 Jing Niu1,2†
Jing Niu1,2† Long Xu2,3†
Long Xu2,3† Yun Qian2,3Zhuo Sun2Dongbao Yu4
Yun Qian2,3Zhuo Sun2Dongbao Yu4 Jiandong Huang4Xiaolin Zhou4Yizhong Wang5
Jiandong Huang4Xiaolin Zhou4Yizhong Wang5 Ting Zhang5
Ting Zhang5 Rongrong Ren6Zhengpeng Li6
Rongrong Ren6Zhengpeng Li6 Jialin Yu1,2*
Jialin Yu1,2* Xuefeng Gao2,3*
Xuefeng Gao2,3*- 1Department of Pediatrics, Shenzhen University General Hospital, Shenzhen, China
- 2Shenzhen University Clinical Medical Academy, Shenzhen, China
- 3Department of Gastroenterology and Hepatology, Shenzhen University General Hospital, Shenzhen, China
- 4Shenzhen Hoiracle Bio-Tech Co., Ltd., Shenzhen, China
- 5Department of Gastroenterology, Hepatology, and Nutrition, Shanghai Children’s Hospital, Shanghai Jiao Tong University, Shanghai, China
- 6Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing, China
Temporal development of the human gut microbiome from infancy to childhood is driven by a variety of factors. We surveyed the fecal microbiome of 729 Chinese children aged 0–36 months, aiming to identify the age-specific patterns of microbiota succession, and evaluate the impact of birth mode, gender, geographical location, and gastrointestinal tract symptoms on the shaping of the gut microbiome. We demonstrated that phylogenetic diversity of the gut microbiome increased gradually over time, which was accompanied by an increase in Bacteroidetes and a reduction in Proteobacteria species. Analysis of community-wide phenotypes revealed a succession from aerobic bacteria and anaerobic bacteria to facultative anaerobes, and from Gram-negative to Gram-positive species during gut microbiota development in early childhood. The metabolic functions of the gut microbiome shifted tremendously alongside early physiological development, including an increase in alanine, aspartate, and glutamate metabolism, and a reduction in glutathione, fatty acid, and tyrosine metabolism. During the first year of life, the Bacteroidetes phylum was less abundant in children born by casarean section compared with those delivered vaginally. The Enterococcaceae family, a group of facultative anaerobic microorganisms with pathogenic potential, was predominant in preterm infants. No measurable effect of maternal antibiotic exposure on gut microbiota development was found in the first 3 years of life. The relative abundances of Coriobacteriaceae and Streptococcaceae families, and Megasphaera genus were found to be higher in girls than in boys. Among the three first-tier Chinese cities, children born and fed in Beijing had a higher abundance of Enterococcaceae and Lachnospiraceae families, and Shenzhen children had a higher abundance of Fusobacteriaceae. The families Alcaligenaceae, Bacteroidaceae, and Porphyromonadaceae were more abundant in children with constipation, whereas the relative abundance of the Clostridium genus was higher in those with diarrhea.
Introduction
The development of the human gut microbiome in early life has lasting effects on the host, and aberrancies in this process may affect health in adulthood (Rautava et al., 2012; Arrieta et al., 2014). Gut microbial alterations during early infancy have been shown to affect the risk of childhood obesity (Koleva et al., 2015), type 1 diabetes (Kostic et al., 2015), non-alcoholic fatty liver disease (NAFLD) (Nash et al., 2017), asthma (Arrieta et al., 2015), and allergic disease (Low et al., 2017; Candy et al., 2018). Therefore, an understanding of gut microbiota development in early life could provide insight into how its development impacts immune development, and lead to microbial-based therapeutics that target disease prevention at an early age. Gut microbiota development can be affected by a range of early life events, such as the mode of delivery (cesarean section versus vaginal delivery), maternal antibiotic usage, breast feeding/formula/mixed feeding, and the cession of breast-feeding (Penders et al., 2006; Gronlund et al., 2007; Dominguez-Bello et al., 2010; Ladirat et al., 2013; Azad et al., 2014; Nylund et al., 2014; Voreades et al., 2014). To date, relatively few studies have investigated the developmental patterns of the gut microbiome during early life in large, multicenter populations.
The Environmental Determinants of Diabetes in the Young (TEDDY) study is a collaborative effort between six research centers in the United States and Europe, aimed at uncovering the environmental causes of type I diabetes (Vatanen et al., 2018). The TEDDY study population produced a robust analysis of gut microbiome development in 783 infants from the age of 3 to 48 months. Using metagenomic sequencing, the TEDDY study demonstrated that the infant microbiome, both in terms of its composition and function, underwent dynamic changes in the first year of life that were exclusively dominated by three Bifidobacterium species (B. bifidum, B. breve, or B. longum) or by the Proteobacteria phylum. Individual variations were most significant during the early months, and inter-subject differences contributed to 35% of the microbial taxonomic variation (Vatanen et al., 2018). Moreover, factors including age, geographical location, and feeding type also had strong effects on the composition of the gut microbiome (Vatanen et al., 2018). Despite the dynamic and individualized nature of infant microbiome development, the TEDDY study identified a range of consistent patterns in the changes of microbial metabolic enzymes, which reflected major shifts in microbial structure and diet during infant microbiome development (Vatanen et al., 2018).
Given the importance of gut microbial development, a new perspective proposes to use microbes as clues to understand human postnatal development (Subramanian et al., 2015). However, the evolution of gut microbial development during early life remains poorly understood. Moreover, the infant microbiome in a predominantly Chinese population has never been explored (Bokulich et al., 2016). Here, we profiled the gut microbial development of a large cohort of Chinese infants during the first 36 months of life and evaluated the effect of delivery modes and digestive disorders on the composition of the gut microbial community.
Materials and Methods
Study Population
A total of 1156 children under 3 years of age were enrolled in this study during January 2016 and December 2018. Written informed consent was obtained from parents prior to study. The collection and use of infant fecal samples were approved by the Regional Ethical Review Board of the Shanghai Children’s Hospital. Infant stool samples were collected by parents, enclosed in 2-ml tubes containing 1.2 ml RNAlater (QIAGEN, Hilden, Germany), and delivered to the HRK (Hoiracle Bio-Tech Co., Ltd., Shenzhen, China.)-biotech laboratory within 48 h. All samples were stored at −80°C. Parents were asked to complete questionnaires and provide metadata regarding pregnancy and medical records of the mothers, as well as provide the delivery mode, feeding, and medical records of the infants. Children were classified as having constipation if they had <1 stool/day before the introduction of solid foods or <3 stools/week after the introduction of solid foods. Diarrhea was defined as watery/loose stools that increased in frequency compared to usual. Any subjects who had birth defects, genetic diseases, metabolic disorders (diabetes or cholestasis), or used systemic antibiotics (intravenous, intramuscular, or oral) one month before sample collection were excluded from this study.
Fecal Microbiome Analysis
The QIAamp Fast DNA Stool Mini Kit (QIAGEN, Hilden, Germany) was used to isolate total microbial DNA from the stool samples using a previously reported procedure (Wang et al., 2018). The 16S rRNA V3-V4 hypervariable regions (encompassing approximately 469 bp) were PCR-amplified with dual-indexed PCR primers. The same procedures for DNA extraction and PCR amplification were applied to water samples as a control. The size and quality of the purified amplicons were checked with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States). An equal amount of each amplicon was pooled and sequenced using the Illumina MiSeq platform, producing 300 or 250 bp paired-end reads.
Raw forward and reverse reads were joined and quality trimmed using USEARCH v10.0.240 with the -fastq_filter command and -fastq_maxee 1.0. After filtering, samples having less than 5000 reads were removed, resulting in 729 samples for later analyses. USEARCH’s fastx_uniques command was used to generate a FASTA file containing unique sequences. NOISE3 was performed to remove chimeras in the unique sequences and generate a BIOM table with representative zero-radius operational taxonomic units (OTUs) for subsequent analyses. The greengenes 13_5 reference database was employed for taxonomic assignment (DeSantis et al., 2006).
The OTU counts were normalized by total sum normalization (TSS) using Calypso (version 8.84) (Zakrzewski et al., 2017), and cumulative-sum scaling (CSS) was applied to correct biases introduced by TSS. Data were then log2 transformed to account for the non-normal distribution of taxonomic count data. Gut microbiome alpha diversity was measured using the Shannon, Chao1, and Simpson indices. The beta diversity between samples was calculated through Principal Coordinate Analysis (PCoA) using the Bray-Curtis distance based on genus abundance. Statistical differences in alpha diversity were determined by the Wilcoxon rank-sum test for the comparison of two groups, and one-way ANOVA followed by Tukey’s test for a comparison of three or more groups. An analysis of the composition of microbiomes (ANCOM) was performed on the top 300 most abundant taxa at the phylum and family levels to determine differentially abundant taxa (Mandal et al., 2015). ANCOM accounts for compositionality of microbiota survey data by reducing false discoveries in detecting differentially abundant taxa (Weiss et al., 2017).
KEGG Ortholog functional profiles of microbial communities were predicted using PICRUSt (Langille et al., 2013) based on the OTUs identified by QIIME 1.9 (Caporaso et al., 2010), and the statistical significance was determined by the Linear Discriminant Analysis (LDA) Effect Size (LEfSe) method (Segata et al., 2011). Microbiome phenotypes at the organism-level were predicted and analyzed using BugBase (Ward et al., 2017). The Spearman correlation coefficient was used to demonstrate the associations between the age of children and the gut microbiome composition, as well as the PICRUSt-predicted metabolic pathways.
Results
Study Cohort
As shown in Table 1, 729 children from 27 regions (provinces or cities) across China were selected for analysis, including 317 girls (43.5%) and 412 boys (56.5%), to investigate the evolutionary dynamics of the gut microbiome. The mean age of the children was 11 ± 8.6 months, and the mean gestational age was 39 ± 1.8 weeks. The characteristics of study participants from each region are given in Supplementary Table S1. We classified the children into twelve age groups: from birth to 3 months (n = 65), 4–6 months (n = 95), 7–9 months (n = 131), 10–12 months (n = 130), 13–15 months (n = 91), 16–18 months (n = 65), 20–21 months (n = 36), 22–24 months (n = 30), 25–27 months (n = 11), 28–30 months (n = 22), 31–33 months (n = 27), and 34–36 months (n = 26). 471 children (64.6%) were born via vaginal delivery, and 258 (35.4%) were born by cesarean section. 86 (11.8%) were constipated at the time of sample collection, and 75 (10.29%) had diarrhea.
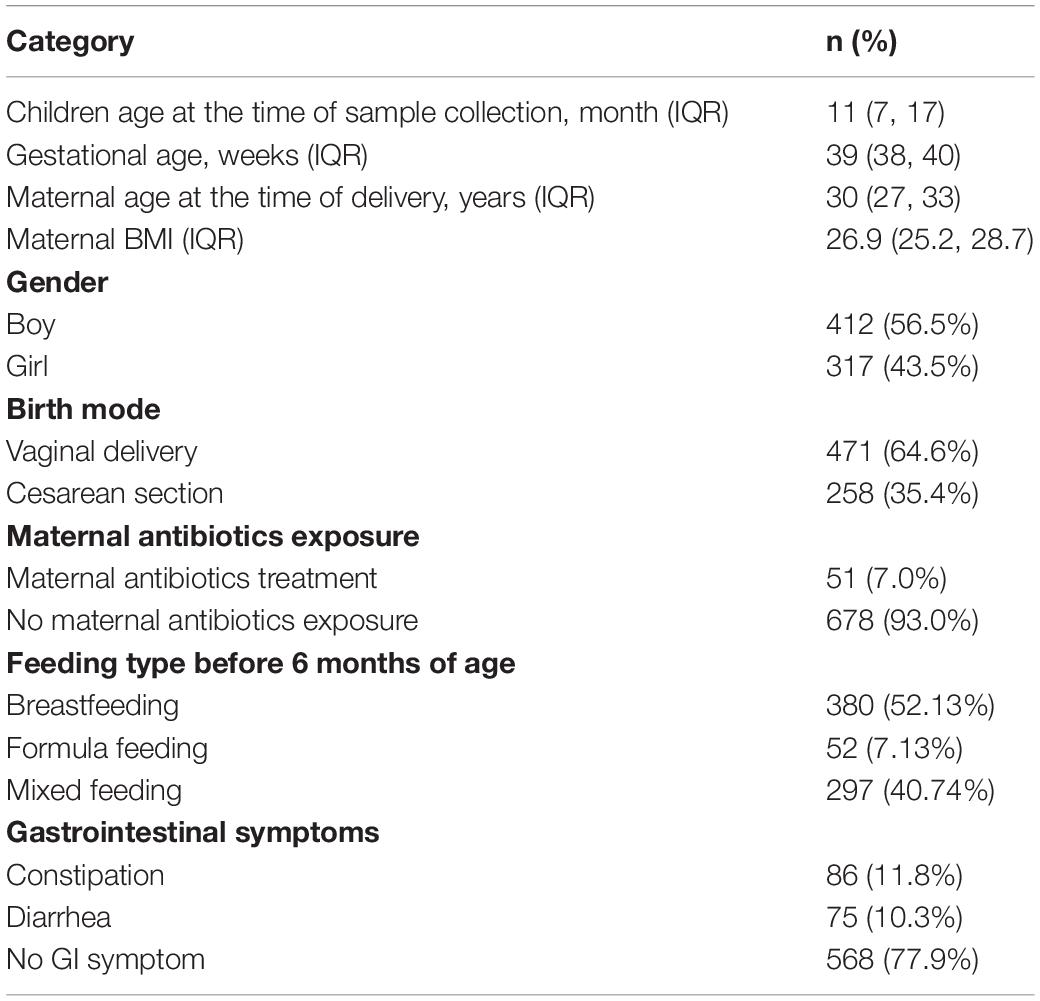
Table 1. Characteristics of the study population.
Dynamics of Gut Microbiome Assembly During the First Three Years of Life
Our data revealed that the gut microbiome alpha diversity increased dramatically from birth to 36 months (Figures 1A–C). A transverse gradient from birth to 36 months was obtained (Figure 1D), indicating progressive changes in the microbial community structure. Comparison of the compositional features of the gut microbiota alongside the age spectrum revealed several characteristic patterns. At the phylum level, Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria were the dominant bacteria across the first 3 years of life (Figure 1E). The abundance of Bacteroidetes increased dramatically from birth to 27 months, and then remained stable until 36 months, while the abundance of Proteobacteria showed an opposite trend, with a significant decrease from birth to 24 months. The relative abundance of Firmicutes remained relatively stable over time (Figure 1E). At the family level, the relative abundances of Bacteroidaceae, Prevotellaceae, and Ruminococcaceae increased progressively from birth to 36 months, and Ruminococcaceae showed a significant positive correlation with age (R = 0.61, Table 2). In contrast, Enterobacteriaceae and Veillonellaceae gradually decreased during the first 3 years of life (Figure 1F). Enterobacteriaceae and Enterococcaceae were negatively correlated with age, with R = −0.59 and −0.53, respectively (Table 2).
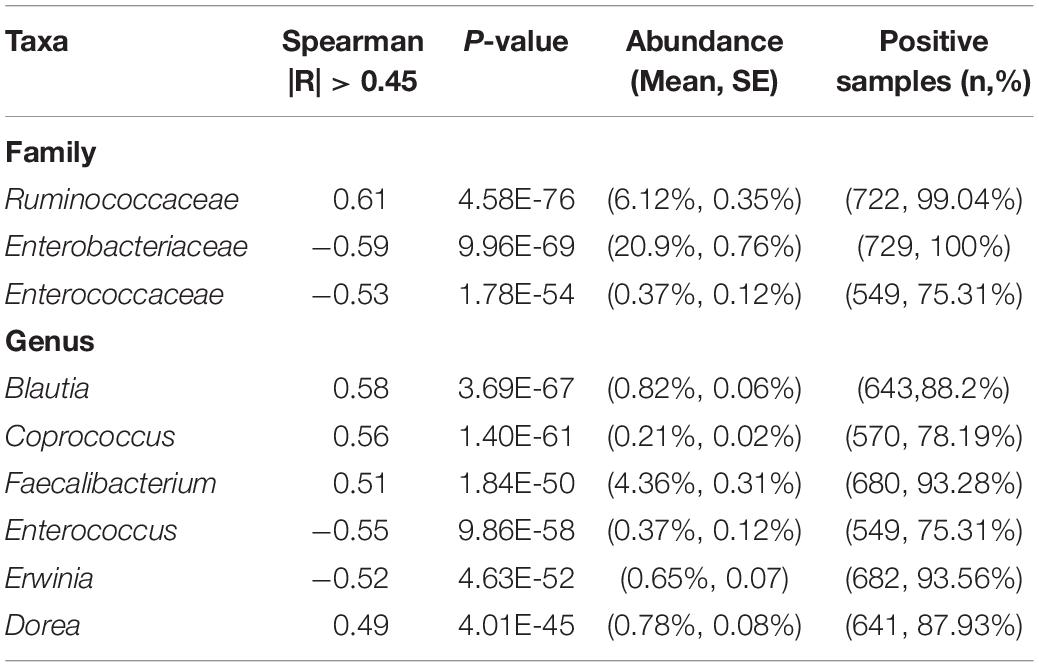
Table 2. Taxa in the gut microbiome that significantly correlate with age.
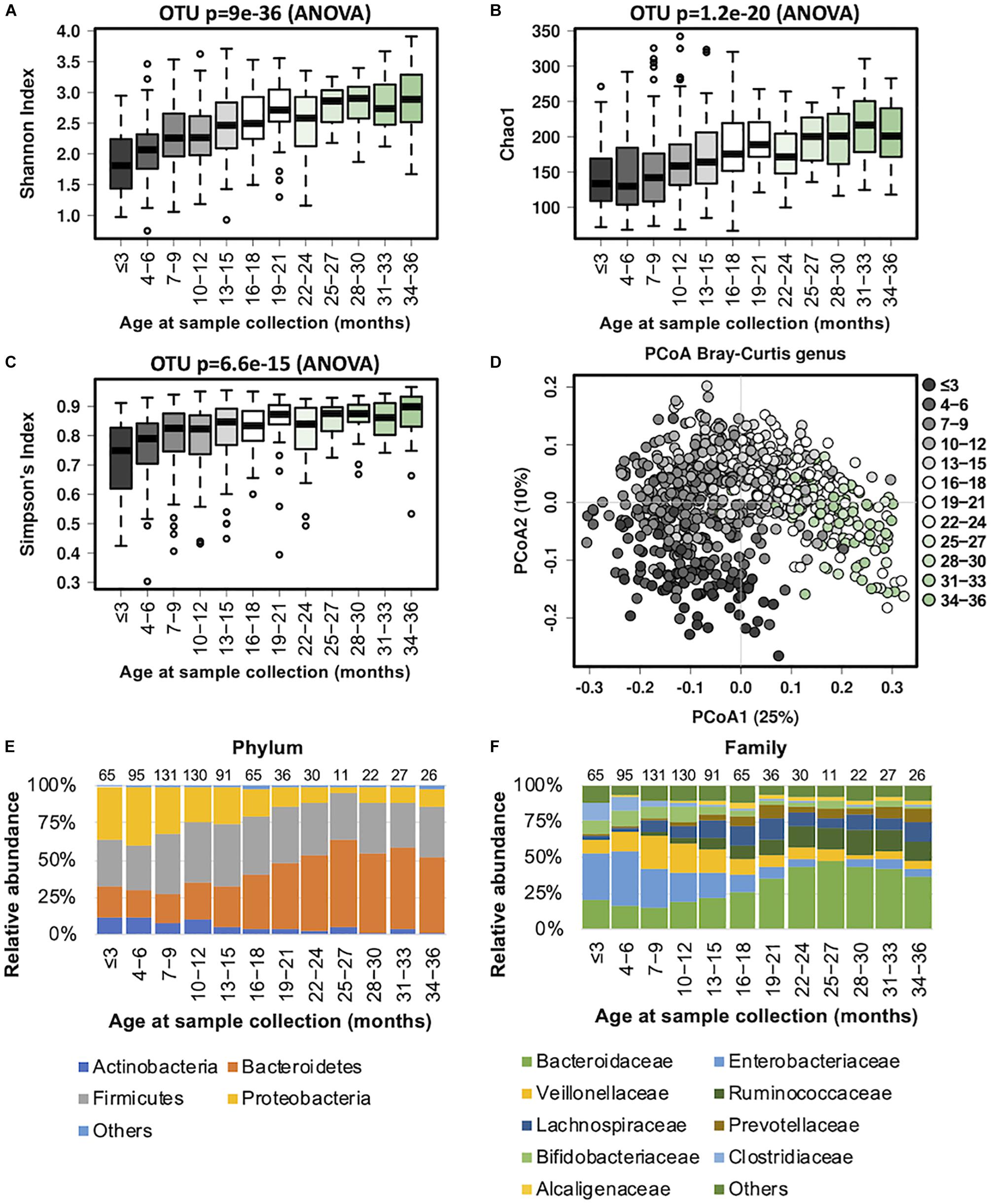
Figure 1. Fecal microbiota diversity and composition for 729 children according to age (0–3 years). Longitudinal alpha diversity from 0 to 36 months of age was measured by (A) Shannon index, (B) Chao1, and (C) Simpson’s index. (D) PCoA of Bray–Curtis distance based on the genus level. Each sample is colored according to age. Evolution of the gut microbiome at the (E) phylum and (F) family levels alongside physiological development. Number of children per age group is provided above bars.
Progressive changes in microbial diversity and abundance of the infant gut microbiome are likely to re-shape the metabolic functions of the host over time. We used BugBase to infer common microbial phenotypes from our 16S rRNA sequencing data, including the anticipated oxygen tolerance and Gram staining of present species. Along the lines of previous reports (Backhed et al., 2015; Bokulich et al., 2016; Chu et al., 2017; Nagpal et al., 2017), our data showed that the early gastrointestinal tract is predominantly populated with facultative anaerobes (such as Enterococcaceae), and progressively shifts to obligate anaerobic microorganisms, resulting in a decreased oxidative stress tolerance (Supplementary Figure S1). The ratio of Gram-negative to Gram-positive bacterial species was higher at the beginning of life, and significantly decreased alongside physical growth. The proportion of mobile element containing, biofilm forming, and pathogenic bacteria also decreased with age.
We also observed that the predicted metabolic functions went through dynamic changes during the first 3 years of child development (Supplementary Figure S2A). The KEGG pathways associated with transcription machinery, streptomycin biosynthesis, oxidative phosphorylation, methane metabolism, alanine, aspartate and glutamate metabolism, drug metabolism other enzymes, and one carbon pool by folate were progressively upregulated over time (with Spearman correlation |R| > 0.45 and p < 0.05; Table 3). In contrast, glutathione metabolism, inorganic ion transport and metabolism, valine, leucine and isoleucine degradation, ubiquinone and other terpenoid quinone, fatty acid metabolism, the phosphotransferase system, the secretion system, propanoate metabolism, the bacterial secretion system, and tyrosine metabolism were downregulated over time. Together, these changes indicate a clear trajectory of gut microbiome development in the first 3 years of life.
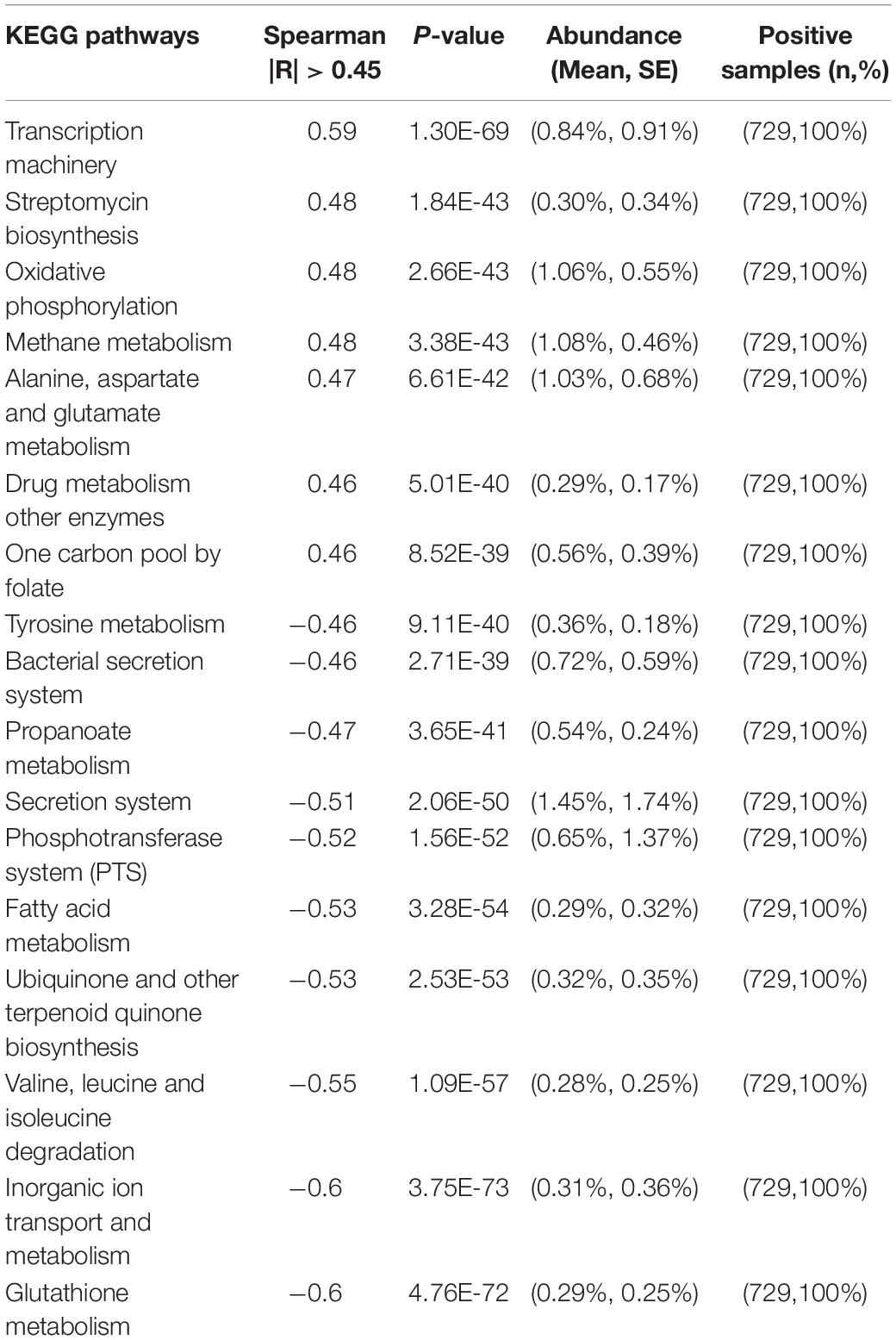
Table 3. Predicted functions of the gut microbiome that significantly correlate with age.
Birth Mode Related Differences in the Gut Microbiome of Young Children
Casarean section (C-section) accounts for 35% of all deliveries in China (35.4% from our study cohort), which is higher than in many other developed countries (Li et al., 2017). Birth mode was shown to be significantly associated with the gut microbiome during the developmental phase (Stewart et al., 2018). We compared the gut microbiome between the two birth modes and found no significant difference in the alpha diversity (Figures 2A–C). PCoA analysis of the beta diversity further indicated that the microbial composition of both modes could be superimposed onto each other (Figure 2D). During the first 18 months of life, the Firmicutes and Proteobacteria phyla predominated in children born via C-section, whereas Bacteroidetes were more abundant in those delivered by vaginal birth (Figure 2E). At the family level, the abundance of Enterococcaceae was higher in C-section-delivered children during the first year of life, whereas Bacteroidaceae was enriched in those born vaginally (Figure 2F). The difference in the gut microbiome composition between the two birth modes diminished after the age of 12 months. Overall, the relative abundance of the Bacteroidaceae family and Bacteroides genus were significantly higher in vaginally born infants compared with those who were born via C-section (ANCOM p < 0.05, Supplementary Table S2).
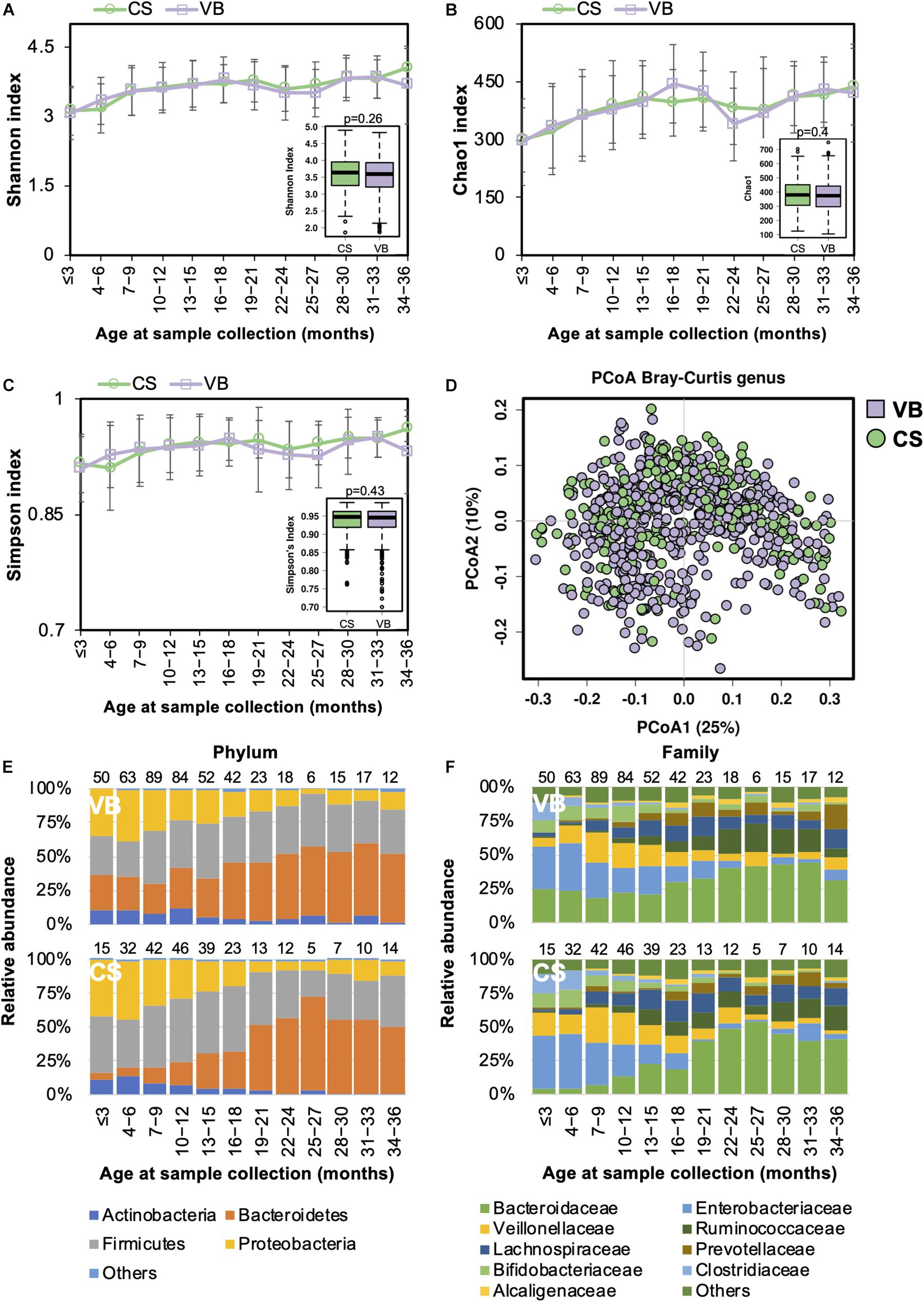
Figure 2. Fecal microbiota diversity and composition for 729 children according to delivery mode. Alpha diversity, which was calculated by (A) Shannon index, (B) Chao1, or (C) Simpson’s index, showed no significant difference between the two birth modes. (D) PCoA showed no clear separation between samples based on the two birth modes. Birth-mode-stratified taxa summary composition of bacterial microbiota at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Functionally, the relative abundance of transporter genes was more abundant in children delivered via C-section (Supplementary Figure S2B).
Gut Microbiome Development Is Similar Between Preterm and Term Children
In preterm infants, microbial colonization is challenging due to organ immaturity, the length of hospital stay, and the use of antibiotics. In our cohort, 51 children were preterm, born with gestational ages less than 37 weeks (Table 1). The complexity of the microbiota increased in both preterm children and in those children born at full term simultaneously (Figures 3A–C). Beta diversity analysis with PCoA revealed that the gut bacterial community structure was not distinguishable between preterm and term children (Figure 3D). During the first 6 months, Enterococcaceae (Proteobacteria phylum), a group of facultative anaerobic microorganisms with pathogenic potential, was predominant in preterm infants (Figures 3E,F). This was in line with previous studies showing that the gut microbiota of preterm infants was impacted by a bloom of opportunistic and potentially pathogenic bacteria (Arboleya et al., 2012; Barrett et al., 2013). Children born preterm showed a lower relative abundance of Blautia genus than those born full term (ANCOM p < 0.05, Supplementary Table S3). No significant difference in the gut microbiota functional profile was detected between children born preterm and those born full term.
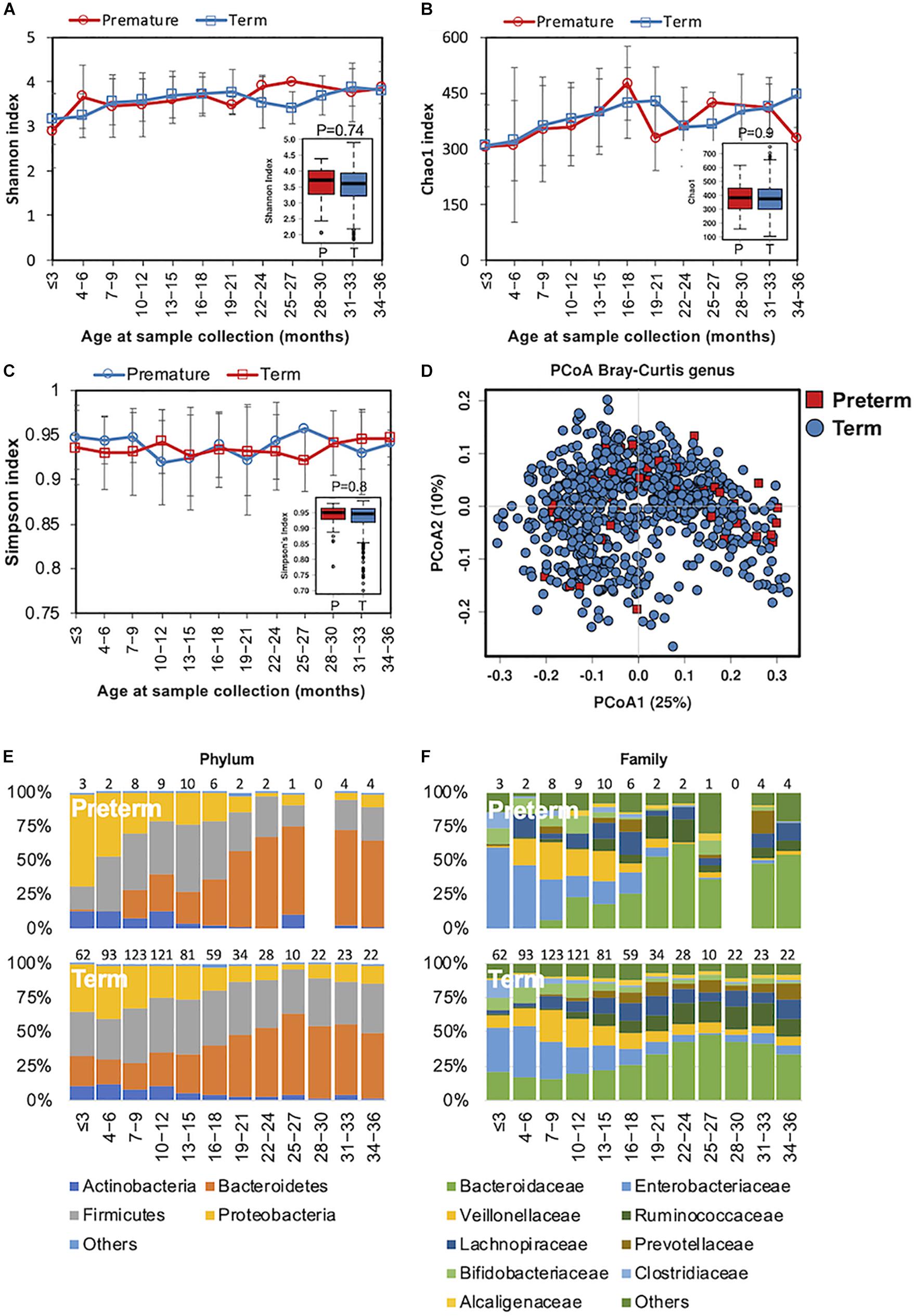
Figure 3. Fecal microbiota diversity and composition for 729 children according to gestational age. Alpha diversity was calculated with (A) Shannon index, (B) Chao1, and (C) Simpson’s index, and showed no significant difference between premature and full-term children. (D) PCoA showed no clear separation between samples from premature and full-term children. Gestational age-stratified taxa summary composition of bacterial microbiota at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Maternal Antibiotic Exposure During Pregnancy May Not Affect the Early Gut Microbiota
Both animal and human studies have shown that antibiotic use by mothers during pregnancy confers an altered microbiota to their offspring (Mueller et al., 2015; Gonzalez-Perez et al., 2016; Kuperman and Koren, 2016). In our study, the mothers of 40 children received antibiotic treatments during pregnancy. We examined if gut microbiota development was influenced by maternal antibiotics treatment (MAT). Alpha diversity in MAT children was not significantly different to those whose mothers had no antibiotic exposure (NA) during pregnancy (Figures 4A–C). The compositional structure of the gut microbiota could not be distinguished by beta diversity analysis with PCoA (Figure 4D). The pattern of gut microbiota development was not disturbed by maternal antibiotic exposure at the phylum or family levels (Figures 4E,F). During the first 3 years of life, there were no significant differences in the gut microbiota composition (at the phylum, family, or genus levels) or functions between children based on maternal antibiotic exposure.
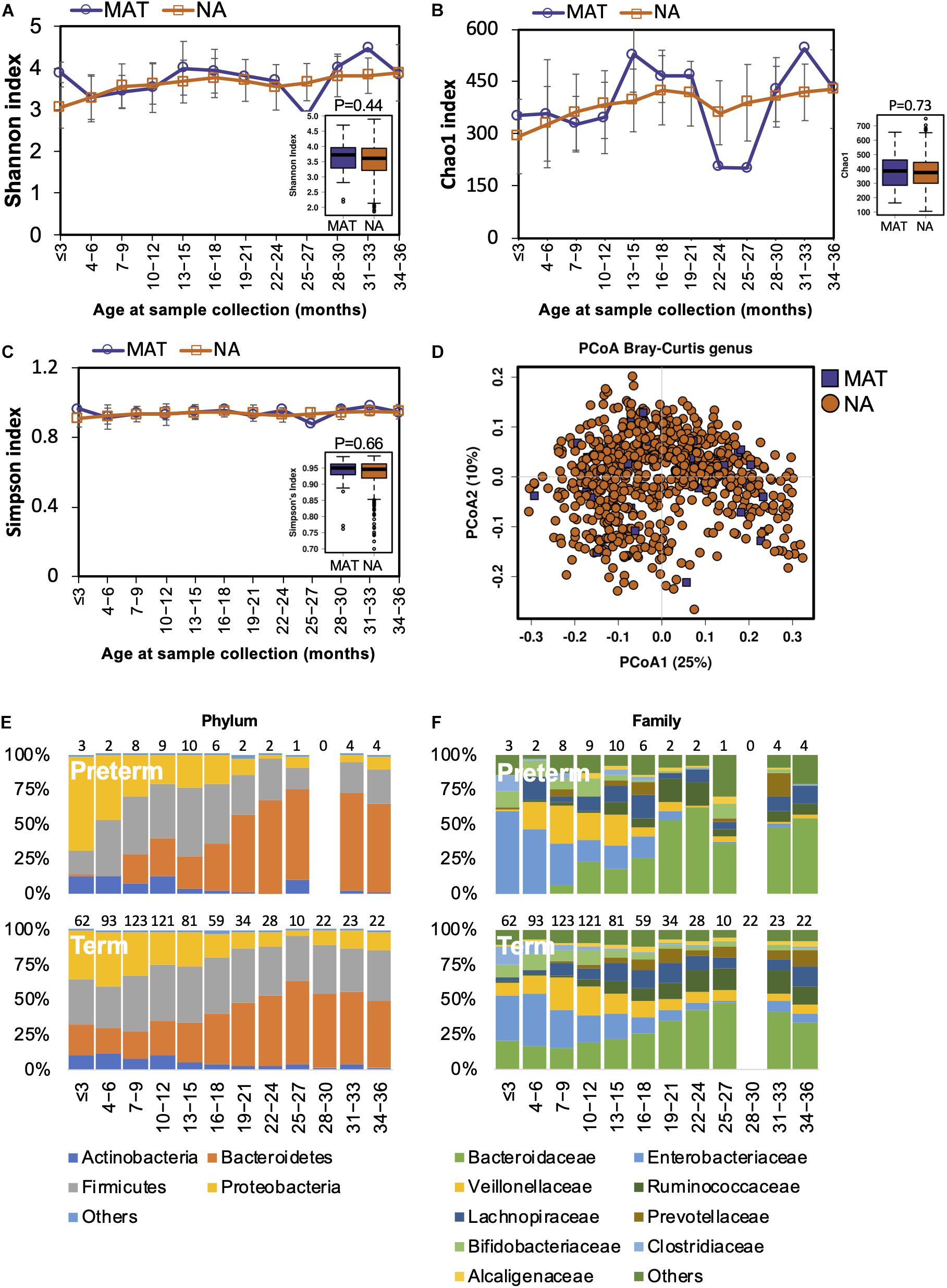
Figure 4. Fecal microbiota diversity and composition for 729 children according to maternal antibiotic exposure. Alpha diversity, which was calculated with (A) Shannon index, (B) Chao1, and (C) Simpson’s index, showed no significant difference between MAT and NA children. (D) PCoA showed no clear separation between samples from MAT and NA children. The gut microbiota development patterns in MAT and NA children at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Gut Microbiome Development in Different Genders
Differences in the gut microbiota of Chinese men and women were reported in our previous study (Gao et al., 2018). In this study, we also tested if the gender-associated differences in the gut microbiota originated from early life. Both alpha and beta diversity were not significantly different between genders (Figures 5A–D). The developmental trajectory of the gut microbiome was similar between genders (Figures 5E,F). Nevertheless, the relative abundances of Coriobacteriaceae and Streptococcaceae families and the Megasphaera genus were more abundant in girls (Supplementary Table S4). Based on the predicted function of metagenomes, no significant difference in the predicted metabolic functions were detected between genders.
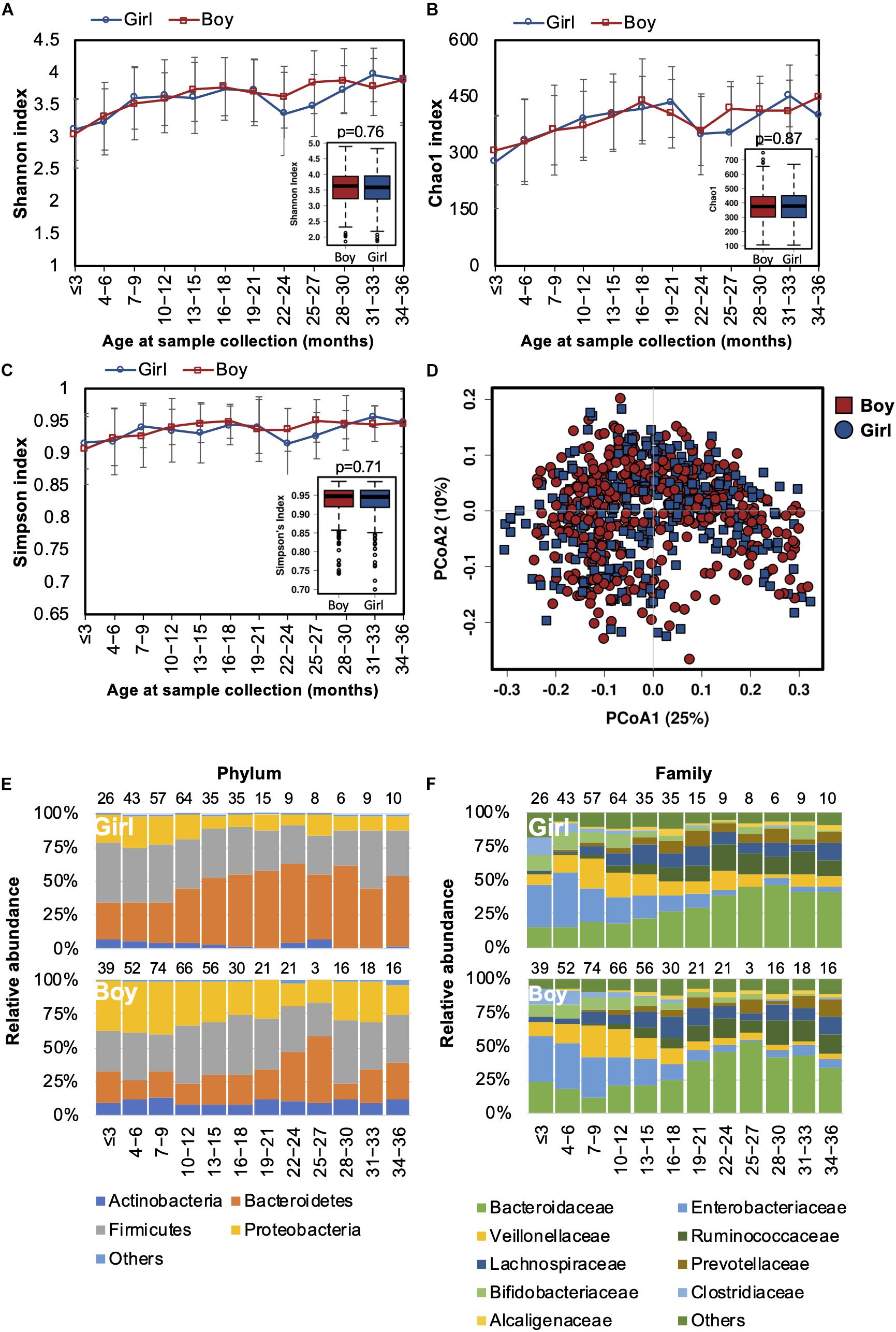
Figure 5. Fecal microbiota diversity and composition for 729 children based on gender. Alpha diversity was calculated with (A) Shannon index, (B) Chao1, or (C) Simpson’s index, and showed no significant difference between genders. (D) PCoA showed no clear separation of the gut microbiota composition between boys and girls. Gender-stratified taxa summary composition of bacterial microbiota at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Geographical Location Affects the Gut Microbiome Composition of Young Children
As lifestyle and dietary patterns play an important role in shaping our gut microbiota, we compared gut microbiota biodiversity and composition of children born and raised in three first-tier cities in China from other distinct geographical areas, including Beijing, Shanghai, and Shenzhen. However, no significant difference in either alpha or beta diversity was detected between geographical groups (Figures 6A–D). The trajectory of gut microbiota development was similar among children from different cities (Figures 6E,F). Nevertheless, significant differences in the relative abundances of the gut microbiome were identified in the children from the three first-tier cities. A higher relative abundance of Lachnospiraceae family was observed in children from Beijing (ANCOM p < 0.05, Supplementary Table S5); the Fusobacteriaceae family (Fusobacterium genus) was more abundant in children from Shenzhen, while the relative abundance of Enterococcaceae was lower in children from Shanghai.
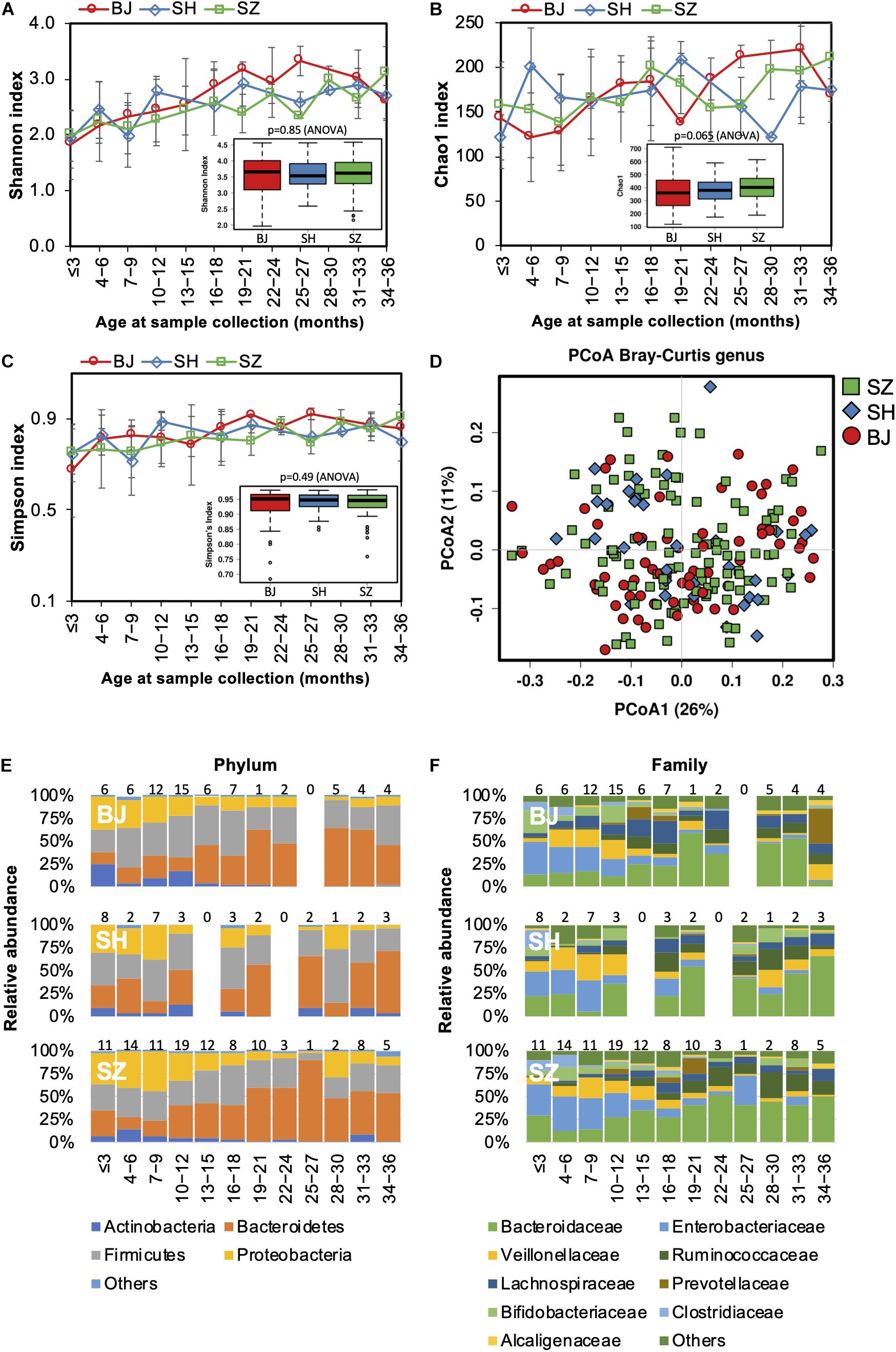
Figure 6. Fecal microbiota diversity and composition for 729 children according to geographical location. Alpha diversity, which was calculated with (A) Shannon index, (B) Chao1, and (C) Simpson’s index, showed no significant difference among the children from three first-tier cities of China. (D) PCoA showed no clear separation among samples of different geographical locations. Gut bacterial microbiota composition at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Geographical variation in the functional profile of the gut microbiome was not detected in our study.
Alterations in the Gut Microbiome Are Associated With Gastrointestinal Disorders in Young Children
Changes in the gut microbiome have been shown to be associated with an increased incidence of gastrointestinal symptoms in early life (Mayer et al., 2014). We next characterized the gut microbiota in infants with constipation (<1 stool/day before the introduction of solid food or <3 stools/week after the introduction of solid food) or diarrhea (watery/loose stools that increase in frequency relative to usual). The bacterial alpha diversity was significantly higher in children with constipation than the alpha diversity in those with diarrhea and without GI symptoms (Figures 7A–C). PCoA results showed no clear separation between samples from children with and without GI symptoms (Figure 7D). Thus, GI disorders did not disturb the overall gut microbiota trajectory (Figures 7E,F). At the family level, however, Alcaligenaceae, Bacteroidaceae, and Porphyromonadaceae were significantly enriched in constipated children, while Enterobacteriaceae and Gemellaceae were diminished in them when compared with those with diarrhea or without GI symptoms (Supplementary Table S6). At the genus level, lower abundances of Gemella and Salmonella, and higher abundances of Bacteroides and Blautia, were observed in children with constipation compared to children without. There was a lower abundance of Bacteroides and a higher abundance of Clostridium in children with diarrhea compared to those with constipation or without GI symptoms.
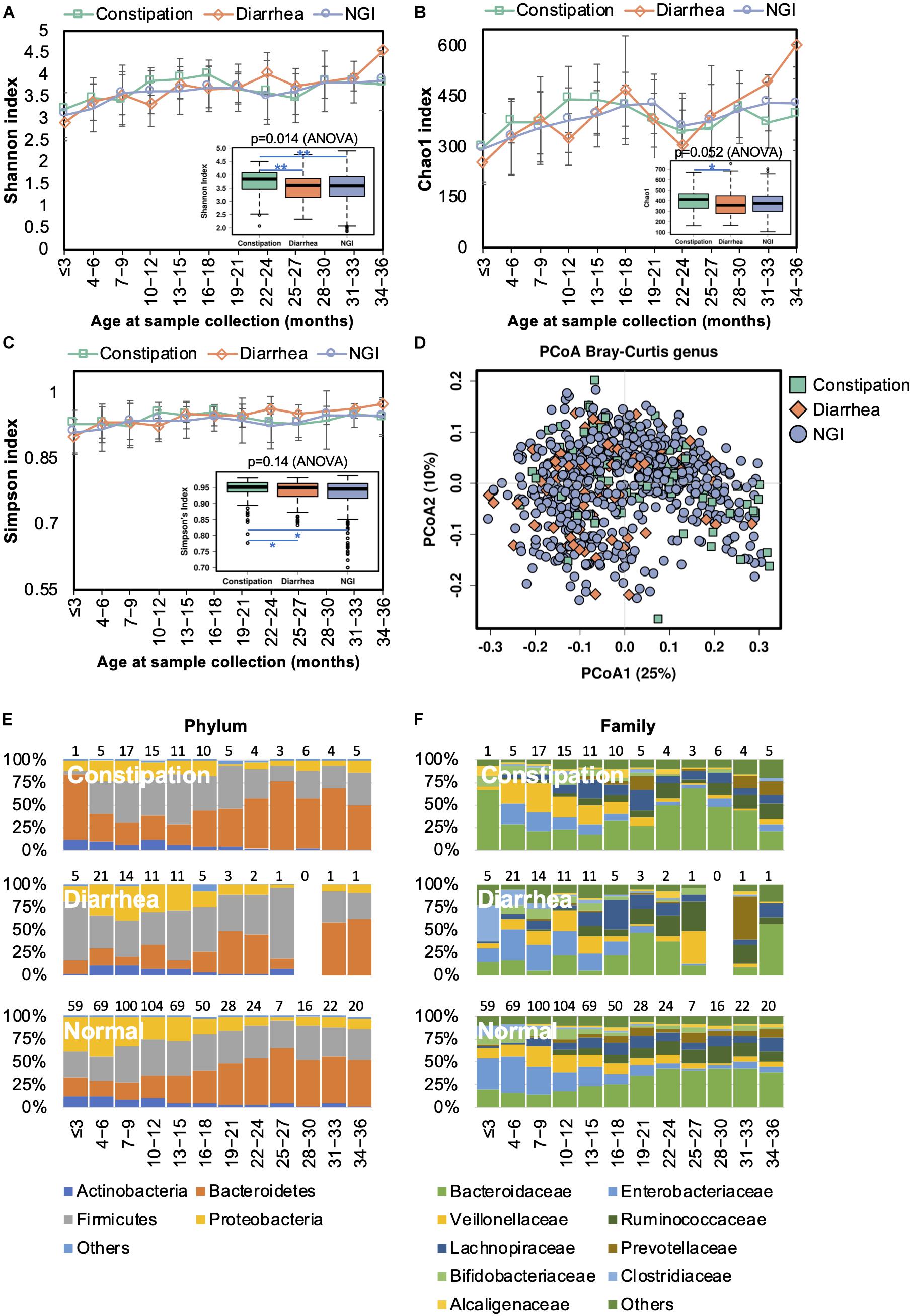
Figure 7. Fecal microbiota diversity and composition for 729 children according to gastrointestinal symptoms. Alpha diversity, which was calculated with (A) Shannon index, (B) Chao1, and (C) Simpson’s index, was found to be higher in children with constipation. (D) PCoA showed no clear separation among samples from children with and without GI symptoms. Gut bacterial microbiota composition at the (E) phylum and (F) family levels. Number of children per age group is provided above bars.
Functional divergence associated with GI symptoms was detected by PICRUSt analysis. Functional genes associated with transporters, ABC transporters, the secretion system, transcription factors, the phosphotransferase system, the two component system, and bacterial motility proteins were significantly upregulated in the gut microbiome of children with diarrhea (Supplementary Figure S2C), whereas ribosome-related genes were enriched in the gut microbiome of children with constipation.
Discussion
The initial colonization of the human gut microbiome is influenced by a wide range of factors that may have long-term physiological consequences (Dominguez-Bello et al., 2010). Consistent with previous studies (Koenig et al., 2011; Yatsunenko et al., 2012), our data shows that the infant gut microbiome undergoes dynamic changes during the first 36 months of life. Newborns have a low microbial diversity, which gradually increases thereafter. The structure of the microbiome community also undergoes dynamic changes during the first 36 months of life. Our data demonstrate that the infant gut microbiome was predominated by facultative anaerobes such as Enterobacteriaceae in the first months of life, which is in line with findings among infants from Japan (Nagpal et al., 2017), United States (Bokulich et al., 2016; Chu et al., 2017), and Europe (Backhed et al., 2015). This is because the newborn gut is a predominantly aerobic environment at birth (Arrieta et al., 2014). Then the gut gradually becomes anaerobic, thereby allowing the colonization of strict anaerobes such as Bifidobacterium (Actinobacteria phylum), Clostridium (Firmicutes phylum), and Bacteroides (Bacteroidetes phylum) (Matamoros et al., 2013). Consistently, we also found that fecal samples collected from infants aged 0–3 years were predominated by the phyla Proteobacteria and Firmicutes, followed by Bacteroidetes and Actinobacteria.
Despite similar developmental patterns in accordance with the timing of food change, the gut microbiota of infants from different countries exhibit different types of microbial composition. These are typified by P-type (abundant in Proteobacteria), A-type (abundant in Actinobacteria), and F-type (abundant in Firmicutes) developmental patterns (Kuang et al., 2016). Most infants from western countries are A-type, with 82% of American, 79% of Canadian, and 54% of Swedish infants being A-type, whereas most of those in Bangladeshi (70%) are F-type (Kuang et al., 2016). It has been reported that Chinese infants (age < 3 months) are predominately P-type (Kuang et al., 2016). Consistently, our data also demonstrated that Chinese infants aged 0–3 months were dominated by Proteobacteria. The establishment of a complex microbiota proceeds faster in developing rather than developed countries due to poor sanitation. For example, Enterobacteriaceae (Gram-negative bacteria) was found to be colonized in Chinese and Pakistani infants during the first weeks of life (Adlerberth et al., 1998), whereas this group of bacteria gradually increased their colonization rate in Swedish infants over the first 6 months of life (Adlerberth et al., 2006).
Although host genetic background accounts for a substantial fraction of the abundance of most common microbiota, the different microbial compositions observed in different studies can readily be explained by that fact that vaginal and skin microbial compositions may vary between mothers from different geographical locations (Huang et al., 2014; Gupta et al., 2017). It was reported that geographic location contributes more to the interindividual differences in the composition of the gut microbiome than other factors (He et al., 2018). In our data, differences in gut microbiota composition have also been observed within China. Among the enrolled subjects, children born and fed in Beijing had higher abundances of families Enterococcaceae and Lachnospiraceae, and the Fusobacterium genus was enriched in children from Shenzhen. Thus, regional variations should be considered when investigating the associations between gut microbiota and infant health and disease.
Diet plays a major role in the shifting of gut microbiota in early life. During the first few months of life, the primary source of nutrition is breast milk or formula. Therefore, the infant intestine at this stage favors the propagation of microbes such as Bifidobacterium that can ferment milk oligosaccharides. Introduction of solid food is a major contributor in the shift of the gut microbiome toward an adult-like structure, which was previously characterized by an increase of Bacteroidetes and the Firmicutes and a decrease in Actinobacteria and Proteobacteria phyla (Arrieta et al., 2014). The rationale behind this change is that the infant diet at this stage contains many polysaccharides that cannot be digested by their immature digestive system, thereby triggering an increase of the aforementioned microbes to facilitate this process. During 12–36 months of life, the infant gut microbiota develops progressively into an adult-like gut microbiota, which is dominated by Firmicutes and Bacteroidetes phyla (these make up >90% of the gut microbial population) (Ringel-Kulka et al., 2013; Voreades et al., 2014).
Previous studies have suggested that early patterns of the infant gut microbiota are impacted by delivery mode (Dominguez-Bello et al., 2010; Lozupone et al., 2013; Matamoros et al., 2013; Arrieta et al., 2014; Jakobsson et al., 2014). A relatively lower gut microbial diversity was previously reported in C-section-delivered infants at 2 years of age (Jakobsson et al., 2014). This decline in biodiversity has been attributed to delayed colonization of Bacteroidetes. Nevertheless, no difference in alpha diversity between the two birth modes was detected in our data. The microbial populations in the infant gut are generally considered to be similar to the microbiota the infant is exposed to at birth (Dominguez-Bello et al., 2010; Lozupone et al., 2013). For infants born by vaginal delivery, the intestinal microbiota resembles their mothers’ vaginal microbiota, which is dominated by Lactobacillus (phylum Firmicutes), Prevotella (phylum Bacteroidetes), or Sneathia (phylum Fusobacteria). In contrast, the gut microbiome of C-section-delivered infants is similar to the mothers’ skin microbiome, which has predominately Staphylococcus (phylum Firmicutes), Corynebacterium (phylum Actinobacteria), and Propionibacterium (phylum Actinobacteria) (Dominguez-Bello et al., 2010). A previous study of Chinese newborns (neonates and 2-month-olds) showed that vaginal delivery results in an enrichment of Bacteroides, Parabacteroides, and Megamonas, whereas C-section delivery led to an enrichment of Prevotella, Streptococcus, and Trabulsiella (Kuang et al., 2016). It is worth noting that Prevotella, which are enriched in vaginally delivered Amerindian infants (Dominguez-Bello et al., 2010), are also found in C-section-delivered Chinese infants (Kuang et al., 2016). Our data also shows that Bacteroidaceae was enriched in the gut of vaginally delivered infants, which was consistent with previous findings (Kuang et al., 2016). Taken together, these results suggest a role for the delivery mode of an infant in structuring the gut microbiota in early life.
Changes in gut microbiota composition have been associated with a variety of GI disorders, such as constipation and diarrhea. Children with functional constipation were shown with a higher fecal level of Clostridium and Bifidobacterium species (Zoppi et al., 1998). In adults with constipation, a depletion of members belonging to the Bacteroides, Roseburia, and Coprococcus phyla in the gut microbiota was observed (Mancabelli et al., 2017). Our data showed a variety of differences in the gut microbiome of children with constipation compared with their counterparts without any GI symptoms, as represented by an increased biodiversity and an increase in the relative abundance of families Alcaligenaceae, Bacteroidaceae, and Porphyromonadaceae. Regarding children with diarrhea, the Clostridium genus, including the pathogenic species, was found to be enriched. All together, these results show the close relationship between gut microbiota and gastrointestinal disorders in children.
Conclusion
With a total of 729 children participating, this is the first comprehensive study of gut microbiome development in young Chinese children. From our results, the evolution of the gut microbiome during early life appears to be defined by shared phylogenetic trajectories. The infant gut microbiome is gradually established during the first 3 years of life. Factors such as birth mode, gestational age, antibiotic exposure, gender, geographical location and GI disorders could affect relative abundances of several groups of gut microbes.
Data Availability Statement
The 16S rRNA sequence data reported in this study have been deposited in the European Nucleotide Archive (ENA) database, under accession number PRJEB35531.
Ethics Statement
The studies involving human participants were reviewed and approved by The Regional Ethical Review Board in Shanghai Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
XG and JY conceived the presented study. JN, ZS, RR, ZL, and YW collected the data. JH and XZ performed the sequencing. XG and DY performed the data analysis. JN and LX wrote the manuscript. YQ, YW, and TZ provided critical feedback and helped shape the research, analysis and manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 81900474, 81571483, and 81971431), Shenzhen key technology R&D program (JSGG20170413152936281), and Shenzhen Science and Technology Innovation Free Exploration Project (JCYJ20170817100735621).
Conflict of Interest
DY, JH, and XZ were employed by Hoiracle Bio-Tech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00439/full#supplementary-material
TABLE S1 | Characteristics of the enrolled children from each region.
TABLE S2 | Significant differences in the gut microbiota composition between two birth modes as determined using ANCOM.
TABLE S3 | Significant differences in the gut microbiota composition between premature and full-term children as determined using ANCOM.
TABLE S4 | Significant differences in the gut microbiota composition between genders as determined by ANCOM.
TABLE S5 | Significant differences in the gut microbiota composition between children from three first-tier cities in China as determined using ANCOM.
TABLE S6 | Significant differences in the gut microbiota composition between children with and without GI symptoms as determined using ANCOM.
FIGURE S1 | Biological phenotypes of gut microbiome change over development. BugBase was employed to predict the proportion of aerobic, anaerobic, facultatively anaerobic, oxidative stress tolerant, Gram-negative, Gram-positive, mobile element containing, biofilm forming, and pathogenic bacteria within the gut microbiome of each age group. Kruskal–Wallis test was implemented to determine significant differences between age groups.
FIGURE S2 | Prediction of metagenome functional content using PICRUSt. (A) The top 40 most abundant metabolism pathways evolved during the first 3 years of age. Metabolism pathways significantly different between children (B) born by vaginal delivery or C-section (CS), and (C) those with or without GI symptoms. The significance was determined by LEFSe, with an LDA score >3.0 and p < 0.05.
References
Adlerberth, I., Jalil, F., Carlsson, B., Mellander, L., Hanson, L. A., Larsson, P., et al. (1998). High turnover rate of Escherichia coli strains in the intestinal flora of infants in Pakistan. Epidemiol. Infect 121, 587–598. doi: 10.1017/s0950268898001484
Adlerberth, I., Lindberg, E., Aberg, N., Hesselmar, B., Saalman, R., Strannegard, I. L., et al. (2006). Reduced enterobacterial and increased staphylococcal colonization of the infantile bowel. an effect of hygienic lifestyle? Pediatr. Res. 59, 96–101. doi: 10.1203/01.pdr.0000191137.12774.b2
Arboleya, S., Binetti, A., Salazar, N., Fernandez, N., Solis, G., Hernandez-Barranco, A., et al. (2012). Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. Ecol. 79, 763–772. doi: 10.1111/j.1574-6941.2011.01261.x
Arrieta, M. C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M., and Finlay, B. (2014). The intestinal microbiome in early life. health and disease. Front. Immunol. 5:427. doi: 10.3389/fimmu.2014.00427
Arrieta, M. C., Stiemsma, L. T., Dimitriu, P. A., Thorson, L., Russell, S., Yurist-Doutsch, S., et al. (2015). Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 7:307ra152. doi: 10.1126/scitranslmed.aab2271
Azad, M. B., Bridgman, S. L., Becker, A. B., and Kozyrskyj, A. L. (2014). Infant antibiotic exposure and the development of childhood overweight and central adiposity. Int. J. Obes 38, 1290–1298. doi: 10.1038/ijo.2014.119
Backhed, F., Roswall, J., Peng, Y., Feng, Q., Jia, H., Kovatcheva-Datchary, P., et al. (2015). Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703. doi: 10.1016/j.chom.2015.04.004
Barrett, E., Kerr, C., Murphy, K., O’Sullivan, O., Ryan, C. A., Dempsey, E. M., et al. (2013). The individual-specific and diverse nature of the preterm infant microbiota. Arch. Dis. Child Fetal Neonatal. Ed. 98, F334–F340. doi: 10.1136/archdischild-2012-303035
Bokulich, N. A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H., et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8:343ra382. doi: 10.1126/scitranslmed.aad7121
Candy, D. C. A., Van Ampting, M. T. J., Oude Nijhuis, M. M., Wopereis, H., Butt, A. M., Peroni, D. G., et al. (2018). A synbiotic-containing amino-acid-based formula improves gut microbiota in non-IgE-mediated allergic infants. Pediatr. Res. 83, 677–686. doi: 10.1038/pr.2017.270
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Chu, D. M., Ma, J., Prince, A. L., Antony, K. M., Seferovic, M. D., and Aagaard, K. M. (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326. doi: 10.1038/nm.4272
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/aem.03006-05
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Gao, X., Zhang, M., Xue, J., Huang, J., Zhuang, R., Zhou, X., et al. (2018). Body mass index differences in the gut microbiota are gender specific. Front. Microbiol. 9:1250. doi: 10.3389/fmicb.2018.01250
Gonzalez-Perez, G., Hicks, A. L., Tekieli, T. M., Radens, C. M., Williams, B. L., and Lamouse-Smith, E. S. (2016). Maternal antibiotic treatment impacts development of the neonatal intestinal microbiome and antiviral immunity. J. Immunol. 196, 3768–3779. doi: 10.4049/jimmunol.1502322
Gronlund, M. M., Gueimonde, M., Laitinen, K., Kociubinski, G., Gronroos, T., Salminen, S., et al. (2007). Maternal breast-milk and intestinal bifidobacteria guide the compositional development of the Bifidobacterium microbiota in infants at risk of allergic disease. Clin. Exp. Allergy 37, 1764–1772. doi: 10.1111/j.1365-2222.2007.02849.x
Gupta, V. K., Paul, S., and Dutta, C. (2017). Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 8:1162. doi: 10.3389/fmicb.2017.01162
He, Y., Wu, W., Zheng, H. M., Li, P., McDonald, D., Sheng, H. F., et al. (2018). Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 24, 1532–1535. doi: 10.1038/s41591-018-0164-x
Huang, B., Fettweis, J. M., Brooks, J. P., Jefferson, K. K., and Buck, G. A. (2014). The changing landscape of the vaginal microbiome. Clin. Lab. Med. 34, 747–761. doi: 10.1016/j.cll.2014.08.006
Jakobsson, H. E., Abrahamsson, T. R., Jenmalm, M. C., Harris, K., Quince, C., Jernberg, C., et al. (2014). Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566. doi: 10.1136/gutjnl-2012-303249
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4578–4585. doi: 10.1073/pnas.1000081107
Koleva, P. T., Bridgman, S. L., and Kozyrskyj, A. L. (2015). The infant gut microbiome. evidence for obesity risk and dietary intervention. Nutrients 7, 2237–2260. doi: 10.3390/nu7042237
Kostic, A. D., Gevers, D., Siljander, H., Vatanen, T., Hyotylainen, T., Hamalainen, A. M., et al. (2015). The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17, 260–273. doi: 10.1016/j.chom.2015.01.001
Kuang, Y. S., Li, S. H., Guo, Y., Lu, J. H., He, J. R., Luo, B. J., et al. (2016). Composition of gut microbiota in infants in China and global comparison. Sci. Rep. 6:36666. doi: 10.1038/srep36666
Kuperman, A. A., and Koren, O. (2016). Antibiotic use during pregnancy. how bad is it? BMC Med. 14:91. doi: 10.1186/s12916-016-0636-0
Ladirat, S. E., Schols, H. A., Nauta, A., Schoterman, M. H., Keijser, B. J., Montijn, R. C., et al. (2013). High-throughput analysis of the impact of antibiotics on the human intestinal microbiota composition. J. Microbiol. Methods 92, 387–397. doi: 10.1016/j.mimet.2012.12.011
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, H. T., Luo, S., Trasande, L., Hellerstein, S., Kang, C., Li, J. X., et al. (2017). Geographic variations and temporal trends in cesarean delivery rates in China, 2008-2014. JAMA 317, 69–76. doi: 10.1001/jama.2016.18663
Low, J. S. Y., Soh, S. E., Lee, Y. K., Kwek, K. Y. C., Holbrook, J. D., Van der Beek, E. M., et al. (2017). Ratio of Klebsiella/Bifidobacterium in early life correlates with later development of paediatric allergy. Benef Microbes 8, 681–695. doi: 10.3920/BM2017.0020
Lozupone, C. A., Stombaugh, J., Gonzalez, A., Ackermann, G., Wendel, D., Vazquez-Baeza, Y., et al. (2013). Meta-analyses of studies of the human microbiota. Genome Res. 23, 1704–1714. doi: 10.1101/gr.151803.112
Mancabelli, L., Milani, C., Lugli, G. A., Turroni, F., Mangifesta, M., Viappiani, A., et al. (2017). Unveiling the gut microbiota composition and functionality associated with constipation through metagenomic analyses. Sci. Rep. 7:9879. doi: 10.1038/s41598-017-10663-w
Mandal, S., Van Treuren, W., White, R. A., Eggesbo, M., Knight, R., and Peddada, S. D. (2015). Analysis of composition of microbiomes. a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26:27663. doi: 10.3402/mehd.v26.27663
Matamoros, S., Gras-Leguen, C., Le Vacon, F., Potel, G., and de La Cochetiere, M. F. (2013). Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 21, 167–173. doi: 10.1016/j.tim.2012.12.001
Mayer, E. A., Savidge, T., and Shulman, R. J. (2014). Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology 146, 1500–1512. doi: 10.1053/j.gastro.2014.02.037
Mueller, N. T., Bakacs, E., Combellick, J., Grigoryan, Z., and Dominguez-Bello, M. G. (2015). The infant microbiome development. mom matters. Trends Mol. Med. 21, 109–117. doi: 10.1016/j.molmed.2014.12.002
Nagpal, R., Tsuji, H., Takahashi, T., Nomoto, K., Kawashima, K., Nagata, S., et al. (2017). Ontogenesis of the gut microbiota composition in healthy, full-term, vaginally born and breast-fed infants over the first 3 years of life. a quantitative Bird’s-Eye View. Front. Microbiol. 8:1388. doi: 10.3389/fmicb.2017.01388
Nash, M. J., Frank, D. N., and Friedman, J. E. (2017). Early microbes modify immune system development and metabolic homeostasis-the “restaurant” hypothesis revisited. Front. Endocrinol. 8:349. doi: 10.3389/fendo.2017.00349
Nylund, L., Satokari, R., Salminen, S., and de Vos, W. M. (2014). Intestinal microbiota during early life - impact on health and disease. Proc. Nutr. Soc. 73, 457–469. doi: 10.1017/S0029665114000627
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. doi: 10.1542/peds.2005-2824
Rautava, S., Luoto, R., Salminen, S., and Isolauri, E. (2012). Microbial contact during pregnancy, intestinal colonization and human disease. Nat. Rev. Gastroenterol. Hepatol. 9, 565–576. doi: 10.1038/nrgastro.2012.144
Ringel-Kulka, T., Cheng, J., Ringel, Y., Salojarvi, J., Carroll, I., Palva, A., et al. (2013). Intestinal microbiota in healthy U.S. young children and adults–a high throughput microarray analysis. PLoS One 8:e64315. doi: 10.1371/journal.pone.0064315
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Stewart, C. J., Ajami, N. J., O’Brien, J. L., Hutchinson, D. S., Smith, D. P., Wong, M. C., et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588. doi: 10.1038/s41586-018-0617-x
Subramanian, S., Blanton, L. V., Frese, S. A., Charbonneau, M., Mills, D. A., and Gordon, J. I. (2015). Cultivating healthy growth and nutrition through the gut microbiota. Cell 161, 36–48. doi: 10.1016/j.cell.2015.03.013
Vatanen, T., Franzosa, E. A., Schwager, R., Tripathi, S., Arthur, T. D., Vehik, K., et al. (2018). The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594. doi: 10.1038/s41586-018-0620-2
Voreades, N., Kozil, A., and Weir, T. L. (2014). Diet and the development of the human intestinal microbiome. Front. Microbiol. 5:494. doi: 10.3389/fmicb.2014.00494
Wang, Y., Gao, X., Ghozlane, A., Hu, H., Li, X., Xiao, Y., et al. (2018). Characteristics of faecal microbiota in paediatric Crohn’s disease and their dynamic changes during infliximab therapy. J. Crohns Colitis 12, 337–346. doi: 10.1093/ecco-jcc/jjx153
Ward, T., Larson, J., Meulemans, J., Hillmann, B., Lynch, J., Sidiropoulos, D., et al. (2017). BugBase predicts organism level microbiome phenotypes. bioRxiv [preprint]. doi: 10.1101/133462.
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27. doi: 10.1186/s40168-017-0237-y
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zakrzewski, M., Proietti, C., Ellis, J. J., Hasan, S., Brion, M. J., Berger, B., et al. (2017). Calypso. a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33, 782–783. doi: 10.1093/bioinformatics/btw725
Keywords: gut microbiome, Chinese children, birth mode, gender, geographical location, gastrointestinal tract symptoms
Citation: Niu J, Xu L, Qian Y, Sun Z, Yu D, Huang J, Zhou X, Wang Y, Zhang T, Ren R, Li Z, Yu J and Gao X (2020) Evolution of the Gut Microbiome in Early Childhood: A Cross-Sectional Study of Chinese Children. Front. Microbiol. 11:439. doi: 10.3389/fmicb.2020.00439
Received: 16 June 2019; Accepted: 02 March 2020;
Published: 03 April 2020.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Xiaoming Bian, University of Georgia, United StatesRavinder Nagpal, Wake Forest School of Medicine, United States
Copyright © 2020 Niu, Xu, Qian, Sun, Yu, Huang, Zhou, Wang, Zhang, Ren, Li, Yu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialin Yu, eXVqaWFsaW40ODZAMTI2LmNvbQ==; Xuefeng Gao, cnlhbi5nYW9pbUBnbWFpbC5jb20=
†These authors have contributed equally to this work