 Jiangang Li1†
Jiangang Li1† Olga V. Mavrodi2,3†
Olga V. Mavrodi2,3† Jinfeng Hou1
Jinfeng Hou1 Chazden Blackmon2
Chazden Blackmon2 Ebrahiem M. Babiker4*
Ebrahiem M. Babiker4* Dmitri V. Mavrodi2*
Dmitri V. Mavrodi2*- 1Key Laboratory of Soil Environment and Pollution Remediation, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, China
- 2Department of Cell and Molecular Biology, School of Biological, Environmental, and Earth Sciences, The University of Southern Mississippi, Hattiesburg, MS, United States
- 3South Mississippi Branch Experiment Station, Mississippi State University, Poplarville, MS, United States
- 4USDA-ARS Southern Horticultural Research Laboratory, Poplarville, MS, United States
Plants are inhabited by millions of parasitic, commensal, and mutualistic microorganisms that coexist in complex ecological communities, and profoundly affect the plant’s productivity, health, and capacity to cope with environmental stress. Therefore, a better understanding of the rhizosphere microbiome may open a yet untapped avenue for the rational exploitation of beneficial plant–microbe interactions in modern agriculture. Blueberries encompass several wild and cultivated species of shrubs of the genus Vaccinium that are native to North America. They are grown commercially for the production of fruits, which are considered a health food due to the rich content of minerals, trace elements, and phenolic compounds with antioxidant, antitumor, and anti-inflammatory properties. Despite a long history of breeding and extensive commercial use, remarkably little is known about the composition and function of the blueberry root microbiome. To address this gap, we employed molecular approaches to characterize and compare microbial communities inhabiting the roots of rabbiteye blueberry (Vaccinium virgatum), Darrow’s blueberry (Vaccinium darrowii), and southern highbush blueberry (SHB; an interspecific hybrid of Vaccinium corymbosum and V. darrowii). Our results revealed that these plant species share a common core rhizobiome, but at the same time differ significantly in the diversity, relative abundance, richness, and evenness of multiple groups of prokaryotic and eukaryotic microorganisms. Although the host signature effects were especially pronounced at the plant species level, we also observed genotype-level variations in the distribution of specific microbial taxa, which suggests that the assembly of the blueberry microbiome is shaped by the plant genotype and modifications associated with the domestication and breeding of members of the Vaccinium genus. We also demonstrated that the studied Vaccinium species differ in the abundance of beneficial rhizobacteria and ericoid mycorrhizal fungi, which play a vital role in their adaptation to soils with low pH and slow turnover of organic matter.
Introduction
Plants are meta-organisms that are inhabited by millions of parasitic, commensal, and mutualistic microorganisms that coexist in complex ecological communities (Vandenkoornhuyse et al., 2015). The plant microbiome profoundly affects the development, health, productivity, and capacity of plants to cope with abiotic and biotic stresses and represents an area of active ongoing research. Most plant-associated microbes are found in contact with, or in immediate proximity to plant roots, where they form part of an extended food web driven by the release of plant rhizodeposits, or exudates (Sørensen and Sessitsch, 2006). Plants and their root-associated microbiome (rhizobiome) have both evolved to use their close association for the mutual benefit. The host plant provides rhizosphere microorganisms with a carbon-rich niche formed by the secretion of exometabolites into the soil that directly surrounds roots (Bulgarelli et al., 2012, 2015; Badri et al., 2013). In return, members of the rhizosphere and root microbiota supply plants with macro- and micronutrients, stimulate organ development, suppress pathogens, and modulate levels of stress phytohormones (Bulgarelli et al., 2015).
The makeup and function of the rhizobiome are strongly influenced by soil properties and by the presence and composition of rhizodeposits (Peiffer et al., 2013, Canarini et al., 2019). Root exudates contain a complex mixture of high- (lysates, mucilages, proteins), and low-molecular-weight (carbohydrates, amino acids, organic and fatty acids, phenolics, sterols) metabolites that supply rhizobacteria with carbon, nitrogen, and energy for growth. The amounts and patterns of rhizodeposition change depending on the plant growth stage, environmental factors, and abiotic stressors (Sasse et al., 2018). The profiles of exudation also vary substantially within and between plant species and it is thought that these differences drive the recruitment of specific taxa from the microbial seed bank of the soil (Lareen et al., 2016; Beattie, 2018). Consistent with the idea that plants actively select and shape their root microbiota, comparative analysis revealed significant variations in the composition of root-associated microbial communities across 30 species of angiosperm plants (Fitzpatrick et al., 2018). Interestingly, the structure and function of the rhizobiome are also significantly affected by the process of domestication, and commercial varieties of barley, maize, lettuce, beet, and agave harbor rhizosphere communities that differ substantially from their counterparts in the closely related species of wild plant (Zachow et al., 2014; Bulgarelli et al., 2015; Cardinale et al., 2015; Szoboszlay et al., 2015; Coleman-Derr et al., 2016). Although the available information is limited, several studies reported that domestication changes the rhizomicrobial diversity and affects the association with beneficial mycorrhizal fungi and nitrogen-fixing rhizobia (Perez-Jaramillo et al., 2016, 2018). Therefore, a better understanding of the impact of plant breeding on the composition and function of the rhizosphere microbiome may open a yet untapped avenue for the rational exploitation of beneficial plant–microbe interactions in modern agriculture.
Blueberries encompass several wild and cultivated species of shrubs of the genus Vaccinium L. that are native to eastern North America (Camp, 1945). They are grown commercially for the production of fruits, which are considered a health food due to the rich content of minerals, trace elements, and phenolic compounds with antioxidant, antitumor, and anti-inflammatory properties (Wang et al., 1997; Lobos and Hancock, 2015; Massarotto et al., 2016). The breeding and selection of blueberries began in the early 1900s (Coville, 1937), and three species, Tetraploid lowbush Vaccinium angustifolium Aiton (2n = 4× = 24), tetraploid highbush Vaccinium corymbosum L. (2n = 4× = 48), and hexaploid rabbiteye Vaccinium virgatum Aiton (2n = 6× = 72), constitute the backbone of the current commercial cultivars (Chavez and Lyrene, 2009). Over the past 70 years, the acreage of the highbush varieties has expanded dramatically due to the introduction of the southern highbush blueberry (SHB) with lower chilling requirements (Retamales and Hancock, 2018). The development of SHB cultivars was initiated by crosses between the tetraploid NHB V. corymbosum L. to Florida’s native diploid blueberry species Vaccinium darrowii Camp, but later, native V. angustifolium and V. virgatum were introduced into breeding programs (Sharpe and Darrow, 1959). These efforts helped to introduce novel adaptation genes and led to the release of several commercial cultivars with improved tolerance to higher soil pH and drought (Finn et al., 1993; Nunez et al., 2015). However, despite significant progress, the expansion of SHB in the Gulf Coast region of the United States is still challenged by fluctuations in temperature, rainfall patterns, UV levels, elevated soil pH, and drought (Lobos and Hancock, 2015).
The ability of the rhizosphere microorganisms to influence plant susceptibility to diseases and fitness in response to water stress, salinization, and soil pollution prompted detailed microbiome studies in numerous crop species (Raaijmakers et al., 2009; Edwards et al., 2015; Mahoney et al., 2017; Pfeiffer et al., 2017). In contrast, most relevant studies in blueberries employed traditional culture-based approaches and focused primarily on the association of Vaccinium spp. with ericoid mycorrhizae. The only comprehensive microbiome study focused on the effect of cultural practices on the structure of rhizosphere microbial communities of wild blueberry V. angustifolium grown in managed and forest sites in Nova Scotia, Canada (Yurgel et al., 2017, 2018). Hence, despite a long history of breeding and extensive commercial use, remarkably little is known about the composition and function of blueberry rhizobiome. We hypothesized that the domestication and long history of breeding introduced changes in the structure of blueberry rhizobiome. We tested this hypothesis by comparing the diversity and abundance of bacteria, fungi, and eukaryotic organisms in microbiomes associated with roots of two genotypes each of the SHB (V. corymbosum Camp), Darrow’s blueberry (V. darrowii Camp), and rabbiteye blueberry (V. virgatum Aiton).
Materials and Methods
Plant Growth Conditions
The study employed three different Vaccinium species, including V. virgatum Aiton (Vg) (2n = 6× = 72), V. corymbosum L. (SHB) (2n = 4× = 48), and V. darrowii Camp (Vd) (2n = 2× = 24) (Table 1). The V. virgatum species was represented by breeding selections MS 1089 and MS 1408, while V. corymbosum was represented by MS 2337 and MS 2276. V. darrowii was represented by the wild-type clone B0008 and breeding selection MS 2230. Plants were grown in a greenhouse in a mixture of pine bark mulch and sand (1:1, v/v) with pH 5.2. Briefly, rooted cuttings of all genotypes were transplanted into 1-gallon pots filled with the potting mix and maintained in a greenhouse under 16 h photoperiod at 22/18°C day/night temperature and drip-irrigated daily with 300 mL of water per pot. Every 15 weeks the pots were fertilized with 1 g of the Osmocote® 14-14-14 slow-release fertilizer (ICL Specialty Fertilizers—North America, Dublin, OH, United States). After about 48 weeks of growth, the plants were transferred in a laboratory, carefully uprooted, and processed for the isolation of rhizosphere soil DNA. The pH of potting mix was monitored with a B-213 Twin pH meter (Horiba Instruments, Irvine, CA, United States).
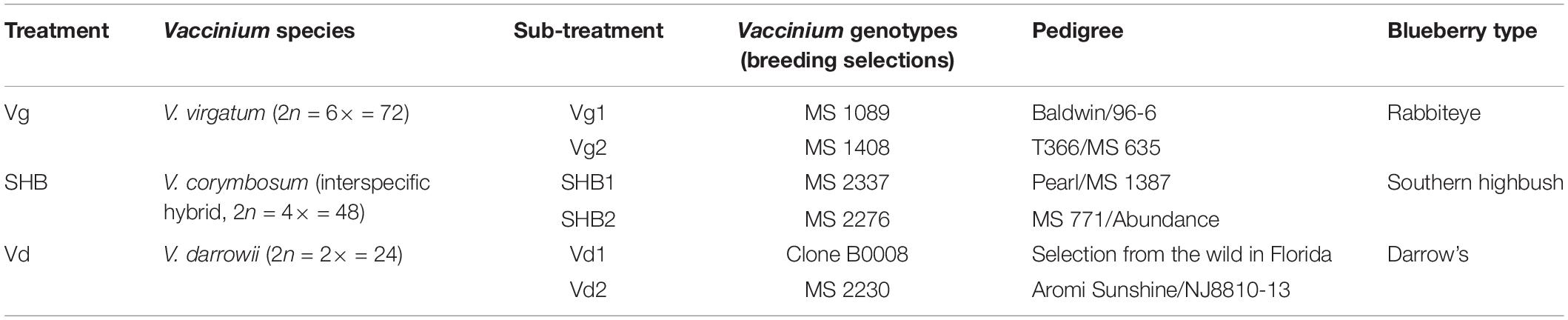
Table 1. Description of species and cultivars of Vaccinium used in the study.
Extraction of Rhizosphere Soil DNA and Sequencing of 16S rRNA, ITS, and 18S rRNA Amplicons
Partial root systems were cut from blueberry plants (n = 6 per Vaccinium genotype), excess soil was removed, and 1 g of excised roots was placed into 50 mL Falcon tubes. Each tube was filled with 20 mL of sterile water and rhizosphere soil was dislodged by a combination of vortexing and treatment in an ultrasonic bath. The resultant soil suspensions were used for the extraction of rhizosphere soil DNA with a DNeasy PowerSoil kit (Qiagen, Germantown, MD, United States). The concentration of the purified DNA was measured using a DNA Quantification kit (Bio-Rad, Hercules, CA, United States) by measuring fluorescence at 460 nm with a Synergy 2 microplate reader (BioTek Instruments, Winooski, VT, United States). The absence of PCR inhibitors in the extracted DNA was verified by PCR with the DreamTaq DNA polymerase (Thermo Fisher Scientific, Waltham, MA, United States) and 16S rRNA-specific universal eubacterial primers 8F and 1492R (Weisburg et al., 1991). The amplification was performed as described by Mavrodi et al. (2018). The samples of purified rhizosphere DNA were shipped for analysis to the Center for Comparative Genomics and Evolutionary Bioinformatics at Dalhousie University1. Barcoded amplicons were generated by PCR with the high-fidelity Phusion DNA polymerase (Thermo Fisher Scientific) and primers targeting the V6–V8 region of bacterial 16S rRNA (forward primer B969F: ACGCGHNRAACCTTACC; reverse primer BA1406R: ACGGGCRGTGWGTRCAA) (Comeau et al., 2011), the internal transcribed spacer (ITS) 2 region of fungi [forward primer ITS86(F): GTGAATCATCGAATCTTTGAA; reverse primer ITS4(R): TCCTCCGCTTATTGATATGC] (Op De Beeck et al., 2014), and the V4 region of eukaryotic 18S rRNA (forward primer: CYGCGGTAATTCCAGCTC; reverse primer: AYGGTATCTRATCRTCTTYG). The purification of amplicons, library preparation, and its sequencing on a MiSeq instrument (Illumina, San Diego, CA, United States) using the MiSeq v3 chemistry (2 × 300 bp) were performed as described by Comeau et al. (2017).
Bioinformatics Analyses
The Illumina sequence data were processed using the Microbiome Helper pipeline (Comeau et al., 2017). The sequence quality was confirmed with the FastQC toolkit2, after which the forward and reverse reads were merged with PEAR v 0.9.10 (Zhang et al., 2014) followed by filtered out the low-quality and chimeric reads with FASTX-Toolkit v 0.0.143 and USEARCH v 6.1 (Edgar et al., 2011), respectively. Operational taxonomic units (OTUs) were characterized by matching to the RDP 16S rRNA database (11 release) (Cole et al., 2014) and the UNITE database (12_11 release) of the fungal ITS sequences (Abarenkov et al., 2010), and SILVA dataset of 18S rRNA sequences (Yilmaz et al., 2014). Reads were subsequently mapped back to OTUs to determine the OTU abundance for each sample, and differences in the community composition/structure in each sample were analyzed with the Quantitative Insights Into Microbial Ecology (QIIME) package (Caporaso et al., 2010). Any OTUs that constituted less than 0.1% of the total OTUs were removed, and the data were normalized by the total sum normalization (TSS) with square root transformation. Differences in alpha diversity between the species of Vaccinium were determined by Duncan’s new multiple range test (P < 0.05). Differences in the diversity and OTU abundance between genotypes of the same Vaccinium species were determined by the two-sample t-test (P < 0.05) or by the Wilcoxon rank-sum test (P < 0.05) and visualized in the heat tree format using Metacoder (Foster et al., 2017). The R package Phyloseq (McMurdie and Holmes, 2013) was used to generate non-metric multidimensional scaling (NMDS) ordination (K = 2) based on Bray–Curtis dissimilarity matrices to visualize the most abundant phyla from different Vaccinium species and genotypes. The amount of variation in the composition of bacterial, fungal, and eukaryotic communities that could be explained by Vaccinium species and genotypes was estimated by the permutational multivariate analysis of variance (PERMANOVA) following the calculation of a Bray–Curtis dissimilarity matrix with 999 permutations. Network analyses and identification of differentially enriched microbial taxa using the linear discriminant analysis effect size (LEfSe) method were carried out in Calypso (Zakrzewski et al., 2017).
Community Level Physiological Profiling (CLPP)
In order to perform the CLPP analysis, 1–3 g of plant roots were gently shaken to remove excess soil and placed into 50 mL conical centrifuge tubes. Nine parts of phosphate-buffered saline (PBS) were added to each tube, and root-associated bacteria were dislodged by vortexing and treatment in a sonicating bath (1 min each). All root washes were further diluted 10-fold with PBS and inoculated into EcoPlates (100 μL per well) containing 31 different carbon sources (Biolog, Hayward, CA, United States). The inoculated EcoPlates were incubated for a week at 23°C in the dark, and patterns of the C source utilization were scored daily by measuring absorbance at 590 nm with a Synergy 2 microplate reader (BioTek Instruments). Functional diversity and CLPP similarity indices were calculated based on readings obtained on the sixth day of incubation. The entire experiment was repeated twice.
Data Availability
Sequences generated in this project were deposited in the NCBI sequence read archive under accession numbers PRJNA577971 and PRJNA578171.
Results
Composition of Rhizobiomes Associated With V. virgatum, V. corymbosum, and V. darrowii
The rhizosphere communities were characterized via high-throughput sequencing of 16S, ITS2, and 18S amplicons generated using DNA extracted from the rhizosphere of two genotypes each of V. virgatum (Vg), V. corymbosum (SHB), and V. darrowii (Vd) species of blueberry (Table 1). In order to profile the bacterial part of the rhizobiome, pools of 16S amplicons were processed to remove low-quality reads, chimeras, and samples with a low depth of coverage. The resultant dataset of 2,096,766 high-quality reads (median reads per sample = 60,990) was rarified to an even depth of 21,000 reads and binned into OTUs at 97% sequence identity. We observed OTUs from 15 bacterial phyla with an average relative abundance of above 0.1%, and seven of these phyla (Proteobacteria, Acidobacteria, Actinobacteria, Verrucomicrobia, Bacteroidetes, Planctomycetes, and Chloroflexi) collectively accounted for 96.7% of all sequencing reads (Figure 1A). Despite the fact that Vaccinium species harbored overall similar rhizobacterial communities, we observed several differences in the abundance of individual phyla. For example, samples from SHB were characterized by higher levels of Cyanobacteria, whereas Vd had a higher abundance of Actinobacteria, Acidobacteria, and Gammaproteobacteria (Kruskal–Wallis, PFDR < 0.05). In contrast, Vg had a higher abundance of Beta- and Deltaproteobacteria, Gemmatimonadetes, Chlorobi, Bacteroidetes, Verrucomicrobia, Fibrobacteres, and Spirochaetes (Kruskal–Wallis, PFDR < 0.05). Genotypes of the same Vaccinium species were not significantly different (PFDR > 0.05) in the relative abundance of prevalent bacterial phyla (data not shown). Core microbiome analysis with the detection threshold of 70% sample prevalence and minimum relative abundance of 0.01% identified several bacterial families, including Hyphomicrobiaceae, Acetobacteriaceae, Rhodospirillaceae, Koribacteriaceae, Sinobacteriaceae, Acidobacteriaceae, Chitinophagaceae, Solibacteriaceae, and Opitutaceae (Supplementary Figure S1A). Many of these dominant core microbiome families belonged to Alphaproteobacteria (especially Rhizobiales), as well as to Acidobacteria and Verrucomicrobia.
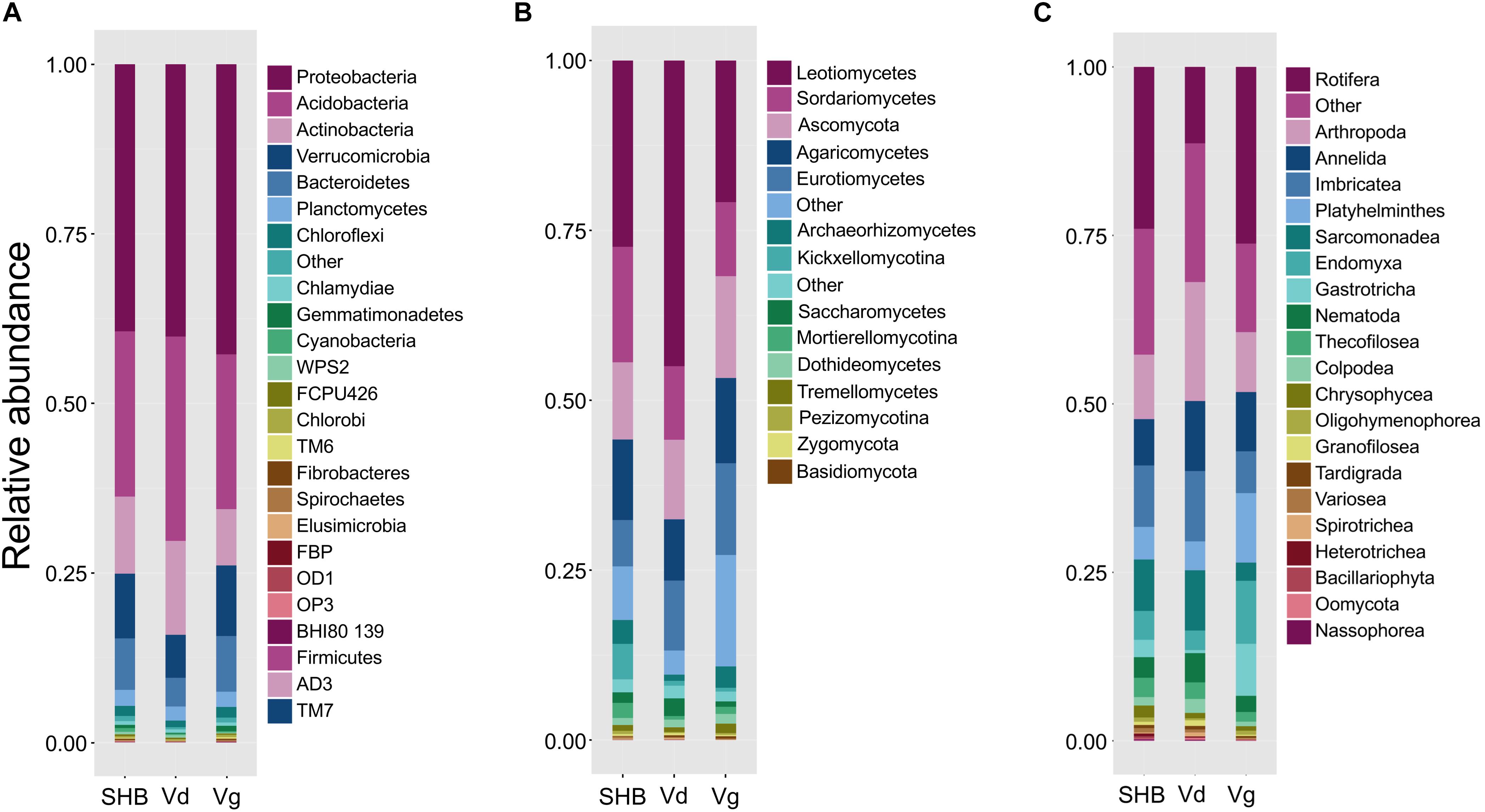
Figure 1. The relative abundance of major bacterial (phylum level) (A), fungal (class level) (B), and non-fungal eukaryotic (class level) (C) taxa present in the rhizosphere of different blueberry species.
The analysis of the fungal communities was performed using a dataset of 1,770,655 high-quality ITS reads (median reads per sample = 50,066), which was obtained after the removal of low-quality and chimeric sequences, as well as samples with the low depth of coverage. The ITS dataset was further rarified to an even depth of 8,000 reads and binned into OTUs at 97% sequence identity. At the phylum level, most of the OTUs were classified as Ascomycota (72.4%) and Basidiomycota (12.4%), whereas at the class level the communities were dominated by Leotiomycetes (30.6%), Sordariomycetes (12.9%), Agaricomycetes (11.2%), Eurotiomycetes (10.4%), Archaeorhizomycetes (2.5%), Saccharomycetes (1.6%), Dothideomycetes (1.2%), and Tremellomycetes (1.0%) (Figure 1B). Several groups of these fungi varied significantly between the studied blueberry species, including Zygomycota, Rozellomycota, Kickxellomycotina, and Archaeorhizomycetes, which were more abundant in the rhizosphere of V. corymbosum (SHB) (Kruskal–Wallis, PFDR < 0.05). In contrast, V. darrowii (Vd) had higher abundance of Ascomycota, Leotiomycetes, and Saccharomycetes, whereas samples from V. virgatum (Vg) had higher abundance of Lecanoromycetes (Kruskal–Wallis, PFDR < 0.05). The core microbiome analysis identified Leotiaceae, Trichocomaceae, Hypocreaceae, Hyaloscyphaceae, Herpotrichiellaceae, Archaeorhizomycetaceae, Saccharomycetaceae, Mortierellaceae, Cryptococcaceae, Cordycipitaceae, and Amphisphaeriaceae as the dominant fungal families of the Vaccinium rhizobiome (Supplementary Figure S1B). Interestingly, the first and third most abundant fungal genera of the core microbiome were Pezoloma and Hyaloscypha, which are prominent ericoid mycorrhizal symbionts of the Ericaceae plants, including species of the genus Vaccinium.
As with the 16S and ITS amplicons, the 18S-based analysis of eukaryotic communities was initiated by the removal of low-quality sequences, chimeras, and low-coverage samples followed by binning into OTUs at 97% identity. The resultant dataset of 443,016 high-quality reads (median reads per sample = 13,262) was processed further to filter out the unassigned OTUs and plant-derived Archaeplastida sequences and then normalized to a depth of 1,459 reads. A largest proportion of 18S OTUs belonged to fungi (54.9%), Metazoa (26.1%), Cercozoa (11%), and Alveolata (1.0%) (data not shown). In agreement with results of the ITS profiling, most fungal reads were belonged to Ascomycota, followed distantly by Basidiomycota, Kickxellomycotina, and Chytridiomycota. The non-fungal OTUs collectively accounted for 42.8% of the 18S reads and were dominated by Rotifera (9.4%), Arthropoda (6.6%), Cryptomycota (5.3%), Platyhelminthes (3.5%), Imbricatea (3.3%), Annelida (3.2%), Endomyxa (3.0%), Sarcomonadea (2.5%), Gastrotricha (2.0%), and Nematoda (1.3%) (Figure 1C). The core part of the eukaryotic rhizobiome was dominated by several families of Cercozoa, including Trinematidae, Euglyphidae, Leptophryidae, Vampyrellidae, Sandonidae, Allapsidae, and Cercomonadidae (Supplementary Figure S1C). Other dominant core microbiome groups included arthropods (Arachnida and Maxillopoda), chrysophytes, and catenulid flatworms. Although the studied Vaccinium species harbored similar eukaryotic rhizosphere communities, we observed significantly higher levels of Sarcomonadea in V. darrowii (Vd), while Rotifera, Endomyxa, and Gastrotricha were more abundant in Vg samples (Kruskal–Wallis, PFDR < 0.05).
Diversity of Rhizosphere Microbial Communities
The alpha diversity of rhizosphere microbial communities was assessed by calculating Chao1 and Shannon indices. The Chao1 index is as an abundance-based coverage estimator of richness, whereas the Shannon index takes into account both richness and evenness. For rhizobacteria, both metrics indicated significant differences between the studied Vaccinium species, with SHB and Vg harboring communities of significantly higher diversity compared to Vd (Kruskal–Wallis; P < 0.05) (Figures 2A,B). The comparison of Chao1 and Shannon indices also revealed significant variation in the richness and evenness of fungal communities, which were higher in V. virgatum (Vg) compared to V. corymbosum (SHB) and V. darrowii (Vd) (Figures 2C,D). In contrast, the eukaryotic alpha diversity estimated using Chao1 and Shannon metrics showed that differences between the Vaccinium species were not significant (Kruskal–Wallis; P < 0.05) (Figures 2E,F). Similarly, no significant differences in the alpha diversity of rhizosphere-dwelling bacteria, fungi, and eukaryotes were detected between different genotypes of the same Vaccinium species (Wilcoxon rank-sum test, P > 0.05).
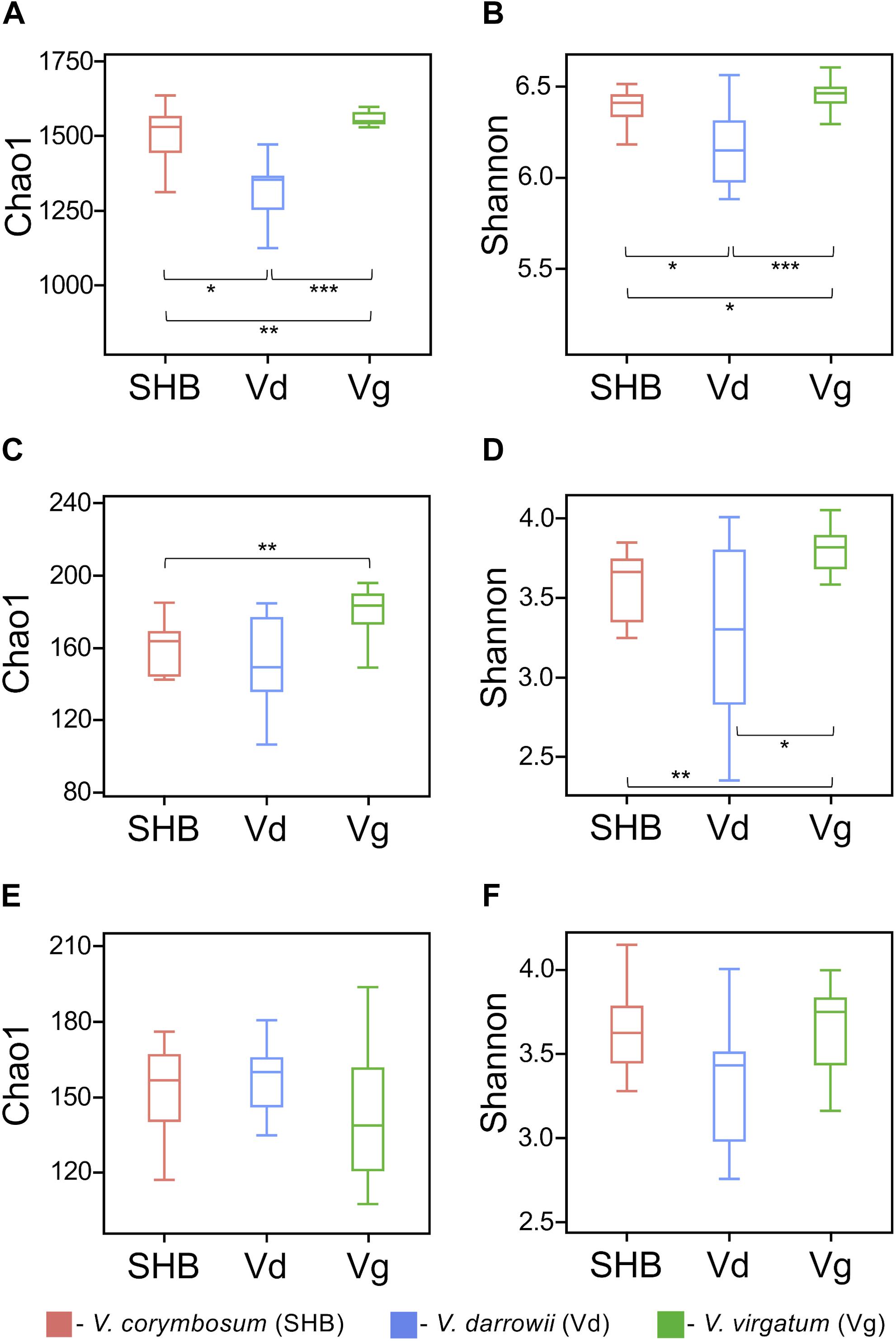
Figure 2. Alpha diversity (Chao1 and Shannon indices) of bacteria (A,B), fungi (C,D), and non-fugal eukaryotes (E,F) in the rhizosphere of studied blueberry species. Significant differences between V. corymbosum, V. darrowii, and V. virgatum are indicated in each figure panel (*P < 0.05, **P < 0.01, ***P < 0.001).
The NMDS analysis based on Bray–Curtis dissimilarities revealed that bacterial and fungal communities of the studied Vaccinium species clustered distinctively in the ordination space (Figure 3). These results were confirmed by the analysis of similarity (ANOSIM) test, which showed significant differences in the composition of bacterial (R = 0.605, P = 0.001), fungal (R = 0.613, P = 0.001), as well as eukaryotic (R = 0.548, P = 0.001) communities across the three Vaccinium species (Supplementary Table S1). Finally, the permutational multivariate ANOVA (Adonis) also demonstrated that bacterial (R2 = 0.33, P < 0.001), fungal (R2 = 0.28, P < 0.001), and eukaryotic (R2 = 0.22, P < 0.001) components of the rhizobiome were significantly differentiated by Vaccinium species. Interestingly, some variation in the composition of rhizobacterial and fungal communities was also significantly associated with genotypes of Vaccinium species (Adonis test: bacteria R2 = 0.16, P < 0.001; fungi R2 = 0.26, P < 0.001; eukaryotes R2 = 0.42, P < 0.001). A particularly pronounced genotype effect was observed in V. darrowii, where the Vd1 and Vd2 bacterial and fungal communities were clearly separated by results of the ANOSIM test (bacteria R = 0.70, P = 0.008; fungi R = 0.96, P = 0.004; eukaryotes R = 0.69, P = 0.014) (Supplementary Table S1).
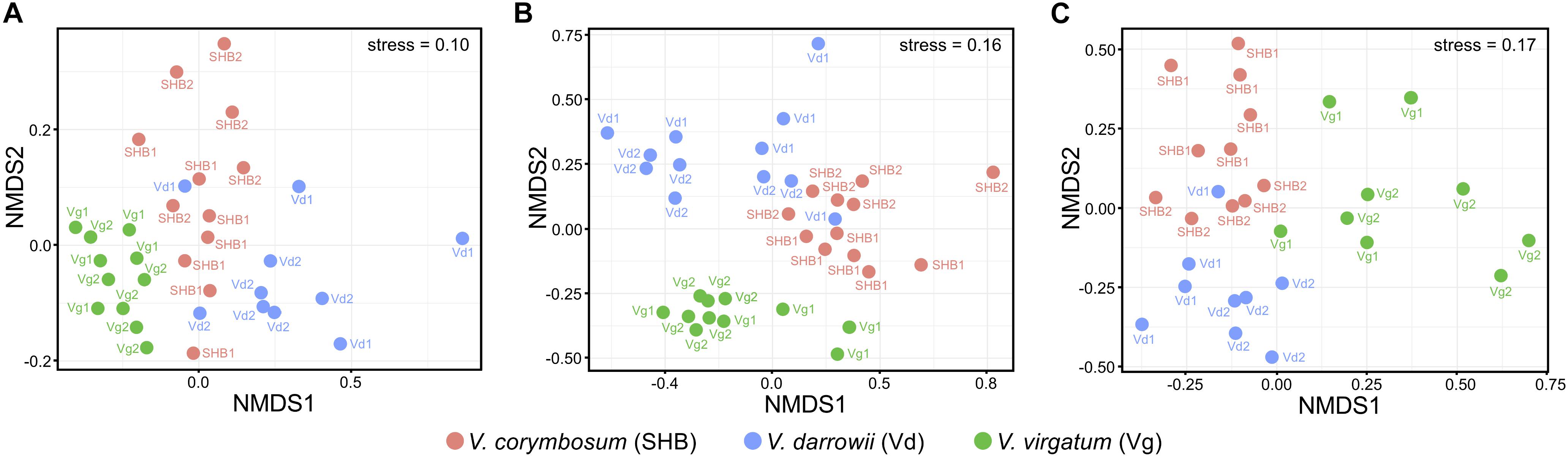
Figure 3. Ordination of bacterial (A), fungal (B), and eukaryotic (C) components of the blueberry rhizobiome by the non-metric multidimensional scaling (NMDS) analysis based on Bray–Curtis dissimilarity matrices. Samples from different Vaccinium genotypes are color-coded and labeled as follows: Vg1, V. virgatum MS 1089; Vg2, V. virgatum MS 1408; SHB1, V. corymbosum MS 2337; SHB1, V. corymbosum MS 2276; Vd1, V. darrowii B0008; Vd2, V. darrowii MS2230.
Finally, we compared the catabolic potential of rhizosphere microbial communities based on the utilization of 31 different carbon substrates present in Biolog EcoPlates. Results of the principal component analysis (PCA) readily distinguished catabolic profiles of the three Vaccinium rhizobiomes, with the first two principal components (PC1 and PC2) accounting for 45 and 19% of the total variance (Supplementary Figure S2A). The hierarchical clustering analysis of absorbance values suggested a broader range of metabolic activity in Vg and SHB samples, which resulted in the utilization (OD590 >0.1) of 27 different carbon sources (Supplementary Figure S2B). In contrast, only 21 substrates were metabolized in EcoPlates inoculated with the Vd root washes. Rhizosphere microorganisms from the three species of Vaccinium actively consumed several carbohydrates (D-galacturonic acid, N-acetyl-D-glucosamine, D-mannitol, D-cellobiose, D-glucosaminic acid), L-asparagine, L-phenylalanine, pyruvic acid methyl ester, γ-hydroxybutyric acid, Tween 40, and Tween 80. On the other hand, the studied microbiomes differed in the capacity to metabolize glycogen, β-methyl-D-glucoside, α-D-lactose, 2-hydroxybenzoic acid, α-cyclodextrin, and itaconic acid.
Differences Between Rhizobiomes of V. virgatum, V. corymbosum, and V. darrowii
Differences in the composition of rhizobiomes of different Vaccinium species were evaluated at the family and genus level by calculating LEfSe scores that indicate the degree of consistent difference in relative abundance between treatments (Segata et al., 2011). Overall, our results revealed both plant species-level and genotype-level differences in the composition of bacterial, fungal, and eukaryotic communities associated with blueberry roots. A total of 25 distinct bacterial biomarkers were identified using the LDA threshold score of ≥ 3.0, most of which were Proteobacteria (especially Alphaproteobacteria) and Actinobacteria (Figure 4A). The SHB-enriched phylotypes belonged to the proteobacterial and actinobacterial families Ectothiorhodospiraceae, Sphingomonadaceae, Steroidobacter, Gaiellaceae, Nocardiaceae, and Streptomycetaceae, as well to the Cytophagaceae (Bacteroidetes) and Nostocaceae (Cyanobacteria). The Vd rhizobiome was characterized by the abundance of several Actinobacteria (Conexibacter, Actinospicaceae, Mycobacterium, Salinispora, and Patulibacteraceae) and Proteobacteria (Sinobacteracea, Acetobacteraceae, Beijerinckiaceae, and Aquicella), and by the presence of Acidobacteriaceae (Acidobacteria) and Sphingobacteriaceae (Bacteroidetes). The identified Vg-specific phylotypes were taxonomically diverse and included members of Proteobacteria (Hyphomonadaceae, Syntrophobacteraceae, Hyphomicrobium), Verrucomicrobia (Opitutus), Spirochaetes, and Chloroflexi (Ktedonobacteraceae). We also observed genotype-level differences in the distribution of individual families and genera associated with roots of V. corymbosum (SHB), V. darrowii (Vd), and V. virgatum (Vg) (Supplementary Figure S3A).
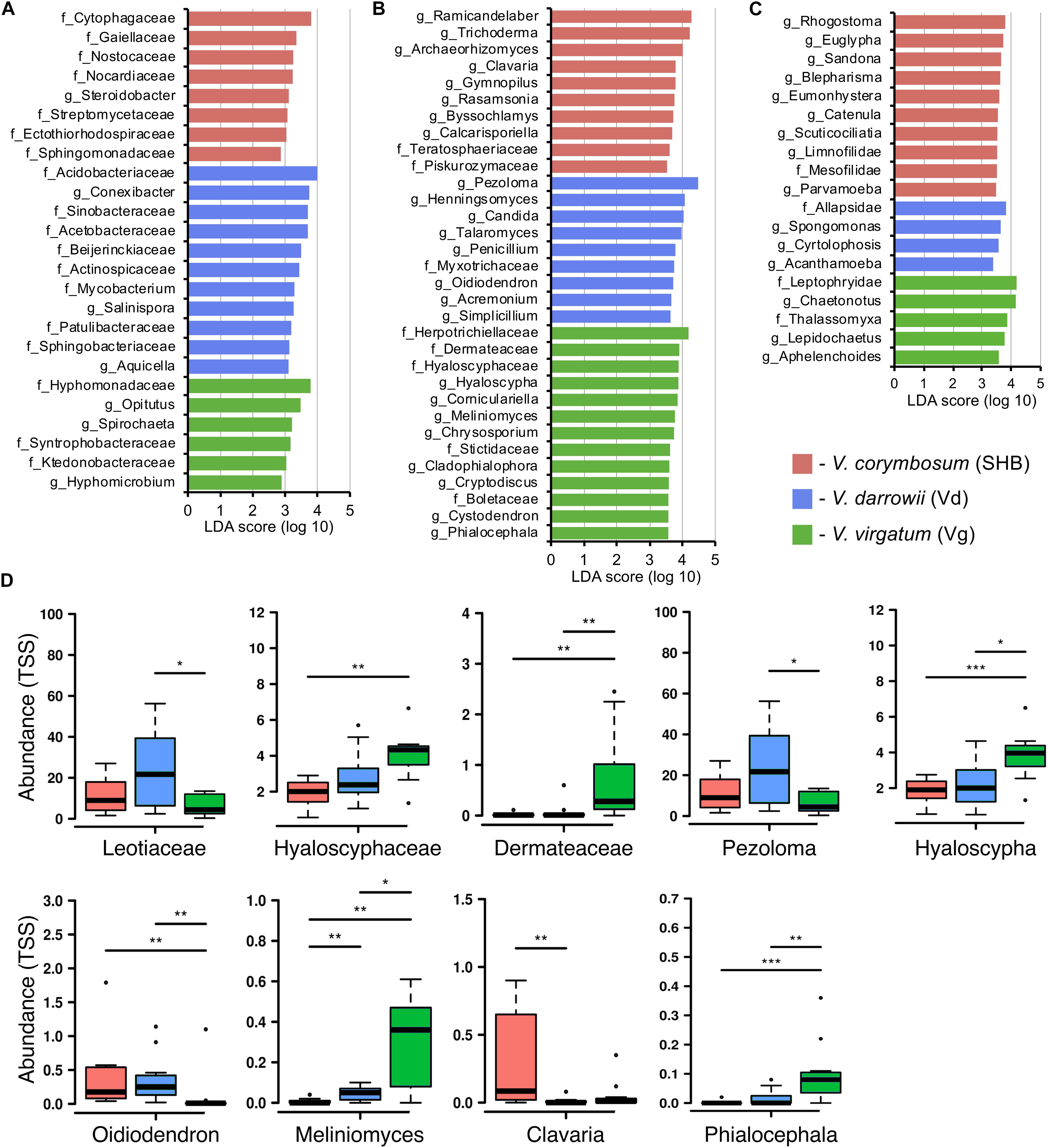
Figure 4. LEfSe analysis of differentially abundant (LDA threshold score ≥ 3.0) families and genera of bacteria (A), fungi (B), and eukaryotes (C) between Vaccinium species. (D) Ericoid mycorrhizal fungi with significant differences (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001) in the total sum normalized (TSS) transformed abundance.
The analysis of fungal communities revealed 32 distinct biomarkers that were differentially distributed among rhizobiomes of the studied blueberry species (Figure 4B). The rhizobiome of V. corymbosum (SHB) was enriched in diverse ascomycetes (Trichoderma, Archaeorhizomyces, Rasamsonia, Byssochlamys, and Teratosphaeriaceae) and basidiomycetes of the orders Agaricales (Clavaria, Gymnopilus) and Filobasidiales (Piskurozymaceae). In contrast, the V. virgatum (Vg)-specific fungi included an abundance of ascomycetes from the orders Helotiales, Ostropales, and Chaetothyriales, and were particularly enriched in ericoid mycorrhizae (EM) (Phialocephala, Meliniomyces, Hyaloscypha) and endophytic taxa (Cystodendron, Corniculariella) (Figure 4D). Some closely related EM fungi (i.e., Pezoloma, Oidiodendron, and Myxotrichaceae) were also specifically associated with roots of V. darrowii (Vd) along with the members of Eurotiales (Penicillium, Talaromyces), Hypocreales (Acremonium, Simplicillium), and Candida yeast (Saccharomycetales). We identified multiple fungal taxa that were differentially distributed between genotypes of three Vaccinium spp., including several species of EM fungi and plant-beneficial endophytes Verruconis, Gongronella, Pestalotiopsis, and Cladophialophora (Supplementary Figure S3B).
The LEfSe analysis revealed 19 eukaryotic biomarkers that were differentially enriched in the rhizosphere of V. corymbosum, V. virgatum, and V. darrowii (Figure 4C). Over half of these taxa were represented by amoeboids and flagellates of the phylum Cercozoa, which were specifically associated with roots of SHB (Rhogostoma, Euglypha, Sandona, Limnofilidae, Mesofilidae), Vd (Allapsidae, Spongomonas), or Vg (Thalassomyxa, Leptophryidae). Other differentially enriched groups of protists included ciliates (Blepharisma, Scuticociliatia, Cyrtolophosis) and amoebas (Parvamoeba, Acanthamoeba), which were present in V. corymbosum and V. darrowii. Finally, 18S-based profiling also identified certain gastrotrichs (Chaetonotus, Lepidochaetus), microscopic nematodes (Eumonhystera, Aphelenchoides), and flatworms (Catenula) as specific biomarkers associated with SHB and Vg. Although there were fewer genotype-level differences in the distribution of eukaryotic biomarkers compared to bacteria or fungi, we identified several genera of ciliates, filose amoebas, and flagellates that differed in abundance between the two genotypes of V. corymbosum (Supplementary Figure S3C). Collectively, the LEfSe results were in agreement with differences in the relative abundance of bacteria, fungi, and eukaryotes estimated by the non-parametric Wilcoxon rank-sum test and visualized in the form of differential heat trees (Supplementary Figure S4). The family-level analysis of rhizosphere microbial communities also revealed the presence of complex co-occurrence networks. The bacterial network contained two distinct clusters of co-occurring taxa (Figure 5A). The first cluster contained some Actinobacteria (Mycobacteriaceae, Conexibacteraceae), Acidobacteria (Acidobacteriaceae, Koribacteraceae), Alpha- (Sinobacteraceae, Beijerinckiacea), and Gammaproteobacteria (Acetobacteraceae) associated predominantly with V. darrowii. The second cluster was associated with V. virgatum and was comprised of Alphaproteobacteria (Rhizobiaceae), Bacteroidetes (Chitinophagaceae, Cytophagaceae), Deltaproteobacteria (Syntrophobacteraceae, Haliangiaceae), and Verrucomicrobia (Opitutaceae). Although the fungal network was overall more diffuse, it still contained a distinct group of co-occurring taxa (Boletaceae, Hyaloscyphaceae, Plectosphaerellace, Dermateaceae, Herpotrichiellacea, Chaetosphaeriaceae, Helotiaceae) found in the rhizosphere of V. virgatum (Figure 5B). Finally, the eukaryotic network included a large group of positively correlated taxa that were associated with roots of V. corymbosum and V. darrowii, as well as a much smaller cluster of Rotifera, Gastrotricha, and Leptophryidae that were associated with V. virgatum (Figure 5C).
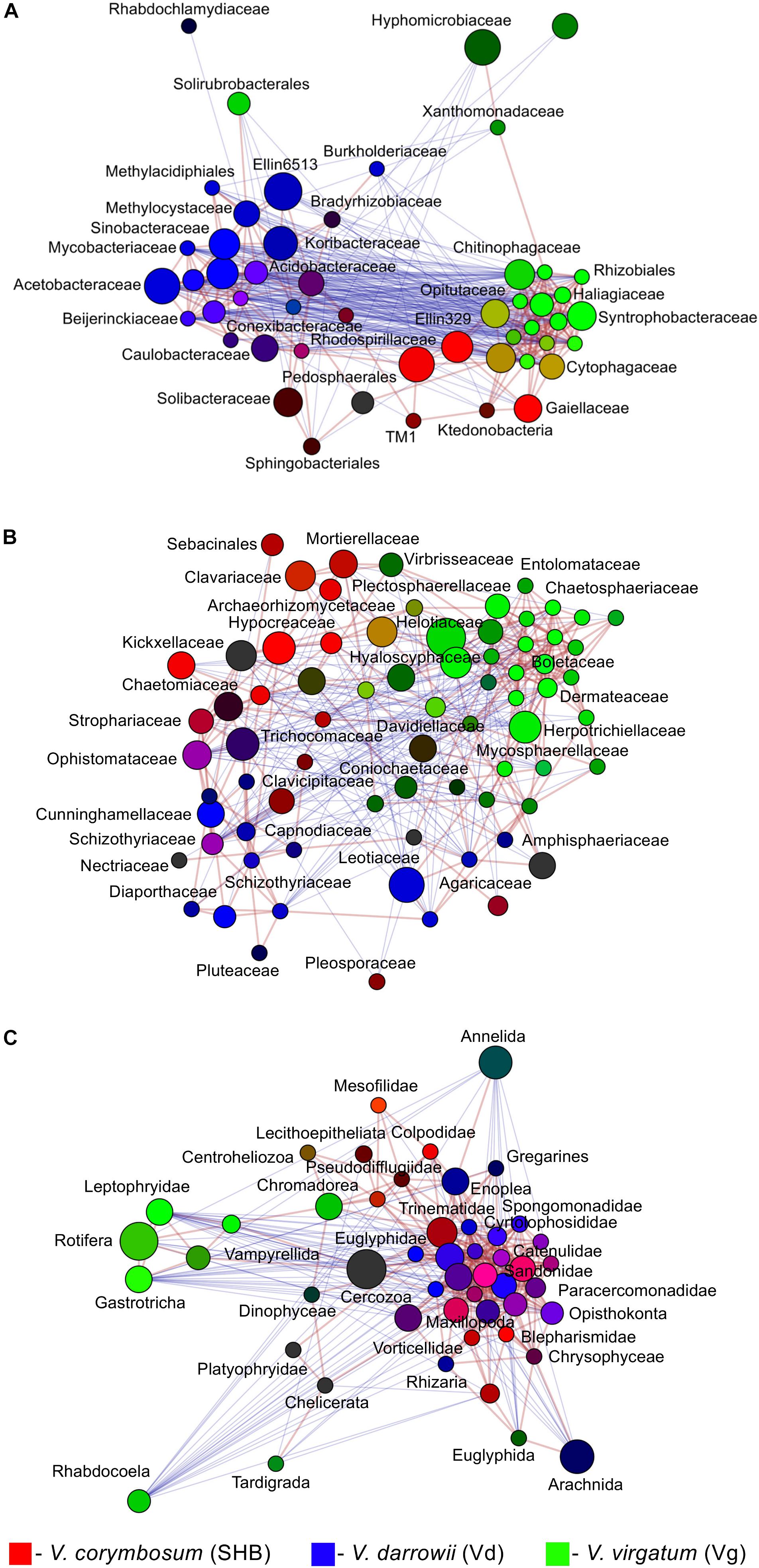
Figure 5. Network analysis illustrating significant co-occurrence patterns among bacterial (A), fungal (B), and eukaryotic (C) taxa and Vaccinium species. The analysis was performed using Spearman correlations. Network edges represent statistically significant positive (red) and negative (blue) correlations (FDR < 0.05). Taxa are represented by nodes colored based on their association with the studied blueberry species [red, V. corymbosum (SHB), blue, V. darrowii (Vd), green, V. virgatum (Vg)]. The relative brightness (i.e., the color gradient) of each node reflects the significance level of the association.
Discussion
To our knowledge, this is the first study that compares side-by-side rhizosphere microbial communities of SHB (V. corymbosum interspecific hybrid), Darrow’s blueberry (V. darrowii), and rabbiteye blueberry (V. virgatum). Our results revealed that these species share a common core rhizobiome, which is similar in composition to that of the lowbush blueberry V. angustifolium (Yurgel et al., 2017). At the same time, the rhizosphere communities of SHB, Vd, and Vg differed significantly in the diversity, relative abundance, richness, and evenness of multiple groups of prokaryotic and eukaryotic microorganisms (Figure 2 and Supplementary Figure S4). Although the host signature effects were especially pronounced at the plant species level, we also observed genotype-level variations in the distribution of specific microbial taxa, which suggests that the assembly of the blueberry microbiome is shaped by the plant genotype and modifications associated with the domestication and breeding of members of the Vaccinium genus. Overall, V. corymbosum (SHB) and V. darrowii (Vd) harbored more similar microbial communities compared to V. virgatum (Vg), a finding that aligns well with the fact that southern highbush blueberries were developed by hybridizing the northern highbush blueberry V. corymbosum with several other Vaccinium species, including V. darrowii (Hancock, 2006). Although the impact of plant domestication on the root-associated microorganisms is poorly understood, several recent studies reported that the domestication is associated with compositional changes in the rhizosphere microbiome (Kiers et al., 2007; Zachow et al., 2014; Bulgarelli et al., 2015; Cardinale et al., 2015; Perez-Jaramillo et al., 2016, 2017). Interestingly, a meta-analysis of microbiome studies in barley, lettuce, common bean, and Arabidopsis revealed that members of the Bacteroidetes were more abundant in roots of wild plant relatives, while Proteobacteria and Actinobacteria were consistently associated with the rhizosphere of corresponding domesticated accessions (Perez-Jaramillo et al., 2018). We also identified these phyla among differentially distributed microbial groups and demonstrated that Bacteroidetes were associated with V. virgatum, whereas Actinobacteria were more abundant in V. darrowii. However, plants used in this study were grown under controlled conditions. It will be interesting to address the effect of domestication on the blueberry rhizobiome by including in future studies field-grown plants of wild accessions of V. angustifolium, V. darrowii, V. arboreum, V. myrsinites, and V. myrtilloides.
The analysis of differentially distributed taxa revealed several genera of ericoid mycorrhizal (EM) fungi, which are important mutualistic endosymbionts of blueberries and other plants of the Ericaceae family (Grelet et al., 2017). Ericoid mycorrhizae enhance the fitness and productivity of the Ericaceae plants and play a vital role in their adaptation to soils with low pH and slow turnover of organic matter. Mycorrhizal fungi supply their host plants with nitrogen, phosphate, and other nutrients, which are mobilized via the breakdown of complex organic compounds by fungal exoenzymes (Kariman et al., 2018). Several studies also reported that EM colonization has positive effects on the tolerance of plants to heavy metals (Daghino et al., 2016). Ericoid mycorrhizae interact with their plant hosts by forming hyphal coils in the epidermal cells of specialized hair roots (Read, 1996). The EM fungi are closely related to Dark Septate Endophytes, a group of fungi with melanized hyphae that form in the root tissue loose loops instead of dense ericoid coils (Vohnik and Albrechtova, 2011). While earlier studies limited the capacity to form ericoid mycorrhizae to a small number of fungi, the advent of molecular methods revealed that hair roots colonized by EM harbor diverse assemblages of ascomycetes of the orders Helotiales, Erysiphales (Oidiodendron spp.), and some basidiomycetes, such as members of the Sebacinales and Clavaria spp. (Leopold, 2016).
Our results revealed an overall enrichment of the blueberry rhizobiome in various EM fungi, as well as the differential abundance of Hyaloscyphaceae (Hyaloscypha) and dark septate endophytes (Meliniomyces, Phialocephala) on roots of V. virgatum. We also observed higher levels of Leotiaceae (Pezoloma) and Oidiodendron in V. darrowii and an abundance of the EM basidiomycete Clavaria in the rhizosphere of V. corymbosum. The differential distribution of several EM taxa and plant-beneficial endophytes was also observed at the genotype level. Although the host preference/specificity effects in the interaction of the Ericaceae plants with EM fungi are poorly understood, Scagel and Yang (2005) demonstrated that highbush blueberry cultivars that fruit early in the season had higher levels of mycorrhizal infection than cultivars fruiting later in the growing season. Similarly, Sun et al. (2011) demonstrated that communities of EM fungi in Rhododendron decorum varied with the host genotype, which was in contrast to non−EM communities that were shaped by geography. That study concluded that genetic make−up of the host plant represents a significant “driving force” shaping communities of ericoid mycorrhizae. Our findings agree with earlier studies that demonstrated the genotype-specific variation in the sensitivity of blueberries to ericoid mycorrhizae and hint at the possibility that the genetics of blueberry plants plays an important role in the recruitment of certain EM fungi from the seed bank of soil microorganisms.
The comparison of rhizosphere communities of SHB, Vd, and Vg revealed several groups of bacteria that may exert beneficial effects on the health and productivity of blueberries. The three compared rhizobiomes had an abundance of free-living diazotrophs from Bradyrhizobiaceae, Methylocystaceae, Burkholderiaceae, and Frankiaceae, which may represent another adaptation to poor soils typical of natural habitats of blueberries and other Ericaceae plants. The microbiome of V. darrowii also had higher levels of nitrogen-fixing Beijerinckiaceae, which are often found in the plant rhizosphere where they exchange ammonium for the photosynthetically fixed carbon. We further identified several groups of bacteria that in other crop systems had been implicated in the suppression of soilborne pathogens. For example, the rhizobiome of V. virgatum had an increased abundance of Opitutus, and the decline of Opitutaceae in soil was implicated in the increased susceptibility of cotton to infection with Fusarium oxysporum f. sp. vasinfectum (Li et al., 2015). The rhizosphere communities of V. corymbosum and V. darrowii had higher levels of Sphingobacteriaceae, Sphingomonadaceae, Cytophagaceae, Gaiellaceae, and several other Actinobacteria (Figure 4). Gaiellaceae is a recently discovered family that belongs to a deep lineage of the class Actinobacteria and consists of a single genus with only one cultured species, Gaiella occulta (Albuquerque et al., 2011). These aerobic chemoorganotrophs have been found worldwide in the rhizosphere of corn, rice, soybean, hemp, and sudex (Eo et al., 2015; Sun et al., 2018; Wang et al., 2019). Members of Gaiellaceae, together with Sphingomonadaceae, and Streptomycetaceae were associated with the soil suppressiveness to F. oxysporum f. sp. cubense, a causative agent of the Panama disease of banana (Xue et al., 2015). The differentially abundant SHB and Vd taxa were also identified among key microbial groups in the Dutch soil suppressive to the plant pathogen Rhizoctonia solani AG2-2IIIB (Chapelle et al., 2016). That study employed metagenomic and metatranscriptomic techniques to characterize the transcriptional changes in the rhizobiome of sugar beet plants grown in the presence of the fungal pathogen in a Rhizoctonia-suppressive soil. The analysis identified members of the Sphingobacteriaceae, Sphingomonadaceae, and Cytophagaceae among rhizobacteria that responded to the presence of pathogens by upregulation of stress-related genes. A follow-up study by van der Voort et al. (2016) used heat treatment of soil to examine the contribution of different rhizobacterial taxa to the suppression of R. solani. The authors suggested that different Actinobacteria, including members of the Streptomycetaceae and Mycobacteriaceae, may contribute to the disease protection since their decrease coincided with the loss of pathogen suppression. Interestingly, Actinobacteria were also identified as hub taxa in the rhizosphere microbiome of lowbush blueberry V. angustifolium, suggesting their crucial role in the structure of microbial communities of blueberries (Yurgel et al., 2018).
The assembly of rhizosphere microbiome is influenced by the interplay between soil properties (structure, pH, moisture, salinity, organic matter), climate, and human practices (Cordovez et al., 2019). For example, the mycorrhizal colonization in blueberries is modulated by the application of fertilizers, and increased N fertilization can reduce the colonization of roots by ericoid mycorrhizae (Sadowsky et al., 2012). However, given that our study was performed under controlled conditions, it is likely that the observed differences in the structure of rhizosphere microbiomes of the studied Vaccinium species are driven by a combination of host effects and microbial interactions. The recruitment of soil microorganisms by plant species and genotypes is mediated by the root morphology and secretion of exudates, which serve as nutrients and signals for the rhizosphere microorganisms (Sasse et al., 2018). A recent elegant study by Zhalnina et al. (2018) demonstrated that the rhizosphere microbiome of wild oat (Avena barbata) is recruited via the metabolic synchronization of the root exudation and microbial substrate utilization traits.
Although currently unknown, it is plausible that similar processes govern the assembly of distinct microbial communities in the rhizosphere of different species of blueberry. The observed differences in the composition of microbial communities of V. corymbosum, V. darrowii, and V. virgatum are also likely shaped by mutualistic and competitive interactions between different members of the rhizobiome. The co-occurrence analysis of bacteria, fungi, and protozoa revealed complex networks with numerous positive and negative correlations, similar to those described in the rhizosphere of wild and managed V. angustifolium (Yurgel et al., 2018). Interestingly, our analysis also revealed that microbial communities of SHB, Vd, and Vg differ in the abundance of various predatory protists, which feed on bacteria and fungi and have recently emerged as a key factor that shapes the rhizosphere microbiome and selects for plant-beneficial functional traits (Gao et al., 2019).
Conclusion
Our study is the first to compare the rhizosphere microbial communities of the SHB, Darrow’s blueberry, and rabbiteye blueberry using the modern culture-independent approaches. Our results revealed an extensive diversity of pro- and eukaryotic microorganisms inhabiting the blueberry rhizosphere and demonstrated that the studied species of Vaccinium differ in the abundance of beneficial rhizobacteria and EM fungi. Cultivated blueberries were initially selected by crossing several wild relatives to improve the yield and fruit quality. However, the rising popularity of this commodity has expanded the production into areas that present a challenging environment for blueberry plants (Retamales and Hancock, 2018). Blueberries, like other members of the Ericaceae family, rely on their microbiomes for the protection against abiotic stresses and survival in soils that are low in nutrients. In particular, mycorrhizal symbiotic partners improve the uptake of soil nutrients, efficacy of fertilizers, and protect plants from the metal toxicity in acidic soils (Scagel, 2005; Scagel and Yang, 2005; Vega et al., 2009; Caspersen et al., 2016). Therefore, we suggest that species- and genotype-specific differences in the structure and function of rhizobiome represent an important facet in the adaptation of blueberry cultivars to soil and climate conditions.
The rhizosphere microbiome is shaped by exudates and root morphology, both of which are influenced by plant genotype (Sasse et al., 2018). There is a growing consensus that the traditional plant breeding should be expanded to include plant genotype × environment × microbiome interactions (Wei and Jousset, 2017). Such microbiome-supported breeding should be performed in the absence of excessive fertilizers and pesticides and complemented by the microbial community profiling and screening for metabolites that mediate interactions with key pathogenic or beneficial species (Wille et al., 2018). We reason that similar experimental approaches can be employed in blueberries to harness microbial communities that improve the disease resistance, tolerance to heat and drought, and vigor to thrive in soils with low organic matter content.
Data Availability Statement
Sequences generated in this project were deposited in the NCBI sequence read archive under accession numbers PRJNA577971 and PRJNA578171.
Author Contributions
DM, OM, and EB conceived the research project. EB provided blueberry plants. OM and JL extracted soil DNA. OM and CB conducted the Biolog EcoPlate profiling. DM, JL, OM, and JH conducted the microbiome analysis. DM, JL, OM, and EB wrote the manuscript. All authors contributed to the manuscript revision.
Funding
This study was funded by cooperative agreement 58-6062-8-002 from the US Department of Agriculture (USDA-ARS) Southern Horticultural Laboratory. The authors acknowledge support from the Mississippi INBRE, funded by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant P20GM103476. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. Jake Schaefer of the USM School of Biological, Environmental, and Earth Sciences for consultations with data analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00370/full#supplementary-material
Footnotes
- ^ http://cgeb-imr.ca/
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- ^ http://hannonlab.cshl.edu/fastx_toolkit/download.html
References
Abarenkov, K., Henrik Nilsson, R., Larsson, K. H., Alexander, I. J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi – recent updates and future perspectives. New. Phytol. 186, 281–285. doi: 10.1111/j.1469-8137.2009.03160.x
Albuquerque, L., Franca, L., Rainey, F. A., Schumann, P., Nobre, M. F., and Da Costa, M. S. (2011). Gaiella occulta gen. nov., sp. nov., a novel representative of a deep branching phylogenetic lineage within the class Actinobacteria and proposal of Gaiellaceae fam. nov. and Gaiellales ord. nov. Syst. Appl. Microbiol. 34, 595–599. doi: 10.1016/j.syapm.2011.07.001
Badri, D. V., Chaparro, J. M., Zhang, R., Shen, Q., and Vivanco, J. M. (2013). Application of natural blends of phytochemicals derived from the root exudates of Arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 288, 4502–4512. doi: 10.1074/jbc.M112.433300
Beattie, G. A. (2018). Metabolic coupling on roots. Nat. Microbiol. 3, 396–397. doi: 10.1038/s41564-018-0139-1
Bulgarelli, D., Garrido-Oter, R., Munch, P. C., Weiman, A., Droge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren Van Themaat, E., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Camp, W. H. (1945). The North American blueberries with notes on other groups of vacciniaceae. Brittonia 5, 203–275. doi: 10.2307/2804880
Canarini, A., Kaiser, C., Merchant, A., Richter, A., and Wanek, W. (2019). Root exudation of primary metabolites: mechanisms and their roles in plant responses to environmental stimuli. Front. Plant Sci. 10:157. doi: 10.3389/fpls.2019.00157
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cardinale, M., Grube, M., Erlacher, A., Quehenberger, J., and Berg, G. (2015). Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 17, 239–252. doi: 10.1111/1462-2920.12686
Caspersen, S., Svensson, B., Håkansson, T., Winter, C., Khalil, S., and Asp, H. (2016). Blueberry-soil interactions from an organic perspective. Sci. Hortic. Amsterdam 208, 78–91. doi: 10.1016/j.scienta.2016.04.002
Chapelle, E., Mendes, R., Bakker, P. A., and Raaijmakers, J. M. (2016). Fungal invasion of the rhizosphere microbiome. ISME J. 10, 265–268. doi: 10.1038/ismej.2015.82
Chavez, D. J., and Lyrene, P. M. (2009). Interspecific crosses and backcrosses between diploid Vaccinium darrowii and tetraploid southern highbush blueberry. J. Amer. Soc. Hort. Sci. 134, 273–280. doi: 10.21273/JASHS.134.2.273
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., Mcgarrell, D. M., Sun, Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Coleman-Derr, D., Desgarennes, D., Fonseca-Garcia, C., Gross, S., Clingenpeel, S., Woyke, T., et al. (2016). Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 209, 798–811. doi: 10.1111/nph.13697
Comeau, A. M., Douglas, G. M., and Langille, M. G. (2017). Microbiome helper: a custom and streamlined workflow for microbiome research. mSystems 2:e00127-16. doi: 10.1128/mSystems.00127-16
Comeau, A. M., Li, W. K., Tremblay, J. E., Carmack, E. C., and Lovejoy, C. (2011). Arctic ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS One 6:e27492. doi: 10.1371/journal.pone.0027492
Cordovez, V., Dini-Andreote, F., Carrion, V. J., and Raaijmakers, J. M. (2019). Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 73, 69–88. doi: 10.1146/annurev-micro-090817-062524
Coville, F. V. (1937). “Improving the wild blueberry,” in Yearbook of Agriculture. (Washington, DC: U.S. Department of Agriculture), 559–574. Available online at: https://naldc.nal.usda.gov/download/IND43893573/PDF (accessed February 16, 2020).
Daghino, S., Martino, E., and Perotto, S. (2016). Model systems to unravel the molecular mechanisms of heavy metal tolerance in the ericoid mycorrhizal symbiosis. Mycorrhiza 26, 263–274. doi: 10.1007/s00572-015-0675-y
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Edwards, J., Johnson, C., Santos-Medellin, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Eo, J., Park, K. -C., and Kim, M. -H. (2015). Plant-specific effects of sunn hemp (Crotalaria juncea) and sudex (Sorghum bicolor x S. bicolor var. sudanense) on the abundance and composition of soil microbial community. Agr. Ecosys. Environ. 213, 86–93. doi: 10.1016/j.agee.2015.07.025
Finn, C. E., Luby, J., Rosen, C. J., and Asher, P. (1993). Blueberry germplasm screening at several soil pH regimes I: plant survival and growth. J. Am. Soc. Hort. Sci. 118, 377–382. doi: 10.21273/JASHS.118.3.377
Fitzpatrick, C. R., Copeland, J., Wang, P. W., Guttman, D. S., Kotanen, P. M., and Johnson, M. T. J. (2018). Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. U.S.A. 115, E1157–E1165. doi: 10.1073/pnas.1717617115
Foster, Z. S., Sharpton, T. J., and Grunwald, N. J. (2017). Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 13:e1005404. doi: 10.1371/journal.pcbi.1005404
Gao, Z., Karlsson, I., Geisen, S., Kowalchuk, G., and Jousset, A. (2019). Protists: puppet masters of the rhizosphere microbiome. Trends Plant Sci. 24, 165–176. doi: 10.1016/j.tplants.2018.10.011
Grelet, G., Martino, E., Dickie, I. A., Tajuddin, R., and Artz, R. (2017). “Ecology of ericoid mycorrhizal fungi: what insight have we gained with molecular tools and what’s missing?,” in Molecular Mycorrhizal Symbiosis, ed. F. Martin, (Hoboken, NJ: John Wiley & Sons), 405–419. doi: 10.1002/9781118951446.ch22
Hancock, J. F. (2006). Highbush blueberry breeders. Hort. Sci. 41, 20–21. doi: 10.21273/HORTSCI.41.1.20
Kariman, K., Barker, S. J., and Tibbett, M. (2018). Structural plasticity in root-fungal symbioses: diverse interactions lead to improved plant fitness. PeerJ 6:e6030. doi: 10.7717/peerj.6030
Kiers, E. T., Hutton, M. G., and Denison, R. F. (2007). Human selection and the relaxation of legume defences against ineffective rhizobia. Proc. Biol. Sci. 274, 3119–3126. doi: 10.1098/rspb.2007.1187
Lareen, A., Burton, F., and Schafer, P. (2016). Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 90, 575–587. doi: 10.1007/s11103-015-0417-8
Leopold, D. R. (2016). Ericoid fungal diversity: challenges and opportunities for mycorrhizal research. Fungal Ecol. 24, 114–123. doi: 10.1016/j.funeco.2016.07.004
Li, X., Zhang, Y., Ding, C., Jia, Z., He, Z., Zhang, T., et al. (2015). Declined soil suppressiveness to Fusarium oxysporum by rhizosphere microflora of cotton in soil sickness. Biol. Fertil. Soils 51, 935–946. doi: 10.1007/s00374-015-1038-8
Lobos, G. A., and Hancock, J. F. (2015). Breeding blueberries for a changing global environment: a review. Front. Plant. Sci. 6:782. doi: 10.3389/fpls.2015.00782
Mahoney, A. K., Yin, C., and Hulbert, S. H. (2017). Community structure, species variation, and potential functions of rhizosphere-associated bacteria of different winter wheat (Triticum aestivum) cultivars. Front. Plant Sci. 8:132. doi: 10.3389/fpls.2017.00132
Massarotto, G., Barcellos, T., Garcia, C. S., Brandalize, A. P., Moura, S., and Schwambach, J., et al. (2016). Chemical characterization and cytotoxic activity of blueberry extracts (cv. Misty) cultivated in Brazil. J. Food Sci. 81, H2076–H2084. doi: 10.1111/1750-3841.13385
Mavrodi, O. V., Jung, C. M., Eberly, J. O., Hendry, S. V., Namjilsuren, S., Biber, P. D., et al. (2018). Rhizosphere microbial communities of Spartina alterniflora and Juncus roemerianus from restored and natural tidal marshes on Deer Island, Mississippi. Front. Microbiol. 9:3049. doi: 10.3389/fmicb.2018.03049
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Nunez, G. H., Olmstead, J. W., and Darnell, R. L. (2015). Rhizosphere acidification is not part of the strategy I iron deficiency response of Vaccinium arboreum and the southern highbush blueberry. Hort. Sci. 50, 1064–1069. doi: 10.21273/HORTSCI.50.7.1064
Op De Beeck, M., Lievens, B., Busschaert, P., Declerck, S., Vangronsveld, J., and Colpaert, J. V. (2014). Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS One 9:e97629. doi: 10.1371/journal.pone.0097629
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Perez-Jaramillo, J. E., Carrion, V. J., Bosse, M., Ferrao, L. F. V., De Hollander, M., Garcia, A. A. F., et al. (2017). Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 11, 2244–2257. doi: 10.1038/ismej.2017.85
Perez-Jaramillo, J. E., Carrion, V. J., De Hollander, M., and Raaijmakers, J. M. (2018). The wild side of plant microbiomes. Microbiome 6:143. doi: 10.1186/s40168-018-0519-z
Perez-Jaramillo, J. E., Mendes, R., and Raaijmakers, J. M. (2016). Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 90, 635–644. doi: 10.1007/s11103-015-0337-7
Pfeiffer, S., Mitter, B., Oswald, A., Schloter-Hai, B., Schloter, M., Declerck, S., et al. (2017). Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 93:fiw242. doi: 10.1093/femsec/fiw242
Raaijmakers, J. M., Paulitz, T. C., Steinberg, C., Alabouvette, C., and Moenne-Loccoz, Y. (2009). The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 321, 341–361. doi: 10.1007/s11104-008-9568-6
Read, D. J. (1996). The structure and function of the ericoid mycorrhizal root. Ann. Bot. 77, 365–374. doi: 10.1006/anbo.1996.0044
Sadowsky, J. J., Hanson, E. J., and Schilder, A. M. C. (2012). Root colonization by ericoid mycorrhizae and dark septate endophytes in organic and conventional blueberry fields in Michigan. Int. J. Fruit Sci. 12, 169–187. doi: 10.1080/15538362.2011.619346
Sasse, J., Martinoia, E., and Northen, T. (2018). Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci. 23, 25–41. doi: 10.1016/j.tplants.2017.09.003
Scagel, C. F. (2005). Inoculation with ericoid mycorrhizal fungi alters fertilizer use of highbush blueberry cultivars. Hortscience 40, 786–794. doi: 10.21273/HORTSCI.40.3.786
Scagel, C. F., and Yang, W. Q. (2005). Cultural variation and mycorrhizal status of blueberry plants in NW Oregon commercial production fields. Int. J. Fruit Sci. 5, 85–111. doi: 10.1300/J492v05n02_10
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Sharpe, R. H., and Darrow, G. M. (1959). Breeding blueberries for the Florida climate. Proc. Florida State Hort. Soc. 72, 308–311.
Sørensen, J., and Sessitsch, A. (2006). “Plant-associated bacteria – lifestyle and molecular interactions,” in Modern Soil Microbiology, ed. J. D. Van Elsas, J. K. Jansson, and J. T. Trevors, (Boca Raton, FL: CRC Press), 211–236.
Sun, G., Yang, Z., Kosch, T., Summers, K., and Huang, J. (2011). Evidence for acquisition of virulence effectors in pathogenic chytrids. BMC Evol. Biol. 11:195. doi: 10.1186/1471-2148-11-195
Sun, W., Xiao, E., Krumins, V., Haggblom, M. M., Dong, Y., Pu, Z., et al. (2018). Rhizosphere microbial response to multiple metal(loid)s in different contaminated arable soils indicates crop-specific metal-microbe interactions. Appl. Environ. Microbiol. 84, e701–e718. doi: 10.1128/AEM.00701-18
Szoboszlay, M., Lambers, J., Chappell, J., Kupper, J. V., Moe, L. A., and Mcnear, D. H. (2015). Comparison of root system architecture and rhizosphere microbial communities of Balsas teosinte and domesticated corn cultivars. Soil Biol. Biochem. 80, 34–44. doi: 10.1016/j.soilbio.2014.09.001
van der Voort, M., Kempenaar, M., Van Driel, M., Raaijmakers, J. M., and Mendes, R. (2016). Impact of soil heat on reassembly of bacterial communities in the rhizosphere microbiome and plant disease suppression. Ecol. Lett. 19, 375–382. doi: 10.1111/ele.12567
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Vega, A. R., Garciga, M., Rodriguez, A., Prat, L., and Mella, J. (2009). Blueberries mycorrhizal symbiosis outside the boundaries of natural dispersion for Ericaceous plants in Chile. Acta Hortic. 810, 665–672. doi: 10.17660/ActaHortic.2009.810.88
Vohnik, M., and Albrechtova, J. (2011). The co-occurrence and morphological continuum between ericoid mycorrhiza and dark septate endophytes in roots of six European Rhododendron species. Folia Geobot. 46, 373–386. doi: 10.1007/s12224-011-9098-5
Wang, H., Cao, G., and Prior, R. L. (1997). Oxygen radical absorbing capacity of anthocyanins. J. Agric. Food Chem. 45, 304–309. doi: 10.1021/jf960421t
Wang, W., Luo, X., Chen, Y., Ye, X., Wang, H., Cao, Z., et al. (2019). Succession of composition and function of soil bacterial communities during key rice growth stages. Front. Microbiol. 10:421. doi: 10.3389/fmicb.2019.00421
Wei, Z., and Jousset, A. (2017). Plant breeding goes microbial. Trends Plant Sci. 22, 555–558. doi: 10.1016/j.tplants.2017.05.009
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Wille, L., Messmer, M. M., Studer, B., and Hohmann, P. (2018). Insights to plant–microbe interactions provide opportunities to improve resistance breeding against root diseases in grain legumes. Plant Cell Environ. 42, 20–40. doi: 10.1111/pce.13214
Xue, C., Penton, C. R., Shen, Z., Zhang, R., Huang, Q., Li, R., et al. (2015). Manipulating the banana rhizosphere microbiome for biological control of Panama disease. Sci. Rep. 5:11124. doi: 10.1038/srep11124
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Yurgel, S. N., Douglas, G. M., Comeau, A. M., Mammoliti, M., Dusault, A., Percival, D., et al. (2017). Variation in bacterial and eukaryotic communities associated with natural and managed wild blueberry habitats. Phytobiomes 1, 102–113. doi: 10.1094/PBIOMES-03-17-0012-R
Yurgel, S. N., Douglas, G. M., Dusault, A., Percival, D., and Langille, M. G. I. (2018). Dissecting community structure in wild blueberry root and soil microbiome. Front. Microbiol. 9:87. doi: 10.3389/fmicb.2018.01187
Zachow, C., Muller, H., Tilcher, R., and Berg, G. (2014). Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima – ancestor of all beet crops and modern sugar beets. Front. Microbiol. 5:415. doi: 10.3389/fmicb.2014.00415
Zakrzewski, M., Proietti, C., Ellis, J. J., Hasan, S., Brion, M. J., Berger, B., et al. (2017). Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33, 782–783. doi: 10.1093/bioinformatics/btw725
Zhalnina, K., Louie, K. B., Hao, Z., Mansoori, N., Da Rocha, U. N., Shi, S., et al. (2018). Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 3, 470–480. doi: 10.1038/s41564-018-0129-3
Keywords: Vaccinium, blueberry, rhizosphere, microbiome, 16S, ITS, 18S
Citation: Li J, Mavrodi OV, Hou J, Blackmon C, Babiker EM and Mavrodi DV (2020) Comparative Analysis of Rhizosphere Microbiomes of Southern Highbush Blueberry (Vaccinium corymbosum L.), Darrow’s Blueberry (V. darrowii Camp), and Rabbiteye Blueberry (V. virgatum Aiton). Front. Microbiol. 11:370. doi: 10.3389/fmicb.2020.00370
Received: 21 October 2019; Accepted: 19 February 2020;
Published: 12 March 2020.
Edited by:
Eligio Malusà, Instytut Ogrodnictwa, PolandReviewed by:
S. Franz Bender, Agroscope, SwitzerlandPeter Kusstatscher, Graz University of Technology, Austria
Copyright © 2020 Li, Mavrodi, Hou, Blackmon, Babiker and Mavrodi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ebrahiem M. Babiker, RWJyYWhpZW0uQmFiaWtlckBhcnMudXNkYS5nb3Y=; Dmitri V. Mavrodi, ZG1pdHJpLm1hdnJvZGlAdXNtLmVkdQ==
†These authors have contributed equally to this work