 Anming Xu1,2†
Anming Xu1,2† Di Wang1†
Di Wang1† Yichen Ding3Yaqian Zheng1,2Bo Wang1,2
Yichen Ding3Yaqian Zheng1,2Bo Wang1,2 Qing Wei1
Qing Wei1 Shiwei Wang1Liang Yang4
Shiwei Wang1Liang Yang4 Luyan Z. Ma1,2*
Luyan Z. Ma1,2*- 1State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Singapore Centre for Environmental Life Sciences Engineering, Nanyang Technological University, Jurong West, Singapore
- 4School of Medicine, Southern University of Science and Technology, Shenzhen, China
Pseudomonas aeruginosa is an environmental microorganism that can thrive in diverse ecological niches including plants, animals, water, soil, and crude oil. It also one of the microorganism widely used in tertiary recovery of crude oil and bioremediation. However, the genomic information regarding the mechanisms of survival and adapation of this bacterium in crude oil is still limited. In this study, three Pseudomonads strains (named as IMP66, IMP67, and IMP68) isolated from crude oil were taken for whole-genome sequencing by using a hybridized PacBio and Illumina approach. The phylogeny analysis showed that the three strains were all P. aeruginosa species and clustered in clade 1, the group with PAO1 as a representitive. Subsequent comparative genomic analysis revealed a high degree of individual genomic plasticity, with a probable alkane degradation genomic island, one type I-F CRISPR-Cas system and several prophages integrated into their genomes. Nine genes encoding alkane hydroxylases (AHs) homologs were found in each strain, which might enable these strains to degrade alkane in crude oil. P. aeruginosa can produce rhamnolipids (RLs) biosurfactant to emulsify oil, which enables their survival in crude oil enviroments. Our previous report showed that IMP67 and IMP68 were high RLs producers, while IMP66 produced little RLs. Genomic analysis suggested that their RLs yield was not likely due to differences at genetic level. We then further analyzed the quorum sensing (QS) signal molecules that regulate RLs synthesis. IMP67 and IMP68 produced more N-acyl-homoserine lactones (AHLs) signal molecules than that of PAO1 and IMP66, which could explain their high RLs yield. This study provides evidence for adaptation of P. aeruginosa in crude oil and proposes the potential application of IMP67 and IMP68 in microbial-enhanced oil recovery and bioremediation.
Introduction
Microorganisms can survive in many harsh environments, such as glaciers, hot springs, and crude oil. In order to survive in these harsh environments, bacteria usually evolve a series of corresponding functional proteins or metabolic systems through adaptive evolution including gene mutation and horizontal gene transfer (HGT) (Ollivier and Magot, 2005). Crude oil is a complex mixture of thousands of compounds that is difficult to be utilized and even toxic for most of bacteria (Xu et al., 2018). Interestingly, bacteria such as Pseudomonads can survive in such harsh environment. Alkanes are saturated hydrocarbons of different carbon-chain length and structure, which are the major components of crude oil. Although they are chemically inert, most of them can be efficiently degraded by several microorganisms (Rojo, 2009; Sun et al., 2018). It has been reported that more than 60 genera of aerobic bacteria and 5 genera of anaerobic bacteria are able to degrade n-alkanes (Prince, 2005).
Alkane hydroxylases (AHs), which catalyze the first step of alkane biodegradation, are the key enzymes in aerobic degradation of alkanes by bacteria. These enzymes hydroxylate alkanes to alcohols, which are further oxidized to fatty acids and catabolized via the bacterial β-oxidation pathway (Rojo, 2009). The constituent proteins of the AH system include alkane hydroxylases, one or two rubredoxin (rubA, rubA1, and rubA2) and rubredoxin reductase (rubB) (Geissdorfer et al., 1999). Two common AHs in bacteria are the alkane hydroxylase (AlkB)-related AHs and the cytochrome P450 family proteins (Nie et al., 2014a). Furthermore, flavin-binding LC-alkane hydroxylase (almA) and thermophilic soluble LC-alkane hydroxylase (ladA) have also been found to be involved in the hydroxylation of alkanes (Wentzel et al., 2007; Wang and Shao, 2012). A number of bacteria have been found to contain multiple AHs (van Beilen et al., 2006). For example, the co-existence of AlkB and CYP153 was found in Dietzia sp. DQ12-45-1b (Wang et al., 2011; Nie et al., 2014b), and multiple AHs were found in Amycolicicoccus subflavus DQS3-9A1T (Nie et al., 2013). Several Pseudomonas species have also been proved to have the ability to utilize n-alkane (Kok et al., 1989; Yuste et al., 2000; Zhang et al., 2011). Among them, P. putida GPo1 has been extensively studied, which can grow on intermediate chain length n-alkanes by virtue of the OCT plasmid-encoded alkane hydroxylase (Chakrabarty et al., 1973). The OCT plasmid carries the alk regulon and consists of two gene clusters: alkBFGHJKL and alkST (van Beilen et al., 1994, 2001). In recent years, many alkane-degrading P. aeruginosa strains have been isolated from crude oil, including P. aeruginosa N002, P. aeruginosa SJTD-1, and P. aeruginosa DQ8 (Zhang et al., 2011; Liu et al., 2012; Das et al., 2015). Genome sequencing has shown that many of these strains contain homologs of the GPo1 alkane hydroxylases genes, which are located on chromosome in contrast to the plasmid-encoded GPo1 (van Beilen et al., 2001).
Under specific environmental conditions, P. aeruginosa could produce rhamnolipids (RLs), which are a class of glycolipids biosurfactants and mainly composed of 3-(3-hydroxyalkanoyloxy) alkanoic acids (HAA), mono-rhamnolipids (MRLs) and di-rhamnolipids (DRLs) (Deziel et al., 2003; Soberon-Chavez et al., 2005). In crude oil environment, RLs emulsify crude oil; which is always considered to have prominent ability to solubilize various hydrocarbons for the biodegradation (Beal and Betts, 2000; Noordman and Janssen, 2002). In P. aeruginosa, the biosynthesis of RLs is positively regulated by RhlI/RhlR quorum sensing (QS) system that directly activates the transcription of rhlAB, which encodes the enzymes catalyzing the first step of RLs biosynthesis. QS is a cell density-dependent communication mechanism which enables bacteria to coordinate cooperative behaviors in response to the accumulation of self-produced autoinducer signals (N-acyl-homoserine lactones, AHLs) in their local environment (Passador et al., 1993; Davies et al., 1998; Daniels et al., 2004). In P. aeruginosa, QS depends on two AHLs regulatory circuits, Las and Rhl. In the Las system, LasI directs the synthesis of N-(3-oxododecanoyl)-homoserine lactone (3-oxo-C12-HSL), which interacts with the transcriptional regulator LasR to control target genes, such as rhlI/rhlR. RhlI is also a signal synthase which generates another signal molecular, N-butanoyl-homoserine lactone (C4-HSL). Subsequently, C4-HSL combines the signal receptor RhlR to form a RhlR/C4-HSL complex that activates the transcription of several genes, including the rhlAB operon (Pearson et al., 1997).
In our previous work, we reported the identification of three rhamnolipid-producing Pseudomonas isolates from crude oil (named as IMP66, IMP67, and IMP68), which exhibited sigificant different phenotypes in RLs and pyocyanin production (Das et al., 2014). The 16s rRNA analysis has suggested that two of them (IMP67 and IMP68) are P. aeruginosa and IMP66 is identified as Pseudomonas sp. Pyr41 (Das et al., 2014). In this study, we performed the comparative genomic analysis for these three Pseudomonas strains to further understand their genetic background. Our analysis indicated that all three strains belong to P. aeruginosa species. Subsequent analysis showed that these three isolates exhibited a high degree of individual genome plasticity along with abundant numbers of alkane hydroxylase encoding genes, conferring survival advantages for P. aeruginosa strains living in the crude oil environment. Moreover, the strains with high yield of RLs produce large amounts of QS signal molecules. This study illustrates the environmental adaptability of P. aeruginosa, and proposes potential applications of these strains in bioremediation as well as in microbial-enhanced oil recovery.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
IMP66, IMP67, and IMP68 were isolated from the crude oil of Karamay W#8805, Xin-Jiang province, China. Unless otherwise indicated, P. aeruginosa strains were grown in LB without sodium chloride (LBNS) or basal salt medium (BSM, K2HPO4 3.815 g, KH2PO4 0.5 g, (NH4)2HPO4 0.825g, KNO3 1.2625 g, Na2SO4 0.2 g, CaCl2 0.02 g, FeCl3 0.002 g, MgCl2 0.02 g/l) at 37°C. Culture was supplemented with 300 μg/ml carbenicillin to select and maintain plasmid-carrying P. aeruginosa. A plasmid-borne fusion of the QS-controlled rhlAB promoter sequence (423 bp) and GFP coding sequence was used to assess the promoter activity of rhlAB in this study. Primers PrhlAB_F (5′-cgccagagcgtttcgac-3′) and PrhlAB_R (5′-ttcacacctcccaaaaattttcgaac-3′) were used to generate an amplicon of the rhlAB promoter by I-5TM High-Fidelity DNA Polymerase (Tsingke Biological Technology). The PCR product was purified and ligated into pPROBE-AT vector (Miller et al., 2000) through Gibson assembly (Gibson et al., 2010). Recombinant plasmid pTH22 (rhlAB:gfp) was then transformed into Escherichia coli DH5α and subsequently into P. aeruginosa for determination of the transcription of rhlAB.
Genome Assembly and Annotation
Genomic DNA was isolated from an overnight broth culture using the Wizard genomic DNA purification kit (Promega, Madison, WI, United States). A spectrophotometer (NanoDrop 2000 UV-Vis; Thermo Scientific, MA, United States) was used to determine the DNA quantity and ensure the DNA quality was suitable for sequencing. Whole-genome sequencing was performed using the long-read PacBio SMRT sequencing (Pacific Bioscience) platform and the Illumina HiSeq sequencer. Genome sequences were de novo assembled using HGAP assembly protocol, which is available with the SMRT Analysis packages and accessed through the SMRT Analysis Portal version 2.1. The pilon v1.5 was then used to correct the assembly (Walker et al., 2014). Annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAAP)1 (Tatusova et al., 2016). The circular genome map of the three strains including all predicted ORFs with COG functional assignments, rRNA, tRNA, G + C content, and GC skew information were generated using CGview (Stothard and Wishart, 2004).
Phylogenetic Analysis
The evolutionary position of the three strains relative to other pathovars and species of Pseudomonas was determined by multi-locus sequence analysis (MLSA) as described previously (Loper et al., 2012). For phylogenetic tree construction, ten conserved housekeeping genes (acsA-aroE-dnaE-guaA-gyrB-mutL-ppsA-pyrC-recA-rpoB) from 51 closely related Pseudomonas spp. were retrieved from Pseudomonas Genome Database (Winsor et al., 2016). Maximum Likelihood analysis was performed for 100 bootstrap replications in RAxML. The final phylogenetic tree was constructed using the iTOLs software (Letunic and Bork, 2016).
In order to detect the strain-level genome content of the three strains, a rapid large-scale prokaryote pan genome analysis by Roary was performed to identify the conserved (core) and non-conserved (accessory) genes (Page et al., 2015), and then created a concatenated core genome for subsequent phylogenetic analysis. 90 closely related Pseudomonas spp. retrieved from Pseudomonas Genome Database (Winsor et al., 2016) were used for the core-genome estimating by Roary bacterial genome analysis pipeline. A cut-off with 90% identity was used and the core genes were defined as those presented in 99% of isolates to generate a multi-FASTA alignment file. These data were then used as the input to the next iteration. RAxML was then run over the final multi-FASTA alignment to provide a high-quality phylogenetic tree in newick format. We used a generalized time reversible (GTR) model with gamma (Y) heterogeneity across nucleotide sites and 100 bootstrap replicates. The final phylogenetic tree was constructed using the iTOLs software (Letunic and Bork, 2016).
Comparative Genome Analysis
The MicroScope platform (Vallenet et al., 2016) provided some of the tools used for the comparative genomic analyses, such as the RGP Finder for determination of regions of genomic plasticity (RGPs). RGPs were defined as DNA segments over 5 kbp possibly related to events of horizontal exchanges, and they were identified using a series of constraints such as G + C% deviation, compositional biases as measured by the tools Alien-Hunter (Vernikos and Parkhill, 2006) and SIGI-HMM (Waack et al., 2006), synteny breaks and proximity to tRNAs. Pan-genome analysis including the genes shared by all these four strains was performed by an “all-against-all” BLAST of the protein sequences of the above genomes using the EDGAR 2.3 software framework (Blom et al., 2016). The genes aligned based on amino acid sequences were used for identifying orthologs. For orthology estimation, EDGAR used a generic orthology threshold calculated from the similarity statistics of the compared genomes, and then generated the veen diagram.
AHLs Extraction and Quantification
Extraction and quantification of AHLs were based on a previously described method (Ortori et al., 2011). Briefly, strains were grown in LBNS media for 24 h, 1 ml of bacterial cells were spun down and supernatants were harvested for AHLs extraction. Acidified ethyl acetate was used for AHLs extraction and the extraction was repeated three times. The organic layers were combined and evaporated under vacuumrotary evaporation. Residues were dissolved in 50 μl methanol and subjected to LC-MS/MS analysis using mass spectrometer (QTRAP 6500 System, AB SCIEX). The calibration curve was prepared from a linear dynamic range (from 0.33 to 33.3 μM) of standard 3-oxo-C12-HSL (Sigma-Aldrich, Germany).
Results
Phylogenetic Analysis of Newly Sequenced Strains With the Other Pseudomonas spp.
IMP66, IMP67, and IMP68 were isolated from the crude oil of Karamay W#8805, Xin-Jiang province, China. The complete genomes of these three strains were sequenced and assembled. A previous report suggested that only IMP67, and IMP68 were P. aeruginosa based on 16S rRNA analysis. It has been showed that the phylogenetic tree based on 16S rRNA lacked resolution at intrageneric level (Anzai et al., 2000). We then used the ten house-keeping genes to further analysis the phylogenetic position of these three strains. A phylogenetic tree was plotted using a maximum likelihood (ML) approach based on multi-locus sequence analysis of 54 representative Pseudomonas spp. strains, including 51 stains with complete genome sequence and these three newly sequenced strains (IMP66, IMP67, and IMP68). E. coli K12 was used as an out-group. Four tightly monophyletic groups could be identified from the phylogenetic tree based on the 10 concatenated housekeeping genes (Figure 1, SC1 to SC3, and SC5). Strains IMP66, IMP67, and IMP68 were all clustered together within the group of P. aeruginosa and were all identified as P. aeruginosa strains (Figure 1).
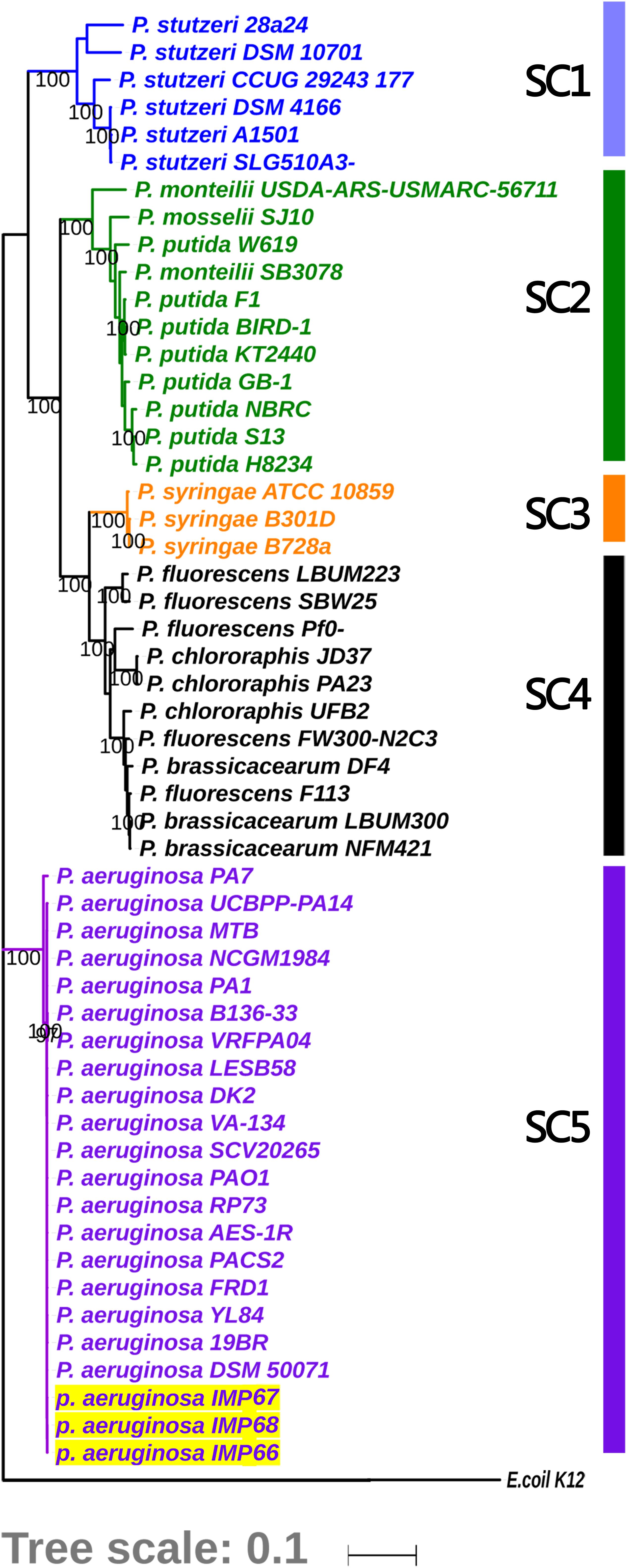
Figure 1. Phylogenetic analysis for depicting the relationships of these three newly sequenced strains with Pseudomonas spp. The tree is based on concatenated alignments of ten core housekeeping genes: acsA, aroE, dnaE, guaA, gyrB, mutL, ppsA, pyrC, recA, and rpoB, and was generated using the RAxML package. Five sequence clusters (SCs) are identified and labeled on the edge of the phylogeny. Clades corresponding to 4 dominant different monophyletic SCs (SC1 to SC3 and SC5) are shown in different colors, while clade for all the strains in the polyphyletic clade (SC4) comprising of the “unclustered” sequences is colored in black. Bootstrap supporting over 90% are labeled for the major nodes. Strains sequenced in this study are shown by yellow shading, which fall into SC5 with P. aeruginosa. The Escherichia coli K-12 genome was used as an out-group control.
In order to investigate the evolutionary history of these three newly sequenced strains, we reconstructed the phylogeny of the genus using concatenated core genomes from 90 P. aeruginosa isolates, including representative P. aeruginosa strains with completely genomes (such as PAO1, PA14, and PA7), IMP66, IMAP67, and IMP68, as well as several crude oil-isolated P. aeruginosa strains (Pb18, M8A1, M8A4, M28A1, SJTD-1, DQ8). There were 51,389 SNPs identified in the 820 core genes of the 90 isolates. P. aeruginosa strains were divided into three major clades (Figure 2A), which was consistent with the previous reports (Freschi et al., 2015; Castañeda-Montes et al., 2018). Interestingly, all crude oil isolates were consistently distributed in clade 1, which was represented by strain PAO1 and contained the strains from a wide range of sources, including natural environment and clinical settings (Figure 2A). Although most of the strains from crude oil were randomly distributed within clade 1, strain IMP66, IMP67, and IMP68 were clustered together (Figure 2B), which was consistent with the results of housekeeping gene analysis (Figure 1). Taken together, our data suggested that these three P. aeruginosa strains were closely related in the evolutionary process.
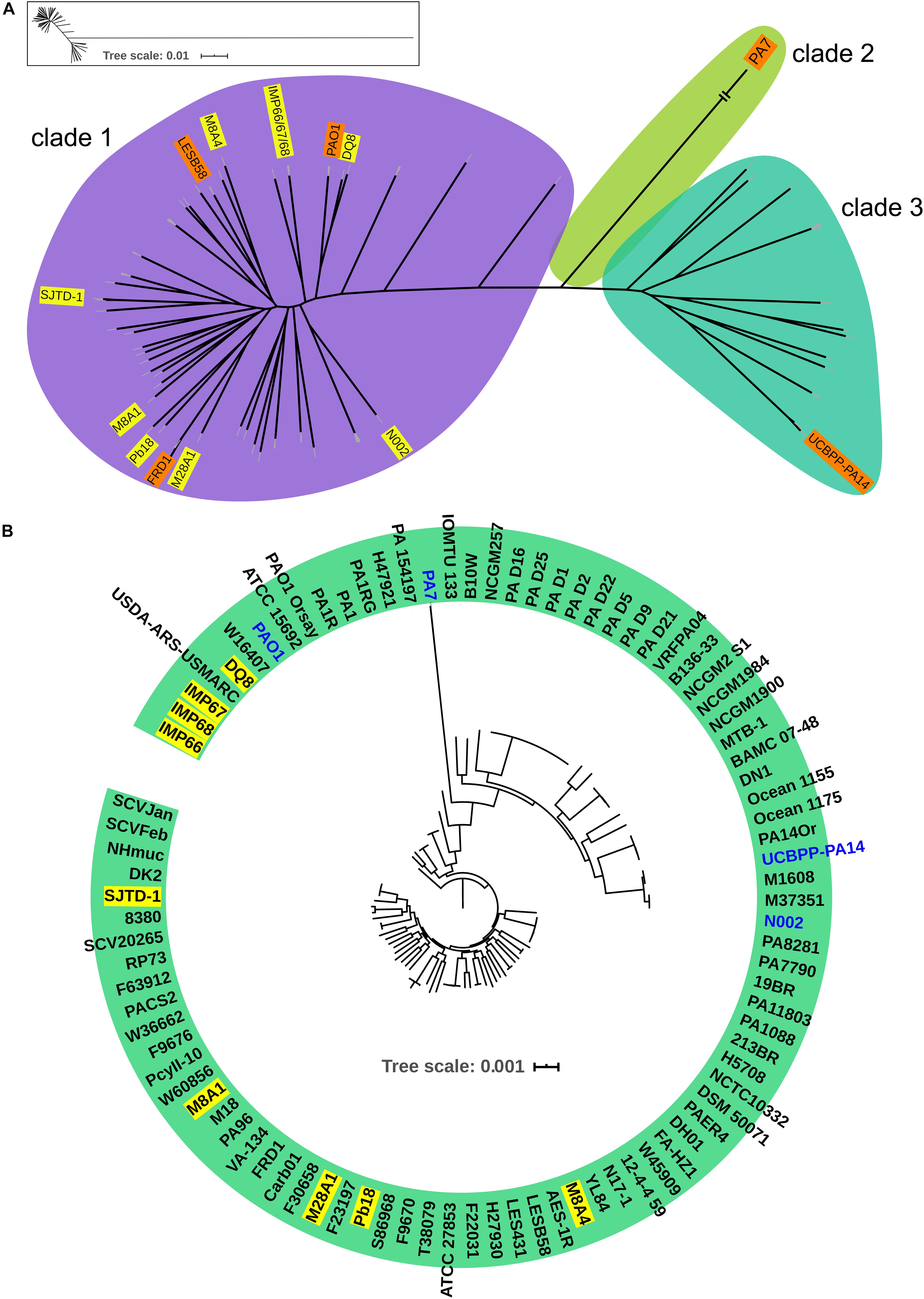
Figure 2. Phylogeny of P. aeruginosa population structure. Unrooted (A) and circular (B) maximum likelihood trees of three newly sequenced crude oil isolates combined with 87 representive P. aeruginosa isolates from diverse niches are generated from multiple-sequence alignment with 51,389 SNPs from 820 universally conserved (core) genes. The phylogenetic trees are constructed using RAxML with GTR + γ nucleotide substitution model and 100 bootstrap replicates. Strains are divided into three major clades. Clade 2 is contracted for visualization purpose. A framed miniature of the true appearance of this tree is presented. Representive crude oil sourced P. aeruginosa strains are highlighted in yellow.
Comparative Genomic Analysis of IMP66, IMP67, IMP68, and PAO1
IMP66, IMP67, and IMP68 were similar in genome size, with a single chromosome of about 6.5 Mb (Table 1 and Figure 3A). The three strains had the same G + C% content of 66.4% (Table 1), which was consistent with other P. aeruginosa strains. The total numbers of coding sequences (CDSs) in the three strains were close to 6100, which were much higher than that of PAO1. We also performed a general comparison of the genomic features including genome size, GC content, and predicted number of CDSs among several crude oil sourced strains and well-studied strain PAO1 (Table 1). The results showed that most of the crude oil isolates harbored bigger genome size than PAO1 strain except SJTD-1. However, SJTD-1 possessed about 200 more CDSs than PAO1 even with similar genome size. These results suggested that genome expansions might confer survival advantages for these isolates living in crude oil.
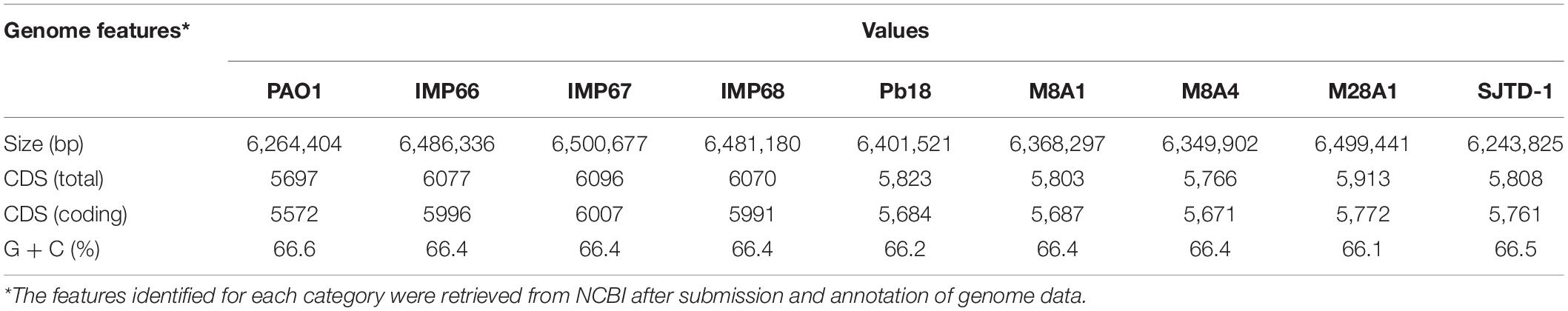
Table 1. General genome properties comparison of the completely sequenced crude oil isolates with laboratory strains.
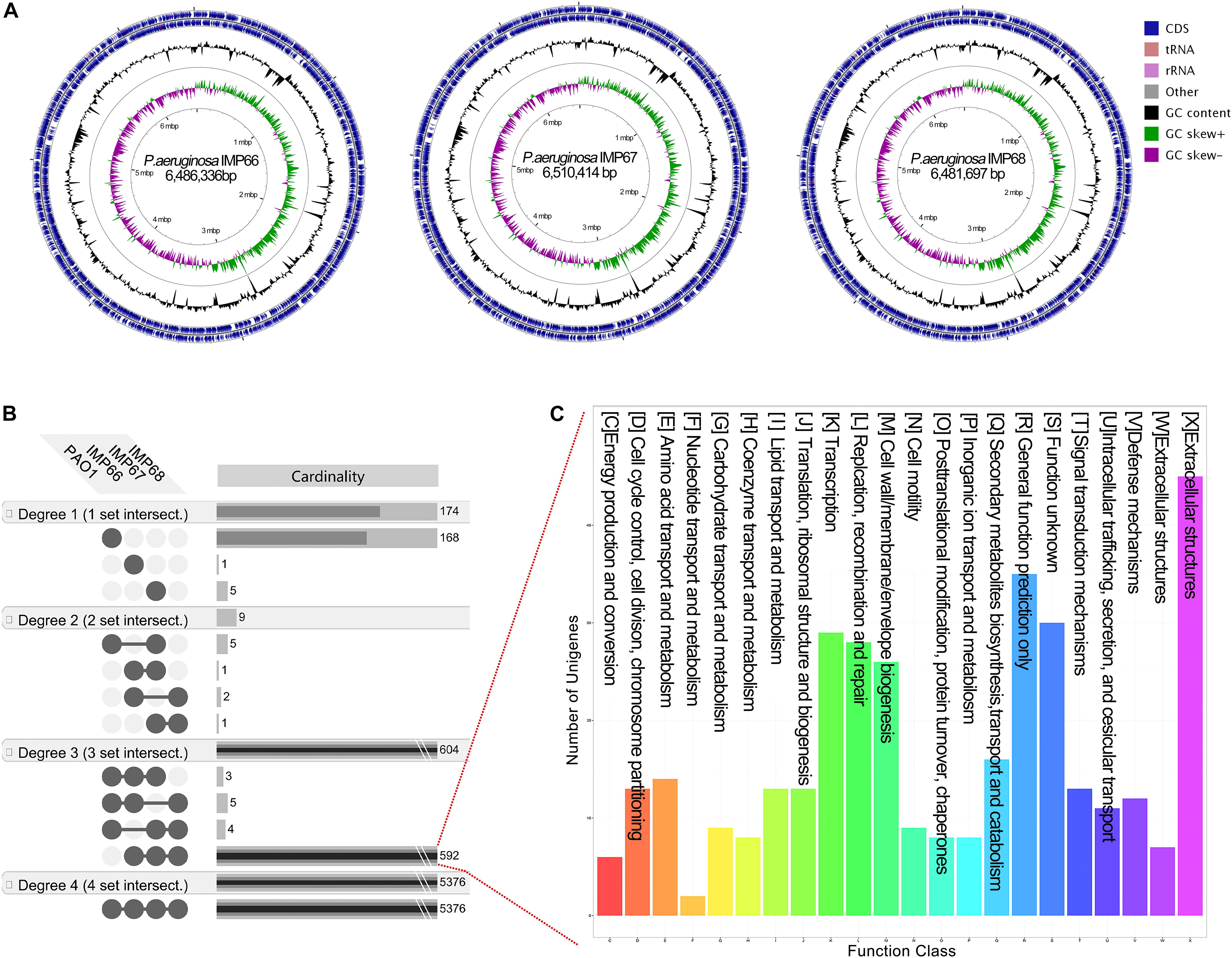
Figure 3. (A) Circular chromosome map of P. aeruginosa IMP66, IMP67, and IMP68. Pan-genome analysis of three crude-oil sourced P. aeruginosa isolates and PAO1. (B) The unique and shared genes among P. aeruginosa IMP66, IMP67, IMP68 and PAO1 are performed using the EDGAR software platform based on the orthology analysis. The results are visualized in four degrees. Degree one represents the distinct singletons harbored by each single strain; degree two represents the genes shared by every two strains; degree three represents the genes shared by every three strains; degree four is the number of core genes shared by all the four strains. (C) COG annotation of all 592 exclusive CDSs shared by these three P. aeruginosa isolates.
We then analyzed the group of shared and unique genes between IMP66, IMP67, IMP68, and PAO1. All the shared and unique genes were determined and visualized by EDGAR platform. The four bacteria shared a conserved core genome comprising of 5,376 CDSs. PAO1 possessed 168 distinct singletons, which was the largest number of singletons among these four strains. Interestingly, there were 592 unique genes shared by IMP66, IMP67, and IMP68 (Figure 3B). The largest portion (46 genes) of the 592 unique genes belonged to mobilomes (prophages and transposons, category X) according to the COG annotation. Other major unique genes were in category K (transcription), category L (replication, recombination, and repair), and category M (cell wall/membrane/envelope biogenesis), respectively. A small portion of genes were related to energy production and conversion, amino acid transport, and metabolism, etc (Figure 3C). The mobilome genes are commonly associated with HGT as they often carry transposons, phage fragments, and IS elements. These results suggested that HGT might commonly occur in these three strains. To further investigate HGT events in these three strains, the MicroScope platform was used to compare the genomes of IMP66, IMP67, IMP68 with PAO1. We selected IMP66 genome as the representative reference (the same results were obtained when using IMP67 or IMP68) of these three strains against the query genome of strain PAO1. A total of 23 conserved RGPs were identified in IMP66 (Figure 4A and Table 2). Most of these regions contained the sequences related to transposable elements and were flanked by genes coding for tRNAs or phage-structures (Table 2), indicating the possible horizontal gene acquisition. Although most of these regions encoded proteins of unknown functions, some RGPs with important genomic information were identified. RGP9 carried an insertion sequence at its 5′-end (IS3 family, DBX28_13665) and harbored CDSs with predicted functions that belonging to type I-F CRISPR-Cas system. RGP10 is a 48.9 kb genomic element with 56 CDSs including the genes for IS3 family transposases and a possible alkane degradation cluster with a long-chain alkane hydroxylase (almA, DBX28_14785). A noteworthy plasticity region with an aromatic ring-hydroxylating dioxygenase (DBX28_20755) was found in RGP11 (Table 2), which might provide competitive advantages for these three strains living in crude-oil areas. A large number of RGPs detected in IMP66, IMP67, and IMP68 indicated a complex exchange pattern of genetic material segments that occurred during the chromosome reshaping process throughout the evolutionary history.
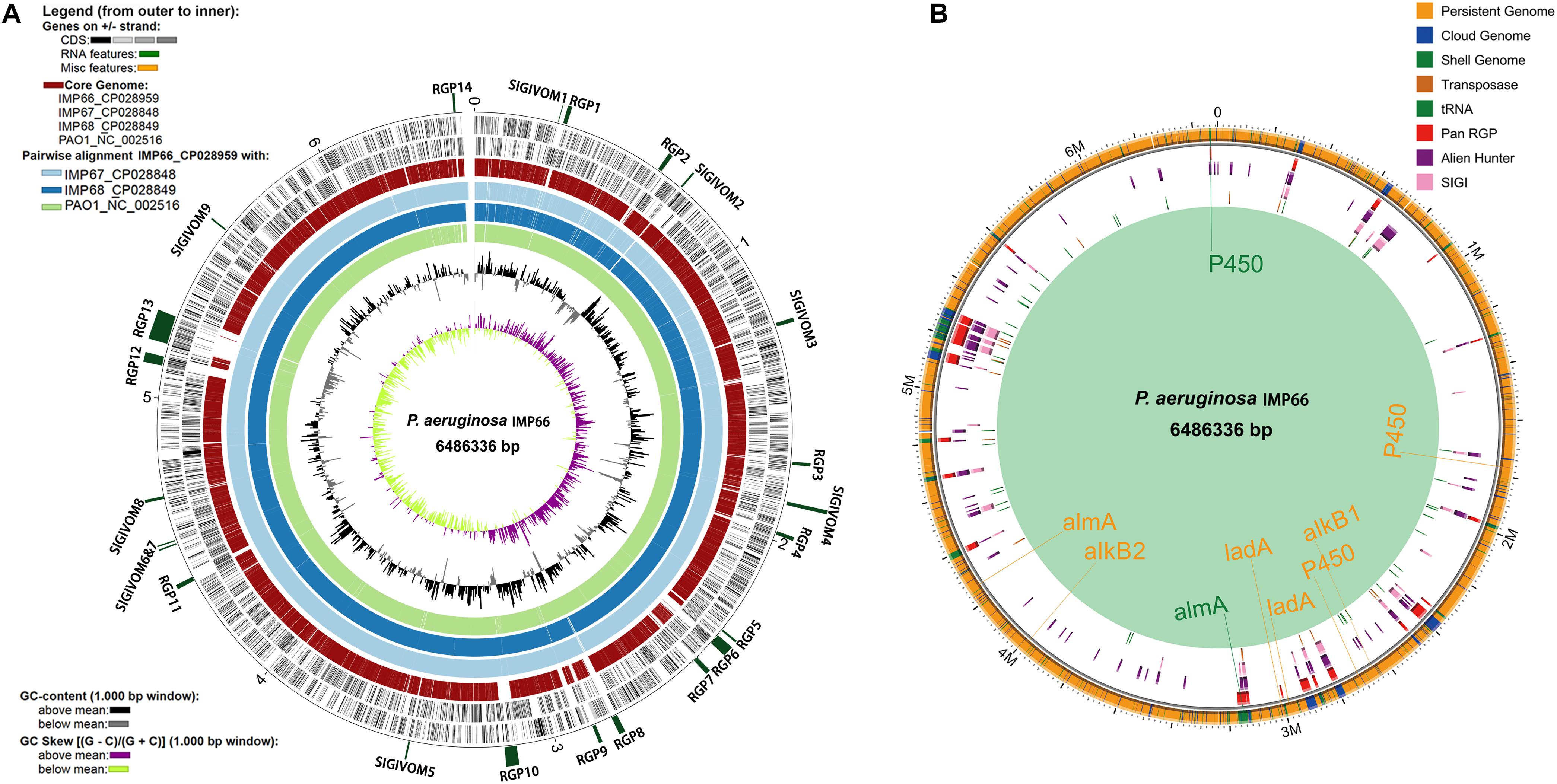
Figure 4. Whole genome comparison of IMP66, IMP67, IMP68, and PAO1. (A) Analysis of regions of genomic plasticity among IMP66, IMP67 and IMP68 (IMP66 was used as a representative of the three strains). The innermost ring represents the G + C skew of IMP66, followed by its G + C content. Light green, dodger blue and light blue rings that follow depict BLASTN comparisons between the genome of IMP66 and those of strains PAO1, IMP67, and IMP68, respectively. The red ring represents the core genome of the four strains. Then the two black rings are the CDSs of IMP66. The outermost interspaced ring (in dark green) represents the localization of the predicted regions of genomic plasticity (RGP) in the IMP66 genome, and the labels of each region follow the ones in Table 2. (B) Regions with different colors represent potentially horizontally transferred genes (HGT) which are gathered in genomic regions (Region of Genomic Plasticity, RGP). All potential alkane hydroxylases were marked at the chromosome with their corresponding colors.
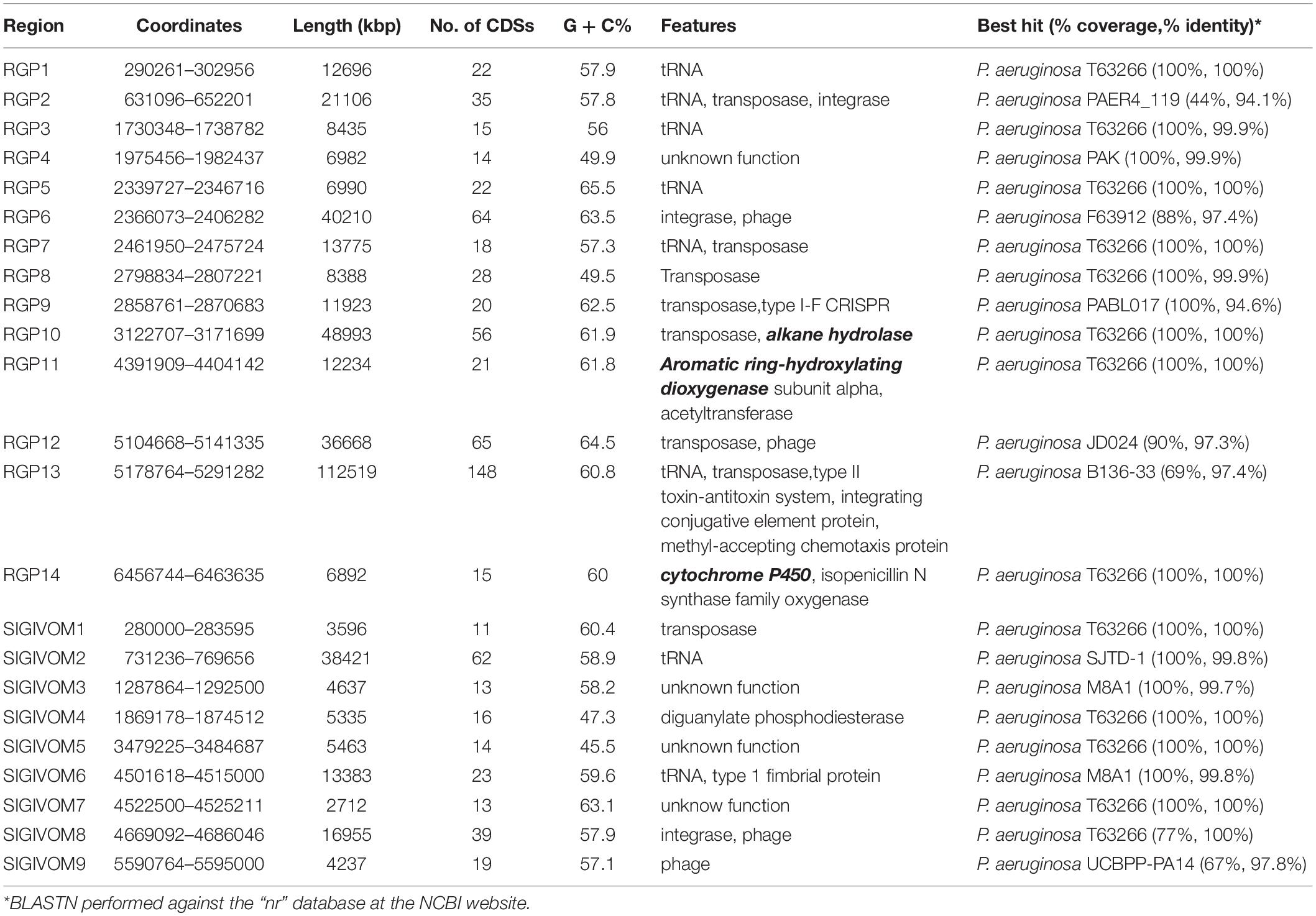
Table 2. Regions of Genomic Plasticity (RGPs) identified in the chromosome of IMP66, IMP67, and IMP68.
Analysis of Genes Involved in Alkane Degradation
Alkane hydroxylases (AHs) catalyzes the first step of alkane degradation and is involved in an AH system that includes electron transport protein rubredoxin and rubredoxin reductase. In the present study, whole-genome screening was used for identification of alkB, P450, almA, and ladA from IMP66, IMP67, and IMP68 through blasting against the reported AHs. We observed that the three strains carried up to 9 AH-encoding genes including two alkB homologous genes, three P450 homologous genes, two almA homologous genes and two ladA homologous genes (Figure 4B). As the “persistent genome” is generally considered to be more stable replicons, it is not surprising that most of the alkane hydroxylase genes were located in the “persistent genome”. Interestingly, two alkane hydroxylases were found in the “shell genome” (an almA gene in RGP10, and a cytochrome P450 gene in RGP14) (Figure 4B and Table 2). RGP10 was later identified to be a possible alkane degradation cluster that contained the genes coding for alkane hydroxylase (almA, DBX28_14785), SDR family oxidoreductase (DBX28_14685, DBX28_14840), phytanoyl-CoA dioxygenase (DBX28_14740, DBX28_14775), NAD(P)/FAD-dependent oxidoreductase (DBX28_14785, DBX28_14815), and NAD-dependent alcohol dehydrogenase (DBX28_14795). Almost all the genes in RGP10 were related to metabolism and energy production (Table 3). These results suggested that these three strains might have the ability to degrade and utilize a broad-spectrum of alkanes for growth.
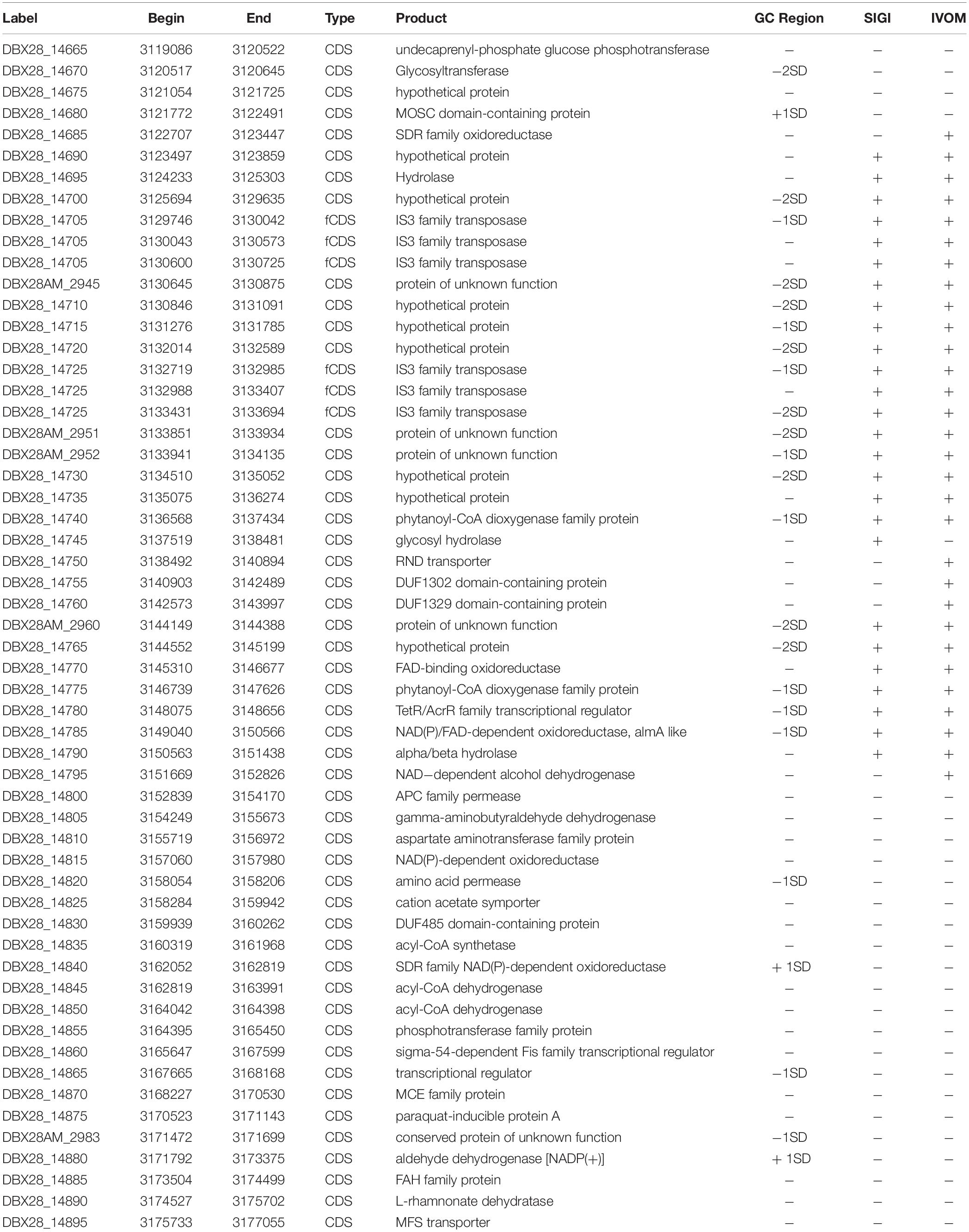
Table 3. Detail information of the probable alkane degradation cluster in RGP10.
Difference in RLs Production Among the Three Strains is Not at Genomic Level
Our previous study described that three strains produced different amounts of RLs. IMP67 and IMP68 produced much larger amounts of RLs than IMP66 (Das et al., 2014). The pan-genome analysis found that IMP67 and IMP68 possessed sereval unique genes compared to IMP66 (Figure 3B). These unique genes belong to type II secretion system, lipopeptide, non-ribosomal peptide synthetase, and hypothetical protein, which were not reported to be directly relevant to the RLs biosynthesis. Moreover, the average nucleotide identity (ANI) value between IMP66 and IMP68 is 99.99%. These results implied that the reason for the different yield of RLs of these strains is unlikely at genomic level.
To find the reason for the difference in RLs production among three strains, we then detected the QS signal molecules (C4-HSL and C12-HSL) by LC-MS/MS quantification, which positively regulate the expression of rhlAB, the two key RLs synthesis genes. After 24 h incubation, IMP67 and IMP68 produced significantly higher (3–10-folds) level of the two QS signal molecules (C4-HSL and C12-HSL) than that of PAO1, yet IMP66 produced trace amount of C4-HSL as well as C12-HSL, which might explain the reason for the less RLs production in IMP66 (Figure 5A). In addition, we used the rhlAB:gfp fluorescence-based reporter plasmid pTH22 to track the intracellular rhlAB transcription level. GFP fluorescence intensity was normalized by the whole cell protein concentration. IMP67 and IMP68 presented higher rhlAB expression than that of IMP66 and PAO1 (Figure 5B), which is correlated with the RLs and QS signal molecule production. Moreover, rhlAB-gfp intensity was significantly improved when IMP67 and IMP68 were grown in the nutrient broth (NB) medium (Figure 5B), which is a known RLs-promoting medium, suggesting that rhlAB transcription is related to the nutrient in the growth medium.
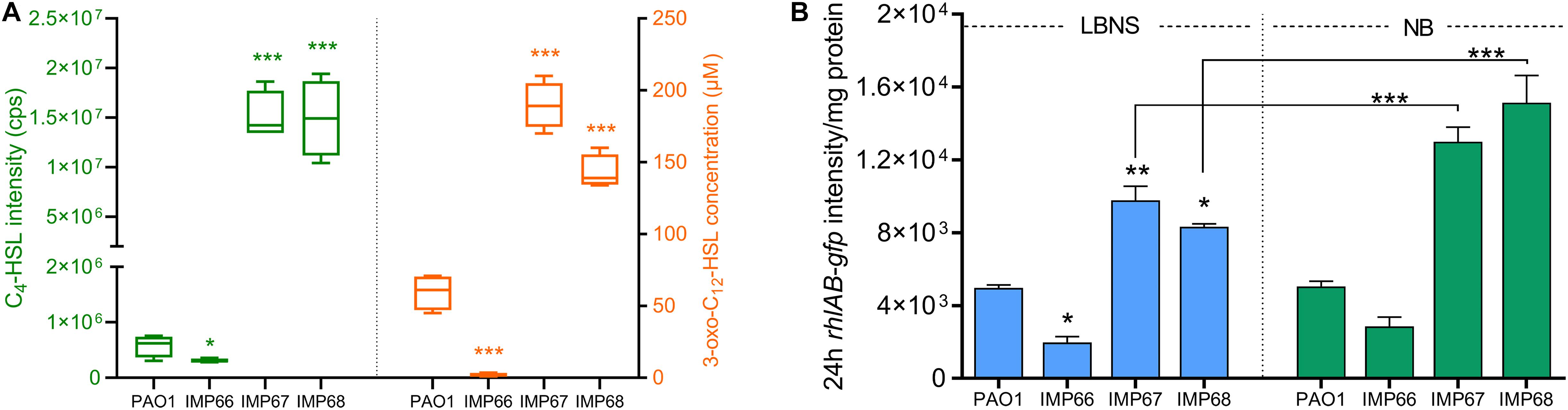
Figure 5. rhlAB transcription and AHLs quantification. (A) The concentration of C4-HSL (right panel) and 3-oxo-C12-HSL (left panel) of these three newly sequenced strains and laboratory strain PAO1. The concentration of C4-HSL is directly displayed using the original peak area due to no standard C4-HSL. (B) The rhlAB transcription level of PAO1, IMP66, IMP67 and IMP68 are indicated by the green fluorescent intensity of plasmid PrhlAB:gfp after 24 h incubation in LBNS or NB broth. Significance determined using Student’s t-test, *p < 0.05; **p < 0.01; ***p < 0.001, error bars represent the SD.
Rhamnolipids can enhance the bioavailability of crude oil, and therefore are crucial for microbial consortium living in the crude oil environment. To investigate whether these three strains possess the ability to produce RLs when grown with crude oil, especially n-alkanes, as the sole carbon source. We used again the rhlAB:gfp to determine the transcription of rhlAB when they cultured with alkane (tetracosane, C24; eicosane, C20) as the sole carbon source. The three strains displayed noticeable growth indicating by total protein concetration, and rhlAB transcription after 72 h incubation in the two types of n-alkanes, while they exhibited similar growth rate and growed better in C20 than C24 (Figures 6A,B). Altogether, our data indicated that high yield of RLs in IMP67 and IMP68 was due to the transcriptional regulation, not at genomic level.
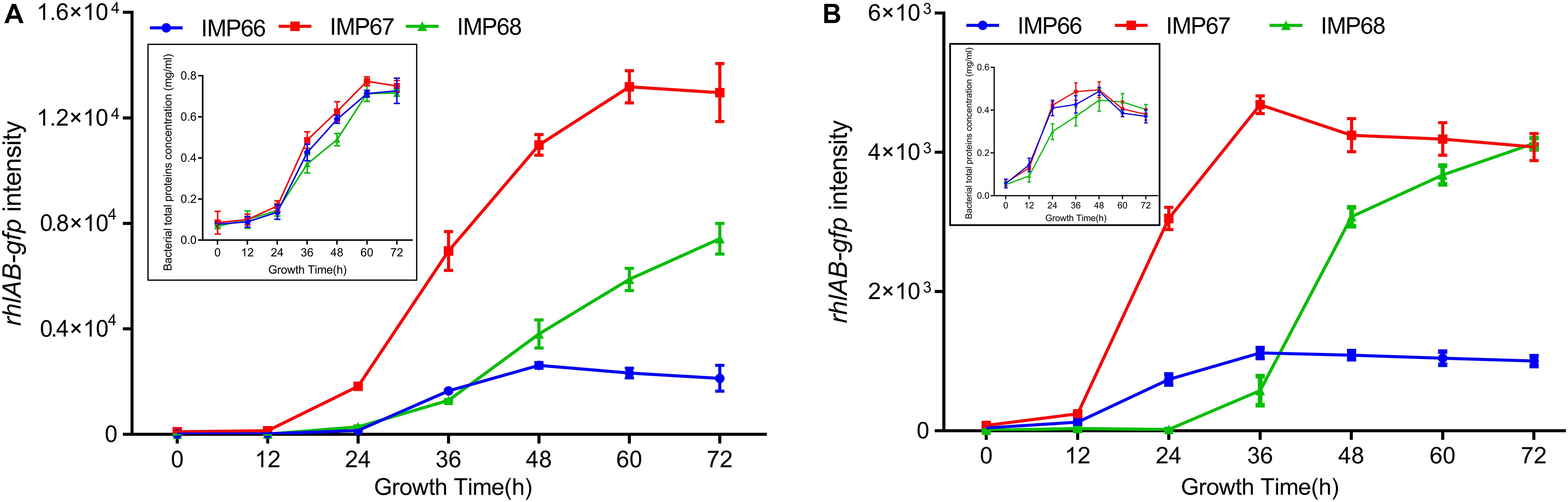
Figure 6. rhlAB transcription of IMP66, IMP67, and IMP68 when grown with n-alkane as the sole carbon source. C20 (A) or C24 (B) was used as the sole carbon source for the growth of IMP66, IMP67, and IMP68 carrying the reporter plasmid PrhlAB:gfp in BSM broth. The expression level of PrhlAB:gfp is monitored and recorded as the flourescent intensity of GFP. The growth curve presented by bacterial total protein concentration was shown as the inset.
Discussion
The environmental microorganism P. aeruginosa possesses great ecological flexibility, which enables it to thrive in diverse ecological niches. Under different environmental settings, P. aeruginosa may use different strategies to survive, such as forming biofilms in high-temperature oilfields (Orphan et al., 2000). It was widely believed that crude oil is a harsh habitat for microbes because of its high toxicity and hydrophobicity. However, growing evidence has revealed the presence of living microbes in crude oil (Yoshida et al., 2005). It has been reported that microbial communities in the crude oil phase were dominated by Pseudomonas, accounting for 96.84–98.87% of the total OTUs (Yamane et al., 2008; Cai et al., 2015), which exhibited significant survival advantages over other genera. In this study, we reported that three P. aeruginosa strains isolated from crude oil, IMP66, IMP67, and IMP68, exhibited excellent capabilities in n-alkane utilization and RLs production, which are two important factors for P. aeruginosa strains to maintain sustainable growth in the crude oil environment. Our work provides a better understanding of the adaptability of P. aeruginosa in crude oil.
Alkanes are the main components of petroleum hydrocarbons, and therefore, alkane degradation ability is critical for microorganisms to survive in petroleum-contaminated environments. Alkane hydroxylases are the key enzymes for aerobic degradation bacteria. Previous reports have identified AHs in several P. aeruginosa strains cultured from petroleum-contaminated environments (Zhang et al., 2011; Liu et al., 2012; Das et al., 2015). Compared to these P. aeruginosa strains, IMP66, IMP67, and IMP68 possess more AHs related genes, especially the coding genes for long-chain alkane monooxygenase, almA, and ladA. The coexistence of multiple alkane hydroxylases including alkB, P450, almA, and ladA in one bacterium is quite common in Marinobacter strains (Sun et al., 2018), Dietziasp. DQ12-45-1b (Nie et al., 2011), Amycolicicoccus subflavus DQS3-9A1T(AEF42720) (Nie et al., 2013), Acinetobacter spp. ADP1 (Barbe et al., 2004), and was also observed in many Alcanivorax isolates (Wang et al., 2010; Wang and Shao, 2012). However, nine AH genes identified in one single chromosome is rare in P. aeruginosa species. These alkane hydroxylases were demonstrated to be complementary and cover expanded substrate ranges, thereby enhancing the adaptive ability of P. aeruginosa in crude oil.
Gene duplication and horizontal gene transfer are common evolutionary processes that generate novel genes or functions for rapid adaptation (Kondrashov et al., 2002; Gogarten and Townsend, 2005). In this study, alkB, P450, almA and ladA paralogs with multiple copies were found in IMP66, IMP67, and IMP68. Moreover, a P450 and an almA gene were found integrated into the chromosome presumably through HGT (Figure 3B). These paralogs may exhibit different functions. For example, alkW1 and alkW2 are two paralogous genes encoding AlkB rubredoxin fusion proteins, which has been reported originally in Dietzia sp. DQ12-45-1b. It was shown that AlkW1 could hydroxylate n-alkanes ranging from C14 to C32, whereas AlkW2 was not expressed in this condition (Nie et al., 2011). Results from the whole genome comparison and pan-genome analysis have indicated that many flanking mobile genetic elements exist in the genomes of IMP66, IMP67, and IMP68. For instance, a 50 kb-long alkane metabolism-related genomic island (RGP10) containing an almA-like (50% identity) alkane monooxygenase along with several IS3 transposases is found in these three strains (Tables 2, 3), suggesting that horizontal gene transfer may occur during their life cycles.
Pseudomonas aeruginosa is previously demonstrated to use QS to regulate the synthesis of RLs, which are critical for P. aeruginosa to emulsify hydrocarbons and make them easier for uptake and assimilation. However, RLs also act as costly extracellular public goods. When the population growth was limited by restricted nutritional conditions, the regulation of QS-controlled public-good related genes may exhibit a metabolic prudent manner (Xavier et al., 2011). This could be a reasonable explanation for the fact that IMP67 and IMP68 expressed huge amount of QS signal molecules (Figure 5A), yet produced just a little higher amount of RLs than PAO1 (Das et al., 2014). Another plausible explanation could be that both RLs and exopolysaccharides share the common sugar precursors catalyzed by AlgC (Olvera et al., 1999; Wang et al., 2014). It could be a tactic for P. aeruginosa to synthese proper amount of RLs even with excess AHLs, and balance the synthesis of RLs and exopolysaccharides.
In conclusion, our results have showed that the alkane degradation ability and the superior AHL synthesis ability are two promoting factors for P. aeruginosa adaption in crude oil environment, which may also confer these three new isolates potential engineering performance in bioremediation of crude-oil pollution as well as microbial-enhanced oil recovery. Moreover, the native type I-F CRISPR-Cas system possessed by IMP66, IMP67, and IMP68 could be further established as a highly efficient, in situ genome-editing technique to modify the strains for future applications in such as industrial RLs production.
Data Availability Statement
The assembled annotated complete genomes used in this study are available at the National Center for Biotechnology Information (NCBI), under the accession code CP028959, CP028848 and CP028849. The other sequenced genomes are abtained from Pseudomonas database (www.pseudomonas.com).
Author Contributions
LM and SW acquired the funding. DW and LM designed the experiments. AX, YD, YZ, BW, and QW carried out the work and made contributions to acquisition of the data. AX wrote the original draft. LM, DW, LY and SW revised and edited the manuscript.
Funding
This work was supported by National Key R&D Program of China (2019YFC1804104 and 2019YFA095501), and the National Natural Science Foundation of China to LM (91951204) and SW (31770512).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Xin-Ping Yang at the Institute of Microbiology, Xinjiang Academy of Agricultural Sciences for providing the IMP66, IMP67, and IMP68 strains. We also thank Dr. Yong Nie at the Department of Energy and Resources Engineering, College of Engineering, Peking University for mining the AHs genes and Mr. Yubiao Qiang for bioinformatics software installation.
Footnotes
References
Anzai, Y., Kim, H., Park, J. Y., Wakabayashi, H., and Oyaizu, H. (2000). Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 50(Pt 4), 1563–1589. doi: 10.1099/00207713-50-4-1563
Barbe, V., Vallenet, D., Fonknechten, N., Kreimeyer, A., Oztas, S., Labarre, L., et al. (2004). Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res. 32, 5766–5779. doi: 10.1093/nar/gkh910
Beal, R., and Betts, W. B. (2000). Role of rhamnolipid biosurfactants in the uptake and mineralization of hexadecane in Pseudomonas aeruginosa. J. Appl. Microbiol. 89, 158–168. doi: 10.1046/j.1365-2672.2000.01104.x
Blom, J., Kreis, J., Spanig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Cai, M., Nie, Y., Chi, C.-Q., Tang, Y.-Q., Li, Y., Wang, X.-B., et al. (2015). Crude oil as a microbial seed bank with unexpected functional potentials. Sci. Rep. 5, 16057. doi: 10.1038/srep16057
Castañeda-Montes, F. J., Avitia, M., Sepúlveda-Robles, O., Cruz-Sánchez, V., Kameyama, L., Guarneros, G., et al. (2018). Population structure of Pseudomonas aeruginosa through a MLST approach and antibiotic resistance profiling of a Mexican clinical collection. Infect. Genet. Evol. 65, 43–54. doi: 10.1016/j.meegid.2018.06.009
Chakrabarty, A. M., Chou, G., and Gunsalus, I. C. (1973). Genetic regulation of octane dissimilation plasmid in Pseudomonas. Proc. Natl. Acad. Sci. U.S.A. 70, 1137–1140. doi: 10.1073/pnas.70.4.1137
Daniels, R., Vanderleyden, J., and Michiels, J. (2004). Quorum sensing and swarming migration in bacteria. FEMS Microbiol. Rev. 28, 261–289. doi: 10.1016/j.femsre.2003.09.004
Das, D., Baruah, R., Roy, A. S., Singh, A. K., Boruah, H. P. D., Kalita, J., et al. (2015). Complete genome sequence analysis of Pseudomonas aeruginosa N002 reveals its genetic adaptation for crude oil degradation. Genomics 105, 182–190. doi: 10.1016/j.ygeno.2014.12.006
Das, P., Yang, X. P., and Ma, L. Z. (2014). Analysis of biosurfactants from industrially viable Pseudomonas strain isolated from crude oil suggests how rhamnolipids congeners affect emulsification property and antimicrobial activity. Front. Microbiol. 5:696. doi: 10.3389/fmicb.2014.00696
Davies, D. G., Parsek, M. R., Pearson, J. P., Iglewski, B. H., Costerton, J. W., and Greenberg, E. P. (1998). The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280, 295–298. doi: 10.1126/science.280.5361.295
Deziel, E., Lepine, F., Milot, S., and Villemur, R. (2003). rhlA is required for the production of a novel biosurfactant promoting swarming motility in Pseudomonas aeruginosa: 3-(3-hydroxyalkanoyloxy)alkanoic acids (HAAs), the precursors of rhamnolipids. Microbiology 149, 2005–2013. doi: 10.1099/mic.0.26154-0
Freschi, L., Jeukens, J., Kukavica-Ibrulj, I., Boyle, B., Dupont, M. J., Laroche, J., et al. (2015). Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 6:1036. doi: 10.3389/fmicb.2015.01036
Geissdorfer, W., Kok, R. G., Ratajczak, A., Hellingwerf, K. J., and Hillen, W. (1999). The genes rubA and rubB for alkane degradation in Acinetobacter sp. strain ADP1 are in an operon with estB, encoding an esterase, and oxyR. J. Bacteriol. 181, 4292–4298. doi: 10.1128/jb.181.14.4292-4298.1999
Gibson, D. G., Glass, J. I., Lartigue, C., Noskov, V. N., Chuang, R.-Y., Algire, M. A., et al. (2010). Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56. doi: 10.1126/science.1190719
Gogarten, J. P., and Townsend, J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679–687. doi: 10.1038/nrmicro1204
Kok, M., Oldenhuis, R., van der Linden, M. P., Raatjes, P., Kingma, J., van Lelyveld, P. H., et al. (1989). The Pseudomonas oleovorans alkane hydroxylase gene. Sequence and expression. J. Biol. Chem. 264, 5435–5441.
Kondrashov, F. A., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2002). Selection in the evolution of gene duplications. Genome Biol. 3:RESEARCH0008.
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–W245. doi: 10.1093/nar/gkw290
Liu, H., Liang, R. B., Tao, F., Ma, C., Liu, Y., Liu, X. P., et al. (2012). Genome sequence of Pseudomonas aeruginosa Strain SJTD-1, a bacterium capable of degrading long-chain alkanes and crude oil. J. Bacteriol. 194, 4783–4784. doi: 10.1128/Jb.01061-12
Loper, J. E., Hassan, K. A., Mavrodi, D. V., Davis, E. W., Lim, C. K., Shaffer, B. T., et al. (2012). Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. Plos Genetics 8:e1002784. doi: 10.1371/journal.pgen.1002784
Miller, W. G., Leveau, J. H., and Lindow, S. E. (2000). Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant Microbe Interact 13, 1243–1250. doi: 10.1094/MPMI.2000.13.11.1243
Nie, Y., Chi, C. Q., Fang, H., Liang, J. L., Lu, S. L., Lai, G. L., et al. (2014a). Diverse alkane hydroxylase genes in microorganisms and environments. Sci. Rep. 4:4968. doi: 10.1038/srep04968
Nie, Y., Fang, H., Li, Y., Chi, C. Q., Tang, Y. Q., and Wu, X. L. (2013). The genome of the moderate halophile Amycolicicoccus subflavus DQS3-9A1(T) reveals four alkane hydroxylation systems and provides some clues on the genetic basis for its adaptation to a petroleum environment. PLoS One 8:e70986. doi: 10.1371/journal.pone.0070986
Nie, Y., Liang, J. L., Fang, H., Tang, Y. Q., and Wu, X. L. (2014b). Characterization of a CYP153 alkane hydroxylase gene in a Gram-positive Dietzia sp. DQ12-45-1b and its “team role” with alkW1 in alkane degradation. Appl. Microbiol. Biotechnol. 98, 163–173. doi: 10.1007/s00253-013-4821-1
Nie, Y., Liang, J., Fang, H., Tang, Y. Q., and Wu, X. L. (2011). Two novel alkane hydroxylase-rubredoxin fusion genes isolated from a Dietzia bacterium and the functions of fused rubredoxin domains in long-chain n-alkane degradation. Appl. Environ. Microbiol, 77, 7279–7288. doi: 10.1128/AEM.00203-11
Noordman, W. H., and Janssen, D. B. (2002). Rhamnolipid stimulates uptake of hydrophobic compounds by Pseudomonas aeruginosa. Appl. Environ. Microbiol. 68, 4502–4508. doi: 10.1128/Aem.68.9.4502-4508.2002
Olvera, C., Goldberg, J. B., Sanchez, R., and Soberon-Chavez, G. (1999). The Pseudomonas aeruginosa algC gene product participates in rhamnolipid biosynthesis. FEMS Microbiol. Lett. 179, 85–90. doi: 10.1111/j.1574-6968.1999.tb08712.x
Orphan, V. J., Taylor, L. T., Hafenbradl, D., and Delong, E. F. (2000). Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl. Environ. Microbiol. 66, 700–711. doi: 10.1128/aem.66.2.700-711.2000
Ortori, C. A., Dubern, J.-F., Chhabra, S. R., Cámara, M., Hardie, K., Williams, P., et al. (2011). Simultaneous quantitative profiling of N-acyl-L-homoserine lactone and 2-alkyl-4 (1H)-quinolone families of quorum-sensing signaling molecules using LC-MS/MS. Anal. Bioanal. Chem. 399, 839–850. doi: 10.1007/s00216-010-4341-0
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Passador, L., Cook, J. M., Gambello, M. J., Rust, L., and Iglewski, B. H. (1993). Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260, 1127–1130. doi: 10.1126/science.8493556
Pearson, J. P., Pesci, E. C., and Iglewski, B. H. (1997). Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 179, 5756–5767. doi: 10.1128/jb.179.18.5756-5767.1997
Prince, R. C. (2005). “The microbiology of marine oil spill bioremediation,” in Petroleum Microbiology, eds B. Ollivier and M. Magot (Washington, D.C: ASM press).
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Soberon-Chavez, G., Lepine, F., and Deziel, E. (2005). Production of rhamnolipids by Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 68, 718–725. doi: 10.1007/s00253-005-0150-3
Stothard, P., and Wishart, D. S. (2004). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Sun, J. Q., Xu, L., Liu, X. Y., Zhao, G. F., Cai, H., Nie, Y., et al. (2018). Functional genetic diversity and culturability of petroleum-degrading bacteria isolated from oil-contaminated soils. Front. Microbiol. 9:1332. doi: 10.3389/fmicb.2018.01332
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vallenet, D., Calteau, A., Cruveiller, S., Gachet, M., Lajus, A., Josso, A., et al. (2016). MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 45, D517–D528. doi: 10.1093/nar/gkw1101
van Beilen, J. B., Funhoff, E. G., van Loon, A., Just, A., Kaysser, L., Bouza, M., et al. (2006). Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral membrane alkane hydroxylases. Appl. Environ. Microbiol. 72, 59–65. doi: 10.1128/AEM.72.1.59-65.2006
van Beilen, J. B., Panke, S., Lucchini, S., Franchini, A. G., Rothlisberger, M., and Witholt, B. (2001). Analysis of Pseudomonas putida alkane-degradation gene clusters and flanking insertion sequences: evolution and regulation of the alk genes. Microbiology 147(Pt 6), 1621–1630. doi: 10.1099/00221287-147-6-1621
van Beilen, J. B., Wubbolts, M. G., and Witholt, B. (1994). Genetics of alkane oxidation by Pseudomonas oleovorans. Biodegradation 5, 161–174. doi: 10.1007/bf00696457
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. doi: 10.1093/bioinformatics/btl369
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, L. P., Wang, W. P., Lai, Q. L., and Shao, Z. Z. (2010). Gene diversity of CYP153A and AlkB alkane hydroxylases in oil-degrading bacteria isolated from the Atlantic Ocean. Environ. Microbiol. 12, 1230–1242. doi: 10.1111/j.1462-2920.2010.02165.x
Wang, S., Yu, S., Zhang, Z., Wei, Q., Yan, L., Ai, G., et al. (2014). Coordination of swarming motility, biosurfactant synthesis, and biofilm matrix exopolysaccharide production in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 80, 6724–6732. doi: 10.1128/AEM.01237-14
Wang, W., and Shao, Z. (2012). Diversity of flavin-binding monooxygenase genes (almA) in marine bacteria capable of degradation long-chain alkanes. FEMS Microbiol. Ecol. 80, 523–533. doi: 10.1111/j.1574-6941.2012.01322.x
Wang, X. B., Chi, C. Q., Nie, Y., Tang, Y. Q., Tan, Y., Wu, G., et al. (2011). Degradation of petroleum hydrocarbons (C6-C40) and crude oil by a novel Dietzia strain. Bioresour. Technol. 102, 7755–7761. doi: 10.1016/j.biortech.2011.06.009
Wentzel, A., Ellingsen, T. E., Kotlar, H. K., Zotchev, S. B., and Throne-Holst, M. (2007). Bacterial metabolism of long-chain n-alkanes. Appl. Microbiol. Biotechnol. 76, 1209–1221. doi: 10.1007/s00253-007-1119-1
Winsor, G. L., Griffiths, E. J., Lo, R., Dhillon, B. K., Shay, J. A., and Brinkman, F. S. (2016). Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653. doi: 10.1093/nar/gkv1227
Xavier, J. B., Kim, W., and Foster, K. R. (2011). A molecular mechanism that stabilizes cooperative secretions in Pseudomonas aeruginosa. Mol. Microbiol. 79, 166–179. doi: 10.1111/j.1365-2958.2010.07436.x
Xu, X., Liu, W., Tian, S., Wang, W., Qi, Q., Jiang, P., et al. (2018). Petroleum hydrocarbon-degrading bacteria for the remediation of oil pollution under aerobic conditions: a perspective analysis. Front. Microbiol. 9:2885. doi: 10.3389/fmicb.2018.02885
Yamane, K., Maki, H., Nakayama, T., Nakajima, T., Nomura, N., Uchiyama, H., et al. (2008). Diversity and similarity of microbial communities in petroleum crude oils produced in Asia. Biosci. Biotechnol. Biochem. 72, 2831–2839. doi: 10.1271/bbb.80227
Yoshida, N., Yagi, K., Sato, D., Watanabe, N., Kuroishi, T., Nishimoto, K., et al. (2005). Bacterial communities in petroleum oil in stockpiles. J. Biosci. Bioeng. 99, 143–149. doi: 10.1263/jbb.99.143
Yuste, L., Corbella, M. E., Turiegano, M. J., Karlson, U., Puyet, A., and Rojo, F. (2000). Characterization of bacterial strains able to grow on high molecular mass residues from crude oil processing. FEMS Microbiol. Ecol. 32, 69–75. doi: 10.1111/j.1574-6941.2000.tb00700.x
Keywords: Pseudomonas aeruginosa, crude oil, alkane hydroxylase, rhamnolipids, N-acyl-homoserine lactones
Citation: Xu A, Wang D, Ding Y, Zheng Y, Wang B, Wei Q, Wang S, Yang L and Ma LZ (2020) Integrated Comparative Genomic Analysis and Phenotypic Profiling of Pseudomonas aeruginosa Isolates From Crude Oil. Front. Microbiol. 11:519. doi: 10.3389/fmicb.2020.00519
Received: 04 December 2019; Accepted: 10 March 2020;
Published: 31 March 2020.
Edited by:
Shan Yi, The University of Auckland, New ZealandReviewed by:
Gloria Soberón-Chávez, National Autonomous University of Mexico, MexicoPing Xu, Shanghai Jiao Tong University, China
Copyright © 2020 Xu, Wang, Ding, Zheng, Wang, Wei, Wang, Yang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luyan Z. Ma, bHV5YW5tYTI3QGltLmFjLmNu
†These authors have contributed equally to this work