
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 May 2020
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00856
 Ling Shen1†
Ling Shen1† Mengting Liu1†
Mengting Liu1† Yan He2†Weaam Hasan Al Anbari1Huaqiang Li1Shuang Lin1Chenwei Chai1Jianping Wang1
Yan He2†Weaam Hasan Al Anbari1Huaqiang Li1Shuang Lin1Chenwei Chai1Jianping Wang1 Zhengxi Hu1*
Zhengxi Hu1* Yonghui Zhang1*
Yonghui Zhang1*Six previously undescribed ophiobolin-type sesterterpenes, namely, bipolatoxins A–F (1–6); and one previously undescribed pimarane-type diterpene, namely, 1β-hydroxy momilactone A (7); together with three known compounds, namely, 25-hydroxyophiobolin I (8), ophiobolin I (9), and ophiobolin A lactone (10); were isolated and identified from the endophytic fungus Bipolaris species TJ403-B1. Their structures with absolute configurations were elucidated on the basis of extensive spectroscopic analyses (including 1D and 2D nuclear magnetic resonance (NMR) and high-resolution electrospray ionization mass spectroscopy data), single-crystal X-ray diffraction analyses, and comparison of experimental circular dichroism data. All compounds (except for 5) were evaluated for antimicrobial potential, which indicated that bipolatoxin D (4) showed significant inhibitory activity against Enterococcus faecalis with a minimum inhibitory concentration (MIC) value of 8 μg/mL, and ophiobolin A lactone (10) showed significant inhibitory activity against Acinetobacter baumannii and E. faecalis with MIC values of 8 and 8 μg/mL, respectively.
Microbial natural products and their derivatives have been an important source of new antibiotics required for the treatment of infectious diseases (Wright, 2017). Since the first antibiotic, penicillin, was discovered from the fungus Penicillium notatum in 1928 (Wright, 2017), multiple classes of anti-infectives have been isolated from a variety of fungi, such as gliotoxin, beauvericin, and roquefortine C (Jakubczyk and Dussart, 2020). However, the rapid evolution of antimicrobial resistance in both hospital and community settings is decreasing the efficacy of our current therapies and causing a serious global public health crisis (Baker, 2015; Brown and Wright, 2016). One strategy to combat antimicrobial resistance is to discover and develop novel antimicrobial drugs that are not subject to existing resistance mechanisms (Demirci et al., 2013; Ziemert et al., 2016). Fungi-derived natural products hold great promise in the search for new therapies.
Ophiobolins, which represent a minor group of sesterterpenes featuring a tricyclic (5-8-5 fused) or tetracyclic (5-8-5-5 spiro-fused) skeleton, are reported to show a broad spectrum of inhibitory activities against nematodes, HMG-CoA reductase, fungi, and bacteria; cytotoxic activity against multiple cancer cells; and anti-inflammatory activity against lipopolysaccharide-induced nitric oxide production (Wang et al., 2013; Liu et al., 2019c), and this member of natural products is widely discovered in the species of Bipolaris. As a part of our ongoing program for exploring new antimicrobial agents from fungi, the fungus Bipolaris species TJ403-B1 attracted our attention and was systematically studied (Liu et al., 2019b, d). Following a further chemical investigation on the EtOAc extract of the title fungus, six previously undescribed ophiobolin-type sesterterpenes, namely, bipolatoxins A–F (1–6); and one previously undescribed pimarane-type diterpene, namely, 1β-hydroxy momilactone A (7); together with three known compounds (8–10); were isolated and identified. Herein, the detailed isolation, structure identification, and antimicrobial activity of these compounds (Figure 1) are described.
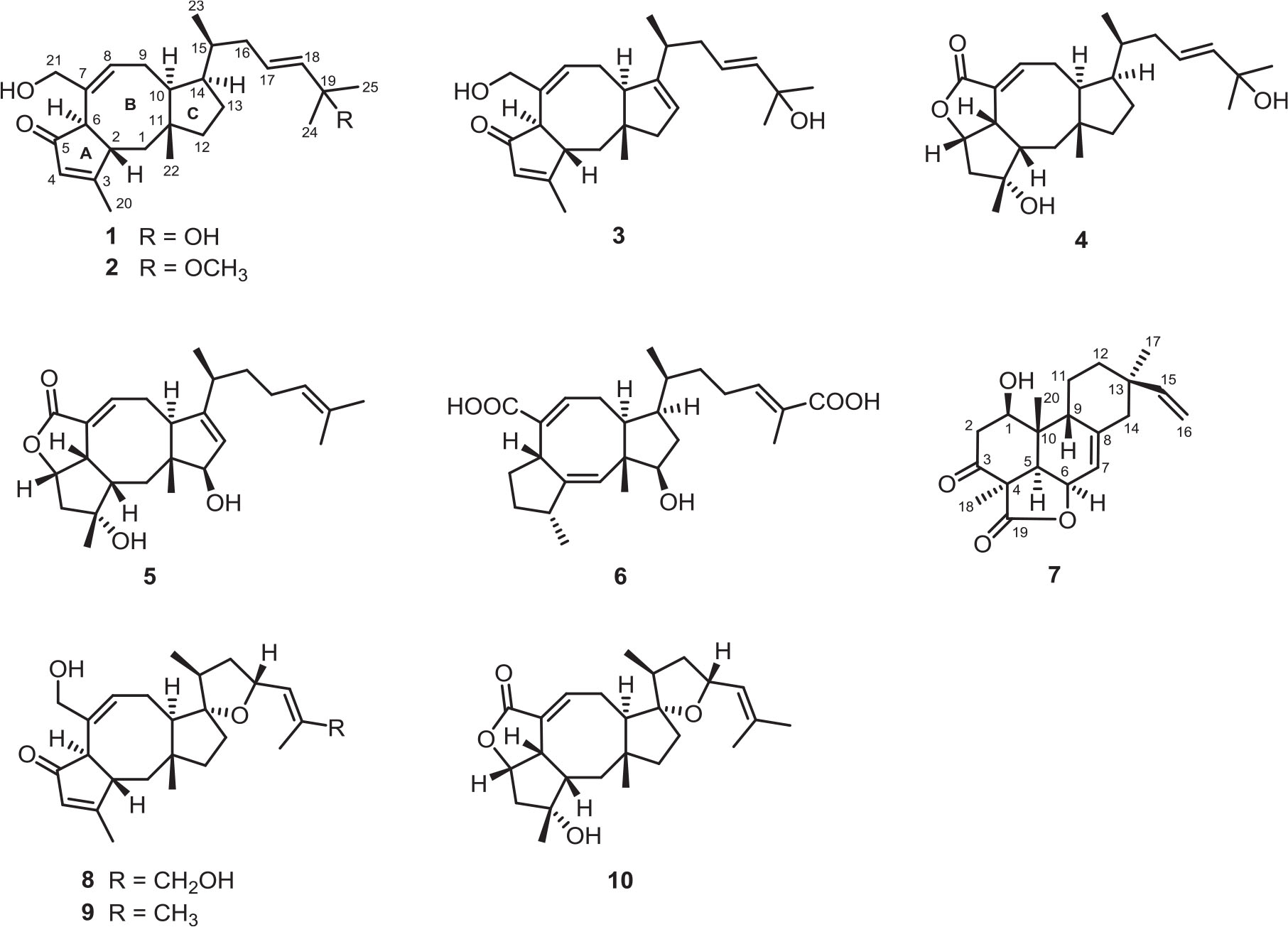
Figure 1. Chemical structures of compounds 1–10.
An X-5 microscopic melting point apparatus (Beijing Tech, Beijing, China) was used, and the reported melting points were uncorrected. Optical rotations were measured in MeOH on a Perkin-Elmer 341 polarimeter (PerkinElmer, Waltham, MA, United States). Infrared (IR) spectra were acquired on a Bruker Vertex 70 FT-IR instrument (Bruker, Karlsruhe, Germany). UV spectra were measured on a Varian Cary 50 spectrometer (Varian, Salt Lake City, UT, United States). Circular dichroism (CD) data were collected from a JASCO J-810 spectrometer (JASCO Co., Ltd., Tokyo, Japan). 1D and 2D nuclear magnetic resonance (NMR) spectra were acquired on a Bruker AM-400, a DRX-600, and a Bruker AM-800 instrument, and the 1H and 13C NMR chemical shifts were referenced to the solvent impurity peaks for methanol-d4 (δH 3.31 and δC 49.0) and acetone-d6 (δH 2.05 and δC 206.3). High-resolution electrospray ionization mass spectroscopy (HRESIMS) was performed on a Thermo Fisher LC-LTQ-Orbitrap XL spectrometer (Thermo Fisher, Palo Alto, CA, United States). Semipreparative high-performance liquid chromatography (HPLC) was performed using a Dionex Ultimate 3000 HPLC (Dionex, Sunnyvale, CA, United States) with an UV detector and an Ultimate XB-C18 (10 × 250 mm, 5 μm, Welch Ultimate XB-C18) column. Silica gel (100–200 mesh and 200–300 mesh; Qingdao Marine Chemical Inc., Qingdao, China), ODS (50 μm; YMC, Kyoto, Japan), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography (CC). Thin-layer chromatography was performed with RP-C18 F254 plates (Merck, Darmstadt, Germany) and silica gel 60 F254 (Yantai Chemical Industry Research Institute, Yantai, China).
The fungal strain in our project was obtained from the leaves of wheat, which was collected from Wuhan City of Hubei Province, China, in May 2016. Sequence data for this fungal strain have been submitted to the DDBJ/EMBL/GenBank under accession no. MH545913. A voucher sample has been preserved in the culture collection center of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China).
The fungal strain was cultured on potato dextrose agar at 28 °C for 5 days to prepare the seed cultures. Then, the agar plugs were inoculated into 450 Erlenmeyer flasks (1 L), previously sterilized by autoclaving, each containing 250 g rice and 250 mL distilled water. All flasks were incubated at 28 °C for 28 days. The fermented rice substrate was extracted five times in 95% aqueous EtOH at room temperature, and the solvent was evaporated under vacuum to afford a residue. The residue was suspended in H2O and successively partitioned with EtOAc to yield a total extract.
The EtOAc extract (300 g) was subjected to RP-C18 silica gel CC with a stepwise gradient of MeOH–H2O (20, 40, 60, 80, and 100%) to afford five major fractions, A–E. Fraction C (40 g) was applied to a silica gel column eluted with petroleum ether–EtOAc (10:0 to 0:1, vol/vol) to furnish eight main fractions, C1–C8. Fraction C4 (11.3 g) was chromatographed on Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and further purified on an RP-C18 silica gel column eluted with MeOH–H2O (40:60 to 60:40, vol/vol) to obtain fractions C4.1–C4.12. Fraction C4.5 (3.0 g) was applied to silica gel CC eluted with stepwise CH2Cl2–MeOH (1:0–100:1, vol/vol) to afford nine fractions (C4.5.1–C4.5.9). Fraction C4.5.2 was repeatedly separated via Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and then separated by semipreparative HPLC (CH3CN–H2O, 73:27, vol/vol, 3.0 mL/min) to afford compound 10 (tR 34.2 min, 11.2 mg). Purification of fraction C4.5.3 by semipreparative HPLC (MeOH–H2O, 80:20, vol/vol, 3.0 mL/min) afforded compound 6 (tR 16.2 min, 4.1 mg). Purification of fraction C4.5.4 by semipreparative HPLC (CH3CN–H2O, 70:30, vol/vol, 3.0 mL/min) afforded compound 7 (tR 11.2 min, 3.2 mg). Fraction C4.5.6 was repeatedly separated via Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and then purified by semipreparative HPLC (MeOH–H2O, 78:22, vol/vol, 3.0 mL/min) to afford compound 4 (tR 31.2 min, 2.2 mg). Fraction C4.5.7 was repeatedly separated via Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and then purified by semipreparative HPLC (MeOH–H2O, 75:25, vol/vol, 3.0 mL/min) to afford compound 5 (tR 30.9 min, 1.0 mg). Fraction C4.5.9 was repeatedly separated via Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and then purified by semipreparative HPLC (CH3CN–H2O, 60:40, vol/vol, 3.0 mL/min) to afford compound 3 (tR 34.9 min, 4.8 mg). Fraction C4.7 (500 mg) was applied to silica gel CC eluted with stepwise CH2Cl2–MeOH (1:0–60:1, vol/vol) to afford six fractions (C4.7.1–C4.7.6). Purification of fraction C4.7.3 by semipreparative HPLC (MeOH–H2O, 77:23, vol/vol, 3.0 mL/min) afforded compound 1 (tR 30.9 min, 6.3 mg). Purification of fraction C4.7.4 by semipreparative HPLC (MeOH–H2O, 83:17, vol/vol, 3.0 mL/min) afforded compound 2 (tR 31.0 min, 4.5 mg). Fraction C5 (4.1 g) was separated on Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1, vol/vol) and further purified by RP-C18 silica gel CC (MeOH–H2O, 40:60 to 60:40, vol/vol) to afford three main fractions, C5.1–C5.3. Compound 8 (tR 34.8 min, 10.6 mg) was purified by semipreparative HPLC (MeOH–H2O, 71:29, vol/vol, 3.0 mL/min) from fraction C5.2. Fraction C6 (2.2 g) was chromatographed on Sephadex LH-20 (CH2Cl2–MeOH, 1:1, vol/vol) and further purified by semipreparative HPLC (MeOH–H2O, 70:30, vol/vol, 3.0 mL/min) to afford compound 9 (tR 39.3 min, 4.5 mg).
Bipolatoxin A (1). Colorless needle crystals; : +70 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 202 (4.11), 229 (4.07) nm; ECD (c 0.18, MeOH) = Δε219 +3.07, Δε313 −0.70; IR νmax = 3,428, 2,935, 1,682, 1,622, 1,459, 1,379, 1,317, 1,227, 1,186, 1,028, 855 cm–1; HRESIMS m/z 409.2729 [M+Na]+ (calcd for C25H38O3Na+, 409.2713). For 1H and 13C NMR data, see Tables 1 and 3.

Table 1. 1H NMR assignments for compounds 1–4 (δ in ppm and J in Hz).
Bipolatoxin B (2). Colorless oil; : +65 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 202 (4.09), 229 (4.02) nm; ECD (c 0.18, MeOH) = Δε218 +1.82, Δε314 −1.14; IR νmax = 3,430, 2,935, 1,684, 1,621, 1,458, 1,378, 1,315, 1,257, 1,171, 1,145, 1,075, 1,027, 977, 857, 815, 616 cm–1; HRESIMS m/z 423.2882 [M+Na]+ (calcd for C26H40O3Na+, 423.2870); 1H and 13C NMR data, see Tables 1 and 3.
Bipolatoxin C (3). Colorless oil; : −3 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 203 (4.14), 229 (3.93) nm; ECD (c 0.18, MeOH) = Δε206 −15.45, Δε222 +1.85, Δε308 −2.30; IR νmax = 3,403, 2,955, 2,933, 2,876, 1,627, 1,516, 1,453, 1,382, 1,343, 1,271, 1,232, 1,172, 1,122, 1,028, 962, 927, 869, 825, 626 cm–1; HRESIMS m/z 407.2550 [M+Na]+ (calcd for C25H36O3Na+, 407.2557); 1H and 13C NMR data, see Tables 1 and 3.
Bipolatoxin D (4). Colorless needle crystals; : +30 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 202 (3.99), 226 (4.07) nm; ECD (c 0.18, MeOH) = Δε223 −10.12, Δε248 +3.93; IR νmax = 3,430, 2,931, 2,851, 1,733, 1,681, 1,638, 1,458, 1,384, 1,315, 1,217, 1,181, 1,150, 1,099, 1,028, 934, 907, 832, 764 cm–1; HRESIMS m/z 425.2653 [M+Na]+ (calcd for C25H38O4Na+, 425.2662); 1H and 13C NMR data, see Tables 1 and 3.
Bipolatoxin E (5). Colorless oil; : −91 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 203 (4.24) nm; ECD (c 0.18, MeOH) = Δε219 −8.49, Δε246 +3.19; IR νmax = 3,427, 2,966, 2,925, 2,853, 1,731, 1,675, 1,631, 1,579, 1,452, 1,384, 1,349, 1,307, 1,290, 1,248, 1,221, 1,130, 1,096, 1,027, 939, 835, 760, 653, 618, 580 cm–1; HRESIMS m/z 423.2500 [M+Na]+ (calcd for C25H36O4Na+, 423.2506); 1H and 13C NMR data, see Tables 2 and 3.
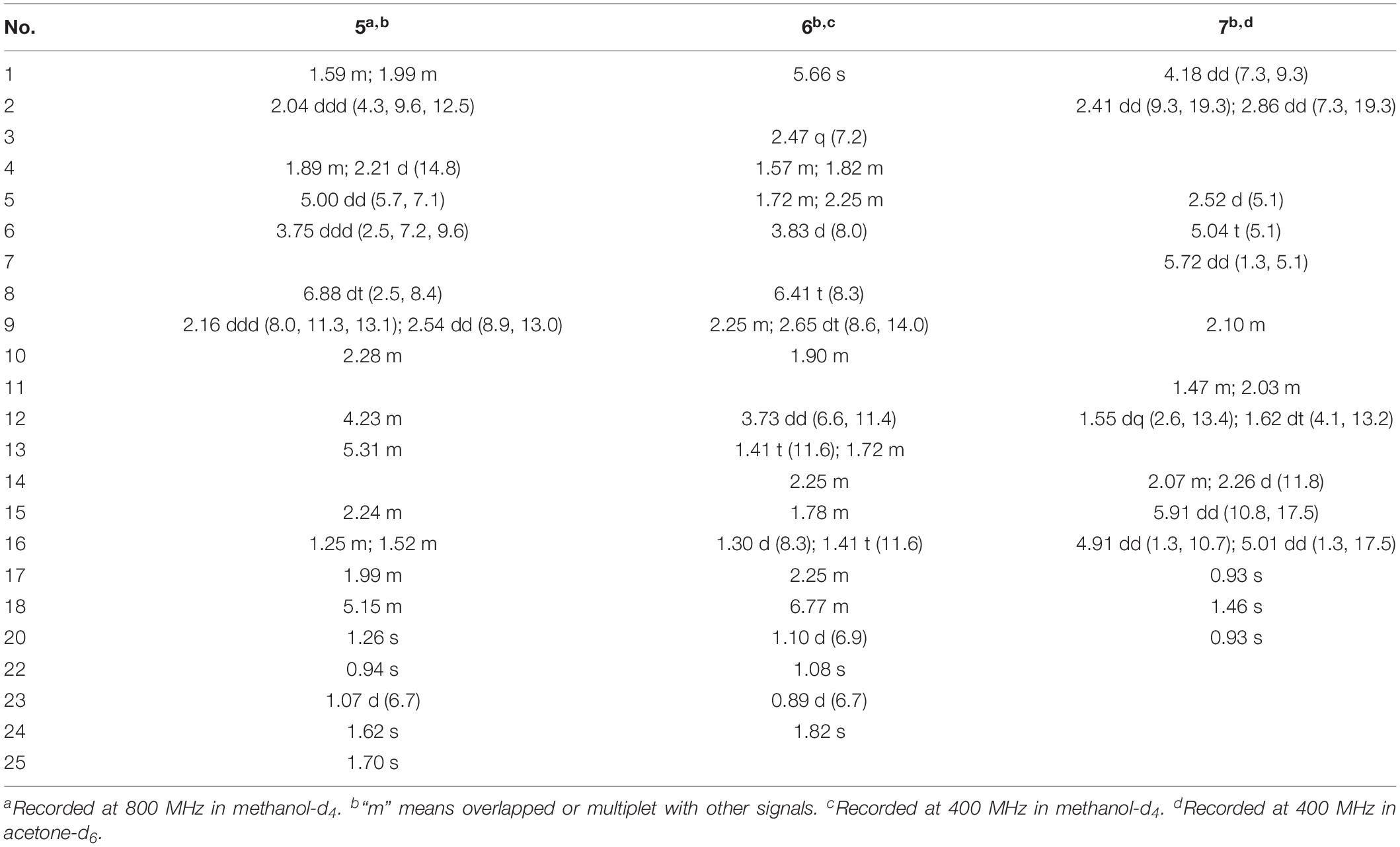
Table 2. 1H NMR assignments for compounds 5–7 (δ in ppm and J in Hz).
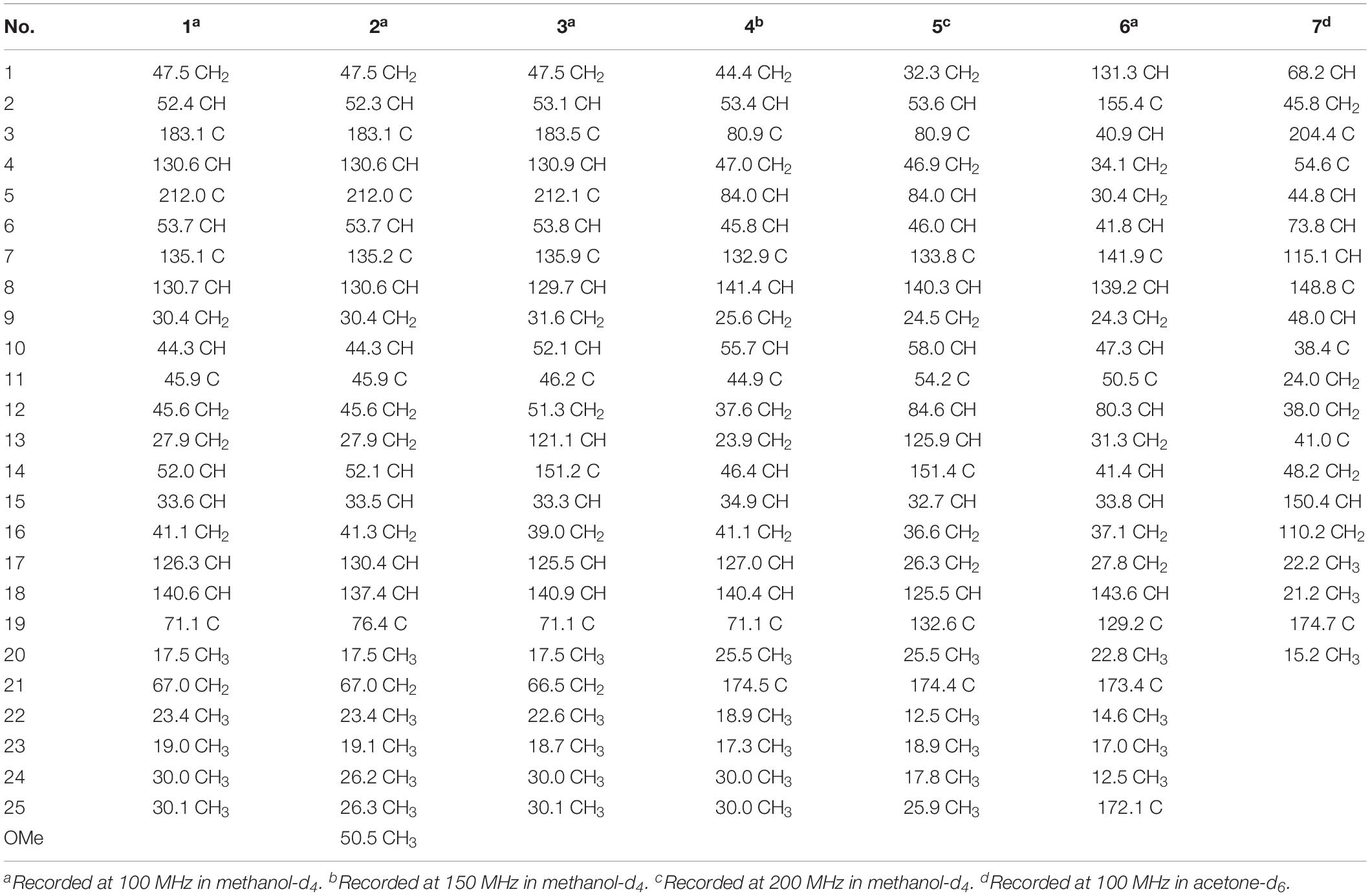
Table 3. 13C NMR assignments for compounds 1–7.
Bipolatoxin F (6). Colorless oil; : −16 (c 0.10, MeOH); UV (MeOH) λmax (log ε) = 204 (4.21) nm; ECD (c 0.18, MeOH) = Δε237 −4.09; IR νmax = 3,435, 2,950, 2,874, 1,687, 1,639, 1,460, 1,452, 1,385, 1,266, 1,203, 1,159, 1,106, 1,027, 664 cm–1; HRESIMS m/z 439.2453 [M+Na]+ (calcd for C25H36O5Na+, 439.2455); 1H and 13C NMR data, see Tables 2 and 3.
1β-Hydroxy momilactone A (7). Colorless oil; : −256 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 203 (4.11) nm; ECD (c 0.18, MeOH) = Δε201 −19.50, Δε293 −1.89; IR νmax = 3,511, 3,431, 3,082, 2,922, 1,756, 1,698, 1,635, 1,457, 1,414, 1,378, 1,338, 1,195, 1,149, 1,093, 988, 908, 864, 772, 747, 647 cm–1; HRESIMS m/z 331.1913 [M+H]+ (calcd for C20H27O4+, 331.1904); 1H and 13C NMR data, see Tables 2 and 3.
The suitable crystals of compounds 1, 4, and 8 were acquired from MeOH–H2O (20:1, vol/vol) at room temperature. The intensity data were recorded on an XtaLAB PRO MM007HF diffractometer (Cu Kα). Using Olex2 (Dolomanov et al., 2009), the structures were solved via direct methods with SHELXL-2014/7 (Sheldrick, 2008). Refinements were executed by the SHELXL-2014/7 refinement package via means of full-matrix least squares on F2, and the anisotropic displacement parameters were applied for all the non-hydrogen atoms. All the hydrogen atoms were placed on the calculated positions and refined by a riding model. The crystallographic data for these structures were deposited in the Cambridge Crystallographic Data Center (CCDC 1971181 for 1, CCDC 1913829 for 4, and CCDC 1913832 for 8). Copies of the data can be obtained free of charge on application to CCDC (Cambridge, United Kingdom; e-mail: ZGVwb3NpQGNjZGMuY2FtLmFjLnVr).
Crystallographic data for compound 1: C25H38O3, M = 386.55, orthorhombic, a = 5.92122(10) Å, b = 15.67980(10) Å, c = 26.2277(2) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 2435.07(5) Å3, T = 100(1) K, space group P212121, Z = 4, μ(Cu Kα) = 0.523 mm–1, 24,079 reflections measured, 4,861 independent reflections (Rint = 0.0218). The final R1 values were 0.0293 [I > 2σ(I)]. The final wR(F2) values were 0.0772 [I > 2σ(I)]. The final R1 values were 0.0296 (all data). The final wR(F2) values were 0.0774 (all data). The goodness of fit on F2 was 1.032. Flack parameter = 0.08(3) (Supplementary Data Sheets S1, S4).
Crystallographic data for compound 4: C25H38O4⋅2H2O, M = 438.58, monoclinic, a = 14.63410(10) Å, b = 6.02160(10) Å, c = 14.70210(10) Å, α = 90.00°, β = 111.0790(10)°, γ = 90.00°, V = 1,208.87(2) Å3, T = 100.00(10) K, space group P21, Z = 2, μ(Cu Kα) = 0.678 mm–1, 25,163 reflections measured, 4,543 independent reflections (Rint = 0.0536). The final R1 values were 0.0337 [I > 2σ(I)]. The final wR(F2) values were 0.0879 [I > 2σ(I)]. The final R1 values were 0.0352 (all data). The final wR(F2) values were 0.0886 (all data). The goodness of fit on F2 was 1.072. Flack parameter = 0.05(10) (Supplementary Data Sheets S2, S4).
Crystallographic data for compound 8: C25H36O4, M = 400.54, monoclinic, a = 11.94134(9) Å, b = 6.14791(4) Å, c = 15.49296(12) Å, α = 90.00°, β = 101.4154(7)°, γ = 90.00°, V = 1114.904(15) Å3, T = 100(2) K, space group P21, Z = 2, μ(Cu Kα) = 0.626 mm–1, 23,169 reflections measured, 4,453 independent reflections (Rint = 0.0341). The final R1 values were 0.0291 [I > 2σ(I)]. The final wR(F2) values were 0.0765 [I > 2σ(I)]. The final R1 values were 0.0301 (all data). The final wR(F2) values were 0.0772 (all data). The goodness of fit on F2 was 1.026. Flack parameter = −0.16(6) (Supplementary Data Sheets S3, S4).
The test strains were acquired from the ATCC: ESBL-producing Escherichia coli (ATCC 35218), Acinetobacter baumannii (ATCC 19606), Pseudomonas aeruginosa (ATCC 15542), Klebsiella pneumoniae (ATCC 700603), methicillin-resistant Staphylococcus aureus (ATCC 43300), Enterococcus faecalis (ATCC 29212), and Candida albicans (ATCC 10231). The reference compounds for the tests were recommended by the National Committee for Clinical Laboratory Standards (Liu et al., 2019a): vancomycin (Sigma, cat #861987), amikacin (Sigma, St. Louis, United States; cat #1019508), ceftriaxone (Sigma, cat #1098184), and fluconazole (Sigma, cat #1271700); all test compounds were ≥95% pure (HPLC, wavelength = 210 nm).
Determination of the minimum inhibitory concentrations (MICs) was conducted according to our previously reported broth microdilution method (Gao et al., 2019; Yang et al., 2019). In short, the inoculum was standardized to approximately 5 × 105 colony-forming units/mL. The plates were incubated at 37 °C for 16 h, and the MIC values were recorded as the lowest concentration of antibiotic, at which no visible microbial growths were observed. Each experiment was performed three times.
GraphPad Prism 5.0 software (GraphPad, San Diego, United States) was used to carry out statistical analysis of the data. The data were expressed as the means ± SD. Values were analyzed with SPSS version 12.0 software (Softonic, Barcelona, Spain) by one-way analysis of variance, and p < 0.05 was considered statistically significant.
The EtOAc extract of the solid medium of Bipolaris species TJ403-B1 was subjected to extensive chromatographic separations over silica gel column, RP-C18 gel column, Sephadex LH-20, and semipreparative HPLC to afford six previously undescribed ophiobolin-type sesterterpenes, namely, bipolatoxins A–F (1–6); and one previously undescribed pimarane-type diterpene, namely, 1β-hydroxy momilactone A (7); along with three known compounds (8–10).
Compound 1 was obtained as colorless needle crystals, and its molecular formula was assigned as C25H38O3, based on the HRESIMS data at m/z 409.2729 ([M+Na]+, calcd for C25H38O3Na+, 409.2713) in conjunction with the NMR data analyses, which was indicative of an index of hydrogen deficiency of seven. The 13C NMR data (Table 3) together with the DEPT spectrum of 1 showed a total of 25 resonances that could be assigned as five methyls, six methylenes (including one oxygenated), nine methines (including four olefinic), and five non-protonated carbons (including one ketone, one oxygenated, and two olefinic). Apart from four indices of hydrogen deficiency attributed to three double bonds (δC 183.1/130.6, 135.1/130.7, 126.3/140.6) and a carbonyl (δC 212.0), the remaining indices of hydrogen deficiency suggested that 1 has a tricyclic ring system.
Further analyses of the HSQC, 1H–1H COSY, and HMBC spectral data established an ophiobolin-type sesterterpene nucleus, structurally related to known ophiobolin Q (Cai et al., 2019) with the major differences being listed as follows: (a) the C-21 formyl group in ophiobolin Q was replaced by a hydroxylated methylene group (C-21, δC 67.0) in 1; and (b) the disappearance of a hydroxylated methylene group (C-18) in ophiobolin Q and the double bond migrated from C-16/C-17 to C-17/C-18 in 1. These conclusions were further supported by the 1H–1H COSY correlations (Figure 2) of H-15 (δH 1.55)/H2-16 (δH 1.71 and 2.20)/H-17 (δH 5.60)/H-18 (δH 5.59) and HMBC correlations from H3-23 (δH 0.90) to C-14 (δC 52.0), C-15 (δC 33.6), and C-16 (δC 41.1) and from H3-24 (δH 1.26) to C-18 (δC 140.6), C-19 (δC 71.1), and C-25 (δC 30.1). The NOESY cross-peaks (Figure 3) of H-6 (δH 3.61)/H-10 (δH 2.71)/H-14 (δH 1.84) and H-2 (δH 2.95)/H3-22 (δH 1.07) indicated that H-6, H-10, and H-14 were all on the same face with α orientations, whereas H-2 and H3-22 were β-oriented. However, for the configuration of C-15, it could not be defined by analysis of the NOE signals. Fortunately, a suitable crystal of 1 was obtained in MeOH–H2O (20:1, vol/vol) at room temperature and then subjected to a single-crystal X-ray diffraction experiment with Cu Kα radiation [Figure 4, Flack parameter = 0.08(3)], which enabled us to establish its absolute configuration as 2S, 6R, 10S, 11R, 14R, and 15S, along with an E geometry of the Δ17,18 double bond. Accordingly, the structure of 1 was defined and named bipolatoxin A. Remarkably, our study further supports that during the cyclization of all ophiobolins AcOS favors a 1,5-H shift (C-8–C-15) to display the 15S configuration of the side chain (Chiba et al., 2013; Narita et al., 2016).
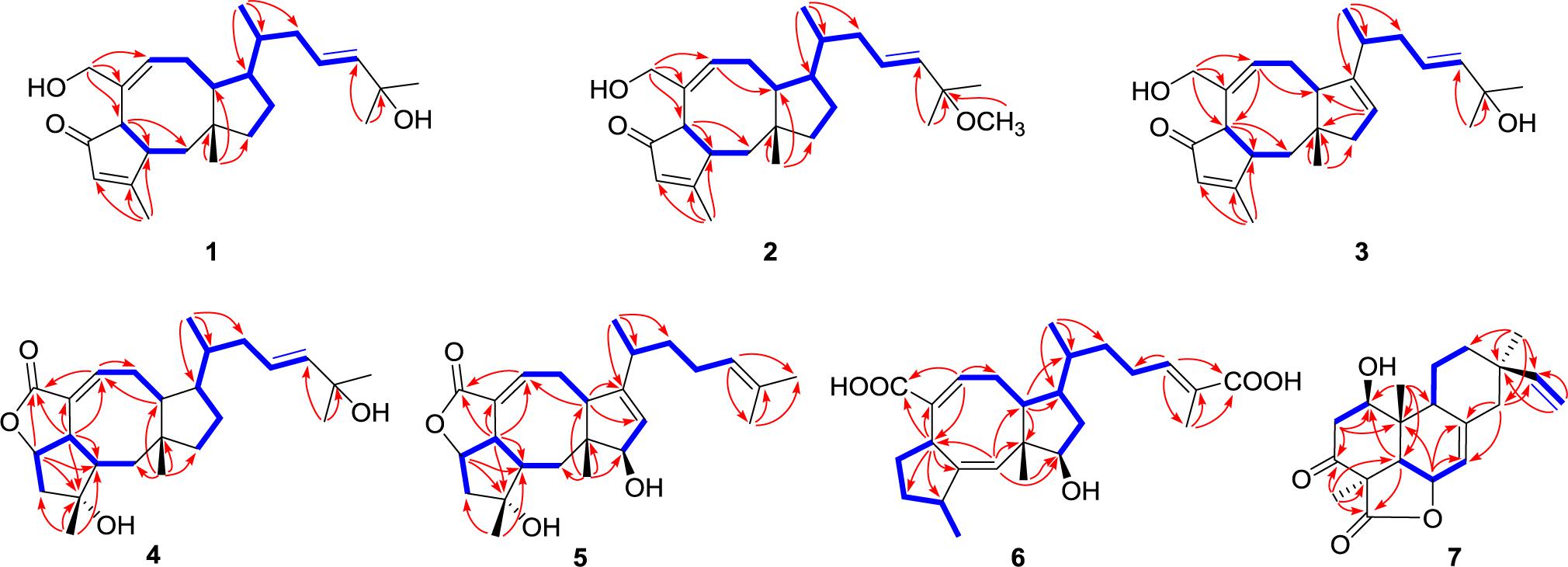
Figure 2. Key HMBC (red lines) and 1H–1H COSY (blue bold lines) correlations of compounds 1–7.
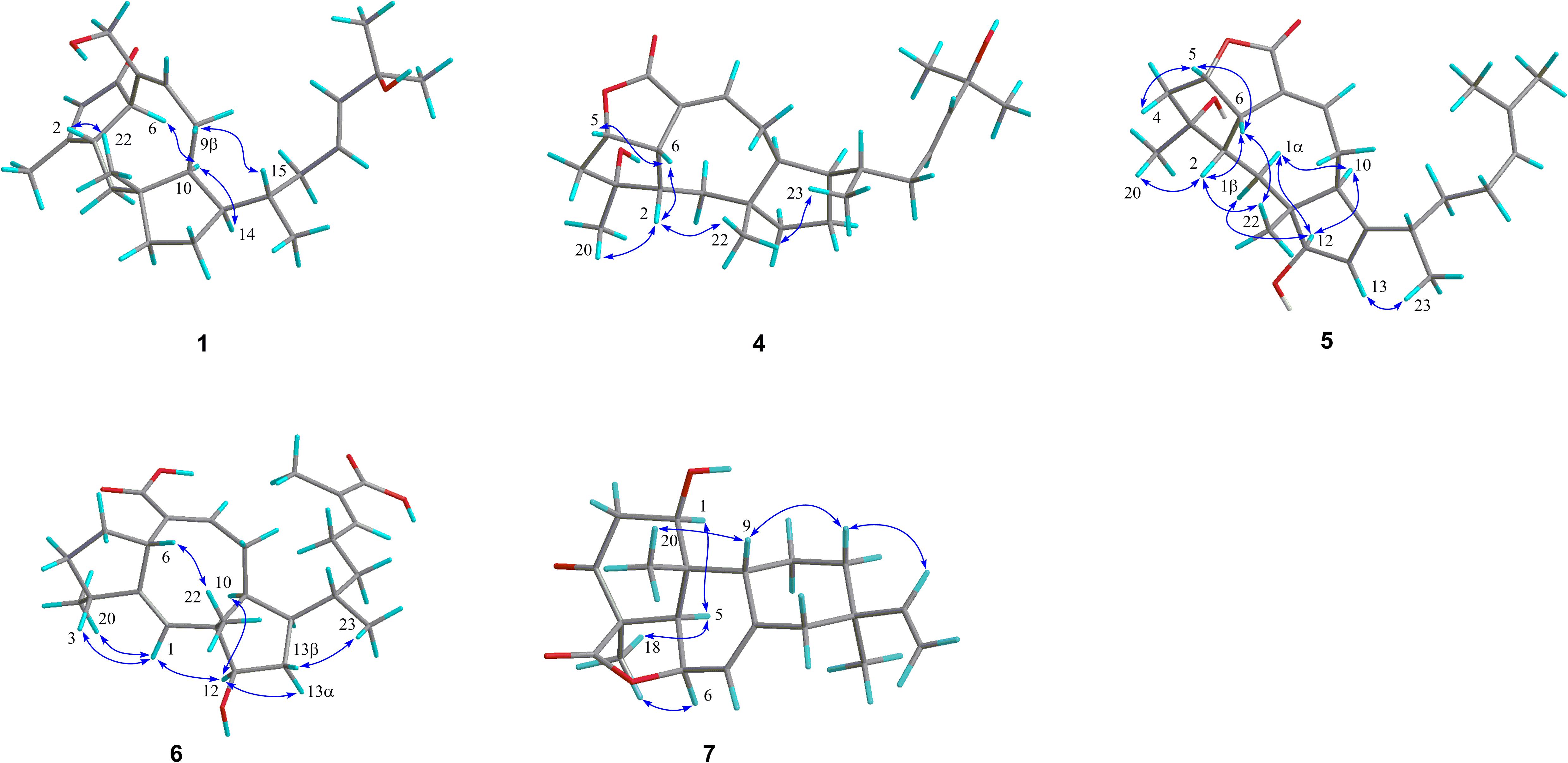
Figure 3. Key NOESY/ROESY correlations of compounds 1 and 4–7.
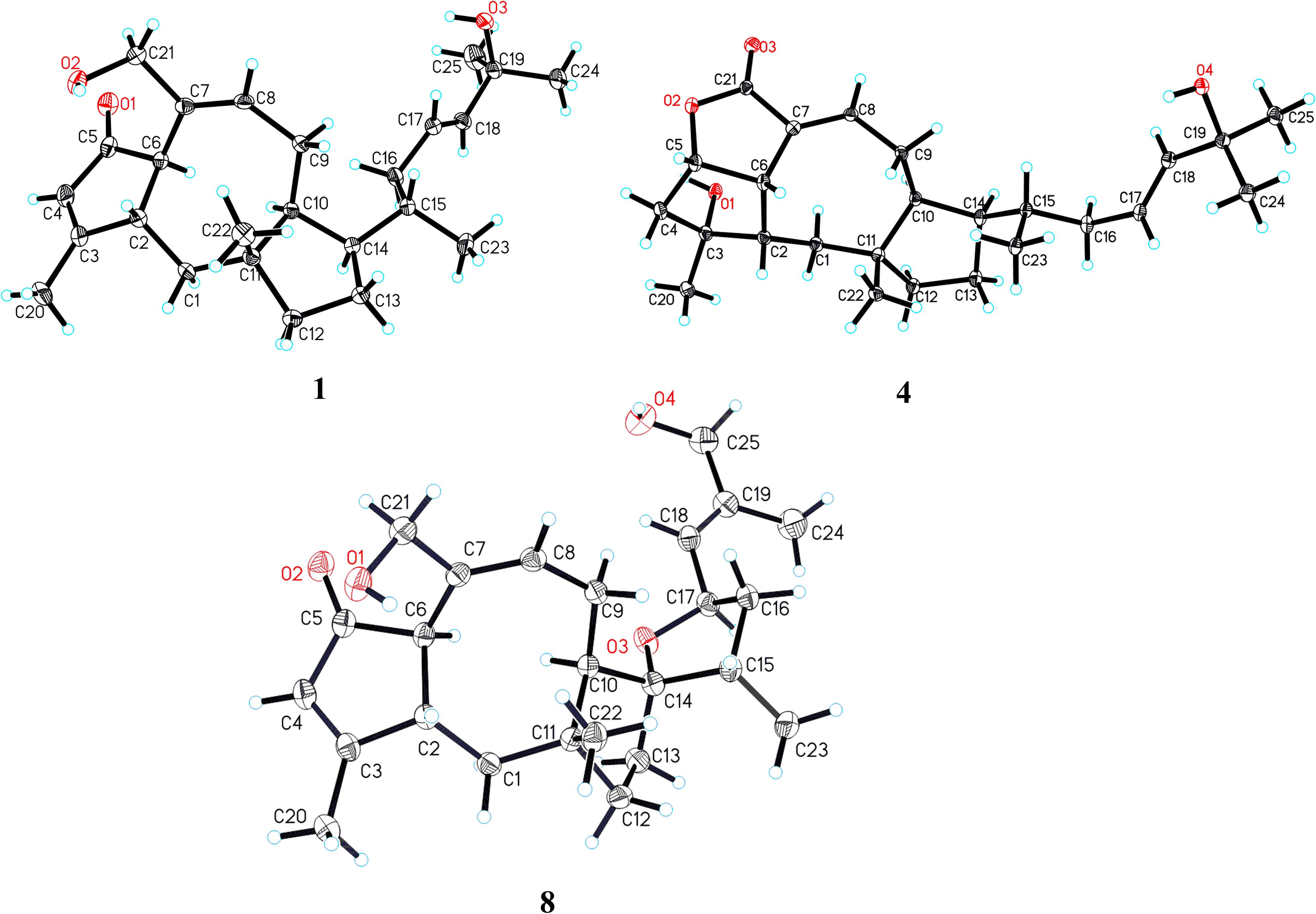
Figure 4. X-ray ORTEP drawings of compounds 1, 4, and 8.
Compound 2 was obtained as a colorless oil, which had a molecular formula of C26H40O3, according to its HRESIMS data at m/z 423.2882 ([M+Na]+, calcd for 423.2870). The 1D NMR data (Tables 1 and 3) showed close similarities to those of 1, except for the C-19 hydroxy group in 1 being replaced by a methoxy group (δC 50.5) in 2, as supported by the key HMBC correlation (Figure 2) from δH 3.14 (3H, s, OMe-19) to C-19 (δC 76.4). The close resemblance of the NOESY data (Supplementary Data Sheet S5) and experimental CD curves (Figure 5) of 1 and 2 suggested their identical absolute configuration. Accordingly, the structure of 2 was defined and named bipolatoxin B.
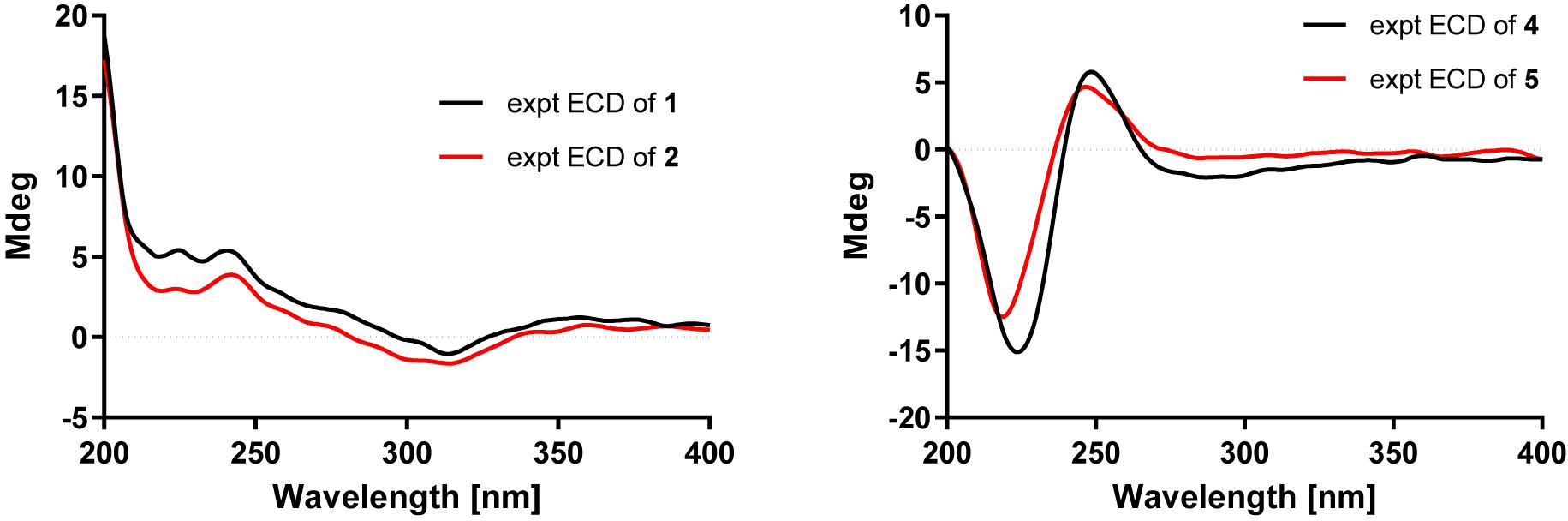
Figure 5. Experimental CD spectra of compounds 1–2 and 4–5.
Compound 3 was also purified as a colorless oil. The HRESIMS analysis of 3 displayed a sodium adduct ion at m/z 407.2550 [M+Na]+ (calcd for 407.2557), suggesting a molecular formula of C25H36O3. Subsequent comparison of the 1H and 13C NMR data (Tables 1 and 3) of 3 with those of 1 suggested that 3 contained an additional double bond (δC 121.1 and 151.2). Further analyses of the HMBC data (Figure 2) of 3 revealed that the double bond was located between C-13 and C-14, as supported by the correlations from H3-23 (δH 1.09) to C-14 (δC 151.2) and from H-13 (δH 5.32) to C-10 (δC 52.1) and C-11 (δC 46.2). The stereochemical configuration of 3 was established to be the same as 1 by their closely resembled NOE data (Supplementary Data Sheet S5) as well as shared biogenesis. Accordingly, the structure of 3 was defined and named bipolatoxin C.
The HRESIMS ion at m/z 425.2653 ([M+Na]+, calcd for 425.2662), together with the 13C NMR and DEPT data for 4, revealed its molecular formula to be C25H38O4, requiring an index of hydrogen deficiency of seven. The 1H and 13C NMR data (Tables 1 and 3) of 4 closely resembled those of ophiobolin X (Zhu et al., 2018), indicating that both compounds shared the same A/B/C rings and corresponding substituents, with the only difference being on the side chain that the conjugated double bonds (C-16/C-17 and C-18/C-19) were replaced by an sp3 methylene (δC 41.1, C-16), a double bond between C-17 (δC 127.0) and C-18 (δC 140.4), and a hydroxylated quaternary carbon (δC 71.1, C-19). These conclusions were further confirmed by the 1H–1H COSY correlations of H-14/H-15 (H3-23)/H2-16/H-17/H-18 and an obvious HMBC correlation from H3-24 to C-19 (Figure 2). The ROESY correlations (Figure 3) of H-5 (δH 4.99)/H-6 (δH 3.72)/H-2 (δH 2.05) and H3-20 (δH 1.21)/H-2 (δH 2.05)/H3-22 (δH 0.99)/H3-23 (δH 0.84) indicated that H-10 and H-14 were α-oriented, whereas H-2, H-5, H-6, H3-20, H3-22, and H3-23 were all β-oriented. After repeated recrystallization in MeOH–H2O (20:1, vol/vol) at room temperature, 4 furnished a crystal suitable for X-ray diffraction analysis (Figure 4). The Flack parameter of 0.05(10) allowed an unambiguous assignment of the complete absolute configurations of all chiral centers as 2S, 3R, 5S, 6S, 10S, 11R, 14R, and 15S, as well as an E geometry of the Δ17,18 double bond. Accordingly, the structure of 4 was defined and named bipolatoxin D.
Compound 5 gave a molecular formula of C25H36O4, as assigned by a sodium adduct ion at m/z 423.2500 ([M+Na]+, calcd for 423.2506) in the HRESIMS analysis. Comparison of its 1D NMR data (Tables 2 and 3) with those of 4 indicated that the double bond from C-17/C-18 in 4 migrated to C-18/C-19 in 5, and an additional double bond (C-13, δC 125.9; C-14, δC 151.4) and an oxygenated methine (δC/H 84.6/4.23, C-12) were present, as supported by the 1H–1H COSY cross-peaks (Figure 2) of H-12 (δH 4.23)/H-13 (δH 5.31), and H2-17 (δH 1.99)/H-18 (δH 5.15), together with HMBC correlations of H3-22 (δH 0.94) with C-12 (δC 84.6) and of H-18 with C-24 (δC 17.8) and C-25 (δC 25.9). Based on the ROESY correlation (Figure 3) between H-10 (δH 2.28) and H-12, the hydroxy group at C-12 was determined to be β-oriented. The similar ROESY data (Figure 3) and experimental CD curves (Figure 5) of 4 and 5 indicated that both compounds shared the identical absolute configuration. Accordingly, the structure of 5 was defined and named bipolatoxin E.
Compound 6, obtained as a colorless oil, gave a molecular formula of C25H36O5 via its (+)-HRESIMS data (m/z 439.2453, [M+Na]+, calcd for 439.2455). The 1H and 13C NMR data (Tables 2 and 3) of 6 were similar to those of 21-dehydroophiobolin U (Zhu et al., 2018), differing in that a ketone carbonyl, a double bond, and an aldehyde carbon signals were absent, and two carboxy groups (δC 172.1 and 173.4) and an additional oxygenated methine (δC/H 80.3/3.73) were present in 6. The HMBC correlations (Figure 2) of H-6 (δH 3.83) and H-8 (δH 6.41) with C-21 (δC 173.4) and of H3-24 (δH 1.82) and H-18 (δH 6.77) with C-25 (δC 172.1) suggested that two carboxy groups should be located at C-21 and C-25, respectively. The HMBC correlations from H3-23 (δH 0.89) to C-14 (δC 41.4), C-15 (δC 33.8) and C-16 (δC 37.1), together with the 1H–1H COSY cross-peaks of H-14 (δH 2.25)/H-15 (δH 1.78)/H2-16 (δH 1.30 and 1.41)/H2-17 (δH 2.25)/H-18 (δH 6.77), suggested that a C-16–C-17 double bond in 21-dehydroophiobolin U was reduced in 6. The HMBC correlations from H-6 to C-2 (δC 155.4) and C-7 (δC 141.9) and from H-1 (δH 5.66) to C-6 (δC 41.8), C-10 (δC 47.3), and C-11 (δC 50.5) indicated that the double bond from C-2/C-6 in 21-dehydroophiobolin U migrated to C-1/C-2 in 6. The 1H–1H COSY correlations of H-12 (δH 3.73)/H2-13 (δH 1.41 and 1.72)/H-14 suggested that an oxygenated methine at δC/H 80.3/3.73 was located at C-12. The NOESY correlations (Figure 3) of H-6/H3-22 (δH 1.08), H-12/H-10 (δH 1.90)/H-14, and H-3 (δH 2.47)/H-1 (δH 5.66) indicated that the HO-12 group and H-6 were β-oriented, whereas H-10, H-14 and H3-20 were α-oriented. Accordingly, the structure of 6 was defined and named bipolatoxin F.
Compound 7 was isolated as a colorless oil, with the molecular formula of C20H26O4, as determined by its HRESIMS data ([M+H]+ ion peak at m/z 331.1913). Its 1H and 13C NMR data (Tables 2 and 3) were similar to those of momilactone A (Germain and Deslongchamps, 2002), with the replacement of an sp3 methylene in momilactone A by an oxygenated sp3 methine (δC/δH 68.2/4.18) in 7, which was supported by analysis of its 2D NMR data (Figure 2). In the 1H–1H COSY spectrum, the correlation of H-1 (δH 4.18)/H2-2 (δH 2.41 and 2.86), together with HMBC correlations from H-5 (δH 2.52) and H3-20 (δH 0.93) to C-1 (δC 68.2), suggested that a hydroxylated methine was located at C-1. In the NOESY spectrum (Figure 3), the cross-peaks of H-1/H-5/H3-18 suggested that the OH-1 group should be β-oriented. Accordingly, the structure of 7 was established as shown and named 1β-hydroxy momilactone A.
The three known compounds were identified as 25-hydroxyophiobolin I (8) (Sugawara et al., 1987), ophiobolin I (9) (Sugawara et al., 1987), and ophiobolin A lactone (10) (Li et al., 1995), by comparison of their NMR data and specific rotations with literature. Remarkably, it is the first time that the absolute structure of compound 8 was defined by the single-crystal X-ray diffraction analysis (Figure 4).
Because of the limited amount of compound 5, only compounds 1–4 and 6–10 were evaluated for the antimicrobial activity against six drug-resistant microbial pathogens, including ESBL-producing E. coli, A. baumannii, P. aeruginosa, K. pneumoniae, methicillin-resistant S. aureus (MRSA), E. faecalis, and one fungus C. albicans. As shown in Table 4, except for compound 8, all the other test compounds showed antimicrobial activity against certain microbial pathogens (MIC = 8–64 μg/mL), of which compound 4 showed significant inhibitory activity against E. faecalis with an MIC value of 8 μg/mL, and compound 10 showed significant inhibitory activity against A. baumannii and E. faecalis with MIC values of 8 and 8 μg/mL, respectively.
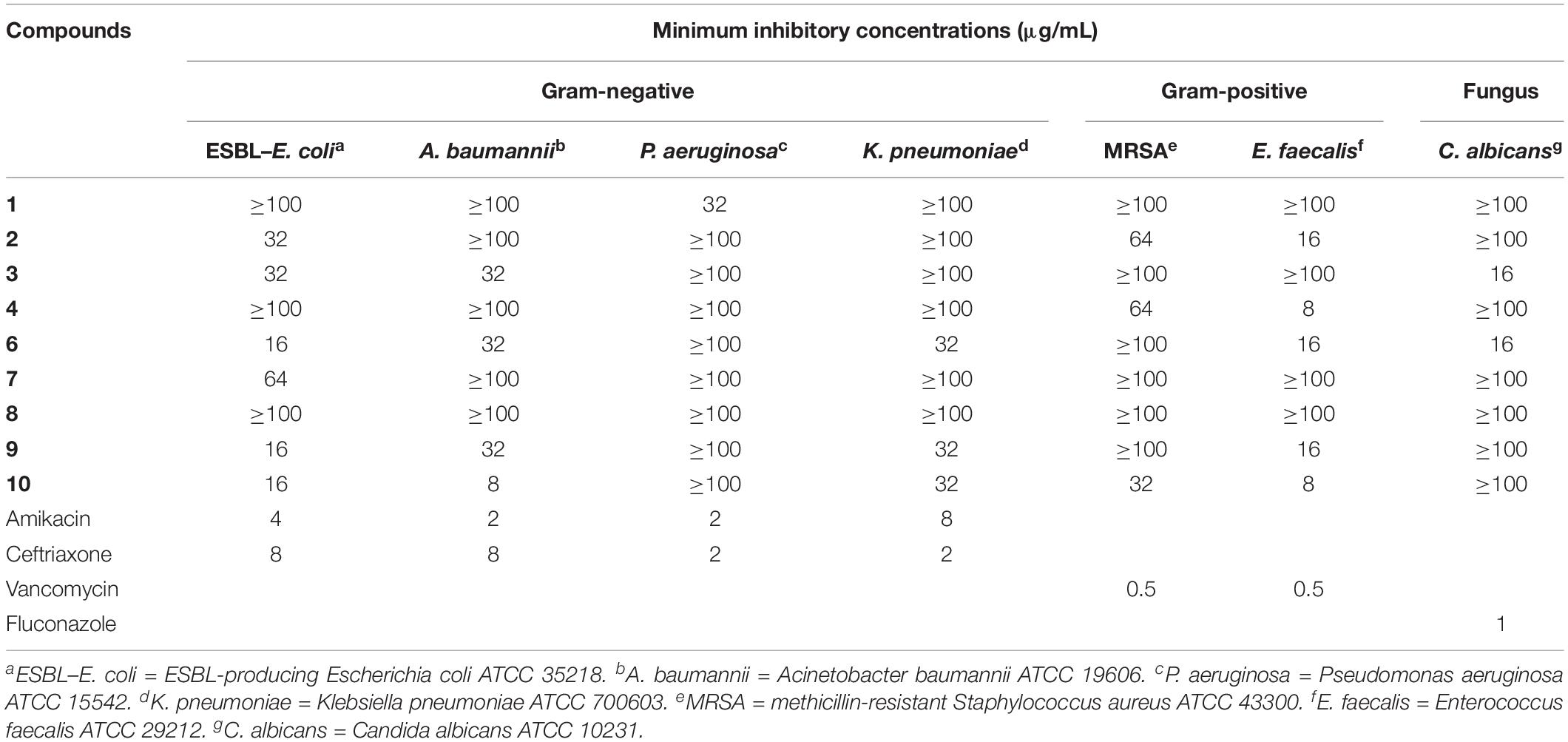
Table 4. Antimicrobial activity of compounds 1–4 and 6–10.
A total of 10 secondary metabolites (1–10), incorporating six new ophiobolin-type sesterterpenes (1–6) and one new pimarane-type diterpene (7), were isolated and identified from the solid cultures of fungus Bipolaris species TJ403-B1. The antimicrobial activity assay revealed that compound 4 showed significant inhibitory activity against E. faecalis with an MIC value of 8 μg/mL, and 10 showed significant inhibitory activity against A. baumannii and E. faecalis with MIC values of 8 and 8 μg/mL, respectively. Our current work not only replenishes new members to ophiobolin-type sesterterpenes, but also furnishes potential antimicrobial lead compounds that are necessary to check further for their synergistic and efflux pump inhibition properties.
The crystallographic data for these structures were deposited in the Cambridge Crystallographic Data Centre (CCDC 1971181 for 1, CCDC 1913829 for 4, and CCDC 1913832 for 8). Direct links: 1. https://www.ccdc.cam.ac.uk/structures/search?access=referee &searchdepnums=1971181&searchauthor=Zhengxi; 2. https://www.ccdc.cam.ac.uk/structures/search?access=referee&searchdepnums=1913829&searchauthor=hu; 3. http://www.ccdc.cam.ac. uk/services/structures?access=referee&searchdepnums=1913832 &searchauthor=hu.
LS and ML contributed to the extraction, isolation, identification, and manuscript preparation. YH contributed to the antimicrobial activity test. WA contributed to the fungal isolation and fermentation. HL, SL, CC, and JW advised and assisted Shen’s experiments. ZH guided Shen’s experiments and wrote the manuscript. YZ designed the experiments and revised the manuscript.
We greatly acknowledge the financial supports from the National Science Fund for Distinguished Young Scholars (No. 81725021), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), the Program for Changjiang Scholars of Ministry of Education of the People’s Republic of China (No. T2016088), the National Natural Science Foundation of China (Nos. 21702067, 81573316, and 81803387), the Hubei Provincial Natural Science Foundation of China (Grant No. 2018CFB152), the Academic Frontier Youth Team of HUST, and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Analytical and Testing Center at Huazhong University of Science and Technology for CD, IR and single-crystal X-ray diffraction analyses.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00856/full#supplementary-material
DATA SHEET S1 | CheckCIF report of 1.
DATA SHEET S2 | CheckCIF report of 4.
DATA SHEET S3 | CheckCIF report of 8.
DATA SHEET S4 | X-ray crystallographic data (CIFs) of 1, 4, and 8.
Baker, S. (2015). A return to the pre-antimicrobial era. Science 347, 1064–1066. doi: 10.1126/science.aaa2868
Brown, E. D., and Wright, G. D. (2016). Antibacterial drug discovery in the resistance era. Nature 529, 336–343. doi: 10.1038/nature17042
Cai, R., Jiang, H., Mo, Y., Guo, H., Li, C., Long, Y., et al. (2019). Ophiobolin-type sesterterpenoids from the mangrove endophytic fungus Aspergillus sp. ZJ-68. J. Nat. Prod. 82, 2268–2278. doi: 10.1021/acs.jnatprod.9b00462
Chiba, R., Minami, A., Gomi, K., and Oikawa, H. (2013). Identification of ophiobolin f synthase by a genome mining approach: a sesterterpene synthase from Aspergillus clavatus. Org. Lett. 15, 594–597. doi: 10.1021/ol303408a
Demirci, H., Murphy, F. I. V., Murphy, E., Gregory, S. T., Dahlberg, A. E., and Jogl, G. (2013). A structural basis for streptomycin-induced misreading of the genetic code. Nat. Commun. 4:1355. doi: 10.1038/ncomms2346
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K., and Puschmann, H. (2009). OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 42, 339–341. doi: 10.1107/S0021889808042726
Gao, W., Chai, C., He, Y., Li, F., Hao, X., Cao, F., et al. (2019). Periconiastone A, an antibacterial ergosterol with a pentacyclo[8.7.0.01,5.02,14.010,15]heptadecane system from Periconia sp. TJ403-rc01. Org. Lett. 21, 8469–8472. doi: 10.1021/acs.orglett.9b03270
Germain, J., and Deslongchamps, P. (2002). Total synthesis of (±)-momilactone A. J. Org. Chem. 67, 5269–5278. doi: 10.1021/jo025873l
Jakubczyk, D., and Dussart, F. (2020). Selected fungal natural products with antimicrobial properties. Molecules 25:911. doi: 10.3390/molecules25040911
Li, E., Clark, A. M., Rotella, D. P., and Hufford, C. D. (1995). Microbial metabolites of ophiobolin A and antimicrobial evaluation of ophiobolins. J. Nat. Prod. 58, 74–81. doi: 10.1021/np50115a009
Liu, M., He, Y., Shen, L., Al Anbari, W. H., Li, H., Wang, J., et al. (2019a). Asperteramide A, an unusual N-phenyl-carbamic acid methyl ester trimer isolated from the coral-derived fungus Aspergillus terreus. Eur. J. Org. Chem. 2019, 2928–2932. doi: 10.1002/ejoc.201900383
Liu, M., He, Y., Shen, L., Hu, Z., and Zhang, Y. (2019b). Bipolarins A–H, eight new ophiobolin-type sesterterpenes with antimicrobial activity from fungus Bipolaris sp. TJ403-B1. Chin. J. Nat. Med. 17, 935–944. doi: 10.1016/S1875-5364(19)30116-5
Liu, M., Sun, W., Shen, L., He, Y., Liu, J., Wang, J., et al. (2019d). Bipolarolides A–G: Ophiobolin-derived sesterterpenes with three new carbon skeletons from Bipolaris sp. TJ403-B1. Angew. Chem. Int. Ed. 58, 12091–12095. doi: 10.1002/anie.201905966
Liu, M., Sun, W., Shen, L., Hao, X., Al Anbari, W. H., Lin, S., et al. (2019c). Bipolaricins A–I, ophiobolin-type tetracyclic sesterterpenes from a phytopathogenic Bipolaris sp. fungus. J. Nat. Prod. 82, 2897–2906. doi: 10.1021/acs.jnatprod.9b00744
Narita, K., Chiba, R., Minami, A., Kodama, M., Fujii, I., Gomi, K., et al. (2016). Multiple oxidative modifications in the ophiobolin biosynthesis: P450 oxidations found in genome mining. Org. Lett. 18, 1980–1983. doi: 10.1021/acs.orglett.6b00552
Sheldrick, G. M. (2008). A short history of SHELX. Acta Cryst. A 64, 112–122. doi: 10.1107/S0108767307043930
Sugawara, F., Strobel, G., Strange, R. N., Siedow, J. N., Van Duyne, G. D., and Clardy, J. (1987). Phytotoxins from the pathogenic fungi Drechslera maydis and Drechslera sorghicola. Proc. Natl. Acad. Sci. U.S.A. 84, 3081–3085. doi: 10.1073/pnas.84.10.3081
Wang, Q. X., Yang, J. L., Qi, Q. Y., Bao, L., Yang, X. L., Liu, M. M., et al. (2013). 3-Anhydro-6-hydroxy-ophiobolin A, a new sesterterpene inhibiting the growth of methicillin-resistant Staphylococcus aureus and inducing the cell death by apoptosis on K562, from the phytopathogenic fungus Bipolaris oryzae. Bioorg. Med. Chem. Lett. 23, 3547–3550. doi: 10.1016/j.bmcl.2013.04.034
Wright, G. D. (2017). Opportunities for natural products in 21st century antibiotic discovery. Nat. Prod. Rep. 34, 694–701. doi: 10.1039/C7NP00019G
Yang, B., He, Y., Lin, S., Zhang, J., Li, H., Wang, J., et al. (2019). Antimicrobial dolabellanes and atranones from a marine-derived strain of the toxigenic fungus Stachybotrys chartarum. J. Nat. Prod. 82, 1923–1929. doi: 10.1021/acs.jnatprod.9b00305
Zhu, T., Lu, Z., Fan, J., Wang, L., Zhu, G., Wang, Y., et al. (2018). Ophiobolins from the mangrove fungus Aspergillus ustus. J. Nat. Prod. 81, 2–9. doi: 10.1021/acs.jnatprod.7b00335
Keywords: Bipolaris species TJ403-B1, ophiobolin-type sesterterpenes, pimarane-type diterpene, structure elucidation, antimicrobial activity
Citation: Shen L, Liu M, He Y, Al Anbari WH, Li H, Lin S, Chai C, Wang J, Hu Z and Zhang Y (2020) Novel Antimicrobial Compounds as Ophiobolin-Type Sesterterpenes and Pimarane-Type Diterpene From Bipolaris Species TJ403-B1. Front. Microbiol. 11:856. doi: 10.3389/fmicb.2020.00856
Received: 30 December 2019; Accepted: 09 April 2020;
Published: 29 May 2020.
Edited by:
Santi M. Mandal, Indian Institute of Technology Kharagpur, IndiaReviewed by:
Steven W. Polyak, University of South Australia, AustraliaCopyright © 2020 Shen, Liu, He, Al Anbari, Li, Lin, Chai, Wang, Hu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengxi Hu, aHp4NjE2QDEyNi5jb20=; Yonghui Zhang, emhhbmd5aEBtYWlscy50am11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.