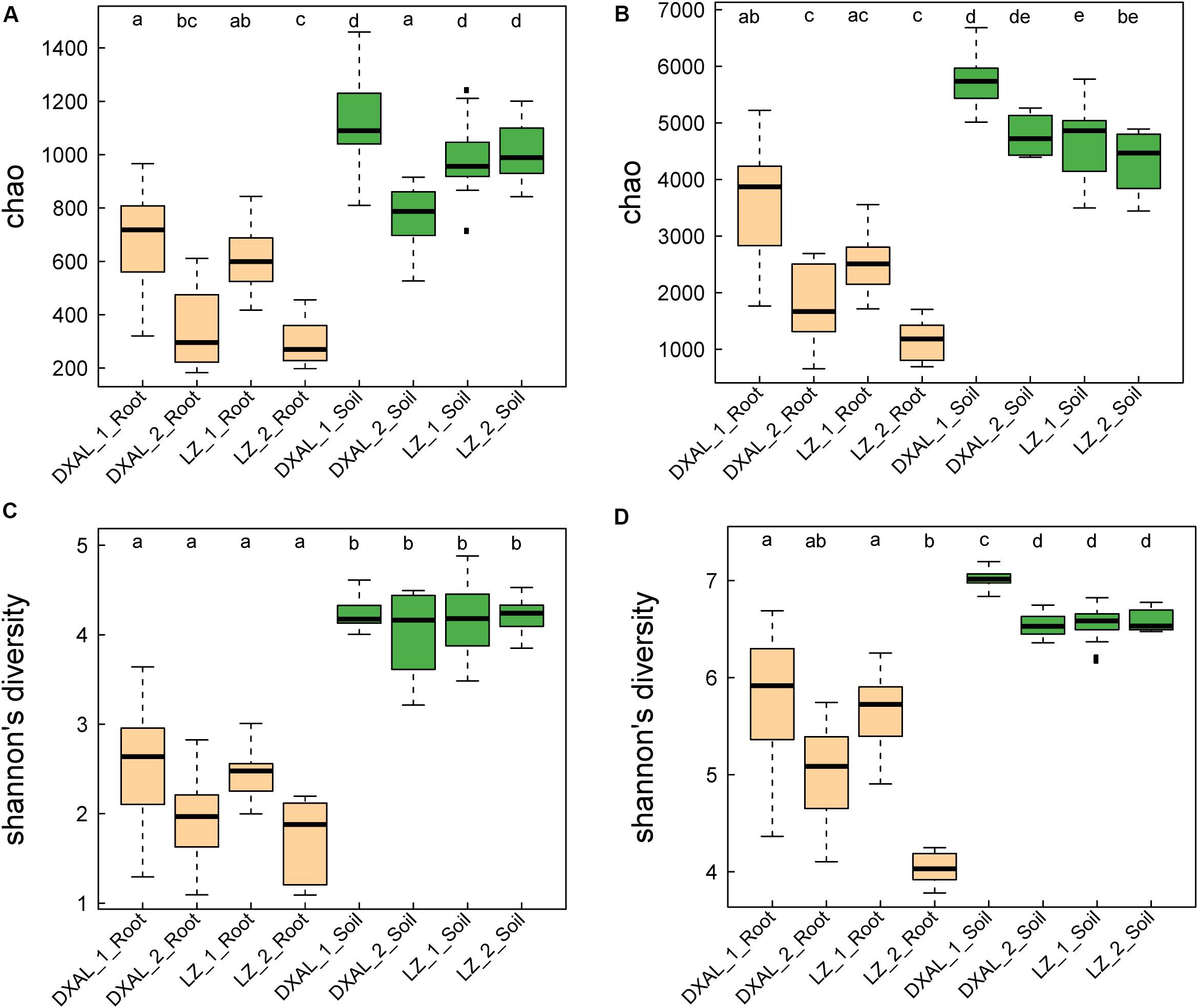
Figure 1. Species richness and diversity of root-associated microbial communities. (A) Microbial richness index comparison between each group based on the Chao index using the ITS data. (B) Microbial diversity index comparison between each group based on the Shannon index using the ITS data. (C) Microbial richness index comparison between each group based on the Chao index using the 16S rRNA gene data. (D) Microbial diversity index comparison between each group based on the Shannon index using the 16S rRNA gene data. Significant differences are depicted with letters (P < 0.05, Kruskal–Wallis with Dunn’s post hoc test). DXAL: Greater Khingan Mountains, LZ: Linzhi, 1: vegetative growth stage, 2: reproductive growth stage.
To examine between-sample variation (β-diversity) and to investigate patterns of separation between microbial communities, PCoAs based on WUF and UUF distances were performed. In both the WUF and UUF PCoAs, fungal and bacterial communities cluster along the first principal coordinate based on the geographical location and development stage, the second factor explaining fungal and bacterial communities was slightly different (Figure 2A; WUF, 2B UUF; 2C WUF, 2D, UUF). In fungal community, compartment was the second factor using WUF metric, while not obvious by UUF metric (Figure 2B); however, in bacterial community, compartment was the second factor in all metric (Figures 2C,D). Interestingly, the PCoA result of the bacterial communities, some samples (include root and soil samples) in the DXAL at reproductive growth stage were close to LZ samples. Additionally, the data for clustering of samples were computed by PERMANOVA with all distance, which revealed that site, compartment and developmental stage comprise the largest contribution to variation for the microbiome data in both fungal and bacterial communities, it was consistent with the PCoA results [R2 of 0.709 and 0.644 (WUF); 0.462 and 0.429 (UUF), respectively, P-value < 0.001 for all tests; Supplementary Table S4]. Whereas according to the single factor, site was the strongest factor shaping these microbiome communities, followed by compartment or plant development stage [R2 of 0.272, 0.204, and 0.091 (WUF); 0.203, 0.063, and 0.072 (UUF); 0.303, 0.076 in fungal community; R2 of 0.244, 0.226, and 0.078 (WUF); 0.177, 0.089, and 0.059 (UUF) in bacterial community, for site, compartment, and stage, respectively, P-value < 0.001 for all tests; Supplementary Table S4]. Together, these results implying that microbial communities vary significantly between plant host biogeography, compartments and developmental stages also impact G. conopsea microbial community composition.
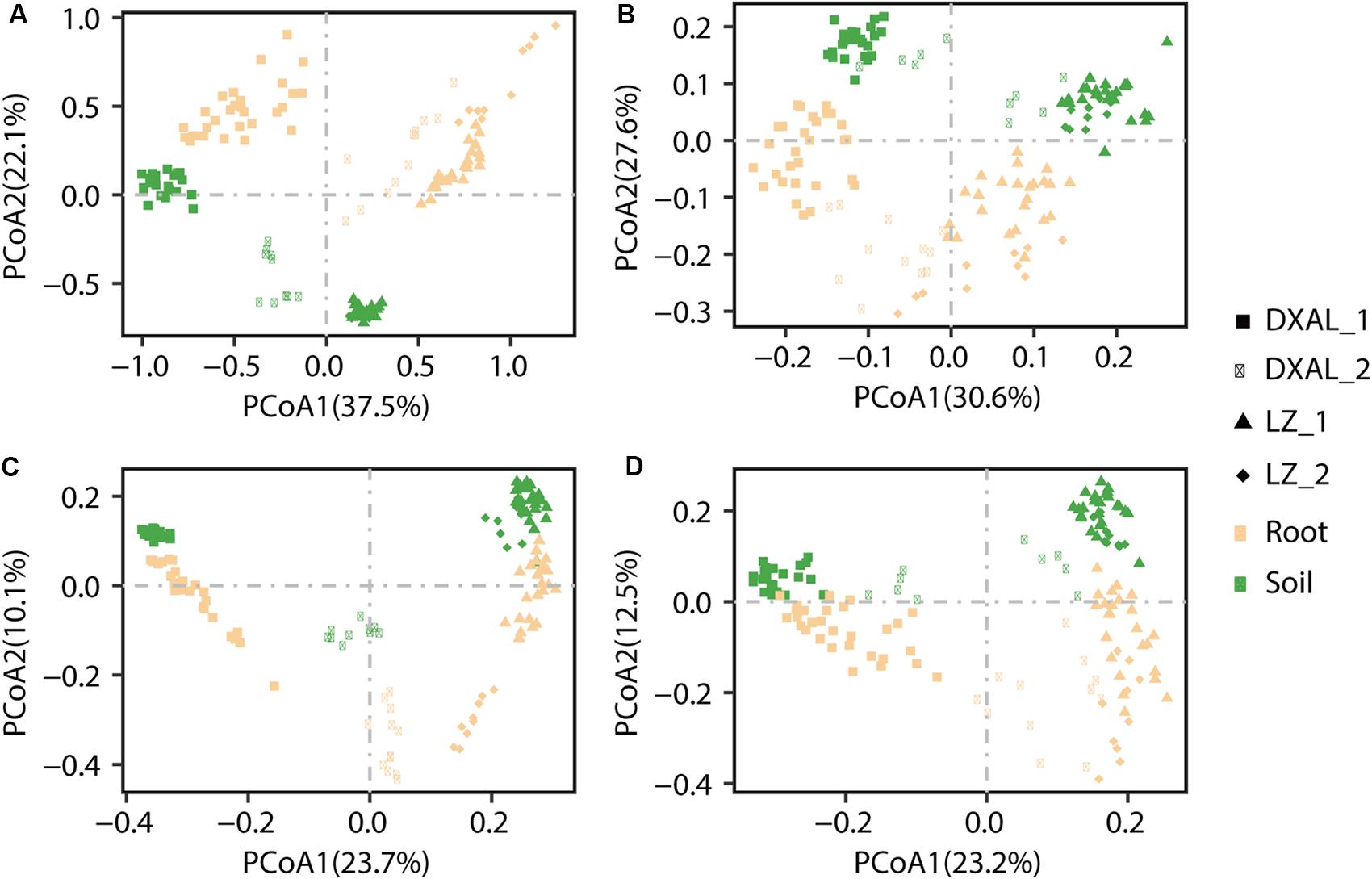
Figure 2. PCoA of microbial community composition among different sample types of G. conopsea. (A) PCoA using the WUF distances at the OTU level using fungal community’s data. (B) PCoA using the UUF distances at the OTU level using fungal community’s data. (C) PCoA using the WUF distances at the OTU level using bacterial community’s data. (D) PCoA using the UUF distances at the OTU level using bacterial community’s data. Samples are color coded according to the sites, compartments, and developmental stages are depicted with different symbols. DXAL: Greater Khingan Mountains, LZ: Linzhi, 1: vegetative growth stage, 2: reproductive growth stage.
A totally of 17/39 different phyla and 65/122 different classes were identified in fungal/bacterial communities, respectively. As expected, there were obvious differences in the proportions of various phyla and classes among the compartments that are consistent with each site and development stage (Figure 3 and Supplementary Figure S1). The dominant (RA > 1%) fungal phyla found in the samples included Ascomycota (>50%), Basidiomycota, Mortierellomycota and Chytridiomycota, whereas prokaryotic phyla, included Proteobacteria (>28%), Bacteroidetes, Acidobacteria, Actinobacteria, Verrucomicrobia, Chloroflexi, TM7 and Planctomycetes (Supplementary Table S5). At lower taxonomic ranks, Leotiomycetes, Agaricomycetes, Dothideomycetes, Sordariomycetes, Eurotiomycetes, Tremellomycetes, and Mortierellomycetes were the dominant fungal classes found in both compartments across two sites, Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Saprospirae, Gammaproteobacteria, Sphingobacteriia, Actinobacteria, Acidobacteria-6, Thermoleophilia, TM7-1, Acidimicrobiia, Solibacteres, Cytophagia, and Spartobacteria were the main bacterial classes (Supplementary Table S6). The root had a significantly greater proportion of Leotiomycetes and Tremellomycetes (q-value < 0.05) than soil, whereas Agaricomycetes, Glomeromycetes, Lobulomycetes, Rhizophydiomycetes, and Spizellomycetes (q-value < 0.05) were largely depleted in the root in comparison with the soil in fungal communities (Supplementary Table S7). In the bacterial communities, Alphaproteobacteria, Methylacidiphilae, Sphingobacteriia and TM7_3 (q-value < 0.01) were more abundant in root, while Acidobacteria_6, AT_s54, EC1113, Gemm_1, Gemm_2, Gemm_5, Gemmatimonadetes, iii1_8, ML635J_21, Nitrospira, OM190, Phycisphaerae, PRR_12, RB25, S035, S085, SJA_28, SM2F11, TA18, TK10, TK17, and TM1 (q-value < 0.01) was more prevalent in soil (Supplementary Table S8). The decrease in relative abundance of these classes among compartments was coincident with the detection that the microbial diversity reduced from soil to root.
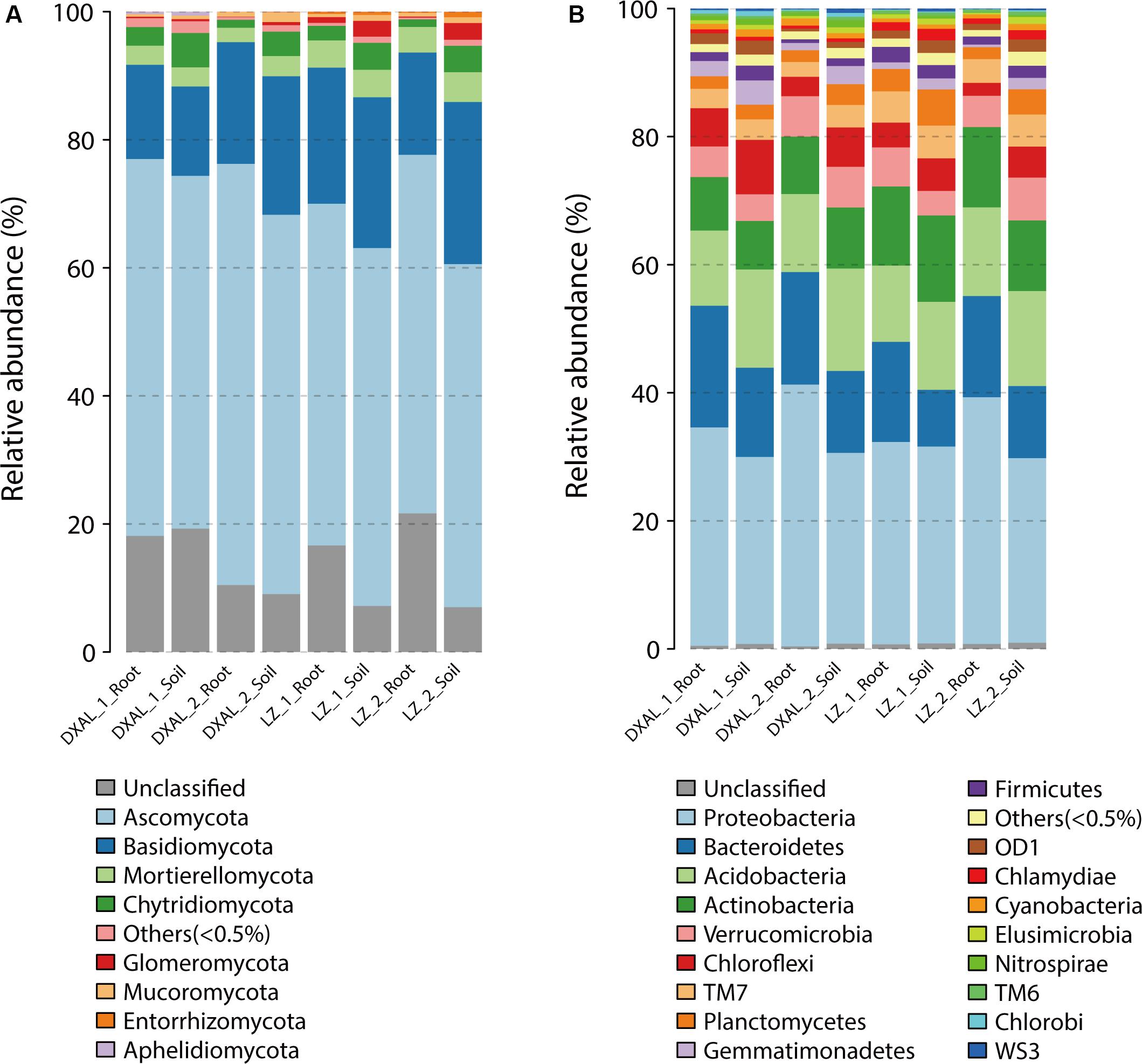
Figure 3. Microbial community composition among different sample types of G. conopsea at phylum level. (A) Relative abundance of fungal taxa of each group. (B) Relative abundance of bacterial taxa of each group. DXAL: Greater Khingan Mountains, LZ: Linzhi, 1: vegetative growth stage, 2: reproductive growth stage.
As microbes that are consistently present across samples likely provide critical ecological functions (Shade and Handelsman, 2012), the core fungal/bacterial communities were analyzed in each group. We defined 390/718 core root fungal OTUs and 1190/1314 core soil fungal OTUs from LZ and DXAL microbiota throughout the two growth stages, respectively. Regarding fungal functional group (guild), all core OTU were classify based on FunGuild assignment. Since the whole genome information of fungi is still relatively scarce, the functional annotation information of fungi is less comprehensive. There were only 38.24% (910/2380) core OTU have reliable functional annotation information. From the trophic mode level, saprotroph was most dominant trophic mode, followed by symbiotroph and pathotroph (Figure 4A and Supplementary Table S9). Undefined_Saprotroph, Plant_Pathogen, Endophyte, Ectomycorrhizal and Animal_Pathogen-Undefined_Saprotroph were the top five high-abundance guilds (Figure 4B and Supplementary Table S9). The Growth_Morphology mainly focus on Microfungus, Agaricoid, Facultative_Yeast, Clavarioid, Yeast, Facultative_Yeast-Microfungus, Dark_Septate_Endophyte, Corticioid, Gasteroid, Thallus, and Pezizoid (Figure 4C and Supplementary Table S9).
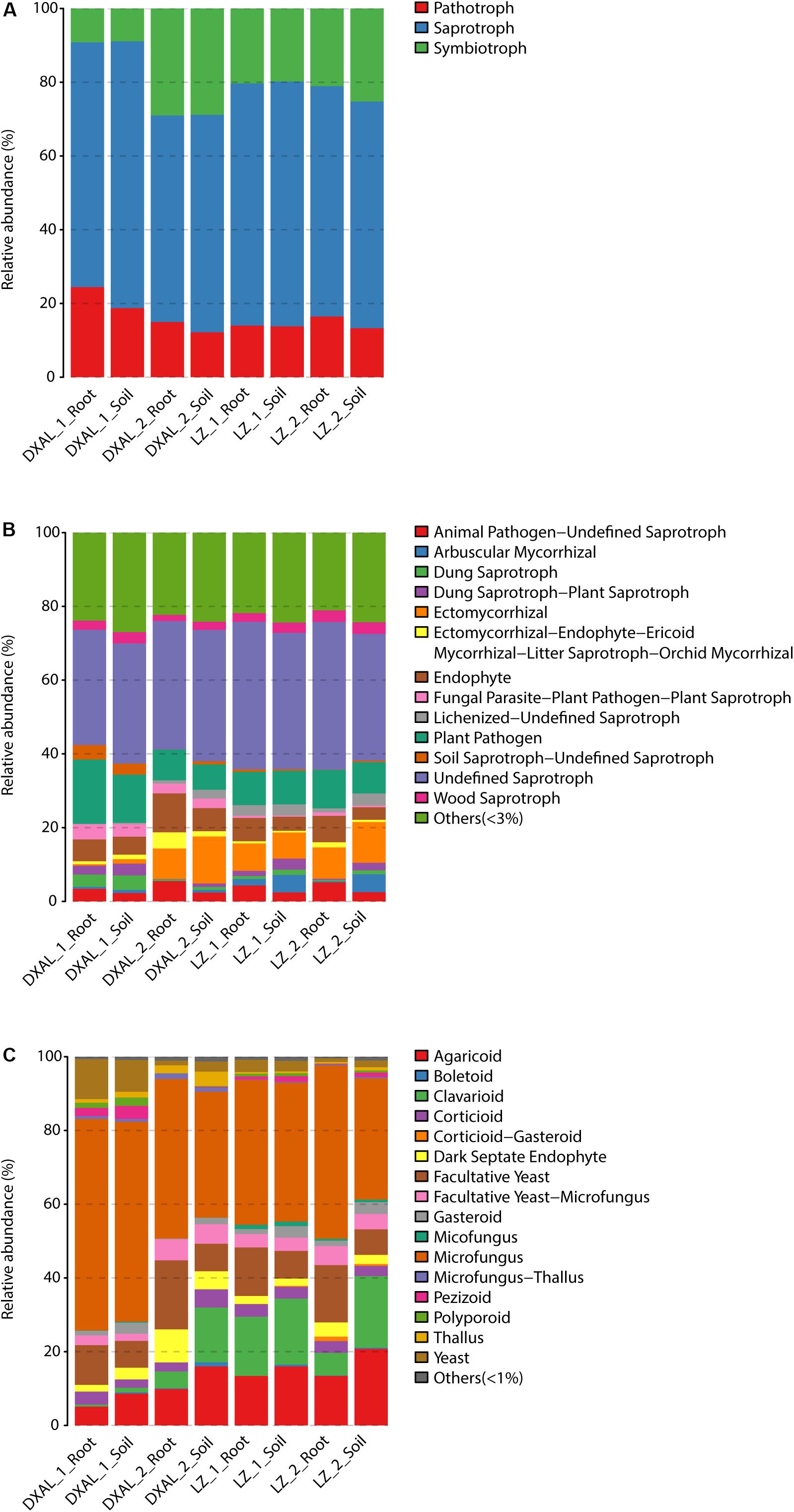
Figure 4. Compositions of fungal functional group (guild) inferred by FUNGuild. (A) At the trophic mode level. (B) At guilds level. (C) At Growth_Morphology level. DXAL: Greater Khingan Mountains, LZ: Linzhi, 1: vegetative growth stage, 2: reproductive growth stage.
For the bacterial core OTUs, 1,417/3,099 core root OTUs and 4,403/5,920 core soil OTUs from LZ and DXAL microbiota were obtained throughout the two growth stages, respectively, using the aforementioned method. In order to gain insight into functional changes within G. conopsea bacterial microbiota, the KEGG pathways and modules enriched in root or soil microbiomes were analyzed (Figure 5 and Supplementary Table S10). The samples from root displayed higher potential for plant nutrition, such as transfer of amino acids and sugars, stress resistance, Aromatics degradation, Drug efflux transporter/pump, Drug resistance, Mineral and organic ion transport system, Pathogenicity, Peptide and nickel transport system, Plant pathogenicity, Phosphate and amino acid transport system, Saccharide, polyol, and lipid transport system and Symbiosis, whereas the modules for Energy metabolism, Nucleotide and amino acid metabolism and Genetic information processing (Aminoacyl tRNA, Aromatic amino acid metabolism, ATP synthesis, Carbon fixation, Cofactor and vitamin biosynthesis, Methane metabolism, Nucleotide sugar, Proteasome, Ribosome and RNA polymerase) were enriched in soil samples (Figure 5 and Supplementary Table S10). Nutrition is of great importance in shaping the root microbiota, the root containing various plant-derived compounds that would be likely source of nutrients for microbes. Consistent with this, the modules that including transport system responsible for transferring phosphate, amino acid, peptide, nickel, Saccharide, polyol, and lipid were enriched in the core root microbiome. Symbiosis is widely thought to act an important role in plant growth owing to its function in plant biodiversity and nutrient supply (van der Heijden et al., 2016), and was observed significantly enriched in root samples compared with soil (Figure 5). The soil microbiome exhibited increased potential for Carbon fixation, consistent with the enrichment of Cyanobacteria in soil samples (Supplementary Table S5).

Figure 5. Alterations in bacteria microbial functional modules in each compare group. Dashed lines indicate a reporter score of 1.96, corresponding to 95% confidence in a normal distribution. DXAL_1: DXAL_1_Root vs. DXAL_1_Soil, DXAL_2: DXAL_2_Root vs. DXAL_2_Soil, LZ_1: LZ_1_Root vs. LZ_1_Soil, LZ_2: LZ_2_Root vs. LZ_2_Soil. DXAL: Greater Khingan Mountains, LZ: Linzhi, 1: vegetative growth stage, 2: reproductive growth stage.
To investigate how plant geographical locations might affect the microbiome, the core OTU that significantly different in abundance in each group were used for further analysis, and the fungal and bacterial community compositions from two plants growth locations were compared (Supplementary Tables S11, S12). The fungal community composition was notably different between two sites at the vegetative growth stage in both root and soil samples, Leotiomycetes, Agaricomycetes, Eurotiomycetes, and Mortierellomycetes were more abundant in LZ core fungal OTUs, whereas Sordariomycetes, Dothideomycetes, and Tremellomycetes were more prevalent in DXAL core fungal OTUs (Figures 6A,B, P < 0.05). The fungal community compositions among samples at the reproductive growth stage were more similar in two sites. In the root samples, Mortierellomycetes was more abundant in LZ, whereas Dothideomycetes and Eurotiomycetes were more prevalent in DXAL (Figure 6C), as for the soil samples, the relative abundance of Sordariomycetes, Dothideomycetes and Eurotiomycetes were higher in DXAL, whereas that of Agaricomycetes was lower in DXAL (Figure 6D).
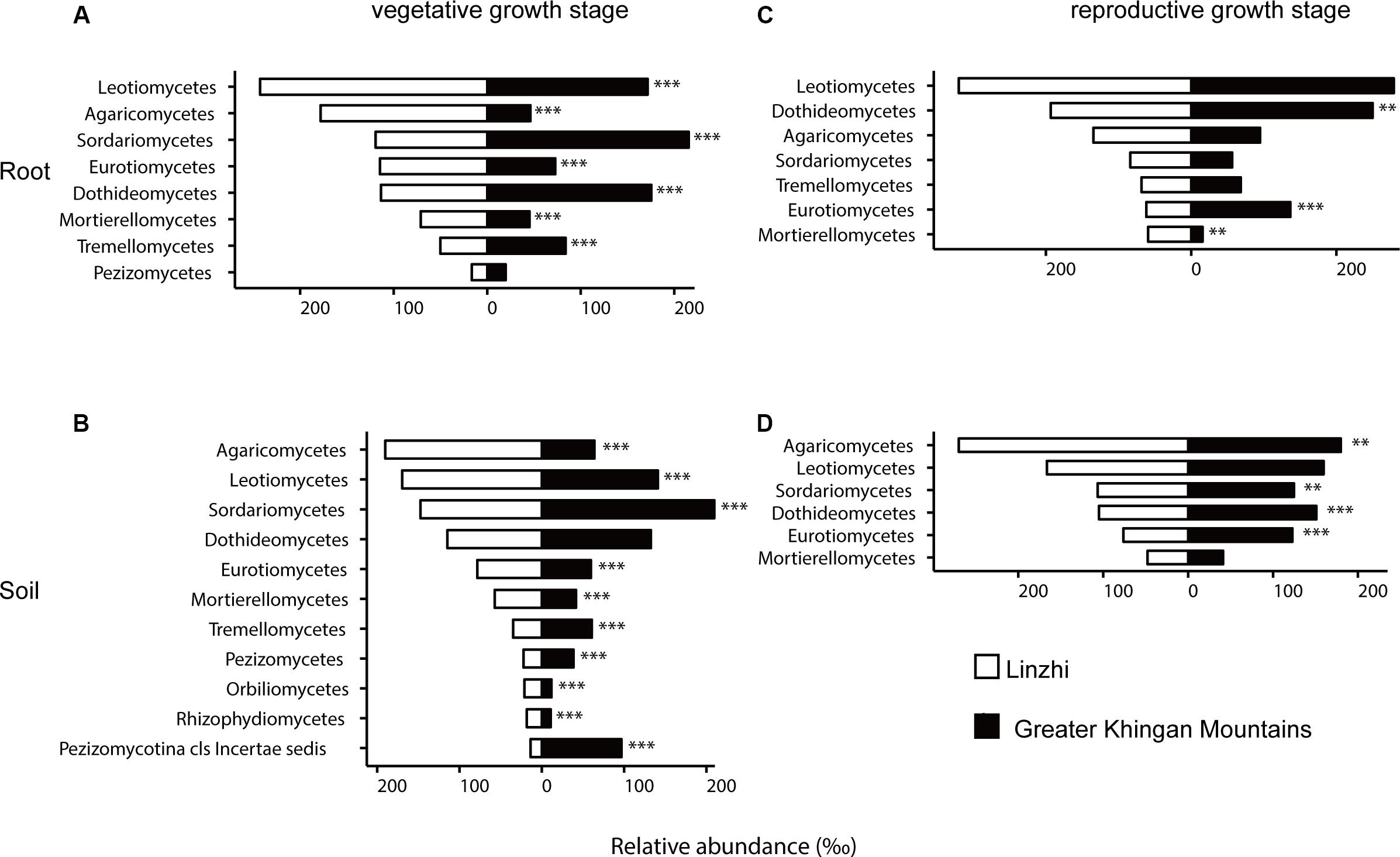
Figure 6. Side-by-side comparison of the relative abundance (‰) of classes which made up at least > 10‰ of the total fungal core microbiome community across different sites. (A) Relative abundance (‰) of the classes detected in root core OTUs of the indicated plant growth sites at vegetative growth stage. (B) Relative abundance (‰) of the classes detected in soil core OTUs of the indicated plant growth sites at vegetative growth stage. (C) Relative abundance (‰) of the classes detected in root core OTUs of the indicated plant growth sites at reproductive growth stage. (D) Relative abundance (‰) of the classes detected in soil core OTUs of the indicated plant growth sites at reproductive growth stage. Asterisks indicate significant differences occur between Linzhi and Greater Khingan Mountains [Benjamini–Hochberg false discovery-rate (FDR) adjusted P-value, ∗ represent < 0.05, ∗∗ represent < 0.01, ∗∗∗ represent < 0.001] in each compartment.
For the taxonomic structure in the bacterial communities, Proteobacteria, Bacteroidetes, Acidobacteria, Actinobacteria, Verrucomicrobia, Chloroflexi, TM7, Firmicutes, and Planctomycetes as dominating phyla in root bacterial communities at the vegetative growth stage, these phyla in relative abundance were significantly different between two sites (Figure 7A). The families belonging to the three dominant (Proteobacteria, Bacteroidetes, and Acidobacteria) phyla were listed, which contained 15 families and belonged to 8 classes (Figure 7B). Rhodospirillaceae, Hyphomicrobiaceae and Rhizobiaceae (Alphaproteobacteria), Sinobacteraceae (Gammaproteobacteria), Sphingomonadaceae (Bacteroidetes) are significantly enriched in LZ compared to DXAL at the vegetative growth stage, whereas Comamonadaceae and Oxalobacteraceae (Betaproteobacteria), Xanthomonadaceae (Gammaproteobacteria), Sphingomonada ceae (Alphaproteobacteria), Cytophagaceae (Cytophagia), Flavobacteriaceae (Flavobacteriia), Ellin6075 (Chloracidobacteria) were more abundant in DXAL (Figure 7B). As for the reproductive growth stage, the relative abundance in Proteobacteria, Acidobacteria, Bacteroidetes, Actinobacteria and Verrucomicrobia were significantly different between two sites (Figure 7C). We noted that most classes belonging to the Proteobacteria, were over-representation in LZ, such as Rhizobiaceae and Beijerinckiaceae (Alphaproteobacteria), Xanthomonadaceae (Gammaproteobacteria), Oxalobacteraceae and Burkholderiaceae (Betaproteobacteria), besides, Acidobacteriaceae (Acidobacteriia), and Sphingobacteriaceae (Sphingobacteriia) also more dominated in LZ; whereas Comamonadaceae (Betaproteobacteria), Caulobacteraceae (Alphaproteobacteria), Bdellovibrionaceae (Deltaproteobacteria), Cytophagaceae (Cytophagia), Chitinophagaceae (Saprospirae) were significantly enriched in DXAL (Figure 7D).
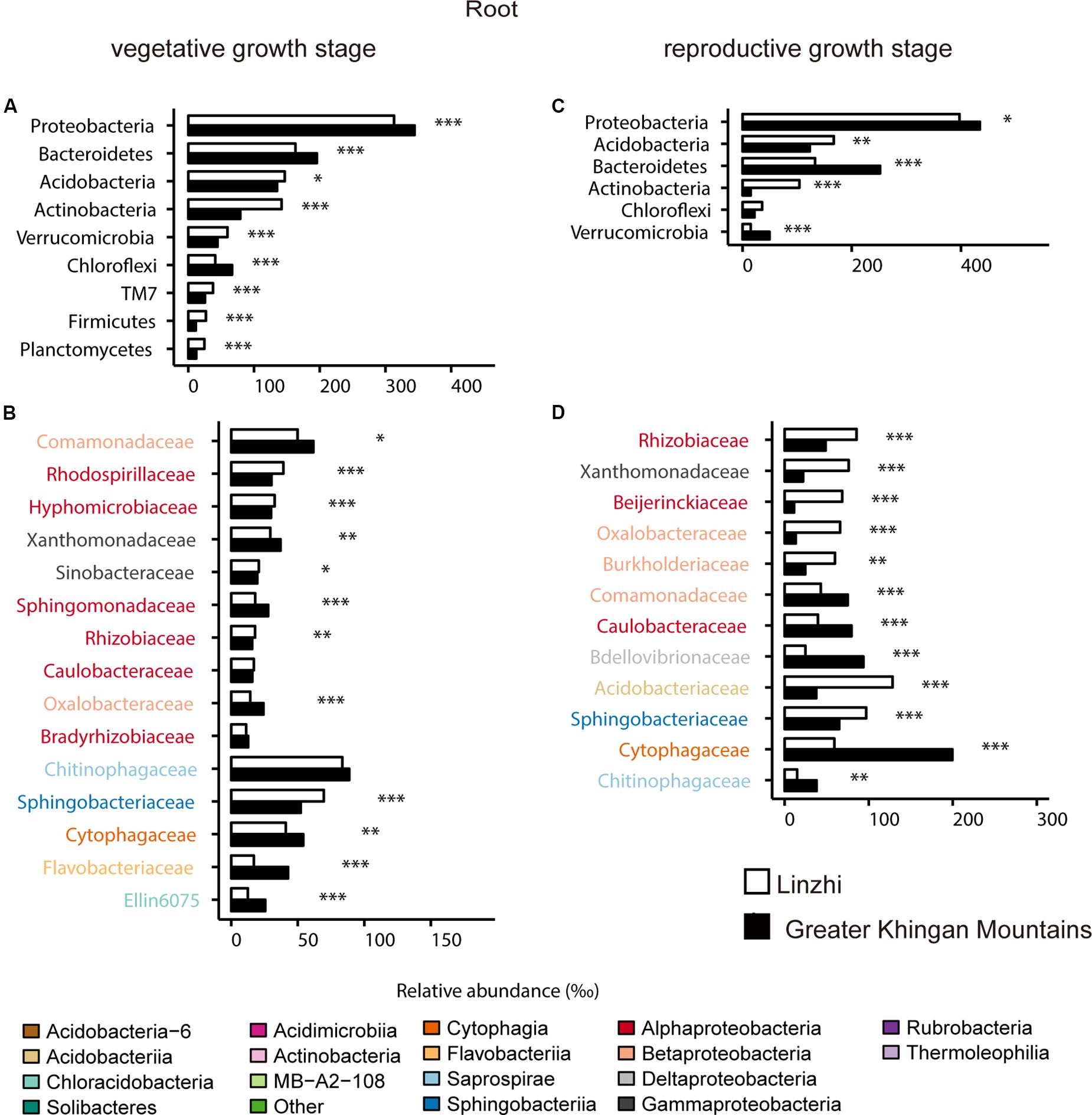
Figure 7. Comparative analyses of bacterial core microbiome across different sites. (A) Relative abundance (‰) of the phyla detected in root core OTUs of the indicated plant growth sites at vegetative growth stage. (B) Relative abundance (‰) of families belonging to the three dominant phyla in the root core OTUs of the indicated plant growth sites at vegetative growth stage. (C) Relative abundance (‰) of the phyla detected in root core OTUs of the indicated plant growth sites at reproductive growth stage. (D) Relative abundance (‰) of families belonging to the three dominant phyla in the root core OTUs of the indicated plant growth sites at reproductive growth stage. Asterisks indicate significant differences [Benjamini–Hochberg false discovery-rate (FDR) adjusted P-value, ∗ represent < 0.05, ∗∗ represent < 0.01, ∗∗∗ represent < 0.001].
Likewise, for the soil samples, the difference of bacterial community composition between two sites was shown in Supplementary Figure S2 and Supplementary Table S12. At vegetative growth stage, the dominant phyla were Proteobacteria, Actinobacteria, and Acidobacteria (Supplementary Figure S2A). Rhodospirillaceae (Alphaproteobacteria), Sinobacteraceae (Gammaproteobacteria), Gaiellaceae (Thermoleophilia), EB1017 and C111 (Acidimicrobiia), RB40 (Acidobacteria-6) were significantly enriched in LZ, whereas Syntrophobacteraceae and Bdellovibrionaceae (Deltaproteobacteria), Sphingomonadaceae (Alphaproteobacteria), Ellin6075 (Chloracidobacteria) were over-present in DXAL (Supplementary Figure S2B); As for the reproductive growth stage, the different phyla was similar with vegetative growth stage (Supplementary Figure S2C), at the family level, the significantly different families were less, Sinobacteraceae and Coxiellaceae (Gammaproteobacteria), Koribacteraceae (Acidobacteriia), Solibacteraceae (Solibacteres), RB40 (Acidobacteria-6), EB1017 (Acidimicrobiia), Nocardioidaceae (Actinobacteria) were dominant families in LZ soil bacterial core OTUs, only Gaiellaceae (Thermoleophilia) was significantly more enriched in DXAL (Supplementary Figure S2D). These results showed microbial composition differed between host biogeography, and the common fungal and bacterial taxa were seen among each location that were roughly in line with other plants (Ottesen et al., 2013; Pinto et al., 2014; Peay et al., 2016).
The previous study revealed that the root-associated microbiomes has been proven to be associated with the plant developmental stages (Lundberg et al., 2012; Skaltsas et al., 2019). To investigate the relationship between growth stage and the root-associated microbiome, two growth stages in two different growing regions were tested and compared. The difference in community composition of fungi at the class level in the root and soil from two sites under two growth stages are shown in Figure 8 and Supplementary Table S13. In LZ, distribution of taxonomic classes between vegetative growth stage and reproductive growth stage were overall similar: Leotiomycetes and Agaricomycetes make up the majority of the plant microbiota. The distribution of Agaricomycetes, Eurotiomycetes, Mortierellomycetes, and Pezizomycetes significantly showed a decreasing trend from vegetative growth stage to reproductive growth stage among root samples, only Orbiliomycetes was showed an increasing trend (Figure 8A). Eurotiomycetes, Sordariomycetes, Dothideomycetes, and Tremellomycetes were enriched at vegetative growth stage in the soil compartment, while Agaricomycetes, Glomeromycetes, and Mortierellomycetes were enriched at reproductive growth stage (Figure 8B). As for DXAL, Sordariomycetes and Mortierellomycetes were over-present in vegetative growth stage in the root compartment, whereas Leotiomycetes, Dothideomycetes, and Eurotiomycetes were over-present in reproductive growth stage (Figure 8C). Sordariomycetes and Tremellomycetes were more dominant in vegetative growth stage in the soil compartment, whereas Leotiomycetes, Agaricomycetes, Eurotiomycetes, Dothideomycetes, and Mortierellomycetes were highly persistent in reproductive growth stage (Figure 8D).
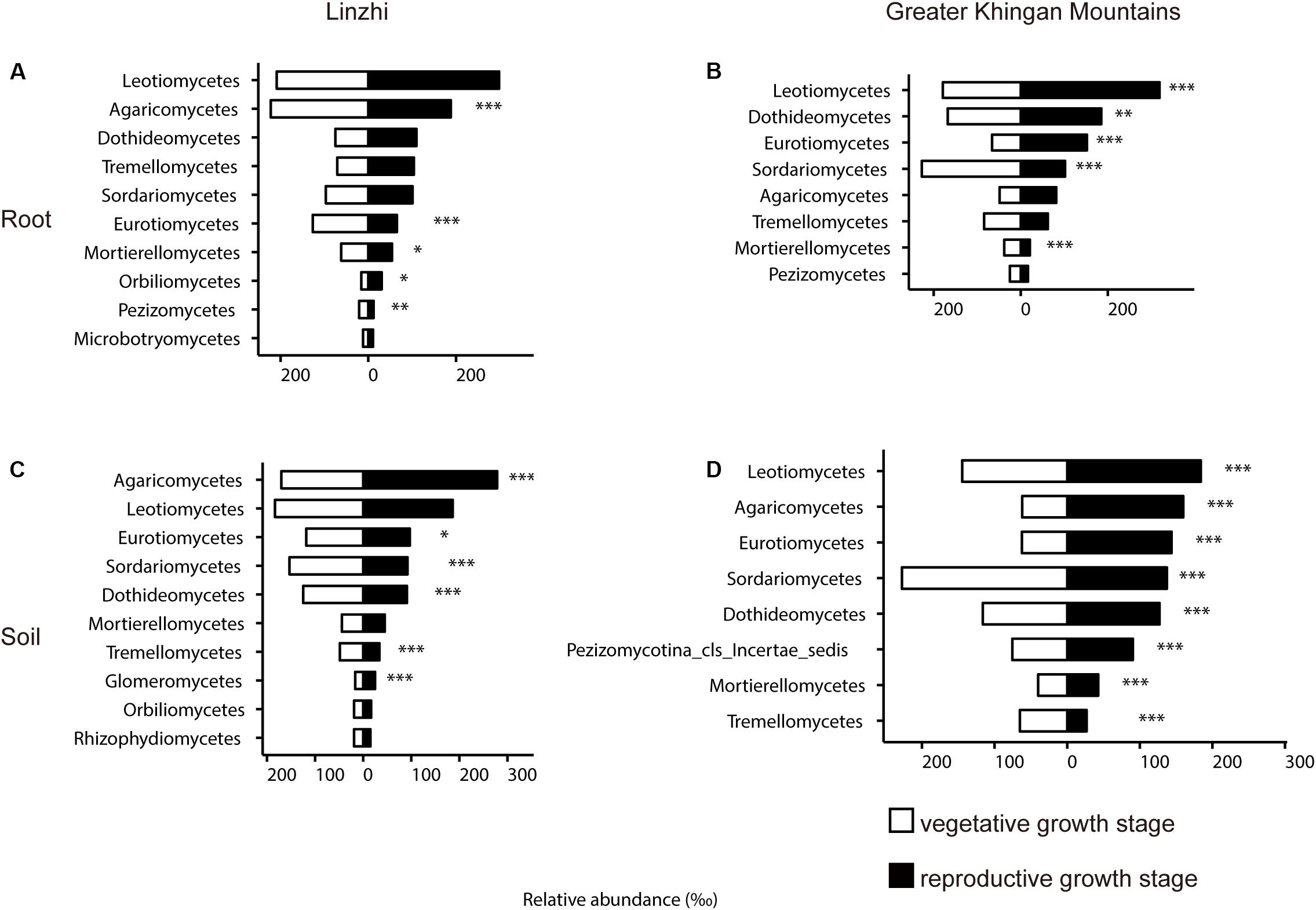
Figure 8. Side-by-side comparison of the relative abundance (‰) of classes which made up at least > 10‰ of the total fungal core microbiome community across different developmental stages. (A) Relative abundance (‰) of the classes detected in root core OTUs of the indicated developmental stages in Linzhi. (B) Relative abundance (‰) of the classes detected in soil core OTUs of the indicated plant growth sites in Linzhi. (C) Relative abundance (‰) of the classes detected in root core OTUs of the indicated plant growth sites in Greater Khingan Mountains. (D) Relative abundance (‰) of the classes detected in soil core OTUs of the indicated plant growth sites in Greater Khingan Mountains. Asterisks indicate significant differences occur between vegetative growth stage and reproductive growth stage [Benjamini–Hochberg false discovery-rate (FDR) adjusted P-value, ∗ represent < 0.05, ∗∗ represent < 0.01, ∗∗∗ represent < 0.001] in each compartment.
For the difference of bacterial community composition between growth stage at two sites was shown in Figure 9, Supplementary Figure S3 and Supplementary Table S14, the differential core OTUs involved phyla are almost similar at two sites in the root compartment, such as Proteobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi and TM7, except Bacteroidetes and Firmicutes in LZ, and Gemmatimonadetes in DXAL (Figures 9A,C). These dominate phyla were further analyzed at the family level. At root samples, whether in LZ or DXAL, relative abundance of most families was higher in reproductive growth stage, for example, Rhizobiaceae, Rhodospirillaceae and Caulobacteraceae (Alphaproteobacteria), Xanthomonadaceae and Sinobacteraceae (Gammaproteobacteria), Comamonadaceae (Betaproteobacteria), Sphingobacteriaceae (Sphingobacteriia), at the vegetative growth stage, Hyphomicrobiaceae (Alphaproteobacteria), Ellin6075 (Chloracidobacteria), Chitinophagaceae (Saprospirae), Cytophagaceae (Cytophagia) were more abundant in LZ (Figure 9B), and Flavobacteriaceae (Flavobacteriia), Ellin6075 (Chloracidobacteria) were more prevent in DXAL (Figure 9D). For the soil samples, the differential core OTUs involved phyla were similar like root samples, but the significantly enriched families in three dominant phyla were different from root. Whether in LZ or DXAL, the relative abundance of most families was higher in the vegetative growth stage (Supplementary Figure S3 and Supplementary Table S14). Consistent with previous observations, microbial composition differed between different development stages, it suggested that the overall recruitment of OTUs is governed by a set of factors that include developmental stage.
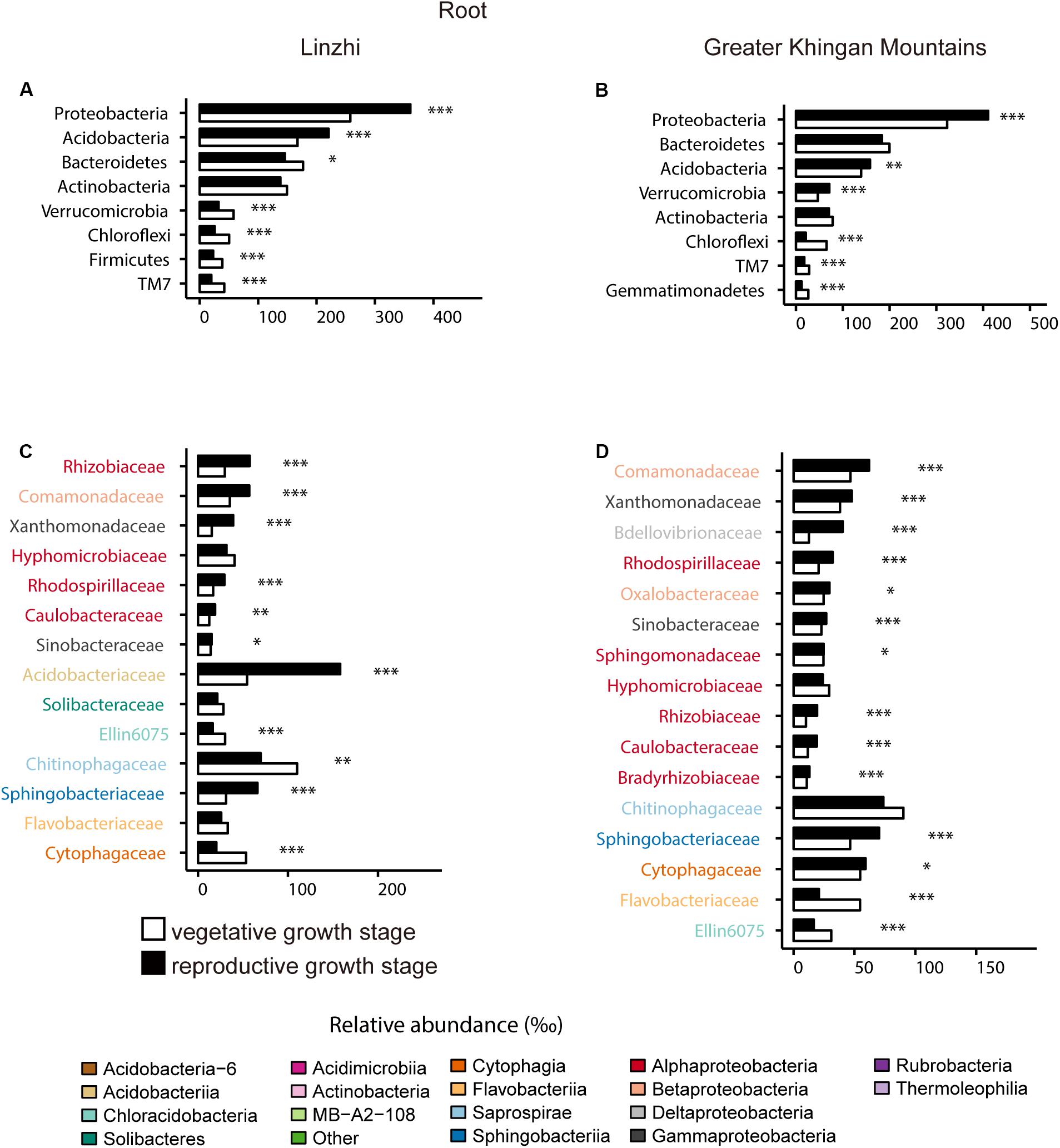
Figure 9. Comparative analyses of bacterial core microbiome across different developmental stages. (A) Relative abundance (‰) of the phyla detected in root core OTUs of the indicated developmental stages in Linzhi. (B) Relative abundance (‰) of families belonging to the three dominant phyla in the root core OTUs of the indicated developmental stages in Linzhi. (C) Relative abundance (‰) of the phyla detected in root core OTUs of the indicated plant growth sites in Greater Khingan Mountains. (D) Relative abundance (‰) of families belonging to the three dominant phyla in the root core OTUs of the indicated plant growth sites in Greater Khingan Mountains. Asterisks indicate significant differences [Benjamini–Hochberg false discovery-rate (FDR) adjusted P-value, ∗ represent < 0.05, ∗∗ represent < 0.01, ∗∗∗ represent < 0.001].
In the present study, we performed geographical location and developmental stage investigation on the taxonomic characters of G. conopsea root-associated microbiomes to gain deeper insight into plant driven taxa and their features in each habitat. Earlier researches on root microbiome have mainly focused on other orchids (Leake and Cameron, 2012; Selosse and Martos, 2014; Xing et al., 2015; Fochi et al., 2017; Voyron et al., 2017). Due to the decreased cost of amplicon-based sequencing, it makes the studies of microbiomes in large-scale and global possible. Recently, Chen and colleagues analyzed the root and rhizosphere microbiomes in wheat by combining the effect of developmental stages and nitrogen fertilization using a amplicon sequencing approach, their results implied that both developmental stages and nitrogen input affect the structure of root microbial community in wheat (Chen S. et al., 2019). Here, we analyzed the factors driving changes in the composition of root-associated microbial community structure using amplicon sequencing approach. This is the first time that simultaneously examine the root-associated microbiomes that includes both fungi and bacteria in G. conopsea.
As revealed by the alpha-diversity index, there was a significant difference in species diversity between corresponding soil and root in G. conopsea, it is consistent with the universal reports of microbial colonization (Beckers et al., 2017; Duran et al., 2018). Soil serves as one of the richest microbial ecosystems on Earth, providing ideal habitats for various microbial lineages (Wei and Ashman, 2018). The species diversity of plant compartments (root) was significantly lower, mainly owing to that, the colonization pattern and formation of a stable microbiome within root were more complicated than expected, such as the response of plant’s the innate immune system to microbial colonization. The decrease of species diversity and richness from soil to plant root showed that only a small part of fungal/bacterial microbes could maintain symbiotic associations with its host plant and ultimately form dominant microbial assemblages in root.
According to the PCoA analysis, notably variation in structure of fungal/bacterial communities was further uncovered regarding compartment, plant geographical location and developmental stage. The results revealed that the most important factor determining fungal/bacterial microbiome dynamics in G. conopsea were the geographical locations of plant. This pattern was in agreement with PERMANOVA results according to UniFrac distances. Our result was consistent with the findings reported by Duran et al. (2018), in which the host biogeography was the key factor shaping the composition of root fungal community. It was also in accord with previous study that a general increase in differentiation with increasing geographical distance in G. conopse-associated fungal communities (Stark et al., 2009; Xing et al., 2020). However, the result of bacterial microbiome was different from the study of other plant, such as rice (Edwards et al., 2015), Arabidopsis (Duran et al., 2018), millet (Jin et al., 2017), Cycas panzhihuaensis (Zheng and Gong, 2019), which suggested that the compartment was the main factor shaping the composition of the bacterial microbiome. It may relate to the growth characteristics of orchids, which fully or partially relies on mycorrhizal fungi for requisite nutrients; meanwhile, some bacteria, such as the mycorrhiza helper bacteria (MHB) were found to be commonly occurring in ectomycorrhiza and in arbuscular mycorriza associations, which assist mycorrhiza formation (Frey-Klett and Garbaye, 2005; Faria et al., 2013; Esposito-Polesi et al., 2017; Li et al., 2017). Nonetheless, very little is known about the composition, structure, and functional activity of bacterial microbiome associated with orchid, let alone the information in G. conopsea. Thus, the major factors affecting the composition of bacterial community in G. conopsea requires further study. In general, these results indicated that G. conopse are not specialists and have a significant amount of geographical variance in their fungal/bacterial communities.
In our study, the samples were derived from two different altitude places approximately 5,466 kilometers away. LZ is in the southern part of Tibet, the altitude is 3600 m, and has a semi-humid tropical climate, whereas DXAL belongs to the north of Heilongjiang province, the altitude is 496 m and has a cold temperate continental monsoon climate. The variation of altitude and climate conditions leaded to the dissimilarity of local microbiota, finally giving rise to the distinct and highly diverse root-associated fungal/bacterial microbiomes. LZ belongs to the region of the Qinghai-Tibet Plateau, which is also characterized with low oxygen, reduced pathogen incidence, low temperature, and high levels of UV radiation (Qiao et al., 2016a, b). For plants, the plateau environment of high altitude, low pressure, low oxygen, strong radiation is undoubtedly harsh plant growth. Due to its unique natural and geographical environment, the microbial composition of G. conopsea in LZ is of great value. As shown in the results, Leotiomycetes, Agaricomycetes, Eurotiomycetes, and Mortierellomycetes were more abundant in LZ core fungal OTUs than DXAL in both root and soil samples at the vegetative growth stage, at family level, it includes Helotiaceae, Dermateaceae, Vibrisseaceae, Myxotrichaceae and Herpotrichiellaceae, which belongs to Dark septate endophytes (DSEs). DSEs belong to Ascomycota and widely distributed group of fungal root colonizers (Rodriguez et al., 2009). Previous researches revealed that DSEs can promote plant growth, possibly through nitrogen mineralization and absorption, and protection from pathogens (Mullen et al., 1998; Mandyam and Jumpponen, 2005; Newsham, 2011), DSEs have also been shown to positively influence plant drought resistance via enhancing nutrient and water uptake, plant growth, and/or improving plant resistance to oxidative stress (Perez-Naranjo, 2009; Santos et al., 2017). Many studies have shown that the plants that can live in the plateau are closely related to DSEs, which can enhance the growth of host plants and absorption of nutrients such as phosphorus (Jumpponen et al., 1998; Jumpponen, 2001). Arbuscular mycorrhiza (AM) is one member of an ancient and common mutualistic symbiosis, formed between Glomeromycota fungi and many plants, in which fungi supply plants of nutrients obtained from soil in exchange for lipids and carbohydrates (Delaux et al., 2015; Rich et al., 2017). In our results, Ambisporaceae, Archaeosporaceae, Acaulosporaceae, Gigasporaceae, Glomeraceae and Paraglomeraceae significantly enriched in LZ soil samples, most of these fungi are barely found in DXAL. In addition, Clavariaceae, Russulaceae, Leotiaceae and Dictyosporiaceae were more dominate in LZ root samples, whereas these families were rare in DXAL root samples. It indicated that these fungal taxa may play certain roles in adapting to specific environments in LZ. To summarize, these results implied that it might exist a different pool of fungal taxa in each site and the plant organized its microbiomes from these pools. However, although the microbial composition varied greatly from LZ to DXAL, the dominant taxa were similar, and just the relative abundance of taxa was different, implying that the symbiotic microorganisms of G. conopsea still have certain specificity.
As for the bacterial communities, Acidobacteria and Actinobacteria were significantly abundant in LZ root samples at two developmental stages, while Actinobacteria, Planctomycetes and TM7 were enriched in LZ soil samples at two developmental stages. Actinobacteria were enriched in LZ from both root and soil samples compared with DXAL. Actinobacteria belongs to gram-positive saprophytic organisms and play an important role in plant development (Verma et al., 2009). Moreover, Actinobacteria have strong colonization ability and can live in a variety of soil, and spore formation which allows them to be able to survive longer at some extreme environments, such as irradiation, and drought (Qin et al., 2011; Yandigeri et al., 2012; Palaniyandi et al., 2013). Acidobacteria are abundant under various conditions with participation in sulfur cycling (Sanchez-Andrea et al., 2011; Serkebaeva et al., 2013; Urbanova and Barta, 2014), have the property that functionality of Acidobacterial dissimilatory sulfur pathways and degradation of cellulose produces acetic acid and hydrogen under anoxic condition (Pankratov et al., 2012; Hausmann et al., 2018). This result was consistent with the characteristics of Acidobacteria and the special environment of LZ.
In addition to fungi, the roots of G. conopsea in wild also harbor a wide variety of bacteria, dominated by Proteobacteria, Bacteroidetes, Acidobacteria, Actinobacteria. The structure of root-associated microbiomes was significantly affected by geographical location in G. conopsea. Nevertheless, some taxa were observed in both locations, implying that these microbiomes have a widespread distributions that are not completely restricted to environmental conditions. Besides, the developmental stage and compartment were also factors contributing to microbiome variation. The richness of both fungal and bacterial communities was lower at reproductive growth stage than vegetative growth stage in the root samples from two locations, indicating that this species may be dependent on interaction with more fungi or bacteria during this early growth period. However, further investigations are needed to assess the variation in microbial community composition of root in G. conopsea, including samples from multiple different growth locations and more growth stages. Besides, we eagerly anticipate future studies that integrating multiple factors to explore the differentiation of microbial community composition in G. conopsea, such as variability on the genetic background and ploidy level. Overall, our study offers new clues to understand the microbiomes associated with two different locations and growth stages of an orchid species. Hopefully, these efforts could lay a basis for future microbiome-assisted G. conopsea cultivation.
All raw sequencing reads were deposited in the NCBI SRA database with the accession numbers PRJNA598558 (ITS data) and PRJNA598739 (16S data).
ZM conceived and supervised the project. ML collected samples and designed the experiment and wrote and revised the manuscript. ML and LL analyzed the data. HX, XX, and ZZ helped with sample treatment. LL provided comments and suggests on the results.
This research was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” of China (No. 2017ZX09301060), National Key Research and Development Program of China (No. 2018YFC1708004), Leading Talent Project of National Ethnic Affairs Commission of China (No. MZR20001), and the Fundamental Research Funds for the Central Universities of China (No. CZJ19001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge Weiwu Chen, Jiumei Ciwang, and Qunfang Wan for their help with sampling. We thank friends at BGI for discussions. This manuscript has been released as a pre-print at Research Square (Lin et al., 2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01257/full#supplementary-material
Arditti, J., and Ghani, A. K. A. (2000). Numerical and physical properties of orchid seeds and their biological implications. New Phytol. 145, 367–421. doi: 10.1046/j.1469-8137.2000.00587.x
Bai, Y., Muller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Beckers, B., Op, D. B. M., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5:25. doi: 10.1186/s40168-017-0241-2
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berendsen, R. L., Vismans, G., Yu, K., Song, Y., et al. (2018). Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 12, 1496–1507. doi: 10.1038/s41396-018-0093-1
Chen, S., Waghmode, T. R., Sun, R., Kuramae, E. E., Hu, C., and Liu, B. (2019). Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization. Microbiome 7:136. doi: 10.1186/s40168-019-0750-2
Chen, Y., Gao, Y., Song, L., Zhao, Z., Guo, S., and Xing, X. (2019). Mycorrhizal fungal community composition in seven orchid species inhabiting song mountain, beijing, china. Sci. China Life Sci. 62, 838–847. doi: 10.1007/s11427-018-9471-x
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., et al. (2018). The populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8
Curtis, J. T. (1939). The relation of specificity of orchid mycorrhizal fungi to the problem of symbiosis. Am. J. Bot. 26, 390–399. doi: 10.1002/j.1537-2197.1939.tb09292.x
Delaux, P. M., Radhakrishnan, G. V., Jayaraman, D., Cheema, J., et al. (2015). Algal ancestor of land plants was preadapted for symbiosis. Proc. Natl. Acad. Sci. U.S.A. 112, 13390–13395. doi: 10.1073/pnas.1515426112
Duran, P., Thiergart, T., Garrido-Oter, R., Agler, M., et al. (2018). Microbial interkingdom interactions in roots promote arabidopsis survival. Cell 175, 973–983. doi: 10.1016/j.cell.2018.10.020
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than blast. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate otu sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). Uchime improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Edwards, J., Johnson, C., Santos-Medellin, C., Lurie, E., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Esposito-Polesi, N. P., de Abreu-Tarazi, M. F., de Almeida, C. V., Tsai, S. M., and de Almeida, M. (2017). Investigation of endophytic bacterial community in supposedly axenic cultures of pineapple and orchids with evidence on abundant intracellular bacteria. Curr. Microbiol. 74, 103–113. doi: 10.1007/s00284-016-1163-0
Ettema, C. H., and Wardle, D. A. (2002). Spatial soil ecology. Trends Ecol. Evol. 17, 177–183. doi: 10.1016/s0169-5347(02)02496-5
Fadrosh, D. W., Ma, B., Gajer, P., Sengamalay, N., et al. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6
Faria, D. C., Dias, A. C., Melo, I. S., and de Carvalho, C. F. (2013). Endophytic bacteria isolated from orchid and their potential to promote plant growth. World J. Microbiol. Biotechnol. 29, 217–221. doi: 10.1007/s11274-012-1173-4
Fochi, V., Chitarra, W., Kohler, A., Voyron, S., et al. (2017). Fungal and plant gene expression in the tulasnella calospora-serapias vomeracea symbiosis provides clues about nitrogen pathways in orchid mycorrhizas. New Phytol. 213, 365–379. doi: 10.1111/nph.14279
Frey-Klett, P., and Garbaye, J. (2005). Mycorrhiza helper bacteria: a promising model for the genomic analysis of fungal-bacterial interactions. New Phytol. 168, 4–8. doi: 10.1111/j.1469-8137.2005.01553.x
Gao, Y., Zhao, Z., Li, J., Liu, N., Jacquemyn, H., Guo, S., et al. (2020). Do fungal associates of co-occurring orchids promote seed germination of the widespread orchid species Gymnadenia conopsea? Mycorrhiza 30, 221–228. doi: 10.1007/s00572-020-00943-1
Hausmann, B., Pelikan, C., Herbold, C. W., Kostlbacher, S., Albertsen, M., Eichorst, S. A., et al. (2018). Peatland acidobacteria with a dissimilatory sulfur metabolism. ISME J. 12, 1729–1742. doi: 10.1038/s41396-018-0077-1
Hultén, E., and Fries, M. (1986). Atlas of North European Vascular Plants (North of the Tropic of Cancer). Königstein: Koeltz Scientific Books.
Jacquemyn, H., Brys, R., Lievens, B., and Wiegand, T. (2012). Spatial variation in below-ground seed germination and divergent mycorrhizal associations correlate with spatial segregation of three co-occurring orchid species. J. Ecol. 100, 1328–1337. doi: 10.1111/j.1365-2745.2012.01998.x
Jacquemyn, H., Waud, M., Brys, R., Lallemand, F., Courty, P. E., Robionek, A., et al. (2017). Mycorrhizal associations and trophic modes in coexisting orchids: an ecological continuum between auto- and mixotrophy. Front. Plant Sci. 8:1497. doi: 10.3389/fpls.2017.01497
Jin, T., Wang, Y., Huang, Y., Xu, J., Zhang, P., Wang, N., et al. (2017). Taxonomic structure and functional association of foxtail millet root microbiome. Gigascience 6:gix089. doi: 10.1093/gigascience/gix089
Jumpponen, A. (2001). Dark septate endophytes - are they mycorrhizal? Mycorrhiza 11, 207–211. doi: 10.1007/s005720100112
Jumpponen, A., Mattson, K. G., and Trappe, J. M. (1998). Mycorrhizal functioning of Phialocephala fortinii with Pinus contorta on glacier forefront soil: interactions with soil nitrogen and organic matter. Mycorrhiza 7, 261–265. doi: 10.1007/s005720050190
Kristiansen, K. A., Taylor, D. L., Kjoller, R., Rasmussen, H. N., and Rosendahl, S. (2001). Identification of mycorrhizal fungi from single pelotons of Dactylorhiza majalis (orchidaceae) using single-strand conformation polymorphism and mitochondrial ribosomal large subunit DNA sequences. Mol. Ecol. 10, 2089–2093. doi: 10.1046/j.0962-1083.2001.01324.x
Leake, J. R., and Cameron, D. D. (2012). Untangling above- and belowground mycorrhizal fungal networks in tropical orchids. Mol. Ecol. 21, 4921–4924. doi: 10.1111/j.1365-294X.2012.05718.x
Li, O., Xiao, R., Sun, L., Guan, C., Kong, D., and Hu, X. (2017). Bacterial and diazotrophic diversities of endophytes in dendrobium Catenatum determined through barcoded pyrosequencing. PLoS One 12:e184717. doi: 10.1371/journal.pone.0184717
Lin, M., Xiong, H., Xiang, X., Zhou, Z., Liang, L., and Mei, Z. (2020). Root-associated microbiomes of Gymnadenia conopsea under the combined effect of plant geographical location and developmental stage. Res. Square [Preprint]. doi: 10.21203/rs.2.21631/v1
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Magoc, T., and Salzberg, S. L. (2011). Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mandyam, K., and Jumpponen, A. (2005). Seeking the elusive function of the root-colonising dark septate endophytic fungi. Stud. Mycol. 53, 173–189. doi: 10.3114/sim.53.1.173
Masuhara, G., and Katsuya, K. (1994). In situ and in vitro specificity between Rhizoctonia spp. and Spiranthes sinensis (Persoon) Ames, var. amoena (M. Bieberstein) Hara (Orchidaceae). New Phytol. 127, 711–718. doi: 10.1111/j.1469-8137.1994.tb02974.x
McCormick, M. K., and Jacquemyn, H. (2014). What constrains the distribution of orchid populations? New Phytol. 202, 392–400. doi: 10.1111/nph.12639
McCormick, M. K., Whigham, D. F., and Canchani-Viruet, A. (2018). Mycorrhizal fungi affect orchid distribution and population dynamics. New Phytol. 219, 1207–1215. doi: 10.1111/nph.15223
Mullen, R. B., Schmidt, S. K., and Jaeger, C. H. (1998). Nitrogen uptake during snowmelt by the snow buttercup, Ranunculus adoneus. Arct. Alp. Res. 30, 121–125.
Newsham, K. K. (2011). A meta-analysis of plant responses to dark septate root endophytes. New Phytol. 190, 783–793. doi: 10.1111/j.1469-8137.2010.03611.x
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Ottesen, A. R., Gonzalez, P. A., White, J. R., Pettengill, J. B., Li, C., Allard, S., et al. (2013). Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol. 13:114. doi: 10.1186/1471-2180-13-114
Palaniyandi, S. A., Yang, S. H., Zhang, L., and Suh, J. W. (2013). Effects of actinobacteria on plant disease suppression and growth promotion. Appl. Microbiol. Biotechnol. 97, 9621–9636. doi: 10.1007/s00253-013-5206-1
Pankratov, T. A., Kirsanova, L. A., Kaparullina, E. N., Kevbrin, V. V., and Dedysh, S. N. (2012). Telmatobacter bradus gen. Nov., Sp. Nov., A cellulolytic facultative anaerobe from subdivision 1 of the Acidobacteria, and emended description of Acidobacterium capsulatum kishimoto et al. 1991. Int. J. Syst. Evol. Microbiol. 62, 430–437. doi: 10.1099/ijs.0.029629-0
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Peay, K. G., Kennedy, P. G., and Talbot, J. M. (2016). Dimensions of biodiversity in the earth mycobiome. Nat. Rev. Microbiol. 14, 434–447. doi: 10.1038/nrmicro.2016.59
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Perez-Naranjo, J. C. (2009). Dark Septate and Arbuscular Mycorrhizal Fungal Endophytes in Roots of Prairie Grasses. Ph.D. dissertation, The University of Saskatchewan, Saskatoon.
Perkins, A., Masuhara, G., and McGee, P. A. (1995). Specificity of the associations between Microtis parviflora (Orchidaceae) and its mycorrhizal fungi. Aust. J. Bot. 43, 85–91.
Pinto, C., Pinho, D., Sousa, S., Pinheiro, M., Egas, C., and Gomes, A. C. (2014). Unravelling the diversity of grapevine microbiome. PLoS One 9:e85622. doi: 10.1371/journal.pone.0085622
Qiao, Q., Huang, Y., Qi, J., Qu, M., Jiang, C., Lin, P., et al. (2016a). The genome and transcriptome of Trichormus sp. NMC-1: insights into adaptation to extreme environments on the Qinghai-Tibet Plateau. Sci. Rep. 6:29404. doi: 10.1038/srep29404
Qiao, Q., Wang, Q., Han, X., Guan, Y., Sun, H., Zhong, Y., et al. (2016b). Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci. Rep. 6:21729. doi: 10.1038/srep21729
Qin, S., Xing, K., Jiang, J. H., Xu, L. H., and Li, W. J. (2011). Biodiversity, bioactive natural products and biotechnological potential of plant-associated endophytic actinobacteria. Appl. Microbiol. Biotechnol. 89, 457–473. doi: 10.1007/s00253-010-2923-6
Qin, Y., Druzhinina, I. S., Pan, X., and Yuan, Z. (2016). Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 34, 1245–1259. doi: 10.1016/j.biotechadv.2016.08.005
Rasmussen, H. N., Dixon, K. W., Jersakova, J., and Tesitelova, T. (2015). Germination and seedling establishment in orchids: a complex of requirements. Ann. Bot. 116, 391–402. doi: 10.1093/aob/mcv087
Rich, M. K., Nouri, E., Courty, P., and Reinhardt, D. (2017). Diet of arbuscular mycorrhizal fungi: Bread and butter? Trends Plant Sci. 22, 652–660. doi: 10.1016/j.tplants.2017.05.008
Rodriguez, R. J., White, J. J., Arnold, A. E., and Redman, R. S. (2009). Fungal endophytes: diversity and functional roles. New Phytol. 182, 314–330. doi: 10.1111/j.1469-8137.2009.02773.x
Sanchez-Andrea, I., Rodriguez, N., Amils, R., and Sanz, J. L. (2011). Microbial diversity in anaerobic sediments at Río Tinto, a naturally acidic environment with a high heavy metal content. Appl. Environ. Microbiol. 77, 6085–6093. doi: 10.1128/AEM.00654-11
Santos, S., Silva, P., Garcia, A. C., Zilli, J. E., and Berbara, R. (2017). Dark septate endophyte decreases stress on rice plants. Braz. J. Microbiol. 48, 333–341. doi: 10.1016/j.bjm.2016.09.018
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Selosse, M. A., and Martos, F. (2014). Do chlorophyllous orchids heterotrophically use mycorrhizal fungal carbon? Trends Plant Sci. 19, 683–685. doi: 10.1016/j.tplants.2014.09.005
Serkebaeva, Y. M., Kim, Y., Liesack, W., and Dedysh, S. N. (2013). Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS One 8:e63994. doi: 10.1371/journal.pone.0063994
Sessitsch, A., Hardoim, P., Doring, J., Weilharter, A., Krause, A., Woyke, T., et al. (2012). Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol. Plant Microbe Interact. 25, 28–36. doi: 10.1094/MPMI-08-11-0204
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Skaltsas, D. N., Badotti, F., Vaz, A., Silva, F., Gazis, R., Wurdack, K., et al. (2019). Exploration of stem endophytic communities revealed developmental stage as one of the drivers of fungal endophytic community assemblages in two Amazonian hardwood genera. Sci. Rep. 9:12685. doi: 10.1038/s41598-019-48943-2
Stark, C., Babik, W., and Durka, W. (2009). Fungi from the roots of the common terrestrial orchid Gymnadenia conopsea. Mycol. Res. 113, 952–959. doi: 10.1016/j.mycres.2009.05.002
Taylor, D. L., and McCormick, M. K. (2008). Internal transcribed spacer primers and sequences for improved characterization of basidiomycetous orchid mycorrhizas. New Phytol. 177, 1020–1033. doi: 10.1111/j.1469-8137.2007.02320.x
Tesitelova, T., Jersakova, J., Roy, M., Kubatova, B., Tesitel, J., Urfus, T., et al. (2013). Ploidy-specific symbiotic interactions: divergence of mycorrhizal fungi between cytotypes of the Gymnadenia conopsea group (Orchidaceae). New Phytol. 199, 1022–1033. doi: 10.1111/nph.12348
Tsavkelova, E. A., Cherdyntseva, T. A., Botina, S. G., and Netrusov, A. I. (2007). Bacteria associated with orchid roots and microbial production of auxin. Microbiol. Res. 162, 69–76. doi: 10.1016/j.micres.2006.07.014
Turner, T. R., James, E. K., and Poole, P. S. (2013). The plant microbiome. Genome Biol. 14:209. doi: 10.1186/gb-2013-14-6-209
Urbanova, Z., and Barta, J. (2014). Microbial community composition and in silico predicted metabolic potential reflect biogeochemical gradients between distinct peatland types. FEMS Microbiol. Ecol. 90, 633–646. doi: 10.1111/1574-6941.12422
van der Heijden, M. G., de Bruin, S., Luckerhoff, L., van Logtestijn, R. S., and Schlaeppi, K. (2016). A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 10, 389–399. doi: 10.1038/ismej.2015.120
Verma, V. C., Gond, S. K., Kumar, A., Mishra, A., Kharwar, R. N., and Gange, A. C. (2009). Endophytic Actinomycetes from Azadirachta indica A. Juss.: isolation, diversity, and anti-microbial activity. Microb. Ecol. 57, 749–756. doi: 10.1007/s00248-008-9450-3
Voyron, S., Ercole, E., Ghignone, S., Perotto, S., and Girlanda, M. (2017). Fine-scale spatial distribution of orchid mycorrhizal fungi in the soil of host-rich grasslands. New Phytol. 213, 1428–1439. doi: 10.1111/nph.14286
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Waterman, R. J., Bidartondo, M. I., Stofberg, J., Combs, J. K., Gebauer, G., Savolainen, V., et al. (2011). The effects of above- and belowground mutualisms on orchid speciation and coexistence. Am. Nat. 177, E54–E68. doi: 10.1086/657955
Waud, M., Busschaert, P., Lievens, B., and Jacquemyn, H. (2016). Specificity and localised distribution of mycorrhizal fungi in the soil may contribute to co-existence of orchid species. Fungal Ecol. 20, 155–165. doi: 10.1016/j.funeco.2015.12.008
Wei, N., and Ashman, T. L. (2018). The effects of host species and sexual dimorphism differ among root, leaf and flower microbiomes of wild strawberries in situ. Sci. Rep. 8:5195. doi: 10.1038/s41598-018-23518-9
Xing, X., Gai, X., Liu, Q., Hart, M. M., and Guo, S. (2015). Mycorrhizal fungal diversity and community composition in a lithophytic and epiphytic orchid. Mycorrhiza 25, 289–296. doi: 10.1007/s00572-014-0612-5
Xing, X., Gao, Y., Zhao, Z., Waud, M., Duffy, K. J., Selosse, M.-A., et al. (2020). Similarity in mycorrhizal communities associating with two widespread terrestrial orchids decays with distance. J. Biogeogr. 47, 421–433. doi: 10.1111/jbi.13728
Xu, J., Zhang, Y., Zhang, P., Trivedi, P., Riera, N., Wang, Y., et al. (2018). The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9:4894. doi: 10.1038/s41467-018-07343-2
Yandigeri, M. S., Meena, K. K., Singh, D., Malviya, N., and Singh, D. P. (2012). Drought-tolerant endophytic actinobacteria promote growth of wheat (Triticum aestivum) under water stress conditions. Plant Growth Regul. 68, 411–420. doi: 10.1007/s10725-012-9730-2
Yuan, J., Zhao, J., Wen, T., Zhao, M., Li, R., Goossens, P., et al. (2018). Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome 6:156. doi: 10.1186/s40168-018-0537-x
Keywords: root-associated microbiomes, biogeography, plant compartment, plant growth stage, 16S rRNA gene, ITS2
Citation: Lin M, Xiong H, Xiang X, Zhou Z, Liang L and Mei Z (2020) The Effect of Plant Geographical Location and Developmental Stage on Root-Associated Microbiomes of Gymnadenia conopsea. Front. Microbiol. 11:1257. doi: 10.3389/fmicb.2020.01257
Received: 24 January 2020; Accepted: 18 May 2020;
Published: 18 June 2020.
Edited by:
Suha Jabaji, McGill University, CanadaReviewed by:
Yang Song, Utrecht University, NetherlandsCopyright © 2020 Lin, Xiong, Xiang, Zhou, Liang and Mei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifeng Liang, bGxmZ3oxOTkxQG91dGxvb2suY29t; Zhinan Mei, bWVpemhpbmFuQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.