 Yufei Lin
Yufei Lin Xiaohong Dong†
Xiaohong Dong† Dawei Rao
Dawei Rao Lihua Zhang
Lihua Zhang Kun Yang
Kun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 March 2020
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00352
This article is part of the Research Topic Antimicrobial Resistance As a Global Public Health Problem: How Can We Address It? View all 50 articles
We sequenced the whole genomes of three mcr-1-positive multidrug-resistant E. coli strains, which were previously isolated from the environment of egret habitat (polluted river) and egret feces. The results exhibit high correlation between antibiotic-resistant phenotype and genotype among the three strains. Most of the mobilized antibiotic resistance genes (ARGs) are distributed on plasmids in the forms of transposons or integrons. Multidrug-resistant (MDR) regions of high homology are detected on plasmids of different E. coli isolates. Therefore, horizontal transfer of resistance genes has facilitated the transmission of antibiotic resistance between the environmental and avian bacteria, and the transfer of ARGs have involved multiple embedded genetic levels (transposons, integrons, plasmids, and bacterial lineages). Inspired by this, systematic metadata analysis was performed for the available sequences of mcr-1-bearing plasmids. Among these plasmids, IncHI2 plasmids carry the most additional ARGs. The composition of these additional ARGs varies according to their geographical distribution. The phylogenetic reconstruction of IncI2 and IncX4 plasmids provides the evidence for their multiregional evolution. Phylogenetic analysis at the level of mobile genetic element (plasmid) provides important epidemiological information for the global dissemination of mcr-1 gene. Highly homologous mcr-1-bearing IncI2 plasmids have been isolated from different regions along the East Asian-Australasian Flyway, suggesting that migratory birds may mediate the intercontinental transportation of ARGs.
Horizontal gene transfer (HGT) plays an important role in the global dissemination of antibiotic resistance, while the mobilization of ARGs is the first and also the most important evolutionary step for their horizontal transfer. The discovery of any new mobilized resistance gene always attracts great attention of the researchers. As evidenced by a number of studies, these mobilized resistance genes have been increasingly spreading all over the world (Stokes and Gillings, 2011). Typical cases include the discovery of blaKPC, blaNDM, and mcr-1 genes. The blaKPC gene was first identified in 1996, and now it can be detected in many regions of the world (Munoz-Price et al., 2013). Its mobilization is related to a 10 kb Tn3-based mobile transposon Tn4401 (Naas et al., 2008; Cuzon et al., 2011). The blaNDM gene was first identified in 2008 in a K. pneumoniae isolate recovered from a Swedish patient, who had previously been hospitalized in New Delhi, India (Yong et al., 2009). Thereafter, this resistance gene was detected in different regions of the world (Dortet et al., 2014). Its mobilization is associated with an ISAba125 composite transposon Tn125 (Poirel et al., 2012). The mcr-1 gene was first discovered in China (Liu et al., 2016), and the same ARG was then reported in various regions of the world (Wang et al., 2018). Its mobilization is mediated by an ISApl1 composite transposon Tn6330 (Snesrud et al., 2016; Li et al., 2017).
Although the effect of HGT on the dissemination of antibiotic resistance has been realized, the spread of specific resistant bacterial clones is also widely concerned (Munoz-Price et al., 2013; Matamoros et al., 2017). In early 1990s, it was found that the moving of transposons between carrier replicons (plasmids) resulted in the spread of ARGs between bacterial species (Liebert et al., 1999). Recently, it has been described as a nested Russian doll-like mobility of ARGs, which once again drew the attention of researchers (Sheppard et al., 2016; Wang et al., 2018; Hernando-Amado et al., 2019). These studies clearly explain the mechanism of the horizontal transfer of resistance genes at multiple genetic levels (transposons, integrons, plasmids, bacterial lineages and bacterial species) (Wang et al., 2018). In our previous study (Wu et al., 2018), we demonstrated that wild birds could transport antibiotic resistance from contaminated river to the surrounding environment, and the spread of antibiotic resistance was not mainly due to the transfer of resistant bacterial clones. Among those resistant E. coli isolates in the aforementioned work, no strong correlation was observed between strain genotypes (repetitive-element PCR genotyping) and their drug-resistance patterns. Therefore, we concluded that the horizontal transfer of resistance genes was the main mechanism of resistance transmission in that process. Nonetheless, there was no direct sequencing evidence at the time. In this study, we sequenced the whole genomes of three mcr-1-positive multidrug-resistant E. coli strains isolated in the previous work to pursue the evidence for horizontal transfer of resistance genes. Inspired by the sequencing results, we systematically analyzed mcr-1-bearing plasmid sequences downloaded from GenBank database. We tried to obtain epidemiological information of the mcr-1 gene by analyzing its global dissemination at multiple levels of mobile genetic elements (MGEs), especially at the plasmid level.
In our previous study, we found that the wild birds (egrets) could mediate the environmental transmission of antibiotic resistance in local area, from the polluted Jin River to birds’ nightly habitat, Wangjianglou Park (Wu et al., 2018). Some of the E. coli strains isolated in that work exhibited colistin resistance (n = 6), among which three isolates were proved to be mcr-1-positive and multidrug-resistant. Two of the three strains were isolated from the river (Jin River) polluted by antibiotic-resistant bacteria and the third one was isolated from egret feces. Three E. coli isolates were respectively labeled as W5-6, W2-5, and BE2-5.
The minimum inhibitory concentrations (MICs) of 10 antibiotics against the three E. coli isolates has previously been determined (Wu et al., 2018) via a modified broth micro-dilution method as per ISO 20776-1:2006 CLSI using 96-well microtiter plates (Clinical and Laboratory Standards Institute [CLSI], 2019). The only modification in our method is in the step of inoculum preparation. Typically, the E. coli isolates were first grown in LB-broth-loaded 96 well plate at 37°C overnight to reach the stationary phase. The cell cultures were then 103 fold diluted and used as inoculum. When inoculating the testing 96-well plates containing antibiotics of different concentrations, a 48-pin replicator was used to improve the efficiency of experimental operation. The MIC endpoints were determined as the lowest concentration, at which there was no visible growth after 20 h of incubation at 37°C. Duplicate tests for each antibiotic concentration were conducted. Positive and negative controls were conducted in antibiotic-free LB to ensure the growth of environmental E. coli isolates under lab conditions and sterility of the assay, respectively. Quality control of the procedure was conducted by using the susceptive E. coli standard strain ATCC 25922.
The whole genome DNA was extracted from fresh bacterial cell mass recovered from 100 mL LB pure culture containing colistin of 4 μg/mL with the FastDNA® Spin Kit for Soil (MP Biomedicals, France). About 1.2 mL of high-quality DNA was obtained for each E. coli strain. A small amount of each DNA sample was used for the amplification and sequencing of 16S rRNA gene to ensure that the DNA sample was obtained from a pure culture. The presence of mcr-1 gene in the DNA sample was also verified via PCR. The primer pair for amplifying 16S rRNA gene was 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1522R (5′-AAGGAGGTGATCCANCCRCA-3′) and that for mcr-1 was CLR5-F (5′-CGGTCAGTCCGTTTGTTC-3′) and CLR5-R (5′-CTTGGTCGGTCTGTAGGG-3′) (Liu et al., 2016). Thereafter, the remaining bacterial DNA samples were kept frozen (−40°C) until sequencing.
The bacterial genome DNA was detected by agarose gel electrophoresis and quantified by Qubit. The genomes of the three isolates were sequenced using Single Molecule, Real-Time (SMRT) technology performed at Beijing Novogene Bioinformatics Technology Co., Ltd. The low quality reads were filtered by the SMRT 2.3.0 (Berlin et al., 2015), and the filtered reads were de novo assembled to generate contigs without gaps (Koren and Phillippy, 2015). All genome sequences (chromosome and plasmid sequences) of the three E. coli isolates were deposited into GenBank database under the BioProject PRJNA495707 with BioSample numbers SAMN10230266 to SAMN10230268 and accession numbers CP032986 to CP032995.
The strain types (STs) of the three E. coli isolates were determined from their assembled genomes using online MLST service provided by the Center for Genomic Epidemiology1 according to Achtman’s MLST scheme (Wirth et al., 2006; Larsen et al., 2012).
We used GeneMarks to predict ORFs for the three E. coli isolates (Besemer et al., 2001). We used seven databases to predict gene functions. They were GO (Gene Ontology) (Ashburner et al., 2000), KEGG (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa et al., 2004, 2006), COG (Clusters of Orthologous Groups) (Tatusov et al., 2003), NR (Non-Redundant Protein Database) (Li et al., 2002), TCDB (Transporter Classification Database) (Saier et al., 2014), Swiss-Prot (Bairoch and Apweiler, 2000), and TrEMBL (Magrane and UniProt, 2011), respectively. A whole genome BLAST search (E-value less than 10–5, minimal alignment length percentage larger than 40%) was performed against above seven databases (Altschul et al., 1990). ARGs were annotated via BLAST searching the ORFs against the Comprehensive Antibiotic Resistance Database (CARD) (Liu and Pop, 2009; McArthur et al., 2013). We set the thresholds of e-value and “best identity” at 10–5 and 80%, respectively.
The discovery of the mobilized colistin resistance gene, mcr-1, is in the era of rapid development of next-generation sequencing technology. During only 4 years, hundreds of sequences of mcr-1-bearing plasmids have been uploaded to the database, which provides a good chance for metadata analysis of these plasmids. We retrieved the GenBank database with keywords “mcr-1” (or mcr1) and “plasmid” in June 2018. From the hit entries, the circular sequences were manually picked and recorded the metadata. Identification of the plasmid incompatibility group was performed for downloaded plasmid sequences via the CGE online services PlasmidFinder v1.32 (Carattoli et al., 2014). ResFinder3 was used to determine other acquired resistance genes on these plasmids (Zankari et al., 2012).
Most of the published mcr-1-bearing plasmids belong to three main incompatibility groups, IncHI2, IncI2, and IncX4 (Li et al., 2017; Matamoros et al., 2017). According to the respective characteristics of the three incompatibility plasmid groups, we performed a targeted analysis. The analysis routine is illustrated in Supplementary Figure S1.
The original sequencing data obtain more than 50 × coverage of the whole genome for the three E. coli isolates. Chromosomes and plasmids of the three isolates are all assembled in circular contigs with no gaps. Three E. coli isolates are of different strain types (Table 1). The number, size and Inc-types of plasmids are also different among the three isolates (Table 1). E. coli W5-6 contains 3 plasmids. The other two isolates both carry 2 plasmids. The plasmid pMCR_W5-6 gives the biggest size (241 kbp) among all plasmids. The numbers of genes predicted by GeneMarks are 4898, 4834, and 4576 for W5-6, W2-5, and BE2-5, respectively. The annotation of the three genomes with the COG database clusters these genes into 23 classes with annotation rate of 89.6, 90.7, and 93.8%, respectively (Supplementary Figure S2). Genomes of all the three E. coli strains, especially the strain W5-6, contain a considerable number of MGEs (Mobilome in Supplementary Figure S2).
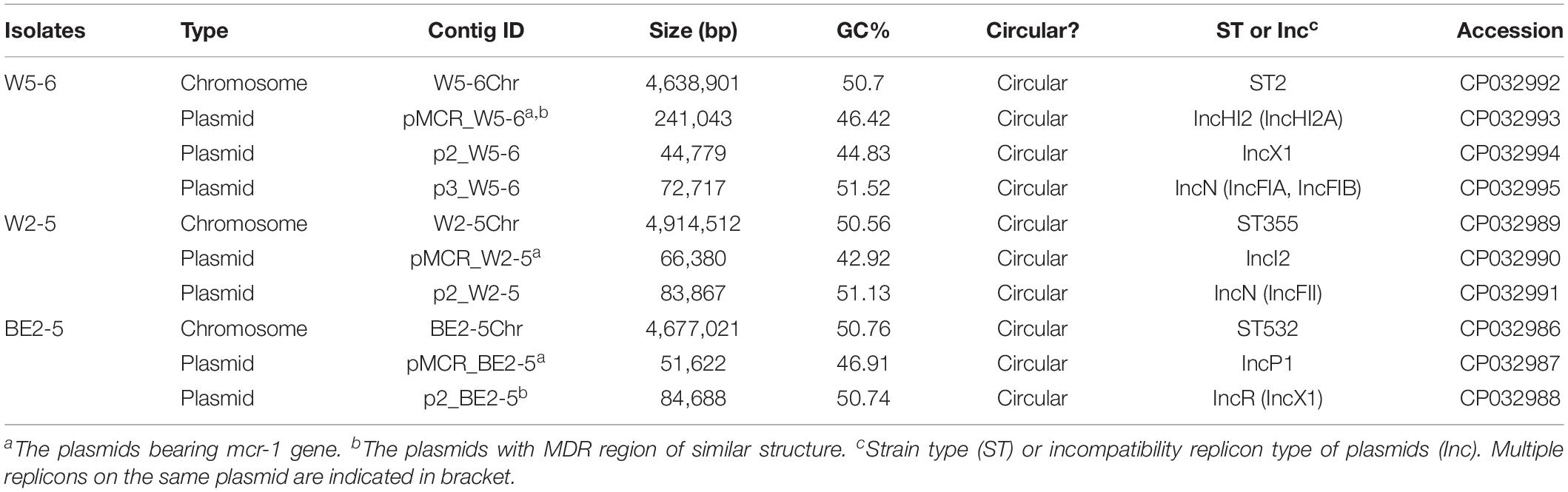
Table 1. Genome assembly results for three E. coli isolates.
All the three strains show multidrug resistance. The E. coli W5-6 demonstrates the highest level of drug resistance among the three isolates. It shows resistance against 9 out of 10 tested antibiotics. The other two E. coli isolates of lower resistance levels are resistant to 7 antibiotics (Table 2).
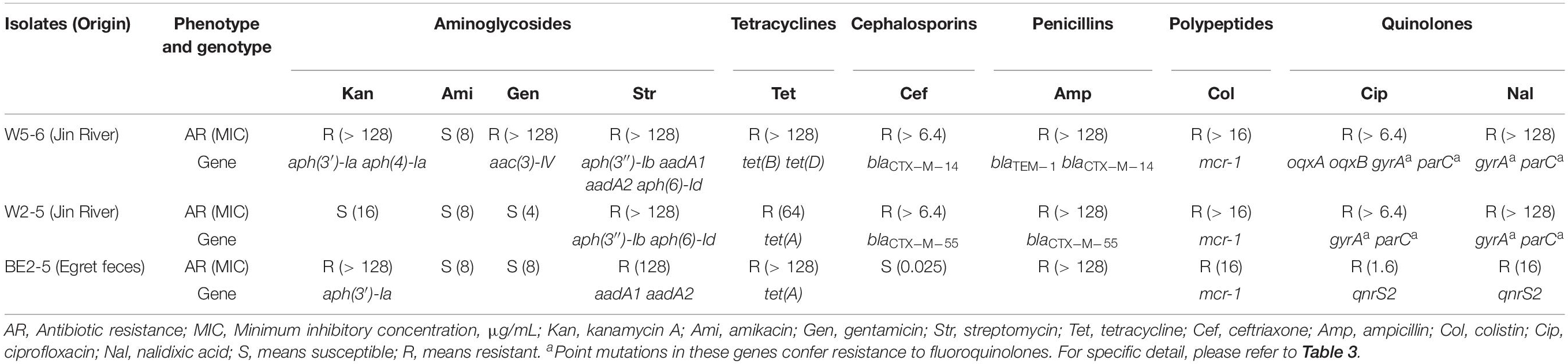
Table 2. Antibiotic-resistant phenotype and corresponding resistance genes or genetic mutations in three E. coli isolates.
Eighty-five putative ARGs (via BLAST against the CARD database) are annotated in the whole genome of E. coli W5-6, 20 of which are on plasmids. The numbers of ARGs on chromosomes of E. coli W2-5 and BE2-5 are 65 and 66, respectively. The numbers of ARGs on plasmids of these two isolates are 3 and 11, respectively (Table 3). Fifty-two ARGs are common among all the three E. coli isolates. However, most of these shared ARGs (n = 44, 84.6%) are efflux pump genes, and they are distributed on their chromosomes, which implies that efflux pump genes are highly conserved in the same bacterial species. These efflux pump genes do not appear to be significantly associated with the antibiotic-resistant phenotype of the three strains. Although these efflux pump genes may be just involved in some detoxification process (Martinez et al., 2009), we still list them below (Table 3) for reference. The only non-efflux-pump common ARG with a definite resistant phenotype among the three isolates is the colistin resistance gene mcr-1. The mcr-1 gene is located on the plasmids of different Inc-types in the three E. coli strains. The plasmids pMCR_W5-6, pMCR_W2-5, and pMCR_BE2-5 are of the IncHI2, IncI2, and IncP1-type, respectively (Table 1). The mcr-1 sequence on plasmid pMCR_W2-5 lost the downstream ISApl1 (Supplementary Figure S3).
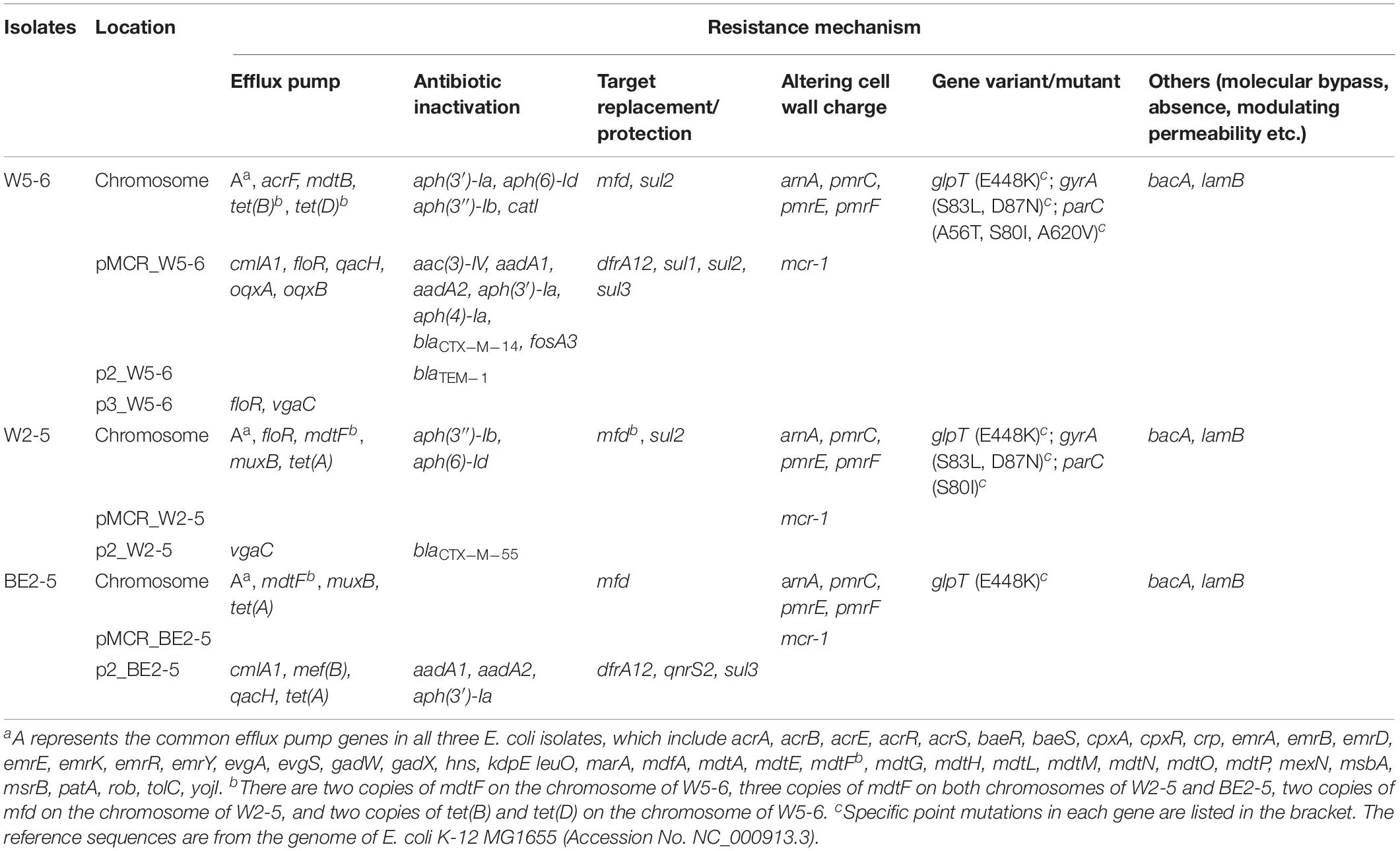
Table 3. CARD database annotated ARGs in three E. coli isolates.
For Aminoglycosides, resistance genes encoding three types of antibiotic inactivation enzymes are detected among the three E. coli isolates, aminoglycoside acetyltransferases [aac(3)-IV], phosphotransferase [aph(3′)-Ia, aph(4)-Ia, aph(3″)-Ib and aph(6)-Id] and nucleotidyltransferase (aadA1 and aadA2) genes. E. coli W5-6 possesses all the three kinds of resistance genes (Table 2), and exhibits the highest resistance level to aminoglycosides. It is resistant to three out of four tested aminoglycosides: kanamycin A, gentamicin and streptomycin (Table 2). E. coli W2-5 has the ARGs, which render resistance to streptomycin [aph(3″)-Ib and aph(6)-Id], and exhibits resistance to this antibiotic. E. coli BE2-5 does not have these two ARGs, while it has aph(3′)-Ia, which enables it to resist against kanamycin A (McArthur et al., 2013). For tetracycline, we detected corresponding ARGs in the genomes of all the three E. coli isolates either on chromosome [tet(A), tet(B) and tet(D)] or on plasmid [tet(A)], which is in good agreement with their resistance to this antibiotic (Tables 2, 3). For β-lactams, different β-lactamase genes are detected on the plasmids of E. coli W5-6 (blaCTX–M–14 on pMCR_W5-6 and blaTEM–1 on p2_W5-6) and W2-5 (blaCTX–M–55 on p2_W2-5), while no β-lactamase gene is detected for E. coli BE2-5, as it is susceptible to ceftriaxone (Table 2). For quinolones, genes encoding subunits of efflux pump complex conferring resistance to fluoroquinolone (oqxA and oqxB) (Kim et al., 2009) are detected on the plasmid (pMCR_W5-6) of E. coli W5-6. Point mutations in gyrA and parC genes conferring resistance to fluoroquinolones are detected in E. coli W5-6 and W2-5 (Oram and Fisher, 1991; Tankovic et al., 1996). In E. coli BE2-5, qnrS2 gene is detected on its plasmid (p2_BE2-5), which confers it weak resistance to ciprofloxacin and nalidixic acid (Gay et al., 2006; Table 2). Besides above ARGs conferring resistance to the tested antibiotics, resistance genes against sulfonamides (sul2 etc.), chloramphenicol (cmlA1), fosfomycin (fosA3), and trimethoprim (drfA12) are also detected among the three E. coli isolates. Point mutation in glpT gene conferring resistance to fosfomycin is detected in all three E. coli isolates (Takahata et al., 2010; Table 3). Overall, the antibiotic-resistant genotype is in good agreement with the resistant phenotype among the three E. coli isolates. The only exception is that no corresponding resistance gene is detected to explain the unexpected resistance of E. coli BE2-5 against ampicillin.
Almost all the ARGs detected in the avian E. coli (BE2-5) are found in two other environmental strains (W2-5 and W5-6, Table 3), which is consistent with our previous view that the antibiotic resistance (resistant bacteria or resistance genes) is mainly transferred from the polluted river (Jin River) to the wild bird (egret) (Wu et al., 2018). Most importantly, highly homologous MDR regions are detected on two plasmids of different Inc-types (pMCR_W5-6 and p2_BE2-5) in E. coli W5-6 and BE2-5, respectively (Figures 1A,B). The sizes of the MDR regions are around 12,800 bp (Figure 1D). Although the size and overall structure of the two plasmids are different (Figures 1A,B), the composition of the ARGs and MGE sequences (gene sequences encoding transposase and integrase) and their arrangement in the MDR regions are almost the same (Figure 1C and Supplementary Figure S4). The only difference is that the MDR region on the plasmid pMCR_W5-6 missing an efflux pump gene mef(B) in comparison to the plasmid p2_BE2-5. On the plasmid pMCR_W5-6, there even remains a 33 nt 3′ fragment of the mef(B) gene. This provides sequencing evidence for gene deletion and recombination during the evolution of the MDR region. The shared ARGs includes the genes conferring resistance to aminoglycosides [aadA1, aadA2 and aph(3′)-Ia], sulfonamides (sul3), trimethoprim (drfA12) and chloramphenicols (cmlA1). The shared MGE sequences, especially the IS26 sequences that are detected more than 5 copies on both plasmids (Figures 1A,B and Supplementary Figure S4), should play an important role during the formation of the MDR regions, and provide conditions for the relocation of these ARGs. Since the highly homologous MDR regions are detected on the plasmids of different Inc-types and in the E. coli of different strain types, the evolution of the MDR regions must have involved the process of HGT.
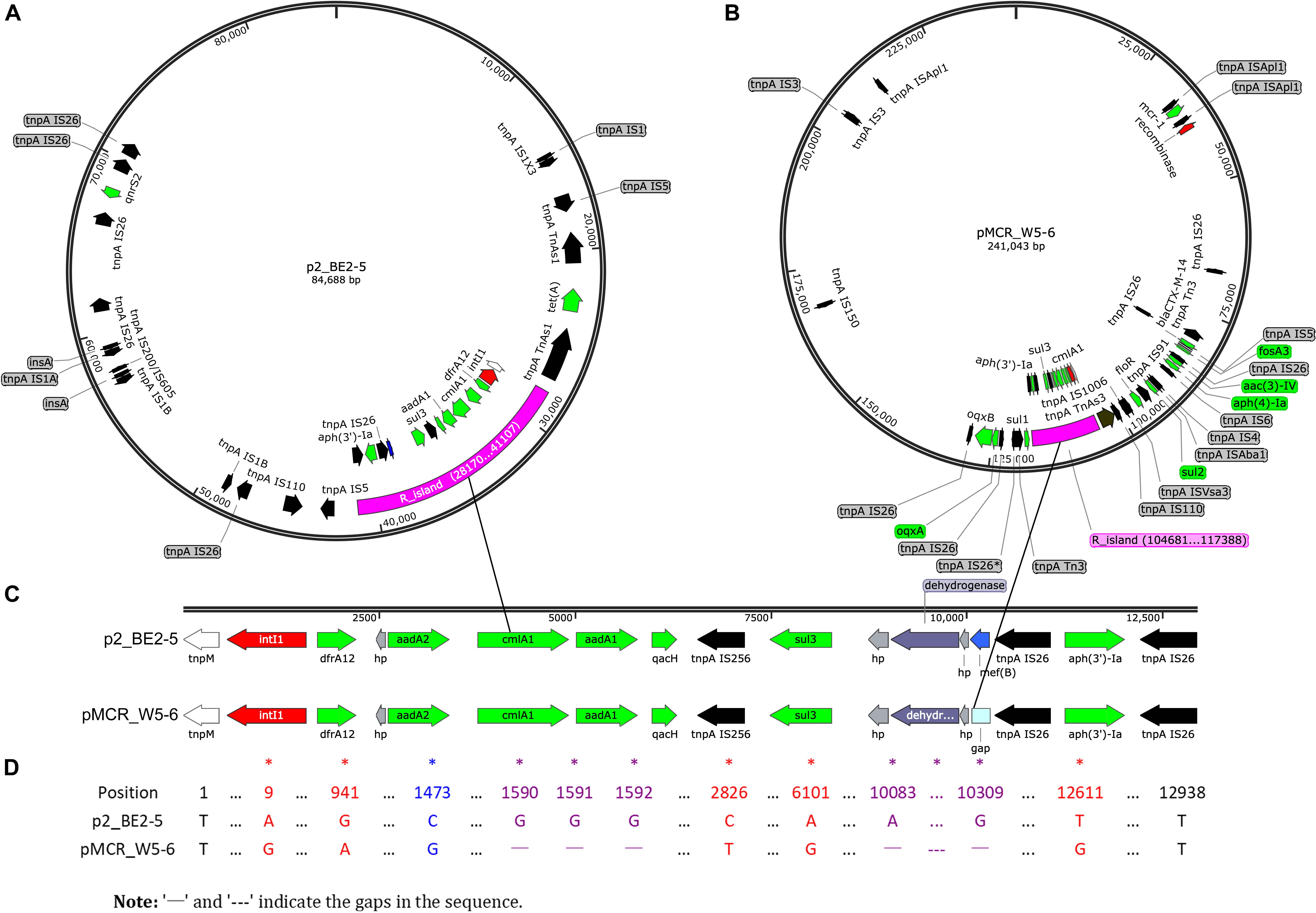
Figure 1. Sequencing evidence for horizontal transfer of ARGs between the environmental (polluted Jin River) and avian (egret) E. coli. (A,B) Exhibit the distribution of ARGs (green arrows) and MGEs on plasmids p2_BE2-5 and pMCR_W5-6, respectively. The MDR regions (R_island) of the similar structure are indicated as purple fragments on both plasmids. Their structures in detail are illustrated in (C), and the sequence alignment of two R_islands is exhibited in (D). The MDR region is composed of one integron (red arrows indicate site-specific integrase/recombinase genes) and one IS26_aph(3′)-Ia_IS26 composite transposon (black arrows for transposase genes, tnpA IS26 etc.). The recombination event (mef(B) gene) in the MDR region is indicated in blue (A,C, on plasmid p2_PE2-5). The mismatches between sequences were indicated with asterisks in (D). A BRIG-Easyfig version of this figure is shown in supporting information as Supplementary Figure S4.
The identical drug-resistant transposon containing aph(3′)-Ia gene is also found on the chromosome (W5-6Chr) and plasmids (pMCR_W5-6 and p2_BE2-5) of different bacteria. Similarly, the resistance gene [aph(3′)-Ia] is also flanked with transposase genes (tnpA IS26) on both sides (Supplementary Figure S5). The ARGs on other plasmids are illustrated in Supplementary Figure S6. Most of these ARGs also show close correlation with MGEs.
The spread of antibiotic resistance is closely related to the horizontal transfer of resistance genes, regardless in the clinical or in a natural environment, which is highly polluted with antibiotics or antibiotic resistant bacteria (Stokes and Gillings, 2011). In our case, Jin River (an urban river in Chengdu, Sichuan, China) is highly polluted with antibiotic resistant bacteria, and the avian inhabitants (egrets) there are also highly affected. The resistance genes conferring antibiotic-resistant phenotype in the three E. coli isolates are mainly distributed on plasmids or on chromosome, but in forms of transposons (Supplementary Figure S5). The resistance genes on plasmids are also closely related to MGE sequences (transposase and integrase genes on plasmids as shown in Figure 1 and Supplementary Figure S6). From environmental (polluted river water) and avian (egret) E. coli of different strain types, we have detected almost identical MDR regions on the plasmids of different incompatibility types. ARGs, which are flanked by MGE sequences, present as transposons or integrons in the MDR regions. Identical drug-resistance transposon [IS26-aph(3′)-Ia-IS26] is also found on chromosome and plasmids of these host bacteria. These results indicate that HGT plays a crucial role in the environmental dissemination of antibiotic resistance, and the transfer of ARGs must involve multiple embedded genetic levels (transposons, integrons, plasmids, and bacterial lineages), which is so-called the nested “Russian doll” model of genetic mobility (Sheppard et al., 2016).
The metadata of 228 circular mcr-1-bearing plasmids were collected, as shown in Supplementary Table S1. The geographical distribution of plasmids of different incompatibility groups is depicted in Supplementary Figure S7. It is worth noting that the map outlines only the types of plasmid replicons on different continents. Some plasmids carry two or more (up to five) replicons (Supplementary Table S1). Therefore, a plasmid may have been counted more than once. The result still reflects the local diversity of plasmid types. Asia contributes the most types of mcr-1-bearing plasmids (13 out of 16). The other two continents in the Northern Hemisphere, Europe and North America also exhibit high diversity of plasmid types, 9 and 6, respectively. On the contrary, the regions in the Southern Hemisphere show poor plasmid diversity (Supplementary Figure S7). Sampling bias may have led to this discrepancy. However, twenty-eight mcr-1-bearing plasmids recovered from South America give only two plasmid types, IncX4 and IncI2. Up to some extent it reflects that the Southern Hemisphere (at least South America) suffers fewer invasions of various mcr-1-bearing plasmids. Among these mcr-1-bearing plasmids, the numbers of IncI2, IncX4 and IncHI2 plasmids are ranked at the top 3. Their detection rates in various Enterobacteriaceae hosts on different continents are illustrated in Figure 2A. IncI2 plasmids exhibit the most diverse bacterial hosts (in 6 unique host species of Enterobacteriaceae) and the most extensive geographical distribution (in all continents).
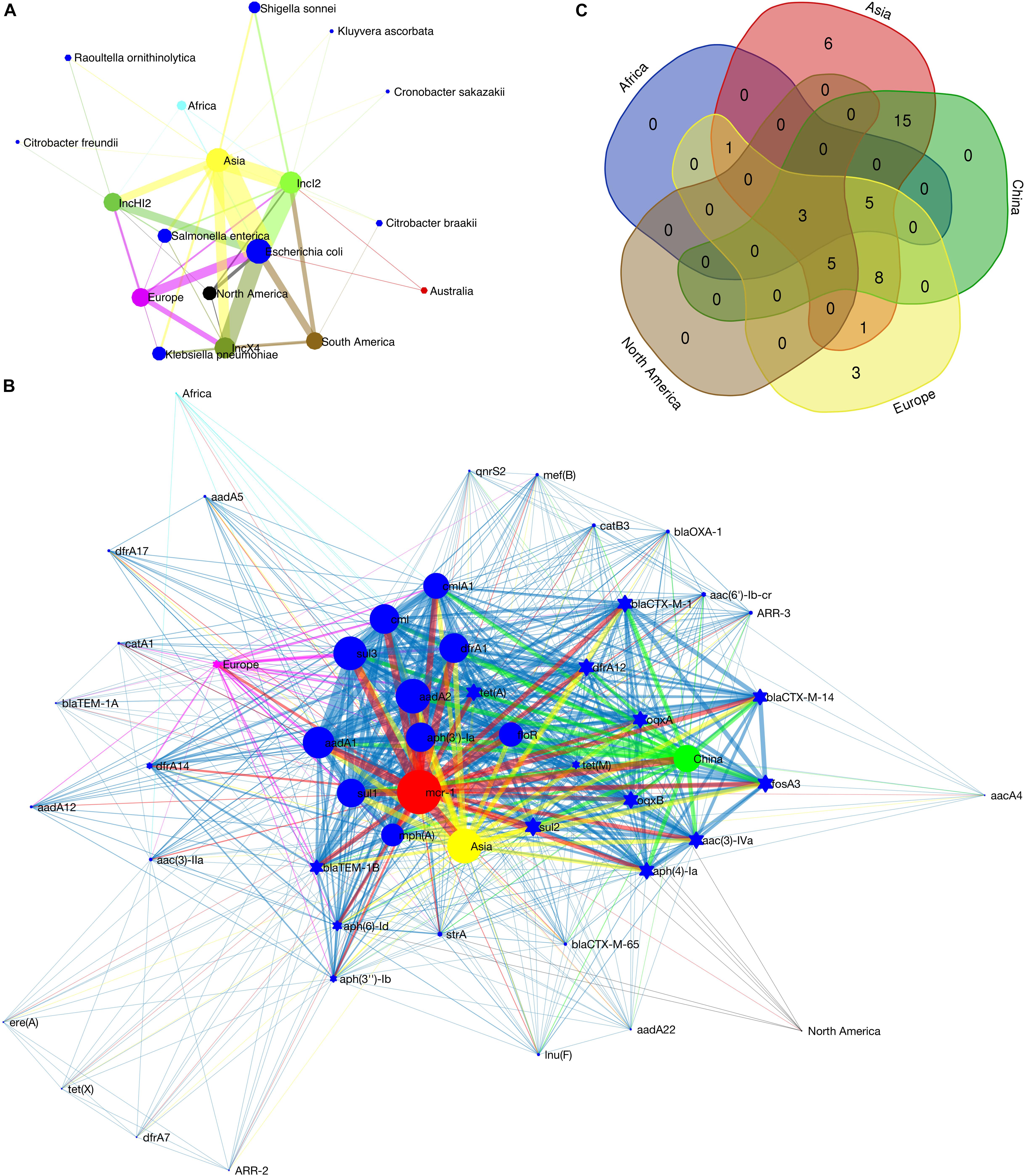
Figure 2. Detection rate of mcr-1-bearing plasmids in various bacterial host species on different continents (A), co-occurrence network of mcr-1 with additional ARGs on IncHI2 plasmids (B) and Venn diagram showing the numbers of shared and unique additional ARGs on IncHI2 plasmids at different regions (C). In (A), the size of the nodes (plasmid type, bacterial species or continent) is proportional to their detection rate (log-transformed), and the width of the edge between two nodes is proportional to the detection rate of one node (plasmid type or bacterial species) from another (bacterial species and/or continent). Blue nodes indicate bacterial species, green of different saturations the plasmid types and the nodes of other colors the continents. In (B), the size of the nodes (ARG or geographical region) indicates the frequency of their occurrence, and the width of the edges is proportional to the co-occurrence rate between two nodes (between two ARGs or between an ARG and its location). Blue nodes indicate additional ARGs, red the mcr-1 gene and the nodes of other colors the geographical regions. Ten most frequently occurred ARGs are shown as blue circles.
The IncHI2-type plasmid is the most diverse plasmid and harbors a large MDR region (Li et al., 2017). Therefore, it is considered as a genetic element mediating the transmission of MDR genes. A wide range of resistance genes and MGEs can be found in a Mosaic MDR region of IncHI2-type plasmids (Li et al., 2017). We recorded the additional ARGs on each IncHI2 plasmid (Supplementary Table S1). Co-occurrence network analysis, showing the correlation between these ARGs and their geographic distribution, was performed in Matlab_R2016a. The composition of ARGs in this MDR region varies according to their geographical distribution (Figure 2B). Such an MDR region is also discovered in our sequenced IncHI2-type plasmid, pMCR_W5-6 (Figure 1B). Unlike other ARGs concentrated in such an MDR region, mcr-1 gene is located in another location on the plasmid away from the MDR region (Supplementary Table S1 also lists the interval between mcr-1 and other ARGs). It has a unique transposon structure, ISApl1-mcr-1-orf-ISApl1 (Snesrud et al., 2016; Li et al., 2017). The insertion of mcr-1-bearing transposon in such type of plasmid should be a late independent event. The exact time of the insertion event is hard to be determined, but the backbone structure of the IncHI2-type plasmid at the time of mcr-1 insertion can be speculated. The ARGs with high occurrence frequency should be the original backbone structure of this type of plasmids (Figure 2B). Besides mcr-1, two other ARGs, floR and aadA2, are common all over the world on this type of plasmids. It has been reported that the resistance (floR) to florfenicol – a veterinary drug – is commonly associated with mcr-1 (Matamoros et al., 2017; Shen et al., 2018), which supports the animal origin of the mobilized mcr-1 gene (Matamoros et al., 2017; Wang et al., 2018). The mcr-1-bearing IncHI2 plasmids recovered from Asia (mainly from China) possess the most diverse ARGs (Figure 2C), which must be due to the extensive use of antibiotics in this area (Zhang et al., 2015). The three unique ARGs on European IncHI2 plasmids are aadA12, blaTEM1A and catA1. This may be attributed to regional evolution events in Europe.
The IncX4-type plasmids are the most conserved and the smallest (mostly around 33 kb) ones among all types of mcr-1-bearing plasmids (Li et al., 2017). The sequences of IncX4-type plasmids were adjusted from the same start point and aligned with the multiple sequence alignment program MAFFT4 (Katoh et al., 2017). The aligned sequences were used to construct a phylogenetic tree in MEGA7 (Kumar et al., 2016). The IncI2-type plasmids contain a site-specific recombination system, the shufflon. The shufflon generates variants of the PilV protein, a minor component of the thin pilus. The shufflon is one of the most difficult regions for de novo genome assembly, because of its structural diversity even in a single bacterial colony (Sekizuka et al., 2017). Therefore, the shufflon structure affects the alignment of the plasmid sequences, and thus affects the phylogenetic reconstruction. For this type of plasmid (n = 92), we excised the shufflon region from each plasmid sequence and used the remaining part for phylogenetic analysis as we did for IncX4-type plasmids. From the phylogenetic trees of the two types of plasmid (Supplementary Figures S8, S9A), we detected the genotypic clusters of a single geographical origin. Typically, the plasmid clusters of South America are observed in both phylogenetic trees, which indicates relatively seldom exchange of mcr-1-bearing plasmids between South America and other geographical regions. Geographical barriers may result in such a regional evolution. Meanwhile, Asia shows the closest communication with other geographical regions, and the interconnection between Europe and North America is relatively high. In addition, according to the phylogenetic trees of the two types of plasmids, the intercontinental exchange of IncI2 plasmids seems more frequent than that of IncX4 plasmids. The diversity of bacterial hosts of IncI2 plasmids (Figure 2A) may have facilitated their transportation among different continents, which can also explain why IncI2 plasmids occupy the largest share of the mcr-1-bearing plasmids (41% of 229 mcr-1-bearing plasmids).
The shufflons extracted from IncI2 plasmids were carefully annotated the structure. Typical shufflon structures are illustrated in Figure 3. There is a pair of inverted sfx repeats (IRL: GKGCCAATCCGGTNBSTGG and IRR: CCASVNACCGGATTGGCMC) at both ends of each shufflon segment, and also a highly conserved inverted or direct repeat sequence (GGAGGCCA) between adjacent shufflon segments. The shufflon structure also exhibits regional disparity (Supplementary Table S2), which is in good agreement with the clustering mode of IncI2 plasmids in the phylogenetic tree (Supplementary Figure S9B). IncI2-type plasmids isolated from Asia contain almost all kinds of shufflon structures. The shufflon rearrangement is closely related to plasmid transmission to a broad range of the Enterobacteriaceae (Ishiwa and Komano, 2003, 2004). This can explain why the most diverse Enterobacteriaceae hosts bearing mcr-1-positive IncI2 plasmids are detected in Asia (Figure 2A). The IncI2 plasmids isolated from Europe and North America commonly contain the shufflon segment E (except those plasmids with only one shufflon segment), while, to date (June 2018), no IncI2 plasmid from South America has been detected with definite structure of shufflon segment E (Supplementary Table S2). The mcr-1-bearing IncI2 plasmids in Europe and North America may mainly be derived from a plasmid with shufflon segment E, while those in South America may have been originated from one without the segment. Geographical barriers retain this original mark.
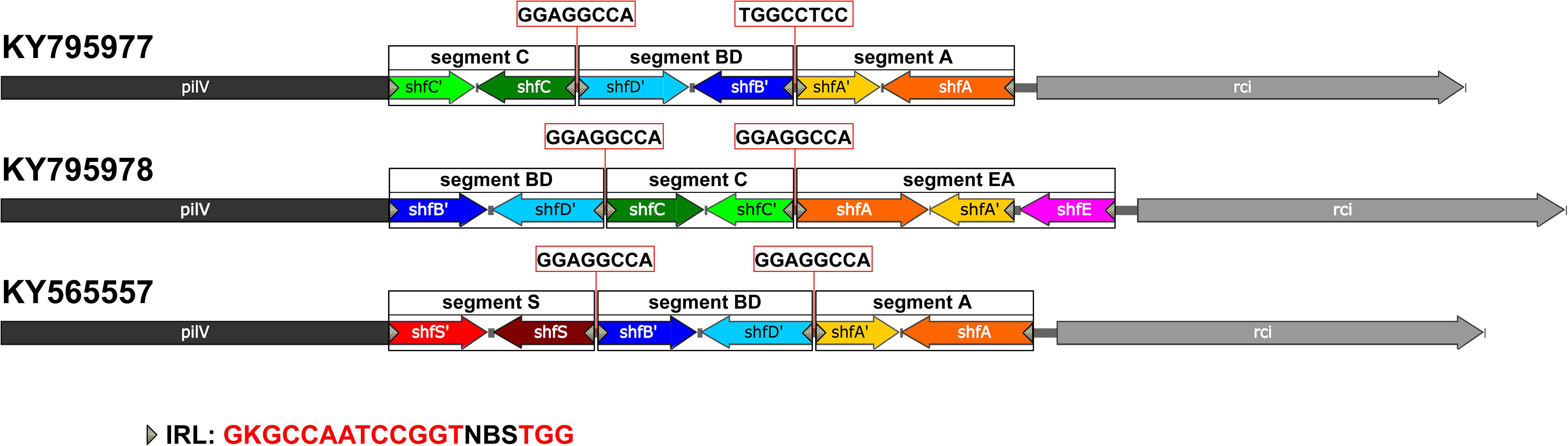
Figure 3. Typical shufflon structures in three representative IncI2 plasmids. Accession numbers is listed beside. The common inverted repeat sequence in shufflon segments is shown at the bottom as IRL. The highly conserved repeat (inverted or direct repeat) sequences between adjacent shufflon segments are shown in red frames.
Some shufflon sequences cannot be annotated as accurate segmental structure, which could be due to sequencing/assembly mistakes (Supplementary Table S2). We also found an unnoticed shufflon segment structure that had never been annotated in the database. It is common and highly conserved in several IncI2 type plasmids (Supplementary Table S2, KY565557, CM008278, CP028153, CP006264, CP007134, FR851304, and CP030766), which cannot be accidental events. Thus we assigned the segment as segment S (Figure 3, KY565557).
The mechanism of Nested Russian Doll-like genetic mobility must be common for the worldwide dissemination of various mobilized ARGs (Dortet et al., 2014; Sheppard et al., 2016; Wang et al., 2018). The blaNDM and blaKPC genes are mainly located on conjugative plasmids of several different incompatibility groups (Dortet et al., 2014; Sheppard et al., 2016), which is similar to the behavior of mcr-1 gene. ARGs may be flanked by different MGE sequences at their mobilization, and exhibit different characteristics in their relocation process. The transfer of mcr-1 is mediated by the insertion sequence ISApl1. The high activity of this MGE enables mcr-1 gene to jump flexibly between different plasmids and between different bacterial species (Supplementary Table S1). At bacterial genome level, no genotypic clustering by geographical origin and isolation source has been observed (Matamoros et al., 2017). While at the transposon level, scientists have predicted the time of the initial mobilization of mcr-1 (Wang et al., 2018). In this work, at the plasmid level (one important genetic level involved in HGT) and according to the analysis routine illustrated in Supplementary Figure S1, we have found that the global spread of mcr-1-bearing plasmids is accompanied by their multiregional evolution. Based on the intrinsic mechanism of HGT, we believe that the analysis of mobilized ARGs at multiple levels of MGEs (transposons, integrons, and plasmids) can give important epidemiological information about their dissemination.
Since we have found sequencing evidence for the horizontal transfer of ARGs between the environmental and avian bacteria, it is possible that migratory birds mediate the intercontinental transportation of the resistance plasmid. Two typical cases were noted at the analysis of IncI2 plasmids. First, the IncI2 plasmid, pJIE3685-1 (KY795978), isolated from Australia is of almost the same size (∼60960 bp) with two plasmids isolated from China (Taiwan), p5CRE51-MCR-1 (CP021176 from a human E. coli), and pP111 (KY120365, from a porcine Salmonella enterica subsp.). The position and the segment composition of the shufflon structure is also the same in the three plasmids (Supplementary Table S2), which indicates that the shufflon of the three plasmids is equivalent. Alignment of the remaining parts of the three plasmids after shufflon excision shows high identity among them with mismatches of only several (∼7 nts out of 59,042 nts) nucleotides (Figure 4A). In addition, another IncI2 plasmid from Australia, pJIE2288-1 (KY795977), is also highly identical (∼8 nts mismatches out of 59,081 nts) to the three plasmids from Asia (Figure 4B), pRYU2912C-1 (AP018412, Japan, from a E. coli), pSh113-m4 (KY363994, Shanghai, China, from a human Shigella sonnei) and pSh069-m6 (KY363995, Shanghai, China, from a human Shigella sonnei). Such close genetic relationships between these plasmids suggest that they do not seem to originate from a common evolutionary ancestor, but rather are duplicate offspring of the same plasmid. Two Australian plasmids are from E. coli strains that were isolated from two clinical patients at New South Wales. Neither patients had ever gone abroad or had taken colistin/polymyxin antimicrobial drugs during hospitalization (Ellem et al., 2017). We noticed that these highly homologous IncI2 plasmids were all found on the same flyway of migratory shorebirds, i.e., the East Asian-Australasian Flyway (Supplementary Figure S7). Many species of migratory shorebirds take extreme long-distance migration between eastern Russia and Oceania. During the migration, they stage once in Eastern Asia (typically the Yellow Sea region) for around 40 days, resting and refueling for the subsequent flight (Battley et al., 2012). There is continental-scale pollution of ARGs at the estuaries along the east coast of China, where the migratory shorebirds pass by Zhu et al. (2017). Migratory shorebirds can be an option for tracking the source of the resistance plasmids.
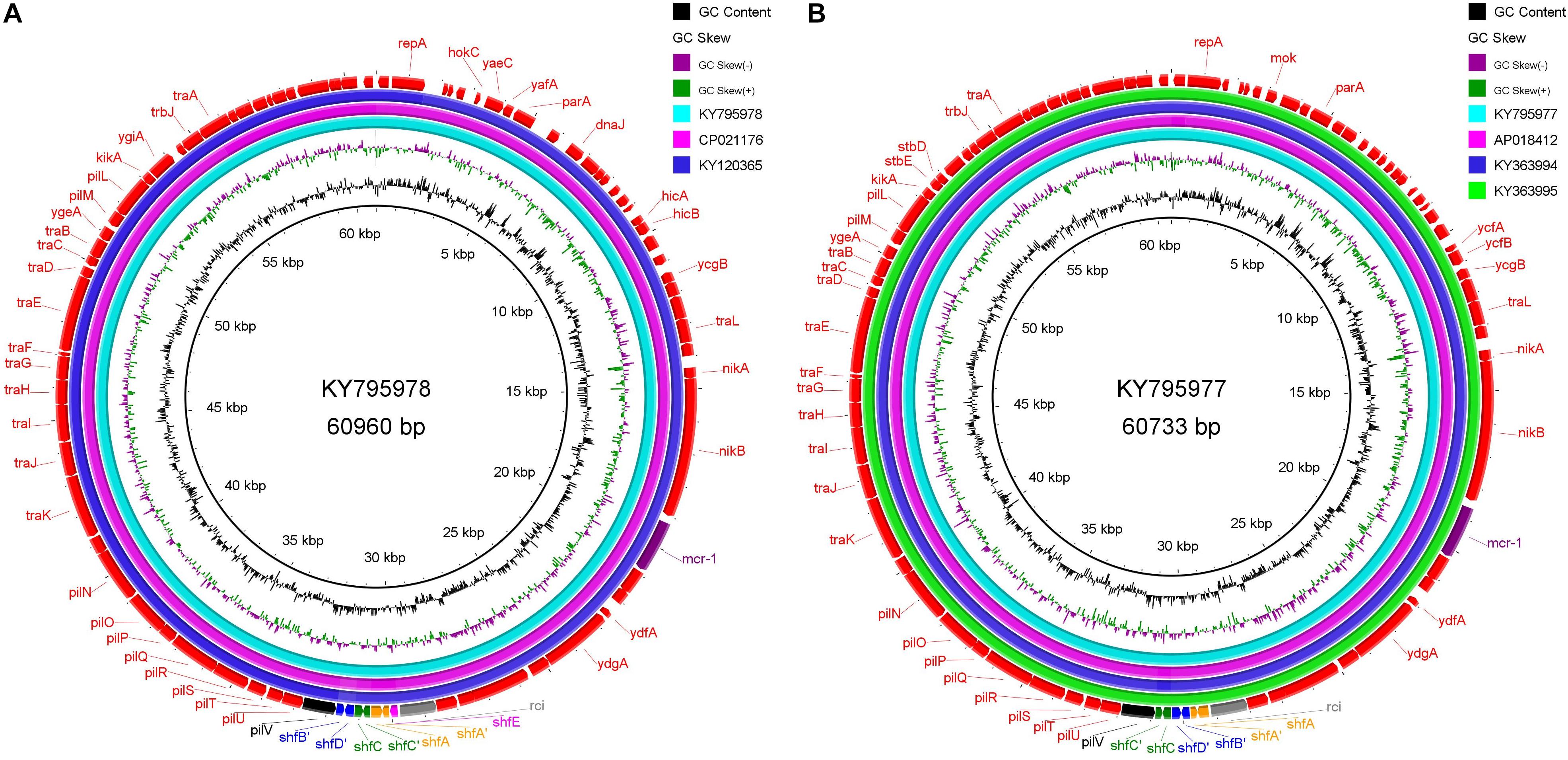
Figure 4. Sequence alignment of IncI2 plasmids with high homology, which were isolated from different regions along the East Asian-Australasian Flyway. (A) KY795978 is used as reference. (B) KY795977 as reference. The alignment was performed using BRIG tool (Alikhan et al., 2011). The outmost arrow ring signifies annotation of the reference sequence. The shufflon structure and the mcr-1 gene are highlighted with outstanding colors.
During the outbreak of avian influenza, migratory birds as potential global spreaders had attracted the attention of scientists (Liu et al., 2005; Normile, 2006; Olsen et al., 2006). However, according to a number of previously reported studies, the risk of migratory birds mediating intercontinental exchange of influenza virus, especially those highly pathogenic ones, is fairly low (Krauss et al., 2007; Langstaff et al., 2009). A sick bird can hardly accomplish the arduous task of long-distance (intercontinental) migration. Nevertheless, migratory shorebirds carrying drug-resistant symbiotic bacteria can cross the Wallace Line and bring ARGs from Asia to Australia (Supplementary Figure S7). Since the discovery of the mobilized colistin resistance gene, several studies have reported the detection of mcr-1-positive E. coli in migratory birds (Liakopoulos et al., 2016; Mohsin et al., 2016; Ruzauskas and Vaskeviciute, 2016; Sellera et al., 2017). Salt tolerant mcr-1-positive E. coli strains have also been isolated from recreational waters of public urban beaches (Fernandes et al., 2017). Therefore, mcr-1-bearing plasmids may be transported, via avian migration, from Eastern Asia to Australia, where the usage of antibiotics is under strict control but is definitely affected by migratory shorebirds. In fact, mcr-1-positive E. coli has recently been isolated from wild bird (silver gull) in Australia (Mukerji et al., 2019). In view of limited data available for bacterial ARGs of migratory birds, it is difficult to discuss the global dissemination of ARGs in the context of avian ecology. Our analysis does not imply that avian migration is the main route for the global dissemination of antibiotic resistance. In today’s highly globalized world, global population mobility and international trade, especially the trade of food animal, must be the main channels (Matamoros et al., 2017; Shen et al., 2018; Hernando-Amado et al., 2019). Even so, the large number of migratory birds and their fixed migration path will have a lasting impact on the receptor environment. In particular, different countries execute different strategies for using antibiotics. Antibiotics used for human in one country may be used as animal feed additives in another. If migratory birds transport antibiotic resistance from the latter environment to the former, the cause originated in one country may have a devastating consequence in another. Nowadays, with the global dissemination of antibiotic resistance, no country can be immune from it. The application of antibiotics should follow the strategy of global unification.
Publicly available datasets were analyzed in this study. This data can be found in GenBank database under the accession numbers CP032986 to CP032995.
KY and JW designed the study. DR isolated the E. coli strains and evaluated their drug resistance. JW and YL prepared the DNA samples for sequencing and analyzed the sequencing data. XD and LZ performed the metadata analysis of the mcr-1-bearing plasmids. YL, JW, and KY constructed the manuscript. YF edited the manuscript and put forward constructive suggestions on it. All authors reviewed, revised, and approved the final report.
We gratefully acknowledge the financial support from the Initiating Research Fund for Talent Introduction of Sichuan University (YJ201355) and the Natural Science Foundation of China (21677104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00352/full#supplementary-material
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/21.13.3093
Battley, P. F., Warnock, N., Tibbitts, T. L., Gill, R. E., Piersma, T., Hassell, C. J., et al. (2012). Contrasting extreme long-distance migration patterns in bar-tailed godwits Limosa lapponica. J. Avian Biol. 43, 21–32. doi: 10.1111/j.1600-048X.2011.05473.x
Berlin, K., Koren, S., Chin, C. S., Drake, J. P., Landolin, J. M., and Phillippy, A. M. (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630. doi: 10.1038/nbt.3238
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1371/journal.pone.0017473
Carattoli, A., Zankari, E., Garcia-Fernandez, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Clinical and Laboratory Standards Institute [CLSI] (2019). Performance Standards for Antimicrobial Susceptibility Testing—Twenty-Ninth Edition: M100. Wayne, PA: CLSI.
Cuzon, G., Naas, T., and Nordmann, P. (2011). Functional characterization of Tn4401, a Tn3-based transposon involved in blaKPC gene mobilization. Antimicrob. Agents Chemother. 55, 5370–5373. doi: 10.1128/AAC.05202-11
Dortet, L., Poirel, L., and Nordmann, P. (2014). Worldwide dissemination of the NDM-type carbapenemases in Gram-negative bacteria. Biomed. Res Int. 2014:249856. doi: 10.1155/2014/249856
Ellem, J. A., Ginn, A. N., Chen, S. C., Ferguson, J., Partridge, S. R., and Iredell, J. R. (2017). Locally acquired mcr-1 in Escherichia coli, Australia, 2011 and 2013. Emerg. Infect. Dis. 23, 1160–1163. doi: 10.3201/eid2307.161638
Fernandes, M. R., Sellera, F. P., Esposito, F., Sabino, C. P., Cerdeira, L., and Lincopan, N. (2017). Colistin-resistant mcr-1-positive Escherichia coli on public beaches, an infectious threat emerging in recreational waters. Antimicrob Agents Chemother. 61:e00234-17. doi: 10.1128/AAC.00234-17
Gay, K., Robicsek, A., Strahilevitz, J., Park, C. H., Jacoby, G., Barrett, T. J., et al. (2006). Plasmid-mediated quinolone resistance in non-Typhi serotypes of Salmonella enterica. Clin. Infect. Dis. 43, 297–304. doi: 10.1086/505397
Hernando-Amado, S., Coque, T. M., Baquero, F., and Martinez, J. L. (2019). Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 4, 1432–1442. doi: 10.1038/s41564-019-0503-9
Ishiwa, A., and Komano, T. (2003). Thin pilus PilV adhesins of plasmid R64 recognize specific structures of the lipopolysaccharide molecules of recipient cells. J. Bacteriol. 185, 5192–5199. doi: 10.1128/jb.185.17.5192-5199.2003
Ishiwa, A., and Komano, T. (2004). PilV adhesins of plasmid R64 thin pili specifically bind to the lipopolysaccharides of recipient cells. J. Mol. Biol. 343, 615–625. doi: 10.1016/j.jmb.2004.08.059
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., et al. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357. doi: 10.1093/nar/gkj102
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kim, H. B., Wang, M., Park, C. H., Kim, E. C., Jacoby, G. A., and Hooper, D. C. (2009). oqxAB encoding a multidrug efflux pump in human clinical isolates of Enterobacteriaceae. Antimicrob. Agents Chemother. 53, 3582–3584. doi: 10.1128/AAC.01574-08
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Krauss, S., Obert, C. A., Franks, J., Walker, D., Jones, K., Seiler, P., et al. (2007). Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Pathog. 3:e167. doi: 10.1371/journal.ppat.0030167
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langstaff, I. G., McKenzie, J. S., Stanislawek, W. L., Reed, C. E., Poland, R., and Cork, S. C. (2009). Surveillance for highly pathogenic avian influenza in migratory shorebirds at the terminus of the East Asian-Australasian flyway. N. Z. Vet. J. 57, 160–165. doi: 10.1080/00480169.2009.36896
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Li, R., Xie, M., Zhang, J., Yang, Z., Liu, L., Liu, X., et al. (2017). Genetic characterization of mcr-1-bearing plasmids to depict molecular mechanisms underlying dissemination of the colistin resistance determinant. J. Antimicrob. Chemother. 72, 393–401. doi: 10.1093/jac/dkw411
Li, W. Z., Jaroszewski, L., and Godzik, A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. doi: 10.1093/bioinformatics/18.1.77
Liakopoulos, A., Mevius, D. J., Olsen, B., and Bonnedahl, J. (2016). The colistin resistance mcr-1 gene is going wild. J. Antimicrob. Chemother. 71, 2335–2336. doi: 10.1093/jac/dkw262
Liebert, C. A., Hall, R. M., and Summers, A. O. (1999). Transposon Tn21, flagship of the floating genome. Microbiol. Mol. Biol. Rev. 63, 507–522. doi: 10.1128/mmbr.63.3.507-522.1999
Liu, B., and Pop, M. (2009). ARDB–antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, J., Xiao, H., Lei, F., Zhu, Q., Qin, K., Zhang, X. W., et al. (2005). Highly pathogenic H5N1 influenza virus infection in migratory birds. Science 309:1206. doi: 10.1126/science.1115273
Liu, Y. Y., Wang, Y., Walsh, T. R., Yi, L. X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
Magrane, M., and UniProt, C. (2011). UniProt knowledgebase: a hub of integrated protein data. Database 2011:bar009. doi: 10.1093/database/bar009
Martinez, J. L., Sanchez, M. B., Martinez-Solano, L., Hernandez, A., Garmendia, L., Fajardo, A., et al. (2009). Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 33, 430–449. doi: 10.1111/j.1574-6976.2008.00157.x
Matamoros, S., van Hattem, J. M., Arcilla, M. S., Willemse, N., Melles, D. C., Penders, J., et al. (2017). Global phylogenetic analysis of Escherichia coli and plasmids carrying the mcr-1 gene indicates bacterial diversity but plasmid restriction. Sci. Rep. 7:15364. doi: 10.1038/s41598-017-15539-7
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mohsin, M., Raza, S., Roschanski, N., Schaufler, K., and Guenther, S. (2016). First description of plasmid-mediated colistin-resistant extended-spectrum beta-lactamase-producing Escherichia coli in a wild migratory bird from Asia. Int. J. Antimicrob. Agents 48, 463–464. doi: 10.1016/j.ijantimicag.2016.07.001
Mukerji, S., Stegger, M., Truswell, A. V., Laird, T., Jordan, D., Abraham, R. J., et al. (2019). Resistance to critically important antimicrobials in Australian silver gulls (Chroicocephalus novaehollandiae) and evidence of anthropogenic origins. J. Antimicrob. Chemother. 74, 2566–2574. doi: 10.1093/jac/dkz242
Munoz-Price, L. S., Poirel, L., Bonomo, R. A., Schwaber, M. J., Daikos, G. L., Cormican, M., et al. (2013). Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 13, 785–796. doi: 10.1016/S1473-3099(13)70190-7
Naas, T., Cuzon, G., Villegas, M. V., Lartigue, M. F., Quinn, J. P., and Nordmann, P. (2008). Genetic structures at the origin of acquisition of the beta-lactamase bla(KPC) gene. Antimicrob. Agents Chemother. 52, 1257–1263.
Normile, D. (2006). Avian influenza. Evidence points to migratory birds in H5N1 spread. Science 311:1225. doi: 10.1126/science.311.5765.1225
Olsen, B., Munster, V. J., Wallensten, A., Waldenstrom, J., Osterhaus, A. D., and Fouchier, R. A. (2006). Global patterns of influenza a virus in wild birds. Science 312, 384–388. doi: 10.1126/science.1122438
Oram, M., and Fisher, L. M. (1991). 4-Quinolone resistance mutations in the DNA gyrase of Escherichia coli clinical isolates identified by using the polymerase chain reaction. Antimicrob. Agents Chemother. 35, 387–389. doi: 10.1128/aac.35.2.387
Poirel, L., Bonnin, R. A., Boulanger, A., Schrenzel, J., Kaase, M., and Nordmann, P. (2012). Tn125-related acquisition of blaNDM-like genes in Acinetobacter baumannii. Antimicrob. Agents Chemother. 56, 1087–1089. doi: 10.1128/AAC.05620-11
Ruzauskas, M., and Vaskeviciute, L. (2016). Detection of the mcr-1 gene in Escherichia coli prevalent in the migratory bird species Larus argentatus. J. Antimicrob. Chemother. 71, 2333–2334. doi: 10.1093/jac/dkw245
Saier, M. H. Jr., Reddy, V. S., Tamang, D. G., and Vastermark, A. (2014). The transporter classification database. Nucleic Acids Res. 42, D251–D258. doi: 10.1093/nar/gkt1097
Sekizuka, T., Kawanishi, M., Ohnishi, M., Shima, A., Kato, K., Yamashita, A., et al. (2017). Elucidation of quantitative structural diversity of remarkable rearrangement regions, shufflons, in IncI2 plasmids. Sci. Rep. 7:928. doi: 10.1038/s41598-017-01082-y
Sellera, F. P., Fernandes, M. R., Sartori, L., Carvalho, M. P., Esposito, F., Nascimento, C. L., et al. (2017). Escherichia coli carrying IncX4 plasmid-mediated mcr-1 and blaCTX-M genes in infected migratory Magellanic penguins (Spheniscus magellanicus). J. Antimicrob. Chemother. 72, 1255–1256. doi: 10.1093/jac/dkw543
Shen, Y., Zhou, H., Xu, J., Wang, Y., Zhang, Q., Walsh, T. R., et al. (2018). Anthropogenic and environmental factors associated with high incidence of mcr-1 carriage in humans across China. Nat. Microbiol. 3, 1054–1062. doi: 10.1038/s41564-018-0205-8
Sheppard, A. E., Stoesser, N., Wilson, D. J., Sebra, R., Kasarskis, A., Anson, L. W., et al. (2016). Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob. Agents Chemother. 60, 3767–3778. doi: 10.1128/AAC.00464-16
Snesrud, E., He, S., Chandler, M., Dekker, J. P., Hickman, A. B., McGann, P., et al. (2016). A model for transposition of the colistin resistance gene mcr-1 by ISApl1. Antimicrob. Agents Chemother. 60, 6973–6976. doi: 10.1128/AAC.01457-16
Stokes, H. W., and Gillings, M. R. (2011). Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 35, 790–819. doi: 10.1111/j.1574-6976.2011.00273.x
Takahata, S., Ida, T., Hiraishi, T., Sakakibara, S., Maebashi, K., Terada, S., et al. (2010). Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 35, 333–337. doi: 10.1016/j.ijantimicag.2009.11.011
Tankovic, J., Perichon, B., Duval, J., and Courvalin, P. (1996). Contribution of mutations in gyrA and parC genes to fluoroquinolone resistance of mutants of Streptococcus pneumoniae obtained in vivo and in vitro. Antimicrob. Agents Chemother. 40, 2505–2510. doi: 10.1128/aac.40.11.2505
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Wang, R., van Dorp, L., Shaw, L. P., Bradley, P., Wang, Q., Wang, X., et al. (2018). The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun. 9:1179. doi: 10.1038/s41467-018-03205-z
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
Wu, J., Huang, Y., Rao, D., Zhang, Y., and Yang, K. (2018). Evidence for environmental dissemination of antibiotic resistance mediated by wild birds. Front. Microbiol. 9:745. doi: 10.3389/fmicb.2018.00745
Yong, D., Toleman, M. A., Giske, C. G., Cho, H. S., Sundman, K., Lee, K., et al. (2009). Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 53, 5046–5054. doi: 10.1128/AAC.00774-09
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, Q. Q., Ying, G. G., Pan, C. G., Liu, Y. S., and Zhao, J. L. (2015). Comprehensive evaluation of antibiotics emission and fate in the river basins of China: source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 49, 6772–6782. doi: 10.1021/acs.est.5b00729
Keywords: antibiotic resistance, intercontinental dissemination, migratory birds, antibiotic resistance gene, horizontal gene transfer
Citation: Lin Y, Dong X, Wu J, Rao D, Zhang L, Faraj Y and Yang K (2020) Metadata Analysis of mcr-1-Bearing Plasmids Inspired by the Sequencing Evidence for Horizontal Transfer of Antibiotic Resistance Genes Between Polluted River and Wild Birds. Front. Microbiol. 11:352. doi: 10.3389/fmicb.2020.00352
Received: 15 September 2019; Accepted: 18 February 2020;
Published: 10 March 2020.
Edited by:
Raffaele Zarrilli, University of Naples Federico II, ItalyReviewed by:
Ruichao Li, Yangzhou University, ChinaCopyright © 2020 Lin, Dong, Wu, Rao, Zhang, Faraj and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Yang, Y29va3lvdW5nQHNjdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.