 Ping Li
Ping Li Xuan Dong
Xuan Dong Xiao-yi Wang
Xiao-yi Wang Ting Du
Ting Du Xin-jun Du
Xin-jun Du Shuo Wang
Shuo Wang- 1State Key Laboratory of Food Nutrition and Safety, Tianjin University of Science and Technology, Tianjin, China
- 2Tianjin Key Laboratory of Food Science and Health, School of Medicine, Nankai University, Tianjin, China
Cronobacter sakazakii is foodborne pathogen that causes serious illnesses such as necrotizing enterocolitis, meningitis and septicemia in infants. However, the virulence determinants and mechanisms of pathogenicity of these species remain unclear. In this study, multilocus sequence typing (MLST) was performed on 34 C. sakazakii strains and two strains with the same sequence type (ST) but distinct adhesion/invasion capabilities were selected for identification of differentially expressed proteins using data-independent acquisition (DIA) proteomic analysis. A total of 2,203 proteins were identified and quantified. Among these proteins, 210 exhibited differential expression patterns with abundance ratios ≥3 or ≤0.33 and P values ≤0.05. Among these 210 proteins, 67 were expressed higher, and 143 were expressed lower in C. sakazakii SAKA80220 (strongly adhesive/invasive strain) compared with C. sakazakii SAKA80221 (weakly adhesive/invasive strain). Based on a detailed analysis of the differentially expressed proteins, the highly expressed genes involved in flagellar assembly, lipopolysaccharide synthesis, LuxS/AI-2, energy metabolic pathways and iron-sulfur cluster may be associated with the adhesion/invasion capability of C. sakazakii. To verify the accuracy of the proteomic results, real-time qPCR was used to analyze the expression patterns of some genes at the transcriptional level, and consistent results were observed. This study, for the first time, used DIA proteomic to investigate potential adhesion/invasion related factors as a useful reference for further studies on the pathogenic mechanism of C. sakazakii.
Introduction
Cronobacter species are rod-shaped, peritrichous, motile, Gram-negative, non-spore-forming facultative anaerobes that are widely present in nature (Iversen and Forsythe, 2003). The genus Cronobacter has been classified into seven species, namely, Cronobacter sakazakii, Cronobacter malonaticus, Cronobacter turicensis, Cronobacter condimenti, Cronobacter muytjensii, Cronobacter dublinensis, and Cronobacter universalis (Iversen et al., 2007, 2008; Joseph et al., 2012; Stephan et al., 2014). Genome analysis has shown that only C. sakazakii and C. malonaticus are associated with severe infections (Forsythe, 2018). Cronobacter spp. can cause necrotizing enterocolitis (NEC), sepsis and meningitis in newborns, low-birth-weight infants (<2.5 kg), immunocompromised newborns and infants under 4 weeks of age with a mortality rate of approximately 27% (Lai, 2001; Friedemann, 2009; Holy and Forsythe, 2014). Furthermore, Cronobacter species can also infect elderly and immunocompromised adults, leading to bacteremia, osteomyelitis, splenic abscess, pneumonia, conjunctivitis, wound infections, and urinary tract infections (Patrick et al., 2014; Blackwood and Hunter, 2016).
Given the adverse conditions associated with Cronobacter, in vitro studies utilizing human-derived cell lines as well as in vivo studies utilizing suckling mice or rats have been employed to explore the mechanism of adhesion/infection of Cronobacter spp. (Townsend et al., 2007; Giri et al., 2012; Quintero-Villegas et al., 2014). Several studies using gene knockouts have demonstrated that outer membrane protein A (OmpA), outer membrane protein X (OmpX) and outer membrane protease Cpa as well as the gene bcsR are crucial virulence factors of Cronobacter (Nair et al., 2009; Kim et al., 2010; Franco et al., 2011; Singh et al., 2015; Gao et al., 2017). Additionally, it has been demonstrated that enterotoxins, lipopolysaccharides (LPSs) and flagella are closely associated with the virulence of Cronobacter (Hotta et al., 1986; Pagotto et al., 2003; Jing et al., 2016). Although the above mentioned genes and factors are relevant for the virulence of Cronobacter, little is known regarding the functions and fundamental mechanisms of other virulence determinants in comparison with those of other major foodborne pathogens. Therefore, further studies on the pathogenesis of Cronobacter are necessary to understand these bacteria and to establish effective strategies for prevention and therapy.
Proteomic techniques are useful tools for investing the mechanisms underlying certain phenotypes or characteristics, such as virulence, desiccation tolerance, and osmotic tolerance (Jiang et al., 2007). Two-dimensional electrophoresis (2-DE) protein analysis couple with MALDI-TOF MS was used to investigate changes in the protein patterns of Enterobacter sakazakii cells in response to osmotic stress, and the result indicated that the genes rpoS and ompC played vital roles in osmotic shock (Riedel and Lehner, 2007). In our previous study, 2-DE was employed to identify virulence-related proteins. A total of 89 proteins were successfully identified and further analysis suggested that 11 of these proteins may be involved in the virulence of this pathogen (Du et al., 2015). Although 2-DE is a powerful proteomics approach for screening and identification of diagnostic markers and therapeutic targets in pathogenic microbes, there are also limitations associated with this technique, such as instability, complexity, and low sensitivity (Orsatti et al., 2009). Iso-baric tags for relative and absolute quantitation (iTRAQ) was used to study the proteome of C. sakazakii cells under dessication. The results indicated 233 differentially expressed proteins involved in trehalose and betaine uptake, which were associated with the ability of C. sakazakii ATCC 29544 to withstand osmotic stress (Hu et al., 2017). Nevertheless, LC-MS/MS coupled with iTRAQ labeling is susceptible to other reported peptide ions, whereas the sample matrix is complex (Minogue et al., 2015). Data-independent acquisition (DIA) is a focused quantitative method for protein mass spectrometry and can be applied to various quantitative proteomics experiments because of its accuracy and high efficiency (Sanda and Goldman, 2016). However, the DIA technique has not been used to screen adhesion/invasion related factors in C. sakazakii.
This study is the first time to investigate proteomic profiles of C. sakazakii using LC-MS/MS coupled with DIA labeling. The objective of this study is to investigate potential adhesion/invasion related factors as a useful reference for elucidation of the mechanism underlying adhesion to and invasion of the host cells by C. sakazakii. At the same time, the results presented herein will promote the understanding of the epidemiology of C. sakazakii.
Materials and Methods
Bacterial Strains and Sequence Typing
Based on the previous results from the adhesion/invasion assay, we selected 34 strains for sequence typing (Du et al., 2016). Based on the manufacturer’s instruction for the DNA Extraction Kit (Omega Bio-Tek, Norcross, GA, United States), genomic DNA from the 34 strains was obtained after overnight cultivation. Primers for seven housekeeping genes (atpD, fusA, glnS, gltB, gyrB, infB, and pps) were used for PCR (Baldwin et al., 2009), and the amplified products were sequenced with the Sanger chain termination method by Suzhou Jinweizhi Biotechnology Co., Ltd. (Suzhou, China). The sequencing results of the seven housekeeping genes were concatenated in a certain order to match with the PubMLST database and used to construct a phylogenetic tree by MEGA 6.0. The relationships among the 34 strains were analyzed via the neighbor-joining statistical method coupled with Tamura’s 3-parameter model (Joseph and Forsythe, 2012).
Bacterial Cell Preparation for Proteomic Analysis and Protein Extraction
Based on the sequence typing results, SAKA80220 and SAKA80221, which showed the same sequence type (ST) but distinct adhesion/invasion capabilities, were selected for proteomic analysis (Ren et al., 2016; Han et al., 2018). These strains, which were stored in LB with 80% glycerol at −80°C, were incubated on LB agar plates for 16 h at 37°C. A single colony was picked and incubated in 10 mL of LB broth at 37°C for 16 h with shaking at 180 rpm. Overnight cultures were transferred to fresh culture medium (diluted 1:100) and grew until the OD600 reached 0.4−0.6. The C. sakazakii cells were then harvested by centrifugation at 5,000 × g for 5 min and washed twice with sterile PBS and once with sterile water. After centrifugation at 5,000 × g for 2 min, the bacterial cells were stored at −80°C until use.
A modified phenol-based protein extraction method was used (Wu et al., 2014). Cell pellets were mixed with 6 mL of protein extraction buffer [500 mM Tris-HCl buffer (pH 8.0), 50 mM EDTA, 700 mM sucrose, 100 mM KCl, 2%β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, and HCl pH 8.0] and ground for 10 min using a precooled mortar on ice. An equal volume of saturated phenol was added, and the samples were thoroughly ground for another 10 min on ice. For better dissolution, the protein solution was ultrasonicated for 5 min at 4°C before centrifugation at 5,500 × g for 10 min. The protein-containing phenol solution was removed into a new ice-cold centrifuge tube, mixed with 6 mL of extraction buffer and centrifuged at 5,000 × g for 10 min at 4°C. The phenol phase was transferred to a new centrifuge tube with ice-cold acetone and stored at −20°C overnight. Next, the samples were centrifuged at 5,000 × g for 10 min at 4°C, and then, the protein pellets were cleaned twice with 2 mL of ice-cold acetone before centrifugation at 5,000 × g for 5 min at 4°C. Finally, the protein concentration was quantified using the Bradford method (Bradford, 1976).
Protein Digestion and Peptide Fractionation
A total of 100 μg of protein from each sample was digested using the FASP method (Wisniewski et al., 2009). Disulfide bonds were cleaved and the sites were blocked using 10 mM dithiothreitol (DTT) and 50 mM iodoacetamide (IAA), respectively. The proteins were then transferred to a 10-kDa filter and cleaned sequentially using 8 M urea and 50 mM NH4HCO3 at 12,000 × g and 20°C. Trypsin was added to each sample at a ratio of 1:50 (mass/mass), and the proteins were digested in 50 mM NH4HCO3 at 37°C for 16 h. A mixed sample was prepared with equal amount of digested peptides from each sample and separated into three fractions using a modified high-pH reversed-phase (High-pH RP) method (Dimayacyac-Esleta et al., 2015). Briefly, a homemade C18 stage tip was cleaned using 80% ACN/H2O after activation with methanol. Then, the stage tip was equilibrated with ammonium hydroxide (pH 10) before peptide loading. A series of ACN/ammonium hydroxide (pH 10) buffers – 6, 9, 12, 15, 18, 21, 25, 30, 35, and 50% – were utilized to elute the peptides.
Mass Spectrometry and Data Analysis
Data-Dependent Acquisition Sample Acquisition and Spectral Library Generation
For generation of the spectral library, peptides from each eluted sample were mixed acquired three times with data dependent acquisition mode using Q Exactive HF (Bremen, Thermo Fisher). The peptide mixtures were separated on an EasyNano LC1000 system (Thermo, San Jose, CA, United States) using a homemade C18 column (3 μm, 75 μm × 15 cm) at a flow rate of 300 nL/min. A 120-min linear gradient was set as follows: 5% B (0.1% FA in 80% acetonitrile/H2O)/95% A (0.1% FA in H2O) to 8% B in 10 min, 8% B to 25% B in 83 min, 25% B to 45% B in 21 min, 45% B to 95% B in 1 min, and maintaining at 95% B for 5 min. For data acquisition, a top 20 scan mode with an MS1 scan range of 400–1,200 m/z was employed, and the other parameters were set as follows: the MS1 and MS2 resolutions were set to 120 and 30 K, respectively; automatic gain control (AGC) target for MS1 and MS2 were set to 3e6 and 1e6, respectively; the isolation window was 2.0 Th; the normalized collision energy was 27 eV; and the dynamic exclusion time was 20 s (Lin et al., 2018).
Data-dependent acquisition (DDA) raw files were searched against the UniProt protein database for C. sakazakii using Proteome Discoverer 2.1 (Thermo, San Jose, CA, United States). The protein sequence was affixed with the iRT fusion protein sequence (Biognosys). The parameter of search engine SequestHT was set as the following: digestion: trypsin; miss cleavages: 2; variable modifications: oxidation (M), deamidated (N, Q); fixed modifications: carbamidomethyl (C); peptide mass tolerance: ± 10 ppm; fragment mass tolerance: ± 0.02 Da; peptide FDR: less than 1%; protein FDR: q value less than 1%. The search results were transferred into a spectral library using Spectronaut 10 (Biognosys, Schlieren, Switzerland). Only high-confidence peptides were used for the generation of the spectral library. Fragment ions within a mass range of 300–1,800 m/z was retained, and peptides with less than three fragment ions were removed.
Data-Independent Acquisition Sample Acquisition and Data Analysis
Each sample treated with the same amount of iRT was analyzed by the DIA method (Muntel et al., 2015). For DIA acquisition, the procedure consisted of one full MS1 scan with a resolution of 60 K using an AGC of 3e6 and a maximum injection time of 20 ms; MS2 scan with a resolution of 30 K using an AGC of 1e6 and a maximum injection time of 45 ms. All the LC conditions were exactly the same as those used for DDA, which are listed above. DIA raw data was processed using Spectronaut 10; the default, which includes: peak detection; dynamic iRT; correction factor 1; interference correction and cross run normalization, enabled; and all peptides were filtered using Q value ≤ 0.01, was used for protein identification and quantitation. Average quantity of fragment ion areas from top three peptides was employed to compare protein abundance between samples. Screening of significantly differentially expressed proteins was performed with abundance ratios ≥3 or ≤0.33, and student test P-value ≤0.05.
Gene Expression Analysis by Real-Time Quantitative PCR
To further validate the different expression patterns of the proteins, we selected five proteins that exhibited much higher expression levels in the strong-adhesive/invasive strain SAKA 80220 and six specific proteins that were only expressed in the strong-adhesive/invasive strain SAKA 80220. Eleven pairs of primers specific for target genes and one pair of primers specific for the 16S rRNA gene, which was used as an internal reference, were designed and synthesized (Table 2). The total RNA was extracted from the strong-adhesive/invasive strain SAKA80220 and weak-adhesive/invasive strain SAKA80221, which were cultured to the logarithmic phase, by utilizing the EZNATM Bacterial RNA Kit (Omega Bio-Tek, Norcross, GA, United States). The RNA was reverse-transcribed to get the cDNA using the PrimeScriptTM RT Reagent Kit (TaKaRa Bio Inc.). The cDNA was further used as a template to perform qRT-PCR using a RealPlex 4 Master Cycler (Eppendorf). The mRNA expression levels of the genes were calculated according to the 2– ΔΔCT method (Du et al., 2018).
Results
Sequence Typing of 34 Strains of C. sakazakii
Multilocus sequence typing (MLST) result indicated that the 34 C. sakazakii strains could be divided into 16 STs. The origin/source and the sequence types of 34 C. sakazakii strains were listed in Table 1, and the phylogenetic tree of the 34 C. sakazakii strains is shown in Figure 1.
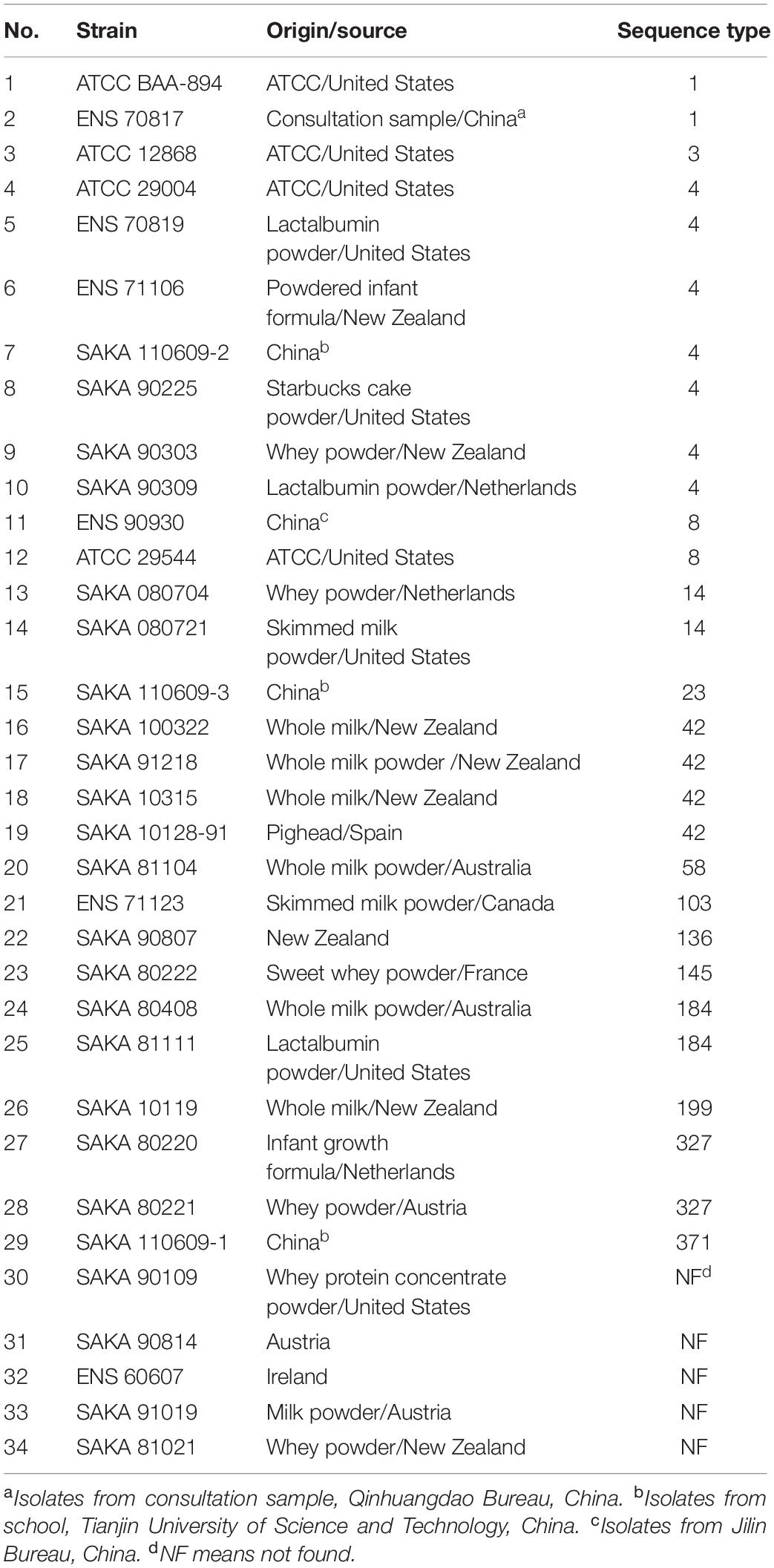
Table 1. The origin/source and the sequence types of 34 C. sakazakii strains.

Table 2. The primers and products for the qRT-PCR of the differentially expressed genes in SAKA80220 and SAKA80221.
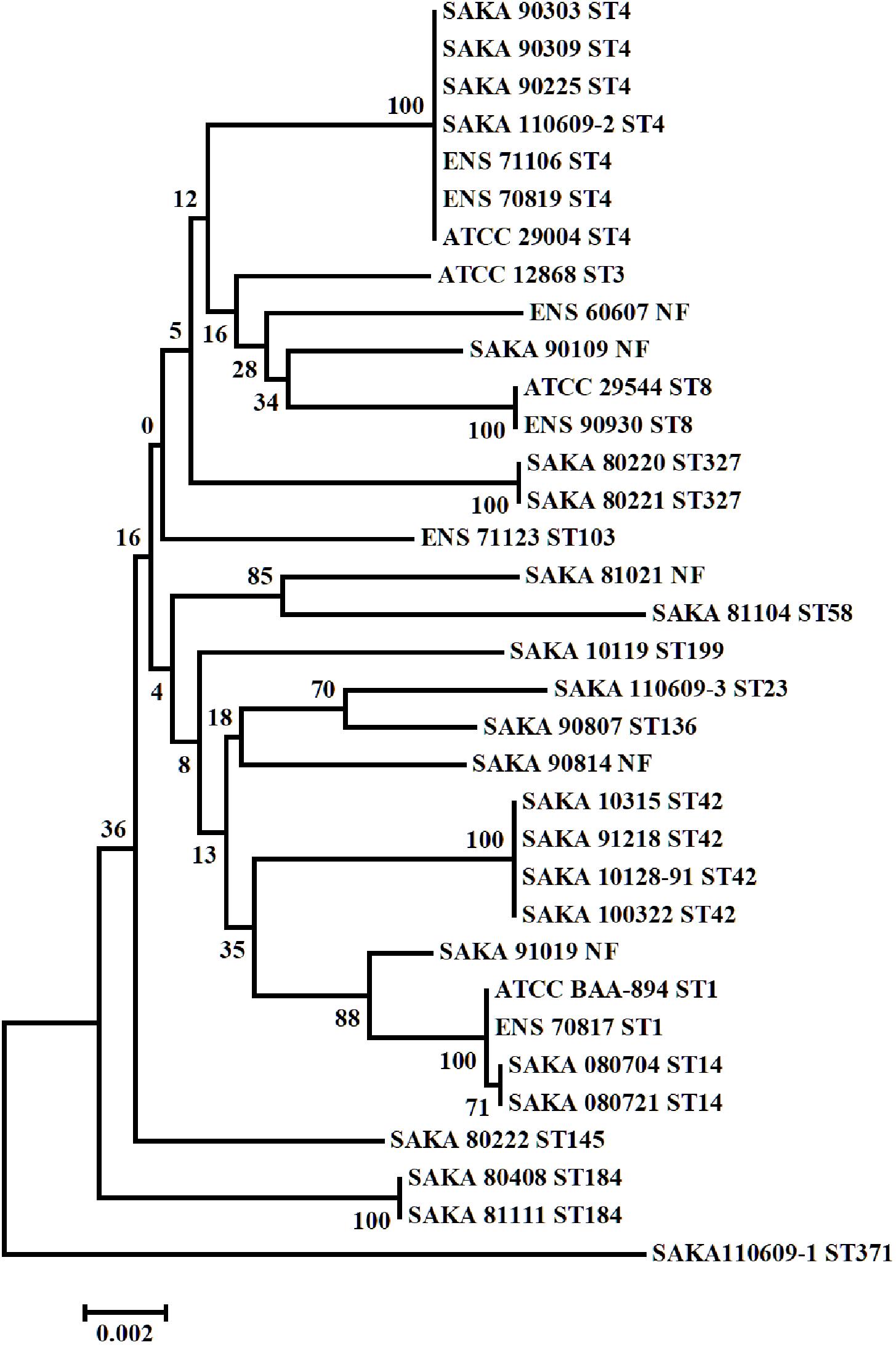
Figure 1. Phylogenetic tree of the 34 C. sakazakii strains.
Among the 16 STs, ST4 included seven strains (7/34) was the dominant type, and ST42 included four strains (4/34), while ST1, ST8, ST14, ST184, and ST327 included two strains (2/34), respectively. Among the remaining strains, eight strains belonged to eight different ST types, and five strains were not found to match the sequence types in the PubMLST database. A strong-adhesive/invasive strain (SAKA80220) and a weak-adhesive/invasive strain (SAKA80221), both belonging to the ST327 type, were selected for proteome analysis using DIA techniques.
Database Construction and Protein Identification
Quantitative DIA analysis was performed on two groups of bacterial samples, and a total of 2,319 proteins were identified for three biological repeats, 2,203 proteins among which were reliably quantified. The result of the correlation analysis between the samples showed that the sample correlation within the group is very high and it close to 1, suggesting a good repeatability of the experiment (Supplementary Figure S1). The results of the cluster analysis indicated that intra-group repeatedly clustered together, suggesting a higher intra-group repeatability (Supplementary Figure S2). Screening of differentially expressed proteins was performed with abundance ratios ≥3 or ≤0.33, and P values ≤0.05. A total of 210 differentially expressed proteins were identified. Of these differentially expressed proteins, 67 were highly expressed in SAKA80220 (strong-adhesive/invasive strain) compared to SAKA80221 (weak-adhesive/invasive strain, ≥3-fold at p ≤ 0.05) and the expression level of 143 proteins were lower in SAKA80220 compared to SAKA80221 (≤0.33-fold at p ≤ 0.05) (Supplementary Table S1). Some of proteins, such as FlgK, WaaC, LptA, LuxS, Fumc, KatG, and YtfE could be considered to be potential factors related to the adhesion and invasion processes. Notably, there were six specific genes encoding electron transport complex proteins (rnfC, rnfD, and rnfG), transcriptional regulators (soxS and ESA_03837), and hypothetical protein (ESA_01856) in the strong-adhesive/invasive strain SAKA80220 (Table 3).
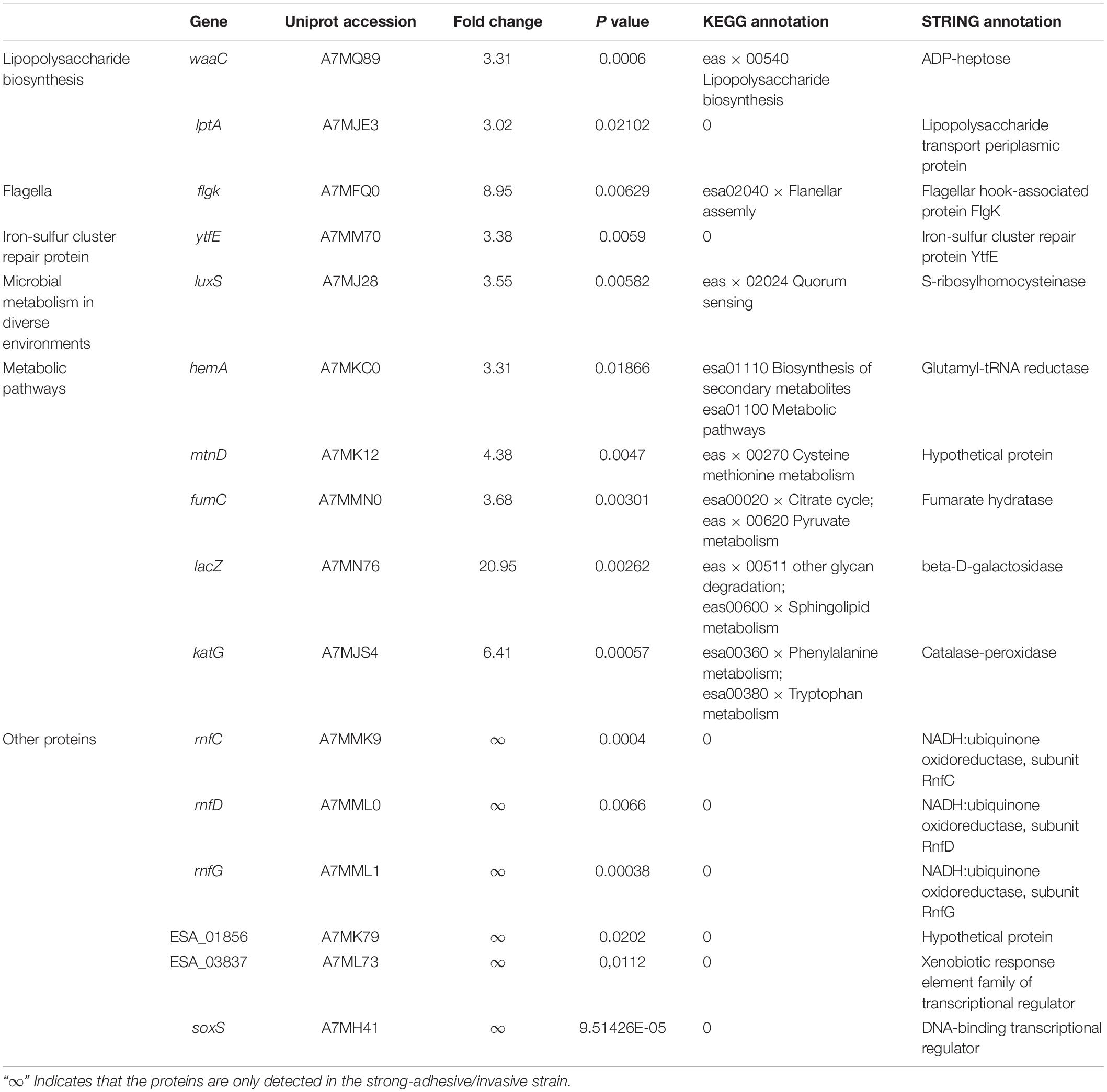
Table 3. The putative adhesion/invasion-related proteins and hypothetical proteins in the strong-adhesive/invasive strain SAKA80220.
Functional Classification of High Expression Differential Proteins in the Strong-Adhesive/Invasive Strain SAKA80220
Among the 67 high expression differential proteins in the strong-adhesive/invasive strain SAKA80220, 48 proteins belonged to specific genomic information categories in UniProt1, and the other 19 were hypothetical or unknown proteins that could not be identified in this database. The summarized GO annotation data of the 48 proteins is shown in Figure 2.
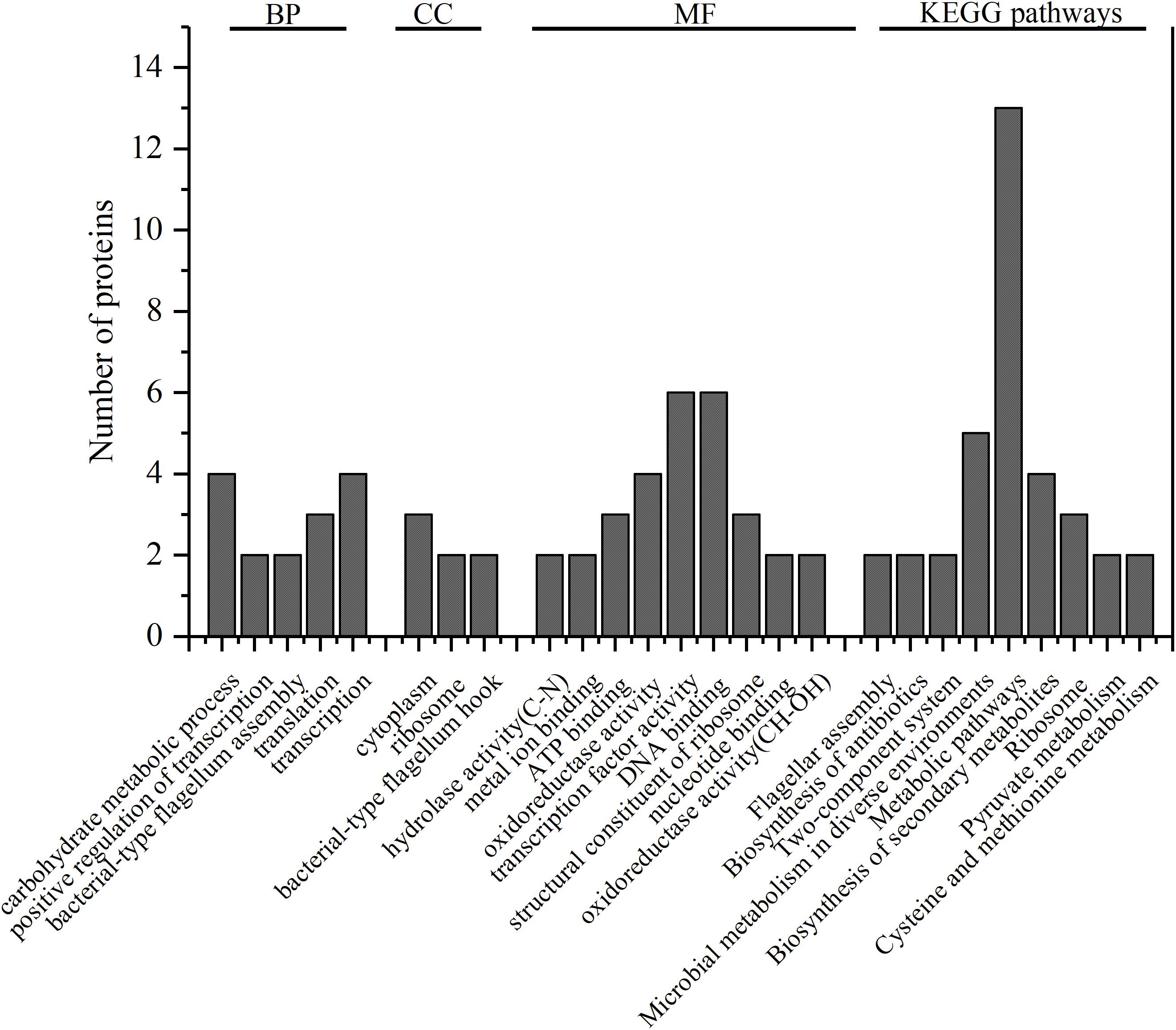
Figure 2. GO annotation of 48 identified proteins. The number of the proteins which significantly enriched categories in biological process (BP), cell component (CC), and molecular function (MF) are shown on the y-axes; KEGG pathways of 47 identified proteins, the number of the proteins in the nine most significantly enriched pathways are shown on the y-axes.
GO annotation showed that these 48 proteins were mainly enriched in the following function categories. In biological processes (BP), 29 proteins were mainly involved in carbohydrate metabolic process, positive regulation of transcription, bacterial-type flagellum assembly, translation and transcription. In the cell component (CC) category, 18 proteins were mostly associated with the cytoplasm, ribosome and bacterial-type flagellum hook. In the molecular function (MF) category, 38 proteins were mainly distributed in hydrolase activity (C–N), metal ion binding, ATP binding, oxidoreductase activity, transcription factor activity, DNA binding, structural constituent of ribosome, nucleotide binding and oxidoreductase activity (CH–OH). Notably, according to GO annotation, a single protein may play more than one role.
KEGG Analysis of Differentially Expressed Proteins
Kyoto Encyclopedia of Genes and Genomes2 (KEGG, http://www.kegg.jp/kegg/tool/map_pathway2.html) analysis provided additional information regarding the enrichment of expressed proteins in each pathway furthermore. In this analysis, 47 out of the 67 proteins were annotated to 31 pathways, and the top 9nine pathways were significantly enriched (Figure 2), namely, flagellar assembly, biosynthesis of antibiotics, two-component system, microbial metabolism in diverse environments, metabolic pathways, biosynthesis of secondary metabolites, ribosome, pyruvate metabolism, and cysteine and methionine metabolism. There was a strong correlation between the enriched proteins in KEGG pathways and the enrichment of proteins in the BP, CC, and MF categories. Furthermore, the detailed analysis of the highly expressed differential proteins and the specific proteins in strong-adhesive/invasive strain SAKA80220 were performed using the protein-protein interaction network analysis database3 (STRING, http://string-db.org).
STRING Analysis of Distinct Proteins
The STRING database was used to comprehensively investigate the interactions among the various proteins. The detailed protein interactions are shown in Figure 3. The line width is positively correlated with the change extent. The following proteins exhibited high numbers of interactions: flagellar proteins; 50s ribosomal subunit protein; LPS biosynthesis proteins; proteins associated with microbial metabolism in diverse environments; proteins associated with nucleotide metabolism and DNA replication; and hypothetical proteins.
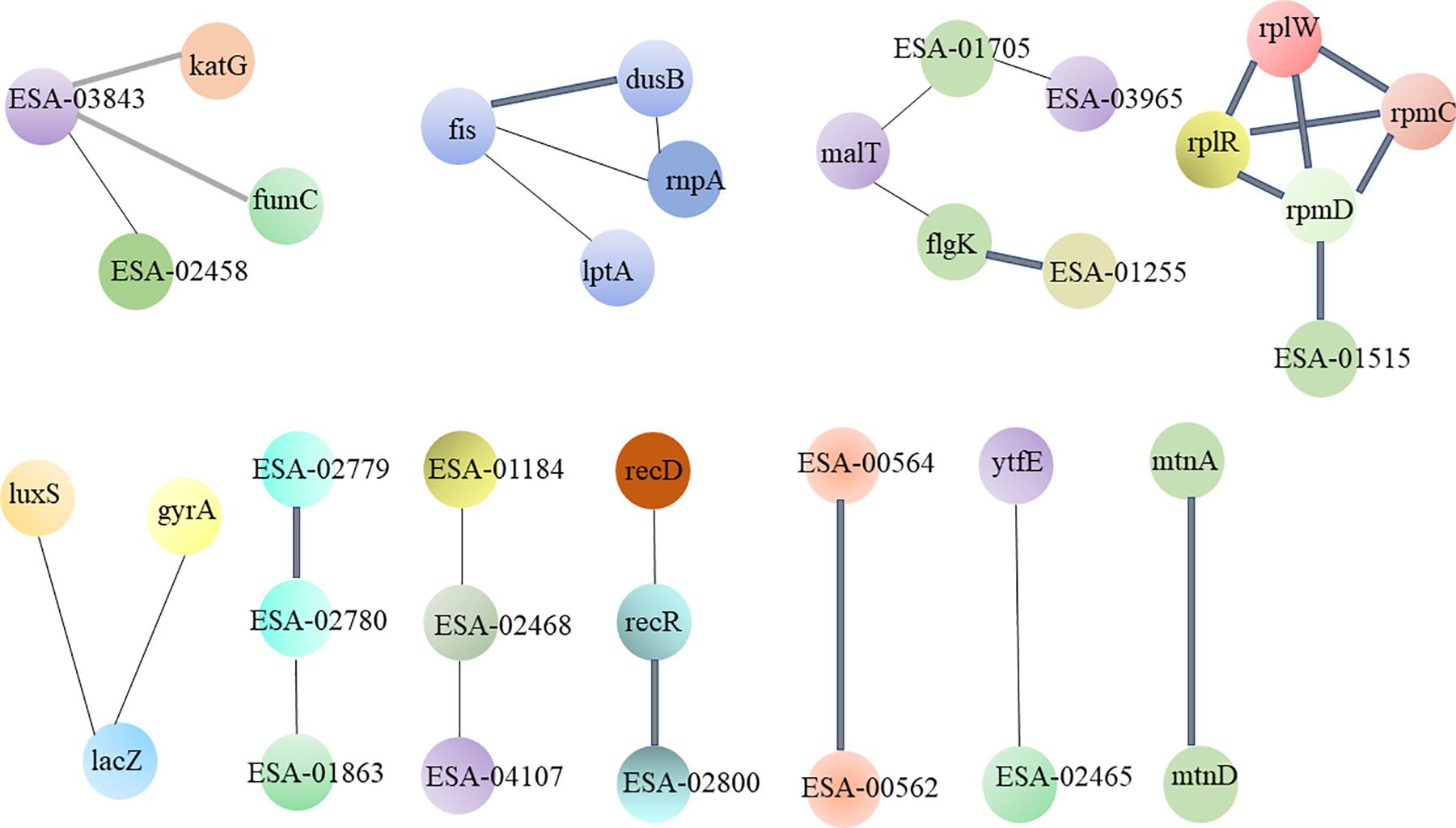
Figure 3. The interacted network of protein-protein was analyzed by STRING. These differential proteins which are high expression in strong-adhesive/invasive strain SAKA80220 compared to weak-adhesive/invasive strain SAKA80221 are mainly concentrated in: flagellar proteins; 50s ribosomal subunit protein; LPS biosynthesis proteins and hypothetical proteins.
Validation of Gene Expression by Quantitative Real-Time qPCR
To investigate the relationships between protein expression and the corresponding mRNA expression patterns, real-time qPCR was used to analyze the dynamic transcriptional expression of 11 representative high expression proteins with 16S rRNA as the reference gene. The results demonstrated that a majority of the genes exhibited coincident protein and mRNA expression patterns, except for the gene of fumc(ESA_01966)and soxS (ESA_00920), which did not show changes in expression between the strains with distinct adhesion/invasion capabilities (Figure 4).
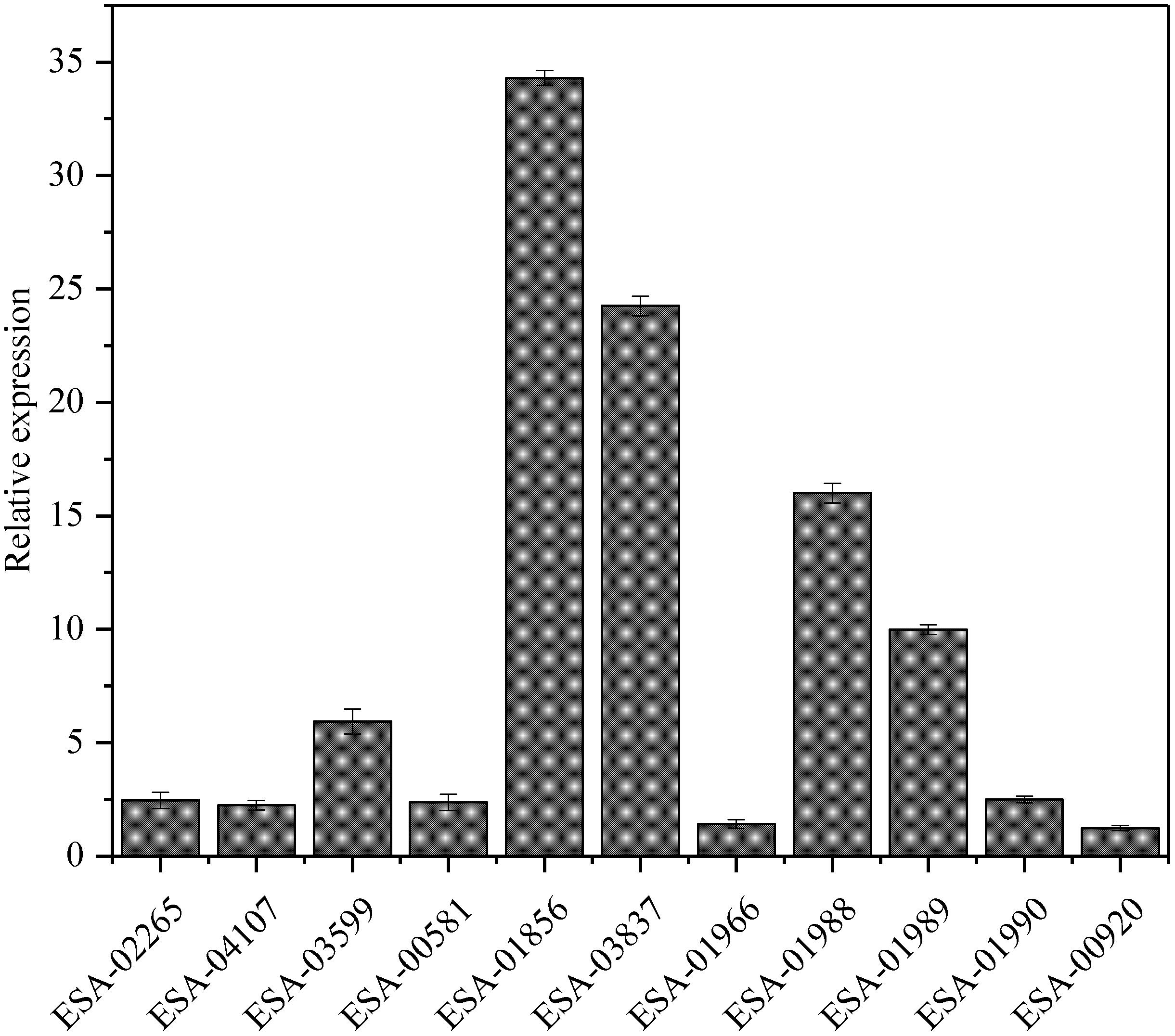
Figure 4. Real-time qPCR analysis of transcription levels of 11 genes in the two strains.
Discussion
Based on our previous results (Du et al., 2016) from adhesion/invasion analysis coupled with sequence typing, we selected the strong-adhesive/invasive strain SAKA80220 and the weak-adhesive/invasive strain SAKA80221 as strains that exhibited distinct adhesion/invasion capabilities but were close in evolutionary relationship for comparative proteomics analysis. By LC-MS/MS coupled with DIA, a total of 2,203 proteins were identified, among which, 67 proteins were high expression in the strong-adhesive/invasive strain SAKA80220 compared to the weak-adhesive/invasive strain SAKA80221 (≥3-fold at p ≤ 0.05). According to the STRING results, some proteins with unknown functions and proteins associated with the flagellar assembly, lipopolysaccharide synthesis, LuxS/AI-2, energy metabolic pathways, iron-sulfur cluster exhibited distinct biosynthesis in the two strains.
The flagellum is the primary organ associated with bacterial motility and is composed of three main parts: basal body, hook, and filament. The role of the flagellum hook is to connect the basal body and filament (Morimoto and Minamino, 2014). The flagellar hook-associated protein FlgK, encoded by the flgK (ESA_02265) gene, is regarded as an important virulence factor for invasion in other microorganisms, such as Escherichia coli and Salmonella enterica (Gauger et al., 2007; Shah et al., 2011). In addition, some studies have shown that mutation of flgK leads to reduced motility and virulence-related enzyme activity in bacteria, in turn leading to decreased pathogenicity (Pallen et al., 2005; Terashima et al., 2017). In this study, the expression of the FlgK protein in the strong-adhesive/invasive strain SAKA80220 was 8.95-fold higher than that in the weak-adhesive/invasive strain SAKA80221, which is consistent with the mRNA levels observed by real-time qPCR. Taken together, all the studies indicate that the flgK gene is responsible for improvement of flagellar motility, thereby enhancing the ability of bacteria to adhere to and invasion of the host cells.
Lipopolysaccharides are the main functional and structural components of the cell surfaces of Gram-negative bacteria and have also been shown to be the main virulence factors (Raetz and Whitfield, 2002). LPSs are composed of lipid A, core oligosaccharides and O-antigens. Lipid A represents the hydrophobic component of LPSs located in the outer leaflet of the outer membrane, while core polysaccharides and O-antigen repeats are displayed on the surfaces of bacterial cells (Wang and Quinn, 2010). In our study, two proteins were differently expressed in the strong-adhesive/invasive strain and the weak-adhesive/invasive strain which were related to LPS biosynthesis and translocation, including waaC (ESA_04107) and lptA (ESA_03599). WaaC encodes heptose-transferase I, as a key gene in the lipopolysaccharide biosynthesis pathway. In E. coli, the heptose-transferase I WaaC transfers the first core heptose to Kdo2-lipid A, which is the main active component of endotoxin (Moreau et al., 2008). In this study, WaaC was expressed much higher levels in the strong-adhesive/invasive strains than in the weak-adhesive/invasive strains (3.31-fold). And the high expression of WaaC may increase LPS synthesis and thereby enhance the ability of bacteria to adhere to and invasion of the host cells. In Gram-negative bacteria, LPS is biosynthesized in the cytoplasm, and then, translocated to the periplasmic side of the inner membrane. During translocation, the periplasmic LPS transport protein LptA forms a bridge between the inner membrane and the outer membrane via interactions with the other LPS transport proteins LptB–LptG, thereby facilitating the transfer of LPS from the inner membrane to the outer membrane. It was also reported that LptA may act as a periplasmic chaperone for LPS transport across the periplasm because LptA plays a role in periplasmic localization and has been shown to bind both LPS and lipid A in vitro (Sperandeo et al., 2007, 2011; Tran et al., 2008). In this study, the expression of the LptA protein in the strong-adhesive/invasive strain was 3.02 times that in the weak-adhesive/invasive strain, indicating the involvement of this protein in pathogenesis. Our results proved that the biosynthesis and translocation of LPS are closely associated with the adhesion/invasion capability of C. sakazakii.
S-ribosylhomocysteinase (LuxS, ESA_00581) is involved in the synthesis of the signal molecule autoinducer 2 (AI-2), which is secreted by bacteria and used to respond to both cell density and metabolism (Ye et al., 2016a, b). Many studies have indicated the importance of LuxS/AI-2 in BP of Gram-negative and Gram-positive bacteria, including antibiotic production, biofilm formation, and carbohydrate metabolism (Ma et al., 2017). Notably, the AI-2 plays an important role in controlling virulence factors during early growth of Edwardsiella (Han et al., 2010). Compared to the wild-type, the luxS-deleted mutants of E. coli SE15 showed dramatically decreased biofilm formation (Kang et al., 2016). The Actinobacillus pleuropneumoniae luxS mutant showed reduced adhesion ability, at the same time, AI-2 could increase adhesion and biofilm formation of A. pleuropneumoniae independent of LuxS (Li et al., 2011). In this study, the expression of LuxS protein in the strong-adhesive/invasive strain was 3.55 times higher than that in the weak-adhesive/invasive strain, which is consistent with the observed mRNA levels. According to accumulating experimental evidence regarding the role of LuxS/AI-2, LuxS/AI-2-mediated quorum sensing may play crucial roles in the formation of biofilms and generation of adhesion/invasion related factors in C. sakazakii.
Another differentially expressed protein which was found is the catalase-peroxidase KatG. Catalase-peroxidase is widely found in animals, plants and microorganisms, such as animal livers and cells, organelles of plants, as well as bacteria. The biological functions of catalase-peroxidase mainly include decomposition of hydrogen peroxide, inhibition of cellular oxidation stimulation and cell senescence (Song et al., 2005). In this study, the expression of the KatG protein in the strong-adhesive/invasive strain SAKA80220 was 6.41-fold higher than that in the weak-adhesive/invasive strain SAKA80221. In the KEGG pathway, KatG was predicted as a catalase-peroxidase involved in the tryptophan metabolism and phenylalanine metabolism of C. sakazakii. By comparing the protein sequence of KatG in C. sakazakii and E. coli, the similarity between the two proteins was high (84.55%). It’s reported that the expression of catalase-peroxidase KatG in E. coli can increase its tolerance to oxidative stress (Kang et al., 2011). By comparing the protein sequence of KatG in C. sakazakii and Mycobacterium tuberculosis, we found that the similarity between the two proteins was 57.71%. The isoniazid resistance of Mycobacterium tuberculosis is closely related to the deletion and mutation of katG gene. When the katG locus is mutated, the isoniazid cannot be activated and converted into an effective bactericidal form. These mutant strains showed reduced virulence when challenged mice, and the introduction of exogenous katG gene into these strains can restore virulence (Ng et al., 2004). Therefore, the high expression of KatG protein may be associated with the high adhesion/invasion capability of C. sakazakii.
To obtain sufficient nutrients and energy from host cells, bacteria must develop multitudinous enzymes and metabolic pathways. Therefore, the demand for nutrients is considered to be the driving factor for the evolution of virulence (Gorke and Stulke, 2008; Eisenreich et al., 2010). Interestingly, proteomics results have indicated that the expression of the β-galactosidase LacZ in the strong-adhesive/invasive strain was 20.95-fold higher than that in the weak-adhesive/invasive strain. It is well known that cells activate the expression of the lactose operon when glucose levels are very low in the metabolic pathway of E. coli, and then, the E. coli cells allow the β-galactosidase to catabolize lactose to galactose and glucose to meet metabolic energy requirements. The lacZ gene was expressed at significantly high levels in the strong-adhesive/invasive strain, perhaps due to initiation of the lactose metabolic pathway to meet energy needs to maintain cellular activity in the adverse environment of the host cells. Fumarase is an important enzyme in the tricarboxylic acid (TCA) cycle, catalyzing the reversible hydration/dehydration of fumarate to L-malate (Shimoyama et al., 2007). Several studies have indicated that E. coli seemed to utilize highly responsive regulatory elements to adapt the levels of the fumarase enzymes (fumA, fumB, and fumC) under different cell growth conditions for energy generation and that the fumC (ESA_01966) gene is affected by the type of carbon source used for cell growth (Park and Gunsalus, 1995; Tseng et al., 2001). In this study, the expression of fumarate hydratase (fumC) in the strong-adhesive/invasive strain was 3.68 times higher than that in the weak-adhesive/invasive strain. The FumC protein may be associated with adverse environments and virulence during pathogenic infection.
In this experiment, the expression level of iron–sulfur cluster repair protein YtfE in the strong-adhesive/invasive strain SAKA80220 was 3.38 times that of the weak-adhesive/invasive strain SAKA80221. YtfE is an important diiron protein and contains a non-heme dual-core iron center, which has a bridging ligand linking μ-oxo and μ-carboxylate and six histidine residues coordinated to iron ions. One role of YtfE is to reverse the nitrosation damage by directly or indirectly releasing nitric oxide (NO) from the nitrosated protein into the cytoplasm in E. coli (Balasiny et al., 2018). As we all known, NO is a key signaling and defense molecule in biological systems (Corker and Poole, 2003). Therefore, in this experiment, the high expression of YtfE protein may be benefit to improve the ability of C. sakazakii to adhere to host cells.
Furthermore, notably, six specific proteins are only expressed in the strong-adhesive/invasive strain SAKA80220, including three electron transport complex proteins. ESA_01988, ESA_01989, and ESA_01990, which act as part of the membrane complex involved in electron transport. The high expression of these proteins indicates that the metabolic activity of the strong-adhesive/invasive strain SAKA80220 was relatively high and the additional stored energy might be helpful for adhesion to or invasion of the host cells. In addition to the genes mentioned above, two genes encoding transcriptional regulators (soxS and ESA_03837) and a gene encoding hypothetical protein (ESA_01856) were also identified in the current study. Sequence alignment showed that these proteins are widely present in Cronobacter and share high identity (85−100%) among different Cronobacter species. However, ESA_01856 and ESA_03837 were not found to be similar in other bacteria. Therefore, analysis of these proteins with unknown functions would be helpful for comprehension of the basis of the pathogenicity of Cronobacter. Further studies are required to confirm the functions of the hypothetical proteins, as the differential expression demonstrates that some proteins may play vital roles in the adhesion to and invasion of the host cells by C. sakazakii.
In this study, MLST was performed to analyze 34 C. sakazakii strains and two strains with the same ST but with distinct adhesion/invasion capabilities were selected for identification of differentially expressed proteins using DIA proteomic analysis. A total of 210 proteins exhibited differential expression patterns with abundance ratios ≥3 or ≤0.33 and P values ≤0.05, 67 proteins were high expression in the strong-adhesive/invasive strain SAKA80220 compared with the weak-adhesive/invasive strain SAKA80221. These proteins, which may impact the adhesion/invasion capability, were mainly associated with the flagellar assembly, lipopolysaccharide synthesis, LuxS/AI-2, energy metabolic pathways and iron–sulfur cluster. Furthermore, the different expression proteins were further ascertained by real-time qPCR at the mRNA level. This research provided a useful reference for further studies on the pathogenic mechanism of C. sakazakii.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD018508. Other datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
XD and XW mainly contributed to the designing of the experiments, performing, and writing of the manuscript. PL, TD, XD, and SW devoted their efforts to conceive and design the work. XD and PL made a great contribution in designing and performing the experiments and modifying of the later manuscript. All authors agreed to be accountable for the content of the work and contributed to the conception of the study.
Funding
This work was supported by grants from the Ministry of Science and Technology of China (Project No. 2018YFC1603803) and Tianjin Science and Technology Commission (Project No. 18JCZDJC34300).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01239/full#supplementary-material
FIGURE S1 | The correlation analysis between the protein samples of the strong-adhesive/invasive strain SAKA80220 and weak-adhesive/invasive strain SAKA80221.
FIGURE S2 | The cluster analysis of the protein samples of the strong-adhesive/invasive strain SAKA80220 and weak-adhesive/invasive strain SAKA80221.
TABLE S1 | The detailed information of the differentially expressed proteins in the two strains with distinct virulence.
Footnotes
- ^ https://david.ncifcrf.gov/tools.jsp
- ^ http://www.kegg.jp/kegg/tool/map_pathway2.html
- ^ http://string-db.org
References
Balasiny, B., Rolfe, M. D., Vine, C., Bradley, C., Green, J., and Cole, J. (2018). Release of nitric oxide by the Escherichia coli YtfE (RIC) protein and its reduction by the hybrid cluster protein in an integrated pathway to minimize cytoplasmic nitrosative stress. Microbiology 164, 563–575. doi: 10.1099/mic.0.000629
Baldwin, A., Loughlin, M., Caubilla-Barron, J., Kucerova, E., Manning, G., Dowson, C., et al. (2009). Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticusreveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC. Microbiol. 9:223. doi: 10.1186/1471-2180-9-223
Blackwood, B. P., and Hunter, C. J. (2016). Cronobacter spp. Microbiol. Spectr. 4:EI10-00022015. doi: 10.1128/microbiolspec.EI10-0002-2015
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Corker, H., and Poole, R. K. (2003). Nitric oxide formation by Escherichia coli. Dependence on nitrite reductase, the NO-sensing regulator Fnr, and flavohemoglobin Hmp. J. Biol. Chem. 278, 31584–31592. doi: 10.1074/jbc.M303282200
Dimayacyac-Esleta, B. R., Tsai, C. F., Kitata, R. B., Lin, P. Y., Choong, W. K., Lin, T. D., et al. (2015). Rapid High-pH reverse phase stage tip for sensitive small-scale membrane proteomic profiling. Anal. Chem. 87, 12016–12023. doi: 10.1021/acs.analchem.5b03639
Du, X. J., Han, R., Li, P., and Wang, S. (2015). Comparative proteomic analysis of Cronobacter sakazakii isolates with different virulences. J. Proteomics 128, 344–351. doi: 10.1016/j.jprot.2015.08.013
Du, X. J., Wang, X. Y., Dong, X., Li, P., and Wang, S. (2018). Characterization of the desiccation tolerance of Cronobacter sakazakii strains. Front. Microbiol. 9:2867. doi: 10.3389/fmicb.2018.02867
Du, X. J., Zhang, X., Li, P., Xue, R., and Wang, S. (2016). Screening of genes involved in interactions with intestinal epithelial cells in Cronobacter sakazakii. AMB Expr. 6:74. doi: 10.1186/s13568-016-0246-4
Eisenreich, W., Dandekar, T., Heesemann, J., and Goebel, W. (2010). Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 8, 401–412. doi: 10.1038/nrmicro2351
Forsythe, S. (2018). Microbial source tracking of Cronobacter spp. Adv. Appl. Microbiol. 103, 49–101. doi: 10.1016/bs.aambs.2018.01.004
Franco, A. A., Kothary, M. H., Gopinath, G., Jarvis, K. G., Grim, C. J., Hu, L., et al. (2011). Cpa, the outer membrane protease of Cronobacter sakazakii, activates plasminogen and mediates resistance to serum bactericidal activity. Infect. Immun. 79, 1578–1587. doi: 10.1128/IAI.01165-10
Friedemann, M. (2009). Epidemiology of invasive neonatal Cronobacter (Enterobacter sakazakii) infections. Eur. J. Clin. Microbiol. Infect. Dis. 28, 1297–1304. doi: 10.1007/s10096-009-0779-4
Gao, J. X., Li, P., Du, X. J., Han, Z. H., Xue, R., Liang, B., et al. (2017). A negative regulator of Cellulose biosynthesis, bcsR, affects biofilm formation, and adhesion/invasion ability of Cronobacter sakazakii. Front. Microbiol. 8:1839. doi: 10.3389/fmicb.2017.01839
Gauger, E. J., Leatham, M. P., Mercado-Lubo, R., Laux, D. C., Conway, T., and Cohen, P. S. (2007). Role of motility and the flhDC operon in Escherichia coli MG1655 colonization of the mouse intestine. Infect. Immun. 75, 3315–3324. doi: 10.1128/IAI.00052-07
Giri, C. P., Shima, K., Tall, B. D., Curtis, S., Sathyamoorthy, V., Hanisch, B., et al. (2012). Cronobacter spp. (previously Enterobacter sakazakii) invade and translocate across both cultured human intestinal epithelial cells and human brain microvascular endothelial cells. Microb. Pathog. 52, 140–147. doi: 10.1016/j.micpath.2011.10.003
Gorke, B., and Stulke, J. (2008). Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624. doi: 10.1038/nrmicro1932
Han, Y., Li, X., and Qi, Z. (2010). Detection of different quorum-sensing signal molecules in a virulent Edwardsiella tarda strain LTB-4. J. Appl. Microbiol. 108, 139–147. doi: 10.1111/j.1365-2672.2009.04405.x
Han, Z. M., Deng, M. W., Yuan, A. Q., Wang, J. H., Li, H., and Ma, J. C. (2018). Vertical variation of a black soil’s properties in response to freeze-thaw cycles and its links to shift of microbial community structure. Sci. Total. Environ. 625, 106–113. doi: 10.1016/j.scitotenv.2017.12.209
Holy, O., and Forsythe, S. (2014). Cronobacter spp. as emerging causes of healthcare-associated infection. J. Hosp. Infect. 86, 169–177. doi: 10.1016/j.jhin.2013.09.011
Hotta, T., Yoshida, N., Yoshikawa, T., Sugino, S., and Kondo, M. (1986). Lipopolysaccharide-induced colitis in rabbits. Res. Exp. Med. 186, 61–69. doi: 10.1007/BF01851834
Hu, S., Yu, Y., Wu, X., Xia, X., Xiao, X., and Wu, H. (2017). Comparative proteomic analysis of Cronobacter sakazakii by iTRAQ provides insights into response to desiccation. Food. Res. Int. 100, 631–639. doi: 10.1016/j.foodres.2017.06.051
Iversen, C., and Forsythe, S. (2003). Risk profile of Enterobacter sakazakii, an emergent pathogen associated with infant milk formula. Trends. Food. Sci. Technol. 14, 443–454. doi: 10.1016/S0924-2244(03)00155-9
Iversen, C., Lehner, A., Mullane, N., Bidlas, E., Cleenwerck, I., Marugg, J., et al. (2007). The taxonomy of Enterobacter sakazakii: proposal of a new genus Cronobacter gen. nov. and descriptions of Cronobacter sakazakii comb. nov. Cronobacter sakazakii subsp. sakazakii, comb. nov., Cronobacter sakazakii subsp. malonaticus subsp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov. and Cronobacter genomospecies 1. BMC. Evol. Biol. 7:64. doi: 10.1186/1471-2148-7-64
Iversen, C., Mullane, N., McCardell, B., Tall, B. D., Lehner, A., Fanning, S., et al. (2008). Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Microbiol. 58, 1442–1447. doi: 10.1099/ijs.0.65577-0
Jiang, Y. Q., Yang, B., Harris, N. S., and Deyholos, M. K. (2007). Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 58, 3591–3607. doi: 10.1093/jxb/erm207
Jing, C. E., Du, X. J., Li, P., and Wang, S. (2016). Transcriptome analysis of Cronobacter sakazakii ATCC BAA-894 after interaction with human intestinal epithelial cell line HCT-8. Appl. Microbiol. Biotechnol. 100, 311–322. doi: 10.1007/s00253-015-7053-8
Joseph, S., Desai, P., Ji, Y. M., Cummings, C. A., Shih, R., Degoricija, L., et al. (2012). Comparative analysis of genome sequences covering the seven Cronobacter species. PLoS One 7:e49455. doi: 10.1371/journal.pone.0049455
Joseph, S., and Forsythe, S. J. (2012). Insights into the emergent bacterial pathogen Cronobacter spp. generated by multilocus sequence typing and analysis. Front. Microbiol. 3:397. doi: 10.3389/fmicb.2012.00397
Kang, S., Kim, J., Hur, J. K., and Lee, S. S. (2016). CRISPR-based genome editing of clinically important Escherichia coli SE15 isolated from indwelling urinary catheters of patients. J. Med. Microbiol. 66, 18–25. doi: 10.1099/jmm.0.000406
Kang, Z., Wang, Y., Gu, P., Wang, Q., and Qi, Q. (2011). Engineering Escherichia coli for efficient production of 5-aminolevulinic acid from glucose. Metab. Eng. 13, 492–498. doi: 10.1016/j.ymben.2011.05.003
Kim, K., Kim, K. P., Choi, J., Lim, J. A., Lee, J., Hwang, S., et al. (2010). Outer membrane proteins A (OmpA) and X (OmpX) are essential for basolateral invasion of Cronobacter sakazakii. Appl. Environ. Microbiol. 76, 5188–5198. doi: 10.1128/AEM.02498-09
Lai, K. K. (2001). Enterobacter sakazakii Infections among neonates, infants, children, and adults. Medicine 80, 113–122. doi: 10.1097/00005792-200103000-00004
Li, L., Xu, Z. F., Zhou, Y., Li, T., Sun, L., Chen, H., et al. (2011). Analysis on Actinobacillus pleuropneumoniae LuxS regulated genes reveals pleiotropic roles of LuxS/AI-2 on biofilm formation, adhesion ability and iron metabolism. Microb. Pathog. 50, 293–302. doi: 10.1016/j.micpath.2011.02.002
Lin, L., Zheng, J. X., Yu, Q., Chen, W. D., Xing, J. C., Chen, C. X., et al. (2018). High throughput and accurate serum proteome profiling by integrated sample preparation technology and single-run data independent mass spectrometry analysis. J. Proteomics 174, 9–16. doi: 10.1016/j.jprot.2017.12.014
Ma, Y. P., Hao, L., Ke, H., Liang, Z., Ma, J., Liu, Z., et al. (2017). LuxS/AI-2 in Streptococcus agalactiae reveals a key role in acid tolerance and virulence. Res. Vet. Sci. 115, 501–507. doi: 10.1016/j.rvsc.2017.07.032
Minogue, C. E., Hebert, A. S., Rensvold, J. W., Westphall, M. S., Pagliarini, D. J., and Coon, J. J. (2015). Multiplexed quantification for data-independent acquisition. Anal. Chem. 87, 2570–2575. doi: 10.1021/ac503593d
Moreau, F., Desroy, N., Genevard, J. M., Vongsouthi, V., Gerusz, V., Le Fralliec, G., et al. (2008). Discovery of new Gram-negative antivirulence drugs: structure and properties of novel E. coli WaaC inhibitors. Bioorg. Med. Chem. Lett. 18, 4022–4026. doi: 10.1016/j.bmcl.2008.05.117
Morimoto, Y. V., and Minamino, T. (2014). Structure and function of the bi-directional bacterial flagellar motor. Biomolecules 4, 217–234. doi: 10.3390/biom4010217
Muntel, J., Xuan, Y., Berger, S. T., Reiter, L., Bachur, R., Kentsis, A., et al. (2015). Advancing urinary protein biomarker discovery by Data-Independent acquisition on a quadrupole-orbitrap mass spectrometer. J. Proteome. Res. 14, 4752–4762. doi: 10.1021/acs.jproteome.5b00826
Nair, M. K., Venkitanarayanan, K., Silbart, L. K., and Kim, K. S. (2009). Outer membrane protein A (OmpA) of Cronobacter sakazakii binds fibronectin and contributes to invasion of human brain microvascular endothelial cells. Foodborne. Pathog. Dis. 6, 495–501. doi: 10.1089/fpd.2008.0228
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D., and McKinney, J. D. (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302. doi: 10.1111/j.1365-2958.2004.04078.x
Orsatti, L., Forte, E., Tomei, L., Caterino, M., Pessi, A., and Talamo, F. (2009). 2-D Difference in gel electrophoresis combined with Pro-Q diamond staining: a successful approach for the identification of kinase/phosphatase targets. Electrophoresis 30, 2469–2476. doi: 10.1002/elps.200800780
Pagotto, F. J., Nazarowec-White, M., Bidawid, S., and Farber, J. M. (2003). Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo. J. Food. Prot. 66, 370–375. doi: 10.4315/0362-028x-66.3.370
Pallen, M. J., Penn, C. W., and Chaudhuri, R. R. (2005). Bacterial flagellar diversity in the post-genomic era. Trends Microbiol. 13, 143–149. doi: 10.1016/j.tim.2005.02.008
Park, S. J., and Gunsalus, R. P. (1995). Oxygen, iron, carbon, and superoxide control of the fumarase fumA and fumC genes of Escherichia coli: role of the arcA, fnr, and soxR gene products. J. Bacteriol. 177, 6255–6262. doi: 10.1128/jb.177.21.6255-6262.1995
Patrick, M. E., Mahon, B. E., Greene, S. A., Rounds, J., Cronquist, A., Wymore, K., et al. (2014). Incidence of Cronobacter spp. infections, United States, 2003-2009. Emerg. Infect. Dis. 20, 1520–1523. doi: 10.3201/eid2009.140545
Quintero-Villegas, M. I., Wittke, A., and Hutkins, R. (2014). Adherence inhibition of Cronobacter sakazakii to intestinal epithelial cells by lactoferrin. Curr. Microbiol. 69, 574–579. doi: 10.1007/s00284-014-0623-7
Raetz, C. R. H., and Whitfield, C. (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700. doi: 10.1016/B978-012373944-5.00196-6
Ren, G. M., Xu, X. H., Qu, J. J., Zhu, L. P., and Wang, T. T. (2016). Evaluation of microbial population dynamics in the co-composting of cow manure and rice straw using high throughput sequencing analysis. World J. Microbiol. Biotechnol. 32:101. doi: 10.1007/s11274-016-2059-7
Riedel, K., and Lehner, A. (2007). Identification of proteins involved in osmotic stress response in Enterobacter sakazakii by proteomics. Proteomics 7, 1217–1231. doi: 10.1002/pmic.200600536
Sanda, M., and Goldman, R. (2016). Data independent analysis of IgG glycoforms in samples of unfractionated human plasma. Anal. Chem. 88, 10118–10125. doi: 10.1021/acs.analchem.6b02554
Shah, D. H., Zhou, X., Addwebi, T., Davis, M. A., Orfe, L., Call, D. R., et al. (2011). Cell invasion of poultry-associated Salmonella enterica serovar Enteritidis isolates is associated with pathogenicity, motility and proteins secreted by the type III secretion system. Microbiology 157, 1428–1445. doi: 10.1099/mic.0.044461-0
Shimoyama, T., Rajashekhara, E., Ohmori, D., Kosaka, T., and Watanabe, K. (2007). MmcBC in Pelotomaculum thermopropionicum represents a novel group of prokaryotic fumarases. FEMS. Microbiol. Lett. 270, 207–213. doi: 10.1111/j.1574-6968.2007.00665.x
Singh, N., Goel, G., and Raghav, M. (2015). Insights into virulence factors determining the pathogenicity of Cronobacter sakazakii. Virulence 6, 433–440. doi: 10.1080/21505594.2015.1036217
Song, Z., Marzilli, L., Greenlee, B. M., Chen, E. S., Silver, R. F., Askin, F. B., et al. (2005). Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J. Exp. Med. 201, 755–767. doi: 10.1084/jem.20040429
Sperandeo, P., Cescutti, R., Villa, R., Di Benedetto, C., Candia, D., Dehò, G., et al. (2007). Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 189, 244–253. doi: 10.1128/JB.01126-06
Sperandeo, P., Villa, R., Martorana, A. M., Samalikova, M., Grandori, R., Dehò, G., et al. (2011). New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J. Bacteriol. 193, 1042–1053. doi: 10.1128/JB.01037-10
Stephan, R., Grim, C. J., Gopinath, G. R., Mammel, M. K., Sathyamoorthy, V., Trach, L. H., et al. (2014). Re-examination of the taxonomic status of Enterobacter helveticus, Enterobacter pulveris and Enterobacter turicensis as members of the genus Cronobacter and their reclassification in the genera franconibacter gen. nov. and Siccibacter gen. nov. as Franconibacter helveticus comb. nov., Franconibacter pulveris comb. nov. and Siccibacter turicensis comb. nov., respectively. Int. J. Syst. Evol. Microbiol. 64, 3402–3410. doi: 10.1099/ijs.0.059832-0
Terashima, H., Kawamoto, A., Morimoto, Y. V., Imada, K., and Minamino, T. (2017). Structural differences in the bacterial flagellar motor among bacterial species. Biophys. Physicobiol. 14, 191–198. doi: 10.2142/biophysico.14.0_191
Townsend, S., Barron, J. C., Loc-Carrillo, C., and Forsythe, S. (2007). The presence of endotoxin in powdered infant formula milk and the influence of endotoxin and Enterobacter sakazakii on bacterial translocation in the infant rat. Food Microbiol. 24, 67–74. doi: 10.1016/j.fm.2006.03.009
Tran, A. X., Trent, M. S., and Whitfield, C. (2008). The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 283, 20342–20349. doi: 10.1074/jbc.M802503200
Tseng, C. P., Yu, C. C., Lin, H. H., Chang, C. Y., and Kuo, J.-T. (2001). Oxygen- and growth rate-dependent regulation of Escherichia coli fumarase (FumA, FumB, and FumC) activity. J. Bacteriol. 183, 461–467. doi: 10.1128/jb.183.2.461-467.2001
Wang, X. Y., and Quinn, P. J. (2010). Lipopolysaccharide: biosynthetic pathway and structure modification. Prog. Lipid Res. 49, 97–107. doi: 10.1016/j.plipres.2009.06.002
Wisniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Wu, X. L., Xiong, E. H., Wang, W., Scali, M., and Cresti, M. (2014). Universal sample preparation method integrating trichloroacetic acid/acetone precipitation with phenol extraction for crop proteomic analysis. Nat. Protoc. 9, 362–374. doi: 10.1038/nprot.2014.022
Ye, Y. W., Jiao, R., Gao, J. N., Li, H., Ling, N., Wu, Q. P., et al. (2016a). Proteins involved in responses to biofilm and planktonic modes in Cronobacter sakazakii. LWT Food Sci. Technol. 65, 1093–1099. doi: 10.1016/j.lwt.2015.09.039
Keywords: C. sakazakii, adhesion/invasion capability, sequence type, DIA proteomic, real-time qPCR
Citation: Li P, Dong X, Wang X, Du T, Du X and Wang S (2020) Comparative Proteomic Analysis of Adhesion/Invasion Related Proteins in Cronobacter sakazakii Based on Data-Independent Acquisition Coupled With LC-MS/MS. Front. Microbiol. 11:1239. doi: 10.3389/fmicb.2020.01239
Received: 18 February 2020; Accepted: 14 May 2020;
Published: 09 June 2020.
Edited by:
Haifeng Zhao, South China University of Technology, ChinaReviewed by:
Angelika Lehner, University of Zurich, SwitzerlandBen Davies Tall, United States Food and Drug Administration, United States
Julio Parra-Flores, University of the Bío Bío, Chile
Copyright © 2020 Li, Dong, Wang, Du, Du and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-jun Du, eGpkdUB0dXN0LmVkdS5jbg==; Shuo Wang, cy53YW5nQHR1c3QuZWR1LmNu; d2FuZ3NodW9AbmFua2FpLmVkdS5jbg==
†These authors have contributed equally to this work