 Inès Khemiri
Inès Khemiri Faiza Tebbji
Faiza Tebbji Adnane Sellam
Adnane Sellam- 1CHU de Québec Research Center, Université Laval, Quebec City, QC, Canada
- 2Department of Microbiology, Infectious Diseases and Immunology, Faculty of Medicine, Université Laval, Quebec City, QC, Canada
Copper homeostasis is an important determinant for virulence of many human pathogenic fungi such as the highly prevalent yeast Candida albicans. However, beyond the copper transporter Ctr1, little is known regarding other genes and biological processes that are affected by copper. To gain insight into the cellular processes that are modulated by copper abundance in C. albicans, we monitored the global gene expression dynamic under both copper depletion and excess using RNA-seq. Beyond copper metabolism, other different transcriptional programs related to fungal fitness such as stress responses, antifungal sensitivity, host invasion and commensalism were modulated in response to copper variations. We have also investigated the transcriptome of the mutant of the copper utilization regulator, mac1, and identified potential direct targets of this transcription factor under copper starvation. We also showed that Mac1 was required for the invasion and adhesion to host cells and antifungal tolerance. This study provides a framework for future studies to examine the link between copper metabolism and essential functions that modulate fungal virulence and fitness inside the host.
Introduction
The redox properties of copper (Cu) make this trace element crucial for biological systems as it serves as an essential cofactor of enzymes that function in many biological processes including iron acquisition, antioxidative defense, and energy metabolism (Festa and Thiele, 2011). Cu is also toxic for the cell and its accumulation should be tightly monitored and kept at homeostatic levels. In the context of host-pathogen interaction, Cu is thought to be elemental for what is known as nutritional immunity of host cells during fungal infections (Hood and Skaar, 2012; Samanovic et al., 2012; Djoko et al., 2015). Cu is differentially distributed across different anatomical sites of the human body where it is mostly abundant in skeleton and bone marrow, skeletal muscle, liver, brain, and blood (Linder et al., 1998). Host cells seems to use both Cu-sequestration and Cu-poisoning to limit fungal pathogens from proliferating in different niches. In the case of the meningitis causing agent Cryptococcus neoformans, Cu is available at limiting concentrations in the brain interstitium while it is highly abundant in lung airways (Ballou and Wilson, 2016). For the highly prevalent human pathogenic yeast Candida albicans, Cu availability is highly dynamic for the same anatomical niche within the host. For instance, upon earlier phase of kidney colonization, C. albicans is confronted by high levels of Cu which is considered as a host-imposed Cu-poisoning strategy (Mackie et al., 2016; Culbertson et al., 2020). This earlier Cu spike is followed by a rapid sequestration by renal tissues to probably limit fungal growth as C. albicans activates its own Cu utilization machinery to promote its fitness.
Our current knowledge on regulatory mechanisms of Cu homeostasis of eukaryotic cells came mainly from works in the model yeast Saccharomyces cerevisiae. When Cu becomes limiting, S. cerevisiae promotes Cu import through the activation of the transcription factor Mac1 that induces the transcription of the high affinity Cu membrane transporters Ctr1 and Ctr3 (González et al., 2008). Transcript levels of the ferric reductase Fre1 and Fre7 that reduces Cu (Cu2+→Cu+) prior to its uptake by Ctr1 is also activated by Mac1 (van Bakel et al., 2005). Response to Cu excess is primarily controlled by the transcription factor Cup2 that activates different Cu-chaperones such as Cup1-1, Crs5 and Ccc2 in addition to the Cu-transporting P-type ATPase that are required for Cu tolerance (González et al., 2008).
Pathogenic fungi are exposed to a labile pool of Cu within the human host and has consequently evolved a tight regulatory control to ensure cellular homeostasis of this trace element. As in S. cerevisiae, C. albicans uses Mac1 to mediate the activation of both the Cu transporter Ctr1 and the ferric reductase Fre7 under Cu starvation (Marvin et al., 2003; Woodacre et al., 2008). Furthermore, C. albicans uses the P-type ATPase Crp1 that function as a Cu extrusion pump to survive in high Cu environments (Weissman et al., 2000). C. albicans has an ortholog of Cup2 that is required for Cu tolerance (Homann et al., 2009), however, its role as a transcriptional modulator of Cu detoxification has not been explored so far. Interestingly, under Cu excess, C. albicans activates the Cu-dependent superoxide dismutase Sod1 (Cu-Sod1) to neutralize the superoxide anion while it uses the Mn-requiring Sod3 (Mn-Sod3) under Cu limitation (Li et al., 2015). As Sod enzymes use metals as cofactors to convert superoxide to oxygen and hydrogen peroxide, C. albicans shifts metal co-factors for superoxide dismutase depending on Cu abundance in the colonized niches. C. neoformans depends on the Cu-transporters Ctr1 and Ctr4 for Cu uptake and on the Cu-metallothioneins MT1 and MT2 for Cu detoxification (Ding et al., 2011, p. 99). In this pathogenic fungus, both Cu uptake and detoxification are governed by the same transcriptional regulator Cuf1 (Ding et al., 2011, p. 99; Garcia-Santamarina et al., 2018). In Aspergillus fumigatus, intracellular Cu uptake relies on the Cu-transporters ctrA2 and ctrC that are both under the control of the transcriptional factor Afmac1 (Cai et al., 2017). Alteration of either uptake or detoxification processes in C. albicans, A. fumigatus, C. neoformans and the dimorphic fungus Histoplasma capsulatum impairs fungal virulence and fitness (Waterman et al., 2007; Ding et al., 2013; Sun et al., 2014; Mackie et al., 2016; Cai et al., 2017) suggesting that Cu metabolism might be a promising therapeutic targeted to treat fungal infections.
While Cu homeostasis is an important virulence determinant in C. albicans (Mackie et al., 2016), the impact of Cu availability on the transcriptome of this important human pathogen remain unexplored. Furthermore, beyond the Cu transporter Ctr1, little is known regarding other genes or biological process that are affected by Cu abundance or modulated by Mac1 at the genome level in C. albicans. To gain insight into the cellular processes that are modulated by Cu abundance in C. albicans, we monitored the global gene expression dynamic under both Cu depletion and excess using RNA-seq. Our data uncovered that, in addition to Cu utilization genes, other different cellular processes related to fungal fitness were modulated in response to Cu fluctuations. We have also investigated the transcriptome of mac1 mutant and identified potential direct targets of this transcription factor under Cu starvation. We also showed that Mac1 was required for the invasion and the adhesion to human enterocytes and antifungal tolerance. Thus, this study provides a framework for future studies to examine the link between Cu metabolism and essential functions that modulate fungal virulence and fitness inside the host.
Materials and Methods
Fungal Strains and Media
Candida albicans was routinely maintained at 30°C on YPD (1% yeast extract, 2% peptone, 2% dextrose, with 50 mg/ml uridine). The C. albicans WT strain SN250 (ura3/:imm434/URA3 iro1:IRO1/iro1:imm434 his1:hisG/his:hisG leu2:CdHIS1/leu2:CmLEU2 arg4/arg4) (Noble et al., 2010) used in this study derives from the SC5314 clinical strain. mac1 (mac1:LEU2/mac1:HIS1) deletion mutant come from the transcription factor deletion collection (Homann et al., 2009) that was constructed in SN125 (his1/his1, leu2/leu2, arg4/arg4, URA3/ura3:imm434, IRO1/iro1:imm434).
To build the mac1 complemented strain, the MAC1 gene was reintegrated into the null mutant mac1 strain using pDUP3 plasmid (Gerami-Nejad et al., 2013). Briefly, the MAC1 locus containing its endogenous promoter [(−678,0) intergenic region] was amplified by PCR using primers containing flanking sequences homologous to pDUP3. The resulting pDUP3-MAC1 construct was digested by SfiI and integrated into the NEU5L genomic site of the mac1 strain as previously described (Gerami-Nejad et al., 2013) using lithium acetate transformation (Wilson et al., 2000). Transformants were selected on YPD plates supplemented with 200 μg/ml nourseothricin and correct integration was verified by PCR. Primers used for MAC1 cloning in pDUP3 plasmid and for the diagnosis of pDUP3-MAC1 integration are listed in the Supplementary Table S1.
Growth Inhibition Assays
All chemicals used in this study were provided by Sigma-Aldrich (St. Louis, MO, United States). Working stock solutions of Copper (II) Sulfate (CuSO4; 532 mM), Bathocuproinedisulfonic acid disodium salt (BCS; 8 mM), Bathophenanthrolinedisulfonic acid (BPS; 8 mM) and fluconazole (3.3 mM) were prepared using Milli-Q sterile water. Amphotericin B and miconazole stock solutions were prepared using dimethyl sulfoxide (DMSO; Sigma-Aldrich) at a concentration of 1 mM and 2 mM, respectively.
For growth assays in liquid YPD medium, overnight cultures of C. albicans were resuspended in fresh YPD medium at an OD600 of 0.05 and added to a flat-bottom 96-well plate in a total volume of 100 μl per well in addition of the tested compounds. For each experiment, a compound-free positive growth control and a cell-free negative control were included. Growth assay curves were performed in triplicate in 96-well plates using a Sunrise plate-reader (Tecan) at 30°C under constant agitation with OD600 readings taken every 10 min for 48 h.
For spot dilution assays, overnight cultures were diluted to an OD600 of 0.1 and fivefold serial dilutions were prepared in distilled water. A total of 4 μl of each dilution were spotted on YP-agar with different carbon sources (glucose, maltose, glycerol, and ethanol) at 2% or on YPD plates with the different antifungals: amphotericin B (1 μg/ml), miconazole (0.5 μg/ml), and fluconazole (0.5 μg/ml). Plates were incubated at 30°C for 3 days and imaged using the SP-imager system.
Expression Analysis by RNA-Seq and qPCR
Overnight cultures of mac1 mutant and WT (SN250) strains were diluted to an OD600 of 0.1 in 40 ml of fresh YPD-uridine medium and grown at 30°C under agitation (200 rpm) to an OD600 of 0.4. Cultures were then either left untreated or exposed to either CuSO4 (2 mM) or BCS (400 μM), and incubated at 30°C for 30 min. For each condition, a total of two biological replicates were considered for RNA-seq analysis. Cells were then harvested by centrifugation at 3,000 × g for 5 min and the pellets were quick-frozen and stored at −80°C. Total RNA was extracted using an RNAeasy purification kit (Qiagen) and glass bead lysis in a Biospec Mini 24 bead-beater. Total RNA was eluted, assessed for integrity on an Agilent 4200 TapeStation System prior to cDNA library preparation. The NEBNext UltraTM II RNA Library Prep Kit for Illumina was used to construct the RNA-seq library following the manufacturer’s instruction. The quality, quantity and the size distribution of the libraries were determined using an Agilent Bioanalyzer. A 2 × 100 paired-end sequencing of cDNAs were performed using an Illumina Novaseq6000 sequencing system.
For qPCR confirmation experiments, a total of three biological and three assay replicates were performed. cDNA was synthesized from 1 μg of total RNA using High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). The mixture was incubated at 25°C for 10 min, 37°C for 120 min and 85°C for 5 min. Two units per microliter of RNAse H (NEB) was added to remove RNA and samples were incubated at 37°C for 20 min. qPCR was performed using a LightCycler 480 Instrument (Roche Life Science) for 40 amplification cycles with the PowerUp SYBR Green master mix (Applied Biosystems). The reactions were incubated at 50°C for 2 min, 95°C for 2 min and cycled for 40 times at 95°C, 15 s; 54°C, 30 s; 72°C, 1 min. Fold-enrichment of each tested transcripts was estimated using the comparative ΔΔCt method. To evaluate the gene expression level, the results were normalized using Ct values obtained from Actin (ACT1, C1_13700W_A). Primer sequences used for this analysis are summarized in Supplementary Table S1.
RNA-Sequencing Data Analysis
Adaptor sequences and low quality score bases (Phred score < 30) were first trimmed using Trimmomatic (Bolger et al., 2014). The resulting reads were aligned to the C. albicans reference assembly SC5314 from Ensembl1, using STAR (Dobin et al., 2013). Read counts are obtained using HTSeq (Anders et al., 2015) and are represented as a table which reports, for each sample (columns), the number of reads mapped to a given gene (rows). For all downstream analyses, we excluded lowly expressed genes with an average read count lower than 1 across all samples, resulting in 5,942 genes in total. The R package limma (Ritchie et al., 2015) was used to identify differences in gene expression levels between treated and non-treated samples. Nominal p-values were corrected for multiple testing using the Benjamini–Hochberg method. Differentially expressed transcripts in the Supplementary Tables S2, S4 were identified using a false-discovery rate (FDR) of 5% and twofold enrichment cut-off. Gene ontology (GO) analysis was performed using GO Term Finder of the Candida Genome Database (Skrzypek et al., 2017). The GSEA Pre-Ranked tool2 (Subramanian et al., 2005) was used to determine statistical significance of correlations between the characterized transcriptomes with a ranked gene list or GO biological process terms as described by Sellam et al. (2014). All RNA-seq raw data are available in the Supplementary Table S5 and also at Gene Expression Omnibus (GEO) with the accession number GSE147697.
HT-29 Adherence and Damage Assay
Damage to the human colon epithelial cell line HT-29 was assessed using a lactate dehydrogenase (LDH) cytotoxicity detection kitPLUS (Roche), which measures the release of the LDH enzyme in the growth medium. HT-29 cells were grown in 96-well plate as monolayers in McCoy’s medium supplemented with 10% FBS at 1 × 104 cells per well and incubated at 37°C with 5% CO2 overnight. HT-29 cells were then infected with C. albicans cells at MOI cell:yeast of 1:2 in the presence or the absence of BCS (100 μM or 800 μM), for 24 h at 37°C with 5% CO2. HT-29 cells were also incubated with 100 μM or 800 μM BCS alone to rule out any toxic effect of this compound on HT-29 cells. No discernable growth defect of HT-29 cells was noticed with both BCS concentrations. Following incubation, 100 μl of supernatant was removed from each experimental well and LDH activity in this supernatant was determined by measuring the absorbance at 490 nm (OD490) following the manufacturer’s instructions.
For the adherence assay, C. albicans cells were co-incubated with HT-29 cells at 37°C and 5% CO2 for 1 h. Non-adherent cells were removed by rinsing five times with 1 ml PBS and cells were then fixed with 4% paraformaldehyde. HT-29 cells were permeabilized with 0.5% Triton X-100 and adherent fungal cells were stained with 2 μM calcofluor white during 30 min in the dark at room temperature. Adherent cells were visualized using Cytation 5 high-content microscope with 20× magnification and DAPI filter. For each well, at least 10 fields were photographed.
Results
Global Transcriptional Responses to Copper Depletion and Repletion in C. albicans
To gain insight into the cellular processes that are modulated by Cu abundance in the pathogenic yeast C. albicans, we monitored the global gene expression dynamic under both Cu depletion (400 μM BCS) and excess (2 mM CuSO4) using RNA-seq. Transcripts associated with both Cu utilization and detoxification were identified by comparing the transcriptional profiles of WT cells treated with BCS and CuSO4, respectively, to that of non-treated WT cells. In response to Cu excess, C. albicans activated transcripts associated with Cu detoxification and transport including the cytosolic small chaperone ATX1, the metallothionein CUP1, the P-type ATPase CCC2 and the Cu efflux pump CRP1 (Figures 1A,B). We also found that genes of Cu uptake such as the Cu transporter CTR1, the transcription factor MAC1 and the cupric reductase FRE7 were downregulated (Figure 1A). Transcripts involved in iron uptake, and both cytosolic and mitochondrial ribosome biogenesis were activated whereas those related to glycolysis, energy and, NAD and hexose/glucose metabolisms were repressed (Figure 1B). Genes related to drug response and detoxification such as the MFS transporter MDR1, as well as its transcriptional activator MRR1, together with other MFS (FLU1, NAG3, NAG4, TPO3, TPO4), ABC (MLT1), and MaTE (ERC1, ERC3) transporters were significantly induced. Among repressed transcripts, we also found many genes of the ergosterol biosynthesis pathway (ERG1, ERG3, ERG11, ARE2) in addition to their bona fide transcriptional regulator Upc2 (Supplementary Table S2).
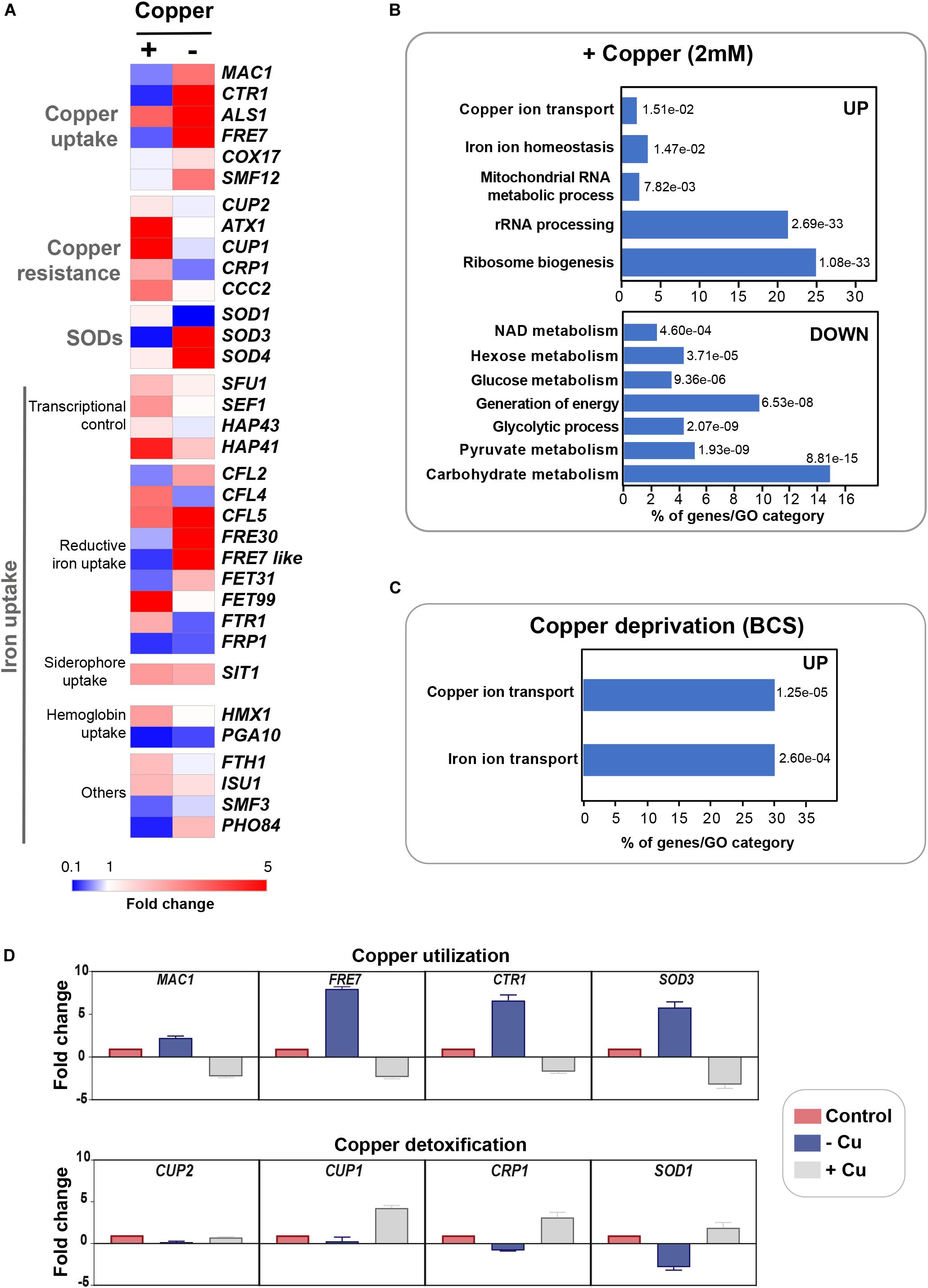
Figure 1. Genome-wide transcriptional profiling of C. albicans to Cu variations by RNA-seq. (A) Heat map visualization of the transcript levels of the Cu homeostasis pathway in C. albicans in response to Cu excess and deprivation. C. albicans WT cells were exposed to either 2 mM CuSO4 or 400 μM BCS, and incubated at 30°C for 30 min. Transcripts associated with both Cu utilization (copper “–”) and detoxification (copper “+”) were identified by comparing the transcriptional profiles of WT cells treated with BCS and CuSO4, respectively, to that of non-treated WT cells. (B,C) Gene function and biological process enriched in the transcriptional profiles of C. albicans growing under Cu excess (B) and limitation (C). (D) qPCR validation of RNA-seq data. Transcript levels of both Cu utilization (MAC1, CTR1, FRE7, and SOD3) and detoxification (CUP1, CRP1, SOD1, and CUP2) genes were assessed and fold-changes were calculated using the comparative ΔΔCt method. Data were normalized using Ct values obtained from actin gene in each condition.
Under Cu deprivation, as in other fungi, C. albicans upregulated genes required for Cu internalization including the Cu transporter Ctr1 and the ferric reductases Fre7, Fre30, and orf19.7077 that reduce Cu to facilitate its uptake by Ctr1 (Figures 1A,C and Supplementary Table S2). Other transcripts including the manganese transporter SMF12 and the ABC transporter SNQ2 were also activated. We also found that under Cu depletion, C. albicans upregulated the Mn-dependant superoxide dismutase SOD3 (Mn-SOD3) and repressed the Cu-dependant SOD1 (Cu-SOD1). Reversely, under Cu excess, Cu-SOD1 were activated and Mn-SOD3 repressed. This switch from Cu to Mn cofactors of Sods is an adaptative strategy for C. albicans cells to resist to reactive oxygen species (ROS) inside the host when Cu become limiting (Li et al., 2015). Taken together, our data suggest that Cu abundance did not affect exclusively Cu detoxification and utilization pathways but also other biological processes that might require this essential micronutrient to fulfill their functions.
qPCR confirmed gene expression alterations as shown by RNA-seq for both Cu utilization (MAC1, CTR1, FRE7, and SOD3) and detoxification (CUP1, CRP1, SOD1, and CUP2) genes under both Cu deprivation and excess, respectively (Figure 1D).
Gene Set Enrichment Analysis of the C. albicans Cu-Sensitive Transcriptomes
Gene set enrichment analysis (GSEA) was used to mine the transcriptomes associated with both Cu starvation and excess, to uncover potential resemblance with C. albicans genome annotations and other experimental large-scale omics data (Subramanian et al., 2005; Sellam et al., 2014; Supplementary Table S3). Figures 2A,C summarize the significant correlations of the Cu-responsive transcriptomes with either functional GO categories, C. albicans transcriptional signatures in different physiological conditions or in specific mutant background, and also with genome-wide promoter occupancies data. For both Cu availability conditions, GSEA recapitulated most of the GO correlations as identified by the conventional methodology. Cells growing in Cu excess exhibited a transcriptional profile similar to that of different mutants of transcription factors such as upc2, efg1, and ace2 and to mutants of different signaling pathways including Ras1-cAMP-PKA (ras1) and the AGC protein kinase Sch9 pathways in addition to a significant correlation with a set of genes with protomers enriched in Cat8 binding motif (Figures 2A,B and Supplementary Table S3).
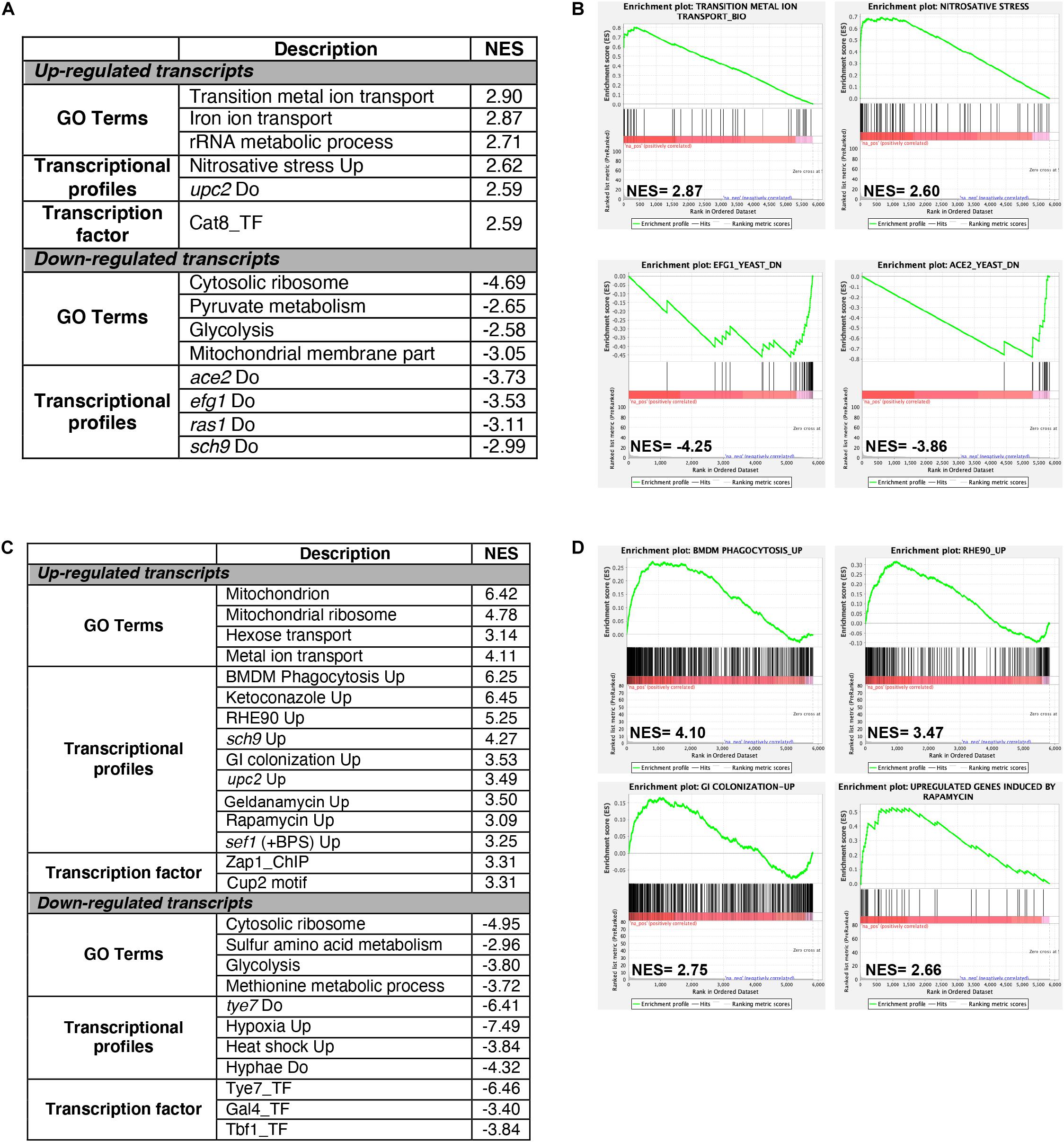
Figure 2. Gene set enrichment analysis of RNA-seq data. RNA-seq data of Cu excess (A,B) and deprivation (C,D) were analyzed using gene set enrichment analysis (GSEA). Relevant correlations between C. albicans transcriptome under Cu excess (A) and limitation (C) and other gene sets and functions are summarized. The complete GSEA correlations are listed in Supplementary Table S3. Graphs of GSEA for both Cu abundance conditions were also shown (B,D). NES, normalized enrichment score.
Under Cu deprivation, upregulated transcripts were significantly similar to the C. albicans transcriptional programs expressed during the colonization of the murine gut (Pierce et al., 2013), and the interaction with host cells including the human oral epithelial cells (Spiering et al., 2010) and the bone marrow-derived mouse macrophages (Marcil et al., 2008). This similarity suggests that, during the interaction with the host, C. albicans might be confronted by a Cu-deprived environment that the host might generate as an immune strategy to sequester this essential metal. Transcriptional similarities were also observed with the transcriptomes associated with fungal fitness and virulence traits including persistence under hypoxia and biofilm formation or during exposures to antifungal molecules (ketoconazole, rapamycin, and geldanamycin) (Figures 2C,D and Supplementary Table S3). As in other human pathogenic fungi, C. albicans might connect the control of fitness and virulence attributes to the Cu cellular adaptive machinery as a cue to promote either commensalism or pathogenicity (Ballou and Wilson, 2016).
Mac1-Mediated Control of Copper Deprivation
Prior to assessing the contribution of the transcription factor Mac1 to the global response of C. albicans to Cu deprivation, we first tested and confirmed the growth defect of mac1 mutant under different conditions including Cu deprivation, utilization of non-fermentable carbon sources and iron chelation as previously reported (Marvin et al., 2004, p. 1; Homann et al., 2009; Khamooshi et al., 2014; Figure 3). To identify Mac1-dependant transcripts associated with Cu utilization, we compared the transcriptional profile of mac1 cells exposed to BCS to that of non-treated mac1 cells. Overall, our RNA-seq data showed that the inducibility of genes of Cu uptake and utilization including CTR1, ALS1, FRE17, COX17 in addition to the Mn-SOD3 and many iron uptake transcripts (CFL2, CFL5, FRE30, FET31, and orf19.7077) were lost in mac1 mutant under Cu depleted conditions (Figure 4A). This data confirmed the role of Mac1 as the bona fide transcriptional regulator of Cu homeostasis in C. albicans.
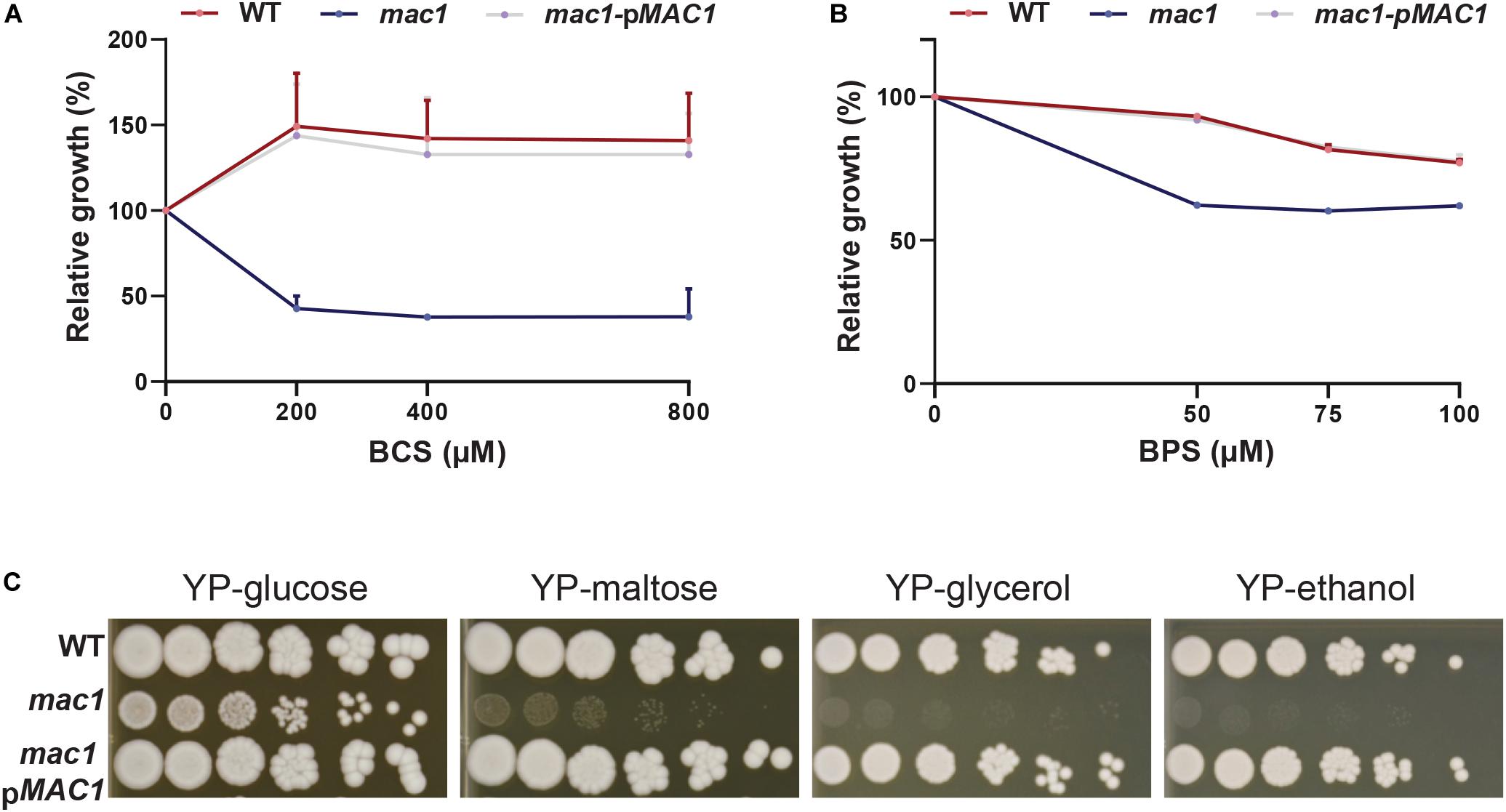
Figure 3. Phenotypic characterization of mac1 mutant. (A,B) The C. albicans WT, mac1 and the mac1 complemented strains were grown at 30°C in YPD supplemented with different concentrations of BCS or BPS. OD600 nm reading was taken after 48 h of incubation. ODs measurements for each concentration of BCS and BPS are the mean of triplicate. (C) mac1 is required for the utilization of alternative and non-fermentable carbon sources. The C. albicans WT, mac1 and the mac1 complemented strains were serially diluted, spotted on media with the indicated carbon sources and incubated for 3 days at 30°C.
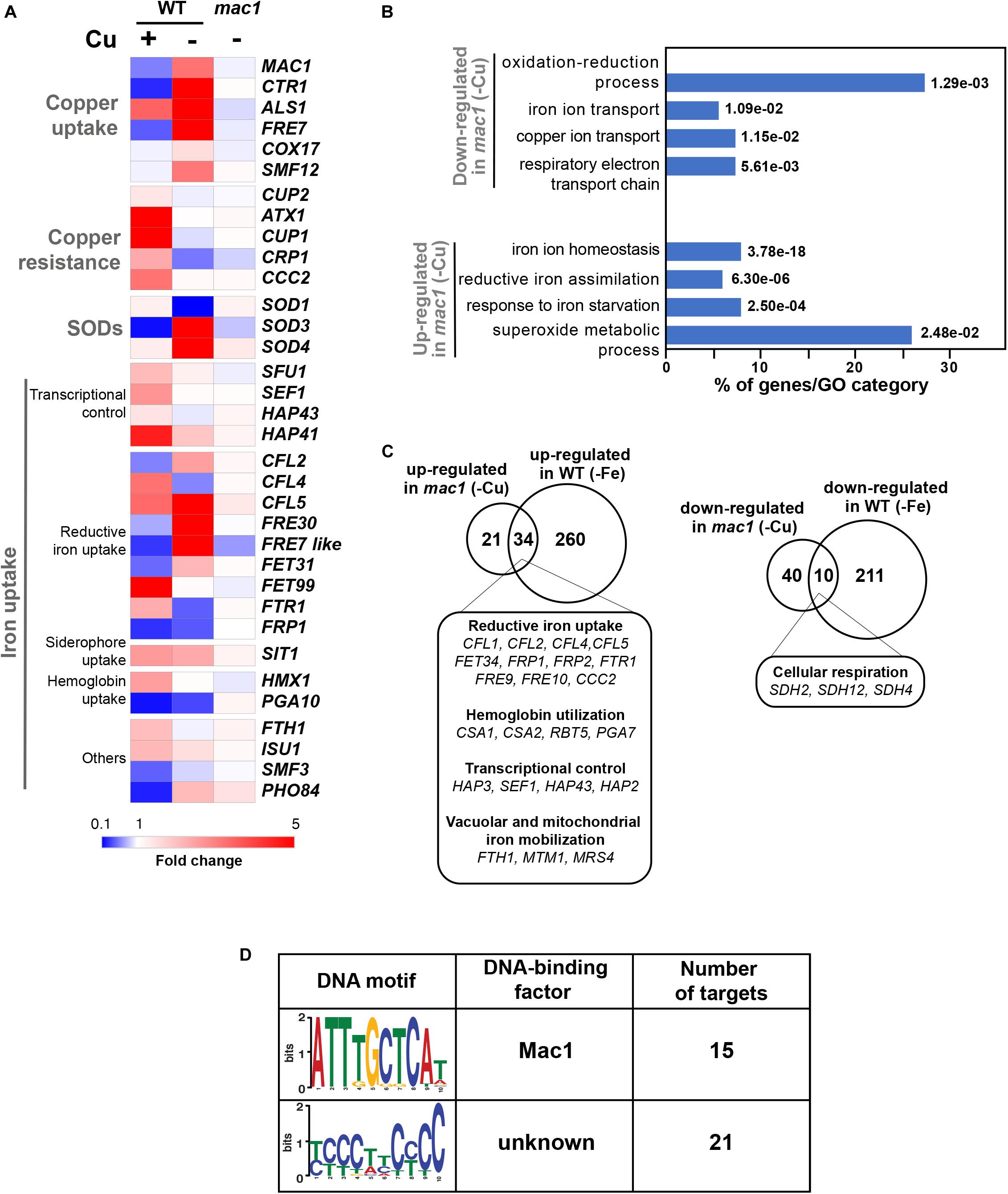
Figure 4. The Mac1-dependant control of Cu deprivation transcriptome. (A) Transcriptional alterations of Cu homeostasis genes in both WT and mac1. Heatmap visualization was presented as in Figure 1A for the WT and includes mac1 RNA-seq data. As the WT strain, mac1 mutant were exposed to 400 μM BCS, and incubated at 30°C for 30 min. Mac1-dependant transcripts associated with Cu utilization were identified by comparing the transcriptional profile of mac1 cells exposed to BCS to that of non-treated mac1 cells. (B) Gene ontology analysis of upregulated and downregulated transcripts in mac1 in response to BSC. The p-values were calculated using hypergeometric distribution. (C) Comparison of the Cu- and the iron-depletion transcriptomes of C. albicans (from Chen et al., 2011). Venn diagrams summarize the similarity between up- and downregulated transcripts under both iron and Cu starvations. Functional gene categories enriched in both conditions are indicated. (D) De novo prediction of DNA cis-regulatory motif enriched in the 50 gene promoters that mac1 failed to activate using MEME analysis tool (http://meme-suite.org).
Other cellular processes were also altered in mac1 mutant as compared to the WT under Cu deprivation (Figure 4B and Supplementary Table S4) including the repression of respiratory electron transport chain genes and the upregulation of iron homeostasis processes (Figures 4A–C). This transcriptional signature is reminiscent of an iron deprived environment that is most likely due to the fact that many iron uptake and utilization proteins depend on Cu for their functionality (Knight et al., 2002). Accordingly, a total of 34 among the 55 upregulated transcripts in mac1 were also upregulated in C. albicans cells under iron starvation as previously shown (Chen et al., 2011) and most of those genes are under the control of the Sfu1-Sef1-Hap43 iron transcriptional control axis (Figure 4C). Of note, we found that the transcriptional level of Sef1 and the CCAAT-binding factors Hap3, Hap1, and Hap43 were activated in mac1 mutant.
We also analyzed the 50 gene promoters that mac1 failed to activate for potential consensus binding motifs and identified two enriched mini-sequences one of which is the well-known Mac1 binding site (McDaniels et al., 1999; Garcia-Santamarina et al., 2018; Figure 4D). Mac1 binding motif was found in the promoters of Ctr1 and Sod3, as previously reported (Woodacre et al., 2008; Li et al., 2015), in addition to the ferric reductases Fre30 and Fre7 (Table 1). Mac1-regulatory motif was also found in 5′ cis-regulatory regions of genes that are not related directly to Cu metabolism such as electron transport chain (Sdh2, Sdh4, Cyb2), nitrosative stress response (Yhb1) and, manganese (Smf12) and iron transport (Ftr1) genes.
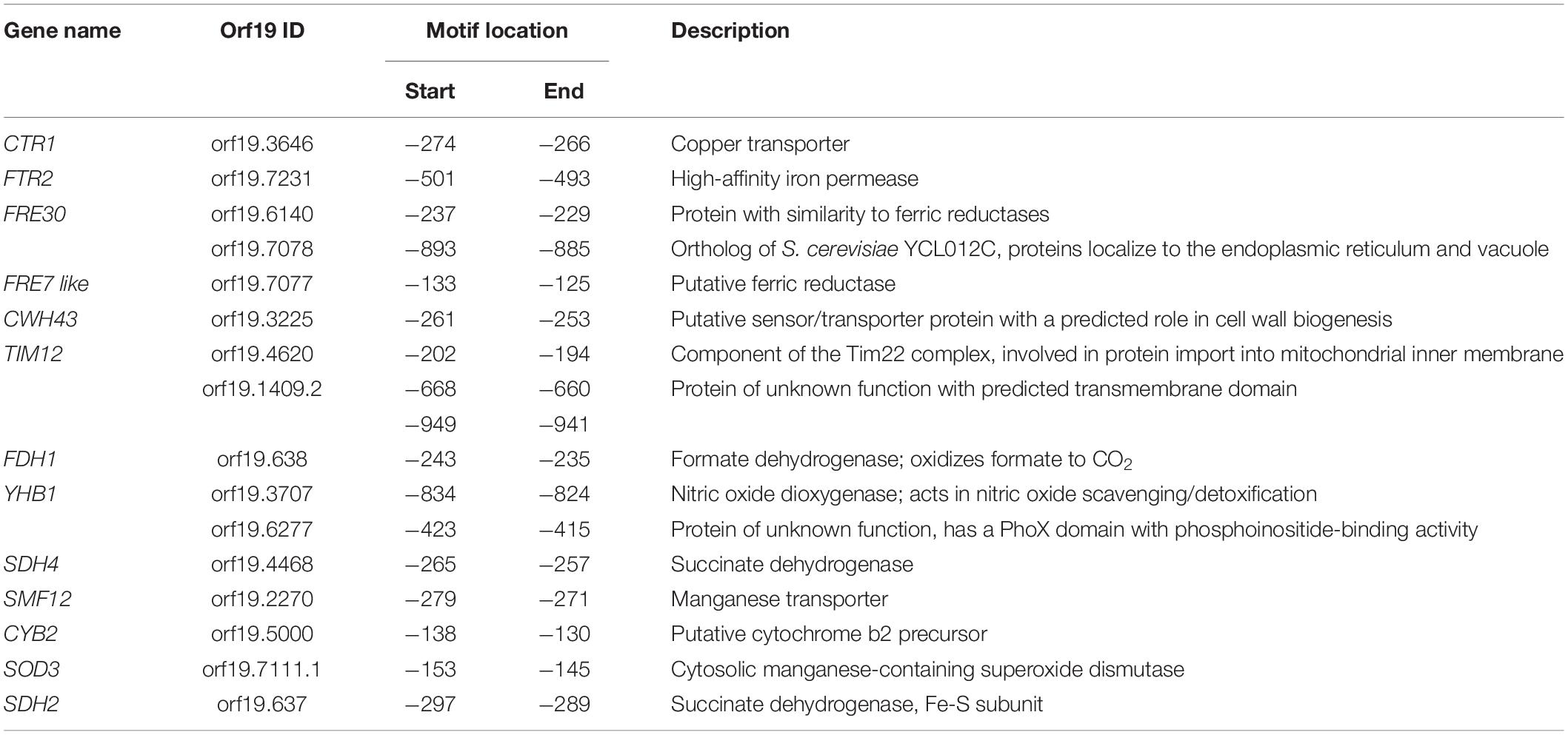
Table 1. List of genes with putative Mac1 cis-regulatory motif.
Copper Modulates Antifungal Sensitivity and Virulence
Gene set enrichment analysis of the C. albicans transcripts modulated by Cu limitation uncovered a significant correlation with the transcriptional program expressed when cells are challenged with the azole antifungal, ketoconazole (Figure 2C). This led to hypothesize that both stresses affect similar cellular processes in C. albicans or that Cu homeostasis modulates drug sensitivity. We found that genetic inactivation of MAC1 rendered C. albicans cells hypersensitive to both polyene (amphotericin B) and azole (fluconazole and miconazole) antifungals (Figure 5A). Antifungal sensitivity of mac1 was reversed by supplementing the growth medium by CuSO4 which suggests that Cu homeostasis modulates antifungal tolerance in C. albicans (Figure 5B).
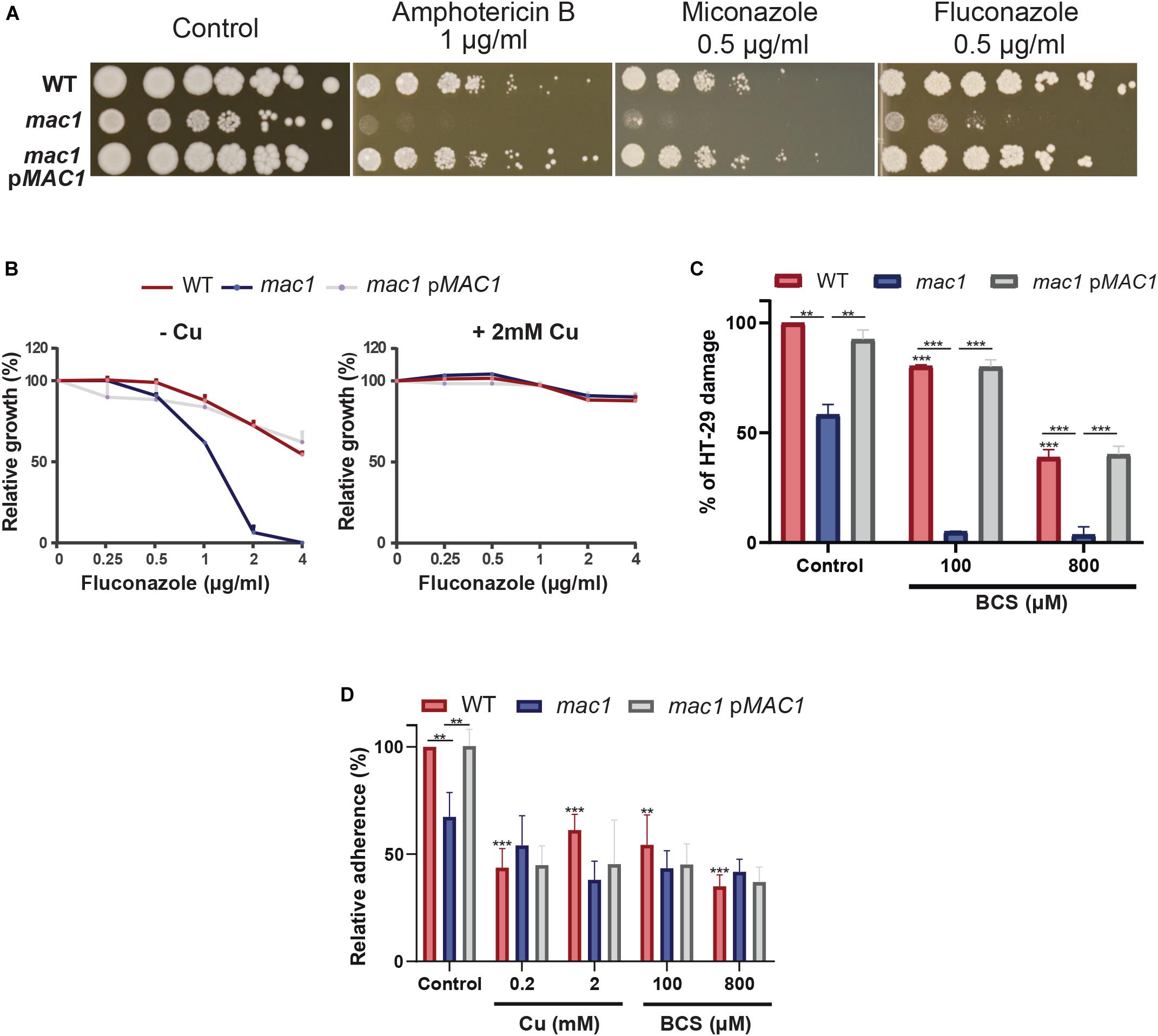
Figure 5. Copper modulates antifungal sensitivity and host invasion. (A) Mac1 modulates antifungal sensitivity. C. albicans WT, mac1 and mac1 complemented strains were serially diluted, spotted on media with different antifungals (amphotericin B, miconazole, and fluconazole) and incubated for 3 days at 30°C. (B) Cu supplementation restore mac1 growth inhibition by fluconazole. The C. albicans WT, mac1 and the mac1 complemented strains were grown at 30°C on YPD with different concentrations of fluconazole with or without 2 mM CuSO4. OD600 reading was taken after 48 h of incubation. (C) MAC1 inactivation and Cu depletion attenuate damage of the human colon epithelial HT-29 cells. HT-29 cell damage was assessed using the lactate dehydrogenase (LDH) release assay and was calculated as percentage of LDH activity in cell infected by mac1 and the revertant strains, with or without BCS, relatively to cells infected by the WT (SN250) strain. At least four biological replicates were obtained for each experiment. (D) mac1 mutant had a reduced adherence to HT-29 cells. C. albicans WT, mac1 and mac1 complemented strains were co-incubated with HT-29 for 1 h at 37°C and 5% CO2. Fungal cells were stained with 2 μM calcofluor white and counted. Data are presented as means ± SD from at least three independent experiments performed in triplicate. The statistical difference for each condition vs. WT control or between conditions was determined by two-tailed Student’s t-test. **p < 0.01, ***p < 0.005.
As the transcriptional profile of C. albicans cells under Cu deprivation is similar to that experienced during the interaction with the human host (Figures 2C,D), we tested whether Mac1 is required to damage the HT-29 human enterocytes using the LDH release assay. mac1 mutant exhibited a reduced ability to damage HT-29 cells as compared to the WT and the revertant strains (Figure 5C). HT-29 damage by mac1 mutant was further attenuated when both C. albicans and host cells were cocultured in the presence of BCS. Interestingly, Cu chelation exerts a protective antifungal activity since supplementation of the culture medium with 100 and 800 μM of BCS prevented 20 and 62% of HT-29 damage by the C. albicans WT strain, respectively. Together these data suggest that Cu homeostasis in C. albicans and the availability of this metal in host niches is essential for fungal virulence.
The adherence of mac1 mutant to the HT-29 enterocytes was also tested and the obtained data indicate a significant reduction of mac1 attachment as compared to the WT and the complemented strain (Figure 5D). Moreover, adding or depriving Cu from the growth medium reduced significantly the WT adherence to the HT-29. mac1 adherence defect to HT-29 was not reverted by Cu supplementation or phenocopied by Cu chelation in the WT strain (Figure 5D). This suggests that Mac1 might control other biological processes independently from its bona fide role as Cu metabolism modulator.
Discussion
So far, the global response of C. albicans to Cu availability remain unexplored. Previous works have focused only on known Cu metabolic genes that are homologous to S. cerevisiae such as the Cu transporter Ctr1 (Marvin et al., 2004, 1), the transcription factor Mac1 (Woodacre et al., 2008) and the Cu efflux pump Crp1 (Weissman et al., 2000). Here, we explored the genome-wide transcriptional response of C. albicans to elevated Cu or Cu deprivation in order to unbiasedly assess cellular processes associated with fungal fitness that are modulated by Cu availability (Figure 6).
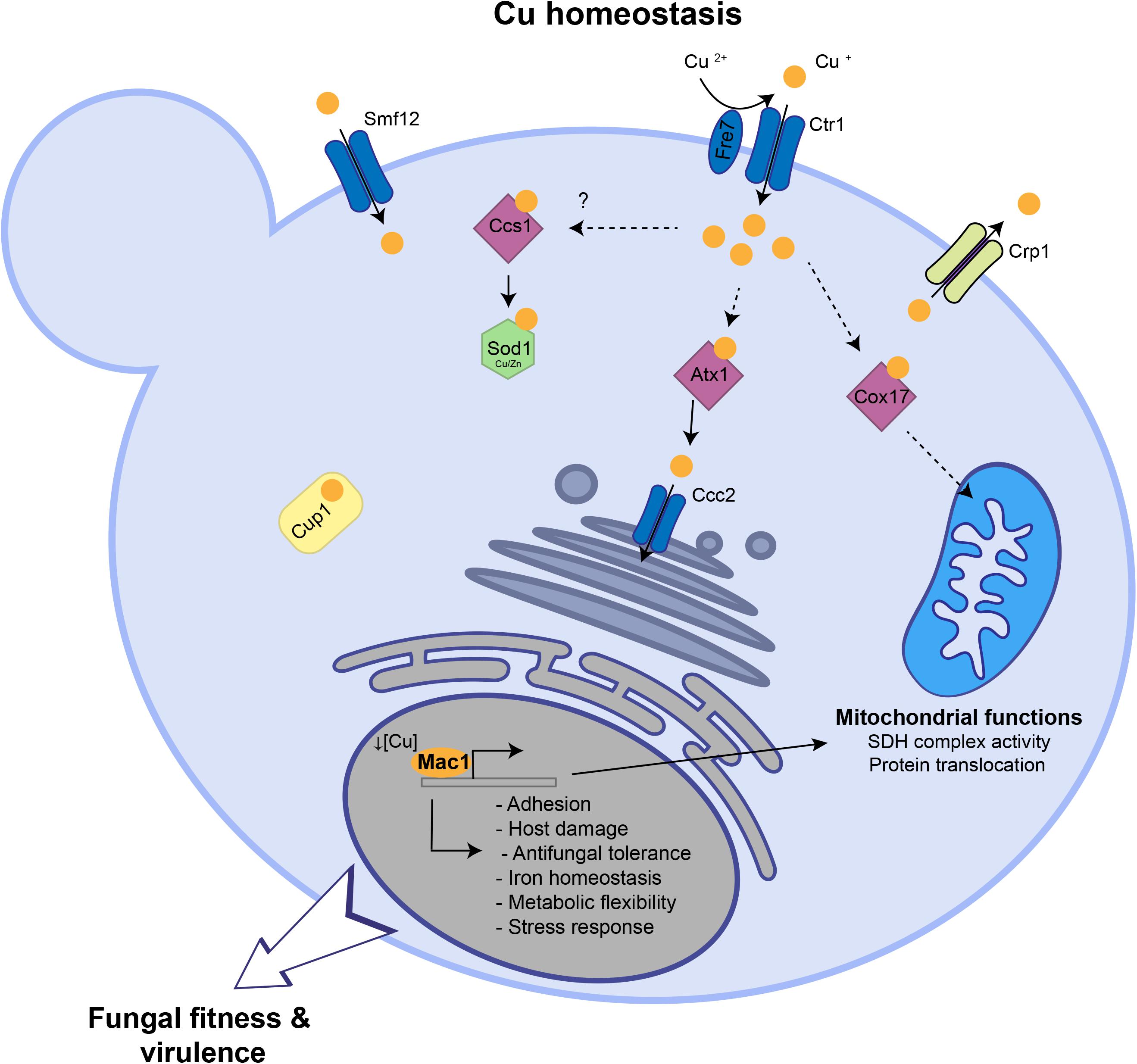
Figure 6. Summary of Cu homeostasis and Cu-modulated processes in C. albicans. Under Cu excess, transcripts associated with Cu detoxification and transport including the cytosolic small chaperone ATX1, the metallothionein CUP1, the P-type ATPase CCC2 and the Cu efflux pump CRP1 are upregulated. Under Cu deprivation, C. albicans activated genes required for Cu uptake including the transporter Ctr1 and the ferric reductases Fre7 that reduce Cu to facilitate its uptake by Ctr1. Other transcripts including the Cu metallochaperone COX17, the manganese transporter SMF12, with a potential role in Cu uptake, and the superoxide dismutase SOD3 were activated. Key C. albicans fitness and virulence processes that are modulated by Cu abundance and Mac1 activity were shown.
Our RNA-seq data recapitulated the bona fide fungal response to Cu. When grown under limiting concentrations, C. albicans activate Cu utilization genes such as the Cu transporter Ctr1 while under Cu excess genes involved in Cu detoxification (Crp1) and relocalization (Atx1, Cup1, Ccc2) were induced. As in other fungi (van Bakel et al., 2005; Rustici et al., 2007; Garcia-Santamarina et al., 2018), we also found that iron utilization genes were differentially modulated by Cu depletion which might reflect a situation of iron deficiency as well. Indeed, iron uptake depends on the multicopper ferroxidases and consequently Cu deprivation engender a subordinate iron depletion in C. albicans cells. Intriguingly, upon Cu excess, transcript levels of the transcription factor Sef1 and its direct target genes related to iron uptake and utilization such as the high affinity iron permease Ftr1, the ferric reductase Cfl5, the siderophore transporter Sit1 and the heme oxygenase Hmx1 were upregulated. This transcriptional signature reflects a defect in iron internalization as was also noticed under Cu limitation. This is similar to what was observed in yeast and mammalian cells where Cu overload deplete intracellular iron levels (Arredondo et al., 2004; Jo et al., 2008). This phenomenon could be explained by the fact that, as eukaryotic cells try to detoxify Cu by efflux, this essential metal become limiting for the Cu-dependent iron uptake machinery which led to iron deficiency.
The main goal of the current study was to uncover, beyond the Cu homeostatic routes, other biological processes that are modulated by Cu. Mining the C. albicans Cu transcriptome uncovered significant similarities with transcriptional programs activated by this opportunistic yeast in different niches within the host or during the interaction with immune cells. This highlight the importance of Cu for both fungal fitness and survival to immune response. Accordingly, our data showed that depriving C. albicans cells from Cu or inactivating the master transcriptional regulator of Cu uptake MAC1 both led to decreased damage and adherence to human enterocytes. Virulence defect in these conditions could be attributed to the fact that Cu is important for C. albicans to express its virulence factors such as metabolic flexibility and stress resistance. As shown previously (Marvin et al., 2004, p. 1; Mackie et al., 2016) and confirmed by our work, a homeostatic level of Cu is essential for C. albicans to metabolize different carbon sources, an important attribute for this yeast to colonize diverse niches with contrasting nutrients (Miramon and Lorenz, 2017; Burgain et al., 2019). Furthermore, as Sod1 requires Cu for the disproportionation of superoxide into peroxide and oxygen, Cu depletion might deteriorate the ability of C. albicans to resist the oxidative burst killing by phagocytic immune cells (Kang et al., 2002; Frohner et al., 2009).
At the genome level, our data uncovered that C. albicans cells grown in BCS exhibited a transcriptional pattern similar to that of cells challenged with ketoconazole. This suggest that Cu starvation, as azoles, might lead to sterol depletion. In accordance with that, we have shown that the Cu deficient mutant mac1 was hypersensitive to the ergosterol inhibitors (miconazole, fluconazole, and amphotericin B) as did sterol deficient mutants such as upc2 (Silver et al., 2004; MacPherson et al., 2005; Hoot et al., 2011). The opposite is also possible where sterol decrease might lead to Cu deprivation. However, a recent study had shown that ergosterol depletion by fluconazole do not affect the intracellular amount of Cu in C. albicans (Hunsaker and Franz, 2019). The growth defect of mac1 mutant in the presence of fluconazole was reverted by Cu supplementation suggesting that Cu might correct ergosterol depletion by promoting de novo synthesis of this fungal sterol. In a support of such hypothesis, studies in S. cerevisiae has shown that Cu promote ergosterol biosynthesis by promoting the transcription of ERG genes which led to the rescue of growth inhibition by lovastatin, an inhibitor of the ergosterol biosynthetic enzyme Hmg1 (Fowler et al., 2011).
This current investigation provides a rich framework for future work to uncover novel candidates for Cu homeostasis. For instance, under Cu excess, different categories of transporters were induced suggesting their contribution to Cu efflux. Under Cu starvation, the manganese transporter SMF12 were induced and it is a potential target of Mac1 as its promoter has a putative Mac1-binding motif. Thus, in addition to manganese, Smf12 might serve as a Cu transporter as was suggested also in S. cerevisiae (Liu et al., 1997). Furthermore, our data indicate that in addition to the bona fide Cu uptake genes, Mac1 might have other potential new targets and modulate other biological functions (Table 1 and Figure 6) such as the succinate dehydrogenase complex activity (SDH12, SDH2, SDH4), mitochondrial protein translocation and insertion in inner membrane (TIM12) and adhesion (ALS1). Why Mac1 might link Cu metabolism to those functions remains to be determined. Taken together, these assumptions should provide a fertile area for future investigations.
Data Availability Statement
The datasets generated for this study can be found in the datasets generated for this study are available at Figshare: https://figshare.com/s/98f0075055b2737ad1f5.
Author Contributions
IK, FT, and AS contributed to the design, execution, and/or data collection, and analysis of the experiments of this work. AS drafted the manuscript. All authors contributed to revision and final approval of the manuscript.
Funding
Work in Sellam’s group is supported by funds from the Canadian Institutes for Health Research project grant (CIHR, IC118460). AS is a recipient of the Fonds de Recherche du Québec-Santé (FRQS) J2 salary award. IK received Ph.D. scholarships from Universiteì Laval (bourse Pierre-Jacob Durand) and the CHU de Québec Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00935/full#supplementary-material
Footnotes
- ^ http://fungi.ensembl.org/Candida_albicans_sc5314_gca_000182965/Info/Index
- ^ http://www.broadinstitute.org/gsea/
References
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Arredondo, M., Cambiazo, V., Tapia, L., González-Agüero, M., Núñez, M. T., Uauy, R., et al. (2004). Copper overload affects copper and iron metabolism in Hep-G2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G27–G32. doi: 10.1152/ajpgi.00297.2003
Ballou, E. R., and Wilson, D. (2016). The roles of zinc and copper sensing in fungal pathogenesis. Curr. Opin. Microbiol. 32, 128–134. doi: 10.1016/j.mib.2016.05.013
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burgain, A., Pic, É, Markey, L., Tebbji, F., Kumamoto, C. A., and Sellam, A. (2019). A novel genetic circuitry governing hypoxic metabolic flexibility, commensalism and virulence in the fungal pathogen Candida albicans. PLoS Pathog. 15:e1007823. doi: 10.1371/journal.ppat.1007823
Cai, Z., Du, W., Zeng, Q., Long, N., Dai, C., and Lu, L. (2017). Cu-sensing transcription factor Mac1 coordinates with the Ctr transporter family to regulate Cu acquisition and virulence in Aspergillus fumigatus. Fungal Genet. Biol. 107, 31–43. doi: 10.1016/j.fgb.2017.08.003
Chen, C., Pande, K., French, S. D., Tuch, B. B., and Noble, S. M. (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135. doi: 10.1016/j.chom.2011.07.005
Culbertson, E., Khan, A., Muchenditsi, A., Lutsenko, S., Sullivan, D., Petris, M., et al. (2020). Changes in mammalian copper homeostasis during microbial infection. Metallomics 12, 416–426. doi: 10.1039/C9MT00294D
Ding, C., Festa, R. A., Chen, Y.-L., Espart, A., Palacios, Ò, Espín, J., et al. (2013). Cryptococcus neoformans copper detoxification machinery is critical for fungal virulence. Cell Host Microbe 13, 265–276. doi: 10.1016/j.chom.2013.02.002
Ding, C., Yin, J., Tovar, E. M. M., Fitzpatrick, D. A., Higgins, D. G., and Thiele, D. J. (2011). The copper regulon of the human fungal pathogen Cryptococcus neoformans H99. Mol. Microbiol. 81, 1560–1576. doi: 10.1111/j.1365-2958.2011.07794.x
Djoko, K. Y., Ong, C. Y., Walker, M. J., and McEwan, A. G. (2015). The role of copper and zinc toxicity in innate immune defense against bacterial pathogens. J. Biol. Chem. 290, 18954–18961. doi: 10.1074/jbc.R115.647099
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Festa, R. A., and Thiele, D. J. (2011). Copper: an essential metal in biology. Curr. Biol. 21, R877–R883. doi: 10.1016/j.cub.2011.09.040
Fowler, D. M., Cooper, S. J., Stephany, J. J., Hendon, N., Nelson, S., and Fields, S. (2011). Suppression of statin effectiveness by copper and zinc in yeast and human cells. Mol. Biosyst. 7, 533–544. doi: 10.1039/C0MB00166J
Frohner, I. E., Bourgeois, C., Yatsyk, K., Majer, O., and Kuchler, K. (2009). Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol. Microbiol. 71, 240–252. doi: 10.1111/j.1365-2958.2008.06528.x
Garcia-Santamarina, S., Festa, R. A., Smith, A. D., Yu, C.-H., Probst, C., Ding, C., et al. (2018). Genome-wide analysis of the regulation of Cu metabolism in Cryptococcus neoformans. Mol. Microbiol. 108, 473–494. doi: 10.1111/mmi.13960
Gerami-Nejad, M., Zacchi, L. F., McClellan, M., Matter, K., and Berman, J. (2013). Shuttle vectors for facile gap repair cloning and integration into a neutral locus in Candida albicans. Microbiology 159, 565–579. doi: 10.1099/mic.0.064097-0
González, M., Reyes-Jara, A., Suazo, M., Jo, W. J., and Vulpe, C. (2008). Expression of copper-related genes in response to copper load. Am. J. Clin. Nutr. 88, 830S–834S. doi: 10.1093/ajcn/88.3.830S
Homann, O. R., Dea, J., Noble, S. M., and Johnson, A. D. (2009). A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 5:e1000783. doi: 10.1371/journal.pgen.1000783
Hood, M. I., and Skaar, E. P. (2012). Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537. doi: 10.1038/nrmicro2836
Hoot, S. J., Smith, A. R., Brown, R. P., and White, T. C. (2011). An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 55, 940–942. doi: 10.1128/AAC.00995-10
Hunsaker, E. W., and Franz, K. J. (2019). Candida albicans reprioritizes metal handling during fluconazole stress. Metallomics 11, 2020–2032. doi: 10.1039/C9MT00228F
Jo, W. J., Loguinov, A., Chang, M., Wintz, H., Nislow, C., Arkin, A. P., et al. (2008). Identification of genes Involved in the toxic response of Saccharomyces cerevisiae against iron and copper overload by parallel analysis of deletion mutants. Toxicol. Sci. 101, 140–151. doi: 10.1093/toxsci/kfm226
Kang, S.-O., Oh, J.-H., Yim, H.-S., Rhie, G., Huh, W.-K., and Hwang, C.-S. (2002). Copper- and zinc-containing superoxide dismutase (Cu/ZnSOD) is required for the protection of Candida albicans against oxidative stresses and the expression of its full virulence. Microbiology 148, 3705–3713. doi: 10.1099/00221287-148-11-3705
Khamooshi, K., Sikorski, P., Sun, N., Calderone, R., and Li, D. (2014). The Rbf1, Hfl1 and Dbp4 of Candida albicans regulate common as well as transcription factor-specific mitochondrial and other cell activities. BMC Genomics 15:56. doi: 10.1186/1471-2164-15-56
Knight, S. A. B., Lesuisse, E., Stearman, R., Klausner, R. D., and Dancis, A. (2002). Reductive iron uptake by Candida albicans: role of copper, iron and the TUP1 regulator. Microbiology 148, 29–40. doi: 10.1099/00221287-148-1-29
Li, C. X., Gleason, J. E., Zhang, S. X., Bruno, V. M., Cormack, B. P., and Culotta, V. C. (2015). Candida albicans adapts to host copper during infection by swapping metal cofactors for superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 112, E5336–E5342. doi: 10.1073/pnas.1513447112
Linder, M. C., Wooten, L., Cerveza, P., Cotton, S., Shulze, R., and Lomeli, N. (1998). Copper transport. Am. J. Clin. Nutr. 67, 965S–971S. doi: 10.1093/ajcn/67.5.965S
Liu, X. F., Supek, F., Nelson, N., and Culotta, V. C. (1997). Negative control of heavy metal uptake by the Saccharomyces cerevisiae BSD2 gene. J. Biol. Chem. 272, 11763–11769. doi: 10.1074/jbc.272.18.11763
Mackie, J., Szabo, E. K., Urgast, D. S., Ballou, E. R., Childers, D. S., MacCallum, D. M., et al. (2016). Host-imposed copper poisoning impacts fungal micronutrient acquisition during systemic Candida albicans infections. PLoS One 11:e0158683. doi: 10.1371/journal.pone.0158683
MacPherson, S., Akache, B., Weber, S., De Deken, X., Raymond, M., and Turcotte, B. (2005). Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob. Agents Chemother. 49, 1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005
Marcil, A., Gadoury, C., Ash, J., Zhang, J., Nantel, A., and Whiteway, M. (2008). Analysis of PRA1 and its relationship to Candida albicans- macrophage interactions. Infect. Immun. 76, 4345–4358. doi: 10.1128/IAI.00588-07
Marvin, M. E., Mason, R. P., and Cashmore, A. M. (2004). The CaCTR1 gene is required for high-affinity iron uptake and is transcriptionally controlled by a copper-sensing transactivator encoded by CaMAC1. Microbiol. Read. Engl. 150, 2197–2208. doi: 10.1099/mic.0.27004-0
Marvin, M. E., Williams, P. H., and Cashmore, A. M. (2003). The Candida albicans CTR1 gene encodes a functional copper transporter. Microbiol. Read. Engl. 149, 1461–1474. doi: 10.1099/mic.0.26172-0
McDaniels, C. P. J., Jensen, L. T., Srinivasan, C., Winge, D. R., and Tullius, T. D. (1999). The yeast transcription factor Mac1 binds to DNA in a modular fashion. J. Biol. Chem. 274, 26962–26967. doi: 10.1074/jbc.274.38.26962
Miramon, P., and Lorenz, M. C. (2017). A feast for Candida: metabolic plasticity confers an edge for virulence. PLoS Pathog. 13:e1006144. doi: 10.1371/journal.ppat.1006144
Noble, S. M., French, S., Kohn, L. A., Chen, V., and Johnson, A. D. (2010). Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 42, 590–598. doi: 10.1038/ng.605
Pierce, J. V., Dignard, D., Whiteway, M., and Kumamoto, C. A. (2013). Normal adaptation of Candida albicans to the murine gastrointestinal tract requires Efg1p-dependent regulation of metabolic and host defense genes. Eukaryot. Cell 12, 37–49. doi: 10.1128/EC.00236-12
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rustici, G., van Bakel, H., Lackner, D. H., Holstege, F., Wijmenga, C., Bahler, J., et al. (2007). Global transcriptional responses of fission and budding yeast to changes in copper and iron levels: a comparative study. Genome Biol. 8:R73. doi: 10.1186/gb-2007-8-5-r73
Samanovic, M. I., Ding, C., Thiele, D. J., and Darwin, K. H. (2012). Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11, 106–115. doi: 10.1016/j.chom.2012.01.009
Sellam, A., van het Hoog, M., Tebbji, F., Beaurepaire, C., Whiteway, M., and Nantel, A. (2014). Modeling the transcriptional regulatory network that controls the early hypoxic response in Candida albicans. Eukaryot. Cell 13, 675–690. doi: 10.1128/EC.00292-13
Silver, P. M., Oliver, B. G., and White, T. C. (2004). Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3, 1391–1397. doi: 10.1128/EC.3.6.1391-1397.2004
Skrzypek, M. S., Binkley, J., Binkley, G., Miyasato, S. R., Simison, M., and Sherlock, G. (2017). The Candida genome database (CGD): incorporation of assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 45, D592–D596. doi: 10.1093/nar/gkw924
Spiering, M. J., Moran, G. P., Chauvel, M., Maccallum, D. M., Higgins, J., Hokamp, K., et al. (2010). Comparative transcript profiling of Candida albicans and Candida dubliniensis identifies SFL2, a C. albicans gene required for virulence in a reconstituted epithelial infection model. Eukaryot. Cell 9, 251–265. doi: 10.1128/EC.00291-09
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Sun, T.-S., Ju, X., Gao, H.-L., Wang, T., Thiele, D. J., Li, J.-Y., et al. (2014). Reciprocal functions of Cryptococcus neoformans copper homeostasis machinery during pulmonary infection and meningoencephalitis. Nat. Commun. 5:5550. doi: 10.1038/ncomms6550
van Bakel, H., Strengman, E., Wijmenga, C., and Holstege, F. C. P. (2005). Gene expression profiling and phenotype analyses of S. cerevisiae in response to changing copper reveals six genes with new roles in copper and iron metabolism. Physiol. Genomics 22, 356–367. doi: 10.1152/physiolgenomics.00055.2005
Waterman, S. R., Hacham, M., Hu, G., Zhu, X., Park, Y.-D., Shin, S., et al. (2007). Role of a CUF1/CTR4 copper regulatory axis in the virulence of Cryptococcus neoformans. J. Clin. Invest. 117, 794–802. doi: 10.1172/JCI30006
Weissman, Z., Berdicevsky, I., Cavari, B.-Z., and Kornitzer, D. (2000). The high copper tolerance of Candida albicans is mediated by a P-type ATPase. Proc. Natl. Acad. Sci. U.S.A. 97, 3520–3525. doi: 10.1073/pnas.97.7.3520
Wilson, R. B., Davis, D., Enloe, B. M., and Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16, 65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508<3.0.CO;2-M
Keywords: Candida albicans, RNA-seq, copper metabolism, fungal fitness, antifungal sensitivity
Citation: Khemiri I, Tebbji F and Sellam A (2020) Transcriptome Analysis Uncovers a Link Between Copper Metabolism, and Both Fungal Fitness and Antifungal Sensitivity in the Opportunistic Yeast Candida albicans. Front. Microbiol. 11:935. doi: 10.3389/fmicb.2020.00935
Received: 11 February 2020; Accepted: 20 April 2020;
Published: 19 May 2020.
Edited by:
Laure Ries, University of São Paulo, BrazilReviewed by:
Changbin Chen, Institut Pasteur of Shanghai (CAS), ChinaDelma S. Childers, University of Aberdeen, United Kingdom
Copyright © 2020 Khemiri, Tebbji and Sellam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adnane Sellam, QWRuYW5lLnNlbGxhbS4xQHVsYXZhbC5jYQ==; YWRuYW5lLnNlbGxhbUBnbWFpbC5jb20=