
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 26 March 2020
Sec. Microbial Symbioses
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00450
This article is part of the Research Topic Advances In The Understanding of The Commensal Eukaryota And Viruses Of The Herbivore Gut View all 10 articles
 Rosalind A. Gilbert1,2*
Rosalind A. Gilbert1,2* Eleanor M. Townsend3
Eleanor M. Townsend3 Kathleen S. Crew1
Kathleen S. Crew1 Thomas C. A. Hitch4
Thomas C. A. Hitch4 Jessica C. A. Friedersdorff5
Jessica C. A. Friedersdorff5 Christopher J. Creevey6
Christopher J. Creevey6 Phillip B. Pope7,8
Phillip B. Pope7,8 Diane Ouwerkerk1,2
Diane Ouwerkerk1,2 Eleanor Jameson3*
Eleanor Jameson3*The rumen contains a multi-kingdom, commensal microbiome, including protozoa, bacteria, archaea, fungi and viruses, which enables ruminant herbivores to ferment and utilize plant feedstuffs that would be otherwise indigestible. Within the rumen, virus populations are diverse and highly abundant, often out-numbering the microbial populations that they both predate on and co-exist with. To date the research effort devoted to understanding rumen-associated viral populations has been considerably less than that given to the other microbial populations, yet their contribution to maintaining microbial population balance, intra-ruminal microbial lysis, fiber breakdown, nutrient cycling and genetic transfer may be highly significant. This review follows the technological advances which have contributed to our current understanding of rumen viruses and drawing on knowledge from other environmental and animal-associated microbiomes, describes the known and potential roles and impacts viruses have on rumen function and speculates on the future directions of rumen viral research.
The presence of viruses in the rumen of domesticated herbivores was first noted in the 1960s (Adams et al., 1966; Hoogenraad et al., 1967) and it was quickly recognized that these viruses were not just those ingested by chance from feed and water. Instead, the majority of viruses in the rumen are those which either co-exist, or actively replicate and predate on, the dense microbial populations of the rumen. While viruses are also thought to infect the eukaryote populations of the rumen (protozoa and fungi), the viruses infecting rumen bacteria, often referred to as bacteriophage or phages, are the most well characterized. More recently, with the increasing focus on enteric methane emissions and rumen methanogenesis, viruses infecting methanogenic archaea (archaeal viruses or archaeaphage) have also been investigated.
Viruses of rumen microbes are an important focus for study, both as a key player in our understanding of rumen microbial community function and as a potential alternative to antibiotics in the face of a potential post-antibiotic era, as either a prophylaxis or treatment for problematic microbes. This review aims to summarize current knowledge of rumen virus populations, encompassing the traditional methods used to study rumen virus populations, as well as new and emerging technologies. We also draw on findings from other, better studied, microbial ecosystems (other animals, water, soils and microbial biofilms) to provide a fresh perspective and improve our understanding of the roles viral populations have in the rumen microbiome.
Developments in rumen virus research have been closely aligned with the technologies available for their study (Table 1). While the existence of viruses infecting bacteria were first noted in the early 1900’s (Twort, 1915; d’Herelle, 1918), it was the development of electron microscopy (EM) in the 1930’s (Haguenau et al., 2003) that showed that phages were actually virus particles and enabled rumen viral populations to be observed and enumerated (Hoogenraad et al., 1967; Paynter et al., 1969). Advances in methodology for the in vitro culture of rumen microbes (Hungate, 1969) enabled the cultivation of obligately anaerobic bacteria from the rumen and consequently the isolation of virus particles and determination of their biological characteristics. The advent of molecular biology enabled the preliminary genetic characterization of virus isolates (Klieve et al., 1991) providing an alternative method to TEM for virus particle enumeration and the determination of virus population dynamics (Klieve and Swain, 1993).

Table 1. Applications, benefits and limitations of technologies used for the study of rumen viruses.0
In the last 10 years, advances in DNA sequencing has led to the more rapid and accurate detection of viral sequences in prokaryote genome sequences and the sequencing of complete phage genomes (Leahy et al., 2010; Gilbert et al., 2017). A combination of high-throughput DNA sequencing and bioinformatic analysis has facilitated viral metagenomics: the culture-independent analysis of all the viral genetic material in a rumen sample. This has allowed the taxonomic classification of viral populations and determination of the relative abundance of individual viral taxonomic groups (Berg Miller et al., 2012). Sequence analysis of rumen sample RNA (metatranscriptomics) has also been used to identify RNA virus populations (Hitch et al., 2019). In addition, alternative technologies are being applied for the isolation of virus particles (for example, chromatography) and enabling the expression of novel viral proteins in vitro (Figura et al., 2011; Krajacic et al., 2017; Altermann et al., 2018). The emerging field of proteomics, is also providing fresh insights into the roles viruses and the proteins produced by viruses, may play within the rumen microbial ecosystem (Solden et al., 2018).
Electron microscopy is a large research field, with applications in materials science as well as biology. The combination of electron microscopy and specialized staining techniques, enables the visualization of virus particle structures at nanometer scale, which could not otherwise be distinguished using conventional light microscopy. Several different electron microscopy technologies exist including scanning electron microscopy (SEM), transmission electron microscopy (TEM), high voltage electron microscopy, immunoelectron microscopy (IEM) and cryoelectron microscopy (cryoEM) with or without shadowing and three dimensional image reconstruction (reviewed by Ackermann, 2012; Romero-Brey and Bartenschlager, 2015). Of these technologies, TEM is the most widely used technique for the visualization of all types of viruses (Ackermann and Prangishvili, 2012; Popov et al., 2019).
The first use of TEM to visualize virus particles within rumen fluid, was published in 1967 (Hoogenraad et al., 1967). This study initially intended to determine the fate of bacterial cell walls within the rumen of sheep and involved direct examination of rumen samples by TEM. The rumen fluid was filtered through muslin and stained for negative contrast using potassium phosphotungstate. This methodology, however, revealed the presence of large numbers of free virus particles (icosahedral particles and tailed phages) and as the microbes were not removed prior to TEM, the attachment of phage particles to intact bacterial cells, including cocci and spirochetes, was also observed. This study was therefore pioneering not only in its use of the relatively new technique of TEM, but also in its hypothesis of what we now know to be true, that “phage may be a constant feature of the bacterial population of the rumen” (Hoogenraad et al., 1967).
Other studies then followed, employing TEM to conduct morphological surveys of rumen fluid collected from various ruminant species, including sheep, cattle and reindeer (Paynter et al., 1969; Ritchie et al., 1970; Tarakanov, 1972; Klieve and Bauchop, 1988). These surveys sought to enumerate the numbers of different viral morphotypes present, with results ranging from 6 (Paynter et al., 1969), 26 (Klieve and Bauchop, 1988), and more than 40 distinct morphotypes (Ritchie et al., 1970). In all the species of ruminants examined, the most abundant viral morphotypes were those with polyhedral heads and tubular tails, which are now classified within the viral order of tailed phages infecting bacteria, the Caudovirales. This viral order currently contains five families, the Myoviridae, Siphoviridae, Podoviridae, Ackermannviridae, and Herelleviridae, the last two being recently added (Adriaenssens et al., 2012; Barylski et al., 2020a, b). Of these, the short non-contractile tailed phages (family Podoviridae), long contractile-tailed phages (family Myoviridae) and many long non-contractile tailed phages (family Siphoviridae) were observed. These early surveys also sought to quantitate the size of the rumen viral populations using TEM to count the numbers of particles present. While counts varied, possibly as a consequences of differences in animal species as well as differences in sample preparation and counting methodology, virus particle counts ranged from 5 × 107 particles per mL bovine rumen fluid (Paynter et al., 1969) to greater than 109 particles per mL bovine rumen fluid (Ritchie et al., 1970). It was also noted that the numbers of phages in crude rumen fluid could exceed bacterial numbers ca. 2 to 10:1 (Ritchie et al., 1970).
Images from a more recent TEM morphological survey of a sample obtained from an anaerobic fermentation system started with an inoculum of bovine rumen fluid (Figure 1) shows virus particle types representative of those observed in previous studies. This survey also includes examples of non-tailed viral particles and while it can sometimes be difficult to distinguish the small tail structures of the Podoviridae, these particles could belong to other viral families infecting bacteria such as Tectiviridae, Corticoviridae, and Microviridae (Ackermann and Prangishvili, 2012). It is now known that morphological surveys may have technical limitations and operator bias can result in errors in identification (Ackermann, 2013). For example, tail-less virus-like-particles may be confused with tailed phages which have lost their tail structures either naturally, with some phages being more susceptible to tail loss in the environment than others, or as a result of particle concentration and TEM sample preparation (Williamson et al., 2012). In addition, filamentous phages (Inoviridae) can be associated with rumen bacteria (Klieve et al., 1989) and have been observed by morphological survey (Klieve and Bauchop, 1988), however, these long, thin viral particles can be difficult to distinguish by TEM, due to their resemblance to fragments of phage tails and extracellular structures of bacteria (fine pili and flagella).
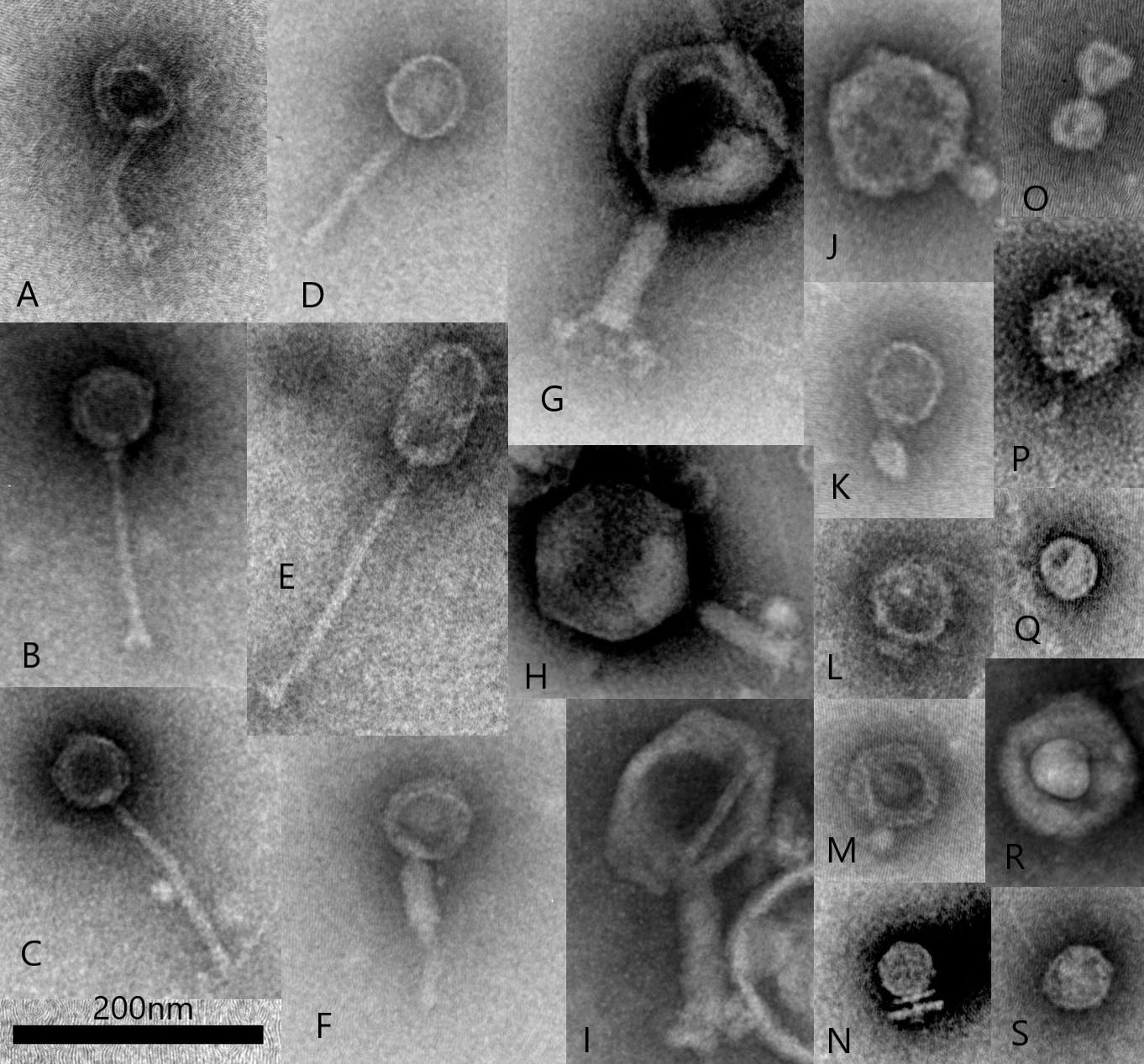
Figure 1. Transmission electron microscopy illustrating examples of common rumen virus morphotypes including Siphoviridae (A–E), Myoviridae (F–I), Podoviridae (J–O) and other virus-like-particles including potentially non-tailed phages (P–S), such as Corticoviridae and Microviridae (scale bar 200 nm). All virus particles were purified from a single sample collected from an in vitro fermentation system (Infors) started with bovine rumen fluid. Virus particles were fixed with 2.5% glutaraldehyde and stained with 1% ammonium molybdate pH 7.0 and viewed with a JEOL JEM1400 Transmission Electron Microscope. Micrographs were captured using a Gatan Orius CCD camera.
The use of TEM for estimating rumen viral numbers and population diversity has been largely superseded in favor of metagenomics and molecular methods. Viral genome sequencing can be used to predict viral particle morphology, as specific viral genes are conserved between morphological groups of viruses (Kristensen et al., 2013; Iranzo et al., 2016; Krupovic et al., 2018). However TEM is still widely recognized in virology as an important tool for the detection and morphological characterization of new virus isolates (Cuervo and Carrascosa, 2018; Popov et al., 2019) including those predicted from metagenomics (Shkoporov et al., 2018). The majority of publications to date describing the isolation of phages from the rumen, have used TEM in order to classify new isolates and verify the intact nature of phage particles (Brailsford and Hartman, 1968; Iverson and Millis, 1976a; Styriak et al., 1994; Cheong and Brooker, 2000). Electron microscopy can also be used for visualizing tail fibers involved in host-receptor binding with immunogold labeling, virus:host cell attachment and infection, intracellular viral multiplication and virus particle assembly and release (Ackermann, 1998; Kaelber et al., 2017; Letarov and Kulikov, 2017). TEM and other new, emerging forms of electron microscopy, can therefore be expected to have a continuing, important role in the study of rumen-associated viruses.
In the 1980’s and 1990’s there were rapid technological advancements in the field of molecular biology enabling: the purification of genetic material (DNA and RNA); charge and size-based separation of nucleic acids (electrophoresis; blotting techniques); the use of restriction enzymes for site-specific cutting of DNA and gene mapping; DNA recombination (DNA cloning and genetic transformation); and DNA sequencing techniques (reviewed by Brody and Kern, 2004; Loenen et al., 2014; Heather and Chain, 2016). Rumen microbiologists readily adopted these technologies (Orpin et al., 1988; Flint, 1997) and the way in which rumen phage isolates and rumen viral populations were studied, changed accordingly. Molecular biology was utilized to characterize new phage isolates, with less emphasis on using culture-based methods to understanding phage growth, survival in rumen fluid and host range, and more focus on genome length and restriction enzyme mapping. These analyses were conducted on phage isolates found to specifically infect several strains of rumen bacteria, including Fusobacterium necroforum (Tamada et al., 1985), Selenomonas ruminantium (Lockington et al., 1988), Lactobacillus plantarum (Nemcova et al., 1993), Bacteroides ruminicola (Klieve et al., 1991; Gregg et al., 1994), and Ruminococcus albus (Klieve et al., 2004). These phage isolates were mainly classified within the viral families Myoviridae and Siphoviridae however, some examples of Podoviridae and Inoviridae were also characterized in this way (Klieve et al., 2004). At this time, whole genome sequencing was technically very challenging and costly and therefore was not routinely undertaken. These studies, however, provided the first insights into the genetic characteristics and unique attributes of the phages infecting rumen bacteria.
Molecular biology also provided new methodologies that could be used for enumerating and describing viral populations occurring within a rumen sample showing a link between populations of rumen bacteria and virus populations (Klieve and Swain, 1993). These methods could also be applied to the analysis of relatively large numbers of rumen fluid samples derived from in vivo experiments (Klieve and Gilbert, 2005). The pulsed field gel electrophoresis (PFGE) technique was used following the purification and concentration of virus particles from relatively small volumes (40–60 mL) of rumen fluid and extraction of viral DNA using agarose-embedding techniques, to enable intact genome lengths to be preserved. Separation of DNA by PFGE then produced a population profile, based on viral genome length (Klieve and Swain, 1993). In addition, as an alternative to TEM viral particle counts, a DNA-based method was developed for the estimation of total viral numbers, based on a combination of conventional DNA extraction and electrophoresis techniques, and laser densitometry (Klieve and Swain, 1993). DNA virus population estimates obtained using this technique were slightly higher than those reported using TEM, with estimates of between 3 × 109 to 1.6 × 1010 particles per mL of ovine rumen fluid reported (Klieve and Swain, 1993).
Using these techniques (Swain et al., 1996a; Swain, 1999) the rumen viral population was found to (1) have variation in viral populations occurring between individual animals maintained on the same diet, as well as variation between different ruminant species (sheep, goats, cattle); (2) result in blooms of phage activity evidenced by sudden increases or bursts of phage particles; (3) have patterns of diurnal (within day) variation viral populations, in sheep fed once a day, with phage numbers showing blooms of phage replication (lysis events) and similar patterns of variation to those previously observed for bacterial populations in once-a-day fed sheep (Leedle et al., 1982); and (4) be affected by plant compounds found in the ruminant diet (Klieve et al., 1998). For example, the hydrolyzable tannin, tannic acid reduced rumen virus populations when added to sheep prior to feeding to give an intra-ruminal concentration of 0.1% wt/vol., suppressing by as much as 50% the increase in phage numbers normally occurring 8–12 h after feeding (Swain et al., 1996b). The mechanism of this reduction was attributed to the protein-binding activity of the tannic acid, and it was hypothesized that viral particles freely circulating in the rumen were being bound by the tannic acid and removed from the liquid phase (Swain, 1999).
These early molecular biology methods provided information about virus genome length (Hallewell et al., 2014), and enabled estimation of the size and relative length profiles of virus populations (Swain et al., 1996a). However, these methods were unable to deliver any DNA or RNA sequence information about the virus populations observed. Only a few studies conducted at this time were able to provide relatively short, gene-specific sequence information (Gregg et al., 1994). Being able to obtain sufficient amounts of genetic information to be able to infer viral taxonomic classification, was achieved only after significant technological advances in DNA sequencing and the emergence of the field of metagenomics.
Sequencing is an important method for understanding virus populations, and there is a wide variety of tools now at our disposal that makes it increasingly easy to generate virus-specific sequence datasets (referred to as viromes). Metagenomics was initially termed as the study of the collective genomes of all microbial species within an environmental sample (Handelsman et al., 1998) and with the advent of next generation sequencing (NGS), direct sequencing of DNA from environmental samples became possible (Escobar-Zepeda et al., 2015). Microbial community sequencing can be subdivided into two major sequencing methods; (i) shotgun metagenomic sequencing and (ii) amplicon sequencing. Shotgun metagenomic sequencing involves fragmentation of the total environmental DNA for sequencing, while amplicon sequencing includes PCR amplification and sequencing of specific genomic regions of interest. For bacterial taxonomy the 16S rRNA gene is used (McGovern et al., 2018) and can be described as metataxonomics (Marchesi and Ravel, 2015). Whilst the communities of bacteria, archaea, protozoa and fungi can be assessed using both amplicon and shotgun sequencing, the viral component can only be studied using shotgun metagenomics, due to the lack of a universally conserved gene across all viral species (Sullivan et al., 2008; Jameson et al., 2011; Wylie et al., 2015). Shotgun sequencing is highly effective in studying virus populations, which have relatively small genomes (∼20–300 kbp) (Wylie et al., 2015). Whilst DNA based sequencing technologies are most commonly undertaken, this approach excludes the potential study of RNA based viruses, which require total RNA metatranscriptomic sequencing for their detection (Hitch et al., 2019).
The marine environment is an easy system on which to develop viral sequencing tools; it is simple to collect liters of water without negatively impacting the environment being sampled, there is no inhibitory chemistry to prevent sequencing and there is minimal debris and organic matter for viruses to adsorb to and be lost. As a result, marine viral communities were one of the very first environments profiled using metagenomic approaches (Breitbart et al., 2002). Through the use of differential filtration, density-dependent gradient centrifugation, shotgun cloning and sequencing, Breitbart et al. revealed a highly diverse, previously unknown marine viral populations and opened the way for the characterization of viral communities in other environments.
Rumen virology is currently lagging behind marine and even human intestinal virology, with fewer studies using metagenomics to describe viral populations (Table 2). These studies have varied in the procedures used for sample preparation, however, most of the recent studies have included steps for the concentration of viral particles prior to DNA extraction and shot-gun sequencing to generate a viral-specific (virome) dataset. This is in contrast to the early metagenome studies, which generated viromes by extracting virus-only sequences from whole-rumen metagenome datasets (Dinsdale et al., 2008; Willner et al., 2009). As the available sequencing technologies have progressed from Pyrosequencing and Ion Torrent sequencing systems to the more high-throughput Illumina technologies, the rumen virome datasets obtained have increased in sequencing depth and provide more comprehensive sequence coverage. To date the rumen virome of several ruminant species have been examined, including sheep, goats, buffalo and moose, with cattle, including dairy cows and steers being the most frequently investigated (Table 2). Most studies use limited numbers of animals and experimental conditions, such as diet, have remained constant (Berg Miller et al., 2012; Ross et al., 2013).

Table 2. Summary of published rumen viral metagenomics studies including the type of ruminant used for each study (breed description provided in publication), number of individual animals sampled and the DNA sequencing technologies used.
Currently only one report has used metagenomics to investigate the effect of changing diets on rumen viral populations which showed the total digestible nutrients was the predominant ecological driver of both bacterial and viral response to dietary change (Anderson et al., 2017). In this study, diversity analysis of both the bacterial and viral communities identified that samples clustered based on diet rather than host, confirming that dietary changes led to consistent changes within both communities (Anderson et al., 2017). This study represented a significant advance in our understanding of rumen viral populations. Diet has been shown in many studies, to greatly influence the relative abundance of rumen microbial species (reviewed by Huws et al., 2018). As viral populations are intrinsically linked to the populations of their microbial hosts (see Lessons to be Learned from Viruses in Other Environments), any changes in the animal diet and consequent availability of specific nutrients for microbial growth in the rumen, can also be expected to cause significance changes the relative abundance of viral populations. While early molecular biology studies eluded to the effects of animal diet on rumen microbial and associated viral populations (Swain et al., 1996a), the advent of viral metagenomics has verified and enhanced our understanding of diet-induced changes on rumen viral populations.
In terms of viral taxonomy the rumen virome varies greatly between individual animals of the same ruminant species and a subset of nearly ubiquitous viral genomic fragments have been identified and proposed to make up a core rumen virome (Anderson et al., 2017). It can also be speculated that these viruses are likely to predate on the bacterial species core to the rumen (Henderson et al., 2015) including the bacterial genera: Prevotella, Butyrivibrio, Ruminococcus and more generally, the families Lachnospiraceae, Ruminococcaceae and Bacteroidales. Metagenomic studies of the rumen have all revealed that the rumen virome is dominated by Siphoviridae (32–36%), Myoviridae (24–32%), and Podoviridae (12–16%), all bacteriophage families belonging to the order Caudovirales (Lefkowitz et al., 2018). As we expect the most abundant rumen viruses to be bacteriophages and public databases are dominated by these, this makes their identification on the basis of genetic homology easier. It is likely that the viral sequencing and identification methods currently adopted, bias against the less common viral groups we anticipate in the rumen, such as archaeal viruses, mycoviruses and other eukaryote viruses.
The dominance of DNA based sequencing methods prevents the study of RNA based viruses including the Totiviridae which infect host-associated protozoa (Grybchuk et al., 2018) and the Partitiviridae, which may exist within the rumen and infect fungi (Hitch et al., 2019). Under these assumptions we may be missing vital functions and relationships. Lower abundance viral families have been identified in the rumen, which have been putatively assigned as large DNA viruses (Mimiviridae, Phycodnaviridae), filamentous lysogenic viruses of bacteria and archaea (Inoviridae), archaeal viruses (Bicaudaviridae) and animal viruses (including; Baculoviridae, and Adenoviridae) (Berg Miller et al., 2012; Parmar et al., 2016; Anderson et al., 2017). Although they have been detected in rumen virome datasets, the animal viruses identified often represent lineages which would not normally infect rumen microbes. These taxonomic assignments are based on sequence homology to databases of viral sequences available at the time. As both the number of phage genomes in Genbank and publications containing novel viral taxonomies are increasing exponentially, the continuing accumulation of new genetic information, will assist the accuracy of future viral taxonomic classification.
Another application of metagenomics to the study of rumen virus populations involves examining clustered regularly interspaced short palindromic repeats (CRISPR) sequences. The identification of CRISPR-cas arrays and prophages within the cellular component of the rumen microbiome allows for the possible identification of viral-host interactions (Berg Miller et al., 2012). Whilst prophage insertions represent whole viral genomes and are therefore easier to identify and characterize, CRISPR arrays incorporate only a short 25–65 bp region of viral DNA (Shmakov et al., 2017). Despite these short insertions, recent advances have separated and annotated these spacer regions against viral datasets, allowing for viral-host interactions to be identified and studied. Data sets such as the Hungate 1000 rumen collection contain a wealth of data on prophages and viral interactions. The Hungate 1000 collection yielded identifiable CRISPR arrays in over 60% of the isolates (Seshadri et al., 2018). CRISPR arrays can also provide clues to the frequency of aborted lytic viral infections (Sanguino et al., 2015; Edwards et al., 2016). Although these methods have been applied to the rumen microbiome, larger datasets are required to gather enough data for meaningful insights on phage infection (Berg Miller et al., 2012).
Prophages identified from bacterial genomes and microbial metagenome sequencing can help shed light on prophage diversity and the lifestyles of viruses in the rumen. Computational approaches to identify prophages and free viruses in metagenomic data vary depending on the type of data to be analyzed and the prior information available for the virus family of interest. If existing genomic data exists for that viral family, then the direct alignment of reads to the genomes can allow inferences of viral abundances and types in the sample, termed an “aligner” approach (Roux et al., 2015). Another approach, which can utilize reads directly from the sequencer, involves “binning” (separation) of sequences into groups based on characteristics inferred from their sequences. This can be comparing their sequence similarity to known viral sequences (for example Zhao et al., 2017), to more complex approaches which calculate frequency profiles of short strings of DNA, termed “kmers” (for example Willner et al., 2009). This latter approach is based on the observation that different organisms can be identified by the kmer profiles of their genomes and is widely used in metagenomic data analysis (Ren et al., 2017). The resulting “bins” can then be individually assembled into what is hoped to be the genome of a single virus, and these assumptions can be tested by subsequent gene prediction or sequence similarity searches to compare against databases such has those hosted by the NCBI. There is no consensus on which approach or tool is best. The rate at which new tools are emerging (for a current list of tools see Zheng et al., 2019), suggests that using a multi-tool approach may allow balancing of the strengths and weaknesses of each (Anderson et al., 2017). An array of techniques have also been successfully employed on human gut metagenome sequence data to obtain a snapshot of virus communities (Nielsen et al., 2014; Ogilvie and Jones, 2015).
In terms of gene function, the rumen virome has been shown to be relatively consistent across animals of the same ruminant species (Ross et al., 2013). Assigning function to putative viral open reading frames (ORFs) is also difficult, as there is a high percentage of unknown and uncharacterized viral genes (Yooseph et al., 2007). Kyoto Encyclopedia of Genes and Genomes (KEGG) based annotation has suggested that 50–70% of the functional reads within the rumen virome are involved in viral replication (KEGG pathways: nucleotide metabolism, replication and repair), whilst the remaining reads were functionally diverse and included viral structural genes (head and tail proteins). A subset viral genes have been identified as auxiliary metabolic genes (AMGs), which redirect host metabolism toward reactions favorable to phage replication (Anderson et al., 2017). In this way AMGs encompassed in virome datasets represent one way rumen viral populations may influence the metabolic potential of the rumen microbiome.
The first report of culture-based, rumen viral isolation used rumen and non-rumen strains of Serratia spp. (facultative anaerobes, family Enterobacteriaceae) to isolate phages, with the intention of demonstrating that phages were prevalent in the rumen of cattle (Adams et al., 1966). As well as successfully isolating phages from rumen fluid, this study also investigated host-specificity (or host range), that is, the ability of these phage isolates to infect multiple strains of bacteria. In this way the study established that phages sourced from the rumen could have limited host-specificity, preferentially infecting Serratia host strains sourced from the rumen and being unable to infect non-ruminant Serratia strains. Correspondingly, the rumen Serratia were not susceptible to infection by Serratia phages isolated from soil, water and sewage (Adams et al., 1966). This study was the first to demonstrate that not only were phages prevalent in the rumen, a finding subsequently confirmed using TEM (Hoogenraad et al., 1967), it also demonstrated that the phages found in the rumen differed from those present in other environments.
This primary investigation was followed by numerous studies conducted in laboratories across the globe (Iverson and Millis, 1976a; Tyutikov et al., 1980; Hazlewood et al., 1983; Tamada et al., 1985; Klieve and Bauchop, 1991; Nemcova et al., 1993; Styriak et al., 1994; Ambrozic et al., 2001). The majority of these studies were conducted in the 1980’s and 1990’s, with very few phage isolations reported in the literature beyond this era. To date, most of the viruses isolated from the rumen have been phages infecting rumen bacteria with only a single, preliminary isolation of a rumen-derived archaeal virus being reported (Baresi and Bertani, 1984).
Bacterial hosts strains used for the isolation of rumen phages have been reviewed elsewhere (Gilbert and Klieve, 2015) and include genera classified within various common phyla associated with the rumen, including Firmicutes (Ruminococcus, Lactobacillus, Eubacterium, Selenomonas, Quinella, and Streptococcus), Bacteroidetes (Bacteroides), Proteobacteria (Serratia), and Actinobacteria (Bifidobacterium). The majority of phages infecting rumen bacteria have a typical tailed morphology (Gilbert and Klieve, 2015) and although originally classified according to the Bradley scheme (Bradley, 1967), the following examples are listed according to the modern international committee on taxonomy of viruses (ICTV) classification scheme (King et al., 2012). Rumen phage isolates with Siphovirus morphology include phage F4 which can infect five strains of Streptococcus bovis (Styriak et al., 1994; Nigutova et al., 2008); and a long-tailed phage infecting Prevotella (Bacteroides) brevis strain GA33 (Ambrozic et al., 2001). An example of a rumen phage isolate with Myoviridae morphology is phage FnP1 infecting Fusobacterium necrophorum strain FnP1 which interestingly also infected 12 other F. necrophorum strains of biovars A and B but none of the Bacteroides strains tested (n = 13) (Tamada et al., 1985). A rumen phage isolate with Podoviridae morphology is phage 2BV which infects Streptococcus bovis (Iverson and Millis, 1976a). Reports of phage isolates infecting rumen bacteria with a non-tailed morphotypes have been relatively infrequent, for example, the non-tailed, filamentous Inoviridae phages φRa01 and φRa03 infect R. albus AR67 (Klieve et al., 2004).
To isolate phages from the rumen, the majority of culture-based studies have used double-layer agar plates for the detection of clearing zones (plaques) within bacterial monolayers (Klieve, 2005). A combination of anaerobic techniques (Hungate et al., 1964) and rumen fluid-based growth medium with modifications are used to ensure the survival and growth of anaerobic bacterial host strains in vitro (Klieve et al., 1989). Culture-based plating techniques have also served as important tools for determining the biological properties of phage isolates, particularly those infecting rumen-associated Streptococcus species. Biological properties determined in this way include: viral particle viability in rumen fluid; the development of phage resistance; and host range (Iverson and Millis, 1977; Klieve and Bauchop, 1991; Klieve et al., 1999).
Culture-based plating methods tend to favor the isolation of phages which undergo the lytic cycle of replication. Several different phage replication lifestyles exist in nature and the terminology to describe these lifestyles or cycles, may vary (Hobbs and Abedon, 2016). However, when phages only follow the lytic cycle of replication, phage particle attachment and infection of the host cell leads directly to replication and culminates in the bursting, or lysis, of the host cell to release the progeny phage particles. True lytic phages cannot adopt the alternative, lysogenic lifestyle of replication, as they lack the functional genes required for control of replication and/or integration of phage DNA into the host genome. These phage therefore cannot form a dormant, heritable genetic state (prophage) or establish what is sometimes referred to as chronic infection (reviewed by Weinbauer, 2004; Howard-Varona et al., 2017).
While the isolation of phages from the rumen originally began as a way to determine the extent of viral population diversity, phage isolation was later driven by (1) the need to isolate phages for use in potential biocontrol (phage therapy) of problematic bacteria, such as the bacteria implicated in the development of ruminal acidosis, Streptococcus bovis (since re-classified as S. equinus) (Tarakanov, 1994); and (2) the requirement for molecular tools (vectors) for the stable transformation of rumen bacteria (Lockington et al., 1988; Gregg et al., 1994). As phage therapy is traditionally based on the use of viable lytic phages, several groups established collections of phages infecting S. bovis strains (Iverson and Millis, 1976a; Tarakanov, 1976; Klieve and Bauchop, 1991; Styriak et al., 1994; Klieve et al., 1999). The development of cloning vectors for the genetic transformation of rumen bacteria, however, involved utilizing either phage replication or integration genes, such as those present in lysogenic phages (Gregg et al., 1994; Cheong and Brooker, 1998).
The bacterial genera that were selected for genetic transformation for example Ruminococcus, Bacteroides, and Butyrivibrio, were either those known to be actively involved in the enzymatic breakdown of feed, or examples of common rumen bacteria (Flint, 1997; Wong et al., 2003). Therefore, although many lysogenic phages infecting S. bovis were identified (Tarakanov, 1974, 1976; Iverson and Millis, 1976b), only a few phages were genetically characterized (Styriak et al., 2000, 2005). Collectively, however, the numerous isolations of lysogenic phages from the rumen, as well as experiments to chemically induce lysogenic phages from rumen bacteria (Klieve et al., 1989) established, in the absence of DNA sequencing technologies, that a high proportion of rumen bacteria harbor viable lysogenic phages within their genomes.
As DNA sequencing technologies have improved, the whole genome sequencing of bacteria has become more cost-effective and convenient (Didelot et al., 2012). In addition, as bioinformatics methods for genome assembly have become more refined, the detection of integrated prophages has become increasingly possible (Roux et al., 2015; Arndt et al., 2016). Many bacterial and archaeal genomes have been shown to contain fragments or remnants of integrated prophages (Krupovic et al., 2011). For prophage sequences to be considered “intact” (Zhou et al., 2011) and representative of a potentially viable phage, they must contain a full complement of phage genes. These genes are usually clustered into structural and function-related modules including DNA replication and transcriptional regulation, head and tail proteins, DNA and particle packaging, translocation and host cell lysis (Brussow et al., 2004; Roux et al., 2015). While culture-based assessments are required to verify prophage viability, the detection of viral sequences in microbial genome sequences has revolutionized the rate at which novel viruses can be detected.
To date, intact prophages have been detected in the genome sequences of many genera of rumen bacteria (Berg Miller et al., 2012; Gilbert and Klieve, 2015) although the prophage genomes and their homology to known phage sequences, have not been described in detail. Recently prophage sequences detected in the bacterial genome sequences of the ovine rumen isolates Streptococcus equinus 2B (∼42 kb, designated φSb2Bpro1) and Bacteroides ruminicola ss brevis AR29 (∼35 kb, designated φB2bAR29pro1) were described (Gilbert et al., 2017). The latter prophage had been previously examined, with some genetic and biological properties determined, this prophage formed tailed phage particles with a Siphovirus morphology (Klieve et al., 1989; Seet, 2005).
Integrated viral sequences have also been detected in methanogenic archaea isolated from the rumen, including a ∼40 kb prophage of Methanobrevibacter ruminantium M1 and a 37 kb prophage of Methanobacterium formicicum BRM9 (Attwood et al., 2008; Kelly et al., 2014), although the ability of these prophages to produce intact viral particles has not been reported. The recent international collaborative genome sequencing project, the Hungate 1000 project (Seshadri et al., 2018), analyzed 410 cultured bacterial and archaeal genomes together with their reference genomes and identified virus genes, categorizing these genes according to Pfam numbers (for example pfam03354 Phage Terminase, pfam04860 Phage portal protein and pfam05105 Bacteriophage holin family) and compared them to virus genes found in human intestinal isolates. In this way the study showed that the prophage genes identified differed between the microbes of the herbivore gut and those of the human gut.
Recently whole genome sequences of lytic phages infecting rumen bacterial isolates of Streptococcus, Bacteroides, and Ruminococcus were reported (Gilbert et al., 2017). These phages were of Siphovirus (phages φBrb01 and φBrb02 infecting B. ruminicola and phage φSb01 infecting S. equinus) and Podovirus morphology (phages φRa02 and φRa04 infecting R. albus). Of these phage genomes, the φSb01 sequence had the most genetic homology to other Streptococcus phages isolated from non-rumen environments, enabling a more comprehensive annotation of genome and identification of functional modules. In contrast, the Podoviruses φRa02 and φRa04 were more distantly related to known phages, with the few functional genes for which identity could be predicted, all related to genes previously shown to be highly conserved in Podovirus genomes (Iranzo et al., 2016), such as head-tail connector proteins, DNA polymerase and Podovirus encapsidation proteins. Despite this lack of gene annotation, the identified highly conserved phage genes (head-tail connector protein for Podoviridae and phage terminase large subunit (terL) for Siphoviridae) could be employed in phylogenetic analysis to enable the identification of closely related phages. On this basis, φRa02 and φRa04 which infect a Ruminococcus host, classified within with the gram positive bacterial phylum Firmicutes, were found to be more highly related to Podoviruses infecting other genera of Firmicutes (for example Bacillus, Clostridium, and Streptococcus), than Podoviruses infecting the gram negative phylum Proteobacteria (for example Yersinia, Pseudomonas, and Vibrio) (Gilbert et al., 2017).
Obtaining the complete genome sequences for rumen virus isolates has many benefits, providing insights into virus:host interactions and facilitating the discovery of novel viral proteins. Most significantly, increasing the number and types of rumen-specific viral genome sequences will greatly enhance the accuracy of viral classification and bioinformatic analysis of viral metagenomics datasets, providing reference sequences and novel viral genes to which meaningful sequence homology can be conferred. Experimental studies with viral isolates is anticipated to remain vital for the determination of important biological properties, such as the determination of virus:host specificity (host range); the characterization of novel viral proteins; determining immunological interactions; and ascertaining viral particle transfer and survival in the gastrointestinal tract and environment.
Metaproteomics is the analysis of the entire protein content of a given microbial community at a specific time point (Wilmes and Bond, 2004). Like many other ‘omic’ technologies, metaproteomics has experienced rapid development in the past 10 years (Kunath et al., 2019), and is quickly becoming commonplace for the analyses of complex microbiome samples (Hagen et al., 2017; Borton et al., 2018; Heyer et al., 2019). Within the rumen microbiome, metaproteomics is a relatively new field, with only sparse datasets generated in recent years (Deusch et al., 2017; Snelling and Wallace, 2017; Hart et al., 2018; Solden et al., 2018). However, multi-omic approaches that combine both metagenome and metaproteome data are now enabling genome-centric resolution that links active metabolic functions to explicit microbial populations in the rumen (Naas et al., 2018; Solden et al., 2018).
Using functional meta-omics to characterize viral taxonomy and function has quickly shown the impact viruses have on ecosystem processes (Emerson et al., 2018). Metaproteomic analysis of viral communities has been used in bioreactors (Heyer et al., 2019) and ocean communities (Brum et al., 2016), which highlighted the presence of newly annotated functions and indications of phage–host interactions causing cell infection. Within the rumen of Alaskan moose, Solden et al. (2018) used a combined meta-omic approach to identify 1,907 viral metagenomics contigs, which clustered into 810 viral populations. This lead to the detection of 148 viral genera, including 35 previously detected from the cow rumen, although the vast majority (75%) were determined to represent previously unknown viral genera.
The moose metaproteome detected a total of 64 viral proteins from 53 different viral contigs, with most proteins (80%) having no known functions, whereas the remainder were largely structural proteins, such as capsids. Against expectations, very few auxiliary metabolic genes (Emerson et al., 2018) were detected, contrary to previous rumen virus findings (Anderson et al., 2017). Examinations of virus–host interaction dynamics, predicted microbial genome hosts, including direct associations between viruses and some of the most active carbohydyrate-degrading and sugar-fermenting populations sampled from the moose rumen (Solden et al., 2018). In particular, numerically dominant Prevotella-affiliated populations that play critical roles in hemicellulose degradation were predicted to be infected, highlighting that rumen viruses are likely to be affecting carbon cycling in a top–down manner, whereby changes in the structure and function of the primary fiber degraders may possibly reverberate through the lower trophic levels of the rumen microbiome (Solden et al., 2018). Much more targeted analyses are required to see if such an effect is indeed the case, or if other versatile populations with similar saccharolytic machinery are capable of either supplementing or contributing to, any newly presented niches in such a scenario.
The study of viral proteins often involves the over-production of the protein of interest in vitro. In this way sufficient quantities of protein can be generated to enable analysis using protein chemistry techniques for determination of biological properties. Understanding the biological properties of viral-encoded proteins can lead to an improved understanding of how viruses specifically infect, replicate and interact with host cells and viral proteins have been extensively studied for some well-known, historically important phages for example, the Escherichia coli phages lambda (Casjens and Hendrix, 2015) and T4 (Miller et al., 2003); and the Phi29 phages infecting Bacillus (Meijer et al., 2001).
The use of viral proteins as new antimicrobials is an area of growing interest, providing an alternative approach to phage therapy, which was traditionally based on the use of whole phage particles to reduce specific microbes (reviewed by Fenton et al., 2010; Abedon, 2012; Shen et al., 2012). Traditionally the use of antibiotics in agriculture was preferred over phage-based therapies for the control of problematic microbes and improving feed efficiency (Russell and Houlihan, 2003). In recent years however, there has been increasing community and regulatory pressure to reduce antibiotic use in agriculture, as well as a drive to reduce enteric methane emissions from ruminant livestock (Hristov et al., 2013; Cameron and McAllister, 2016). One of the antibiotic-free approaches to reduce enteric methane emission harnessed the potential of phage-encoded enzymes to specifically target and lyse rumen methanogens (Klieve and Hegarty, 1999; Leahy et al., 2013). Previously phage-encoded enzymes found in methanogen genomes had only been applied to increase the permeability of methanogen cell walls in order increase the penetration of microscopy stains (Nakamura et al., 2006). Genome sequencing of the rumen methanogen, M. ruminantium M1, however, revealed the presence of a prophage gene related to those usually responsible for hydrolyzing the pseudomurein cell walls of methanogens: the endoisopeptidase peiR (Attwood et al., 2008) and the potential use of this enzyme was recognized (Leahy et al., 2010).
Recently the in vitro expression of peiR was reported (Altermann et al., 2018). In this study, initial experiments were conducted to produce this protein using traditional gene cloning, transformation, protein expression in a commercially-available E. coli expression strain (Schofield et al., 2015), with the final purified protein employed in lytic activity assays. To further improve the stability and enable practical delivery of the enzyme, an alternative protein expression strategy was adopted. This approach involved the peiR gene being fused to the polyhydroxyalkanoate (PHA) synthase gene which was then expressed in E. coli to create PHA bionanoparticles with the lytic enzyme being displayed on the outer surface of the bionanoparticle (Altermann et al., 2018). Using this combination of techniques, sufficient quantities were produced to allow testing for cell-wall degrading effects against several strains of Methanobrevibacter, which are usually the most highly abundant methanogen genera found in the rumen (St-Pierre and Wright, 2012), and other methanogenic genera (Methanobacterium, Methanosphaera, Methanospirillum, and Methanonosarcina). This study therefore, successfully demonstrated for the first time how viral genes, identified by whole genome sequencing of rumen microbes, can be successfully produced using modern protein expression technologies and developed into a product for activity testing. This study also showed how protein expression techniques are integral to the development of new enzybiotics approaches for the modulation of rumen microbial populations.
Technologies for studying viruses in environmental samples are constantly being developed, and older technologies are being revisited. New technologies, which could be applied in the context of the rumen, include those being developed to address long-standing biological questions. For example, it is currently difficult to link the extensive populations of viruses identified in viral metagenomes, to their respective microbial hosts (Koskella, 2019). In addition, an older technique, being revisited in the context of the rumen, is the use of Chromatography for size-based virus particle fractionation.
Techniques are evolving that enable us to link viruses to their potential hosts using metagenomic information alone, such as CRISPR-spacer regions (Berg Miller et al., 2012). Hi-C or 3C is another promising technique developed to cross-link DNA in genomes that is distant on the DNA strand, but comes into close physical proximity, due to folding and chromosome architecture (Dekker et al., 2002; Lajoie et al., 2015). This method has the potential to cross-link viral and host DNA during infection to identify viral hosts prior to lysis (Marbouty et al., 2017). Single-cell viral tagging is a further tool that has been tested and proven to work on viruses from the human gut and involves fluorescently labeling phage particles, allowing these to attach to hosts, then sorting and sequencing these pairs (Willner and Hugenholtz, 2013; Džunková et al., 2019).
Chromatography is a traditional chemistry technique for the separation of mixtures, and has the potential be applied for the purification of viruses from rumen samples. Currently the use of chromatography in rumen research has been limited to metabolomic and proteomic studies with only a few publications combining chromatography and virus protein research (Schofield et al., 2015; Altermann et al., 2018). Given the proteinaceous nature of virus structures, particularly phages, implementing protein chromatography methods to separate or isolate whole phages directly should be possible. Despite the size difference and more complicated morphology of phages compared to proteins, it is expected that phages would act like proteins in chromatography systems (Oslizlo et al., 2011). Indeed, chromatography for viral purification is not novel, with literature dating back to 1957, where the coliphages T1, T2r and T2r+ were purified from host lysates using ECTEOLA cellulose ion exchange chromatography (Creaser and Taussig, 1957). This emerging technology for separating bacteriophages from environmental samples such as rumen fluid, involves the use of coil column separation and high performance counter current chromatography (HPCCC) (Hu and Pan, 2012; Berthod and Faure, 2015); utilizing the size differences of phages and combining this with the centrifugal forces applied in the HPCCC to separate viruses (Friedersdorff et al., 2018). Although this approach is in its infancy, it is anticipated that depending on the resolution of the CCC technique used, viral fractions based on particle size may be obtained from rumen fluid, enabling the study of less-understood viral morphotypes, for example, viral families which form small, non-tailed icosahedral particles.
Viruses play a pivotal role in microbial ecology and evolution, contributing to global nutrient cycles, pathogenicity and antimicrobial resistance, and influence animal host immune systems (Salmond and Fineran, 2015). The roles viruses play in aquatic, terrestrial and animal-associated environments are vitally important and viruses, particularly phages, have been acknowledged as being responsible for significant bacterial mortality (Clokie et al., 2011). Our understanding of rumen virus ecology is currently inadequate, we must therefore learn from studies undertaken for other environments, where viruses have been shown to affect microbial populations by maintaining microbial diversity; cycling key nutrients; acting as mobile genetic elements to facilitate genetic exchange; infecting and replicating in a host-specific manner (limited host range); interacting with microbial biofilms (Figure 2) and the immune systems of animal hosts.
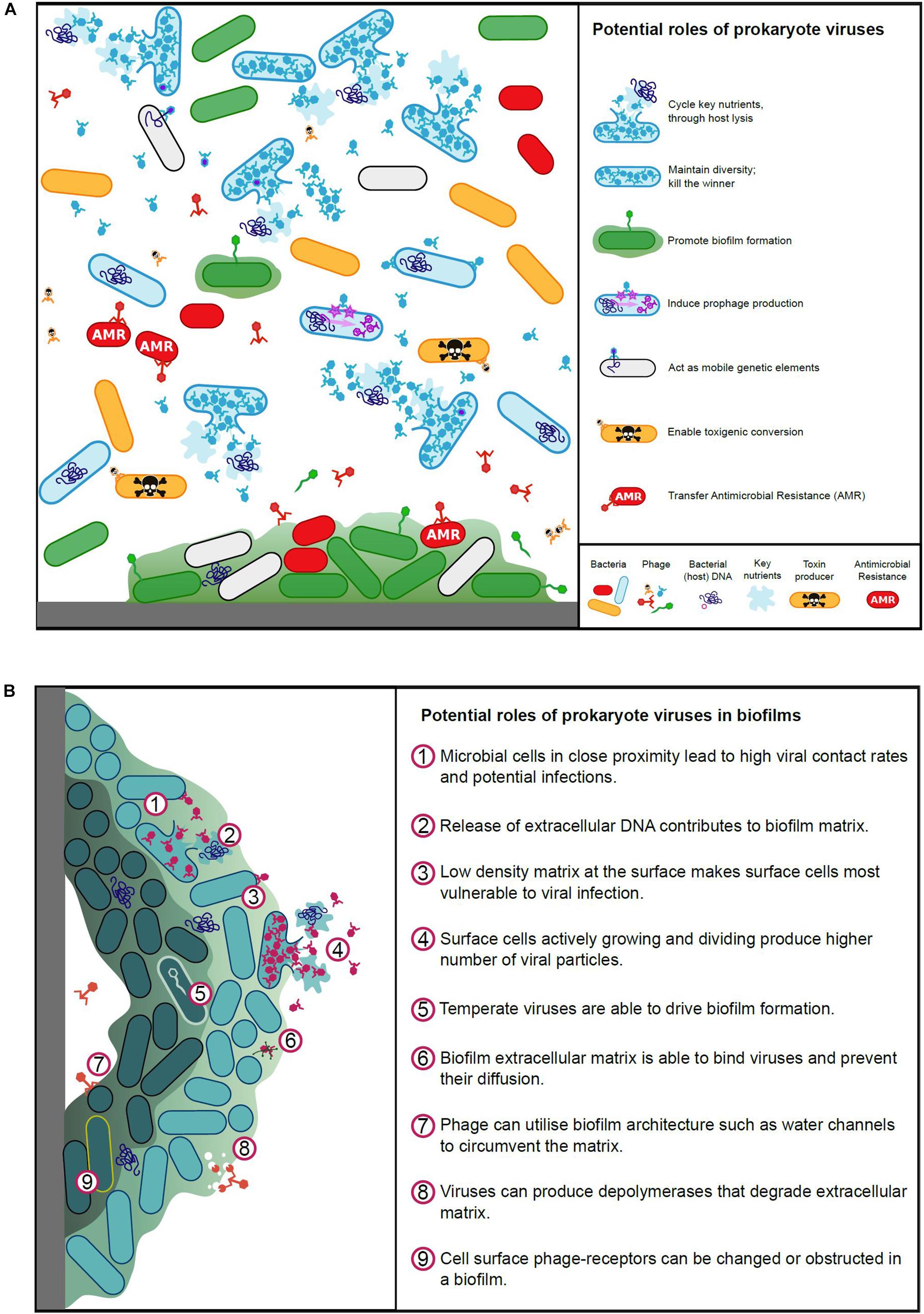
Figure 2. (A) Potential roles of prokaryote viruses in microbial ecology and evolution. (B) Potential roles of prokaryote viruses in biofilms.
Viruses limit the growth of bacteria in aquatic environments, accounting for an estimated 10–80% of bacterial mortality (reviewed by Weinbauer, 2004). This relentless pressure on bacterial communities shapes their diversity, community structure, function, and temporal dynamics (Letarov and Kulikov, 2009). In the marine environment, the model produced by Thingstad (2000) predicts that viral predation modulates microbial populations via a microbial feedback loop described as the “kill the winner” strategy (i.e., targeting and reducing the size of the most abundant microbial host species). This allows viruses and bacteria to coexist in an equilibrium, thus ensuring microbial diversity. Furthermore, mesocosm experiments by Sandaa et al. (2009) support this model, showing that viruses act as a regulating force, preventing fast growing bacteria from outcompeting slower growers. Viruses have also been shown to control algal blooms (phytoplankton and/or cyanobacteria) occurring in both marine and freshwater environments (reviewed by Weinbauer, 2004).
Within gut-associated microbial communities, viruses are also likely to play a role in modulating microbial populations using predation and microbial feedback loops, particularly during the stages of microbial succession occurring in early life development and at times of microbial community disturbance, e.g., dietary change, dysbiosis and antibiotic use (Lozupone et al., 2012). For example, the “kill the winner” strategy has been observed in the horse gut, where E. coli and coliphages co-exist (Golomidova et al., 2007). However, in contrast to the microbial communities found in aquatic microbial ecosystems, the near-theoretical-maximum cell densities combined with the animal immune system lead to temperate phages predominating in animal guts, with the immune system also helping to prevent the over-proliferation of individual gut microbes. In the human gut, phage therapy has been shown to eliminate bacterial pathogens, rather than regulating their numbers, due to the added selective pressure of the immune system (reviewed by Sulakvelidze, 2005).
Microbial hosts have evolved numerous defense mechanisms to prevent phages from killing the winner. These mechanisms target every stage of infection from receptor binding to phage assembly. These include: (1) modification of phage receptors or hosts only display them during part of life cycle; (2) superinfection exclusion, often associated with lysogenic phages to protect their hosts; (3) bacteriophage exclusion (BREX) is a similar host-encoded defense system that prevents phage DNA replication; (4) restriction modification enzymes to degrade foreign DNA, defense island system associated with restriction–modification (DISARM) is more complex, they are found in bacteria and archaea and restricts phage DNA; (5) methylation and CRISPR/Cas elements block phage replication (6), abortive infection/programmed cell death (Abi). In addition bacteria can hijack phages to utilize them as gene transfer agents (GTA) and phage-inducible chromosomal islands (PICI) which repackage host DNA into phage structures (for review see Rostøl and Marraffini, 2019).
For each host defense system microbes have evolved, however, viruses appear to have developed a countermeasure, such as the anti-restriction, anti-CRISPR and anti-toxin systems (Dolgin, 2019; Rollie et al., 2020). While these viral countermeasure systems have been largely described for genera not normally found in the rumen, for example, Pseudomonas and Mycobacterium (Dedrick et al., 2017; Landsberger et al., 2018). Given the extensive microbial and viral diversity observed in the rumen, and the ecological and evolutionary pressures rumen microbial communities experience, these viral countermeasures are likely to exist and play a role in enabling viral replication and persistence in the rumen.
In aquatic systems viruses have been shown to impact on microbial mortality, causing episodes of microbial lysis, contributing to the recycling of nutrients (Fuhrman and Noble, 1995; Steward et al., 1996; Weinbauer and Höfle, 1998). Viral lysis results in the release of nutrients in the form of dissolved organic matter (DOM) (Shibata et al., 1997). In low nutrient aquatic systems DOM is an essential source of carbon and growth limiting nutrients that is quickly reabsorbed by the microbial community (Bratbak et al., 1990; Proctor and Fuhrman, 1990).
The gut (and rumen) represent much more productive systems than the open ocean, with microbial densities the highest of any studied environment, reaching ∼1010–1011 mL–1 in the rumen (Russell and Hespell, 1981). Due to this high microbial density, these microbes are in constant competition for nutrients and may be subject to control mechanisms, such as virus predation, that maintain stability and cycle nutrients (Lozupone et al., 2012). Due to the microbial competition, nutrients such as iron and vitamin B12 (cobalamin), are also limiting to microbial growth in the human gut (Deriu et al., 2013; Degnan et al., 2014).
Viruses infecting microbial populations in various environments have been shown to act as mobile genetic elements, acting as important vectors of horizontal gene transfer and guiding the evolution and diversification of bacteria (Wommack and Colwell, 2000; Koskella and Brockhurst, 2014). Through either generalized transduction (packaging only host bacterial DNA into virus particles) or specialized transduction (mis-packaging host DNA along with virus DNA) viruses contribute to bacterial genetic variability, introducing changes to the genetic make-up of otherwise genetically homogeneous bacterial strains, for example E. coli strains causing urinary tract and bloodstream infections (Frost et al., 2005; Ben Zakour et al., 2016). Conversely, the pangenome of dsDNA viruses and prophages infecting a broad range of hosts shows an extensive network of genetic similarity for a subset of viruses, suggestive of a common virus genetic pool and modules of function-related viral genes, resulting in mosaic virus genomes (Hendrix et al., 1999).
Virus genetic exchange from the gut microbiome may have a direct impact on the animal, as well as their bacterial hosts, and virus integrases have been shown to mediate chromosomal integration in human cells (Groth et al., 2000). Lysogenic phages and prophages can influence host phenotype through phage-encoded gene expression, a process termed lysogenic conversion (Brabban et al., 2005). Qin et al. (2010) found that prophages exert a strong influence on the functioning of the human gut microbiome and contribute an astonishing 5% of the conserved microbiome functions observed. Viral DNA can also be incorporated into the genomes of prokaryotes via CRISPR elements: adaptive immune elements that protect against virus infection through targeted nucleases (Barrangou et al., 2007; Koonin et al., 2017). The host-encoded CRISPR spacers can also recombine with incoming virus DNA to perform specialized transduction (Varble et al., 2019).
One of the most studied attributes of viral genetic exchange is their contribution to the development of antibiotic resistance. Antibiotic resistance is a natural bacterial survival strategy, however, the pervasive use of antibiotics in farming and human health-care has led to the development of multidrug-resistant bacterial pathogens (Balcazar, 2014). Antibiotic resistance genes (ARGs) can be subject to virus-mediated horizontal gene transfer (Parsley et al., 2010). Both gram-positive (Ubukata et al., 1975; Mazaheri Nezhad Fard et al., 2011; Goh et al., 2013) and gram-negative (Blahova et al., 1992; Willi et al., 1997; Oliver et al., 2005) bacteria have been shown to carry virus-encoded ARGs. The diversity of ARGs is lower in pathogenic bacteria and environments contaminated with anthropogenic antibiotics, than in unpolluted environments (Muniesa et al., 2013). It has been postulated by Muniesa et al. (2013) that viruses could allow dissemination of these rare ARGs from the natural environment to the human microbiome. A few studies have begun to examine the prevalence and spread of virus-ARGs. In sewage waters, ARGs were detected in virus DNA (Muniesa et al., 2004; Marti et al., 2014). Virus-encoded putative ARGs are also prevalent in healthy human feces, being present in 77% of samples (Minot et al., 2011; Quirós et al., 2014) and have a high carriage rate of in cattle, pigs and poultry (Pereira et al., 1997; Colomer-Lluch et al., 2011).
However, ARGs identified by bioinformatic searches of viral metagenomes of the human and mouse gut may overestimate ARG abundance, as during experimental testing only a few of the predicted ARGs could confer antibiotic resistance in vitro (Enault et al., 2017). Experimental testing is vital to determine the functionality of virus-encoded putative ARGs with low sequence identity to bacterial genes. For example, experimental testing has been used to establish the function of virus-encoded β-lactamases identified in the genomes of human gut viruses (Ogilvie et al., 2013). Interestingly, a high proportion of ARGs are carried on plasmids (Bennett, 2008), meaning that even if viral integration-mediated horizontal gene transfer of ARGs is irrelevant, viruses could still facilitate the spread of ARGs, through the release of intact bacterial plasmids following virus-mediated bacterial lysis (Keen et al., 2017).
Another well-studied example of virus-mediated genetic transfer between bacterial genera, is the movement of virus-encoded toxin genes and the evolution of toxin-producing bacteria. A variety of virus-mediated extracellular toxin genes have been identified (listed in Table 3). Conversely, bacteria which develop resistance to phages, rather than allowing the integration of prophage DNA, may become less virulent as a consequence (Leon and Bastias, 2015). In addition to toxicity genes, a number of phage-encoded accessory genes are directly related to pathogenicity and can be expressed alongside toxins. Phage-encoded genes can span an even wider range of functions that increase the fitness of their bacterial hosts including modification of host cell antigens and antibiotic resistance genes (Table 3).
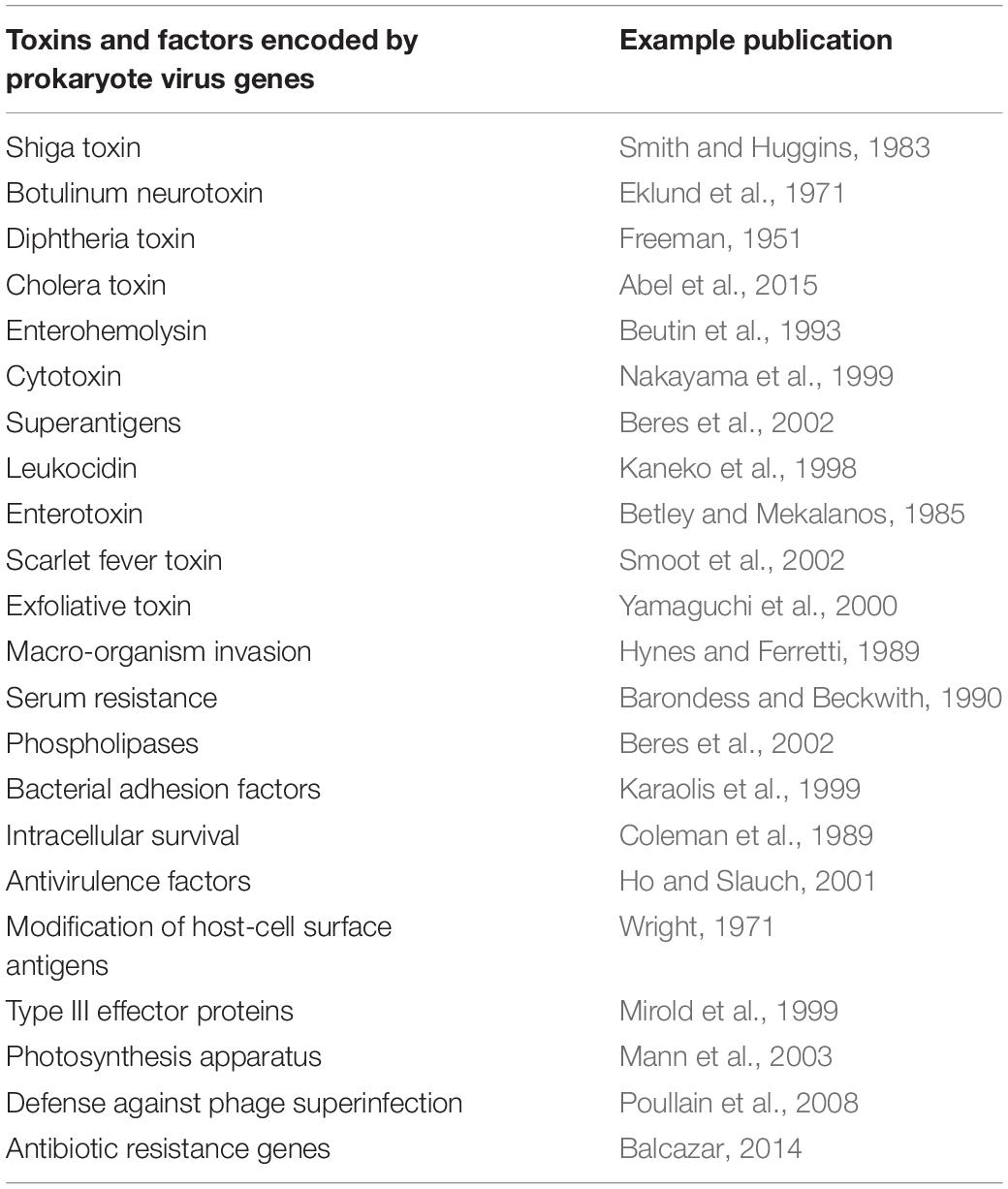
Table 3. Examples of genes and other factors conveyed by prokaryote viruses which enhance the virulence, pathogenicity and functional capacity of the infected host.
In recent years, studies of the human and mouse gastrointestinal tract have provided insights into the establishment of viral populations within the developing gut, and of the interactions occurring between the viruses infecting commensal intestinal microbes and the animal itself. Viruses are rapidly recruited to the human gut within the first few months of life, leading to a diverse viral population dominated by viruses infecting the microbes of the gut, particularly Caudovirales, which then declines in diversity over the following 2 years (Lim et al., 2016). This commensal viral community is an important component of the gut microbiome and plays a role in intestinal inflammation, stimulating a low-level immune response without causing clinical symptoms (Norman et al., 2014).
Growing evidence suggests that viruses have a role in immune defense within the gut mucosa, where they can influence the innate and adaptive immune system (Barr et al., 2015). A number of viruses possess adhesin proteins, with immunoglobulin-like domains, located on the capsid, collar whiskers or tail shaft that are not involved in host attachment, but can facilitate virus attachment to the gut mucosa (Fraser et al., 2007; Barr et al., 2013). Hypervariable regions collocated in either the virus tail fibers and/or immunoglobulin-like genes might help these viruses to evade the adaptive immune system of the gut (Doulatov et al., 2004; Minot et al., 2012). These features allow viruses to attach and persist in the gut mucosa at high concentrations, where they can outnumber bacteria (Barr et al., 2013). Silveira and Rohwer (2016) proposed that the survival strategy employed by virus changes within the mucosa from a lysogenic strategy or “piggyback-the-winner” at the top of the mucosa where hosts are plentiful, to a lytic or “kill-the-winner” strategy deeper in the mucosal layer, where bacterial hosts are scarce.
Although the immune system clears the vast majority of viruses effectively, phages have been shown to be actively transported by transcytosis out of the gut across epithelial cells (Nguyen et al., 2017). This probably accounts for the discovery of phages, delivered to the gastrointestinal tract of mammals for therapy, being found in the blood stream and systemic tissue, where they can trigger both the innate and adaptive immune system (Wolochow et al., 1966; Wolf et al., 1981; Duerr et al., 2004). The translocation of viruses is not always equal among viruses or individuals (Bruttin and Brussow, 2005; Górski et al., 2006).
Biofilms are a community of microorganisms aggregated together within a self-produced matrix (Zobell and Anderson, 1936; Caron, 1987). Biofilms are ubiquitous in nature and most bacteria as well as a number of fungal and yeast species, have the potential to form a biofilm. The biofilms that develop are often poly-microbial and inter-kingdom communities (Swidsinski et al., 2013; Smith et al., 2016; Raghupathi et al., 2018). Within biofilms viruses are often viewed as a predator of bacteria and can therefore offer an alternative to antimicrobial agents against recalcitrant biofilms. The efficacy of viruses against in vitro monospecies biofilms has been shown, e.g., in E. coli (Doolittle et al., 1995). Not only are viruses are able to prevent biofilm formation (Hibma et al., 1997) and disrupt mature biofilms (Sillankorva et al., 2004; Soni and Nannapaneni, 2010), but they demonstrate synergy with antimicrobial treatments (Sharma et al., 2005; Bedi et al., 2009). The anti-biofilm activity results from not only lysing cells, but also degrading the matrix using depolymerases (Hughes et al., 1998).
Biofilms represent a rich resource for viral production, but the physical nature of biofilms limits the ability of viruses to exploit it (Figure 2B). The biofilm environment brings cells close together and structural elements, such as water channels, allow virus particles rapid access to bacterial cells (Wood et al., 2000; Donlan, 2009). The varying metabolic states within a biofilm, along with the matrix itself however, can pose challenges for viral infection (Doolittle et al., 1996; Hanlon, 2007). To combat this, viruses often produce disruptive enzymes to enable access within the matrix (Hughes et al., 1998; Hanlon et al., 2001). Bacteria at the periphery of a biofilm are most vulnerable to virus adsorption, whilst the dense structure at the center of the biofilm can entrap viruses (Abedon, 2016; Bull et al., 2018; Vidakovic et al., 2018). Hydrophobicity and electrostatic charges are important for viral binding, these can be disrupted within biofilms, and furthermore the aggregation of bacteria may obstruct virus surface receptors (Nordstrom and Forsgren, 1974; Sutherland et al., 2004).
To add to this complexity, viruses has been shown to promote biofilm formation for a number of bacterial genera, such as Bacillus and Actinomyces, via spontaneous induction of prophage and release of extracellular DNA, which contributes to the extracellular matrix (Schuch and Fischetti, 2009; Carrolo et al., 2010; Wang et al., 2010; Shen et al., 2018). Davies et al. (2016) have also demonstrated that temperate viruses can drive the evolution and adaptation of cells within a biofilm through insertional inactivation of genes. Furthermore, viruses may enhance biofilms through promoting host phenotypic variants and viral particles can physically support the biofilm (Rice et al., 2009). As in any natural system, viruses and biofilms have an ill-defined and complex relationship, with the two entities, being simultaneously at war and peace.
The role and impact of viruses within the rumen microbial ecosystem has been an area of interest for many years, however, relatively few studies have been conducted to directly address this. Several important aspects of rumen virus populations and the biology of individual viral isolates have been established and on the basis of these findings, the effects of viral activity in the rumen can be inferred (Table 4). Tailed viruses of prokaryotes, classified within the viral order Caudovirales, have been frequently shown to dominate the rumen viral community. These viruses are always present in high numbers, actively replicating in the dense bacterial populations of the rumen and consequently these prokaryote viruses (bacteriophages) are the most studied and best understood.

Table 4. Roles and characteristics of virus populations within the rumen, as suggested by previous studies of rumen virus populations, rumen microbes (bacteria and archaea) and viruses isolated from the rumen.
Historically, the primary impact of viral populations in the rumen has been their contribution to bacterial cell lysis (Jarvis, 1968; Nolan and Leng, 1972; Swain et al., 1996a). The final stages of viral replication results in the accumulation of intact viral particles inside the cells and fragmentation (lysis) of the cell membrane, causing cell death and the release of progeny viral particles. Lysis also releases remnants of the microbial cell, including cell wall fragments as well as proteins and nucleic acids, which may then be taken up and utilized by other rumen microbes, a process often described as intra-ruminal recycling (Firkins et al., 1992; Hartinger et al., 2018) (Figure 2A). The recycling of nutrients amongst microbes was initially regarded as being a negative consequence of viral activity, limiting the amount of microbial protein carried through to the lower intestine where it can be absorbed by the animal (Leng and Nolan, 1984). A more recent study using proteomics (Solden et al., 2018), suggested that virus-mediated cell lysis also releases intact microbial enzymes, including those involved in carbohydrate breakdown, which in turn facilitates and enhances extracellular feed breakdown within the rumen. This study re-enforces the initial hypotheses that virus populations significantly impact on microbial lysis and nutrient recycling within the rumen, but in contrast, has also provided an alternative, more positive perspective on the long standing question of whether viral-mediated microbial lysis is detrimental or beneficial to rumen function.
The second major impact of rumen viral populations is that they modulate microbial populations, notably dominant bacterial populations, causing viral blooms. These viral blooms were first observed in studies monitoring phage population numbers over a 24 h period in once-a-day fed sheep (Swain et al., 1996a). As virus particles, even those sourced from the rumen, are degraded by rumen fluid (Orpin and Munn, 1974; Tarakanov, 1976) there must be constant viral replication to replenish and sustain viral numbers. Blooms of lytic activity may reflect top–down or “kill the winner” activity, with dominant microbial populations being targets of viral predation, however this has never been shown experimentally for the rumen. Given the functional redundancy of rumen microbial populations, whereby multiple microbial species can perform the same function (Henderson et al., 2015; Weimer, 2015), it can be anticipated that blooms of lytic phage activity are unlikely to impact on overall rumen efficiency, with the niche made available by the selective removal of dominant genera, being rapidly filled by a bacterial population with similar fermentative capacity. This turn-over of microbial populations, however, represents the way viruses can drive and maintain microbial diversity in the rumen.
The third proposed impact of rumen viral populations is their capacity to act as mobile genetic elements (MGE). In this way, viruses can pick up and transfer genetic material between the microbes which they infect and replicate in. The most notable examples include transducing phages, which readily package the host DNA into phage particles (Brussow et al., 2004), and lysogenic phages which form a stable heritable state with the host cell (Weinbauer, 2004; Touchon et al., 2017). Of these, lysogenic phages infecting rumen bacteria and archaea have been most frequently reported (see section “Culture-Based Viral Isolations and Genome Sequencing”). It has also been suggested in a bioinformatics study that lysogenic phages can transfer auxiliary metabolic genes (AMGs) between rumen bacteria, resulting in metabolic re-programming (Anderson et al., 2017). In other microbial environments virus-mediated genetic transfer also contributes to the development of antimicrobial resistance and toxigenic conversion (see section “Virus-Mediated Genetic Exchange”).
Although several rumen genera have been shown to produce antimicrobial proteins, for example bacteriocins produced by Streptococcus and Ruminococcus (Chen and Weimer, 2001; Azevedo et al., 2015), there have been no investigations into the extent of toxin production by rumen microbes and the contribution of viruses to toxigenic conversion is not understood. Rumen microbes have been shown to demonstrate antimicrobial resistance (Mantovani and Russell, 2001; Benahmed et al., 2014) and the use of antibiotics in feed supplements can contribute to the development of this resistance (Russell and Houlihan, 2003; Cameron and McAllister, 2016). The contribution of viruses to the development of antimicrobial resistance in the rumen has not been investigated. Most of the more abundant rumen microbial genera are not recognized as important zoonotic pathogens and if virus-mediated toxigenic conversion does occur, given the lack of published data, it appears to have had little impact on ruminant or human health. Instead the lower intestine of ruminants represents more of a public health concern, because the intestine can harbor zoonotic and highly pathogenic E. coli strains, particularly strains that have undergone toxigenic conversion following infection by Shiga-toxigenic phages (Brussow et al., 2004; Bonanno et al., 2016).
The other potential impacts of viruses on microbial populations are in accordance with observations of viral populations in other environments, but have not been extensively investigated. These impacts include: (1) Virus activity facilitating the formation of biofilms, with bacteria changing their physical growth habit, forming clumps and layers of extracellular polysaccharide in response to phage infection and the development of phage resistance (Klieve and Bauchop, 1991). (2) The ruminant gastrointestinal tract may never be free of viruses as commensal microbes and their associated viruses may simultaneously colonize the ruminant gut during early life development. In addition, viruses may be readily introduced into the rumen with between-animal transfer of viruses occurring via aerosols and saliva from close contact (e.g., maternal transfer, especially pre-weaning) and from shared feed and water sources, as well as housing and farm infrastructure (Figure 3). (3) Viruses infecting rumen bacteria may interact with the cells of the rumen wall, eliciting an immune response. For all these potential roles, further investigations and technological advances are required, e.g., to link specific virus and host populations, to provide both experimental evidence and new insights, into how rumen viral populations contribute to the rumen microbiome, fermentation efficiency and overall ruminant growth and condition.
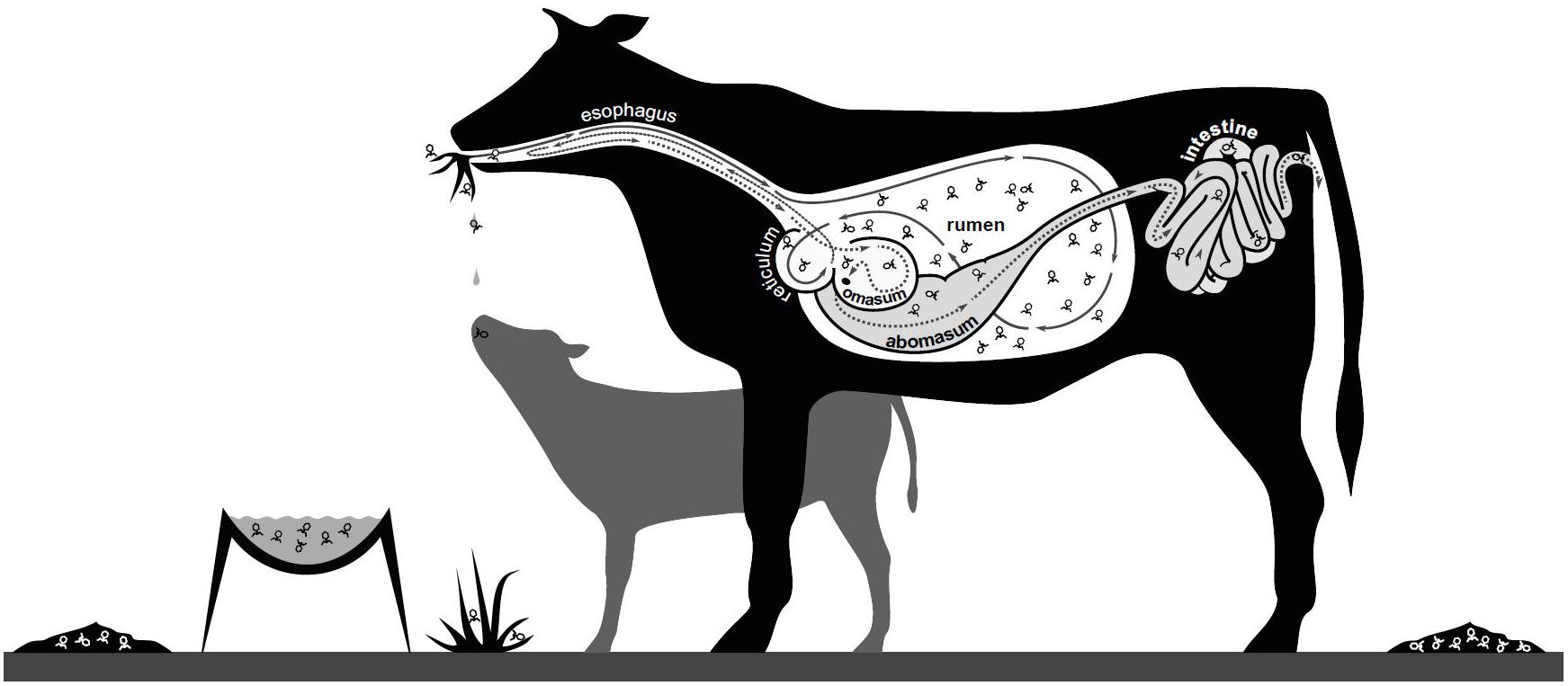
Figure 3. Potential mechanisms for spread of rumen viruses. Viruses may be spread by aerosols, saliva and fecal shedding, with viral particles moving between animals (maternal and within herd interactions) and their environment (water, feed, housing and infrastructure).
Our ability to explore microbial communities has expanded with our ability to sequence microbes quickly and cheaply from any given environment. This presents an amazing opportunity to investigate viruses without the requirement to grow them, or their host. Techniques are evolving that enable us to link viruses to their potential hosts using metagenomic information alone; such as matching CRISPR-spacer regions in hosts to homologous sequences in their viruses which also provides clues to the frequency of viral interactions (Berg Miller et al., 2012; Sanguino et al., 2015; Edwards et al., 2016), or, through linking physically close genomic information by proximity ligation, such as infected host genomes and viral progeny prior to release (Marbouty et al., 2017). Our ability to interpret metagenomic sequence data gives us a tantalizing insight into virus-host community dynamics, but a deeper understanding of functionality is hindered by a lack of basic knowledge of the roles and functions of the organisms in the rumen. There is a dearth of basic knowledge of rumen viruses and that is critical to understanding their function; such as prophage induction, auxiliary genes and functional roles, lytic virus virulence, replication rate, host range and burst size. The future of rumen viral work will need to focus on two areas: answering outstanding questions (Table 5) and developing practical applications.

Table 5. Future rumen virus research: outstanding questions for further investigation.
To address the outstanding questions, rumen viral research will need to be carried out on three fronts, standard laboratory based in vitro work, coupled with in vivo rumen studies to inform in silico computer modeling which will generate testable hypotheses. Advances in sequencing and proteomics provide vast quantities of data that enable us to characterize, understand and model the role of rumen viruses. Basic knowledge of the rumen microbiome as a whole, including viruses, is essential to allow us to explore the rumen microbiome through modeling (Louca and Doebeli, 2017). Modeling of whole microbial communities opens the possibility to understand community functionality and dynamics (Widder et al., 2016). Rumen modeling has largely concentrated on the impacts of nutrition on emissions and efficiency rather than on the microbes and their functions (Ellis et al., 2009; Mills et al., 2014; Gregorini et al., 2015). A recent study examined a simple rumen microbiome consisting of representatives of three microbial groups: Bacteroidetes, Firmicutes, and Archaea, plus their associated viruses (Islam et al., 2019). This simple model seeks to identify novel metabolic interplay between these microbes, to demonstrate metabolite exchange and the potential importance of viral-encoded auxiliary metabolic genes. Modeling is one tool to understand rumen function and opens the door to explore the use of viruses as alternatives to antibiotics to increase feed efficiency, maintain robust animal health and lower methane production.
The current widespread use of antibiotics as feed additives can be viewed as unsustainable (Economou and Gousia, 2015) and phage therapy shows promise as a targeted approach to control bacterial populations and promote animal health. Natural viruses have been used through phage therapy to remove problematic bacteria, archaea and their biofilms, from plants, animals and food crops, to humans, aquaculture and poultry (Donlan, 2009; Monk et al., 2010; Sarker et al., 2016). More recently engineered viruses have been used clinically to treat human patients with multi-antibiotic resistant infections (Dedrick et al., 2019), however this has been on a compassionate basis, as phage therapy is not currently authorized as a medicinal product. Temperate viruses that integrate into their host genomes also have the potential to confer positive genetic traits, but tend to be avoided due to the negative traits they can also confer (Monteiro et al., 2019). To develop effective phage therapies, we still need to generate rumen-specific information on viral dose size and viral particle longevity, to overcome potential viral inactivation by factors such as tannins, antibodies, macrophages, and clearance rates, as well as undertake basic viral characterization of adsorption efficiency, latency and burst size (Ly-Chatain, 2014). Phage therapy has been applied to ruminants with varying results (Gill et al., 2006; Johnson et al., 2008), however, this has been aimed primarily at the lower intestine (Dini et al., 2012) and has yet to be widely exploited in the rumen.
Phage therapy using intact viruses is highly host specific and presents regulatory issues, the use of virus-derived products or “enzybiotics” could directly replace conventional antimicrobials (O’Mahony et al., 2011; Shen et al., 2012; Louca and Doebeli, 2017). Mechanisms for the delivery and release of viral material from the host often involves enzymes (lysins/holins, peptidases, depolymerases, tail spike proteins), which can be repurposed as enzybiotics (Murphy et al., 1995; Andres et al., 2012; São-José, 2018). Indeed, enzybiotics have been tested in animals (O’Flaherty et al., 2009) and targeted nanoparticles displaying enzybiotics have been demonstrated to reduce methanogenesis by rumen-associated archaea (Altermann et al., 2018). Virus-derived enzybiotics are appealing, due to their scalability and ease of use within existing guidelines, enzybiotics could represent the future direction of phage research.
The rumen microbial community is integral in maintaining ruminant health, feed efficiency and is responsible for methanogenesis. Viruses are numerous, ubiquitous components of microbial communities, including those found in the rumen and have been overlooked in the past due to the technical difficulties experienced in their study. From the moment viral particles were first visualized it was clear that rumen viruses deserved investigating. However, direct study was limited to observations and indirect study proved problematic. This has led to rumen viruses being either ignored or overlooked in favor of more rewarding studies. Our current understanding shows that rumen viruses are not only numerically important, but are rapidly turned over. Within the rumen microbiome viruses enable nutrient cycling, genetic exchange and appear to promote functional stability. We can also extrapolate from other environments their likely influence on the rumen, but these require experimental testing.
Fortunately, new technologies are emerging that allow the scientific community to answer many of the fundamental questions pertaining to viruses; what viruses are present, how abundant are they and how do viruses interact with microbes, influence microbial community function and what impact do rumen microbial viruses have on the whole animal? Whole microbiome modeling, metagenome sequencing and metaproteomics are powerful and exciting tools that will enable us to answer these questions and test our predictions. Viruses offer the chance to understand, model and manipulate complex rumen communities. The future potential of viral applications are many and varied and require a more integrative research effort. Viruses are an intrinsic part of rumen microbial communities and are no more or less important than any other component. It is vital that viruses be incorporated into well-designed, large-scale studies that encompass all components of the rumen microbiome for both fundamental and hypothesis driven research. With affordable, powerful tools at our disposal we have a fantastic opportunity to explore rumen viruses and provide practical applications, based on sound knowledge of the whole ruminant.
RG and EJ conceived and designed the manuscript. RG and EJ wrote the majority of the manuscript with ET, TH, JF, CC, and PP contributing specialist sections to the manuscript. EJ, ET, RG, and KC contributed to the figures and tables. RG prepared the rumen viral fraction. KC undertook the electron microscopy. DO critically reviewed the manuscript and contributed to the table improvement. All authors approved the final version of the manuscript.
EJ and ET are funded by a Warwick Integrative Synthetic Biology (WISB) early career fellowship awarded to EJ, funded jointly by BBSRC and EPSRC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, SN, declared a shared affiliation, with no collaboration, with several of the authors, RG and DO, to the handling editor at the time of review.
The authors acknowledge the University of Warwick for assisting with publication costs. The authors also acknowledge and recognize Dr. Athol Klieve for his pioneering contributions to rumen phage research.
Abedon, S. T. (2012). “Phage-therapy best practices,” in Bacteriophages in Health and Disease, eds P. Hyman and S. T. Abedon (Wallingford: CAB International), 256–272. doi: 10.1079/9781845939847.0256
Abedon, S. T. (2016). Bacteriophage exploitation of bacterial biofilms: phage preference for less mature targets? FEMS Microbiol. Lett. 363:fnv246. doi: 10.1093/femsle/fnv246
Abel, S., Abel Zur, Wiesch, P., Davis, B. M., and Waldor, M. K. (2015). Analysis of bottlenecks in experimental models of infection. PLoS Pathog. 11:e1004823. doi: 10.1371/journal.ppat.1004823
Ackermann, H. W. (1998). Tailed bacteriophages: the order caudovirales. Adv. Virus Res. 51, 135–201. doi: 10.1016/s0065-3527(08)60785-x
Ackermann, H. W. (2012). Bacteriophage electron microscopy. Adv. Virus Res. 82, 1–32. doi: 10.1016/B978-0-12-394621-8.00017-0
Ackermann, H.-W. (2013). Sad state of phage electron microscopy. Please shoot the messenger. Microorganisms 2, 1–10. doi: 10.3390/microorganisms2010001
Ackermann, H. W., and Prangishvili, D. (2012). Prokaryote viruses studied by electron microscopy. Arch. Virol. 157, 1843–1849. doi: 10.1007/s00705-012-1383-y
Adams, J. C., Gazaway, J. A., Brailsford, M. D., Hartman, P. A., and Jacobson, N. L. (1966). Isolation of bacteriophages from the bovine rumen. Experentia 22, 717–718. doi: 10.1007/BF01901335
Adriaenssens, E. M., Ackermann, H.-W., Anany, H., Blasdel, B., Connerton, I. F., Goulding, D., et al. (2012). A suggested new bacteriophage genus: “Viunalikevirus”. Arch. Virol. 157, 2035–2046. doi: 10.1007/s00705-012-1360-5
Altermann, E., Schofield, L. R., Ronimus, R. S., Beatty, A. K., and Reilly, K. (2018). Inhibition of rumen methanogens by a novel archaeal lytic enzyme displayed on tailored bionanoparticles. Front. Microbiol. 9:2378–2378. doi: 10.3389/fmicb.2018.02378
Ambrozic, J., Ferme, D., Grabnar, M., Ravnikar, M., and Avgustin, G. (2001). The bacteriophages of ruminal prevotellas. Folia Microbiol. 46, 37–39. doi: 10.1007/bf02825881
Anderson, C. L., Sullivan, M. B., and Fernando, S. C. (2017). Dietary energy drives the dynamic response of bovine rumen viral communities. Microbiome 5:155. doi: 10.1186/s40168-017-0374-3
Andres, D., Roske, Y., Doering, C., Heinemann, U., Seckler, R., and Barbirz, S. (2012). Tail morphology controls DNA release in two Salmonella phages with one lipopolysaccharide receptor recognition system. Mol. Microbiol. 83, 1244–1253. doi: 10.1111/j.1365-2958.2012.08006.x
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Attwood, G. T., Kelly, W. J., Altermann, E. H., and Leahy, S. C. (2008). Analysis of the Methanobrevibacter ruminantium draft genome: understanding methanogen biology to inhibit their action in the rumen. Aust. J. Exp. Agric. 48, 83–88. doi: 10.1071/ea07269
Azevedo, A. C., Bento, C. B. P., Ruiz, J. C., Queiroz, M. V., and Mantovani, H. C. (2015). Distribution and genetic diversity of bacteriocin gene clusters in rumen microbial genomes. Appl. Environ. Microbiol. 81, 7290–7304. doi: 10.1128/AEM.01223-15
Balcazar, J. L. (2014). Bacteriophages as vehicles for antibiotic resistance genes in the environment. PLoS Pathog. 10:e1004219. doi: 10.1371/journal.ppat.1004219
Baresi, L., and Bertani, G. (1984). Isolation of a bacteriophage for a methanogenic bacterium. Abstracts Ann. Meeting American Soc. Microbiol. 28, 133.
Barondess, J. J., and Beckwith, J. (1990). A bacterial virulence determinant encoded by lysogenic coliphage lambda. Nature 346, 871–874. doi: 10.1038/346871a0
Barr, J. J., Auro, R., Furlan, M., Whiteson, K. L., Erb, M. L., Pogliano, J., et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776. doi: 10.1073/pnas.1305923110
Barr, J. J., Auro, R., Sam-Soon, N., Kassegne, S., Peters, G., Bonilla, N., et al. (2015). Subdiffusive motion of bacteriophage in mucosal surfaces increases the frequency of bacterial encounters. Proc. Natl. Acad. Sci. U.S.A. 112, 13675–13680. doi: 10.1073/pnas.1508355112
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Barylski, J., Enault, F., Dutilh, B. E., Schuller, M. B., Edwards, R. A., and Gillis et al. (2020a). Analysis of Spounaviruses as a case study for the overdue reclassification of tailed phages. Syst. Biol. 69, 110–123. doi: 10.1093/sysbio/syz036
Barylski, J., Kropinski, A. M., Alikhan, N.-F., and Adriaenssens, E. M. Ictv Report. (2020b). ICTV Virus taxonomy profile: Herelleviridae. J. Gen. Virol. doi: 10.1099/jgv.1090.001392 [Epub ahead of print].
Bedi, M., Verma, V., and Chhibber, S. (2009). Amoxicillin and specific bacteriophage can be used together for eradication of biofilm of Klebsiella pneumoniae B5055. World J. Microbiol. Biotech. 25, 1145–1151. doi: 10.1007/s11274-009-9991-8
Ben Zakour, N. L., Alsheikh-Hussain, A. S., Ashcroft, M. M., Khanh Nhu, N. T., Roberts, L. W., Stanton-Cook, M., et al. (2016). Sequential acquisition of virulence and fluoroquinolone resistance has shaped the evolution of Escherichia coli ST131. mBio 7, e347-16. doi: 10.1128/mBio.00347-16
Benahmed, F. H., Gopinath, G. R., Harbottle, H., Cotta, M. A., Luo, Y., Henderson, C., et al. (2014). Draft genome sequences of Streptococcus bovis strains ATCC 33317 and JB1. Genome Ann. 2, e1012–e1014. doi: 10.1128/genomeA.01012-14
Bennett, P. M. (2008). Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 153(Suppl. 1), S347–S357. doi: 10.1038/sj.bjp.0707607
Beres, S. B., Sylva, G. L., Barbian, K. D., Lei, B., Hoff, J. S., Mammarella, N. D., et al. (2002). Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. U.S.A. 99, 10078–10083. doi: 10.1073/pnas.152298499
Berg Miller, M. E., Yeoman, C. J., Chia, N., Tringe, S. G., Angly, F. E., and Edwards et al. (2012). Phage–bacteria relationships and CRISPR elements revealed by a metagenomic survey of the rumen microbiome. Environ. Microbiol. 14, 207–227. doi: 10.1111/j.1462-2920.2011.02593.x
Berthod, A., and Faure, K. (2015). “Separations with a liquid stationary phase: countercurrent chromatography or centrifugal partition chromatography,” in Analytical Separation Science, eds V. Pino, J. L. Anderson, A. Berthod, and A. M. Stalcup (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 1177–1206. doi: 10.1002/9783527678129.assep046
Betley, M. J., and Mekalanos, J. J. (1985). Staphylococcal enterotoxin A is encoded by phage. Science 229, 185–187. doi: 10.1126/science.3160112
Beutin, L., Stroeher, U. H., and Manning, P. A. (1993). Isolation of enterohemolysin (Ehly2)-associated sequences encoded on temperate phages of Escherichia coli. Gene 132, 95–99. doi: 10.1016/0378-1119(93)90519-9
Blahova, J., Hupkova, M., Krcmery, V., and Schafer, V. (1992). Imipenem and cefotaxime resistance: transduction by wild-type phages in hospital strains of Pseudomonas aeruginosa. J. Chemother. 4, 335–337. doi: 10.1080/1120009x.1992.11739187
Bonanno, L., Petit, M.-A., Loukiadis, E., Michel, V., and Auvray, F. (2016). Heterogeneity in induction level, infection ability, and morphology of Shiga toxin-encoding phages (Stx phages) from dairy and human Shiga toxin-producing Escherichia coli O26:H11 isolates. Appl. Environ. Microbiol. 82, 2177–2186. doi: 10.1128/AEM.03463-15
Borton, M. A., Hoyt, D. W., Roux, S., Daly, R. A., Welch, S. A., Nicora, C. D., et al. (2018). Coupled laboratory and field investigations resolve microbial interactions that underpin persistence in hydraulically fractured shales. Proc. Natl. Acad. Sci. U.S.A. 115, E6585–E6594. doi: 10.1073/pnas.1800155115
Brabban, A. D., Hite, E., and Callaway, T. R. (2005). Evolution of foodborne pathogens via temperate bacteriophage-mediated gene transfer. Foodborne Pathog. Dis. 2, 287–303. doi: 10.1089/fpd.2005.2.287
Bradley, D. E. (1967). Ultrastructure of bacteriophages and bacteriocins. Bacteriol. Rev. 31, 230–314. doi: 10.1128/mmbr.31.4.230-314.1967
Brailsford, M. D., and Hartman, P. A. (1968). Characterisation of Streptococcus durans bacteriophages. Can. J. Microbiol. 14, 397–402. doi: 10.1139/m68-063
Bratbak, G., Heldal, M., Norland, S., and Thingstad, T. F. (1990). Viruses as partners in spring bloom microbial trophodynamics. Appl. Environ. Microbiol. 56, 1400–1405. doi: 10.1128/aem.56.5.1400-1405.1990
Breitbart, M., Salamon, P., Andresen, B., Mahaffy, J. M., Segall, A. M., Mead, D., et al. (2002). Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U.S.A. 99:14250. doi: 10.1073/pnas.202488399
Brody, J. R., and Kern, S. E. (2004). History and principles of conductive media for standard DNA electrophoresis. Anal. Biochem. 333, 1–13. doi: 10.1016/j.ab.2004.05.054
Brum, J. R., Ignacio-Espinoza, J. C., Kim, E.-H., Trubl, G., Jones, R. M., Roux, S., et al. (2016). Illuminating structural proteins in viral “dark matter” with metaproteomics. Proc. Natl. Acad. Sci. U.S.A. 113, 2436–2441. doi: 10.1073/pnas.1525139113
Brussow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/mmbr.68.3.560-602.2004
Bruttin, A., and Brussow, H. (2005). Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob. Agents Chemother. 49, 2874–2878. doi: 10.1128/aac.49.7.2874-2878.2005
Bull, J. J., Christensen, K. A., Scott, C., Jack, B. R., Crandall, C. J., and Krone, S. M. (2018). Phage-bacterial dynamics with spatial structure: self organization around phage sinks can promote increased cell densities. Antibiotics (Basel) 7:8. doi: 10.3390/antibiotics7010008
Cameron, A., and McAllister, T. A. (2016). Antimicrobial usage and resistance in beef production. J. Anim. Sci. Biotechnol. 7:68. doi: 10.1186/s40104-016-0127-3
Caron, D. A. (1987). Grazing of attached bacteria by heterotrophic microflagellates. Microb. Ecol. 13, 203–218. doi: 10.1007/BF02024998
Carrolo, M., Frias, M. J., Pinto, F. R., Melo-Cristino, J., and Ramirez, M. (2010). Prophage spontaneous activation promotes DNA release enhancing biofilm formation in Streptococcus pneumoniae. PLoS One 5:e15678. doi: 10.1371/journal.pone.0015678
Casjens, S. R., and Hendrix, R. W. (2015). Bacteriophage lambda: Early pioneer and still relevant. Virology 47, 310–330. doi: 10.1016/j.virol.2015.02.010
Chen, J., and Weimer, P. (2001). Competition among three predominant ruminal cellulolytic bacteria in the absence or presence of non-cellulolytic bacteria. Microbiol. 147, 21–30. doi: 10.1099/00221287-147-1-21
Cheong, J. P., and Brooker, J. D. (1998). Lysogenic bacteriophage M1 from Selenomonas ruminantium: isolation, characterisation and DNA sequence analysis of the integration site. Microbiology 144, 2195–2202. doi: 10.1099/00221287-144-8-2195
Cheong, J. P., and Brooker, J. D. (2000). Isolation of a virulent bacteriophage from a Propionibacterium species in the sheep rumen. Aust. J. Agric. Res. 51, 119–123.
Clokie, M. R. J., Millard, A. D., Letarov, A. V., and Heaphy, S. (2011). Phages in nature. Bacteriophage 1, 31–45. doi: 10.4161/bact.1.1.14942
Coleman, D. C., Sullivan, D. J., Russell, R. J., Arbuthnott, J. P., Carey, B. F., and Pomeroy, H. M. (1989). Staphylococcus aureus bacteriophages mediating the simultaneous lysogenic conversion of beta-lysin, staphylokinase and enterotoxin A: molecular mechanism of triple conversion. J. Gen. Microbiol. 135, 1679–1697. doi: 10.1099/00221287-135-6-1679
Colomer-Lluch, M., Jofre, J., and Muniesa, M. (2011). Antibiotic resistance genes in the bacteriophage DNA fraction of environmental samples. PLoS ONE 6:e17549. doi: 10.1371/journal.pone.0017549
Creaser, E. H., and Taussig, A. (1957). The purification and chromatography of bacteriophages on anion-exchange cellulose. Virol. 4, 200–208. doi: 10.1016/0042-6822(57)90057-0
Cuervo, A., and Carrascosa, J. L. (2018). Observation of bacteriophage ultrastructure by cryo-electron microscopy. Methods Mol. Biol. 1693, 43–55. doi: 10.1007/978-1-4939-7395-8_5
Davies, E. V., James, C. E., Williams, D., O’brien, S., Fothergill, J. L., Haldenby, S., et al. (2016). Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. U.S.A. 113:8266. doi: 10.1073/pnas.1520056113
Dedrick, R. M., Guerrero-Bustamante, C. A., Garlena, R. A., Russell, D. A., Ford, K., Harris, K., et al. (2019). Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733. doi: 10.1038/s41591-019-0437-z
Dedrick, R. M., Jacobs-Sera, D., Bustamante, C. A., Garlena, R. A., Mavrich, T. N., Pope, W. H., et al. (2017). Prophage-mediated defence against viral attack and viral counter-defence. Nature Microbiol. 2:16251. doi: 10.1038/nmicrobiol.2016.251
Degnan, P. H., Barry, N. A., Mok, K. C., Taga, M. E., and Goodman, A. L. (2014). Human gut microbes use multiple transporters to distinguish vitamin B12 analogs and compete in the gut. Cell Host Microbe 15, 47–57. doi: 10.1016/j.chom.2013.12.007
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 295, 1306–1311. doi: 10.1126/science.1067799
Deriu, E., Liu, J. Z., Pezeshki, M., Edwards, R. A., Ochoa, R. J., and Contreras et al. (2013). Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14, 26–37. doi: 10.1016/j.chom.2013.06.007
Deusch, S., Camarinha-Silva, A., Conrad, J., Beifuss, U., Rodehutscord, M., and Seifert, J. (2017). A structural and functional elucidation of the rumen microbiome influenced by various diets and microenvironments. Front. Microbiol. 8:1605. doi: 10.3389/fmicb.2017.01605
d’Herelle, F. (1918). Technique de la recherche du microbe filtrant bacteriophage (Bacteriophagum intestinale). Comptes Rendus Des Seances De La Societe De Biologie Et De Ses Filiales 81, 1160–1162.
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E. A., and Crook, D. W. (2012). Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13:601. doi: 10.1038/nrg3226
Dini, C., Islan, G. A., De Urraza, P. J., and Castro, G. R. (2012). Novel biopolymer matrices for microencapsulation of phages: enhanced protection against acidity and protease activity. Macromol. Biosci. 12, 1200–1208. doi: 10.1002/mabi.201200109
Dinsdale, E. A., Edwards, R. A., Hall, D., Angly, F., Breitbart, M., Brulc, J. M., et al. (2008). Functional metagenomic profiling of nine biomes. Nature 452, 629–830. doi: 10.1038/nature06810
Dolgin, E. (2019). The secret social lives of viruses. Nature 570, 290–292. doi: 10.1038/s41586-020-1936-2
Donlan, R. M. (2009). Preventing biofilms of clinically relevant organisms using bacteriophage. Trends Microbiol. 17, 66–72. doi: 10.1016/j.tim.2008.11.002
Doolittle, M. M., Cooney, J. J., and Caldwell, D. E. (1995). Lytic infection of Escherichia coli biofilms by bacteriophage T4. Can. J. Microbiol. 41, 12–18. doi: 10.1139/m95-002
Doolittle, M. M., Cooney, J. J., and Caldwell, D. E. (1996). Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Ind. Microbiol. 16, 331–341. doi: 10.1007/bf01570111
Doulatov, S., Hodes, A., Dai, L., Mandhana, N., Liu, M., Deora, R., et al. (2004). Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature 431, 476–481. doi: 10.1038/nature02833
Duerr, D. M., White, S. J., and Schluesener, H. J. (2004). Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J. Virol. Methods 116, 177–180. doi: 10.1016/j.jviromet.2003.11.012
Džunková, M., Low, S. J., Daly, J. N., Deng, L., Rinke, C., and Hugenholtz, P. (2019). Defining the human gut host-phage network through single-cell viral tagging. Nat. Microbiol. 4, 2192–2203. doi: 10.1038/s41564-019-0526-2
Economou, V., and Gousia, P. (2015). Agriculture and food animals as a source of antimicrobial-resistant bacteria. Infect. Drug Resist. 8, 49–61. doi: 10.2147/idr.s55778
Edwards, R. A., McNair, K., Faust, K., Raes, J., and Dutilh, B. E. (2016). Computational approaches to predict bacteriophage–host relationships. FEMS Microbiol. Rev. 40, 258–272. doi: 10.1093/femsre/fuv048
Eklund, M. W., Poysky, F. T., Reed, S. M., and Smith, C. A. (1971). Bacteriophage and the toxigenicity of Clostridium botulinum type C. Science 172, 480–482. doi: 10.1126/science.172.3982.480
Ellis, J. L., Kebreab, E., Odongo, N. E., Beauchemin, K., Mcginn, S., Nkrumah, J. D., et al. (2009). Modeling methane production from beef cattle using linear and nonlinear approaches. J. Anim. Sci. 87, 1334–1345. doi: 10.2527/jas.2007-0725
Emerson, J. B., Roux, S., Brum, J. R., Bolduc, B., Woodcroft, B. J., Jang, H. B., et al. (2018). Host-linked soil viral ecology along a permafrost thaw gradient. Nature Microbiol. 3, 870–880. doi: 10.1038/s41564-018-0190-y
Enault, F., Briet, A., Bouteille, L., Roux, S., Sullivan, M. B., and Petit, M.-A. (2017). Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J. 11:237. doi: 10.1038/ismej.2016.90
Escobar-Zepeda, A., Vera-Ponce, De León, A., and Sanchez-Flores, A. (2015). The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front. Genet. 6:348. doi: 10.3389/fgene.2015.00348
Fenton, M., Ross, P., Mcauliffe, O., O’mahony, J., and Coffey, A. (2010). Recombinant bacteriophage lysins as antibacterials. Bioeng. Bugs 1, 9–16. doi: 10.4161/bbug.1.1.9818
Figura, G., Kopciuch, A., Owczarek, B., Piotrowicz, A., Miernikiewicz, P., Oślizło, A., et al. (2011). Purification of phage display-modified bacteriophage T4 by affinity chromatography. BMC Biotechnol. 11:59. doi: 10.1186/1472-6750-11-59
Firkins, J. L., Weiss, W. P., and Piwonka, E. J. (1992). Quantification of intraruminal recycling of microbial nitrogen using Nitrogen-15. J. Anim. Sci. 70, 3223–3233. doi: 10.2527/1992.70103223x
Flint, H. J. (1997). The rumen microbial ecosystem—some recent developments. Trends Microbiol. 5, 483–488. doi: 10.1016/s0966-842x(97)01159-1
Fraser, J. S., Maxwell, K. L., and Davidson, A. R. (2007). Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr. Opin. Microbiol. 10, 382–387. doi: 10.1016/j.mib.2007.05.018
Freeman, V. J. (1951). Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J. Bacteriol. 61, 675–688. doi: 10.1128/jb.61.6.675-688.1951
Friedersdorff, J., Kingston-Smith, A., Creevey, C., Rooke, D., Bright, C., and West, D. (2018). “Using high performance counter current chromatography for the separation, preparation and purification of bacteriophages from the rumen microbiome” in Proceedings of the The 10th International Conference on Countercurrent Chromatography (Braunschweig: Technische Universität Braunschweig), doi: 10.13140/RG.2.2.12796.90246
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Fuhrman, J. A., and Noble, R. T. (1995). Viruses and protists cause similar bacterial mortality in coastal seawater. Limnol. Oceanogr. 40, 1236–1242. doi: 10.4319/lo.1995.40.7.1236
Gilbert, R. A., Kelly, W. J., Altermann, E., Leahy, S. C., Minchin, C., Ouwerkerk, D., et al. (2017). Toward understanding phage:host interactions in the rumen; complete genome sequences of lytic phages infecting rumen bacteria. Front. Microbiol. 8:2340. doi: 10.3389/fmicb.2017.02340
Gilbert, R. A., and Klieve, A. V. (2015). “Ruminal viruses (Bacteriophages, Archaeaphages),” in Rumen Microbiology: From Evolution to Revolution, eds A. K. Puniya, R. Singh, and D. N. Kamra (Berlin: Springer), 121–141. doi: 10.1007/978-81-322-2401-3
Gill, J. J., Pacan, J. C., Carson, M. E., Leslie, K. E., Griffiths, M. W., and Sabour, P. M. (2006). Efficacy and pharmacokinetics of bacteriophage therapy in treatment of subclinical Staphylococcus aureus mastitis in lactating dairy cattle. Antimicrob. Agents Chemother. 50, 2912–2918. doi: 10.1128/aac.01630-05
Goh, S., Hussain, H., Chang, B. J., Emmett, W., Riley, T. V., and Mullany, P. (2013). Phage phiC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. MBio. 4:e840-13. doi: 10.1128/mBio.00840-13
Golomidova, A., Kulikov, E., Isaeva, A., Manykin, A., and Letarov, A. (2007). The diversity of coliphages and coliforms in horse feces reveals a complex pattern of ecological interactions. Appl. Environ. Microbiol. 73, 5975–5981. doi: 10.1128/aem.01145-07
Górski, A., Ważna, E., Dąbrowska, B.-W., Dąbrowska, K., Świtała-Jeleł, K., and Miȩdzybrodzki, R. (2006). Bacteriophage translocation. Pathog. Dis. 46, 313–319. doi: 10.1111/j.1574-695X.2006.00044.x
Gregg, K., Kennedy, B. G., and Klieve, A. V. (1994). Cloning and DNA sequence analysis of the region containing AttP of the temperate phage phiAR29 of Prevotella ruminicola AR29. Microbiology 140, 2109–2114. doi: 10.1099/13500872-140-8-2109
Gregorini, P., Beukes, P., Waghorn, G., Pacheco, D., and Hanigan, M. (2015). Development of an improved representation of rumen digesta outflow in a mechanistic and dynamic model of a dairy cow. Molly. Ecolog. Model. 313, 293–306. doi: 10.1016/j.ecolmodel.2015.06.042
Groth, A. C., Olivares, E. C., Thyagarajan, B., and Calos, M. P. (2000). A phage integrase directs efficient site-specific integration in human cells. Proc. Natl. Acad. Sci. U.S.A. 97, 5995–6000. doi: 10.1073/pnas.090527097
Grybchuk, D., Akopyants, N. S., Kostygov, A. Y., Konovalovas, A., Lye, L.-F., Dobson, D. E., et al. (2018). Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. U.S.A. 115:E506. doi: 10.1073/pnas.1717806115
Hagen, L. H., Frank, J. A., Zamanzadeh, M., Eijsink, V. G. H., Pope, P. B., Horn, S. J., et al. (2017). Quantitative metaproteomics highlight the metabolic contributions of uncultured phylotypes in a thermophilic anaerobic digester. Appl. Environ. Microbiol. 83:e1955-16. doi: 10.1128/AEM.01955-16
Haguenau, F., Hawkes, P. W., Hutchison, J. L., Satiatjeunematre, B., Simon, G. T., and Williams, D. B. (2003). Key events in the history of electron microscopy. Microsc. Microanal. 9, 96–138. doi: 10.1017/S1431927603030113
Hallewell, J., Niu, Y. D., Munns, K., Mcallister, T. A., Johnson, R. P., Ackermann, H.-W., et al. (2014). Differing populations of endemic bacteriophages in cattle shedding high and low numbers of Escherichia coli O157:H7 bacteria in feces. Appl. Environ. Microbiol. 80, 3819–3825. doi: 10.1128/AEM.00708-14
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5, R245–R249. doi: 10.1016/S1074-5521(98)90108-9
Hanlon, G. W. (2007). Bacteriophages: an appraisal of their role in the treatment of bacterial infections. Int. J. Antimicrob. Agents 30, 118–128. doi: 10.1016/j.ijantimicag.2007.04.006
Hanlon, G. W., Denyer, S. P., Olliff, C. J., and Ibrahim, L. J. (2001). Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 67, 2746–2753. doi: 10.1128/AEM.67.6.2746-2753.2001
Hart, E. H., Creevey, C. J., Hitch, T., and Kingston-Smith, A. H. (2018). Meta-proteomics of rumen microbiota indicates niche compartmentalisation and functional dominance in a limited number of metabolic pathways between abundant bacteria. Sci. Rep. 8:10504. doi: 10.1038/s41598-018-28827-7
Hartinger, T., Gresner, N., and Sudekum, K. H. (2018). Does intra-ruminal nitrogen recycling waste valuable resources? A review of major players and their manipulation. J. Anim. Sci. Biotechnol. 9:33. doi: 10.1186/s40104-018-0249-x
Hazlewood, G. P., Munn, E. A., and Orpin, C. G. (1983). “Temperate bacteriophages of Selenomonas ruminantium and a Fusobacterium sp. isolated from the ovine rumen,” in Abstracts Canadian Society Microbiology 33rd Annual Meeting, Winnipeg, 76.
Heather, J. M., and Chain, B. (2016). The sequence of sequencers: the history of sequencing DNA. Genomics 107, 1–8. doi: 10.1016/j.ygeno.2015.11.003
Henderson, G., Cox, F., Ganesh, S., Jonker, A., Young, W., and Janssen, P. H. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5:14567. doi: 10.1038/srep14567
Hendrix, R. W., Smith, M. C., Burns, R. N., Ford, M. E., and Hatfull, G. F. (1999). Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc. Natl. Acad. Sci. U.S.A. 96, 2192–2197. doi: 10.1073/pnas.96.5.2192
Heyer, R., Schallert, K., Siewert, C., Kohrs, F., Greve, J., Maus, I., et al. (2019). Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 7, 1–17. doi: 10.1186/s40168-019-0673-y
Hibma, A. M., Jassim, S. A., and Griffiths, M. W. (1997). Infection and removal of L-forms of Listeria monocytogenes with bred bacteriophage. Int. J. Food. Microbiol. 34, 197–207. doi: 10.1016/S0168-1605(96)01190-7
Hitch, T. C. A., Edwards, J. E., and Gilbert, R. A. (2019). Metatranscriptomics reveals mycoviral populations in the ovine rumen. FEMS Microbiol. Lett. 366:fnz161. doi: 10.1093/femsle/fnz161
Ho, T. D., and Slauch, J. M. (2001). Characterization of grvA, an antivirulence gene on the gifsy-2 phage in Salmonella enterica serovar typhimurium. J. Bacteriol. 183, 611–620. doi: 10.1128/JB.183.2.611-620.2001
Hobbs, Z., and Abedon, S. T. (2016). Diversity of phage infection types and associated terminology: the problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett. 363:fnw047. doi: 10.1093/femsle/fnw047
Hoogenraad, N. J., Hird, F. J. R., Holmes, I., and Millis, N. F. (1967). Bacteriophages in rumen contents of sheep. J. Gen. Virol. 1, 575–576. doi: 10.1099/0022-1317-1-4-575
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Hristov, A. N., Oh, J., Firkins, J. L., Dijkstra, J., Kebreab, E., Waghorn, G., et al. (2013). Special topics -Mitigation of methane and nitrous oxide emissions from animal operations: I. A review of enteric methane mitigation options. J. Anim. Sci. 91, 5045–5069. doi: 10.2527/jas2013-6583
Hu, R., and Pan, Y. (2012). Recent trends in counter-current chromatography. Trends Anal. Chem. 40, 15–27. doi: 10.1016/j.trac.2012.07.018
Hughes, K. A., Sutherland, I. W., Clark, J., and Jones, M. V. (1998). Bacteriophage and associated polysaccharide depolymerases–novel tools for study of bacterial biofilms. J. Appl. Microbiol. 85, 583–590. doi: 10.1046/j.1365-2672.1998.853541.x
Hungate, R. E. (1969). “A roll-tube method for the cultivation of strict anaerobes,” in Methods in Microbiology, eds J. R. Norris and D. W. Ribbons (Cambridge, MA: Academic Press), 117–132. doi: 10.1016/s0580-9517(08)70503-8
Hungate, R. E., Bryant, M. P., and Mah, R. A. (1964). The rumen bacteria and protozoa. Ann. Rev. Microbiol. 18, 131–166. doi: 10.1146/annurev.mi.18.100164.001023
Huws, S. A., Creevey, C. J., Oyama, L. B., Mizrahi, I., Denman, S. E., Popova, M., et al. (2018). Adressing global ruminant agricultural challenges through understanding the rumen microbiome: past, present and future. Front. Microbiol. 9:2161. doi: 10.3389/fmicb.2018.02161
Hynes, W. L., and Ferretti, J. J. (1989). Sequence analysis and expression in Escherichia coli of the hyaluronidase gene of Streptococcus pyogenes bacteriophage H4489A. Infect. Immun. 57, 533–539. doi: 10.1128/iai.57.2.533-539.1989
Iranzo, J., Krupovic, M., and Koonin, E. V. (2016). The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 7:e978-16. doi: 10.1128/mBio.00978-16
Islam, M. M., Fernando, S. C., and Saha, R. (2019). Metabolic modeling elucidates the transactions in the rumen microbiome and the shifts upon virome interactions. Front. Microbiol. 10:2412. doi: 10.3389/fmicb.2019.02412
Iverson, W. G., and Millis, N. F. (1977). Succession of Streptococcus bovis strains with differing bacteriophage sensitivities in the rumens of two fistulated sheep. Appl. Environ. Microbiol. 33, 810–813. doi: 10.1128/aem.33.4.810-813.1977
Iverson, W. J., and Millis, N. F. (1976a). Characterisation of Streptococcus bovis bacteriophages. Can. J. Microbiol. 22, 847–852. doi: 10.1139/m76-122
Iverson, W. J., and Millis, N. F. (1976b). Lysogeny in Streptococcus bovis. Can. J. Microbiol. 22, 853–857. doi: 10.1139/m76-123
Jameson, E., Mann, N. H., Joint, I., Sambles, C., and Muhling, M. (2011). The diversity of cyanomyovirus populations along a north-south atlantic ocean transect. ISME J. 5, 1713–1721. doi: 10.1038/ismej.2011.54
Jarvis, B. D. W. (1968). Lysis of viable rumen bacteria in bovine rumen fluid. Appl. Microbiol. 16, 714–723. doi: 10.1128/aem.16.5.714-723.1968
Johnson, R. P., Gyles, C. L., Huff, W. E., Ojha, S., Huff, G. R., Rath, N. C., et al. (2008). Bacteriophages for prophylaxis and therapy in cattle, poultry and pigs. Anim. Health Res. Rev. 9, 201–215. doi: 10.1017/s1466252308001576
Kaelber, J. T., Hryc, C. F., and Chiu, W. (2017). Electron cryomicroscopy of viruses at near-atomic resolutions. Ann. Rev. Virol. 4, 287–308. doi: 10.1146/annurev-virology-101416-041921
Kaneko, J., Kimura, T., Narita, S., Tomita, T., and Kamio, Y. (1998). Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage phiPVL carrying panton-valentine leukocidin genes. Gene 215, 57–67. doi: 10.1016/s0378-1119(98)00278-9
Karaolis, D. K., Somara, S., Maneval, D. R. Jr., Johnson, J. A., and Kaper, J. B. (1999). A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature 399, 375–379. doi: 10.1038/20715
Keen, E. C., Bliskovsky, V. V., Malagon, F., Baker, J. D., Prince, J. S., Klaus, J. S., et al. (2017). Novel “Superspreader” bacteriophages promote horizontal gene transfer by Transformation. MBio 8, e02115–e02116. doi: 10.1128/mBio.02115-16
Kelly, W. J., Leahy, S. C., Li, D., Perry, R., Lambie, S. C., and Attwood, G. T. (2014). The complete genome sequence of the rumen methanogen Methanobacterium formicicum BRM9. Stand. Genomic Sci. 9:15. doi: 10.1186/1944-3277-9-15
King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J. (2012). “Order – Caudovirales,” in Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses, eds A. M. Q. King, M. J. Adams, E. B. Carstens, and E. J. Lefkowitz (Amstadam: Elsevier), 39–45. doi: 10.1016/B978-0-12-384684-6.00001-X
Klieve, A. V. (2005). “2.2. Bacteriophages,” in Methods in gut Microbial Ecology for Ruminants, eds H. P. S. Makkar and C. S. McSweeney (Berlin: Springer), 39–46. doi: 10.1007/1-4020-3791-0
Klieve, A. V., Bain, P. A., Yokoyama, M. T., Ouwerkerk, D., Forster, R. J., and Turner, A. F. (2004). Bacteriophages that infect the cellulolytic ruminal bacterium Ruminococcus albus AR67. Lett. Appl. Microbiol. 38, 333–338. doi: 10.1111/j.1472-765X.2004.01493.x
Klieve, A. V., and Bauchop, T. (1988). Morphological diversity or ruminal bacteriophages from sheep and cattle. Appl. Environ. Microbiol. 54, 1637–1641. doi: 10.1128/aem.54.6.1637-1641.1988
Klieve, A. V., and Bauchop, T. (1991). Phage resistance and altered growth habit in a strain of Streptococcus bovis. FEMS Microbiol. Lett. 80, 155–160. doi: 10.1016/0378-1097(91)90587-z
Klieve, A. V., and Gilbert, R. A. (2005). “4.2 Bacteriophage populations,” in Methods in Gut Microbial Ecology for Ruminants, eds H. P. S. Makkar and C. S. McSweeney (Berlin: Springer), 129–137. doi: 10.1007/1-4020-3791-0
Klieve, A. V., Gregg, K., and Bauchop, T. (1991). Isolation and characteristics of lytic phages from Bacteroides ruminicola ss brevis. Curr. Microbiol. 23, 183–187. doi: 10.1007/BF02092277
Klieve, A. V., Heck, G. L., Prance, M. A., and Shu, Q. (1999). Genetic homogeneity and phage susceptibility of ruminal strains of Streptococcus bovis isolated in Australia. Lett. Appl. Microbiol. 29, 108–112. doi: 10.1046/j.1365-2672.1999.00596.x
Klieve, A. V., and Hegarty, R. (1999). Opportunities for biological control of ruminal methanogenesis. Aust. J. Agric. Res. 50, 1315–1319. doi: 10.1071/AR99006
Klieve, A. V., Hudman, J. F., and Bauchop, T. (1989). Inducible bacteriophages from ruminal bacteria. Appl. Environ. Microbiol. 55, 1630–1634. doi: 10.1128/aem.55.6.1630-1634.1989
Klieve, A. V., and Swain, R. A. (1993). Estimating ruminal bacteriophage numbers using pulsed field gel electrophoresis and laser densitometry. Appl. Environ. Microbiol. 59, 2299–2303. doi: 10.1128/aem.59.7.2299-2303.1993
Klieve, A. V., Turner, A. F., and Heck, G. L. (1998). Dietary influences on bacteriophage numbers in the rumen. Proc. Aust. Soc. Anim. Prod. 22:341.
Koonin, E. V., Makarova, K. S., and Wolf, Y. I. (2017). Evolutionary genomics of defense systems in Archaea and Bacteria. Ann. Rev. Microbiol. 71, 233–261. doi: 10.1146/annurev-micro-090816-093830
Koskella, B. (2019). New approaches to characterizing bacteria–phage interactions in microbial communities and microbiomes. Environ. Microbiol. Rep. 11, 15–16. doi: 10.1111/1758-2229.12706
Koskella, B., and Brockhurst, M. A. (2014). Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Krajacic, M., Ravnikar, M., Štrancar, A., and Gutiérrez-Aguirre, I. (2017). Application of monolithic chromatographic supports in virus research. Electrophoresis 38, 2827–2836. doi: 10.1002/elps.201700152
Kristensen, D. M., Waller, A. S., Yamada, T., Bork, P., Mushegian, A. R., and Koonin, E. V. (2013). Orthologous gene clusters and taxon signature genes for viruses of prokaryotes. J. Bacteriol. 195, 941–950. doi: 10.1128/JB.01801-12
Krupovic, M., Cvirkaite-Krupovic, V., Iranzo, J., Prangishvili, D., and Koonin, E. V. (2018). Viruses of archaea: Structural, functional, environmental and evolutionary genomics. Virus Res. 244, 181–193. doi: 10.1016/j.virusres.2017.11.025
Krupovic, M., Prangishvili, D., Hendrix, R. W., and Bamford, D. H. (2011). Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 75, 610–635. doi: 10.1128/MMBR.00011-11
Kunath, B. J., Minniti, G., Skaugen, M., Hagen, L. H., Vaaje-Kolstad, G., Eijsink, V. G. H., et al. (2019). Metaproteomics: sample preparation and methodological considerations. Adv. Exp. Med. Biol. 1073, 187–215. doi: 10.1007/978-3-030-12298-0_8
Lajoie, B. R., Dekker, J., and Kaplan, N. (2015). The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods 72, 65–75. doi: 10.1016/j.ymeth.2014.10.031
Landsberger, M., Gandon, S., Meaden, S., Rollie, C., Chevallereau, A., and Chabas et al. (2018). Anti-CRISPR phages cooperate to overcome CRISPR-Cas immunity. Cell 174, 908–916.e12. doi: 10.1016/j.cell.2018.05.058
Leahy, S. C., Kelly, W. J., Altermann, E., Ronimus, R. S., Yeoman, C. J., and Pacheco et al. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS ONE 5:e8926. doi: 10.1371/journal.pone.0008926
Leahy, S. C., Kelly, W. J., Ronimus, R. S., Wedlock, N., Altermann, E., and Attwood, G. T. (2013). Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal 7(Suppl. 2), 235–243. doi: 10.1017/S1751731113000700
Leedle, J. A., Bryant, M. P., and Hespell, R. B. (1982). Diurnal variations in bacterial numbers and fluid parameters in ruminal contents of animals fed low- or high-forage diets. Appl. Environ. Microbiol. 44, 402–412. doi: 10.1128/aem.44.2.402-412.1982
Lefkowitz, E. J., Dempsey, D. M., Hendrickson, R. C., Orton, R. J., Siddell, S. G., and Smith, D. B. (2018). Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi: 10.1093/nar/gkx932
Leng, R. A., and Nolan, J. V. (1984). Nitrogen metabolism in the rumen. J. Dairy Sci. 67, 1072–1089. doi: 10.3168/jds.S0022-0302(84)81409-5
Leon, M., and Bastias, R. (2015). Virulence reduction in bacteriophage resistant bacteria. Front. Microbiol. 6:343. doi: 10.3389/fmicb.2015.00343
Letarov, A., and Kulikov, E. (2009). The bacteriophages in human- and animal body-associated microbial communities. J. Appl. Microbiol. 107, 1–13. doi: 10.1111/j.1365-2672.2009.04143.x
Letarov, A. V., and Kulikov, E. E. (2017). Adsorption of bacteriophages on bacterial cells. Biochemistry (Mosc) 82, 1632–1658. doi: 10.1134/S0006297917130053
Lim, E. S., Wang, D., and Holtz, L. R. (2016). The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 24, 801–810. doi: 10.1016/j.tim.2016.06.001
Lockington, R. A., Attwood, G. T., and Brooker, J. D. (1988). Isolation and characterisation of a temperate bacteriophage from the ruminal anaerobe Selenomonas ruminantium. Appl. Environ. Microbiol. 54, 1575–1580. doi: 10.1128/aem.54.6.1575-1580.1988
Loenen, W. A., Dryden, D. T., Raleigh, E. A., Wilson, G. G., and Murray, N. E. (2014). Highlights of the DNA cutters: a short history of the restriction enzymes. Nucl. Acids Res. 42, 3–19. doi: 10.1093/nar/gkt990
Louca, S., and Doebeli, M. (2017). Taxonomic variability and functional stability in microbial communities infected by phages. Environ. Microbiol. 19, 3863–3878. doi: 10.1111/1462-2920.13743
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Ly-Chatain, M. H. (2014). The factors affecting effectiveness of treatment in phages therapy. Front. Microbiol. 5:51. doi: 10.3389/fmicb.2014.00051
Mann, N. H., Cook, A., Millard, A., Bailey, S., and Clokie, M. (2003). Marine ecosystems: bacterial photosynthesis genes in a virus. Nature 424:741. doi: 10.1038/424741a
Mantovani, H. C., and Russell, J. B. (2001). Nisin resistance of Streptococcus bovis. Appl. Environ. Microbiol. 67, 808–813. doi: 10.1128/AEM.67.2.808-813.2001
Marbouty, M., Baudry, L., Cournac, A., and Koszul, R. (2017). Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci. Adv. 3:e1602105. doi: 10.1126/sciadv.1602105
Marchesi, J. R., and Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome 3:31. doi: 10.1186/s40168-015-0094-5
Marti, R., Tien, Y.-C., Murray, R., Scott, A., Sabourin, L., and Topp, E. (2014). Safely coupling livestock and crop production systems: how rapidly do antibiotic resistance genes dissipate in soil following a commercial application of swine or dairy manure? Appl. Environ. Microbiol. 80, 3258–3265. doi: 10.1128/aem.00231-14
Mazaheri Nezhad Fard, R., Barton, M. D., and Heuzenroeder, M. W. (2011). Bacteriophage-mediated transduction of antibiotic resistance in Enterococci. Lett. Appl. Microbiol. 52, 559–564. doi: 10.1111/j.1472-765X.2011.03043.x
McGovern, E., Waters, S. M., Blackshields, G., and McCabe, M. S. (2018). Evaluating established methods for rumen 16S rRNA amplicon sequencing with mock microbial populations. Front. Microbiol. 9:1365. doi: 10.3389/fmicb.2018.01365
Meijer, W. J., Horcajadas, J. A., and Salas, M. (2001). Phi29 family of phages. Microbiol. Mol. Biol. Rev. 65, 261–287. doi: 10.1128/MMBR.65.2.261-287.2001
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Ruger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/mmbr.67.1.86-156.2003
Mills, J. A., Crompton, L. A., Ellis, J. L., Dijkstra, J., Bannink, A., Hook, S., et al. (2014). A dynamic mechanistic model of lactic acid metabolism in the rumen. J. Dairy Sci. 97, 2398–2414. doi: 10.3168/jds.2013-7582
Minot, S., Grunberg, S., Wu, G. D., Lewis, J. D., and Bushman, F. D. (2012). Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. U.S.A. 109, 3962–3966. doi: 10.1073/pnas.1119061109
Minot, S., Sinha, R., Chen, J., Li, H. Z., Keilbaugh, S. A., Wu, G. D., et al. (2011). The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 21, 1616–1625. doi: 10.1101/gr.122705.111
Mirold, S., Rabsch, W., Rohde, M., Stender, S., Tschape, H., Russmann, H., et al. (1999). Isolation of a temperate bacteriophage encoding the type III effector protein SopE from an epidemic Salmonella typhimurium strain. Proc. Natl. Acad. Sci. U.S.A. 96, 9845–9850. doi: 10.1073/pnas.96.17.9845
Monk, A. B., Rees, C. D., Barrow, P., Hagens, S., and Harper, D. R. (2010). Under the Microscope, Bacteriophage applications: where are we now? Lett. Appl. Microbiol. 51, 363–369. doi: 10.1111/j.1472-765X.2010.02916.x
Monteiro, R., Pires, D. P., Costa, A. R., and Azeredo, J. (2019). Phage therapy: going temperate? Trends Microbiol. 27, 368–378. doi: 10.1016/j.tim.2018.10.008
Muniesa, M., Colomer-Lluch, M., and Jofre, J. (2013). Could bacteriophages transfer antibiotic resistance genes from environmental bacteria to human-body associated bacterial populations? Mob. Genet. Elements 3:e25847. doi: 10.4161/mge.25847
Muniesa, M., Garcia, A., Miro, E., Mirelis, B., Prats, G., Jofre, J., et al. (2004). Bacteriophages and diffusion of beta-lactamase genes. Emerg Infect. Dis. 10, 1134–1137. doi: 10.4161/mge.25847
Murphy, F. A., Fauquet, C. M., Bishop, D. H. L., Ghabrial, S. A., Jarvis, A. W., and Martelli et al. (1995). Virus Taxonomy, 6th Report of the International Committee on Taxonomy of Viruses. Berlin: Springer-Verlag.
Naas, A. E., Solden, L. M., Norbeck, A. D., Brewer, H., Hagen, L. H., and Heggenes et al. (2018). “Candidatus Paraporphyromonas polyenzymogenes” encodes multi-modular cellulases linked to the type IX secretion system. Microbiome 6:44. doi: 10.1186/s40168-018-0421-8
Nakamura, K., Terada, T., Sekiguchi, Y., Shinzato, N., Meng, X. Y., Enoki, M., et al. (2006). Application of pseudomurein endoisopeptidase to fluorescence in situ hybridization of methanogens within the family Methanobacteriaceae. Appl. Environ. Microbiol. 72, 6907–6913. doi: 10.1128/AEM.01499-06
Nakayama, K., Kanaya, S., Ohnishi, M., Terawaki, Y., and Hayashi, T. (1999). The complete nucleotide sequence of phi CTX, a cytotoxin-converting phage of Pseudomonas aeruginosa: implications for phage evolution and horizontal gene transfer via bacteriophages. Mol. Microbiol. 31, 399–419. doi: 10.1046/j.1365-2958.1999.01158.x
Namonyo, S., Wagacha, M., Maina, S., Wambua, L., and Agaba, M. (2018). A metagenomic study of the rumen virome in domestic caprids. Arch. Virol. 163, 3415–3419. doi: 10.1007/s00705-018-4022-4
Nemcova, R., Styriak, I., Stachova, M., and Kmet, V. (1993). Isolation and partial characterisation of three rumen Lactobacillus plantarum bacteriophages. Microbiologica 16, 177–180.
Nguyen, S., Baker, K., Padman, B. S., Patwa, R., Dunstan, R. A., Weston, T. A., et al. (2017). Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. MBio 8, e1874-17. doi: 10.1128/mBio.01874-17
Nielsen, H. B., Almeida, M., Juncker, A. S., Rasmussen, S., Li, J., Sunagawa, S., et al. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotech. 32, 822–828. doi: 10.1038/nbt.2939
Nigutova, K., Styriak, I., Javorsky, P., and Pristas, P. (2008). Partial characterization of Enterococcus faecalis bacteriophage F4. Folia Microbiol. 53, 234–236. doi: 10.1007/s12223-008-0033-y
Nolan, J. V., and Leng, R. A. (1972). Dynamic aspects of ammonia and urea metabolism in sheep. Br. J. Nutr. 27, 177–194. doi: 10.1079/bjn19720081
Nordstrom, K., and Forsgren, A. (1974). Effect of protein A on adsorption of bacteriophages to Staphylococcus aureus. J. Virol. 14, 198–202. doi: 10.1128/jvi.14.2.198-202.1974
Norman, J. M., Handley, S. A., and Virgin, H. W. (2014). Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 146, 1459–1469. doi: 10.1053/j.gastro.2014.02.001
O’Flaherty, S., Ross, R. P., and Coffey, A. (2009). Bacteriophage and their lysins for elimination of infectious bacteria. FEMS Microbiol. Rev. 33, 801–819. doi: 10.1111/j.1574-6976.2009.00176.x
Ogilvie, L., and Jones, B. (2015). The human gut virome: a multifaceted majority. Front. Microbiol. 6:918. doi: 10.3389/fmicb.2015.00918
Ogilvie, L. A., Bowler, L. D., Caplin, J., Dedi, C., Diston, D., Cheek, E., et al. (2013). Genome signature-based dissection of human gut metagenomes to extract subliminal viral sequences. Nat. Comm. 4:2420. doi: 10.1038/ncomms3420
Oliver, A., Coque, T. M., Alonso, D., Valverde, A., Baquero, F., and Canton, R. (2005). CTX-M-10 linked to a phage-related element is widely disseminated among Enterobacteriaceae in a Spanish hospital. Antimicrob. Agents Chemother. 49, 1567–1571. doi: 10.1128/aac.49.4.4567-1571.2005
O’Mahony, J., Fenton, M., Henry, M., Sleator, R. D., and Coffey, A. (2011). Lysins to kill – a tale of viral weapons of mass destruction. Bioeng. Bugs 2, 306–308. doi: 10.4161/bbug.2.6.16804
Orpin, C. G., Hazlewood, G. P., and Mann, S. P. (1988). Possibilities of the use of recombinant_DNA techniques with rumen micro-organisms. Animal Feed Sci. Technol. 21, 161–174. doi: 10.1016/0377-8401(88)90097-1
Orpin, C. G., and Munn, E. A. (1974). The occurrence of bacteriophages in the rumen and their influence on rumen bacterial populations. Experentia 30, 1018–1020. doi: 10.1007/bf01938983
Oslizlo, A., Miernikiewicz, P., Piotrowicz, A., Owczarek, B., Kopciuch, A., Figura, G., et al. (2011). Purification of phage display-modified bacteriophage T4 by affinity chromatography. BMC Biotechol. 11:59. doi: 10.1186/1472-6750-11-59
Parmar, N. R., Jakhesara, S. J., Mohapatra, A., and Joshi, C. G. (2016). Rumen virome: an assessment of viral communities and thier functions in the rumen of an Indian Buffalo. Curr. Sci. 111, 919–925. doi: 10.18520/cs/v111/i5/919-925
Parsley, L. C., Consuegra, E. J., Kakirde, K. S., Land, A. M., and Harper, W. F. Jr. et al. (2010). Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl. Environ. Microbiol. 76, 3753–3757. doi: 10.1128/aem.03080-09
Paynter, M. J. B., Ewert, D. L., and Chalupa, W. (1969). Some morphological types of bacteriophages in bovine rumen contents. Appl. Microbiol. 18, 942–943. doi: 10.1128/aem.18.5.942-943.1969
Pereira, M. S., Barreto, V. P., and Siqueira-Junior, J. P. (1997). Phage-mediated transfer of tetracycline resistance in Staphylococcus aureus isolated from cattle in Brazil. Microbiology 92, 147–155.
Popov, V. L., Tesh, R. B., Weaver, S. C., and Vasilakis, N. (2019). Electron microscopy in discovery of novel and emerging viruses from the collection of the world reference center for emerging viruses and Arboviruses (WRCEVA). Viruses 11:v11050477. doi: 10.3390/v11050477
Poullain, V., Gandon, S., Brockhurst, M. A., Buckling, A., and Hochberg, M. E. (2008). The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 62, 1–11. doi: 10.1111/j.1558-5646.2007.00260.x
Proctor, L. M., and Fuhrman, J. A. (1990). Viral mortality of marine bacteria and cyanobacteria. Nature 343, 60–62. doi: 10.1038/343060a0
Qin, J. J., Li, R. Q., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, U59–U70. doi: 10.1038/nature08821
Quirós, P., Colomer-Lluch, M., Martinez-Castillo, A., Miro, E., Argente, M., Jofre, J., et al. (2014). Antibiotic resistance genes in the bacteriophage DNA fraction of human fecal samples. Antimicrob. Agents Chemother. 58, 606–609. doi: 10.1128/aac.01684-13
Raghupathi, P. K., Zupancic, J., Brejnrod, A. D., Jacquiod, S., Houf, K., Burmolle, M., et al. (2018). Microbial diversity and putative opportunistic pathogens in dishwasher biofilm communities. Appl. Environ. Microbiol. 84:e2755-17. doi: 10.1128/AEM.02755-17
Ren, J., Ahlgren, N. A., Lu, Y. Y., Fuhrman, J. A., and Sun, F. (2017). VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 5:69. doi: 10.1186/s40168-017-0283-5
Rice, S. A., Tan, C. H., Mikkelsen, P. J., Kung, V., Woo, J., and Tay et al. (2009). The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J. 3, 271–282. doi: 10.1038/ismej.2008.109
Ritchie, A. E., Robinson, I. M., and Allison, M. J. (1970). “Rumen bacteriophage: survey of morphological types,” in Microscopie Électronique, ed. P. Favard (Paris: Societie Francaise de Microscopie Electronique), 333–334.
Rollie, C., Chevallereau, A., Watson, B. N. J., Chyou, T.-Y., Fradet, O., Mcleod, I., et al. (2020). Targeting of temperate phages drives loss of type I CRISPR–Cas systems. Nature 578, 149–153. doi: 10.1038/s41586-020-1936-2
Romero-Brey, I., and Bartenschlager, R. (2015). Viral infection at high magnification: 3D electron microscopy methods to analyze the architecture of infected cells. Viruses 7, 6316–6345. doi: 10.3390/v7122940
Ross, E., Petrovski, S., Moate, P., and Hayes, B. (2013). Metagenomics of rumen bacteriophage from thirteen lactating dairy cattle. BMC Microbiol. 13:242. doi: 10.1186/1471-2180-13-242
Rostøl, J. T., and Marraffini, L. (2019). (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe 25, 184–194. doi: 10.1016/j.chom.2019.01.009
Roux, S., Enault, F., Hurwitz, B. L., and Sullivan, M. B. (2015). VirSorter: mining viral signal from microbial genomic data. Peer J. 3:e985. doi: 10.7717/peerj.985
Russell, J. B., and Hespell, R. B. (1981). Microbial rumen fermentation. J. Dairy Sci. 64, 1153–1169. doi: 10.3168/jds.S0022-0302(81)82694-X
Russell, J. B., and Houlihan, A. J. (2003). Ionophore resistance of ruminal bacteria and its potential impact on human health. FEMS Microbiol. Rev. 27, 65–74. doi: 10.1016/S0168-6445(03)00019-6
Salmond, G. P. C., and Fineran, P. C. (2015). A century of the phage: past, present and future. Nat. Rev. Microbiol. 13, 777–786. doi: 10.1038/nrmicro3564
Sandaa, R. A., Gómez-Consarnau, L., Pinhassi, J., Riemann, L., Malits, A., Weinbauer, M. G., et al. (2009). Viral control of bacterial biodiversity – evidence from a nutrient-enriched marine mesocosm experiment. Environ. Microbiol. 11, 2585–2597. doi: 10.1111/j.1462-2920.2009.01983.x
Sanguino, L., Franqueville, L., Vogel, T. M., and Larose, C. (2015). Linking environmental prokaryotic viruses and their host through CRISPRs. FEMS Microbiol. Ecol. 91:fiv046. doi: 10.1093/femsec/fiv046
São-José, C. (2018). Engineering of phage-derived lytic enzymes: Improving their potential as antimicrobials. Antibiotics (Basel) 7:29. doi: 10.3390/antibiotics7020029
Sarker, S. A., Sultana, S., Reuteler, G., Moine, D., Descombes, P., Charton, F., et al. (2016). Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: a randomized trial in children from Bangladesh. EBioMedicine 4, 124–137. doi: 10.1016/j.ebiom.2015.12.023
Schofield, L. R., Beattie, A. K., Tootill, C. M., Dey, D., and Ronimus, R. S. (2015). Biochemical characterisation of phage pseudomurein endoisopeptidases PeiW and PeiP using synthetic peptides. Archaea 2015:828693. doi: 10.1155/2015/828693
Schuch, R., and Fischetti, V. A. (2009). The secret life of the anthrax agent Bacillus anthracis: bacteriophage-mediated ecological adaptations. PLoS ONE 4:e6532. doi: 10.1371/journal.pone.0006532
Seet, S. G. M. (2005). Genome Sequence of Bacetriophage Phi-AR29: A Basis for Integrative Plasmid Vectors. PhD Thesis, Murdoch University, Perth, WA.
Seshadri, R., Leahy, S. C., Attwood, G. T., Teh, K. H., Lambie, S. C., Cookson, A. L., et al. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechol. 36, 359–367. doi: 10.1038/nbt.4110
Sharma, M., Ryu, J. H., and Beuchat, L. R. (2005). Inactivation of Escherichia coli O157:H7 in biofilm on stainless steel by treatment with an alkaline cleaner and a bacteriophage. J. Appl. Microbiol. 99, 449–459. doi: 10.1111/j.1365-2672.2005.02659.x
Shen, M., Yang, Y., Shen, W., Cen, L., Mclean, J. S., Shi, W., et al. (2018). A linear plasmid-like prophage of Actinomyces odontolyticus promotes biofilm assembly. Appl. Environ. Microbiol. 84:e1263-18. doi: 10.1128/AEM.01263-18
Shen, Y., Mitchell, M. S., Donovan, D. M., and Nelson, D. C. (2012). “Phage-based enzybiotics,” in Bacteriophages in Health and Disease, eds P. Hyman and S. T. Abedon (Wallingford: CAB International), 217–239. doi: 10.1079/9781845939847.0217
Shibata, A., Kogure, K., Koike, I., and Ohwada, K. (1997). Formation of submicron colloidal particles from marine bacteria by viral infection. Mar. Ecol. Prog. Ser. 155, 303–307. doi: 10.3354/meps155303
Shkoporov, A. N., Khokhlova, E. V., Fitzgerald, C. B., Stockdale, S. R., Draper, L. A., Ross, R. P., et al. (2018). ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Comm. 9:4781. doi: 10.1038/s41467-018-07225-7
Shmakov, S. A., Sitnik, V., Makarova, K. S., Wolf, Y. I., Severinov, K. V., and Koonin, E. V. (2017). The CRISPR spacer space is dominated by sequences from species-specific mobilomes. mBio 8:e1397-17. doi: 10.1128/mBio.01397-17
Sillankorva, S., Oliveira, R., Vieira, M. J., Sutherland, I. W., and Azeredo, J. (2004). Bacteriophage Phi S1 infection of Pseudomonas fluorescens planktonic cells versus biofilms. Biofouling 20, 133–138. doi: 10.1080/08927010410001723834
Silveira, C. B., and Rohwer, F. L. (2016). Piggyback-the-winner in host-associated microbial communities. NPJ Biofilms Microbiom. 2:16010. doi: 10.1038/npjbiofilms.2016.10
Smith, H. W., and Huggins, M. B. (1983). Effectiveness of phages in treating experimental Escherichia coli diarrhoea in calves, piglets and lambs. Microbiology 129, 2659–2675. doi: 10.1099/00221287-129-10-3121
Smith, H. W., Huggins, M. B., and Shaw, K. M. (1987). Factors influencing the survival and multiplication of bacteriophages in calves and in their environment. J. Gen. Microbiol. 133, 1127–1135. doi: 10.1099/00221287-133-5-1127
Smith, K., Collier, A., Townsend, E. M., O’Donnell, L. E., Bal, A. M., and Butcher et al. (2016). One step closer to understanding the role of bacteria in diabetic foot ulcers: characterising the microbiome of ulcers. BMC Microbiol. 16:54. doi: 10.1186/s12866-016-0665-z
Smoot, J. C., Barbian, K. D., Van Gompel, J. J., Smoot, L. M., Chaussee, M. S., Sylva, G. L., et al. (2002). Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc. Natl. Acad. Sci. U.S.A. 99, 4668–4673. doi: 10.1073/pnas.062526099
Snelling, T., and Wallace, R. (2017). The rumen microbial metaproteome as revealed by SDS-PAGE. BMC Microbiol. 17:9. doi: 10.1186/s12866-016-0917-y
Solden, L. M., Naas, A. E., Roux, S., Daly, R. A., Collins, W. B., Nicora, C. D., et al. (2018). Interspecies cross-feeding orchestrates carbon degradation in the rumen ecosystem. Nat. Microbiol. 3, 1274–1284. doi: 10.1038/s41564-018-0225-4
Soni, K. A., and Nannapaneni, R. (2010). Removal of Listeria monocytogenes biofilms with bacteriophage P100. J. Food Prot. 73, 1519–1524. doi: 10.4315/0362-028x-73.8.1519
Steward, G., Smith, D., and Azam, F. (1996). Abundance production of bacteria and viruses in the Bering and Chukchi Seas. Mar. Ecol. Prog. Ser. 131, 287–300. doi: 10.3354/meps131287
St-Pierre, B., and Wright, A. D. G. (2012). Diversity of gut methanogens in herbivorous animals. Animal 7, 49–56. doi: 10.1017/S1751731112000912
Styriak, I., Galfi, P., and Kmet, V. (1991). Preliminary observations of interaction between bacteriophages and Streptococcus bovis bacteria on ruminal epithelium primoculture. Vet. Microbiol. 29, 281–287. doi: 10.1016/0378-1135(91)90135-3
Styriak, I., Kmet, V., and Spanova, A. (1989). Isolation and characterisation of two rumen Streptococcus bovis bacteriophages. Microbiologica 12, 317–322.
Styriak, I., Pristas, P., and Javorsky, P. (2000). Lack of GATC sites in the genome of Streptococcus bovis bacteriophage F4. Res. Microbiol. 151, 285–289. doi: 10.1016/s0923-2508(00)00148-0
Styriak, I., Spanova, A., Montagova, H., and Kmet, V. (1994). Isolation and characterisation of a new ruminal bacteriophage lytic to Streptococcus bovis. Curr. Microbiol. 28, 355–358. doi: 10.1007/BF01570201
Styriak, I., Spanova, A., and Zitnan, R. (2005). Partial characterization of two ruminal bacteriophages with similar restriction patterns and different capsids morphology. Archiv. Tierzucht-Arch. Anim. Breed. 48, 572–579. doi: 10.5194/aab-48-572-2005
Sulakvelidze, A. (2005). Phage therapy: an attractive option for dealing with antibiotic-resistant bacterial infections. Drug Discov. Today 10, 807–809. doi: 10.1016/S1359-6446(05)03441-0
Sullivan, M. B., Coleman, M. L., Quinlivan, V., Rosenkrantz, J. E., Defrancesco, A. S., Tan, G., et al. (2008). Portal protein diversity and phage ecology. Environ. Microbiol. 10, 2810–2823. doi: 10.1111/j.1462-2920.2008.01702.x
Sutherland, I. W., Hughes, K. A., Skillman, L. C., and Tait, K. (2004). The interaction of phage and biofilms. FEMS Microbiol. Lett. 232, 1–6. doi: 10.1016/S0378-1097(04)00041-2
Swain, R. A. (1999). Factors Affecting Bacteriophages of the Rumen Ecosystem Ph.D Thesis. University of New England, Armidale, NSW.
Swain, R. A., Nolan, J. V., and Klieve, A. V. (1996a). Natural variability and diurnal fluctuations within the bacteriophage population of the rumen. Appl. Environ. Microbiol. 62, 994–997. doi: 10.1128/aem.62.3.994-997.1996
Swain, R. A., Nolan, J. V., and Klieve, A. V. (1996b). Effect of tannic aicd on the bacteriophage population within sheep rumen. Microbiol. Aust. 17:A87. doi: 10.1128/aem.62.3.994-997.1996
Swidsinski, A., Verstraelen, H., Loening-Baucke, V., Swidsinski, S., Mendling, W., and Halwani, Z. (2013). Presence of a polymicrobial endometrial biofilm in patients with bacterial vaginosis. PLoS ONE 8:e53997. doi: 10.1371/journal.pone.0053997
Tamada, H., Harasawa, R., and Shinjo, T. (1985). Isolation of a bacteriophage in Fusobacterium necrophorum. Jpn. J. Vet. Sci. 47, 483–486. doi: 10.1292/jvms1939.47.483
Tarakanov, B. V. (1972). The electron-microscopy examination of the microflora of reindeer rumen. Mikrobiologia 41, 862–870.
Tarakanov, B. V. (1974). Lysogenic cultures of Streptococcus bovis isolated from rumen of cattle and sheep. Microbiologica 43, 375–377.
Tarakanov, B. V. (1976). Biological properties of Streptococcus bovis bacteriophages isolated from lysogenic cultures and sheep rumen. Microbiologiia 45, 695–700.
Tarakanov, B. V. (1994). Regulation of microbial processes in the rumen by bacteriophages of Streptococcus bovis. Microbiology 63, 373–378. (translated from Mikrobiologiya 363, 657–667).
Thingstad, T. F. (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 45, 1320–1328. doi: 10.4319/lo.2000.45.6.1320
Touchon, M., Moura, De Sousa, J. A., and Rocha, E. P. (2017). Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. doi: 10.1016/j.mib.2017.04.010
Twort, F. W. (1915). An investigation on the nature of ultra-microscopic viruses. Lancet 2, 1241–1243. doi: 10.1016/S0140-6736(01)20383-3
Tyutikov, F. M., Bespalova, I. A., Rebentish, B. A., Aleksanrushkina, N. N., and Krivisky, A. S. (1980). Bacteriophages of Methanotropic bacteria. J. Bacteriol. 144, 375–381.
Ubukata, K., Konno, M., and Fujii, R. (1975). Transduction of drug resistance to tetracycline, chloramphenicol, macrolides, lincomycin and clindamycin with phages induced from Streptococcus pyogenes. J. Antibiot. (Tokyo) 28, 681–688. doi: 10.7164/antibiotics.28.681
Varble, A., Meaden, S., Barrangou, R., Westra, E. R., and Marraffini, L. A. (2019). Recombination between phages and CRISPR-cas loci facilitates horizontal gene transfer in Staphylococci. Nat. Microbiol. 4, 956–963. doi: 10.1038/s41564-019-0400-2
Vidakovic, L., Singh, P. K., Hartmann, R., Nadell, C. D., and Drescher, K. (2018). Dynamic biofilm architecture confers individual and collective mechanisms of viral protection. Nat. Microbiol. 3, 26–31. doi: 10.1038/s41564-017-0050-1
Wang, X., Kim, Y., Ma, Q., Hong, S. H., Pokusaeva, K., Sturino, J. M., et al. (2010). Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1:147. doi: 10.1038/ncomms1146
Weimer, P. J. (2015). Redundancy, resilience, and host specificity of the ruminal microbiota: implications for engineering improved ruminal fermentations. Front. Microbiol. 6:296. doi: 10.3389/fmicb.2015.00296
Weinbauer, M. G. (2004). Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28, 127–181. doi: 10.1016/j.femsre.2003.08.001
Weinbauer, M. G., and Höfle, M. G. (1998). Significance of viral lysis and flagellate grazing as factors controlling bacterioplankton production in a eutrophic lake. Appl. Environ. Microbiol. 64:431. doi: 10.1128/aem.64.2.431-438.1998
Wells, J. E., and Russell, J. B. (1996). Why do many ruminal bacteria die and lyse so quickly? J. Dairy Sci. 79, 1487–1495. doi: 10.3168/jds.S0022-0302(96)76508-6
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 10, 2557–2568. doi: 10.1038/ismej.2016.45
Willi, K., Sandmeier, H., Kulik, E. M., and Meyer, J. (1997). Transduction of antibiotic resistance markers among Actinobacillus actinomycetemcomitans strains by temperate bacteriophages Aaφ23. Cell. Mol. Life Sci. 53, 904–910. doi: 10.1007/s000180050109
Williamson, K. E., Helton, R. R., and Wommack, K. E. (2012). Bias in bacteriophage morphological classification by transmission electron microscopy due to breakage or loss of tail structures. Microbiol. Res. Technol. 75, 452–457. doi: 10.1002/jemt.21077
Willner, D., and Hugenholtz, P. (2013). From deep sequencing to viral tagging: recent advances in viral metagenomics. Bioessays 35, 436–442. doi: 10.1002/bies.201200174
Willner, D., Thurber, R. V., and Rohwer, F. (2009). Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol. 11, 1752–1766. doi: 10.1111/j.1462-2920.2009.01901.x
Wilmes, P., and Bond, P. L. (2004). The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 6, 911–920. doi: 10.1111/j.1462-2920.2004.00687.x
Wolf, J. L., Rubin, D. H., Finberg, R., Kauffman, R. S., Sharpe, A. H., Trier, J. S., et al. (1981). Intestinal M cells: a pathway for entry of reovirus into the host. Science 212, 471–472. doi: 10.1126/science.6259737
Wolochow, H., Hildebrand, G. J., and Lamanna, C. (1966). Translocation of microorganisms across the intestinal wall of the rat: effect of microbial size and concentration. J. Infect. Dis. 116, 523–528. doi: 10.1093/infdis/116.4.523
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/mmbr.64.1.69-114.2000
Wong, C. M., Klieve, A. V., Hamdorf, B. J., Schafer, D. J., Brau, L., Seet, S. G. M., et al. (2003). Family of shuttle vectors for ruminal Bacteroides. J. Mol. Microbiol. Biotechnol. 5, 123–132. doi: 10.1159/000069982
Wood, S. R., Kirkham, J., Marsh, P. D., Shore, R. C., Nattress, B., and Robinson, C. (2000). Architecture of intact natural human plaque biofilms studied by confocal laser scanning microscopy. J. Dent. Res. 79, 21–27. doi: 10.1177/00220345000790010201
Wright, A. (1971). Mechanism of conversion of the Salmonella O antigen by bacteriophage ε34. J. Bacteriol. 105, 927–936. doi: 10.1128/jb.105.3.927-936.1971
Wylie, T. N., Wylie, K. M., Herter, B. N., and Storch, G. A. (2015). Enhanced virome sequencing using targeted sequence capture. Genome Res. 25, 1910–1920. doi: 10.1101/gr.191049.115
Yamaguchi, T., Hayashi, T., Takami, H., Nakasone, K., Ohnishi, M., Nakayama, K., et al. (2000). Phage conversion of exfoliative toxin A production in Staphylococcus aureus. Mol. Microbiol. 38, 694–705. doi: 10.1046/j.1365-2958.2000.02169.x
Yooseph, S., Sutton, G., Rusch, D. B., Halpern, A. L., Williamson, S. J., Remington, K., et al. (2007). The sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol. 5:e16. doi: 10.1371/journal.pbio.0050016
Yutin, N., Kapitonov, V. V., and Koonin, E. V. (2015). A new family of hybrid virophages from an animal gut metagenome. Biol. Direct 10:19. doi: 10.1186/s13062-015-0054-9
Zhao, G., Wu, G., Lim, E. S., Droit, L., Krishnamurthy, S., Barouch, D. H., et al. (2017). VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 503, 21–30. doi: 10.1016/j.virol.2017.01.005
Zheng, T., Li, J., Ni, Y., Kang, K., Misiakou, M. A., Imamovic, L., et al. (2019). Mining, analyzing, and integrating viral signals from metagenomic data. Microbiome 7:42. doi: 10.1186/s40168-019-0657-y
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: A fast phage search tool. Nucl. Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: rumen, virus, phage, metagenomics, proteomics, biofilm, methanogen
Citation: Gilbert RA, Townsend EM, Crew KS, Hitch TCA, Friedersdorff JCA, Creevey CJ, Pope PB, Ouwerkerk D and Jameson E (2020) Rumen Virus Populations: Technological Advances Enhancing Current Understanding. Front. Microbiol. 11:450. doi: 10.3389/fmicb.2020.00450
Received: 26 November 2019; Accepted: 02 March 2020;
Published: 26 March 2020.
Edited by:
Diego P. Morgavi, INRA Centre Auvergne-Rhône-Alpes, FranceReviewed by:
Rajib Saha, University of Nebraska–Lincoln, United StatesCopyright © 2020 Gilbert, Townsend, Crew, Hitch, Friedersdorff, Creevey, Pope, Ouwerkerk and Jameson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalind A. Gilbert, Um9zLkdpbGJlcnRAZGFmLnFsZC5nb3YuYXU=; Eleanor Jameson, RWxlYW5vci5KYW1lc29uQHdhcndpY2suYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.