 David F. Woods
David F. Woods Iwona M. Kozak
Iwona M. Kozak Fergal O’Gara
Fergal O’Gara- 1BIOMERIT Research Centre, School of Microbiology, University College Cork, Cork, Ireland
- 2Human Microbiome Programme, School of Biomedical Sciences, Curtin Health Innovation Research Institute, Curtin University, Perth, WA, Australia
- 3Telethon Kids Institute, Perth Children’s Hospital, Perth, WA, Australia
Traditional food preservation processes are vital for the food industry. They not only preserve a high-quality protein and nutrient source but can also provide important value-added organoleptic properties. The Wiltshire process is a traditional food curing method applied to meat, and special recognition is given to the maintenance of a live rich microflora within the curing brine. We have previously analyzed a curing brine from this traditional meat process and characterized a unique microbial core signature. The characteristic microbial community is actively maintained and includes the genera, Marinilactibacillus, Carnobacterium, Leuconostoc, and Vibrio. The bacteria present are vital for Wiltshire curing compliance. However, the exact function of this microflora is largely unknown. A microbiome profiling of three curing brines was conducted and investigated for functional traits by the robust bioinformatic tool, Tax4Fun. The key objective was to uncover putative metabolic functions associated with the live brine and to identify changes over time. The functional bioinformatic analysis revealed metabolic enrichments over time, with many of the pathways identified as being involved in organoleptic development. The core bacteria present in the brine are Lactic Acid Bacteria (LAB), with the exception of the Vibrio genus. LAB are known for their positive contribution to food processing, however, little work has been conducted on the use of Vibrio species for beneficial processes. The Vibrio genome was sequenced by Illumina MiSeq technologies and annotated in RAST. A phylogenetic reconstruction was completed using both the 16S rRNA gene and housekeeping genes, gapA, ftsZ, mreB, topA, gyrB, pyrH, recA, and rpoA. The isolated Vibrio species was defined as a unique novel species, named Vibrio hibernica strain B1.19. Metabolic profiling revealed that the bacterium has a unique substrate scope in comparison to other closely related Vibrio species tested. The possible function and industrial potential of the strain was investigated using carbohydrate metabolizing profiling under food processing relevant conditions. Vibrio hibernica is capable of metabolizing a unique carbohydrate profile at low temperatures. This characteristic provides new application options for use in the industrial food sector, as well as highlighting the key role of this bacterium in the Wiltshire curing process.
Introduction
The use of microorganisms in processing technologies has a rich and sophisticated narrative throughout history. This ranges from ancient textile techniques, to the production of antimicrobials, sewage treatment and utilizing biotechnology in the construction industry for microbially mediated construction processes and biomaterial production (Fleming, 2001; Dhall et al., 2012; Liu et al., 2014; Stabnikov et al., 2015; Dapurkar and Telang, 2017; Heine et al., 2019). Microbial processing technologies have also been widely implemented in the food industry. Our increased knowledge of microbial processing of food has led to the incorporation and encouragement of specific microbial activities to form a safer, healthier and more flavorsome end products (Singh et al., 2016). These microbiological processes can produce a value-added product in comparison to the raw material inputs. Bacteria have been used in the production of yogurt, in the >1000 varieties of cheese, olives, kefir, wines, kimchi and in sausage meat to name but a few (Sandine and Elliker, 1970; Lücke, 2000; Fleet, 2008; Kearney et al., 2011; Jung et al., 2012; Gul et al., 2015; Papadelli et al., 2015; Fontana et al., 2016; De Pasquale et al., 2019).
Fermented meat products are of particular importance with regards to the maintenance of their nutrients as well as the increase in organoleptic qualities and market value (Fontana et al., 2016). Research into the microbial composition of traditional food processes is a rapidly growing area with particular emphasis put on the genomic potential of microorganisms present within a process. However, the complex dynamics mediated by the microbial milieu during food processing require further investigation and this is particularly evident with traditional meat processes (Bokulich et al., 2012b, 2016). There is a strong need to investigate the overall function of these processes as well as their component parts, to elucidate the activities that the microbes are completing. The definition of normal and abnormal microbial compositions is vital at every stage of bacterial mediated food processing. This goal is essential to understand key issues underpinning organoleptic consistency, problem diagnostics, spoilage investigation and process improvement (Bokulich et al., 2012a, 2016).
Both culture-dependent and culture-independent based techniques have been used to examine the microflora present in meat. Culture-dependent techniques are not commonly applicable to many food-associated niches for complete microbial characterization. This is due to difficulties in designing and reproducing the appropriate growth media and conditions to enable indigenous microbes to grow under laboratory conditions (Amann et al., 1995). However, the culturing of microbes is an important step to gain more insights into bacterial function and for the potential development of inocula (Hugas and Monfort, 1997). With regards to culture-independent techniques Next Generation Sequencing (NGS) technologies have advanced rapidly and have been applied to solve the difficulties of obtaining accurate microbial profiles. The rate of NGS technological advancements has been noted as being significantly higher than the rate of standard computational power advancements based on Moore’s Law (Batt, 2016). The NGS of marker genes, such as the 16S rRNA gene, provides an accurate snapshot of the bacterial content within a food process and has been successfully applied to characterize a number of traditional meat processes (Mayo et al., 2014; Franciosa et al., 2018; Woods et al., 2019). Moreover, NGS sequence datasets could be applied to functional prediction algorithms such as Tax4Fun to aid in the interrogation of the potential metabolic capabilities of traditional bacterial food processes (Aßhauer et al., 2015). Thus, giving a putative overview of a process at a particular time. While Tax4Fun has been predominantly utilized for disease prediction and dysbiosis, traditional food practices can be very intricate and this functional prediction software could illuminate the processing capabilities of the bacteria present (Coffey et al., 2019; Sun et al., 2019; Wang et al., 2019).
Wiltshire curing is a traditional process which yields high-quality cured hams. Wiltshire curing is seen as both culturally and economically important, and there is special recognition given to the maintenance of the bacterial component of the process (Andersen, 2004). Meat is cured in microbially rich brine for 5–7 days at a temperature of 4°C. After approximately 40 days use, brine that is deemed to be spent is replenish with half fresh brine to maintain the microbial component (Andersen, 2004). Curing/processing of meat at lower temperature decreases the propagation of most pathogenic bacteria, which are predominantly mesophilic (Húngaro et al., 2014). Using cold adapted bacteria, referred to as psychrophiles, to process food at lower temperatures would be advantageous and increase the food safety of a product (Durán Quintana et al., 1999). This Wiltshire process has been successfully profiled for its core microbial content (Woods et al., 2019). The core genera identified in a Wiltshire compliant brine were Marinilactibacillus, Carnobacterium, Leuconostoc, and Vibrio. Species of Marinilactibacillus, Carnobacterium, and Leuconostoc are known as lactic acid producing bacteria and these Lactic Acid Bacteria (LAB) strains are often used in food processing (Toffin et al., 2005; Lucena-Padros et al., 2015; Pogacic et al., 2016; Soto Del Rio Mde et al., 2016; Liu et al., 2017; Saraoui et al., 2017). A Vibrio species has previously been identified in a Wiltshire compliant brine with no detrimental effect to the end product (Hinrichsen et al., 1994; Woods et al., 2019). Moreover, an unidentified autochthonous Vibrio species from a Wiltshire brine, was used to inoculate a brine and the meat was cured in this mixture. The Vibrio was shown to improve the organoleptic qualities of the meat specifically with regards to the color and flavor of the cured meat (Petaja et al., 1972).
The genus of Vibrio is one of the most diverse bacterial genera and has more than 142 species defined (Sawabe et al., 2013 2014). Vibrio species are ubiquitous in marine, estuarine and freshwater environments, and have been isolated from a wide variety of organisms including fish, plankton and mammals (Thompson et al., 2004; Reen et al., 2006; Froelich et al., 2013). The genus consists of Gram-negative short rod-shaped halophilic bacteria. The genome of Vibrio is organized into a two-chromosome configuration. The larger chromosome is predominantly between 3.0 and 3.3 Mb and the smaller between 0.8 and 2.4 Mb (Okada et al., 2005).
While much research has been conducted on the pathogenicity and the detrimental cost of notorious Vibrio species such as V. cholerae and V. parahaemolyticus, there is a plethora of non-pathogenic Vibrio species which harbor beneficial and industrially important applications (Makino et al., 2003; Bertuzzo et al., 2010). V. natriegens is an example of an industrially relevant bacteria which has been recognized as a novel candidate for future biotechnological processes due to its high biomass specific consumption rate in industrial fermentation processes (Hoffart et al., 2017). The marine V. costicola has important pharmaceutical relevance, as it is capable of producing the potent antileukemic agent L-Glutaminase and shows great substrate specificity over a wide variety of conditions (Nagendra Prabhu and Chandrasekaran, 1997). The genus has also been recognized for its importance in aquaculture. Investigation into the gut microbiota of healthy fish has seen the predominance of Vibrio species in a wide spectrum of studies, thus potentially identifying Vibrio as a key component of the healthy fish gut microbiome (Egerton et al., 2018).
In order to better understand the contributing effects of the microbial components of a Wiltshire cure, our study investigated the functional possibilities of the microbial content. We first predicted the potential functions of the microbes present in the brine, and at the different time points. We subsequently focused our study on the only non-LAB genus identified as a major component of the core microbiome. To assess the possible contribution of the Vibrio species to the overall organoleptic qualities of traditionally cured Wiltshire compliant products, we sequenced and studied the genome. The overall microbial functional analysis provided an insight into the prospects and opportunities of the total brine. In contrast, the genome was investigated for functionality with regards to meat processing and improvements to organoleptic related food properties. We investigated the metabolism of this Vibrio species and showed a wide range of metabolic capabilities that could be industrially relevant and are related to flavor development. Moreover, the bacteria are able to metabolize industrially relevant molecules at 4°C. Therefore, the investigation into the capabilities of the bacterial genome is vital to further advance food processing and to increase our insight into the complex matter of flavor development.
Materials and Methods
Sampling and Characterization of an Active Wiltshire Brine
To sample and characterize the brine, procedures were followed from Woods et al. (2019). Brine was sampled from a fully Wiltshire compliant facility longitudinally at three timepoints, Day 0 where the brine is composed of half “spent” brine and half newly reconstituted brine, Day 20, where this brine was used for 20 days to cure different meats and Day 40, where the brine was deemed to be “spent.” The brine was sampled in late 2016 from three monitored curing containers. These samples are distinct from the previously characterized brine samples in Woods et al. (2019). To culture the brine microflora, Tryptic Soy Broth (TSB) (Merck) with 1.5% Agar (Merck) (TSA) ± additional 6% NaCl (Sigma) was used, incubated at 23°C for 2 weeks. Unique morphotypes were isolated and stored at −80°C.
To characterize the microbiome of the brine, 1 ml of brine was centrifuged at 13,000 × g for 5 min and the total genomic (g)DNA isolated using the Puregene DNA Extraction kit (QIAGEN). The quantity and quality were assessed by a Qubit 2.0 Fluorometer (Life Technologies) and a 0.5% agarose gel (Sigma), respectively. MWG Eurofins (Ebersberg, Germany) conducted the microbiome analysis from the isolated gDNA, by a tagged 16S rRNA gene PCR of the v3-v5 region and sequenced the amplicons on a Next Gen Illumina MiSeq (V3), 2 × 300 bp. Raw reads were demultiplexed and sequences were assigned an Operational Taxonomic Unit (OTU) in QIIME 1.9.0 (Caporaso et al., 2010). Bar charts were created in Excel. The raw reads are publicly available in the Sequence Read Archive (SRA) of the NCBI Database under Accession numbers; SRR11073122, SRR11073123, SRR11073124, SRR11073125, SRR11073126, SRR11073127, SRR11073128, SRR11073129, SRR11073130, SRR11073131, SRR11073132, SRR11073133, SRR11073134, SRR11073135, SRR11073136, SRR11073137, SRR11073138, SRR11073139, SRR11073140, SRR11073141, SRR11073142, SRR11073143, SRR11073144, SRR11073145, SRR11073146, SRR11073147, and SRR11073148.
Functional Bioinformatic Analysis of a Wiltshire Brine (Tax4Fun)
R package Tax4Fun was used to predict the possible functional capabilities of the microbial communities in the analyzed brine samples (Aßhauer et al., 2015). R version 3.3.2 was used to host the Tax4Fun package. The biom file of the microbiome data of each of the brine samples profiled in this study was converted to a text file in QIIME 1.9.0 (Caporaso et al., 2010) and imported to Tax4Fun (importQIIMEData). The SILVA123 database was downloaded from the Tax4Fun website1 and used to predict functional capabilities. The Tax4Fun result lists were formatted into data frames and saved as Comma-Separated Values (csv) files. This was imported into Microsoft Excel where the data was delineated by a comma. To produce the heatmap, the three technical replicates were averaged and each individual KEGG of the biological replicates was conditionally formatted to a color scale. All relevant data was included when above the overall average of all of the dataset. The data were arranged by the 0_1 sample set for clarity and trend identification. Within sample replicate variance was calculated in Excel.
Amplification of the 16S rRNA Gene for Species Identification
Morphotypes were identified by amplifying the 16S rRNA gene, with the previously published primer set, forward primer 63f (5′-CAG GCC TAA CAC ATG CAA GTC-3′) and reverse primer 1387r (5′-GGG CGG WGT GTA CAA GGC-3′) using the Q5 Hot Start High-Fidelity Polymerase system (New England Biolabs) (Marchesi et al., 1998). The following thermocycler conditions were used, initial activation at 98°C for 30 s and then subjected to 35 cycles of amplification (denaturation at 98°C for 10 s, annealing at 58°C for 30 s and elongation at 72°C for 90 s) followed by a final elongation step at 72°C for 2 min (BIOMETRA T3000). Amplicons were visualized on 0.8% agarose gels stained with SYBR Safe (Thermo Fisher Scientific) and sequenced by Eurofins Genomics2. Consensus sequences were achieved using Unipro UGENE version 1.27 and species identification was assessed using the in silico BLAST (NCBI) algorithm (Altschul et al., 1990; Okonechnikov et al., 2012).
Genome Extraction and Sequencing
Genomic DNA (gDNA) was isolated using the Gentra Puregene DNA Isolation Kit (Qiagen). The gDNA was checked for quantity using a Qubit 2.0 Fluorometer (Life Technologies) and the integrity assessed on a 0.5% agarose gel (Sigma). Library preparation, validation and sequencing was conducted by Eurofins Genetic Services. In brief, the genome was sequenced using the Illumina MiSeq platform with chemistry v3 and 2 × 300 bp paired-end sequencing.
Genome Assembly and Annotation
The assembly of the genome was conducted using Unicycler (Galaxy version 0.4.6.0) for Illumina short reads in the FASTQ format to produce complete and accurate assemblies (Wick et al., 2016). Contigs shorter than 1,000 bp were excluded from the final assembly. Genes were predicted and annotated using the Rapid Annotation Subsystem Technology (RAST version 2.0) software with the Classic RAST algorithm (Aziz et al., 2008; Overbeek et al., 2014). The BLAST function was used for additional annotation of genes.
Bioinformatic Species Definition
Two in silico methods of species definition were used to define Vibrio hibernica strain B1.19 as a novel species. The reference genomes of the following Vibrio species were downloaded in FASTA format from the National Centre for Biotechnology Information (NCBI) website: V. rumoiensis (NZ_AP018685.1, NZ_AP018686.1, NZ_AP018687.1, NZ_AP018688.1), V. litoralis (NZ_AUFZ00000000.1), Vibrio scophthalmi (NZ_CP016307.1, NZ_CP016308.1, NZ_CP016309.1, NZ_CP016310.1NZ_CP016311.1), Aliivibrio fischeri (NC_006840.2, NC_006841.2, NC_006842.1) and V. parahaemolyticus (NC_004603.1, NC_004605.1). The assembled genome of V. hibernica in FASTA format was uploaded to the ANI Calculator3 and compared to the reference genomes of other Vibrio species. The other method used in this study was in silico DNA-DNA Hybridization (DDH) using the Genome-To-Genome Distance Calculator (GGDC). The V. hibernica and reference genomes of other Vibrio species tested were uploaded in a pair-wise comparison to the GGDC website4.
Molecular Phylogenetic Analysis
To infer the phylogenetic history, nucleotide sequences were retrieved and downloaded from NCBI: GenBank5. The House Keeping (HK) genes used for the phylogenetic reconstruction were ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA. Sequence alignments were generated for the HK and 16S rRNA trees using Clustal Omega and trimmed to a consistent length (Sievers et al., 2011). The HK alignments were concatenated into a single alignment. The evolutionary analysis was conducted in MEGA X via the Maximum Likelihood (ML) method using the General Time Reversible model for the HK tree and the Kimura 2-parameter model for the 16S rRNA tree (Kimura, 1980; Nei and Kumar, 2000; Kumar et al., 2018). A discrete Gamma distribution with Invariant (G + I) sites was used to model evolutionary rate differences among sites. The phylogenetic distribution was tested by the Bootstrapping method using 1,000 replicates (Felsenstein, 1985). Nucleotide sequences from Salinivibrio costicola, Enterovibrio coralii, and Photobacterium lutimaris were used as Outgroups for the phylogenetic reconstruction. Details on the HK and 16S rRNA sequences accession numbers are in Supplementary Tables S1, S2.
Phenotypic Analysis of Vibrio hibernica
The strains used for comparative analysis included V. hibernica sp. nova (Vh) strain B1.19, V. litoralis DSM17657 (Vl), A. fischeri MJ11 (Af), V. scophthalmi DSM19140 (Vs), V. parahaemolyticus RIMD 2210633 (Vp) and V. gallaecicus DSM23502 (Vg). The temperature growth range was assessed on TSB and TSA (Merck), Marine Agar (MA) (BD Difco), Brain Heart Infusion (BHI) (Merck) and Nutrient Agar with NaCl (NA) (Peptone (Merck) 5 g/l, Meat Extract (Sigma) 3 g/l and Agar (Sigma) 15g/l at 4°C, 23°C, 30°C, and 37°C. Single colonies were tested for catalase activity with 10% hydrogen peroxide (Sigma) and cultured on Columbia Blood Agar (CBA) (Fannin) at 23°C for 48 h to assay hemolysis. Salt tolerance was conducted in TSB (Merck) supplemented with 0, 3, 6, 10, and 20% additional NaCl (Sigma) and grown at 23°C with agitation (180 rpm) for 24 h. To assess the biochemical profile of the organisms, an API 20NE kit (BioMerieux) was used with API NaCl Medium (BioMerieux). To profile the carbohydrate metabolism range of the bacteria, API 50CH kits (BioMerieux) were used in conjunction with API 50 CHB/E Medium (BioMerieux). Both the API 20NE and API 50CH were used with manufacturers guidelines with the exception of the temperatures used, 4 and 23°C. All assays were completed with three independent biological replicates.
Results
Microbial Profile and Functional Prediction of an Active Wiltshire Cure
The traditional Wiltshire curing process was profiled to determine the microbial signature of an active curing process. The brine was sampled in late 2016 from three monitored curing containers, Day 0, at the initial replenishing phase, where half of the spent brine is replaced with half freshly reconstituted brine, Day 20, where the brine had been used for 20 days and Day 40 where the brine was used for 40 days and thus designated as needing to be replenished to start the cycle anew. Samples were taken in triplicate for each sampling point. The microbial constituents of the meat cure were profiled by microbiome analysis. Marinilactibacillus, Carnobacterium, Leuconostoc, Vibrio, and Photobacterium were the dominant bacteria present. To further assess the consistency and stability of the Wiltshire microflora, a longitudinal microbiome profile of brine from three meat processing containers was conducted. Samples were taken at three time points, with three sampling replicates included. We compared this microbiome, sampled in late 2016, to Wiltshire brine microbiomes that we conducted in early 2016 and in 2014 and this demonstrated a highly similar profile in each (Supplementary Figure S1). Again, the profile signature was dominated with the genera, Marinilactibacillus, Carnobacterium, Leuconostoc, and Vibrio. The Genus Photobacterium was not present as a dominant member in all of the previous studies (Woods et al., 2019). Hence this Genus was not included as a stable dominant microbial member of the Wiltshire brine signature. There was minor variation within the sampling replicates and the microbial diversity percentages between brine samples, however, overall the profiles are highly similar (Supplementary Figure S1).
To further improve our understanding of this food curing process, it was necessary to analyze the function of the microbes within the brine samples. Using the powerful bioinformatic tool Tax4Fun, a functional prediction of the microbial capabilities of a Wiltshire brine was analyzed over time (Figure 1). The three individual cures that were sampled at three time points show similar trends from Day 0 to Day 20 up until Day 40. Within the triplicate samples for Day 0 and Day 20 a similar profile was observed, whereas Day 40 samples showed greater variability when compared. There are two major categories of functional groups, Cluster I where functional enrichment increases from Day 0 to Day 20 and subsequently shows a decrease by Day 40. Within Cluster II, there is a decreased enrichment from Day 0 until Day 20 and an increase in enrichment by Day 40. Metabolic and cellular functions were placed in an overall grouping such as amino acid and carbohydrate metabolism. Within Cluster I there are amino acid metabolism systems such as valine, leucine and isoleucine degradation, D-alanine metabolism, lysine biosynthesis, cysteine and methionine metabolism, alanine, aspartate, and glutamate metabolism. There are also amino acid metabolism systems in Cluster II such as phenylalanine metabolism, valine, leucine, and isoleucine biosynthesis, lysine degradation, phenylalanine, tyrosine and tryptophan biosynthesis, glycine, serine and threonine metabolism and arginine and proline metabolism. There are a number of genes annotated to functions involved in the metabolism of complex carbohydrates, most of these are in Cluster I. These included galactose metabolism, other glycan degradation, the pentose phosphate pathway, glycolysis/gluconeogenesis, starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism. While genetic functions such as the citrate acid cycle (TCA), glyoxylate and dicarboxylate metabolism and glutathione metabolism showed an enrichment in Cluster II. Both functions for ABC transporters and folate biosynthesis are present in Cluster I and show an increase enrichment from Day 0 to Day 20. This analysis provides a functional snapshot of a Wiltshire brine immediately after replenishment (Day 0), during full productive use (Day 20) and toward a spent brine phase (Day 40). It is likely that the functions most important for Wiltshire curing are those that are enriched for in Cluster I on Day 20, thus revealing a predicted functional profile for a fully compliant curing process.
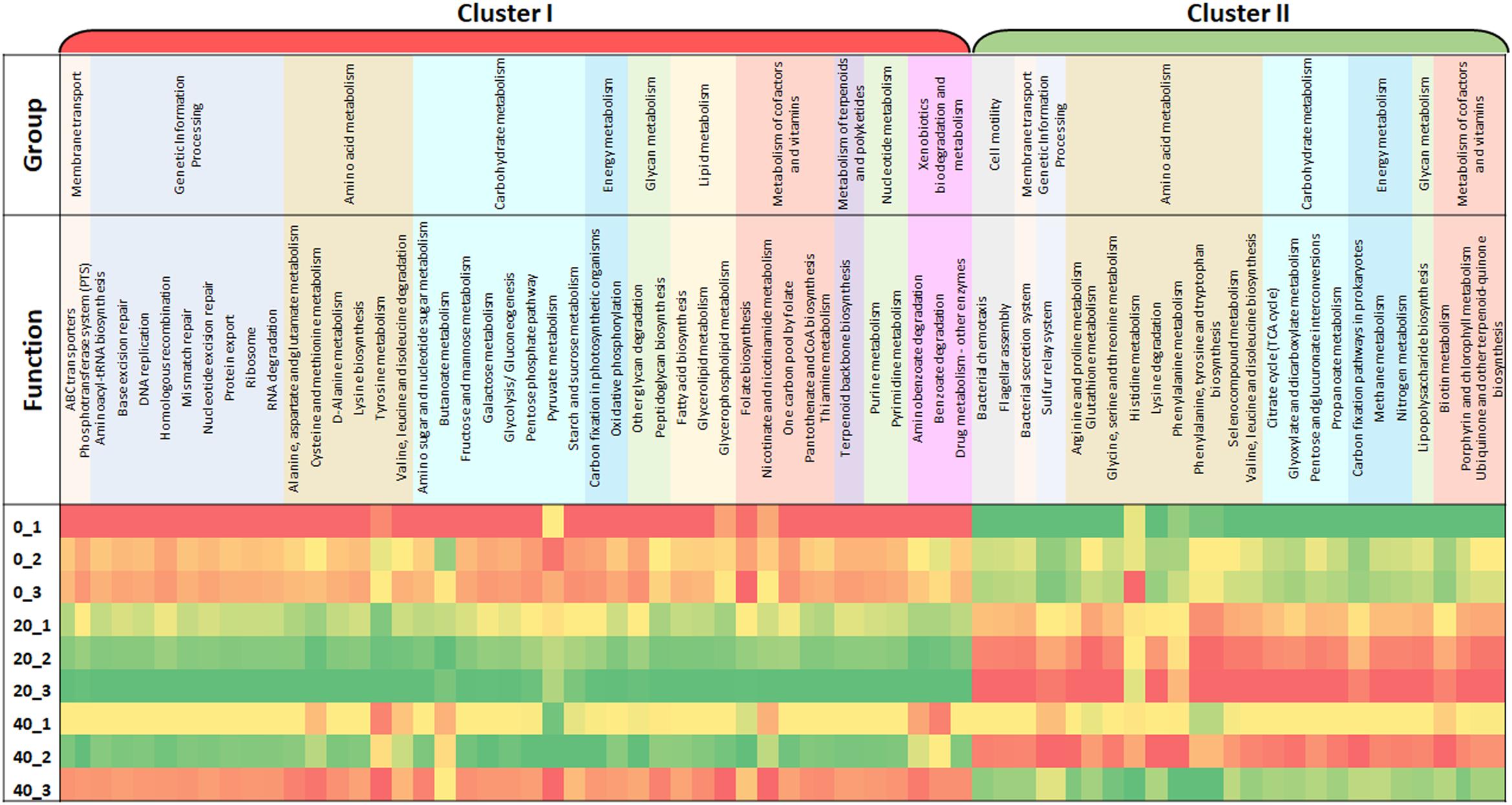
Figure 1. This heat map illustrates changes in functional capabilities of a brine microbiome at three timepoints, immediately after the initial replenishment of the Wiltshire brine (Day 0, denoted _0), during the use of the brine (Day 20) and just before the spent brine is replenished (Day 40). At each timepoint three replicates of brine was sampled to give an average value. Three containers of brine were monitored in tandem Container 1, 2, and 3. Samples are designated as Container Number_Day, for example 0_1 is equivalent to Day 0_Container 1. The data sets cluster into two major sets, Cluster I and II. Cluster I shows a functional enrichment increase from Day 0 to Day 20 and are subsequently selectively decreased for by Day 40, and Cluster II shows a decreased enrichment from Day 0 until Day 20 and selectivity enriched for by Day 40. The color scale ranges from red through yellow and to green, red indicates a low functional enrichment, yellow, a medium functional enrichment and green, a high functional enrichment of the averaged individual KEGG function.
Genomic Components and Characteristics of the Vibrio Strain Isolated From Wiltshire Brine
Initially to investigate the novelty of the Vibrio isolate (hereto named Vibrio hibernica), we sequenced the 16S rRNA marker gene. We analyzed ten independently cultured isolates, randomly selected Vibrio morphotypes from different brine containers on different days. The 16S rRNA marker gene was sequenced in each and showed sequence homology supported by at least three independent reads of each nucleotide. This analysis showed 98% sequence identity with the closest published Vibrio species, V. litoralis (DQ097524.1). The phylogenetic analysis of members of the Vibrio genus was reconstructed using the common genetic marker of the 16S rRNA gene (Supplementary Figure S2). The 16S rRNA gene tree was not supported well with the 1,000 bootstraps. However, this tree gave us an indication of the closely related species (V. litoralis) and Vibrio species from different clades (V. parahaemolyticus, V. scophthalmi, A. fischeri, and V. gallaecicus).
Due to the 16S rRNA gene having sequence novelty, the genome of V. hibernica was sequenced. The genome comprised of 3,425,268 bp with an N50 of >60 kb and an average GC content of 40.6% (Table 1). There is a predicted total of 3,151 protein coding genes within the genome and 57 RNA genes. The data of the genome sequence of V. hibernica strain B1.19 that is studied here is deposited in DDBJ/ENA/GenBank under the accession VHKO00000000. The analysis from the RAST SEED Viewer revealed that there are 481 subsystems present in the genome analysis (Figure 2).

Table 1. General features of the Vibrio hibernica genome.
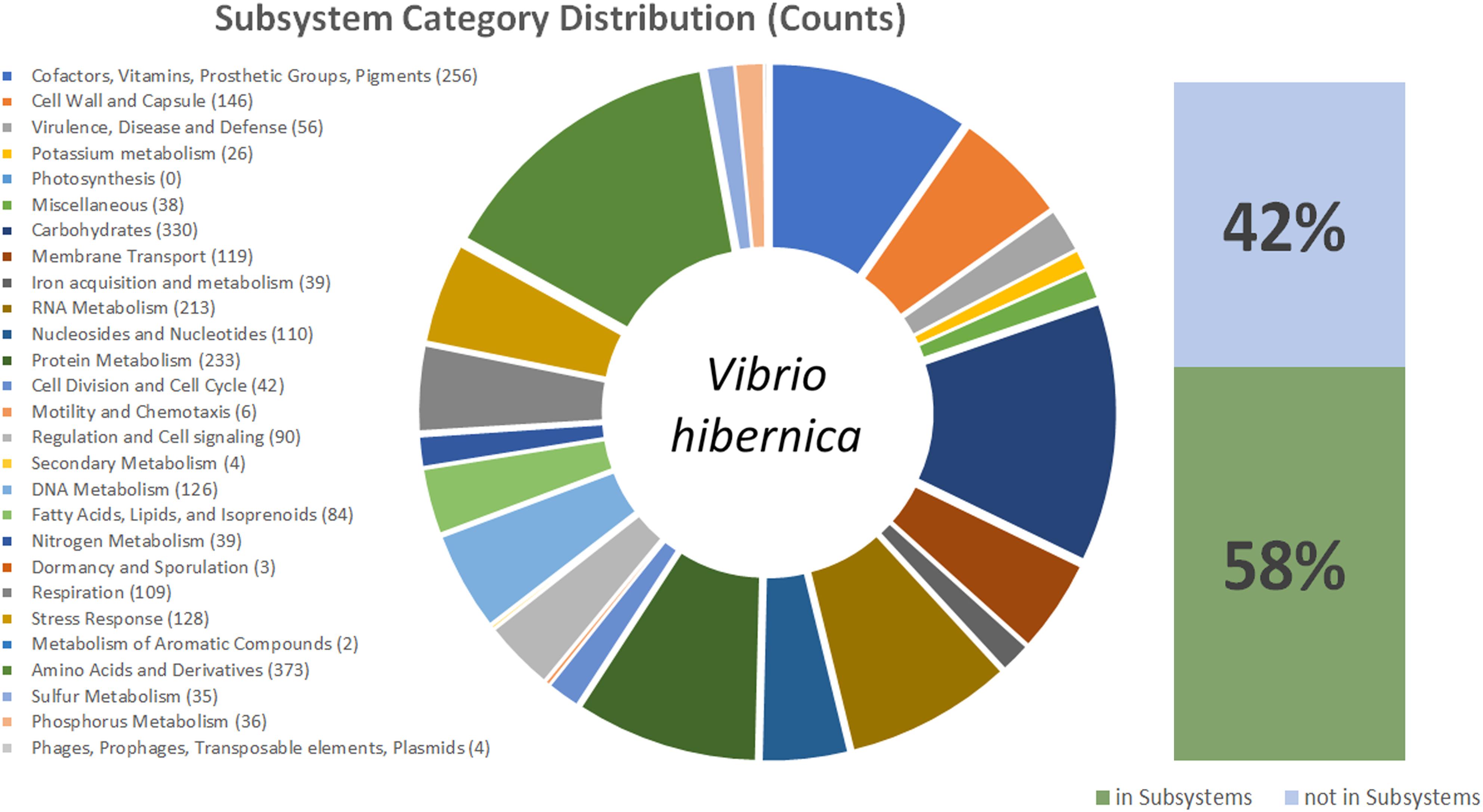
Figure 2. This figure shows a graphical representation of the V. hibernica genome. Annotation was completed using the ClassicRAST annotation scheme. The Subsystem Categories Distributions are listed and in brackets the number of genes assigned to each category. The pie chart illustrates the distribution of genes among the different categories in V. hibernica. Also included are the percentages of the annotated genes of V. hibernica found in Subsystems 42% and not in Subsystems 58%.
Bioinformatic Species Distinction of Vibrio hibernica
To further assess and define V. hibernica as a species, bioinformatic tools were employed. The ANI algorithm measures the nucleotide level genomic similarities between the coding regions of two genomes. When V. hibernica was compared to each of the species tested V. rumoiensis and V. litoralis were shown to be the closest relative based on sequence similarity at 75.77 and 75.61% respectively (Table 2). Using the in silico DNA–DNA Hybridization method to assay species distinction, V. hibernica differed from all of the tested genomes by greater than 1%. Again, the genomes of V. rumoiensis and V. litoralis were the closest with regards the GC% when compared to V. hibernica. V. parahaemolyticus differed from the Wiltshire brine isolate by the most (>4.05% GC difference) (Table 2).
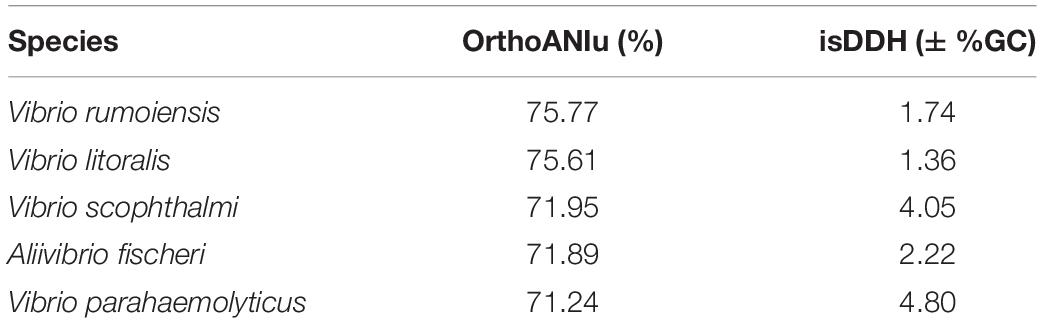
Table 2. Genome comparative analysis of Vibrio hibernica for species definition against closely related Vibrio species.
Phylogenetic Reconstruction of Species of the Genus Vibrio
The 16S rRNA phylogenetic tree provided information on the novelty and evolutionary branching of this Vibrio isolate. However, using the 16S rRNA gene as a phylogenetic indicator can be problematic and could lead to inaccuracies in generating phylogenetic trees. This can arise from such factors as multiple copies of the gene with variable sequences often being present in the genome (Thompson et al., 2009; Kembel et al., 2012; Louca et al., 2018). To combat this difficulty, a phylogenetic analysis of members of the Vibrio genus was reconstructed using eight HK genes (Figure 3). All of the nodes were supported well within the HK tree, with the exception of the discrimination between V. parahaemolyticus and V. azureus. The outgroups used to root the tree clustered together, and importantly clustered distinctly from all of the Vibrio strains present. V. hibernica isolated from Wiltshire brine was distinct from the other biological entities present. It grouped with the other Vibrio species present, not the outgroups, thus further supporting the bacterial assignment as a member of the genus of Vibrio. V. rumoiensis and V. litoralis grouped on a discrete clade with V. hibernica. Both V. rumoiensis and V. litoralis were previously defined as members of the Rumoiensis clade and we propose that our V. hibernica is a member of this clade (Sawabe et al., 2013; Sawabe et al., 2014).
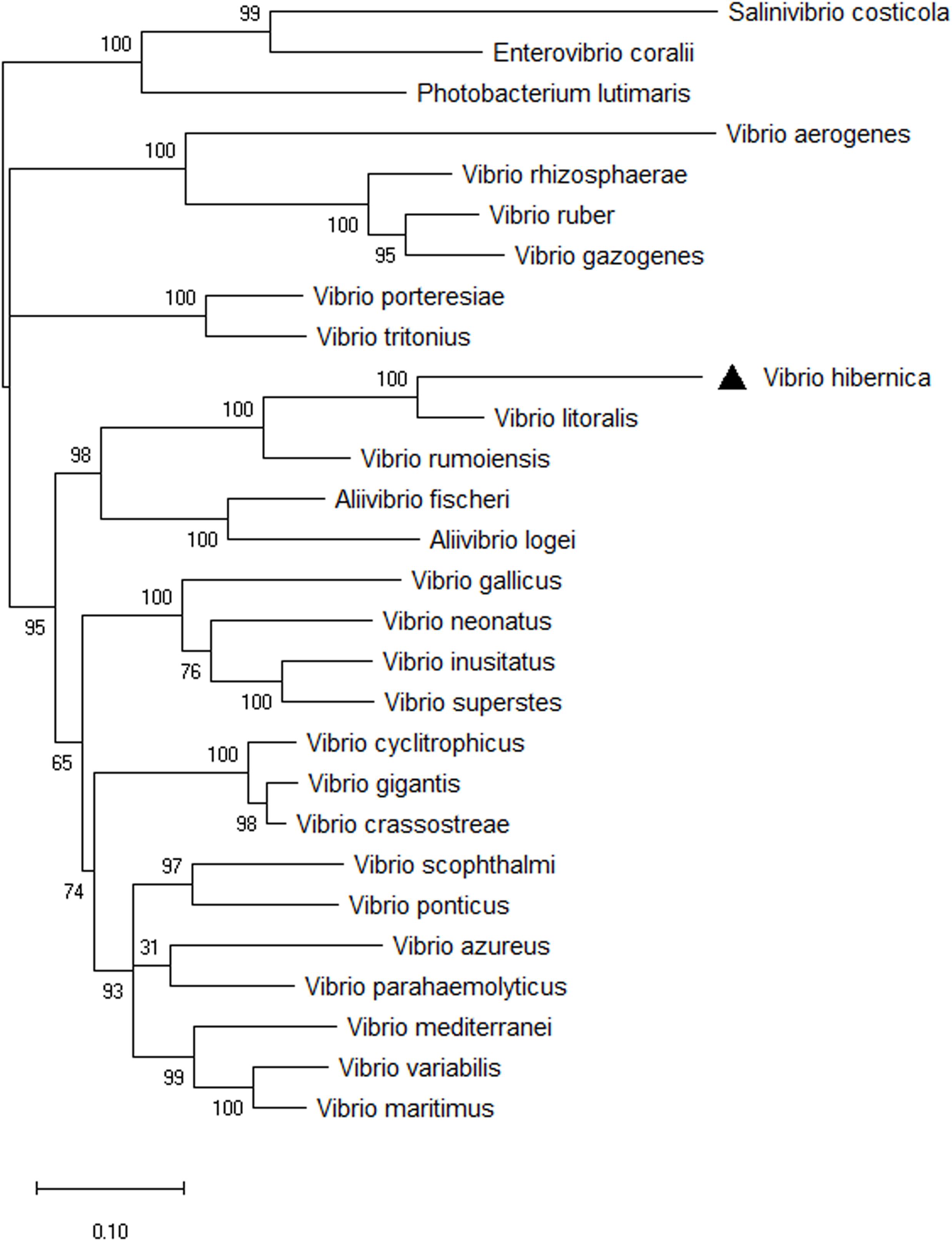
Figure 3. Phylogenetic tree of eight HK genes, ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA from selected Vibrio species. The evolutionary history was inferred using the Maximum Likelihood method based on the General Time Reversible model. A discrete Gamma (+G) distribution was used with rate variation model to allow evolutionarily invariable (+I). The phylogeny was tested using 1,000 Bootstrap Replicates. Three outgroups were used to root the three: Salinivibrio costicola, Enterovibrio coralii, and Photobacterium lutimaris. There was a total of 3,778 positions in the final dataset. V. hibernica is indicated by a black triangle.
Functional Genomics of V. hibernica to Identify Putative Organoleptic Related Properties
The functional genomic potential of V. hibernica was interrogated from its annotated genome to understand the possible role of the microbe in the Wiltshire curing process, as well as any future industrial applications of the strain. Within the genome 58% of the genes were successfully placed in a subsystem (Figure 2). There are 373 genes annotated to be associated with amino acids and their derivatives, this is the largest annotated group within the genome (Figure 2). Within this group there are 12 genes involved in the degradations of methionine. Cysteine, another volatile organic sulfur compound, is a precursor of methionine and there are 20 annotated genes for the biosynthesis of this compound and 7 genes annotated in the production of ribose. The enzymatic machinery for the production of another amino acid, serine, is present in the genome. There are 8 genes present for RNA processing and degradation. Methionine, cysteine, ribose, serine and RNA degradation are all important flavor contributors, especially with regards umami taste (Solms, 1969; Deoda and Singhal, 2003; Gonda et al., 2010). The genome has 15 annotated genes for the biosynthesis of threonine. This is an essential amino acid which cannot be synthesized by humans and must be obtained from our diet.
The subsystem of carbohydrates has the second highest number of annotated features with 330 counts within the genome (Figure 2), with a wide range of sugar metabolism genes, from simple sugars to more complex polysaccharides. The degradation of carbohydrates is one the most important contributors to a food process for improved flavor perception (Weenen and Apeldoorn, 1996). Within this subgroup, central carbohydrate metabolism accounts for most of the features (99), however there are also a number of genes for fermentation (39), monosaccharides (68), di- and oligo-saccharides (31) and aminosugars (13). There are 17 genes responsible for the biosynthesis of heme and siroheme from glycine and L-glutamate. Heme is known for having a pleasing red color and would be specifically beneficial to Wiltshire specific organoleptic traits (Chasco et al., 1996). Genes are present for the biosynthesis of the important osmolyte betaine. There were 56 features identified in the virulence, disease and defense category, these included genes related to resistance to toxic compounds (40) including copper, cobalt, zinc, cadmium, fluoroquinolones, arsenic and tetracycline, as well as efflux pumps. Of the remaining 16 features these included the production of an antibacterial peptide, specifically Colicin and genes involved in invasion and intracellular resistance. One of the most important attributes of the genome is that there are no genes annotated by RAST to be involved in toxin and superantigen production, as well as no genes identified to be present in the category of pathogenicity islands.
There is a large number of genes annotated to be involved in the production of vitamins which are of industrial and market value. Genes (16) are present for the biosynthesis of the essential cofactor vitamin B1 (Thiamin). There are 34 genes annotated to be involved in the biosynthesis of vitamin B7, commonly known as biotin. The complete gene mechanism needed to convert chorismate to menaquinone (vitamin K2), an important vitamin involved in the coagulation of blood, is also present. There are a number of genes annotated for the production of the important coenzyme, ubiquinone, again produced from the compound chorismate. For the biosynthesis of the coenzyme B12, 12 genes are present which are involved in the production of this important compound. There are 38 genes and 58 genes involved in the production of folate and pterines, respectively. As well as vitamins there are genes present that are involved in the production of the antioxidant glutathione. These compounds are all categories as value-add ingredients and when available in a food product can provide an enhanced potential for marketability of a product.
Phenotypic and Biochemical Profiles of Vibrio hibernica With Industrial Relevance
Additional metabolic related assays were performed to further characterize V. hibernica (Vh) as a species, as well as to give practical examples of industrially relevant processes. The strains phenotypic characteristics were compared and contrasted to closely related species. Both API 20NE and 50CH kits were used to test the practical metabolic capabilities of V. hibernica (Vh) and to highlight this isolates distinctiveness. Phenotypically all of the Vibrio species tested were oxidase positive and were able to reduce nitrates to nitrite, an important characteristic of a Wiltshire curing microbe (Table 3) (Andersen, 2004). All strains fermented glucose (GLU) with the exception of V. gallaecicus (Vg). However, only V. parahaemolyticus (Vp) and V. hibernica (Vh) were able to metabolize glucose in the presence of oxygen (GLU) and both were able to assimilate malate. V. gallaecicus (Vg), V. parahaemolyticus (Vp) and V. hibernica (Vh) were each able to hydrolase galactosides to monosaccharides. V. hibernica (Vh) was able to assimilate N-acetyl-glucosamine (NAG) and malate (MLT). The closest phylogenetically related species, V. litoralis (Vl), showed a completely different and limited profile compared to the brine isolate, V. hibernica (Vh).
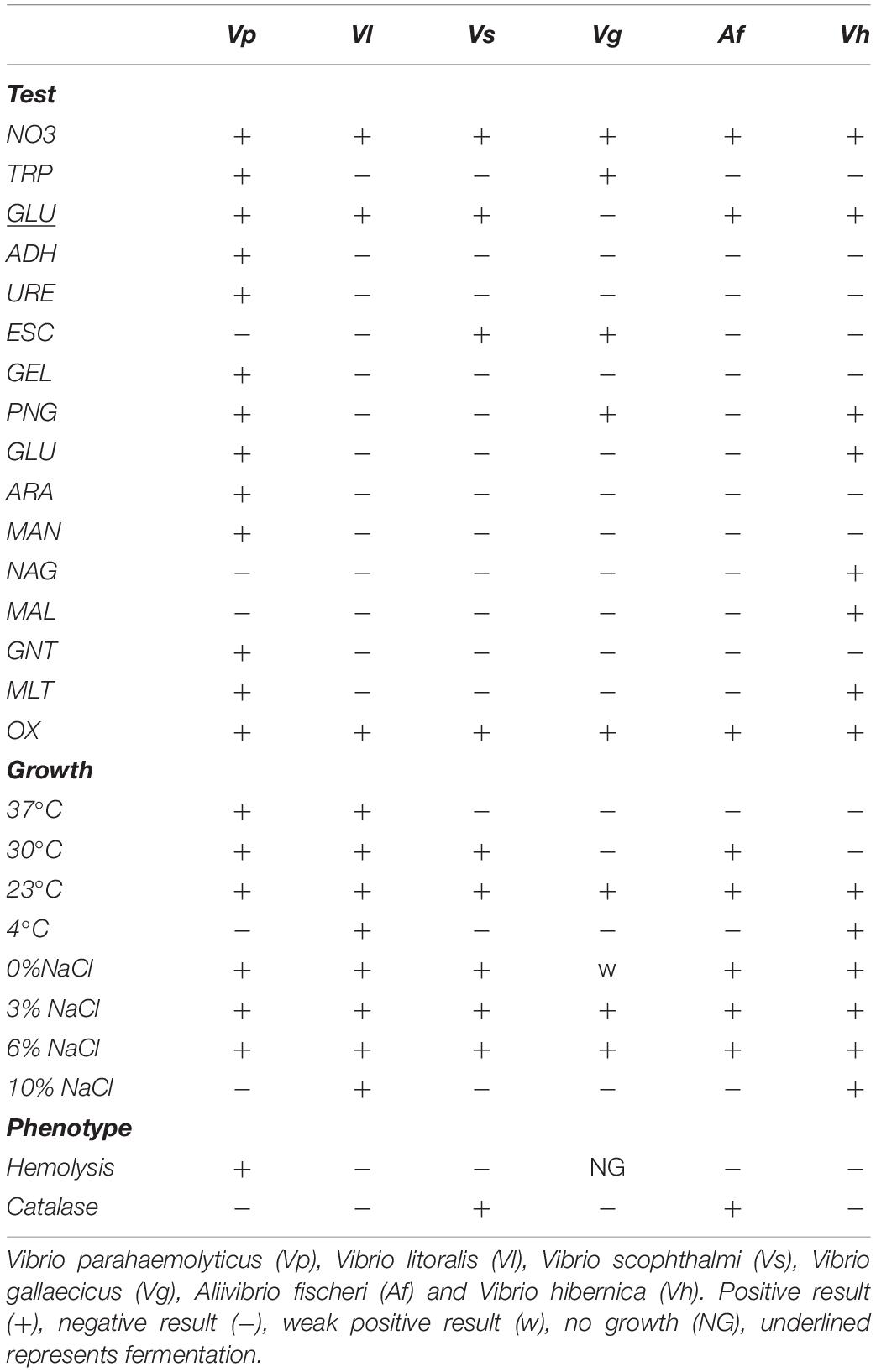
Table 3. Differential biochemical characteristics of examined species of Vibrio in this study.
The growth temperature range was different for each of the species, with V. litoralis (Vl) growing at all of the temperatures tested (Table 3). V. scophthalmi (Vs), V. gallaecicus (Vg), and A. fischeri (Af) showed a limited temperature growth range. V. hibernica (Vh) demonstrated a preference for the lower temperatures and grew at 4°C after 16 h. The V. hibernica (Vh) strain did not grow at the higher temperatures tested unlike the mesophilic pathogen V. parahaemolyticus (Vp). All of the Vibrio strains grew with the addition of 6% NaCl, both V. litoralis (Vl) and the V. hibernica (Vh) strains showed a higher degree of salt tolerance, growing in the presence of 10% added NaCl (Table 3). None of the strains tested were halotolerant to 20% added NaCl. V. hibernica (Vh) did not demonstrate the ability to lyse erythrocytes (β-hemolysis). V. scophthalmi (Vs) and A. fischeri (Af) were both catalase positive in contrast to the brine isolate as well as the other tested strains.
To test the Wiltshire brine isolate’s practical capabilities for carbohydrate metabolism, an API CH50 kit was utilized (Table 4). We assayed V. parahaemolyticus (Vp) and V. litoralis (Vl) for comparative purposes. The other strains used in previous tests could not grow in the media used. Numerous conditions were tested to optimize the assay for growth of these strains; however, they were not culturable under the conditions specified by the manufacturers. This kit confirmed that V. hibernica (Vh) was able to metabolize glucose, malate, maltose, galactose, fructose, sucrose and contained the enzyme β-galactosidase. Genes annotated for the metabolism of these carbohydrates were present in the genome. V. hibernica showed the ability to metabolize D-ribose as noted in the genome annotation and this is an important meat flavor compound. Glycerol was metabolized by V. hibernica at 23°C weakly, however it was strongly metabolized at the lower temperature of 4°C. Genes for other sugar alcohols (D-arabitol and mannitol) are also annotated within the genome and both were utilized by V. hibernica. Strikingly, the V. hibernica isolated from brine was the only Vibrio tested that maintained (or improved) its ability to metabolize flavor development and industrially relevant compounds at 4°C (Table 4). It is important to note that the genome annotation corresponded with the phenotypic profile of V. hibernica. This demonstrates the usefulness of coupling genomic sequencing and annotation with phenotypic characterization, specifically with regards organoleptic qualities.
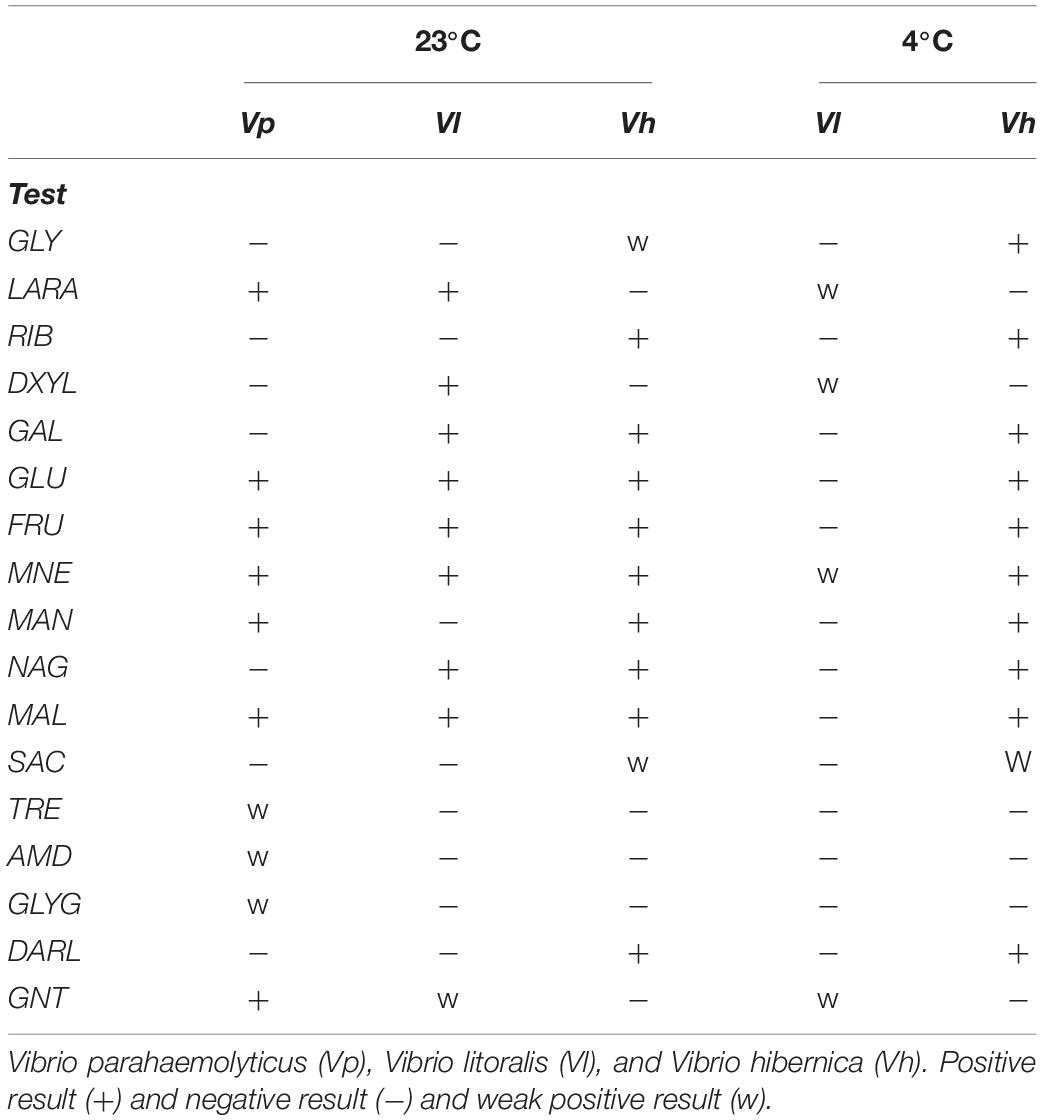
Table 4. Vibrio species carbohydrates metabolism patterns on an API 50 CH kit grown at 23°C and 4°C.
Discussion
The application of bioinformatic (Tax4Fun) and molecular (NGS) tools to food processing related microorganisms is vital for the modernization, standardization, improved safety and optimization of traditional food processes (He et al., 2017; Behera et al., 2018; Woods et al., 2019). One of the key aspects of the traditional food process of Wiltshire curing is the role of the microbial community (Gardner, 1971). There is a compelling need to investigate the function and mechanism of action of the microbial community within Wiltshire brine and particularly how the microbiota contributes to the organoleptic qualities of the final product. This traditional food process maintains its microflora by cycling the brine. Spent brine is replenished with fresh brine in a 50:50 ratio. This brine is then used for the curing (5–7 days) of meat until the brine is defined as spent by an expert technician. This is roughly after approximately 40 days of use, after which the cycle begins anew. To investigate this process, we sampled brine at an initial timepoint (Day 0) which was immediately after replenishing the Wiltshire brine with half fresh cure. The second timepoint (Day 20) was when the brine was in full use and by Day 40 it was toward the spent phase of the brine. Bioinformatic functional community prediction software was applied to the microbiome data sets of three curing containers and tested at the three timepoints. Similar functional profiles were seen within the Day 0 and Day 20 replicates, however, on Day 40 the functionality of microbial community showed greater variability. The food industry strives to maintain product consistency. A functionally stable microbiome is crucial for food processing standardization and this variability highlights the necessity to replenish the brine at Day 40.
Within the two defined clusters (I and II) by the functional prediction analysis, there is a switch between functions from Day 0 to Day 20. Functions such as valine, leucine, and isoleucine biosynthesis which are initially highly enriched, were selected against by Day 20. Functions such as valine, leucine, and isoleucine degradation are enriched for by Day 20. A similar pattern is seen with lysine degradation and biosynthesis. There were a number of systems identified as relating to the metabolism of amino acids. Metabolic functions such as amino acid degradation and synthesis are valuable for the meat curing as they contribute to flavor development (Kirimura et al., 1969; Nishimura and Kato, 1988; Montel et al., 1998). A number of functions have been identified that are enriched for by Day 20, many of which have important flavor enhancing roles. These include the metabolism of amino acids, such as valine, leucine, and isoleucine degradation, cysteine and methionine metabolism and alanine, aspartate and glutamate metabolism (Solms, 1969; Gonda et al., 2010; Liu et al., 2012). Generally complex sugars do not give a sensory perception in humans, therefore the metabolism of complex sugars is an important function facilitating a positive flavor sensory response (Sclafani, 1987; Beauchamp, 2016). The enrichment of genes for the degradation of polysaccharides is highest at Day 20 and this would produce more sensory perceptive simple sugars. Both sugars and amino acids need to be transported into the cell for metabolism and ABC transporters often facilitate this process. It is clear in the functional enrichment, that ABC transporter are also increased by Day 20 (Hosie and Poole, 2001; Schneider, 2001). The use of Tax4Fun not only gives an insight into the potential functional activity within a Wiltshire brine over time but can also be used to provide a functional standard fully optimized signature for an active Wiltshire cure. While Tax4Fun offers an insight into potential metabolic capabilities of a system, follow-on functional studies would be needed, such as expression studies and metabolomics, to fully understand the function of microbial communities in brine.
We have demonstrated that V. hibernica is an integral part of an active Wiltshire brine microbiome. This bacterium differs from other genera found in the core microbial community, as it is not a Lactic Acid Bacteria (LAB). However, the metabolic potential of its genome clearly indicates involvement in organoleptic quality development. The V. hibernica isolated from Wiltshire brine demonstrated a unique phenotypic profile when compared to the other tested Vibrio species. The species was named due to its country of origin, Ireland. The reconstruction of the phylogenetic history of the Vibrio species was far better supported in the HK tree compared to the 16S rRNA gene tree and this result is further strengthened by other studies with similar outcomes (Tanaka et al., 2017). Phylogenic analysis of this species was seen to be distinct from its closest relatives, V. rumoiensis and V. litoralis. Both of these species as well as V. algivorus, V. casei and V. aphrogenes belong to the Rumoiensis clade and we propose V. hibernica be included into this clade (Tanaka et al., 2017). Two of the in silico methods of species definition identified V. hibernica as a novel species. Average nucleotide identity (OrthoANIu) is an improved algorithm of ANI which uses USEARCH as a fast clustering method. ANI has a species boundary cut-off of 95% similarity (Richter and Rossello-Mora, 2009; Tindall et al., 2010; Kim et al., 2014; Yoon et al., 2017). The other method used in this study was in silico DNA-DNA Hybridization (DDH), where the same species cannot differ in the G + C content by more than 1% by the Genome-To-Genome Distance Calculator (GGDC) pairwise comparison (Meier-Kolthoff et al., 2014). When compared to the other Vibrio genomes, V. hibernica showed less than 95% ANI and greater than 1% G + C difference by in silico DDH, thus supporting this bacterium as a new species.
Flavor is one of the most important components of food products and gaining knowledge into the contributions of microbial genomics to flavor and other organoleptic properties is vital. This is especially the case for the advancement and understanding of traditional meat processes such as Wiltshire curing. The annotated V. hibernica genome has a number of genes that could aid in the development of the specific organoleptic properties of Wiltshire cured hams. Many genes in the genome of V. hibernica were annotated as being involved in the biosynthesis and degradation of amino acids and these have an important role in flavor development, specifically with the production of volatile flavor compounds (Nishimura and Kato, 1988). The enzymes involved in the degradation of the essential amino acid methionine are present in the genome, and this degradation pathway is involved in the production of important aroma compounds, particularly volatile sulfur compounds which are associated with desirable flavors in cured meats (Ferchichi et al., 1985; Weimer et al., 1999; Pripis-Nicolau et al., 2004). Other amino acids such as serine and cysteine have an important role in flavor. Serine has previously been reported as having a sweet and umami taste (Kirimura et al., 1969). Cysteine along with ribose can undergo the Maillard reaction, yielding a typical meat-like flavor (Mottram, 1998). The degradation of RNA is also a key component to flavor development and the genome has the genetic potential to do this. Nucleic acid degradation can produce 5′-guanosine monophosphate (5′-GMP) (Nagodawithana et al., 2013). This compound is an important flavor enhancer (Kuninaka, 1960; Ikeda, 2002; Ninomiya, 2015).
The breaking down of complex sugars into more simple sugars is a vital function in the curing/fermenting of Wiltshire cured hams, as there is a consumer preference for di- and mono-saccharide over more complex carbohydrates (Sclafani, 1987). The metabolism of lipids, fatty acid and isoprenoids into smaller compounds also leads to a value-added product with an increased preference for taste, accompanied by an increase in umami and kokumi tastes in a variety of products (Gänzle, 2009; Feng et al., 2014). Genes encoding the osmolyte betaine are also present in the genome. Osmolytes may confer the high salt tolerance of V. hibernica. Moreover, betaine is known to have an enhancing effect on taste receptors responses and improving intestinal function (Kiyohara et al., 1994; Wang et al., 2018). Additional benefits of increased dietary betaine levels have been linked to health benefits, specifically with decreasing the risk of developing several severe diseases (McGregor et al., 2002; Ross et al., 2011; Ying et al., 2013). It is important to note that V. hibernica was capable of growing and metabolizing sugars at 4°C, fermenting meat products and improving the organoleptic properties at this temperature is highly advantageous to the food industry as most detrimental bacteria are mesophilic organisms. Fermentation at lower temperatures though often comes with difficulties such as slow bacterial growth, extended lag phases, decreased industrial processing and increase financial costs (Uchimoto and Cruess, 1952; Charoenchai et al., 1998; Llauradó et al., 2002; Chiva et al., 2012). Thus, V. hibernica and its ability to metabolize sugars at low temperatures would be highly advantageous to a variety of industrial applications.
Another important characteristic of a Wiltshire cured product is the maintenance and depth of color of cured meat products, specifically with regards to the red and iridescent appearance of hams. The machinery for the production of heme is present in the genome of the V. hibernica isolate and this plays a vital role in color development (Chasco et al., 1996; Hugas and Monfort, 1997; Krause et al., 2011). Moreover, heme is currently being added to vegetarian food to produce meat flavored products (Fraser et al., 2018). An important parameter of Wiltshire curing brine is that nitrate reducing bacteria are promoted and maintained in the microbial milieu. The sequenced genome has 39 genes related to nitrate metabolism and the metabolism of nitrates also contributes to the development of the distinct red color of cured meats (Krause et al., 2011).
As well as evidence of genes enhancing organoleptic properties, a number of sequences were annotated as being involved in the production of vitamins and coenzymes. The potential increase in these during the curing process could yield a market for the cured meat product as a value added nutraceutical (Malik, 2007). There are a number of genes present for the biosynthesis of vitamins, such as B1, B2, B7, B9, B12 and K2, as well as antioxidants such as ubiquinone and glutathione. Vitamin B1 is an essential cofactor generally produced by plants, but never by vertebrates (Takusagawa et al., 1998; Rodionov et al., 2002). Vitamins B2 and B7 have important functions with regards to cellular processes (Powers, 2003; Heidker et al., 2016). Vitamin B9 is an essential vitamin necessary for human health and development (Ebara, 2017; Naderi and House, 2018). Vitamin K2 has positive effects on preventing osteoporosis and cardiovascular disease, as well as aiding in blood coagulation (Mahdinia et al., 2017). Studies have already been employed to produce vitamin K2 enriched products by traditional fermentation food processes (Mahanama et al., 2011; Singh et al., 2015). The vitamin B12 is an important cofactor for many enzymes (Cheng et al., 2016; Bridwell-Rabb and Drennan, 2017). Moreover this compound is known for its deep red color, which is a vital characteristic of Wiltshire cured products (Scott et al., 1964). Ubiquinone and glutathione are known for their antioxidant properties (Wang and Hekimi, 2016). Additionally glutathione is known to contribute to the kokumi flavor of a product (Tang et al., 2017). In a world where there is a trend for a decrease in excessive meat consumption, optimizing and increasing the nutritional and vitamin content of the meat is imperative, moreover curing meat has previously been shown to increase the nutritional value of the product (Okoń et al., 2017; Neff et al., 2018). Thus, the manipulation of a microbiome and the individual microbial components to use their full genetic potential would be beneficial to a number of industries. The absence of pathogenicity genes is a vital trait, if the bacteria is to be used in the food industry.
Conclusion
The curing of ham in brine is a traditional artisan process that has been widely utilized in food processing for many years. The curing of meat often has an associated natural microbial diversity that can enhance the quality of the final product. We have investigated an important traditional value-add food process, Wiltshire curing, and delved into the microbial milieu that are an important part of this process. Molecular techniques were coupled with NGS to confirm the persistence of the core microbiome previously identified (Woods et al., 2019). We completed a predicted functional analysis of a fully compliant brine and identified functions that are enriched during each stage in the life cycle of the curing process. A Vibrio species has previously been proven to positively affect the organoleptic qualities in Wiltshire cured hams (Petaja et al., 1972). A new Vibrio isolate was cultured from an active Wiltshire brine with distinct phenotypic characteristics. Moreover, the food associated microbe was phylogenetically and genetically distinct from other published Vibrio species. We have defined this species as Vibrio hibernica strain B1.19, due to its isolation on the island of Ireland. We examined the genome of V. hibernica and highlighted several processes and genes that may contribute to the distinct specific organoleptic qualities of the Wiltshire product. The contribution of the enzymatic action of bacteria in food technology requires further investigation to evaluate the exact process behind this complex science. Our study gives an insight into the value-add processing of meat by a bacterium in a Wiltshire compliant fully functional brine.
Data Availability Statement
The raw reads of the Microbiome dataset are publicly available in the Sequence Read Archive (SRA) of the NCBI Database under Accession numbers; SRR11073122, SRR11073123, SRR11073124, SRR11073125, SRR11073126, SRR11073127, SRR11073128, SRR11073129, SRR11073130, SRR11073131, SRR11073132, SRR11073133, SRR11073134, SRR11073135, SRR11073136, SRR11073137, SRR11073138, SRR11073139, SRR11073140, SRR11073141, SRR11073142, SRR11073143, SRR11073144, SRR11073145, SRR11073146, SRR11073147, and SRR11073148. The data of the genome sequence of V. hibernica that is studied here is deposited in DDBJ/ENA/GenBank under the accession VHKO00000000.
Author Contributions
DW, IK, and FO’G were involved in the conception and authorship of the work. DW and IK both contributed to the experimental work.
Funding
This research was supported in part by grants awarded to FO’G by; Enterprise Ireland (CF-2017-0757-P; IP-2015-0390), the European Commission (FP7-PEOPLE-2013-ITN, 607786; FP7-KBBE-2012-6, CP-TP-312184; FP7-KBBE-2012-6, 311975; OCEAN 2011-2, 287589; EU2020-634486-2015), Science Foundation Ireland (SSPC-3, 12/RC/2275; SSPC-2, 12/RC/2275; 13/TIDA/B2625; 12/TIDA/B2411; 12/TIDA/B2405; 14/TIDA/2438; 15/TIDA/2977; SFI09/RFP/BMT2350), the Department of Agriculture and Food (FIRM 11/F009/MabS; FIRM 13/F/516), the Irish Research Council for Science, Engineering and Technology (GOIPG/2014/647), the Health Research Board/Irish Thoracic Society (MRCG-2014-6, MRCG-2018-16), The Health Research Board (ILP-POR-2019-004), the Department of the Marine (BEAU/BIOD/01), Cystic Fibrosis Foundation, USA (OG1710).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge our colleague Bernie O’Connell for her helpful proofreading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00647/full#supplementary-material
FIGURE S1 | Microbiome characterization of an active Wiltshire brine, sampled from three monitored brine containers at three time point (Day 0, Day 20, and Day 40) in late 2016. The genera are represented as a relative percentage of the sample’s total microbiome. The dominant genera present are Marinilactibacillus, Carnobacterium, Leuconostoc, Vibrio, Photobacterium and genera present in <1% (Other).
FIGURE S2 | Phylogenetic tree of the 16S rRNA genes from selected Vibrio species. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter model. A discrete Gamma (+G) distribution was used with rate variation model to allow evolutionarily invariable (+I). The phylogeny was tested using 1,000 Bootstrap Replicates. Three outgroups were used to root the tree: Salinivibrio costicola, Enterovibrio coralii, and Photobacterium lutimaris. There were a total of 1212 positions in the final dataset. Vibrio hibernica is indicated by a black triangle.
TABLE S1 | Accession numbers for the strains used for the reconstruction of the 16S rRNA phylogenetic tree.
TABLE S2 | Accession numbers for the strains used for the reconstruction of the HK phylogenetic tree.
Footnotes
- ^ http://tax4fun.gobics.de/
- ^ www.eurofinsgenomics.eu
- ^ https://www.ezbiocloud.net/tools/ani
- ^ http://ggdc.dsmz.de/ggdc.php
- ^ http://www.ncbi.nlm.nih.gov/
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/s0022-2836(05)80360-80362
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/mmbr.59.1.143-169.1995
Andersen, H. J. (2004). “Bacon production | wiltshire sides A2 - jensen, werner klinth,” in Encyclopedia Of Meat Sciences, eds C. Devine and M. Dikeman (Oxford: Elsevier), 51–56. doi: 10.1016/b0-12-464970-x/00144-6
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Batt, C. A. (2016). “Technology in food microbiology,” in Reference Module in Food Science (Amsterdam: Elsevier). Available online at: http://www.sciencedirect.com/science/article/pii/B9780081005965034399
Beauchamp, G. K. (2016). Why do we like sweet taste: a bitter tale? Physiol. Behav. 164, 432–437. doi: 10.1016/j.physbeh.2016.05.007
Behera, S. S., Ray, R. C., and Zdolec, N. (2018). Lactobacillus plantarum with functional properties: an approach to increase safety and shelf-life of fermented foods. Biomed. Res. Intern. 2018:9361614. doi: 10.1155/2018/9361614
Bertuzzo, E., Casagrandi, R., Gatto, M., Rodriguez-Iturbe, I., and Rinaldo, A. (2010). On spatially explicit models of cholera epidemics. J. R. Soc. Interf. 7, 321–333. doi: 10.1098/rsif.2009.0204
Bokulich, N. A., Bamforth, C. W., and Mills, D. A. (2012a). Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS One 7:e35507. doi: 10.1371/journal.pone.0035507
Bokulich, N. A., Joseph, C. M. L., Allen, G., Benson, A. K., and Mills, D. A. (2012b). Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS One 7:e36357. doi: 10.1371/journal.pone.0036357
Bokulich, N. A., Lewis, Z. T., Boundy-Mills, K., and Mills, D. A. (2016). A new perspective on microbial landscapes within food production. Curr. Opin. Biotechnol. 37, 182–189. doi: 10.1016/j.copbio.2015.12.008
Bridwell-Rabb, J., and Drennan, C. L. (2017). Vitamin B12 in the spotlight again. Curr. Opin. Chem. Biol. 37, 63–70. doi: 10.1016/j.cbpa.2017.01.013
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Met. 7, 335–336. doi: 10.1038/nmeth.f.303
Charoenchai, C., Fleet, G. H., and Henschke, P. A. (1998). Effects of temperature, pH, and sugar concentration on the growth rates and cell biomass of wine yeasts. Am. J. Enol. Viticul. 49:283.
Chasco, J., Lizaso, G., and Beriain, M. J. (1996). Cured colour development during sausage processing. Meat Sci. 44, 203–211. doi: 10.1016/S0309-1740(96)00092-97
Cheng, Z., Yamamoto, H., and Bauer, C. E. (2016). Cobalamin’s (Vitamin B12) surprising function as a photoreceptor. Trends Biochem. Sci. 41, 647–650. doi: 10.1016/j.tibs.2016.05.002
Chiva, R., Lopez-Malo, M., Salvado, Z., Mas, A., and Guillamon, J. M. (2012). Analysis of low temperature-induced genes (LTIG) in wine yeast during alcoholic fermentation. FEMS Yeast Res. 12, 831–843. doi: 10.1111/j.1567-1364.2012.00834.x
Coffey, M. J., Nielsen, S., Wemheuer, B., Kaakoush, N. O., Garg, M., Needham, B., et al. (2019). Gut microbiota in children with cystic fibrosis: a taxonomic and functional dysbiosis. Sci. Rep. 9:18593. doi: 10.1038/s41598-019-55028-55027
Dapurkar, D., and Telang, M. (2017). A patent landscape on application of microorganisms in construction industry. World J. Microbiol. Biotechnol. 33:138. doi: 10.1007/s11274-017-2302-x
De Pasquale, I., Di Cagno, R., Buchin, S., De Angelis, M., and Gobbetti, M. (2019). Use of autochthonous mesophilic lactic acid bacteria as starter cultures for making pecorino crotonese cheese: effect on compositional, microbiological and biochemical attributes. Food Res. Int. 116, 1344–1356. doi: 10.1016/j.foodres.2018.10.024
Deoda, A. J., and Singhal, R. S. (2003). 5′-Phosphodiesterase (5′-PDE) from germinated barley for hydrolysis of RNA to produce flavour nucleotides. Bioresour. Technol. 88, 245–250. doi: 10.1016/S0960-8524(02)00267-265
Dhall, P., Kumar, R., and Kumar, A. (2012). Biodegradation of sewage wastewater using autochthonous bacteria. Sci. World J. 2012, 861903–861903. doi: 10.1100/2012/861903
Durán Quintana, M. C., García, G. P., and Garrido Fernández, A. (1999). Establishment of conditions for green table olive fermentation at low temperature. Intern. J. Food Microbiol. 51, 133–143. doi: 10.1016/S0168-1605(99)00123-123
Egerton, S., Culloty, S., Whooley, J., Stanton, C., and Ross, R. P. (2018). The gut microbiota of marine fish. Front. Microbiol. 9:873. doi: 10.3389/fmicb.2018.00873
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Feng, Y., Chen, Z., Liu, N., Zhao, H., Cui, C., and Zhao, M. (2014). Changes in fatty acid composition and lipid profile during koji fermentation and their relationships with soy sauce flavour. Food Chem. 158, 438–444. doi: 10.1016/j.foodchem.2014.02.147
Ferchichi, M., Hemme, D., Nardi, M., and Pamboukdjian, N. (1985). Production of methanethiol from methionine by Brevibacterium linens CNRZ 918. J. Gen. Microbiol. 131, 715–723. doi: 10.1099/00221287-131-4-715
Fleet, G. H. (2008). Wine yeasts for the future. FEMS Yeast Res. 8, 979–995. doi: 10.1111/j.1567-1364.2008.00427.x
Fleming, A. (2001). On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bull. World Health Organ. 79, 780–790.
Fontana, C., Bassi, D., Lopez, C., Pisacane, V., Otero, M. C., Puglisi, E., et al. (2016). Microbial ecology involved in the ripening of naturally fermented llama meat sausages. A focus on lactobacilli diversity. Int. J. Food Microbiol. 236, 17–25. doi: 10.1016/j.ijfoodmicro.2016.07.002
Franciosa, I., Alessandria, V., Dolci, P., Rantsiou, K., and Cocolin, L. (2018). Sausage fermentation and starter cultures in the era of molecular biology methods. Int. J. Food Microbiol. 279, 26–32. doi: 10.1016/j.ijfoodmicro.2018.04.038
Fraser, R., Brown, P. O., Karr, J., Holz-schietinger, C., and Cohn, E. (2018). Methods and Compositions for Affecting the Flavor and Aroma Profile of Consumables. United States patent application 20180199606. Redwood City, CA: Impossible Foods Inc. Available online at: http://www.freepatentsonline.com/y2015/0351435.html
Froelich, B., Bowen, J., Gonzalez, R., Snedeker, A., and Noble, R. (2013). Mechanistic and statistical models of total vibrio abundance in the neuse river estuary. Water Res. 47, 5783–5793. doi: 10.1016/j.watres.2013.06.050
Gänzle, M. G. (2009). From gene to function: metabolic traits of starter cultures for improved quality of cereal foods. Intern. J. Food Microbiol. 134, 29–36. doi: 10.1016/j.ijfoodmicro.2009.05.018
Gardner, G. A. (1971). Microbiological and chemical changes in lean Wiltshire bacon during aerobic storage. J. Appl. Bacteriol. 34, 645–654. doi: 10.1111/j.1365-2672.1971.tb02327.x
Gonda, I., Bar, E., Portnoy, V., Lev, S., Burger, J., Schaffer, A. A., et al. (2010). Branched-chain and aromatic amino acid catabolism into aroma volatiles in Cucumis melo L. fruit. J. Exp. Bot. 61, 1111–1123. doi: 10.1093/jxb/erp390
Gul, O., Mortas, M., Atalar, I., Dervisoglu, M., and Kahyaoglu, T. (2015). Manufacture and characterization of kefir made from cow and buffalo milk, using kefir grain and starter culture. J. Dairy Sci. 98, 1517–1525. doi: 10.3168/jds.2014-8755
He, G.-Q., Liu, T.-J., Sadiq, F. A., Gu, J.-S., and Zhang, G.-H. (2017). Insights into the microbial diversity and community dynamics of Chinese traditional fermented foods from using high-throughput sequencing approaches. J. Zhejiang Univer. Sci. B 18, 289–302. doi: 10.1631/jzus.B1600148
Heidker, R. M., Emerson, M. R., and LeVine, S. M. (2016). Intersections of pathways involving biotin and iron relative to therapeutic mechanisms for progressive multiple sclerosis. Discov. Med. 22, 381–387.
Heine, T., Grossmann, C., Hofmann, S., and Tischler, D. (2019). Indigoid dyes by group E monooxygenases: mechanism and biocatalysis. Biol. Chem. 400, 939–950. doi: 10.1515/hsz-2019-2109
Hinrichsen, L. L., Montel, M. C., and Talon, R. (1994). Proteolytic and lipolytic activities of Micrococcus roseus (65), Halomonas elongata (16) and Vibrio sp. (168) isolated from Danish bacon curing brines. Intern. J. Food Microbiol. 22, 115–126. doi: 10.1016/0168-1605(94)90136-90138
Hoffart, E., Grenz, S., Lange, J., Nitschel, R., Müller, F., Schwentner, A., et al. (2017). High substrate uptake rates empower Vibrio natriegens as production host for industrial biotechnology. Appl. Environ. Microbiol. 83:e01614-17.
Hosie, A. H. F., and Poole, P. S. (2001). Bacterial ABC transporters of amino acids. Res. Microbiol. 152, 259–270. doi: 10.1016/S0923-2508(01)01197-1194
Hugas, M., and Monfort, J. M. (1997). Bacterial starter cultures for meat fermentation. Food Chem. 59, 547–554. doi: 10.1016/S0308-8146(97)00005-8
Húngaro, H. M., Peña, W. E. L., Silva, N. B. M., Carvalho, R. V., Alvarenga, V. O., and Sant’Ana, A. S. (2014). “Food microbiology,” in Encyclopedia of Agriculture and Food Systems, ed. N. K. Van Alfen (Oxford: Academic Press), 213–231.
Jung, J. Y., Lee, S. H., Lee, H. J., Seo, H. Y., Park, W. S., and Jeon, C. O. (2012). Effects of Leuconostoc mesenteroides starter cultures on microbial communities and metabolites during kimchi fermentation. Int. J. Food Microbiol. 153, 378–387. doi: 10.1016/j.ijfoodmicro.2011.11.030
Kearney, N., Stack, H. M., Tobin, J. T., Chaurin, V., Fenelon, M. A., Fitzgerald, G. F., et al. (2011). Lactobacillus paracasei NFBC 338 producing recombinant beta-glucan positively influences the functional properties of yoghurt. Intern. Dairy J. 21, 561–567. doi: 10.1016/j.idairyj.2011.03.002
Kembel, S. W., Wu, M., Eisen, J. A., and Green, J. L. (2012). Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 8:e1002743. doi: 10.1371/journal.pcbi.1002743
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64(Pt 2), 346–351. doi: 10.1099/ijs.0.059774-59770
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/bf01731581
Kirimura, J., Shimizu, A., Kimizuka, A., Ninomiya, T., and Katsuya, N. (1969). Contribution of peptides and amino acids to the taste of foods. J. Agric. Food Chem. 17, 689–695. doi: 10.1021/jf60164a031
Kiyohara, S., Yonezawa, H., and Hidaka, I. (1994). “Enhancing effects of betaine on the taste receptor responses to amino acids in the puffer Fugu pardalis,” in Olfaction and Taste XI, eds K. Kurihara, N. Suzuki, and H. Ogawa (Berlin: Springer), 734–738. doi: 10.1007/978-4-431-68355-1_294
Krause, B. L., Sebranek, J. G., Rust, R. E., and Mendonca, A. (2011). Incubation of curing brines for the production of ready-to-eat, uncured, no-nitrite-or-nitrate-added, ground, cooked and sliced ham. Meat Sci. 89, 507–513. doi: 10.1016/j.meatsci.2011.05.018
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kuninaka, A. (1960). Studies on taste of ribonucleic acid derivatives. J. Agric. Chem. Soc. Jpn. 34, 489–492. doi: 10.1271/nogeikagaku1924.34.6_489
Liu, A., Li, X., Pu, B., Ao, X., Zhou, K., He, L., et al. (2017). Use of psychrotolerant lactic acid bacteria (Lactobacillus spp. and Leuconostoc spp.) Isolated from Chinese traditional paocai for the quality improvement of paocai products. J. Agric. Food Chem. 65, 2580–2587. doi: 10.1021/acs.jafc.7b00050
Liu, M., Prakash, C., Nauta, A., Siezen, R. J., and Francke, C. (2012). Computational analysis of cysteine and methionine metabolism and its regulation in dairy starter and related bacteria. J. Bacteriol. 194, 3522–3533. doi: 10.1128/JB.06816-6811
Liu, Y., Ahmed, S., Liu, B., Guo, Z., Huang, W., Wu, X., et al. (2014). Ethnobotany of dye plants in Dong communities of China. J. Ethnobiol. Ethnomed. 10:23. doi: 10.1186/1746-4269-10-23
Llauradó, J., Rozès, N., Bobet, R., Mas, A., and Constantí, M. (2002). Low temperature alcoholic fermentations in high sugar concentration grape musts. J. Food Sci. 67, 268–273. doi: 10.1111/j.1365-2621.2002.tb11396.x
Louca, S., Doebeli, M., and Parfrey, L. W. (2018). Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6:41. doi: 10.1186/s40168-018-0420-429
Lucena-Padros, H., Jimenez, E., Maldonado-Barragan, A., Rodriguez, J. M., and Ruiz-Barba, J. L. (2015). PCR-DGGE assessment of the bacterial diversity in Spanish-style green table-olive fermentations. Int. J. Food Microbiol. 205, 47–53. doi: 10.1016/j.ijfoodmicro.2015.03.033
Lücke, F.-K. (2000). Utilization of microbes to process and preserve meat. Meat Sci. 56, 105–115. doi: 10.1016/S0309-1740(00)00029-22
Mahanama, R., Berenjian, A., Valtchev, P., Talbot, A., Biffin, R., Regtop, H., et al. (2011). Enhanced production of menaquinone 7 via solid substrate fermentation from Bacillus subtilis. Intern. J. Food Eng. 7, 1–23.
Mahdinia, E., Demirci, A., and Berenjian, A. (2017). Production and application of menaquinone-7 (vitamin K2): a new perspective. World J. Microbiol. Biotechnol. 33:2. doi: 10.1007/s11274-016-2169-2162
Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda, T., Tagomori, K., et al. (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361, 743–749. doi: 10.1016/s0140-6736(03)12659-12651
Marchesi, J. R., Sato, T., Weightman, A. J., Martin, T. A., Fry, J. C., Hiom, S. J., et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 64, 795–799. doi: 10.1128/aem.64.2.795-799.1998
Mayo, B., Rachid, C. T. C. C., Alegría, A., Leite, A. M. O., Peixoto, R. S., and Delgado, S. (2014). Impact of next generation sequencing techniques in food microbiology. Curr. Genom. 15, 293–309. doi: 10.2174/1389202915666140616233211
McGregor, D. O., Dellow, W. J., Robson, R. A., Lever, M., George, P. M., and Chambers, S. T. (2002). Betaine supplementation decreases post-methionine hyperhomocysteinemia in chronic renal failure. Kidney Int. 61, 1040–1046. doi: 10.1046/j.1523-1755.2002.00199.x
Meier-Kolthoff, J. P., Klenk, H. P., and Goker, M. (2014). Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64(Pt 2), 352–356. doi: 10.1099/ijs.0.056994-56990
Montel, M. C., Masson, F., and Talon, R. (1998). Bacterial role in flavour development. Meat Sci. 49, S111–S123. doi: 10.1016/S0309-1740(98)90042-90040
Mottram, D. S. (1998). Flavour formation in meat and meat products: a review. Food Chem. 62, 415–424. doi: 10.1016/S0308-8146(98)00076-74
Naderi, N., and House, J. D. (2018). “Chapter five - recent developments in folate nutrition,” in Advances in Food and Nutrition Research, ed. N. A. M. Eskin (Cambridge, MA: Academic Press), 195–213. doi: 10.1016/bs.afnr.2017.12.006
Nagendra Prabhu, G., and Chandrasekaran, M. (1997). Impact of process parameters on l-glutaminase production by marine Vibrio costicola in solid state fermentation using polystyrene as an inert support. Process Biochem. 32, 285–289. doi: 10.1016/S0032-9592(96)00083-80
Nagodawithana, T., Reed, G., and Taylor, S. (2013). Enzymes in Food Processing. Amsterdam: Elsevier.
Neff, R. A., Edwards, D., Palmer, A., Ramsing, R., Righter, A., and Wolfson, J. (2018). Reducing meat consumption in the USA: a nationally representative survey of attitudes and behaviours. Public Health Nutr. 21, 1835–1844. doi: 10.1017/S1368980017004190
Nei, M., and Kumar, S. (2000). Molecular Evolution And Phylogenetics. Oxford: Oxford University Press.
Ninomiya, K. (2015). Science of umami taste: adaptation to gastronomic culture. Flavour 4:13. doi: 10.1186/2044-7248-4-13
Nishimura, T., and Kato, H. (1988). Taste of free amino acids and peptides. Food Rev. Intern. 4, 175–194. doi: 10.1080/87559128809540828
Okada, K., Iida, T., Kita-Tsukamoto, K., and Honda, T. (2005). Vibrios commonly possess two chromosomes. J. Bacteriol. 187, 752–757. doi: 10.1128/JB.187.2.752-757.2005
Okoń, A., Stadnik, J., and Dolatowski, Z. J. (2017). Effect of Lactobacillus acidophilus Bauer and Bifidobacterium animalis ssp. lactis BB12 on proteolytic changes in dry-cured loins. Food Sci. Biotechnol. 26, 633–641. doi: 10.1007/s10068-017-0076-74
Okonechnikov, K., Golosova, O., and Fursov, M. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Papadelli, M., Zoumpopoulou, G., Georgalaki, M., Anastasiou, R., Manolopoulou, E., Lytra, I., et al. (2015). Evaluation of two lactic acid bacteria starter cultures for the fermentation of natural black table olives (Olea europaea L cv Kalamon). Pol. J. Microbiol. 64, 265–271. doi: 10.5604/01.3001.0009.2121
Petaja, E., Laine, J. J., and Niinivaara, F. P. (1972). Einfluss der pokelbakterien auf die eigenschaften gepokelten fleisches. Fleischwirtschaft 3, 680–686.
Pogacic, T., Maillard, M. B., Leclerc, A., Herve, C., Chuat, V., Valence, F., et al. (2016). Lactobacillus and Leuconostoc volatilomes in cheese conditions. Appl. Microbiol. Biotechnol. 100, 2335–2346. doi: 10.1007/s00253-015-7227-7224
Powers, H. J. (2003). Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr. 77, 1352–1360. doi: 10.1093/ajcn/77.6.1352
Pripis-Nicolau, L., de Revel, G., Bertrand, A., and Lonvaud-Funel, A. (2004). Methionine catabolism and production of volatile sulphur compounds by OEnococcus oeni. J. Appl. Microbiol. 96, 1176–1184. doi: 10.1111/j.1365-2672.2004.02257.x
Reen, F. J., Almagro-Moreno, S., Ussery, D., and Boyd, E. F. (2006). The genomic code: inferring Vibrionaceae niche specialization. Nat. Rev. Microbiol. 4, 697–704. doi: 10.1038/nrmicro1476
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodionov, D. A., Vitreschak, A. G., Mironov, A. A., and Gelfand, M. S. (2002). Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 277, 48949–48959. doi: 10.1074/jbc.M208965200
Ross, A. B., Bruce, S. J., Blondel-Lubrano, A., Oguey-Araymon, S., Beaumont, M., Bourgeois, A., et al. (2011). A whole-grain cereal-rich diet increases plasma betaine, and tends to decrease total and LDL-cholesterol compared with a refined-grain diet in healthy subjects. Br. J. Nutr. 105, 1492–1502. doi: 10.1017/s0007114510005209
Sandine, W. E., and Elliker, P. R. (1970). Microbially induced flavors and fermented foods flavor in fermented dairy products. J. Agric. Food Chem. 18, 557–562. doi: 10.1021/jf60170a023
Saraoui, T., Cornet, J., Guillouet, E., Pilet, M. F., Chevalier, F., Joffraud, J. J., et al. (2017). Improving simultaneously the quality and safety of cooked and peeled shrimp using a cocktail of bioprotective lactic acid bacteria. Int. J. Food Microbiol. 241, 69–77. doi: 10.1016/j.ijfoodmicro.2016.09.024
Sawabe, T., Ogura, Y., Matsumura, Y., Feng, G., Amin, A. K., Mino, S., et al. (2014). Corrigendum: updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 5:583. doi: 10.3389/fmicb.2014.00583
Sawabe, T., Ogura, Y., Matsumura, Y., Feng, G., Amin, A. R., Mino, S., et al. (2013). Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 4:414. doi: 10.3389/fmicb.2013.00414
Schneider, E. (2001). ABC transporters catalyzing carbohydrate uptake. Res. Microbiol. 152, 303–310. doi: 10.1016/s0923-2508(01)01201-3
Sclafani, A. (1987). Carbohydrate taste, appetite, and obesity: an overview. Neurosci. Biobehav. Rev. 11, 131–153. doi: 10.1016/S0149-7634(87)80019-80012
Scott, W. M., Burgus, R. C., Hufham, J. B., and Pfiffner, J. J. (1964). Microbial degradation of corrinoids I. Vitamin B12. J. Bacteriol. 88, 581–585. doi: 10.1128/jb.88.3.581-585.1964
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Singh, R., Kumar, M., Mittal, A., and Mehta, P. K. (2016). Microbial enzymes: industrial progress in 21st century. 3 Biotech. 6:174. doi: 10.1007/s13205-016-0485-488
Singh, R., Puri, A., and Panda, B. P. (2015). Development of menaquinone-7 enriched nutraceutical: inside into medium engineering and process modeling. J. Food Sci. Technol. 52, 5212–5219. doi: 10.1007/s13197-014-1600-1607
Solms, J. (1969). [The taste of amino acids, peptides and proteins]. Int. Z. Vitaminforsch. 39, 320–322.
Soto Del Rio Mde, L., Andrighetto, C., Dalmasso, A., Lombardi, A., Civera, T., and Bottero, M. T. (2016). Isolation and characterisation of lactic acid bacteria from donkey milk. J. Dairy Res. 83, 383–386. doi: 10.1017/s0022029916000376
Stabnikov, V., Ivanov, V., and Chu, J. (2015). Construction biotechnology: a new area of biotechnological research and applications. World J. Microbiol. Biotechnol. 31, 1303–1314. doi: 10.1007/s11274-015-1881-1887
Sun, H., You, Z., Jia, L., and Wang, F. (2019). Autism spectrum disorder is associated with gut microbiota disorder in children. BMC Pediatr. 19:516. doi: 10.1186/s12887-019-1896-1896
Takusagawa, F., Fujioka, M., Spies, A., and Schowen, R. (1998). Comprehensive Biological Catalysis. Cambridge, MA: Academic Press.
Tanaka, M., Endo, S., Kotake, F., Al-saari, N., Amin, A. K. M. R., Feng, G., et al. (2017). Vibrio aphrogenes sp. nov., in the Rumoiensis clade isolated from a seaweed. PLoS One 12:e0180053. doi: 10.1371/journal.pone.0180053
Tang, K. X., Zhao, C. J., and Gänzle, M. G. (2017). Effect of glutathione on the taste and texture of Type I sourdough bread. J. Agric. Food Chem. 65, 4321–4328. doi: 10.1021/acs.jafc.7b00897
Thompson, C. C., Vicente, A. C. P., Souza, R. C., Vasconcelos, A. T. R., Vesth, T., Alves, N., et al. (2009). Genomic taxonomy of Vibrios. BMC Evol. Biol. 9:258. doi: 10.1186/1471-2148-9-258
Thompson, F. L., Iida, T., and Swings, J. (2004). Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 68, 403–431. doi: 10.1128/MMBR.68.3.403-431.2004
Tindall, B. J., Rossello-Mora, R., Busse, H. J., Ludwig, W., and Kampfer, P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60(Pt 1), 249–266. doi: 10.1099/ijs.0.016949-16940
Toffin, L., Zink, K., Kato, C., Pignet, P., Bidault, A., Bienvenu, N., et al. (2005). Marinilactibacillus piezotolerans sp. nov., a novel marine lactic acid bacterium isolated from deep sub-seafloor sediment of the Nankai Trough. Int. J. Syst. Evol. Microbiol. 55(Pt 1), 345–351. doi: 10.1099/ijs.0.63236-63230
Uchimoto, D. A. N., and Cruess, W. V. (1952). Effect of temperature on certain products of vinous fermentation. J. Food Sci. 17, 361–366. doi: 10.1111/j.1365-2621.1952.tb16774.x
Wang, F., Wu, J., Wang, Y., Jin, Y., Jiang, X., Qiu, Z., et al. (2019). Gut microbiota functional biomolecules with immune-lipid metabolism for a prognostic compound score in epstein-barr virus-associated gastric adenocarcinoma: a pilot study. Clin. Transl. Gastroenterol. 10:e00074. doi: 10.14309/ctg.0000000000000074
Wang, H., Li, S., Fang, S., Yang, X., and Feng, J. (2018). Betaine improves intestinal functions by enhancing digestive enzymes, ameliorating intestinal morphology, and enriching intestinal microbiota in high-salt stressed rats. Nutrients 10:907. doi: 10.3390/nu10070907
Wang, Y., and Hekimi, S. (2016). Understanding ubiquinone. Trends Cell Biol. 26, 367–378. doi: 10.1016/j.tcb.2015.12.007
Weenen, H., and Apeldoorn, W. (1996). “Carbohydrate cleavage in the maillard reaction,” in Flavour Science, eds A. J. Taylor and D. S. Mottram (Sawston, CA: Woodhead Publishing), 211–216. doi: 10.1533/9781845698232.4.211
Weimer, B., Seefeldt, K., and Dias, B. (1999). Sulfur metabolism in bacteria associated with cheese. Antonie Van Leeuwenhoek 76, 247–261. doi: 10.1023/A:1002050625344
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2016). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Woods, D. F., Kozak, I. M., Flynn, S., and O’Gara, F. (2019). The Microbiome of an Active Meat Curing Brine. Front. Microbiol. 9:3346. doi: 10.3389/fmicb.2018.03346
Ying, J., Rahbar, M. H., Hallman, D. M., Hernandez, L. M., Spitz, M. R., Forman, M. R., et al. (2013). Associations between dietary intake of choline and betaine and lung cancer risk. PLoS One 8:e54561. doi: 10.1371/journal.pone.0054561
Keywords: brine, Wiltshire, genome, Vibrio, next generation sequencing, phylogeny, bioinformatics, food
Citation: Woods DF, Kozak IM and O’Gara F (2020) Microbiome and Functional Analysis of a Traditional Food Process: Isolation of a Novel Species (Vibrio hibernica) With Industrial Potential. Front. Microbiol. 11:647. doi: 10.3389/fmicb.2020.00647
Received: 24 July 2019; Accepted: 20 March 2020;
Published: 09 April 2020.
Edited by:
Javier Carballo, University of Vigo, SpainReviewed by:
Lucilla Iacumin, University of Udine, ItalyAoife Boyd, National University of Ireland Galway, Ireland
Copyright © 2020 Woods, Kozak and O’Gara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fergal O’Gara, Zi5vZ2FyYUB1Y2MuaWU=