 Kendall R. Corbin
Kendall R. Corbin Bridget Bolt
Bridget Bolt Carlos M. Rodríguez López
Carlos M. Rodríguez López- 1Environmental Epigenetics and Genetics Group, Department of Horticulture, College of Agriculture, Food and Environment, University of Kentucky, Lexington, KY, United States
- 2Biosystems and Agricultural Engineering, College of Agriculture, Food and Environment, University of Kentucky, Lexington, KY, United States
Introduction
Traditionally, breeding programs have tapped into two main sources of diversity for the identification of alleles capable of conferring beneficial traits to the crop of interest: (1) The genetic diversity of landraces and wild relatives, and (2) the creation of novel alleles using random or directed mutation approaches.
The generation of mutant populations has been the base for many successful breeding programs, including barley, soybean, tomato, and wheat (Jankowicz-Cieslak et al., 2017). Genetic variability is used to develop novel agronomically beneficial traits but also to determine the putative function of non-characterized genes (reverse epigenetics), and to identify genomic locations responsible for traits of interest (forward epigenetics) in crops (Rodríguez López and Wilkinson, 2015). Genetic variation has been conventionally achieved through insertional mutagenesis (transfer DNA, transposons, and entrapment tagging) (Ram et al., 2019), chemical (i.e., ethyl methanesulfonate, EMS) or ionizing radiation (i.e., gamma ray) treatment (Jankowicz-Cieslak et al., 2017), and more recently through targeted gene editing approaches (TALEN, ZNF, and CRISPR/Cas9) (Wolter et al., 2019).
This approach has been pivotal in transforming food production systems but with an ever-changing environmental landscape and increasing global population, improvement rates fall short of providing food security (Mehrabi et al., 2018). Concerted research efforts have been made to address this pitfall. Advancements in recent years include marker-assisted selection for genes of interest (Karanjalker and Begane, 2016), the development of speed breeding methodologies to shorten generation time (Mehrabi et al., 2018), targeted breeding using directed approaches (as opposed to trial-by-error breeding strategies), reverse-breeding strategies to introduce genetic diversity (ancestral traits) back into commercial crops (Palmgren et al., 2015), and random chemical mutagenesis (Jankowicz-Cieslak et al., 2017). These progressive breeding programs have made strides in increasing crop quality and quantity, but it is increasingly recognized that they do not target all possible sources of phenotypic variability (Rodríguez López and Wilkinson, 2015).
Epigenetic Mechanisms as a Source Variability for Crop Improvement
Epigenetic mechanisms regulate gene expression in response to plant development and environmental stimuli, ultimately affecting the plant's phenotype (Kumar, 2018). The field of applied epigenetics is a rapidly evolving area of research, stimulating new opportunities for the improvement of crop productivity. It is now widely accepted that epigenetic mechanisms have been the source of useful variability during crop varietal selection (Rodríguez López and Wilkinson, 2015; Crisp et al., 2016; Fortes and Gallusci, 2017; Gallusci et al., 2017). An early example of epigenetic breeding demonstrated the potential to improve crop performance and energy use efficiency (an important yield determinant) in a commercially valuable crop, rapeseed (Brassica napus), through recurrent epigenetic selection of isogenic lines (Hauben et al., 2009).
The major epigenetic mechanisms mediating these effects include histone modifications, DNA methylation and small RNA molecules, which act in an interactive, and redundant fashion to affect gene expression (Rodríguez López and Wilkinson, 2015). DNA methylation involves the addition of a methyl group to the 5th carbon of cytosines (forming 5-methylcytosine) by a set of enzymes called DNA methyltransferases. Gene promoter methylation has been associated to transcriptional repression (Kass et al., 1997). Importantly, this classic promoter methylation–gene expression model does not seem to be universal (Anastasiadi et al., 2018). A more complex model has been suggested where the methylation of the promoter and the gene body exerts separate influences on gene expression (Wang et al., 2015). In general, a negative association has been found between gene body methylation (GbM) and gene expression (Anastasiadi et al., 2018; Magris et al., 2019). Nevertheless, GbM has also been linked to higher gene expression in certain gene subclasses (Dubin et al., 2015; Anastasiadi et al., 2018).
Exploiting the relationship between gene DNA methylation and expression through deliberate perturbation of DNA methylation via exogenous interventions, has been proposed as a fast method to generate variability for crop improvement (Rodríguez López and Wilkinson, 2015; Gallusci et al., 2017). This can be achieved by using methods that are analogous to those used in mutation breeding, application of chemical inhibitors of DNA methyltransferases, which causes stochastic genome-wide DNA demethylation (Geyer et al., 2011; Amoah et al., 2012; Browne et al., 2020) and so, generates new variants carrying epi-alleles (defined here, as any of a group of otherwise identical genes that differ in the extent of their methylation). The use of targeted epigenome editing techniques capable of altering DNA methylation or histone modifications in the genes of interest may also be employed (Vojta et al., 2016). The induction of such epialleles lead to changes in gene expression and phenotype. This strategy, similar to mutation breeding, can be used to generate novel, and valuable epigenetic variation for crop improvement (Amoah et al., 2012). Novel epialleles can be inherited, even over multiple rounds of sexual reproduction, (Amoah et al., 2012; Tricker et al., 2013a,b). More importantly, they can become fixed in hybrids, resulting in heritable molecular and physiological phenotypes (Wibowo et al., 2018) without the need for genetic modification.
The Holobiont as a Potential Breeding Target
Thus far, breeding approaches consider the crop as a single species. However, in nature, plants do not exist as an entity, but cohabit with diverse microbes (collectively termed the plant microbiota). The assemblage of the host and the microbiota is referred to as the holobiont, while the term hologenome is used to indicate the entire set of genomes within the holobiont (Figure 1).
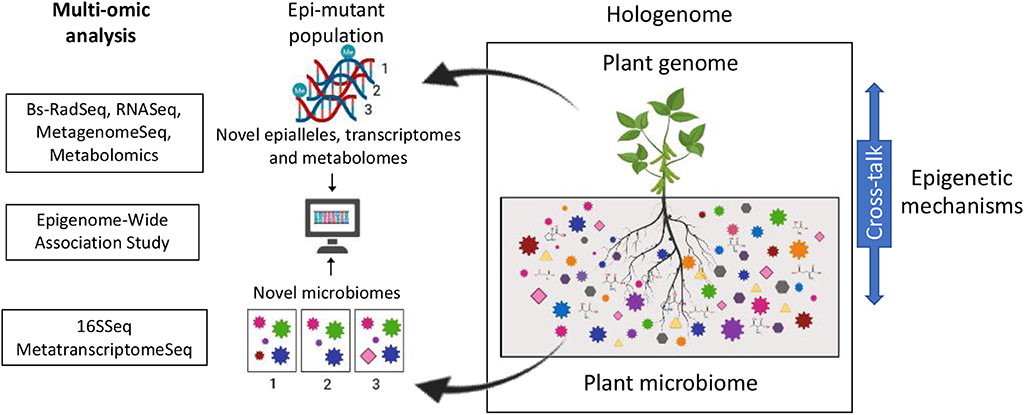
Figure 1. Proposed model for the identification of genes regulating plant-microbe interactions via the stochastic perturbation of DNA methylation patterns in plant populations. Exogenously induced DNA demethylation randomly generates novel epialleles in the plant population. These in turn alter the expression of genes which will alter the plant phenotype (i.e., microbiomes) of the plants carrying such novel alleles. Changes in DNA methylation in the plant population can be assessed using reduced representation approaches such as BsRADSeq. Changes in microbiome composition and functionality can be assessed using 16S sequencing and/or Meta-transcriptome analysis. Association between individual changes in DNA methylation (novel epialleles) and changes in phenotype (novel microbial community composition and functions) will be determined using epigenome wide association studies. Analysis of plant gene expression and metabolite production will be used as a validation step of the identified associations.
Microbial communities provide multiple benefits to their hosts, including better access to nutrients, enhanced growth, and improved tolerance to biotic and abiotic insult (Powell et al., 2015; Harman and Uphoff, 2019). The realization of the importance of microbiotas for crop health, has led to the development of prebiotic and probiotic cocktails intended to enhance the holobiont (Rodriguez et al., 2019). However, their effectiveness has been proven to be highly inconsistent (Ownley et al., 2003). This inconsistency has been attributed to different causes, including the host plant or pathogen genotype (Yang et al., 2018), agricultural practices (Schippers et al., 1990), and loss of activity due to mutation of the biocontrol strain (Duffy and Défago, 2000). These findings highlight the on-going need to better understand the host-microbiota interactions for crop improvement, and suggest non-specific additive cocktails are sub-optimal for general application (Rodriguez et al., 2019).
Interestingly, a handful of recent studies have shown that crop domestication and breeding have inadvertently altered the microbial communities of the target crops (Leff et al., 2017; Chaluvadi and Bennetzen, 2018), suggesting that microbiota composition is a trait that can be bred (Wissuwa et al., 2009). Unfortunately, very little research effort has been invested in understanding host-microbe interactions from a community perspective (Beilsmith et al., 2019). This makes the understanding of what makes a “good microbiota host” critical in the conceptualization of breeding programs aimed at improving productivity, quality and sustainability through the management of the holobiont (Wissuwa et al., 2009).
Drivers of Soil Microbiome Composition
The below-ground microbiota is considered the richer and more functionally active of all the plant's compartments and is consequentially the most intensely studied (Rodriguez et al., 2019). The structural and functional diversity of the plant microbiota fluctuate in response to environmental and host pressures, creating a biological feedback loop (for an extensive review see Vorholt, 2012).
Although abiotic cues, such as soil physical and chemical characteristics, climate, and spatial features, have traditionally considered the main drivers of the plant microbiota composition (Weckert, 2016; Delgado-Baquerizo et al., 2020), it is now well-established that host factors such as genotype (Bulgarelli et al., 2015; Chaluvadi and Bennetzen, 2018), developmental stage (Sugiyama et al., 2014; Wagner et al., 2016) and plant organ (Wagner et al., 2016) contribute to the shaping and maintenance of the plant's microbial communities (Rodriguez et al., 2019).
The Plant as a Driver of Its Own Microbiota
The plant itself plays a key role in shaping the composition and relative abundance of microbial species in their rhizosphere through physical (e.g., root architecture) (Chaluvadi and Bennetzen, 2018; Saleem et al., 2018) and chemical mechanisms (i.e., exudation of small molecules that serve as growth substrates or signals for suitable microbial partners, and as antimicrobials or growth deterrents for other microbes) (Bais et al., 2006). Interestingly, the diversity of the microbial community sharply decreases with proximity to the plant (bulk soil>rhizosphere>endophytic compartment) (Rodriguez et al., 2019). This observed decline in diversity suggests that plants impose a strong selective pressure on their immediate surroundings (Bais et al., 2006).
Historically, the plant's genotype has been recognized as one of the key factors mediating this selectivity. The genotype impacts the microbial community to promote plant growth, improve abiotic stress tolerance, facilitate pathogen defense (Jones et al., 2019). However, Wibowo et al. (2018) showed that genetically identical plants, displaying distinct epigenomes differentially alter their microbiota. Symbionts have been shown to provide beneficial selectable variation to their hosts through the modification of the epigenetic profiles (Gilbert et al., 2010; Gómez-Díaz et al., 2012). Moreover, multiple studies have highlighted the importance of epigenetic mechanisms in regulating the cross-talk between the host and its associated microbiota (Figure 1; Gómez-Díaz et al., 2012; Cheeseman and Weitzman, 2015; Wang et al., 2016; Zhu et al., 2016; Kumar et al., 2018). Enhancing this genome-microbiome communication could be the target for future breeding programs.
Epimutagenesis: a Tool to Identify Genes Regulating Host-Microbiome Interactions
Understanding what makes a plant a good host for its microbiota will be essential to harness the plant-microbiota complex for crop improvement. Identifying the genes that enable plants to regulate the assembly of a beneficial root microbiota is paramount for future breeding programs aimed at a sustainably improving productivity and produce quality of produce. Although, very little is known about the molecular mechanisms regulating the assembly of plant microbiota, multiple studies have pointed at the importance of epigenetic mechanisms regulating the cross-talk between the host and its associated microbiota. We propose that capitalizing on the availability of epimutant populations as a platform for the identification of loci involved in the regulation of plant microbiota assemblies using epigenome wide association studies (EWAS) (Flanagan, 2015; Birney et al., 2016; Jullian Fabres et al., 2017; Figure 1). The rationale behind the use of an EWAS approach resides on the stochastic nature of the epimutations induced by the application of exogenous demethylating agents. This approach would generate a unique set of epialleles in each plant within the epimutant population, which could partially alter the plant's ability to direct the assembly of its microbiota. Identifying the genes regulating host/microbe interactions will provide with valuable targets for breeding aiming at producing crops capable of assembling healthier microbiotas. This in turn, has the potential to aid global efforts in addressing the challenge of feeding a growing population via the development of socially and environmentally responsible agricultural approaches (Mehrabi et al., 2018).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, Hatch Program number 2352987000.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amoah, S., Kurup, S., Rodriguez Lopez, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of brassica rapa for forward and reverse epi-genetics. BMC Plant Biol. 12:193. doi: 10.1186/1471-2229-12-193
Anastasiadi, D., Esteve-Codina, A., and Piferrer, F. (2018). Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin. 11:37. doi: 10.1186/s13072-018-0205-1
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Beilsmith, K., Thoen, M. P. M., Brachi, B., Gloss, A. D., Khan, M. H., and Bergelson, J. (2019). Genome-wide association studies on the phyllosphere microbiome: embracing complexity in host-microbe interactions. Plant J. 97, 164–181. doi: 10.1111/tpj.14170
Birney, E., Smith, G. D., and Greally, J. M. (2016). Epigenome-wide association studies and the interpretation of disease -omics. PLoS Genet. 12:e1006105. doi: 10.1371/journal.pgen.1006105
Browne, L., Mead, A., Horn, C., Chang, K., Celikkol, Z., Henriquez, C., et al. (2020). Experimental DNA demethylation associates with changes in growth and gene expression of oak tree seedlings. G3 (Bethesda). 10, 1019–1028. doi: 10.1534/g3.119.400770
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Chaluvadi, S., and Bennetzen, J. L. (2018). Species-associated differences in the below-ground microbiomes of wild and domesticated setaria. Front. Plant Sci. 9:1183. doi: 10.3389/fpls.2018.01183
Cheeseman, K., and Weitzman, J. B. (2015). Host-parasite interactions: an intimate epigenetic relationship. Cell Microbiol. 17, 1121–1132. doi: 10.1111/cmi.12471
Crisp, P. A., Ganguly, D., Eichten, S. R., Borevitz, J. O., and Pogson, B. J. (2016). Reconsidering plant memory: intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2:e1501340. doi: 10.1126/sciadv.1501340
Delgado-Baquerizo, M., Reich, P. B., Trivedi, C., Eldridge, D. J., Abades, S., Alfaro, F. D., et al. (2020). Multiple elements of soil biodiversity drive ecosystem functions across biomes. Nat. Ecol. Evol. 4, 210–220. doi: 10.1038/s41559-019-1084-y
Dubin, M. J., Zhang, P., Meng, D., Remigereau, M. S., Osborne, E. J., Paolo Casale, F., et al. (2015). DNA methylation in arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 4:e05255. doi: 10.7554/eLife.05255
Duffy, B. K., and Défago, G. (2000). Controlling instability in gacS-gacA regulatory genes during inoculant production of Pseudomonas fluorescens biocontrol strains. Appl. Environ. Microbiol. 66, 3142–3150. doi: 10.1128/AEM.66.8.3142-3150.2000
Flanagan, J. M. (2015). Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol. Biol. 1238, 51–63. doi: 10.1007/978-1-4939-1804-1_3
Fortes, A. M., and Gallusci, P. (2017). Plant stress responses and phenotypic plasticity in the epigenomics era: perspectives on the grapevine scenario, a model for perennial crop plants. Front. Plant Sci. 8:82. doi: 10.3389/fpls.2017.00082
Gallusci, P., Dai, Z., Génard, M., Gauffretau, A., Leblanc-Fournier, N., Richard-Molard, C., et al. (2017). Epigenetics for plant improvement: current knowledge and modeling avenues. Trends Plant Sci. 22, 610–623. doi: 10.1016/j.tplants.2017.04.009
Geyer, K. K., Rodríguez López, C. M., Chalmers, I. W., Munshi, S. E., Truscott, M., Heald, J., et al. (2011). Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni. Nat. Commun. 2:424. doi: 10.1038/ncomms1433
Gilbert, S. F., McDonald, E., Boyle, N., Buttino, N., Gyi, L., Mai, M., et al. (2010). Symbiosis as a source of selectable epigenetic variation: taking the heat for the big guy. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 365, 671–678. doi: 10.1098/rstb.2009.0245
Gómez-Díaz, E., Jordà, M., Peinado, M. A., and Rivero, A. (2012). Epigenetics of host-pathogen interactions: the road ahead and the road behind. PLoS Pathog. 8:e1003007. doi: 10.1371/journal.ppat.1003007
Harman, G. E., and Uphoff, N. (2019). Symbiotic root-endophytic soil microbes improve crop productivity and provide environmental benefits. Scientifica 2019:9106395. doi: 10.1155/2019/9106395
Hauben, M., Haesendonckx, B., Standaert, E., Van Der Kelen, K., Azmi, A., Akpo, H., et al. (2009). Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. U.S.A. 106, 20109–20114. doi: 10.1073/pnas.0908755106
Jankowicz-Cieslak, J., Mba, C., and Till, B. J. (2017). “Mutagenesis for crop breeding and functional genomics,” in Biotechnologies for plant mutation breeding, eds. J. Jankowicz-Cieslak, T. H. Tai, J. Kumlehn, and B. J. Till (Cham: Springer International Publishing), 3–18. doi: 10.1007/978-3-319-45021-6_1
Jones, P., Garcia, B. J., Furches, A., Tuskan, G. A., and Jacobson, D. (2019). Plant host-associated mechanisms for microbial selection. Front. Plant Sci. 10:862. doi: 10.3389/fpls.2019.00862
Jullian Fabres, P., Collins, C., Cavagnaro, T. R., and Rodríguez López, C. M. (2017). A Concise review on multi-omics data integration for terroir analysis in Vitis vinifera. Front. Plant Sci. 8:1065. doi: 10.3389/fpls.2017.01065
Karanjalker, G., and Begane, N. (2016). Breeding perennial fruit crops for quality improvement. Erwerbs-Obstbau 58, 119–126. doi: 10.1007/s10341-015-0264-4
Kass, S. U., Pruss, D., and Wolffe, A. P. (1997). How does DNA methylation repress transcription? Trends Genet. 13, 444–449. doi: 10.1016/S0168-9525(97)01268-7
Kumar, S. (2018). Epigenomics of plant responses to environmental stress. Epigenomes 2, 1–17. doi: 10.3390/epigenomes2010006
Kumar, V., Khare, T., Shriram, V., and Wani, S. H. (2018). Plant small RNAs: the essential epigenetic regulators of gene expression for salt-stress responses and tolerance. Plant Cell Rep. 37, 61–75. doi: 10.1007/s00299-017-2210-4
Leff, J. W., Lynch, R. C., Kane, N. C., and Fierer, N. (2017). Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 214, 412–423. doi: 10.1111/nph.14323
Magris, G., Di Gaspero, G., Marroni, F., Zenoni, S., Tornielli, G. B., Celii, M., et al. (2019). Genetic, epigenetic and genomic effects on variation of gene expression among grape varieties. Plant J. 99, 895–909. doi: 10.1111/tpj.14370
Mehrabi, Z., Ellis, E. C., and Ramankutty, N. (2018). The challenge of feeding the world while conserving half the planet. Nat. Sustain. 1, 409–412. doi: 10.1038/s41893-018-0119-8
Ownley, B. H., Duffy, B. K., and Weller, D. M. (2003). Identification and manipulation of soil properties to improve the biological control performance of phenazine-producing Pseudomonas fluorescens. Appl. Environ. Microbiol. 69, 3333–3343. doi: 10.1128/AEM.69.6.3333-3343.2003
Palmgren, M. G., Edenbrandt, A. K., Vedel, S. E., Andersen, M. M., Landes, X., Østerberg, J. T., et al. (2015). Are we ready for back-to-nature crop breeding? Trends Plant Sci. 20, 155–164. doi: 10.1016/j.tplants.2014.11.003
Powell, J. R., Welsh, A., and Hallin, S. (2015). Microbial functional diversity enhances predictive models linking environmental parameters to ecosystem properties. Ecology 96, 1985–1993. doi: 10.1890/14-1127.1
Ram, H., Soni, P., Salvi, P., Gandass, N., Sharma, A., Kaur, A., et al. (2019). Insertional mutagenesis approaches and their use in rice for functional genomics. Plants 8:310. doi: 10.3390/plants8090310
Rodríguez López, C. M., and Wilkinson, M. J. (2015). Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 6:397. doi: 10.3389/fpls.2015.00397
Rodriguez, P. A., Rothballer, M., Chowdhury, S. P., Nussbaumer, T., Gutjahr, C., and Falter-Braun, P. (2019). Systems biology of plant-microbiome interactions. Mol. Plant 12, 804–821. doi: 10.1016/j.molp.2019.05.006
Saleem, M., Law, A. D., Sahib, M. R., Pervaiz, Z. H., and Zhang, Q. (2018). Impact of root system architecture on rhizosphere and root microbiome. Rhizosphere 6, 47–51. doi: 10.1016/j.rhisph.2018.02.003
Schippers, B., Bakker, A. W., Bakker, P. A. H. M., and Van Peer, R. (1990). Beneficial and deleterious effects of HCN-producing pseudomonads on rhizosphere interactions. Plant Soil 129, 75–83. doi: 10.1007/BF00011693
Sugiyama, A., Ueda, Y., Zushi, T., Takase, H., and Yazaki, K. (2014). Changes in the bacterial community of soybean rhizospheres during growth in the field. PLoS ONE 9:e100709. doi: 10.1371/journal.pone.0100709
Tricker, P., Rodríguez López, C., Hadley, P., Wagstaff, C., and Wilkinson, M. (2013b). Pre-conditioning the epigenetic response to high vapor pressure deficit increases the drought tolerance of Arabidopsis thaliana. Plant Signal. Behav. 8:e25974. doi: 10.4161/psb.25974
Tricker, P. J., López, C. M. R., Gibbings, G., Hadley, P., and Wilkinson, M. J. (2013a). Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int. J. Mol. Sci. 14, 6674–6689. doi: 10.3390/ijms14046674
Vojta, A., Dobrinić, P., Tadić, V., Bočkor, L., Korać, P., Julg, B., et al. (2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615–5628. doi: 10.1093/nar/gkw159
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Wagner, M. R., Lundberg, D. S., Del Rio, T. G., Tringe, S. G., Dangl, J. L., and Mitchell-Olds, T. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7:12151. doi: 10.1038/ncomms12151
Wang, H., Beyene, G., Zhai, J., Feng, S., Fahlgren, N., Taylor, N. J., et al. (2015). CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl. Acad. Sci. U.S.A. 112, 13729–13734. doi: 10.1073/pnas.1519067112
Wang, M., Weiberg, A., Lin, F. M., Thomma, B. P. H. J., Huang, H. D., and Jin, H. (2016). Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2:16151. doi: 10.1038/nplants.2016.151
Weckert, M. (2016). Vineyard terroir - is there a link with soil microbiology? Wine Viticult. J. 31, 39–40.
Wibowo, A., Becker, C., Durr, J., Price, J., Spaepen, S., Hilton, S., et al. (2018). Partial maintenance of organ-specific epigenetic marks during plant asexual reproduction leads to heritable phenotypic variation. Proc. Natl. Acad. Sci. U.S.A. 115, E9145–E9152. doi: 10.1073/pnas.1805371115
Wissuwa, M., Mazzola, M., and Picard, C. (2009). Novel approaches in plant breeding for rhizosphere-related traits. Plant Soil 321, 409–430. doi: 10.1007/s11104-008-9693-2
Wolter, F., Schindele, P., and Puchta, H. (2019). Plant breeding at the speed of light: the power of CRISPR/Cas to generate directed genetic diversity at multiple sites. BMC Plant Biol. 19:176. doi: 10.1186/s12870-019-1775-1
Yang, M., Mavrodi, D. V., Thomashow, L. S., and Weller, D. M. (2018). Differential response of wheat cultivars to Pseudomonas brassicacearum and Take-All Decline Soil. Phytopathology 108, 1363–1372. doi: 10.1094/PHYTO-01-18-0024-R
Keywords: holobiont, crop breeding, sustainable agriculture, microbiome, epigenetics, epimutant populations
Citation: Corbin KR, Bolt B and Rodríguez López CM (2020) Breeding for Beneficial Microbial Communities Using Epigenomics. Front. Microbiol. 11:937. doi: 10.3389/fmicb.2020.00937
Received: 22 February 2020; Accepted: 20 April 2020;
Published: 15 May 2020.
Edited by:
Nikolay Vassilev, University of Granada, SpainReviewed by:
Suresh Kumar, Indian Agricultural Research Institute (ICAR), IndiaLuciano Kayser Vargas, Secretaria Estadual da Agricultura, Pecuária e Irrigação, Brazil
Copyright © 2020 Corbin, Bolt and Rodríguez López. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos M. Rodríguez López, Y21ybzI2NyYjeDAwMDQwO3VreS5lZHU=