 Tomasz Kulik1*
Tomasz Kulik1* Balazs Brankovics2
Balazs Brankovics2 Anne D. van Diepeningen2
Anne D. van Diepeningen2 Katarzyna Bilska1
Katarzyna Bilska1 Maciej Żelechowski1
Maciej Żelechowski1 Kamil Myszczyński1,3
Kamil Myszczyński1,3 Tomasz Molcan4
Tomasz Molcan4 Alexander Stakheev5
Alexander Stakheev5 Sebastian Stenglein6,7
Sebastian Stenglein6,7 Marco Beyer8
Marco Beyer8 Matias Pasquali9
Matias Pasquali9 Jakub Sawicki1
Jakub Sawicki1 Joanna Wyrȩbek1
Joanna Wyrȩbek1 Anna Baturo-Cieśniewska10
Anna Baturo-Cieśniewska10- 1Department of Botany and Nature Protection, University of Warmia and Mazury in Olsztyn, Olsztyn, Poland
- 2Biointeractions & Plant Health, Wageningen Plant Research, Wageningen, Netherlands
- 3Molecular Biology Laboratory, Institute of Animal Reproduction and Food Research, Polish Academy of Sciences, Olsztyn, Poland
- 4Department of Animal Anatomy and Physiology, Faculty of Biology and Biotechnology, University of Warmia and Mazury in Olsztyn, Olsztyn, Poland
- 5Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
- 6National Scientific and Technical Research Council, Godoy Cruz, Argentina
- 7Universidad Nacional del Centro de la Provincia de Buenos Aires, Tandil, Argentina
- 8Department of Environmental Research and Innovation, Agro-Environmental Systems, Luxembourg Institute of Science and Technology, Belval, Luxembourg
- 9Department of Food, Environmental and Nutritional Sciences, University of Milan, Milan, Italy
- 10Laboratory of Phytopathology and Molecular Mycology, Department of Biology and Plant Protection, UTP University of Science and Technology, Bydgoszcz, Poland
Much of the mitogenome variation observed in fungal lineages seems driven by mobile genetic elements (MGEs), which have invaded their genomes throughout evolution. The variation in the distribution and nucleotide diversity of these elements appears to be the main distinction between different fungal taxa, making them promising candidates for diagnostic purposes. Fungi of the genus Fusarium display a high variation in MGE content, from MGE-poor (Fusarium oxysporum and Fusarium fujikuroi species complex) to MGE-rich mitogenomes found in the important cereal pathogens F. culmorum and F. graminearum sensu stricto. In this study, we investigated the MGE variation in these latter two species by mitogenome analysis of geographically diverse strains. In addition, a smaller set of F. cerealis and F. pseudograminearum strains was included for comparison. Forty-seven introns harboring from 0 to 3 endonucleases (HEGs) were identified in the standard set of mitochondrial protein-coding genes. Most of them belonged to the group I intron family and harbored either LAGLIDADG or GIY-YIG HEGs. Among a total of 53 HEGs, 27 were shared by all fungal strains. Most of the optional HEGs were irregularly distributed among fungal strains/species indicating ancestral mosaicism in MGEs. However, among optional MGEs, one exhibited species-specific conservation in F. culmorum. While in F. graminearum s.s. MGE patterns in cox3 and in the intergenic spacer between cox2 and nad4L may facilitate the identification of this species. Thus, our results demonstrate distinctive traits of mitogenomes for diagnostic purposes of Fusaria.
Introduction
Most fungi contain mitochondria, organelles playing a key role in the generation of metabolic energy. Besides, fungal mitochondria have been shown to contribute to diverse cellular and organismal functions including senescence, quiescence, biofilm regulation and hyphal growth (Chatre and Ricchetti, 2014; Calderone et al., 2015; Bartelli et al., 2018; Sandor et al., 2018). They may also be involved in antifungal drug resistance, as well as in fungal virulence and pathogenicity (Shingu-Vazquez and Traven, 2011; Sandor et al., 2018). It is therefore not surprising that mitochondrial structure and function, as well as mitogenomes of fungi, have been studied intensively since the 2000s (Hausner, 2003; Chatre and Ricchetti, 2014).
Mitogenomes are expected to provide new insights for understanding the phylogenetic relationships and evolutionary biology of fungi (Aguileta et al., 2014; Lin et al., 2015; Franco et al., 2017). The reason for this is that fungal mitogenomes are highly divergent among even closely related lineages (Yin et al., 2012; Pogoda et al., 2019). This fact opens entirely new perspectives in diagnostics of fungi. The concept of applying mitogenomes for the identification of species mainly derives from their higher DNA copy number compared with nuclear DNA, and hence higher recovery and amplification success (Santamaria et al., 2009). However, for many fungal lineages, the characterization of mitochondrial DNA (mtDNA) has been largely limited by a low number of mitochondrial sequences in the GenBank database.
Fungal mitogenomes are double-stranded DNA molecules with relatively simple genetic structures often containing a set of 14 protein-coding genes, two rRNA coding genes and a large group of tRNA coding genes. The set of 14 core-genes encoding proteins is involved in the respiratory chain: the apocytochrome b (cob), 3 subunits of the cytochrome c oxidase (cox genes), 7 subunits of the NADH dehydrogenase (nad genes) and 3 components of the ATP synthase (atp genes). The two conserved rRNA genes encode the small (rns) and large (rnl) ribosomal RNA (rRNA) subunits (Bullerwell and Lang, 2005). In addition, fungal mitogenomes harbor a variable number of mobile genetic elements (MGEs) such as introns and associated homing endonucleases (HEGs), which have invaded the mitogenomes throughout the evolution (Basse, 2010; Joardar et al., 2012; Pogoda et al., 2019).
Surveys of intron distribution among different filamentous fungi have pinpointed a surprisingly high variation in MGE content, from the single-intron mitogenome of Fusarium proliferatum (Brankovics et al., 2017) to MGE-rich mitogenome of Rhizoctonia solani containing several dozens of MGEs (Losada et al., 2014). Most of the MGE insertion sites are highly conserved (Hausner, 2003) and occur in mitochondrial protein-coding genes, but certain genes can display remarkably different MGE densities (Hausner, 2003; Yin et al., 2012; Aguileta et al., 2014; Franco et al., 2017). In addition, the same MGEs can be irregularly distributed in evolutionarily distant species and mosaicism in MGE patterns can be found between different populations or even strains of the same species driving large genome size differences among them (Yin et al., 2012; Kolesnikova et al., 2019).
The widespread distribution of MGEs in fungi can be explained by an intron-rich progenitor of major eukaryotic lineages from which extensive and lineage-dependent intron loss has occurred. The most frequently assumed mechanism of intron loss considers the replacement of intron-containing genes with their intronless versions through homologous recombination between intronless cDNA and the corresponding genomic DNA (Hausner, 2003; Yin et al., 2012; Pogoda et al., 2019). In addition to intron/HEG loss, the infective nature of HEGs, which can propagate horizontally between different lineages is often suggested to drive the observed variation in MGE content. The mobility of HEGs is primarily explained by their transfer and site-specific integration, which usually involves three steps: recognition of an intronless allele, cleaving, and insertion of the HEG (Haugen et al., 2005; Yin et al., 2012). In addition, HEGs themselves may propagate over a variety of lineages independently from their host intron, resembling free-standing HEGs, which frequently occur in genomes of phages (Edgell, 2009; Franco et al., 2017). This type of mobility, although not frequently documented, drives variation in size and nucleotide content of introns (Wu and Hao, 2014).
Several recent studies addressed the comprehensive analyses of mitogenomes in important plant pathogenic Fusarium species (Al-Reedy et al., 2012; Fourie et al., 2013; Brankovics et al., 2017, 2018). Mitogenomes of Fusaria pose a typical set of mitochondrial genes with identical gene order. A unique feature apparently common in all Fusarium species is the presence of a large open reading frame with unknown function (LV-uORF) firstly described in mitogenomes of F. graminearum, F. verticillioides and F. solani (Al-Reedy et al., 2012). Fusarium mitogenomes have been shown to vary considerably in size. So far, Fusarium pseudograminearum contains the largest mitogenome of 110,526 bp long (described in this study), while the smallest proved only 30,629 bp long in F. oxysporum (Pantou et al., 2008). Such large size differences are caused primary by the irregular distribution of MGEs (Al-Reedy et al., 2012; Fourie et al., 2013; Brankovics et al., 2018). In F. graminearum sensu stricto (s.s.), MGEs comprise more than half of the mitogenome. Diversity in MGE content contributed to remarkable variation among geographically diverse strains which raised the question of the feasibility of using this type of variation in mitochondrial population genetic studies of Fusarium field populations (Brankovics et al., 2018).
Fusarium graminearum s.s. and F. culmorum are important and closely related plant pathogens exhibiting a mainly necrotrophic lifestyle after a short biotrophic infection stage and can be described as generalists based on their broad host ranges. Fusarium Head Blight (FHB) and Fusarium Crown Rot (FCR) are among the most important diseases of cereals caused by these pathogens. Both have led to major economic losses for the cereal-based feed and food supply chains. Besides these two species, other closely related fungi such as F. cerealis and F. pseudograminearum can be involved in cereal diseases (Leslie and Summerell, 2008).
All four pathogens exhibit a similar toxigenic potential by the production of type B trichothecenes and zearalenone (ZEA). These compounds are nowadays among the most frequently detected mycotoxins in different wheat- and barley-growing regions of the world (Desjardins, 2006). Trichothecenes have been found to induce mycotoxicoses in humans and animals and play a role in the virulence of fungal strains (Desjardins, 2006). ZEA is a non-steroidal estrogen that may cause female reproductive changes due to its strong estrogenic activity (Hueza et al., 2014).
However, despite the aforementioned similarities, all four Fusarium species appear to exhibit considerable variation in distribution and frequency in agroecosystems. The most noticeable, despite their frequent co−existence, is the dramatic prevalence of F. graminearum s.s. in many cropping regions of the world (van der Lee et al., 2015). The emergence of F. graminearum s.s. resulted in the continuous displacement of other FHB causing Fusarium species. The best example illustrating this displacement is the gradual decrease of F. culmorum, which has been traditionally reported as the primary cause of FHB in Northern, Central and Western Europe (Waalwijk et al., 2003; Scherm et al., 2013; Bilska et al., 2018a). F. cerealis causes root rots and seedling blights, but can also be found on corn cobs and as emerging, mycotoxigenic FHB pathogen in temperate regions (Amarasinghe et al., 2015). While the predominance of F. pseudograminearum has been linked to the rather warm and dry environmental conditions (Kazan and Gardiner, 2018).
Even when using a software package designed for patch characterization in images, Fusarium cerealis could neither be distinguished unambiguously from F. sambucinum nor from F. graminearum s. s. based on their morphological spore shape parameters (Dubos et al., 2012). All of them produce similar banana-shaped macroconidia and chlamydospores. F. graminearum s.s. is morphologically identical to F. pseudograminearum, while F. culmorum can easily be confused with F. cerealis (Leslie and Summerell, 2008). More reliable identification of these four species could be achieved by using molecular markers, especially with methods employing species-specific primers in either end point (conventional PCR) or qPCR (Waalwijk et al., 2004; Nicolaisen et al., 2009). A more laborious method of species discrimination is based on multi-locus sequencing (MLST) (Geiser et al., 2004; Wang et al., 2011). However, the major disadvantage of nuclear markers is their reduced detection limits mostly due to the targeted nuclear single-copy genes (Kulik et al., 2015; Bilska et al., 2018b). Amongst the most promising solutions is targeting repeated genomic sequences such as mitochondrial DNA. However, the low number of available mtDNA sequences from Fusaria so far limited the evaluation of mitochondrial DNA as a diagnostic marker.
Today, the growing body of mitogenome data suggests that variation in MGE number can follow the divergence of different fungal taxa (Brankovics et al., 2017; Liang et al., 2017; Fourie et al., 2018), offering great perspectives for future diagnostic purposes. Brankovics et al. (2017) showed that mosaicism in intron patterns can support the separation of three phylogenetic species in F. oxysporum. Fourie et al. (2018) revealed significant differences in intron patterns among species from the Fusarium fujikuroi species complex (FFSC). Beyond Fusarium, recent studies on Colletotrichum mitogenomes indicated that intron content variation could improve cryptic species delimitation within this genus (Liang et al., 2017).
This study demonstrates that the variable landscape of MGEs is the most prominent type of variation among mitogenomes of closely related F. graminearum s.s. and F. culmorum. The major objective of this study was to discover if the pattern of MGE diversity might facilitate the delimitation of these species.
Therefore, we sequenced, assembled and annotated the mitogenomes from geographically diverse strains. Mitogenomes of the studied Fusaria were MGE-rich and most of them were present in all studied strains. Protein homology searches in GenBank database revealed a number of HEG orthologs shared with other fungal species with considerable variation in protein identity scores. Among the total of 53 identified HEGs, one showed species-specific conservation in F. culmorum. F. graminearum s.s. did not harbor unique MGEs throughout the mitogenome, however, we found that MGE patterns in cox3 and in the intergenic spacer between cox2 and nad4L that can serve as potential markers for the identification of this species.
Materials and Methods
Fungal Strains
Supplementary File 1 provides characteristics of all fungal strains used in this study. A more detailed description of the strains can be found in the ToxGen database (Kulik et al., 2017). The mitogenomes of 78 strains of F. graminearum s.s. and 33 strains of F. culmorum were included into the analyses. In addition, 8 strains of F. cerealis and 4 strains of F. pseudograminearum were included for comparison. Moreover, 23 complete mitogenomes of F. graminearum s.s. previously described by Brankovics et al. (2018) and one mitogenome of F. culmorum (Kulik et al., 2016a) were also included.
DNA Extraction and Sequencing
For DNA extraction, fungal strains were incubated on Petri plates (Ø 80 mm) with PDA medium at 24°C for 6 days. DNA from fungal strains was extracted from 0.1 g of mycelium with the use of the Quick-DNA Plant/Seed Miniprep Kit (Zymo Research, Irvine, CA, United States) according to the manufacturer’s protocol.
Whole-genome sequencing was performed at Macrogen (Seoul, South Korea). Genome libraries were constructed using a TruSeq DNA PCR-free library preparation kit (Illumina, San Diego, CA, United States). An Illumina HiSeq X Ten platform was used to sequence the genomes using a paired-end read length of 2 × 150 bp with an insert size of 350 bp.
Assembly and Annotation of Mitogenomes
NOVOPlasty 2.7.2 (Dierckxsens et al., 2017) was used for de novo assembly of fungal mitogenomes from whole-genome data. HEGs were identified using NCBI’s ORF Finder1, Blast+ (v. 2.9.0) (Johnson et al., 2008) and Geneious Prime software (Biomatters Ltd., New Zealand). Annotations were performed using MFannot2, InterPro (Mitchell et al., 2015), CD-Search (Marchler-Bauer and Bryant, 2004) and Geneious Prime software (Biomatters Ltd., New Zealand). Annotation of tRNA genes was improved using tRNAscan-SE (Pavesi et al., 1994).
Complete mitogenomes have been deposited in the NCBI database under the GenBank accession numbers MT036635-757 (Supplementary File 1).
Alignment
Mitogenome comparison included a total of 147 complete mitogenomes. Multiple sequence alignments of the complete mitogenomes were performed with progressive Mauve (Darling et al., 2010) implemented in the Geneious Prime software (Biomatters Ltd., New Zealand) to examine the distribution of MGEs in fungal mitogenomes.
BlastP Analyses
To reveal the distribution and the diversity of HEGs, blastP searches were performed using the BLAST program available at the National Center for Biotechnology Information web page https://www.ncbi.nlm.nih.gov/. Hits were retained only if they had an e-value cut off lower than 0.001 and which covered at least 70% of the query sequence with >60% identity.
Phylogenetic Analysis
HEGs showing higher protein similarity to sequences from distantly related species than to those found in other Fusaria were subjected for phylogenetic analysis. Protein sequences were aligned using the Muscle algorithm implanted in the software package Geneious Prime (Biomatters Ltd., New Zealand). An optimal evolutionary model was estimated using MEGA X (Kumar et al., 2018). The trees were constructed using the Maximum Likelihood method as implemented in Mega X with JTT + G + F model and 2000 bootstrap replicates.
Results
General Features of Fungal Mitogenomes
The mitochondrial genomes of all strains contained a conserved set of genes in the same order and orientation; a set of 14 protein-coding genes, 2 rRNA genes (rns and rnl) and 28 tRNA genes. The four Fusarium species displayed a similar GC content of 31.6–32.0% within their mitogenomes; clearly in the middle limit of the GC content range for fungal mitogenomes (8.4–52.7%, estimated from sequence data deposited in the GenBank database of the NCBI by April 2019).
Mitogenome sizes ranged from of 92.816 bp (F. cerealis strain 64722) to 110.526 bp for (F. pseudograminearum strain CBS 109956) (Supplementary File 1). These sizes of mitogenomes are well in between the current size limits of the fungal mitogenomes ranging from 12 kb to over 235 kb (based on GenBank database by April 2019). To date, the mitogenome of F. pseudograminearum remains the largest among fungi of the genus Fusarium.
Besides genes with functional predictions, Fusarium mitogenomes contained a large open reading frame with unknown function (LV-uORF). This ORF is located in the intergenic spacer between rnl and the nad2 gene (Al-Reedy et al., 2012). The GC content of the LV-uORF is higher than the usual set of mitochondrial genes suggesting its horizontal acquisition by a common ancestor of Fusarium species (Al-Reedy et al., 2012).
Our results showed a highly conserved distribution of LV-uORF in four studied species (Supplementary File 1). We found that LV-uORF is present in all examined strains in the same position and orientation. However, we found considerable variation within LV-uORF mainly due to multiple indel mutations altering its length, which ranged from 4.155 to 6.390 bp (Figure 1). The apparently conserved placement of indels within LV-uORF and their irregular distribution among strains and species suggests that most of these events occurred prior to the divergence of the Fusarium species.
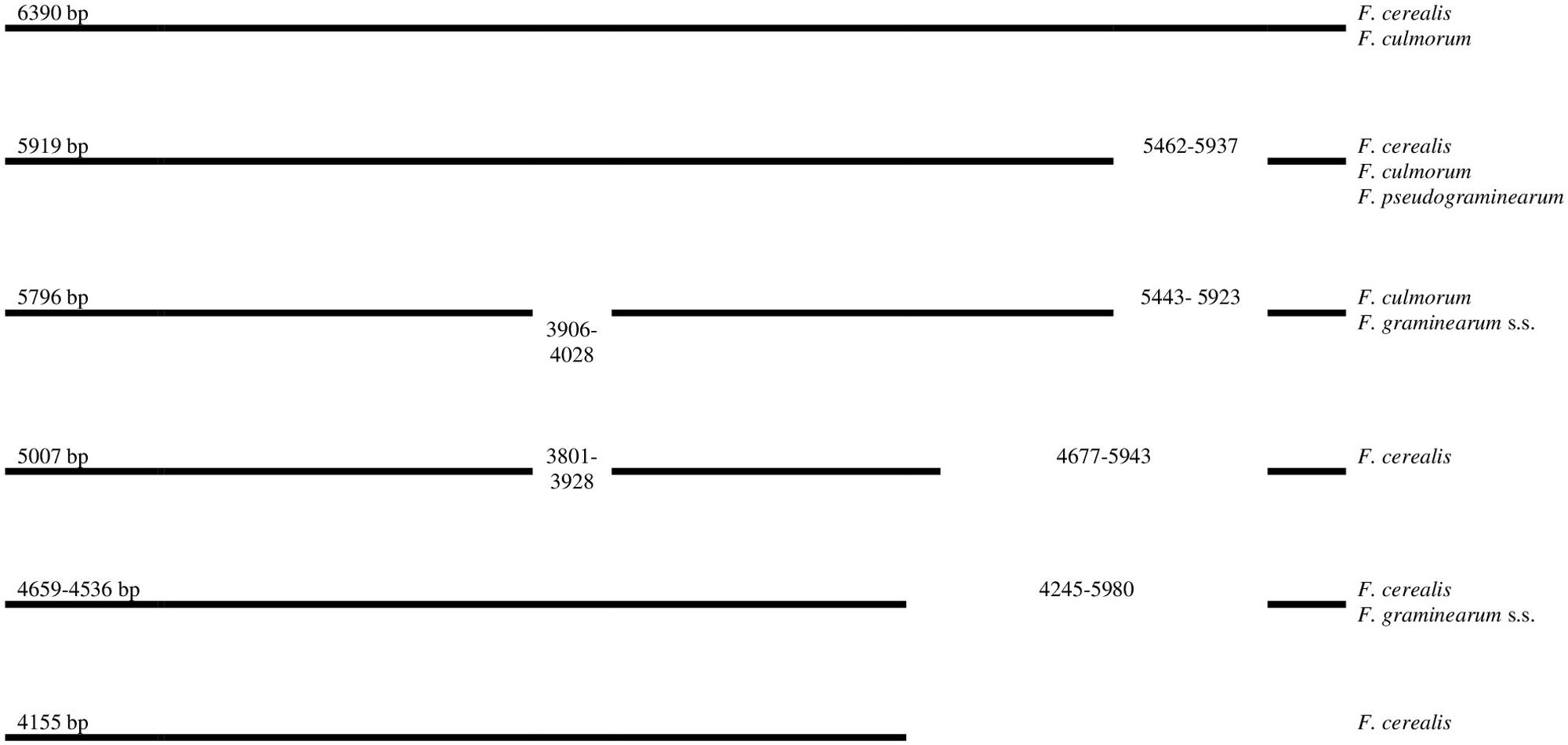
Figure 1. Schematic position of indels in LV-uORF in four Fusarium species. Position of indels (bp) are referred to F. cerealis (GenBank accession number MT036645, CBS 110268 strain).
Mobile Genetic Elements
A total of 47 introns has been identified in the basic set of mitochondrial protein-coding genes of the studied strains (Supplementary Files 1, 2a–6a). All introns belonged to the group I intron family with two exceptions: the first one was iIIcob intron, which was classified to the group II introns and encodes a protein of the reverse transcriptase family (Supplementary File 2a). Notably, the distribution of this intron within studied Fusaria seems to be very limited. We detected its presence in only 2 F. graminearum s.s. strains (16-462-z and INRA-156). The second exception was the i4cox2 intron, which lacked distinguishable features for classification (Supplementary File 3a).
Most introns harbored HEGs, either of the LAGLIDADG or GIY-YIG type, which are determined based on the differences in conserved amino acid motifs. Among 53 distinct HEGs, 36 were assigned as LAGLIDADG endonucleases being present in 9 out of 11 intron-rich genes. GIY-YIG endonucleases appear to be less commonly occurring HEGs in mitogenomes of Fusaria. We identified 15 distinct GIY-YIG endonucleases in only six out of eleven protein-coding genes. However, two HEGs [i4cob HEG (112 aa) and i6cob HEG (472 aa)] could not be precisely predicted through similarity searches with blastP, presumably due to loss of conserved amino acid motifs that reflect these functional differences. We found that orthologs of i4cob HEG present in GenBank are much larger in the other species; e.g., in Podospora comata this HEG is 314 aa in length (YP_009550007.1), while in Neurospora crassa – 477 aa (YP_009126715.1), which suggests deletion events during the divergence of Fusaria. The second i6cob HEG showed the highest identity (73.8 – 75.5%) to unclassified mitochondrial proteins (ATI20208.1, ATI20295.1, ATI20458.1) found in Juglanconis sp. (q-cover = 99-100%, E value = 0).
More than half of the introns (n = 26) identified in Fusaria were located in 3 subunits of the cytochrome c oxidase (cox genes): 17 in cox1, 5 in cox2, and 4 in cox3 (Supplementary Files 2a–6a). Positions of MGEs were highly conserved among different strains, however, with one exception. We found that two different introns icox1a and icox1b can share the same location in cox1 (Supplementary File 4a). Intron icox1a harbored a LAGLIDADG motif and was present in F. cerealis, while icox1b contained a GIY-YIG motif and was found in all strains of F. culmorum, F. graminearum s.s. and F. pseudograminearum. Further analyses of a larger collection of strains could confirm whether the LAGLIDAD HEG found in icox1a is fixed in F. cerealis.
HEGs drive mobility of MGEs, which can move across species, genera or even kingdoms (Koufopanou et al., 2002; Wu and Hao, 2014). We performed comprehensive blastP searches against GenBank to gather information on their distribution and the diversity among species. BlastP analyses revealed that most of the identified HEGs can be found in other Fusarium species as well as in species out of the genus Fusarium, however, with considerable variation in protein identity scores (Supplementary Files 2b–6b). Notably, for most HEGs (49 out of 53), protein identity was greater within than outside the genus Fusarium, which indicates their ancestral acquisition in the genus Fusarium. However, 4 HEGs showed higher similarity to sequences from distantly related species than to those found in the other Fusaria, suggesting their more recent acquisitions from distantly related donors. These include LAGLIDADG HEGs found in introns: i1nad5, i3cox1, i13cox1 and single i14cox1 GIY-YIG HEG. To test their horizontal transfer, we attempted to construct maximum-likelihood (ML) amino acid trees using GenBank records covering at least 85% of the query protein sequence (Figures 2–5).
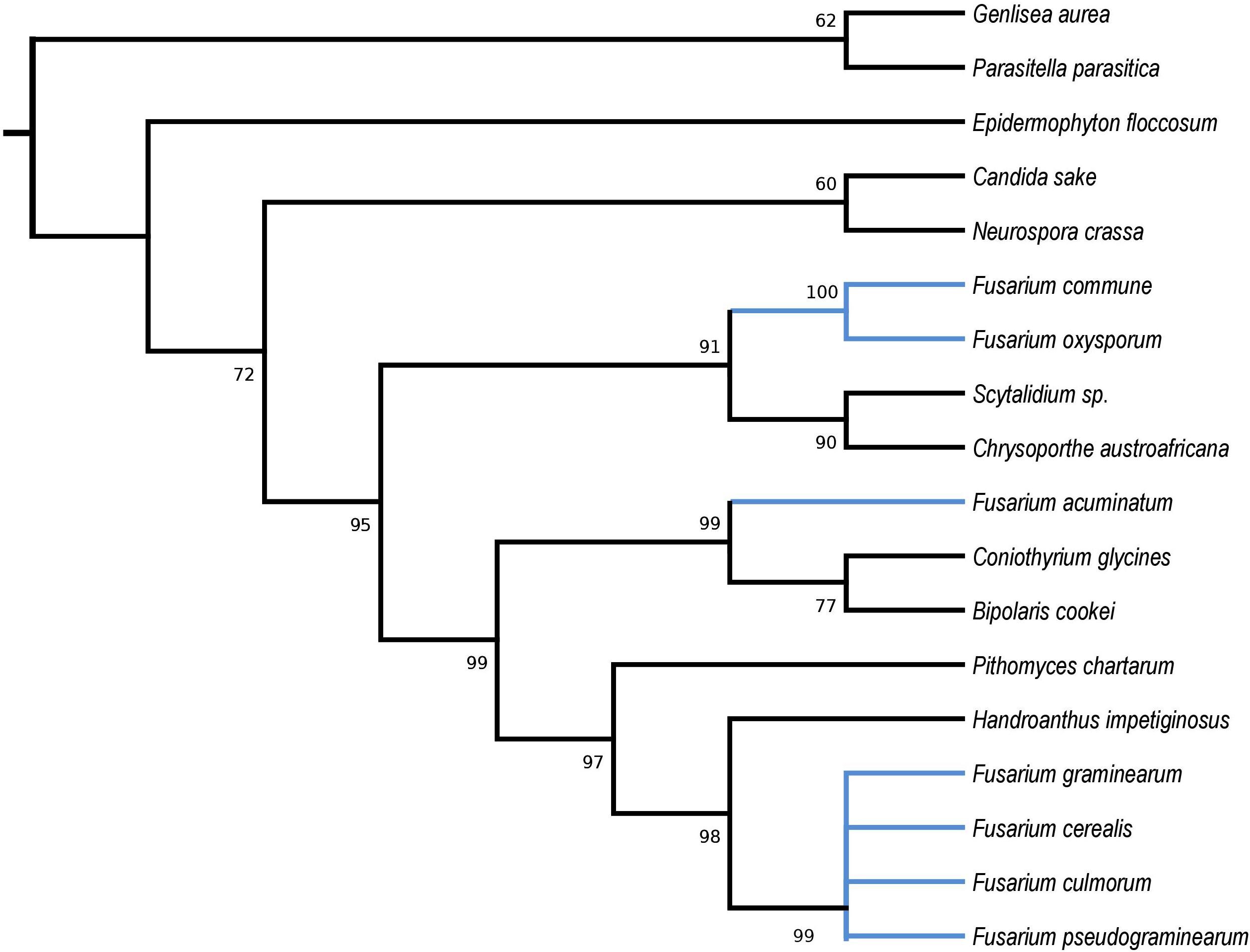
Figure 2. Maximum Likelihood tree inferred from i1nad5 LAGLIDADG HEG. GenBank accession numbers are given in brackets Bipolaris cookie (YP_009445560.1), Candida sake (YP_008475145.1), Chrysoporthe austroafricana (YP_009262028.1), Coniothyrium glycines (YP_009543504.1), Epidermophyton floccosum (YP_313631.1), Fusarium acuminatum (CDL73463.1), Fusarium cerealis (YP_009741100.1), Fusarium commune (YP_009437834.1), Fusarium culmorum (CDL73494.1), Fusarium graminearum (YP_001249315.1), Fusarium oxysporum (ATG24573.1), Fusarium pseudograminearum (CDL72485.1), Genlisea aurea (EPS67323.1), Handroanthus impetiginosus (PIM96989.1), Neurospora crassa (YP_009126712.1), Parasitella parasitica (YP_009059710.1), Pithomyces chartarum (YP_009415186.1), and Scytalidium sp. (QDG01253.1).
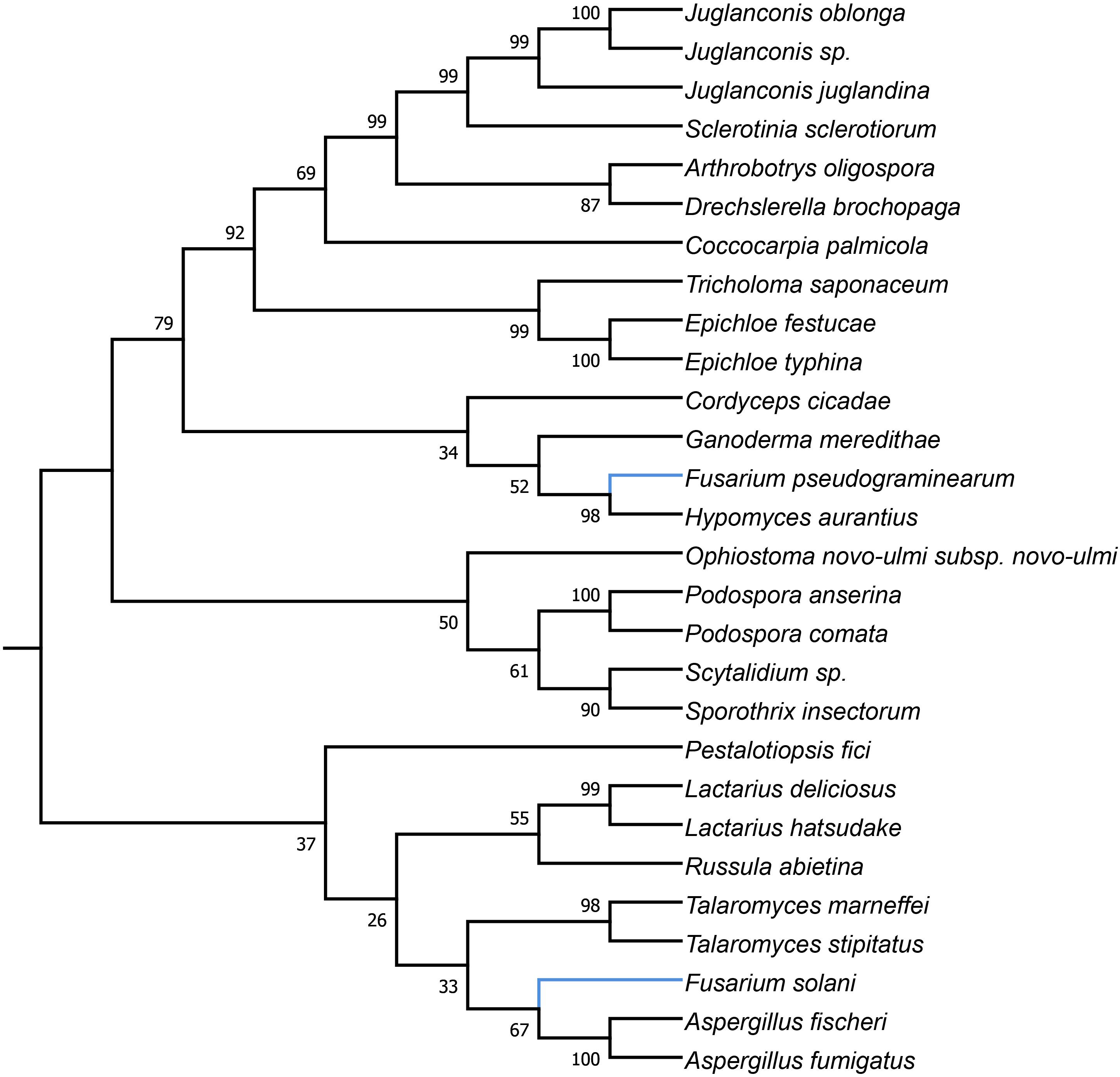
Figure 3. Maximum Likelihood tree inferred from i3cox1 LAGLIDADG HEG. GenBank accession numbers are given in brackets Arthrobotrys oligospora (QID02895.1), Aspergillus fischeri (AFD95944.1), Aspergillus fumigatus (YP_005353060.1), Coccocarpia palmicola (YP_009355407.1), Cordyceps cicadae (YP_009577892.1), Drechslerella brochopaga (QCW06877.1), Drechslerella brochopaga (YP_009568474.1), Epichloe festucae (YP_009327861.1), Epichloe typhina (YP_009327804.1), Fusarium pseudograminearum (QID41639.1), Fusarium solani (YP_005088111.1), Ganoderma meredithae (YP_009129958.1), Hypomyces aurantius (YP_009254043.1), Juglanconis juglandina (ATI20474.1), Juglanconis oblonga (ATI20215.1), Juglanconis sp. (ATI20300.1), Lactarius deliciosus (YP_009498182.1), Lactarius hatsudake (YP_009504192.1), Ophiostoma novo-ulmi (AVD96803.1), Pestalotiopsis fici (YP_009317077.1), Podospora anserina (NP_074927.1), Podospora comata (YP_009550011.1), Russula abietina (YP_009487192.1), Sclerotinia sclerotiorum (YP_009389070.1), Scytalidium sp. (QDG01205.1), Sporothrix insectorum (QGX43780.1), Talaromyces marneffei (NP_943724.1), Talaromyces stipitatus (AFD95918.1), and Tricholoma saponaceum (QIC20272.1).
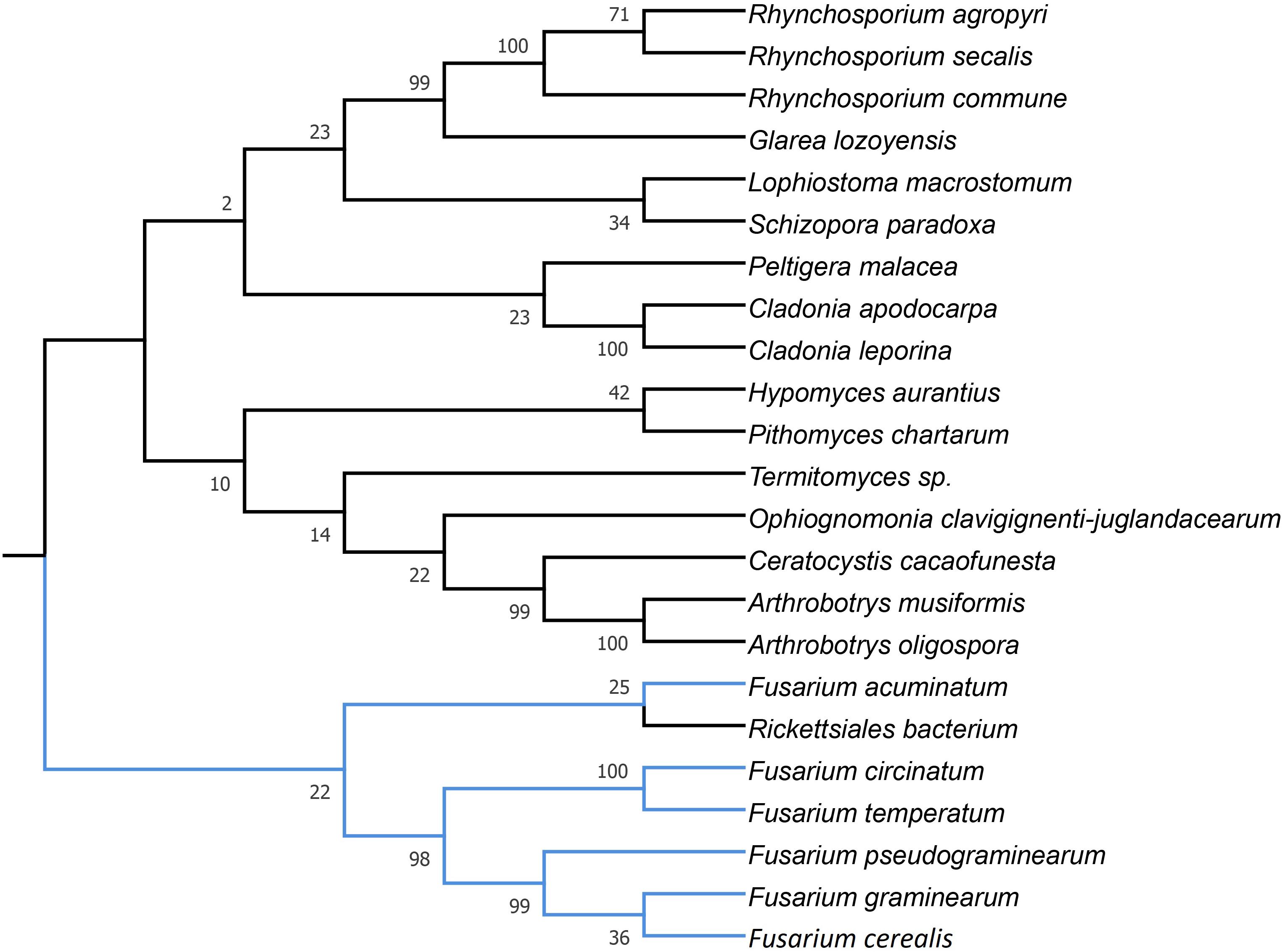
Figure 4. Maximum Likelihood tree inferred from i13cox1 LAGLIDADG HEG. GenBank accession numbers are given in brackets Arthrobotrys musiformis (YP_009574367.1), Arthrobotrys oligospora (QID02793.1), Ceratocystis cacaofunesta (YP_007507075.1), Ceratocystis cacaofunesta (YP_007507075.1), Cladonia apodocarpa (YP_009514117.1), Cladonia leporina (YP_009526663.1), Fusarium acuminatum (CDL73453.1), Fusarium circinatum (YP_008757706.1), Fusarium cerealis (YP_009741121.1), Fusarium graminearum (YP_001249335.1), Fusarium pseudograminearum (CDL72517.1), Fusarium temperatum (AKM98021.1), Glarea lozoyensis (AGN74496.1), Glarea lozoyensis (YP_009306750.1), Hypomyces aurantius (YP_009254047.1), Lophiostoma macrostomum (KAF2647136.1), Ophiognomonia clavigignenti-juglandacearum (ATI20563.1), Peltigera malacea (YP_005351183.1), Pithomyces chartarum (YP_009415166.1), Rhynchosporium agropyri (YP_008965308.1), Rhynchosporium commune (YP_008965356.1), Rhynchosporium secalis (YP_008965429.1), Rickettsiales bacterium (RYE15942.1), Schizopora paradoxa (YP_009690425.1), and Termitomyces sp. (AYE93211.1).
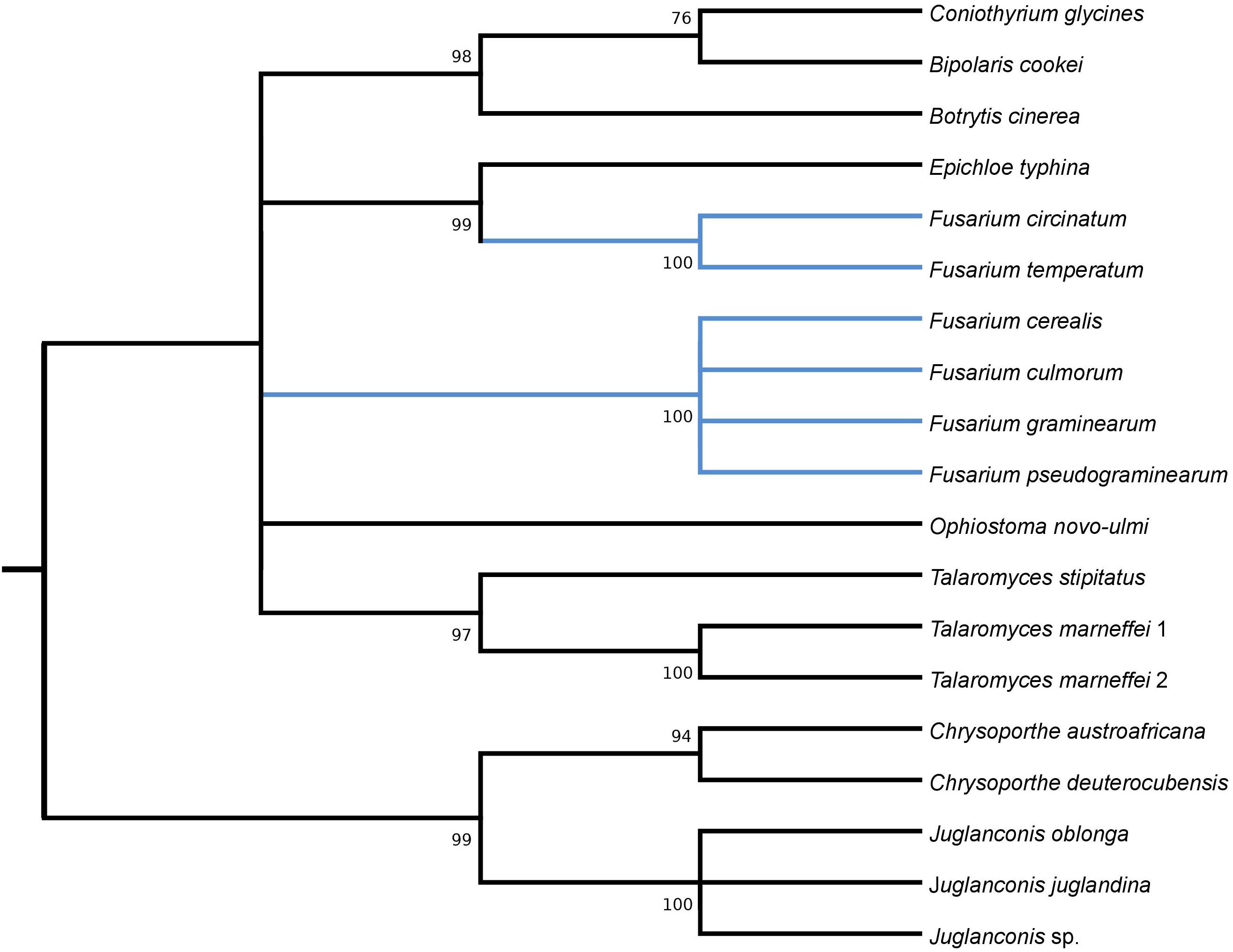
Figure 5. Maximum Likelihood tree inferred from i14cox1 GIY-YIG HEG. GenBank accession numbers are given in brackets Bipolaris cookei (YP_009445523.1), Botrytis cinerea (AGN49010.1), Chrysoporthe austroafricana (YP_009262000.1), Chrysoporthe deuterocubensis (YP_009262086.1), Coniothyrium glycines (YP_009543497.1), Epichloe typhina (YP_009327808.1), Fusarium cerealis (YP_009741122.1), Fusarium culmorum (CDL73523.1), Fusarium graminearum (YP_001249336.1), Fusarium pseudograminearum (QID41650.1), Fusarium circinatum (YP_008757707.1), Fusarium temperatum (AKM98023.1), Juglanconis juglandina (ATI20489.1), Juglanconis oblonga (ATI20223.1), Juglanconis sp. (ATI20312.1), Ophiostoma novo-ulmi (AVD96809.1), Talaromyces marneffei 1 (AFD95988.1), Talaromyces marneffei 2 (NP_943730.1), and Talaromyces stipitatus (AFD95924.1).
Phylogenetic analyses provided strong evidence for horizontal gene transfer in the case of i1nad5 LAGLIDADG HEG (Figure 2). On this particular tree, the LAGLIDADGs from different Fusaria are scattered in three different well-supported clades together with HEGs harbored by more distantly fungi such as Bipolaris cookei, Chrysoporthe austroafricana, Coniothyrium glycines, Pithomyces chartarum, and Scytalidium sp. Maximum Likelihood analyses also provided strong evidence for different evolutionary histories of two i3cox1 LAGLIDADG HEGs present in F. pseudograminearum and in F. solani. Phylogenetic trees depict that both HEGs cluster into a different clades (Figure 3). The HEG found in F. pseudograminearum forms a single cluster with Hypomyces aurantius, while the F. solani HEG shows its closest relationship to orthologs found in Aspergillus spp. However, phylogenetic analysis failed to support a different phylogenetic history of i13cox1 HEG in Fusaria. Many nodes on the tree had low bootstrap support (Figure 4). Supplementary File 4b depicts distribution of this HEG among fungal lineages. It displays high conservation among F. cerealis, F. culmorum, F. graminearum s.s., F. pseudograminearum, F. circinatum and F. temperatum (Identity = 90–100%, q-cover = 89–100%, E-value = 0), but only 61% identity to HEG found in F. acuminatum. BlastP analyses indicated its higher similarity (70–76%) to orthologs present in more distantly species such as Hypomyces aurantius, Ceratocystis cacaofunesta, Peltigera malacea, Arthrobotrys spp., Glarea lozoyensis, Cladonia spp., Termitomyces sp., Rhynchosporium spp. and Pithomyces chartarum. We hypothesize that the incorrect conclusion from this phylogenetic analysis may be likely due to high divergence within groups and a reduced number of taxa in the data set. The evidence for horizontal transfer of HEG between different fungal species is also indicated in the case of the i14cox1 GIY-YIG HEG tree by demonstrating a closer relationship of F. circinatum/F. temperatum to Epichloe typhina than to the remaining Fusaria (Figure 5).
The Majority of MGEs Show Highly Conserved Distribution in Fusarium Strains/Species
Twenty-six introns and 27 HEGs were shared by all fungal strains. The majority of them showed high sequence conservation. Eleven from these introns showed very high similarity with no size variation and pairwise identity ranging from 99.9 to 100%. 12 introns displayed some size variation due to small, strain-dependent indels mostly below 100 bp in size. When excluding indel variation, their nucleotide diversity was very low, with pairwise identity above 99%. The last three introns: the i11cox1, the i4cob intron, and the i1atp6 intron showed huge size variation (Supplementary Files 2a, 4a, 5a). Among these conserved introns, all contained two LAGLIDADGs among which the first was conserved (invariably present in all studied strains), while the second one (optional) was irregularly distributed among the strains, presumably due to independent of introns spread of HEGs. However, the distribution of optional HEGs across multiple strains was low. An optional i11bcox1 LAGLIDADG HEG (318 aa) was found in only one fungal mitogenome (CBS 119173). Only four fungal strains (all F. pseudograminearum) harbored optional LAGLIDADG HEG (387 aa) in the atp6 gene. The occurrence of the third optional i4cob LAGLIDADG HEG (252 aa) was more frequent, but still low, being found in 14% of strains. We also found that the unexceptionally short (112 aa) i4cob HEG displays optional distribution among fungal strains and was more frequent than the other optional HEGs being found in 27% of strains (Supplementary Files 1, 2a).
HEG Content Variation Between in F. culmorum and in F. graminearum s.s.
Among 53 HEGs identified in introns, 26 were optional (irregularly distributed among all strains). We analyzed their distribution in the context of species-specific occurrence in F. culmorum and in F. graminearum s.s. Mitogenomes of F. cerealis and F. pseudograminearum were not analyzed in the context of species-specific distribution of MGEs due to low number of representative strains. We found that among these optional HEGs, one located in i3acox2 intron was fixed in F. culmorum (Supplementary Files 3a,b). We also found another i4cox3 LAGLIDADG HEG in F. culmorum which was absent in four closely related Fusaria, but BlastP searches indicated its presence in other, more distantly F. solani (Supplementary Files 3a,b), however, with 23% difference in protein identity scores. Noteworthy, its nucleotide sequence appears to be highly conserved in F. culmorum fulfilling criterion of diagnostic marker, which should display enough conservation to achieve less variability within species than between species (Raja et al., 2017).
The absence of unique MGEs in F. graminearum s.s. prompted us to search for unique MGE patterns in F. graminearum s.s. Among protein-coding genes, we found that intron content variation in cox3 exhibits unique mosaicism in F. graminearum s.s. Cox3 contains four introns (Supplementary File 3a), among which two: i1cox3 and i2cox3 are present in all species. The third i3cox3 intron is present in F. cerealis and F. pseudograminearum, while the fourth the i4cox3 is uniquely present in F. culmorum. Thus, the lack of these last two introns in F. graminearum s.s. determines a unique MGE pattern in F. graminearum s.s.
Besides HEGs found in protein-coding genes, the mitogenomes of three Fusaria (F. cerealis, F. culmorum, and F. pseudograminearum) contained an approximately 1895 bp insertion between cox2 and nad4L, which is absent in F. graminearum s.s. This insertion harbors two ORFs: ORF 313 (942 bp) and ORF 132 (399 bp), both encoding LAGLIDADG HEGs. From a diagnostic point of view, the absence of these two HEGs in F. graminearum s.s. also appears to be promising diagnostic features separating F. graminearum s.s. from the remaining three species.
Discussion
Highly sensitive and precise detection and quantification of fungi are critical to support diagnosis for disease surveillance. Therefore, this research field has been revolutionized by the advent of molecular markers (Tsui et al., 2011). However, the detection of fungi is still a challenge, because fungal biomass is often present at very low and uneven concentrations in environmental or clinical samples. For that reason, the use of DNA extraction methods facilitating efficient recovery of fungal DNA is highly recommended, however, with even highly efficient extraction techniques, low amounts of fungal DNA are often extracted in the presence of large amounts of background DNA, hampering largely the sensitivity of the assays (Wickes and Wiederhold, 2018).
The use of assays targeting repeated genomic sequences such as internal transcribed spacer (ITS) of the rRNA region (official fungal barcode) provides one of the best solutions to this problem (Yang et al., 2018). Unfortunately, ITS fails to delineate most agriculturally important Fusarium species (Schoch et al., 2012). Consequently, for most Fusaria, diagnostic markers enabling their species-specific identification, have been developed based on single-copy nuclear loci (Nicolaisen et al., 2009; O’Donnell et al., 2015), which can limit successful detection of low amounts of fungal biomass (Kulik et al., 2015; Bilska et al., 2018b).
Targeting mitochondrial DNA could be a viable solution due to its high copy number in fungal cells (Krimitzas et al., 2013). The use of mtDNA for barcoding of fungi has been studied more than a decade ago (Seifert et al., 2007). In one of the first in-depth studies, it was demonstrated that the standard animal barcode cox1 could be highly effective in the resolution of Penicillium species. However, cox1 showed significant diagnostic limitations for Fusarium fungi (Gilmore et al., 2009) mostly due to the presence of paralogous copies of cox1 in studied strains and low species resolution. Besides, one of the major shortcomings of mitogenome-based studies in fungi is the difficulty in obtaining amplicons for further sequencing, mainly due to large indel polymorphism associated with irregular distribution of MGEs. Further study of Santamaria et al. (2009) showed that most mitochondrial genes of Ascomycota fungi contain MGEs, which largely limits their potential use for barcoding purposes.
Emerging next-generation sequencing provides a new opportunity for the rapid characterization of fungal mitogenomes. Mitogenomes can be assembled from the whole genome reads via either de novo assemblies or mapping to a reference genome (Tenaillon et al., 2011) providing an unbiased look into the patterns of MGE diversity. It is worth noting that exploration of this type of diversity on species or population level was previously not possible due to low numbers of complete mitogenomes. In one of the first large-scale studies on budding yeast, Saccharomyces cerevisiae, Wolters et al. (2015) revealed that the distribution of certain MGEs follows species divisions and even exhibits population-specific profiles. Further, but still, very limited studies on plant pathogenic fungi appeared to support this evidence. Despite the observed strain-dependent variation in MGE content, some of these elements appear to show species-specific occurrence in F. oxysporum (Brankovics et al., 2017), Fusarium fujikuroi species complex (FFSC) (Fourie et al., 2018) or more distantly related Colletotrichum spp. (Liang et al., 2017). In this study, we identified a single i3acox2 HEG displaying species-specific conservation in F. culmorum (Supplementary Files 3a,b). A previous study by Bilska et al. (2018b) demonstrated that targeting of this HEG through qPCR technology offers highly specific and sensitive quantification of F. culmorum.
We found that positions of MGEs were highly conserved among different strains. More than half of introns (n = 26) were found in cox genes (Supplementary Files 2a–6a), which is in line with accumulating evidence from various fungal lineages confirming remarkably high density in MGE content in especially cox1 (Sandor et al., 2018; Zubaer et al., 2018; Pogoda et al., 2019). This, however, is notably in contrast to recent findings which showed that other Fusaria display rather low intron density within their cox genes, which can be even intronless in species from F. oxysporum (Brankovics et al., 2017) and Fusarium fujikuroi species complex (FFSC) (Fourie et al., 2018). However, it is worth to note that mitogenomes of the strains analyzed in our study are MGE−rich in comparison with previously studied Fusaria (Brankovics et al., 2017; Fourie et al., 2018). Notably, there were large differences in frequency distribution of MGEs. The vast majority of them occur at relatively higher frequencies compared to others, such as the iIIcob intron or the i11bcox1 LAGLIDADG HEG (318 aa), raising the question on their role in gene regulation and their maintenance through selection (Guha et al., 2018; Zubaer et al., 2018).
We found that among 53 HEGs identified in introns, 9 showed irregular distribution within F. graminearum s.s., and only 4 within F. culmorum (Supplementary Files 2a–6a). The increased variation in HEG content within F. graminearum s.s. may be explained by its recombining reproductive behavior (outcrossing) (Trail, 2009; Talas and McDonald, 2015), which is assumed to favor intron mobility (Hausner, 2003). Unlike in F. graminearum s.s., no evidence of sexual reproduction was found in both F. culmorum and F. cerealis (Leslie and Summerell, 2008; Scherm et al., 2013). Sexual recombination can occur in F. pseudograminearum; however, it was rarely observed under field conditions (Summerell et al., 2001). Previous population genomic analyses by Kelly and Ward (2018) (restricted to North American populations) showed that in F. graminearum s.s., gene content variation in the nuclear genome displays population-specific patterns of conservation. We attempted to find out if variation in the MGE content shows population-specific profiles. To achieve it, we investigated if the distribution of optional HEGs reflects the geographic origin of the strains. We found that such a link does not exist. The same HEG patterns could be found in strains originated from distant geographic locations as in strains isolated from the same and even relatively small geographic locations (e.g., Luxembourg), which is indicative of low mobility of ancestrally acquired MGEs shared by different populations of this phylogenetic species. Our findings are, however, in sharp contrast to previous survey on Saccharomyces cerevisiae showing that the MGEs can be distributed in a population-specific manner (Wolters et al., 2015).
Assuming low mobility of MGEs in Fusaria, it could be speculated that HEG content variation will not follow cryptic divergence of other phylogenetic species from the Fusarium graminearum species complex (FGSC). Until now, only one complete mitogenome of F. gerlachii (Kulik et al., 2016b) is available in the GenBank database. Indeed, analysis of its mitogenome did not reveal unique MGEs separating F. gerlachii from F. graminearum s.s. (Supplementary Files 2b–6b). Besides MGE content variation, we previously showed that SNP polymorphism found in conserved introns can facilitate design of species-specific markers in FGSC. A previously developed mitochondrial-based qPCR assay has been designed based on such a polymorphism for species-specific detection and quantification of F. graminearum s.s. (Kulik et al., 2015). The assay has been demonstrated not to interfere with other phylogenetic species from FGSC, which indicates that this type of polymorphism could be useful in the assessment of cryptic diversity in Fusaria. This issue will need to be fully addressed in our further studies by analysis of mitogenomes from all known cryptic species from FGSC.
Blast alignments are commonly employed to reveal different evolutionary histories of genes (Zhu et al., 2014). In this study, protein homology searches in GenBank database showed that most HEGs found in four Fusarium species are shared with other fungal species (Supplementary Files 2b–6b). This variation was very low when comparing HEG orthologs from closely related F. cerealis, F. culmorum, F. gerlachii, F. graminearum s.s. and F. pseudograminearum, and increased when including other Fusaria. In most cases variation in protein identity scores was highest for HEG orthologs from other fungal genera, which is indicative of an ancestral acquisition of HEGs in the genus Fusarium.
MGEs, when fixed in population, are susceptible to degeneration. To ensure their survival, these elements can spread to new populations or even horizontally across species (Koufopanou et al., 2002; Wu and Hao, 2014). HEGs are well-adapted for horizontal transmission by the maintenance of highly conserved recognition sequences and the conserved nucleotide sites most critical for homing (Koufopanou et al., 2002). However, in this study, we found, that among a total of 53 HEGs, only 4 showed hallmarks of horizontal mobility across different fungi (Figures 2–5). Our analyses also suggest two cross-kingdom horizontal gene transfer events (Figures 2, 4). The first one is indicated by a higher (66.91%) protein identity (q-cover = 88%, E value = 3e-115) of i1nad5 LAGLIDADG HEG (Supplementary Files 6a,b) to the plant ortholog (PIM96989.1) found in Handroanthus impetiginosus than to the other fungal orthologs. The second event is indicated by higher (63.6%) identity of i13cox1 LAGLIDADG HEG (Supplementary Files 4a,b) to ortholog found in Rickettsiales bacterium (RYE15942.1) (q-cover = 94%, E value = 9e-174) than to the other fungal derived orthologs. BlastP searches showed that both HEGs gave no significant BLAST hits in species outside fungi, which suggests a cross-kingdom jump from a fungal donors to the plants (PIM96989.1) and alphaproteobacteria (RYE15942.1).
Overall, our results presented in this study demonstrate that the MGE diversity in fungal mitogenomes may lead to the development of novel diagnostic markers offering improved detection of fungi. Diagnostic markers for pathogenic fungi need to be tested against the range of strains from different geographical origins to ensure their uniformity. The geographically diverse mitogenomes provide valuable resources for the marker design. However, in case of F. cerealis and F. pseudograminearum more strains are needed for robust conclusions. Today, mitochondrial-based qPCR assays are available for both F. culmorum and F. graminearum s.s. (Kulik et al., 2015; Bilska et al., 2018b). qPCR remains a valuable option due to the ability to quantify fungal DNA, especially from difficult food or environmental samples from which extraction of high-molecular-weight DNA is problematic. In addition, the increased sensitivity of mitochondrial-based markers may reduce the incidence of scoring errors, making interpretation qPCR results less error-prone. Besides qPCR, other promising techniques such as loop-mediated isothermal amplification (LAMP) may be also considered (Panek and Frac, 2019). Among a number of molecular methods for the identification of fungi, barcoding DNA provides the best potential for biodiversity monitoring (Begerow et al., 2010). However, it would require multiple group-specific primers for sequencing of MGEs-barcodes, which is nowadays time-consuming and cost-intensive. Whatever the method, the revealed strong evidence for species-specific polymorphism in fungal mitogenomes opens entirely new perspectives to fungal diagnostics, which can be of great benefit in the field of agriculture and food security when highly sensitive detection of fungi is needed.
Conclusion
Mitogenomes of F. culmorum and F. graminearum s.s. are MGE-rich and harbor especially group I introns encoding LAGLIDADG and GIY-YIG HEGs. The majority of mitochondrial MGEs showed a highly conserved distribution in all studied strains. MGEs present in these species can be found in other Fusaria and in other fungal genera with considerable variation in protein identity scores. Intraspecies variation in MGE content is more evident in F. graminearum s.s. than in F. culmorum but is indicative of ancestral mosaicism of HEGs shared by different fungal populations. Among optional HEGs, one exhibits species-specific conservation in F. culmorum. F. graminearum s.s. does not harbor unique MGEs throughout the mitogenome, however, MGE patterns in cox3 and in the intergenic spacer between cox2 and nad4L may support identification of this species.
Data Availability Statement
The datasets generated for this study can be found in the NCBI GenBank database. MT036635, MT036636, MT036637, MT036638, MT036639, MT036640, MT036641, MT036642, MT036643, MT036644, MT036645, MT036646, MT036647, MT036648, MT036649, MT036650, MT036651, MT036652, MT036653, MT036654, MT036655, MT036656, MT036657, MT036658, MT036659, MT036660, MT036661, MT036662, MT036663, MT036664, MT036665, MT036666, MT036667, MT036668, MT036669, MT036670, MT036671, MT036672, MT036673, MT036674, MT036675, MT036676, MT036677, MT036678, MT036679, MT036680, MT036681, MT036682, MT036683, MT036684, MT036685, MT036686, MT036687, MT036688, MT036689, MT036690, MT036691, MT036692, MT036693, MT036694, MT036695, MT036696, MT036697, MT036698, MT036699, MT036701, MT036702, MT036703, MT036704, MT036705, MT036706, MT036707, MT036708, MT036709, MT036710, MT036711, MT036712, MT036713, MT036714, MT036715, MT036716, MT036717, MT036718, MT036719, MT036720, MT036721, MT036722, MT036723, MT036724, MT036725, MT036726, MT036727, MT036728, MT036729, MT036730, MT036731, MT036732, MT036733, MT036734, MT036735, MT036736, MT036737, MT036738, MT036739, MT036740, MT036741, MT036742, MT036743, MT036744, MT036745, MT036746, MT036747, MT036748, MT036749, MT036750, MT036751, MT036752, MT036753, MT036754, MT036755, MT036756, and MT036757.
Author Contributions
TK was responsible for the study design and wrote the manuscript. TK, AD, KB, MŻ, AS, SS, MB, MP, and AB-C contributed the isolation and strain growth. KB and MŻ extracted fungal DNA and prepared a library for whole-genome sequencing. TK and BB assembled the mitogenomes. BB, KM, and TM performed the mitogenome annotation. JS performed the phylogenetic analysis. AD, AS, KB, MB, MP, MŻ, and JS contributed the manuscript editing of relevant sections. JW edited supplementary files.
Funding
This research was funded by the National Science Center, Poland, grant number 2015/19/B/NZ9/01329. MB acknowledges the financial support of the project “Sentinelle” by the Administration des Services Techniques de l’Agriculture of Luxembourg.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01002/full#supplementary-material
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/orffinder/
- ^ http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl
References
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Al-Reedy, R. M., Malireddy, R., Dillman, C. B., and Kennell, J. C. (2012). Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet. Biol. 49, 2–14. doi: 10.1016/j.fgb.2011.11.008
Amarasinghe, C. C., Tittlemier, S. A., and Fernando, W. G. D. (2015). Nivalenol-producing Fusarium cerealis associated with fusarium head blight in winter wheat in Manitoba, Canada. Plant Pathol. 64, 988–995. doi: 10.1111/ppa.12329
Bartelli, T. F., Bruno, D. C. F., and Briones, M. R. S. (2018). Evidence for mitochondrial genome methylation in the yeast Candida albicans: a potential novel epigenetic mechanism affecting adaptation and pathogenicity? Front. Genet. 9:166. doi: 10.3389/fgene.2018.00166
Basse, C. W. (2010). Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 13, 321–326. doi: 10.1016/j.mib.2010.09.003
Begerow, D., Nilsson, H., Unterseher, M., and Maier, W. (2010). Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 87, 99–108. doi: 10.1007/s00253-010-2585-4
Bilska, K., Jurczak, S., Kulik, T., Ropelewska, E., Olszewski, J., Zelechowski, M., et al. (2018a). Species composition and trichothecene genotype profiling of Fusarium field isolates recovered from wheat in Poland. Toxins 10:325. doi: 10.3390/toxins10080325
Bilska, K., Kulik, T., Ostrowska-Kolodziejczak, A., Busko, M., Pasquali, M., Beyer, M., et al. (2018b). Development of a highly sensitive FcMito qPCR assay for the quantification of the toxigenic fungal plant pathogen Fusarium culmorum. Toxins 10:211. doi: 10.3390/toxins10050211
Brankovics, B., Kulik, T., Sawicki, J., Bilska, K., Zhang, H., de Hoog, G. S., et al. (2018). First steps towards mitochondrial pan-genomics: detailed analysis of Fusarium graminearum mitogenomes. PeerJ 6:e5963. doi: 10.7717/peerj.5963
Brankovics, B., van Dam, P., Rep, M., de Hoog, G. S., van der Lee, T. A. J., Waalwijk, C., et al. (2017). Mitochondrial genomes reveal recombination in the presumed asexual Fusarium oxysporum species complex. BMC Genomics 18:735. doi: 10.1186/s12864-017-4116-5
Bullerwell, C. E., and Lang, B. F. (2005). Fungal evolution: the case of the vanishing mitochondrion. Curr. Opin. Microbiol. 8, 362–369. doi: 10.1016/j.mib.2005.06.009
Calderone, R., Li, D. M., and Traven, A. (2015). System-level impact of mitochondria on fungal virulence: to metabolism and beyond. FEMS Yeast Res. 15:fov027. doi: 10.1093/femsyr/fov027
Chatre, L., and Ricchetti, M. (2014). Are mitochondria the Achilles’ heel of the Kingdom Fungi? Curr. Opin. Microbiol. 20, 49–54. doi: 10.1016/j.mib.2014.05.001
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Desjardins, A. E. (2006). Fusarium mycotoxins: chemistry, genetics, and biology. Am. Phytopathol. Soc. 56:337. doi: 10.1111/j.1365-3059.2006.01505.x
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Dubos, T., Pogoda, F., Ronellenfitsch, F. K., Junk, J., Hoffmann, L., and Beyer, M. (2012). Fractal dimension and shape parameters of asexual Fusarium spores from selected species: Which can be distinguished? J. Plant Dis. Prot. 119, 8–14. doi: 10.1007/BF03356413
Edgell, D. R. (2009). Selfish DNA: homing endonucleases find a home. Curr. Biol. 19, R115–R117. doi: 10.1016/j.cub.2008.12.019
Fourie, G., van der Merwe, N. A., Wingfield, B. D., Bogale, M., Tudzynski, B., Wingfield, M. J., et al. (2013). Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics 14:605. doi: 10.1186/1471-2164-14-605
Fourie, G., Van der Merwe, N. A., Wingfield, B. D., Bogale, M., Wingfield, M. J., and Steenkamp, E. T. (2018). Mitochondrial introgression and interspecies recombination in the Fusarium fujikuroi species complex. IMA Fungus 9, 37–48. doi: 10.5598/imafungus.2018.09.01.04
Franco, M. E. E., Lopez, S. M. Y., Medina, R., Lucentini, C. G., Troncozo, M. I., Pastorino, G. N., et al. (2017). The mitochondrial genome of the plant-pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS One 12:e0185545. doi: 10.1371/journal.pone.0185545
Geiser, D. M., Jimenez-Gasco, M. D., Kang, S. C., Makalowska, I., Veeraraghavan, N., Ward, T. J., et al. (2004). FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur. J. Plant Pathol. 110, 473–479. doi: 10.1023/B:EJPP.0000032386.75915.a0
Gilmore, S. R., GrÄfenhan, T., Louis-Seize, G., and Seifert, K. A. (2009). Multiple copies of cytochrome oxidase 1 in species of the fungal genus Fusarium. Mol. Ecol. Resour. 9, 90–98. doi: 10.1111/j.1755-0998.2009.02636.x
Guha, T. K., Wai, A., Mullineux, S. T., and Hausner, G. (2018). The intron landscape of the mtDNA cytb gene among the Ascomycota: introns and intron-encoded open reading frames. Mitochondrial DNA Part A 29, 1015–1024. doi: 10.1080/24701394.2017.1404042
Haugen, P., Simon, D. M., and Bhattacharya, D. (2005). The natural history of group I introns. Trends Genet. 21, 111–119. doi: 10.1016/j.tig.2004.12.007
Hausner, G. (2003). Fungal mitochondrial genomes, plasmids and introns. Appl. Mycol. Biotechnol. 3, 101–131. doi: 10.1016/S1874-5334(03)80009-6
Hueza, I. M., Raspantini, P. C. F., Raspantini, L. E. R., Latorre, A. O., and Gorniak, S. L. (2014). Zearalenone, an estrogenic mycotoxin, is an immunotoxic compound. Toxins 6, 1080–1095. doi: 10.3390/toxins6031080
Joardar, V., Abrams, N. F., Hostetler, J., Paukstelis, P. J., Pakala, S., Pakala, S. B., et al. (2012). Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics 13:698. doi: 10.1186/1471-2164-13-698
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, 5–9. doi: 10.1093/nar/gkn201
Kazan, K., and Gardiner, D. M. (2018). Fusarium crown rot caused by Fusarium pseudograminearum in cereal crops: recent progress and future prospects. Mol. Plant Pathol. 19, 1547–1562. doi: 10.1111/mpp.12639
Kelly, A. C., and Ward, T. J. (2018). Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen. PLoS One 13:e0194616. doi: 10.1371/journal.pone.0194616
Kolesnikova, A. I., Putintseva, Y. A., Simonov, E. P., Biriukov, V. V., Oreshkova, N. V., Pavlov, I. N., et al. (2019). Mobile genetic elements explain size variation in the mitochondrial genomes of four closely-related Armillaria species. BMC Genomics 20:351. doi: 10.1186/s12864-019-5732-z
Koufopanou, V., Goddard, M. R., and Burt, A. (2002). Adaptation for horizontal transfer in a homing endonuclease. Mol. Biol. Evol. 19, 239–246. doi: 10.1093/oxfordjournals.molbev.a004077
Krimitzas, A., Pyrri, I., Kouvelis, V. N., Kapsanaki-Gotsi, E., and Typas, M. A. (2013). A phylogenetic analysis of greek isolates of Aspergillus species based on morphology and nuclear and mitochondrial gene sequences. Biomed Res. Int. 2013:260395. doi: 10.1155/2013/260395
Kulik, T., Abarenkov, K., Buśko, M., Bilska, K., van Diepeningen, A. D., Ostrowska-Kołodziejczak, A., et al. (2017). ToxGen: an improved reference database for the identification of type B-trichothecene genotypes in Fusarium. PeerJ 5:e2992. doi: 10.7717/peerj.2992
Kulik, T., Brankovics, B., Sawicki, J., and van Diepeningen, A. (2016a). The complete mitogenome of Fusarium culmorum. Mitochondrial DNA Part A 27, 2425–2426. doi: 10.3109/19401736.2015.1030626
Kulik, T., Brankovics, B., Sawicki, J., and van Diepeningen, A. D. (2016b). The complete mitogenome of Fusarium gerlachii. Mitochondrial DNA Part A 27, 1895–1896. doi: 10.3109/19401736.2014.971275
Kulik, T., Ostrowska, A., Busko, M., Pasquali, M., Beyer, M., Stenglein, S., et al. (2015). Development of an FgMito assay: a highly sensitive mitochondrial based qPCR assay for quantification of Fusarium graminearum sensu stricto. Int. J. Food Microbiol. 210, 16–23. doi: 10.1016/j.ijfoodmicro.2015.06.012
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Leslie, J. F., and Summerell, B. A. (2008). The Fusarium Laboratory Manual. New York, NY: John Wiley & Sons.
Liang, X. F., Tian, X. L., Liu, W. K., Wei, T. Y., Wang, W., Dong, Q. Y., et al. (2017). Comparative analysis of the mitochondrial genomes of Colletotrichum gloeosporioides sensu lato: insights into the evolution of a fungal species complex interacting with diverse plants. BMC Genomics 18:171. doi: 10.1186/s12864-016-3480-x
Lin, R. M., Liu, C. C., Shen, B. M., Bai, M., Ling, J., Chen, G. H., et al. (2015). Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 15:5. doi: 10.1186/s12866-015-0341-8
Losada, L., Pakala, S. B., Fedorova, N. D., Joardar, V., Shabalina, S. A., Hostetler, J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. doi: 10.1111/1574-6968.12387
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331. doi: 10.1093/nar/gkh454
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Nicolaisen, M., Supronien, S., Nielsen, L. K., Lazzaro, I., Spliid, N. H., and Justesen, A. F. (2009). Real-time PCR for quantification of eleven individual Fusarium species in cereals. J. Microbiol. Methods 76, 234–240. doi: 10.1016/j.mimet.2008.10.016
O’Donnell, K., Ward, T. J., Robert, V., Crous, P. W., Geiser, D. M., and Kang, S. (2015). DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43, 583–595. doi: 10.1007/s12600-015-0484-z
Panek, J., and Frac, M. (2019). Loop-mediated isothermal amplification (LAMP) approach for detection of heat-resistant Talaromyces flavus species. Sci. Rep. 9:5846. doi: 10.1038/s41598-019-42275-x
Pantou, M. P., Kouvelis, V. N., and Typas, M. A. (2008). The complete mitochondrial genome of Fusarium oxysporum: insights into fungal mitochondrial evolution. Gene 419, 7–15. doi: 10.1016/j.gene.2008.04.009
Pavesi, A., Conterio, F., Bolchi, A., Dieci, G., and Ottonello, S. (1994). Identification of new eukaryotic tRNA genes in genomic DNA databases by a multistep weight matrix anaylsis of transcriptional control regions. Nucleic Acids Res. 22, 1247–1256. doi: 10.1093/nar/22.7.1247
Pogoda, C. S., Keepers, K. G., Nadiadi, A. Y., Bailey, D. W., Lendemer, J. C., Tripp, E. A., et al. (2019). Genome streamlining via complete loss of introns has occurred multiple times in lichenized fungal mitochondria. Ecol. Evol. 9, 4245–4263. doi: 10.1002/ece3.5056
Raja, H. A., Miller, A. N., Pearce, C. J., and Oberlies, N. H. (2017). Fungal identification using molecular tools: a primer for the natural products research community. J. Nat. Prod. 80, 756–770. doi: 10.1021/acs.jnatprod.6b01085
Sandor, S., Zhang, Y. J., and Xu, J. P. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Santamaria, M., Vicario, S., Pappada, G., Scioscia, G., Scazzocchio, C., and Saccone, C. (2009). Towards barcode markers in Fungi: an intron map of Ascomycota mitochondria. BMC Bioinformatics 10:S15. doi: 10.1186/1471-2105-10-s6-s15
Scherm, B., Balmas, V., Spanu, F., Pani, G., Delogu, G., Pasquali, M., et al. (2013). Fusarium culmorum: causal agent of foot and root rot and head blight on wheat. Mol. Plant Pathol. 14, 323–341. doi: 10.1111/mpp.12011
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Seifert, K. A., Samson, R. A., DeWaard, J. R., Houbraken, J., Lévesque, C. A., Moncalvo, J. M., et al. (2007). Prospects for fungus identification using CO1 DNA barcodes, with Penicillium as a test case. Proc. Natl. Acad. Sci. U.S.A. 104, 3901–3906. doi: 10.1073/pnas.0611691104
Shingu-Vazquez, M., and Traven, A. (2011). Mitochondria and fungal pathogenesis: drug tolerance, virulence, and potential for antifungal therapy. Eukaryot. Cell 10, 1376–1383. doi: 10.1128/ec.05184-11
Summerell, B. A., Burgess, L. W., Backhouse, D., Bullock, S., and Swan, L. J. (2001). Natural occurrence of perithecia of Gibberella coronicola on wheat plants with crown rot in Australia. Australas. Plant Pathol. 30, 353–356. doi: 10.1071/ap01045
Talas, F., and McDonald, B. A. (2015). Genome-wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genomics 16:996. doi: 10.1186/s12864-015-2166-0
Tenaillon, M. I., Hufford, M. B., Gaut, B. S., and Ross-Ibarra, J. (2011). Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol. Evol. 3, 219–229. doi: 10.1093/gbe/evr008
Trail, F. (2009). For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 149, 103–110. doi: 10.1104/pp.108.129684
Tsui, F., Wagner, M., Cooper, G., Que, J., Harkema, H., Dowling, J., et al. (2011). Probabilistic case detection for disease surveillance using data in electronic medical records. Online J. Public Health Inform. 3:ojhi.v3i3.3793. doi: 10.5210/ojphi.v3i3.3793
van der Lee, T., Zhang, H., van Diepeningen, A., and Waalwijk, C. (2015). Biogeography of Fusarium graminearum species complex and chemotypes: a review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 32, 453–460. doi: 10.1080/19440049.2014.984244
Waalwijk, C., Kastelein, P., de Vries, I., Kerenyi, Z., van der Lee, T., Hesselink, T., et al. (2003). Major changes in Fusarium spp. in wheat in the Netherlands. Eur. J. Plant Pathol. 109, 743–754. doi: 10.1023/a:1026086510156
Waalwijk, C., van der Heide, R., de Vries, I., van der Lee, T., Schoen, C., Costrel-de Corainville, G., et al. (2004). Quantitative detection of Fusarium species in wheat using TaqMan. Eur. J. Plant Pathol. 110, 481–494. doi: 10.1023/b:ejpp.0000032387.52385.13
Wang, H., Xiao, M., Kong, F. R., Chen, S., Dou, H. T., Sorrell, T., et al. (2011). Accurate and practical identification of 20 Fusarium species by seven-locus sequence analysis and reverse line blot hybridization, and an in vitro antifungal susceptibility study. J. Clin. Microbiol. 49, 1890–1898. doi: 10.1128/jcm.02415-10
Wickes, B. L., and Wiederhold, N. P. (2018). Molecular diagnostics in medical mycology. Nat. Commun. 9:5135. doi: 10.1038/s41467-018-07556-5
Wolters, J. F., Chiu, K., and Fiumera, H. L. (2015). Population structure of mitochondrial genomes in Saccharomyces cerevisiae. BMC Genomics 16:451. doi: 10.1186/s12864-015-1664-4
Wu, B. J., and Hao, W. L. (2014). Horizontal transfer and gene conversion as an important driving force in shaping the landscape of mitochondrial introns. G3 4, 605–612. doi: 10.1534/g3.113.009910
Yang, R. H., Su, J. H., Shang, J. J., Wu, Y. Y., Li, Y., Bao, D. P., et al. (2018). Evaluation of the ribosomal DNA internal transcribed spacer (ITS), specifically ITS1 and ITS2, for the analysis of fungal diversity by deep sequencing. PLoS One 13:e206428. doi: 10.1371/journal.pone.0206428
Yin, L. F., Hu, M. J., Wang, F., Kuang, H. H., Zhang, Y., Schnabel, G., et al. (2012). Frequent gain and loss of introns in fungal cytochrome b genes. PLoS One 7:e49096. doi: 10.1371/journal.pone.0049096
Zhu, Q., Kosoy, M., and Dittmar, K. (2014). HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genomics 15:717. doi: 10.1186/1471-2164-15-717
Keywords: Fusarium graminearum sensu stricto, F. culmorum, mitogenome, mobile genetic elements, mitochondrial introns, homing endonucleases
Citation: Kulik T, Brankovics B, van Diepeningen AD, Bilska K, Żelechowski M, Myszczyński K, Molcan T, Stakheev A, Stenglein S, Beyer M, Pasquali M, Sawicki J, Wyrȩbek J and Baturo-Cieśniewska A (2020) Diversity of Mobile Genetic Elements in the Mitogenomes of Closely Related Fusarium culmorum and F. graminearum sensu stricto Strains and Its Implication for Diagnostic Purposes. Front. Microbiol. 11:1002. doi: 10.3389/fmicb.2020.01002
Received: 19 February 2020; Accepted: 24 April 2020;
Published: 25 May 2020.
Edited by:
James Bernard Konopka, Stony Brook University, United StatesCopyright © 2020 Kulik, Brankovics, van Diepeningen, Bilska, Żelechowski, Myszczyński, Molcan, Stakheev, Stenglein, Beyer, Pasquali, Sawicki, Wyrȩbek and Baturo-Cieśniewska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomasz Kulik, dG9tYXN6a3VsaWs3NkBnbWFpbC5jb20=